

Abstract
Chimeric antigen receptor (CAR) tonic signaling, defined as spontaneous activation and release of proinflamatory cytokines by CAR-T cells, is considered a negative attribute because it leads to impaired antitumor effects. Here, we report that CAR tonic signaling is caused by the intrinsic instability of the monoclonal antibody single-chain variable fragment (scFv) to promote self-aggregation and signaling via the CD3ζ chain incorporated into the CAR construct. This phenomenon was detected in a CAR encoding either CD28 or 4–1BB costimulatory endodomains. Instability of the scFv was caused by specific amino acids within the framework regions (FWRs) that can be identified by computational modeling. Substitutions of the amino acids causing instability, or humanization of the FWRs, corrected tonic signaling of the CAR, without modifying antigen specificity, and enhanced the antitumor effects of CAR-T cells. Overall, we demonstrated that tonic signaling of CAR-T cells is determined by the molecular instability of the scFv and that computational analyses of the scFv can be implemented to correct the scFv instability in CAR-T cells with either CD28 or 4–1BB costimulation.
Introduction
Chimeric antigen receptors (CARs), in their original conception, are fusion proteins in which the variable regions of the heavy chain (VH) and light chain (VL) of a monoclonal antibody (Ab) are assembled with a non-cleavable flexible linker to form an antibody with a single-chain variable fragment (scFv), which is fused with signaling molecules of the T-cell receptor (TCR) and costimulatory endodomains (1–3). Remarkable and sustained antitumor activity has been achieved using CD19-specific CAR-T cells, and these cells have been infused in pediatric patients with acute B-cell leukemia (4).
CAR engagement with the targeted antigen expressed by tumor cells promotes rapid activation of the T cells, which is characterized by cytolysis, cytokine secretion, and proliferation (5). However, it has emerged that CARs can cause tonic signaling in T cells, which denotes sustained antigen-independent activation leading to rapid T-cell exhaustion and impaired antitumor activity (6). The first report describing tonic signaling in CAR-T cells attributed this effect to the propensity of certain scFvs to self-aggregate, causing cell surface CAR clustering and consequent signaling (6).
It is well known that VH and VL domains coupled to form scFvs have the tendency to unfold, leading to scFv oligomerization (7). A variety of strategies have been developed in the effort to stabilize scFvs which include engineering disulfide bonds between the VH and VL domains, introduction of charged amino acids within the VH and VL domains, or grafting the complementarity-determining regions (CDRs) into different framework regions (FWRs)(8–10). In the contest of scFvs assembled into a CAR format, the tonic signaling caused by the scFv derived from the murine 14g2a monoclonal antibody (Ab) is specifically attributed to the Ab’s FWRs(6). Remarkably, the engraftment of the CDRs of the scFv derived from the FMC63 monoclonal Ab into the FWRs of the 14g2a Ab is sufficient to cause tonic signaling of the FMC63 scFv when used to generate a CAR (6), indicating that the FWRs play a critical role in causing tonic signaling of scFvs in the CAR format.
Here, we showed that amino acid substitutions into the FWRs can stabilize the scFv and correct the tonic signaling of the CAR. We additionally showed that substitution of murine FWRs with stable human FWRs also prevents antigen-independent activation of CAR-T cells. Thus, CAR tonic signaling is caused by unstable scFvs, and we provide structure-based strategies that can be used to correct tonic signaling.
Materials and Methods
Cell lines
The tumor cell line WM115 and SK-MEL-2 were obtained from ATCC between 2016 and 2018, whereas M14 cells were provided by Dr. Ferrone (Massachusetts General Hospital). The tumor cell lines MDA-MB-468 and MDA-MB-231 were obtained from German Collection of Microorganism and Cell Cultures GmbH (DSMZ, ACC 738 and ACC 732). The 293T cells used for the production of retroviral vectors were obtained from ATCC in early 2000. All cells were maintained in culture with the appropriate media, either RPMI-1640 (Gibco), DMEM (Gibco), or MEM (Gibco) supplemented with 10% FBS (Sigma), 1% L-glutamine (Gibco), and 1% penicillin/streptomycin (Gibco) in a humidified atmosphere containing 5% CO2 at 37°C. WM115 cells were modified to express the fusion protein firefly luciferase and enhanced GFP (eGFP-FFluc)(11). Cells were kept in culture for less than six consecutive months, after which, aliquots from the original expanded vial (cultured for 4 to 6 passages after being received) were used. All tumor cell lines were routinely tested to exclude contamination with Mycoplasma and assessed for the expression of transgenes and tumor markers by flow cytometry to confirm identity. Glioblastoma-derived neurospheres (GBM-NS) were generated as previously described (12). The variable regions of the immunoglobulin genes expressed by the 763.74 hybridoma were amplified with a set of proprietary primers from cDNA generated from the hybridoma cells using a RT-PCR protocol and sequenced using a standard dye-terminator capillary sequencing method (SynBuild, Tempe, Az).
Humanization of the scFv 763.74(A)
Humanization of the murine scFv 763.74(A) was performed by grafting its CDRs into the stable human framework rFW1.4 (13). To design the first humanized variant, six CDRs of the murine 763.74(A) were combined with the unmodified framework regions of the rFW1.4. The sequence of the framework, referred here as rFW1.4, is as follows: EIVMTQSPSTLSASVGDRVIITC *CDRL1* WYQQKPGKAPKLLIY *CDRL2* GVPSRFSGSGSGTEFTLTISSLQPDDFATYYC *CDRL3* FGQGTKLTVLG (GGGGSGGGGSGGGGSGGGGS) EVQLVESGGGLVQPGGSLRLSCTASG *CDRH1* WVRQAPGKGLEWVG *CDRH2* RFTISRDTSKNTVYLQMNSLRAEDTAVYYCAR *CDRH3* WGQGTLVTVSS. Asterisks separate the amino acid sequence of the FWRs from the CDR sequences. A polypeptide linker consisting of (Gly4Ser)4 was used to join the VL and VH chains and shown in round brackets. The scFv 763.74(A) sequence and the human framework rFW1.4 were aligned, and 24 critical murine amino acids were identified as described (37). Twenty six humanized variants were designed by introducing the identified murine amino acid residues back into the human framework regions. Some variants were generated by shuffling humanized heavy and light chains. The numbers of “back to mouse” mutations for all humanized heavy and light chain variants are shown in Supplementary Table S1. The positions of murine amino acids are shown for the four best selected humanized scFvs (h763.74 #2, h763.74 #3, h763.74 #4, and h763.74 #5 [Supplementary Fig. S6B]. DNA sequences of humanized scFvs were optimized for E.coli codon usage and synthesized by DNA2.0 (Newark, USA) and cloned into the expression vector with the IPTG-inducible T7 promoter by DNA2.0. The expression vector had a His(6) fusion tag on the carboxyl terminus. A His(6) fusion tag allowed detection of humanized scFvs in binding studies. Each DNA synthesis at DNA2.0 included a stringent quality control process with 100% accuracy. Additionally, the genes encoding for scFvs were confirmed by sequencing at Microsynth AG (Switzerland).
Expression of scFv fragments
E. coli BL21-DE3 (100 μL, Invitrogen, C600003) transformed with the corresponding expression plasmids (5 ng) containing His-tag (scFv h763.74 #2, h763.74 #3, h763.74 #4 or h763.74 #5) were grown at 37°C in LB medium (VWR Chemicals, Switzerland) containing kanamycin (30 μg/mL; AppliChem GmBH, Germany). Protein expression was initiated by addition of 1 mM isopropyl 1-thio-β-d-galactopyranoside (AppliChem) at an absorbance (A600) of 1. Four hours after induction, E. coli cells were harvested and disrupted by sonication. Inclusion bodies were isolated by repeated washing with 10 mM Tris-HCl (Applichem), 150 mM NaCl (AppliChem), pH 7.3 containing 0.5% N,N-dimethyldodecylamine N-oxide (Sigma-Aldrich) and centrifugation and solubilized at a concentration of 10 mg/mL in the presence of 6 M guanidine HCl (AppliChem) 100 mM Tris-HCl, 1 mM EDTA (Sigma-Aldrich). Solubilized inclusion bodies were reduced by adding 20 mM dithiothreitol (Sigma-Aldrich). Refolding was performed in refolding buffer (4 M urea, 50 mM glycine, 2 mM cysteine, 2 mM cysteine pH 10.0, all chemicals were purchased from AppliChem) overnight at room temperature. After up-concentration and buffer exchange using tangential flow filtration with a 10 kDa cut-off (Centramate T-Series Cassette OSO010T12), scFvs were purified using hydrophobic interaction chromatography followed by size-exclusion chromatography, as described below. ScFv 763.74(B) and h763.74 #5 were expressed in EXPI293 transient expression system (Thermo Fisher) following the manufacturer’s protocol. The Expi293 cells were transfected and incubated for 4 days in Expi293 medium (ThermoFisher) at 37°C, 125 rpm, 8% CO2 atmosphere (5% was also acceptable) with 80% humidity. Cells were harvested by centrifugation at 2000 × g for 15 minutes. Supernatant was collected and then filtered through a 0.22 μm filter, followed by purification on a Ni-Penta™ affinity column (Marvelgent Biosciences).
Purification of scFVs fragments
Refolded scFvs were purified by hydrophobic interaction chromatography (HIC) on an AEKTA Purifier system (GE Healthcare), which was operated with Unicorn software, with a Phenyl Sepharose 6 FF (low sub) XK26/60 column (GE Healthcare). The column was equilibrated with the column volumes of 20 mM Na-Phosphate, 50 mM NaCl (AppliChem), 1M (NH4)2SO4 (Merck), pH 7.0 at 5 mL/min flow rate. Humanized scFv solution was mixed with (NH4)2SO4 to a final concentration of 1.0 M. This solution was loaded on a Phenyl Sepharose 6 FF column at 0.2 mL/min flow rate. After washing with buffer 20 mM Na-Phosphate, 50 mM NaCl, 1M (NH4)2SO4 pH 7.0, the bound scFv was eluted with 100% buffer 20 mM sodium phosphate, 50 mM NaCl pH 7–0 at 5 mL/min flow rate. Fractions containing scFvs were pooled and concentrated with a Vivaspin-20 concentrator (Sartorius) using the Multifuge X3R (Thermo Fisher) at 3800xg.
The polishing step was conducted by size exclusion chromatography on an AEKTA Pure system (GE Healthcare) with a HiLoad 26/60 Superdex 75 prep grade column (GE Healthcare). The column was equilibrated with three column volumes of PBS pH 7.4 (Gibco) at a flow rate of 2.6 mL/min. The scFv sample of 10 mL was fractionated with a HiLoad 26/60 Superdex 75 column at 2.6 mL/min flow rate using a mobile phase buffer PBS pH 7.4 to separate dimers from monomers. Fractions containing monomeric scFvs were analyzed by SDS-PAGE electrophoresis using NuPAGE 12% Bis-Tris Gels (Novex/Invitrogen), and absorbance of samples was measured at 280 nm and 320 nm using a BioSpectrometer basic. ScFv 763.74(B) and h763.74 #5, labeled with His tag in C terminus, were purified by Ni-Penta™ affinity column (Marvelgent Biosciences) and eluted with an imidazole gradient. Protein identity and purity was determined by SDS-PAGE gel.
Binding and titration studies of the scFvs
For binding studies, soluble scFvs were used at 5 μg/mL. For the titration studies, soluble scFvs were used at the specific concentrations reported in the figures. ScFvs were detected either by protein L-biotin (5 mg/mL used at 1:3000; PI-29997, Pierce, Thermo Scientific) and streptavidin-PE (used at 1:2000; BD Pharmingen, cat. no. 55406) or by anti-Myc tag FITC-conjugated (Abcam, ab1394). In some experiments, scFvs were detected by anti-His-biotin (Quiagen, 34440, diluted 1:1000) and SAV-PE. Tumor cell lines (MDA MB231, MDA-MB-468, M14, and SK-MEL-2; 0.2 × 106 cells/sample) were incubated in the presence of 763.74 and control scFvs in PBS with 2% FCS and 5 mM EDTA. Cells were acquired on a FACSAria III or BD LSRFortessa (BD Biosciences) instrument using the FACSDiva software. The EC50 was calculated with Graphpad Prism.
Stability of the scFvs
Humanized scFvs were formulated in PBS pH-7.2 (Gibco) at 1 mg/mL. After 48 hours of storage at 4°C or 37°C, samples were inspected visually, and protein concentration was measured at 280 nm and 320 nm using a BioSpectrometer basic (Eppendorf). The following equation was used to calculate protein concentration: (A280-A320) × Dilution factor × Molecular weight/Extinction coefficient equal to scFv concentration in mg/ml.. Samples were analyzed by size-exclusion chromatography (SEC)-HPLC to determine the percentage of monomers, dimers, and high molecular weight oligomers in relation to total peak area. A TOSOH TSKgel G2000 SWXL column, phase diol, L × I.D. 30 cm × 7.8 mm, 5 μm particle size (Sigma-Aldrich, 08540) was used for size-exclusion chromatography. Five μL of scFvs at 1 mg/mL were loaded. The mobile phase was PBS pH 7.2.
SEC-HPLC
The monomer purity of the samples was analysed using SEC-HPLC. The analysis was performed using an Ultimate 3000 HPLC system (Dionex) and Chromeleon software and UV detection at 280 nm absorbance, TOSOH TSKgel G2000 SWXL column (Sigma-Aldrich) and corresponding Guard column. The column was washed with two column volumes of de-gassed MilliQ water and equilibrated with two column volumes of de-gassed PBS pH 7.2 at a flow rate of 0.3 mL/min. 5 μL of scFvs were injected and separated using a mobile phase containing PBS pH 7.2 at a flow of 0.5 mL/min.
Nanoscale differential scanning fluorometry (nanoDSF)
NanoDSF was performed with the scFvs 763.74(B) and h763.74 (20 μM in PBS) using a Nanotemper Prometheus NT.48. Fluorescence emission at 350 nm and 330 nm was collected from 25°C to 95°C at a ramp rate of 1°C/min. Analysis of melting temperature (Tm) was performed using the PR. ThermControl Software (Nanotemper, https://nanotempertech.com/prometheus-software).
Generation of retroviral supernatants, T-cell isolation, transduction, and in vitro expansion
Retroviral supernatants were prepared by transient transfection of 293T cells and used to transduce T cells (14). Buffy coats from healthy volunteer blood donors were purchased from the Gulf Coast Regional Blood Center (Houston, TX). Peripheral blood mononuclear cells (PBMCs) were isolated by Lymphoprep (Accurate Chemical and Scientific Corporation) density-gradient centrifugation. T cells isolated from PBMCs were cultured in complete T cells medium, consisting of 45% Click’s medium (Irvine Scientific), 45% RPMI-1640 (Hyclone), 10% FBS (Hyclone), 1% L-glutamine (Gibco), and 1% penicillin/streptomycin (Gibco). T cells were activated, transduced, and expanded in complete medium with IL7 (10 ng/mL, PeproTech) and IL15 (5 ng/mL, PeproTech) as previously reported (14).
Xenograft models
Mouse experiments using a melanoma model were performed in accordance with UNC Animal Husbandry and Institutional Animal Care and Use Committee (IACUC) guidelines and were approved by UNC IACUC (ID: 17029). Mouse experiments using the glioblastoma model were performed following directives of Fondazione IRCCS Istituto Neurologico Carlo Besta in Milan in accordance with the Italian Principle of Laboratory Animal Care (D. Lgs. 26/2014) and European Communities Council Directives (86/609/EEC and 2010/63/UE). For the melanoma model, female and male NSG mice (7 – 9 weeks of age, obtained from the UNC Animal Core) were injected subcutaneously (s.c.) with 0.5 × 106 eGFP-FFluc-labeled WM115 tumor cells. Seven days after tumor cell injection (day 0) mice were infused intravenously (i.v.) with 5 × 106 CAR-T cells. Melanoma tumor cell growth was monitored weekly with caliper measurement for s.c. tumors and by bioluminescence (BLI; total flux, photons/second) using the IVIS kinetic in vivo imaging system (PerkinElmer). Mice were sacrificed according to UNC guidelines for tumor growth or occurrence of sign of discomfort. At sacrifice, peripheral blood was collected from heart in 1.7 mL tubes (GeneMate) with 20 μL of 0.5M EDTA (Corning). Spleen and liver were harvested and smashed on cell strainers and washed with 2 mL of PBS. Peripheral blood, spleen, and liver were analyzed to detect the presence of human T cells using the following antibodies: CD3 (APC-H7, clone SK7), CD45 (APC, clone 2D1), PD-1 (PE/Cy7, clone EH12.1) from BD Biosciences, and CAR-specific anti-idiotype by flow cytometry, described below in “Immunophenotyping,” using CountBright absolute counting beads (Invitrogen). For the glioblastoma model, antitumor activity of CAR-T cells was evaluated in CD1 nude mice (CD1-Foxn1nu purchased from Charles Rivers) engrafted with GBM-NS. Five to 6-week-old mice were injected intra-caudate nucleus (i.c.) with 0.1 × 106 GBM-NS in 2 μL 1X PBS. The coordinates, with respect to the bregma, were 0.7 mm post, 3 mm left lateral, 3.5 mm deep, and within the nucleus caudatum. On day 15 after tumor cell injection, CAR-T cells were injected i.c. in 5 μL PBS 1X using the same tumor coordinates. GBM xenograft mice underwent magnetic resonance imaging (MRI). For survival studies, mice were monitored three times a week and euthanized when signs of discomfort appeared in accordance with the institutional guidelines.
GBM explants
GBM xenografts were explanted at different timepoints after CAR-T cell injection. Tumor masses were cut into small pieces using a scalpel. The tissue pieces were enzymatically and mechanically digested, using Human Tumor Dissociation kit (Miltenyi Biotec) in combination with the GentleMACS Dissociator (Miltenyi Biotec). First, tumor tissues were transferred in C-Tubes (Miltenyi Biotec) containing the digestion enzyme mix of the kit, and the GentleMACS Dissociator was used for the mechanical dissociation using h_tumor_1 program and h_tumor_2 program. The dissociated tissue was filtered using Pre-Separation Filters (30 μm, Miltenyi Biotec), and a cell suspension was ready for flow cytometry analysis.
Immunophenotyping
T cells were stained with antibodies against CD3 (APC-H7, clone SK7), CD45RA (PE, clone HI100), CCR7 (FITC, clone 150503), CTLA-4 (BV421, clone BNI3), PD-1 (PE- Cy7, clone EH12.1), LAG3 (PE, clone T47–530), TIM3 (BV711, clone 7D3), and CD45 (APC, clone 2D1) from BD Biosciences. Anti-CD45 (PerCP, clone REA747) and anti-CD69 (APC, clone REA824) were obtained from REAffinity by Miltenyi Biotec. Tumor cells were stained with antibodies against CD276 (BV421, clone 7–517) from BD Biosciences and with anti-763.74 [anti-CSPG4; kindly provided by Dr. Ferrone (Massachusetts General Hospital)], followed by the staining with a secondary rat anti-Mouse (PE, clone X56) from BD Biosciences. The expression of the 763.74(A) and 763.74(B) CARs and the anti-CD19 CAR was assessed using specific anti-idyotipic antibodies [MK2-23 mAb for scFv 763.74(A) and 763.74(B) and 233–4A mAb for anti-CD19 CAR; kindly provided by Dr. Ferrone], followed by the staining with a secondary rat anti-Mouse antibody (PE, clone X56) from BD Biosciences. The expression of the humanized 763.74 CARs was assessed using a biotinylated monoclonal anti-rFW1.4 antibody (generated and biotinylated internally at Cell Medica, Switzerland) followed by the staining with streptavidin protein (PE-conjugated) from Invitrogen (catalog number 12-4317-87). Data acquisition was performed on BD LSRFortessa or Canto II flow cytometer using the BD FACS-Diva software or on a MACSQuant (Miltenyi Biotec). Data analyses was performed with the FlowJo software (Version 9 or 10) or FlowLogic software (Version 7.2, Miltenyi Biotec).
Confocal microscopy
T cells expressing the CAR fused with GFP were fixed with cytofix buffer (BD Biosciences), stained with DAPI (Invitrogen) according to manufacturer’s protocol, washed with PBS, and mounted on glass-bottom microwell dishes (MatTek corporation). Data acquisition was performed on LSM700 Zeiss laser scanning confocal microscope (objective lens 63X/1.4 Plan Apo Oil, pixel size 0.07 μm, pinhole size 1 AU) using ZEN software (ZEISS Microscopy). All groups of images were acquired using the same settings. Data analysis was performed with Fiji software. The samples were scorded by drawing ROIs as shown in Fig.2 and calculating the pixel MFI. The Coefficient of Variation was calculated as standard deviation/average pixel MFI.
Figure 2. Amino acid substitutions in the FWRs of the scFv 763.74(A) reverse the tonic signaling in CAR-T cells.
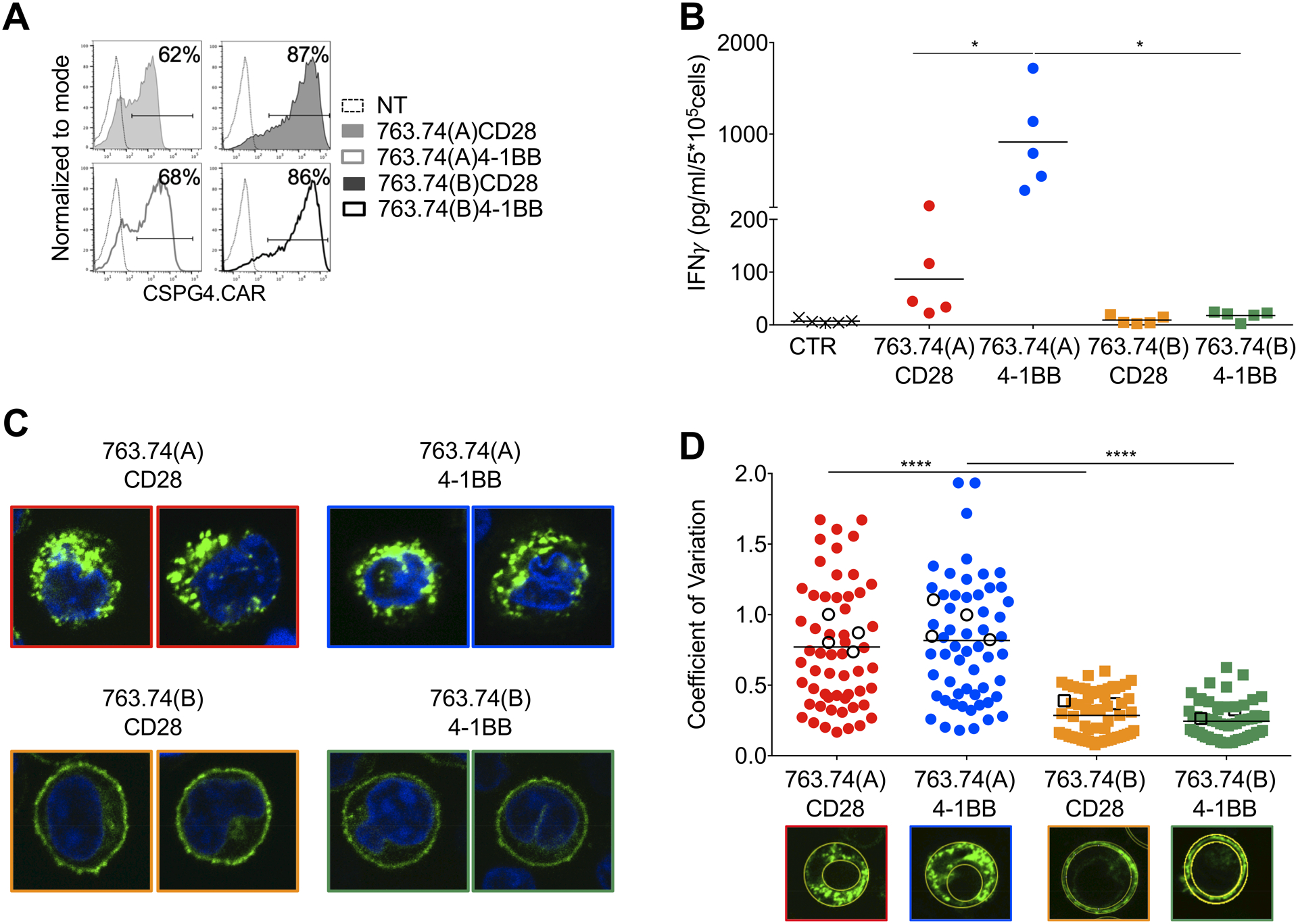
A. Representative flow cytometry plots showing CAR expression in T cells engineered with the scFv 763.74(A) and scFv 763.74(B) CARs encoding either CD28 or 4–1BB endodomains. CAR expression was assessed using an anti-idyotipic antibody followed by the staining with a secondary rat anti-mouse antibody. Non-transduced (NT) T cells were a negative control. B. Quantification of IFNγ released into supernatants by T cells expressing the different CARs without CAR-specific activation after 24 hours. CTR: control CAR-T cells (n=5, mean shown). *P=0.0154 763.74(A) CD28 vs. 4–1BB; *P=0.194 763.74(A) vs. (B)4–1BB, paired t test. C. Representative confocal microscopy imaging showing GFP aggregation in T cells expressing GFP-tagged CARs in which the CARs were obtained using either the scFv 763.74(A) or scFv 763.74(B) and either CD28 or 4–1BB endodomains. Blue staining indicates the DAPI. Shown are representative cells of a single field (Magnification 63X). D. Quantification of the relative variability of GFP-tagged CAR expression using the Coefficient of Variation calculated as standard deviation/average pixel MFI. The standard deviation and average pixel MFI were calculated on the GFP+ areas. White symbols represent the cells shown in Fig. 1F and Fig. 2C (n=60, mean shown). ****P<0.0001, unpaired t-test.
Coculture experiments and ELISAs
For spontaneous IFNγ release assays, 1 × 106 CAR-T cells were plated in 24-well plates in 2 mL of complete T cells media without cytokines. T cells (2 × 104 cells/well) were cocultured with tumor cell lines (M14-wt or WM115, 105 cells/well in 24-well plates), in complete medium, in the absence of cytokines (E:T=1:5). After 5 days of culture, cells were harvested and stained with CD3 (APC-H7, clone SK7 from BD Biosciences) and CD276 (BV421, clone 7–517 from BD Biosciences) monoclonal antibodies to detect T cells and tumor cells, respectively (15). Surface staining was performed in 200 μL of PBS for 20 minutes at 4°C. Residual tumor cells in culture were enumerated by flow cytometry. Data acquisition was performed on BD LSRFortessa or Canto II flow cytometer using the BD FACS-Diva software. Data analyses was performed with the FlowJo software (Version 9 or 10). Culture supernatants were harvested after 24 hours of culture, and IFNγ and IL2 measured using the DuoSet Human IFNγ and IL2 ELISA kit (R&D Systems). For the detection of IFNγ, 10 μL of culture supernatants were used diluted with 90 μL of reagent diluent prepared according to R&D Systems protocol. For the detection of IL2, 100 μL of culture supernatants were used without any dilution. Data acquisition was performed on a Synergy2 microplate reader (BioTek) using Gen5 software (BioTek). IFNγ and IL-2 concentration was calculated using Microsoft Excel. Glioblastoma neurospheres (GBM-NS) were plated at 5 × 105 cells in 24-well plates with T cells at E:T ratio of 1:5 in GBM-NS medium without serum and in the presence of 1X B27 supplement (Thermo Fisher). T cells were maintained in GBM-NS medium for 3 days before plating the cocultures (12). GBM-NS and T cells were collected at different time points following 2, 4, 6, and 24 hours, and residual tumor cells and T cells were measured by flow cytometry based on CSPG4 and CD45 expression, respectively. The activation of CAR-T cells was measured by evaluating the expression of CD69. Data acquisition was performed on a MACSQuant (Miltenyi Biotec). Data analyses was performed with the FlowLogic software (Version 7.2, Miltenyi Biotec).
Computational analysis
To generate the 3D conformation of scFvs, we initially considered the primary sequence of scFv and performed BLAST search against the RCSB database to identify homologous template structures with high sequence similarity. BLASTp (protein BLAST)analysis identified scFv 1696 (16)(resolution 2.70 Å; sequence identify: 70.51%; PDB ID: 1JP5), anthrax-neutralizing single-chain antibody 14b7 (resolution: 1.30 Å; sequence identify: 69.92%; PDB ID: 3ESU), the omalizumab scFv (resolution: 2.30 Å; sequence identify: 72.08%; PDB ID: 6TCS), and the sc-dsFv (single-chain disulfide-stabilized antibody variable fragments) derived from the G6-Fab (resolution: 2.40 Å; sequence identify: 73.50%; PDB ID: 3AUV) as potential templates of scFv 763.74(B), FMC63, h763.74 #2, and h763.74 #5 for homology modeling. We generated 40 models using Modeller-9v19 (https://salilab.org/modeller) (17) and selected the structure with least molecular objective function score as the representative conformation of the scFv. Since Because steric clashes are common in modelled and low-resolution structures, we employed Chiron (https://dokhlab.med.psu.edu/chiron) (18) to optimize the structure of the scFv. Chiron resolves atomic clashes by performing short-DMD (Discrete Molecular Dynamics) simulations (19–21) on protein structure with minimal or no perturbation to the backbone. After the high energy caused by atomic clashes is relaxed by Chiron, the scFv structure was subsequently considered for in silico mutagenesis studies using Eris molecular suite (https://dokhlab.med.psu.edu/eris) (22). The Eris protocol induces mutations in protein and estimates free energies of mutant (ΔGmut) and wild-type (ΔGwt) conformations. Eris performs rapid side-chain repacking and backbone relaxation around the mutated site using Monte-Carlo algorithm and subsequently evaluates ΔGwt and ΔGmut using Medusa force field (22;23). The Eris algorithm then computes change in free energy of protein upon mutation by employing the following formula: ΔΔGmut = ΔGmut − ΔGwt. We evaluated ΔΔGmut values to estimate the stabilizing (ΔΔGmut < 0) or destabilizing (ΔΔGmut > 0) mutations. Eris has been extensively validated and used in designing novel proteins (24–26).
Magnetic Resonance Imaging (MRI).
MRI was performed using a horizontal-bore preclinical scanner (BioSpec 70/20 USR, Bruker, Ettlingen, Germany). The system has a magnetic field strength of 7 T (1H frequency 300 MHz) and a 20 cm bore diameter. The scanner was equipped with an actively shielded gradient system with integrated shims set up to 2nd order. The maximum gradient amplitude was 440 mT/m. All acquisitions were carried out using a cross-coil configuration: a 72 mm linear birdcage coil was used for radiofrequency excitation and a mouse brain surface coil received signal. Mice injected with GBM-NS and treated with CAR-T cells underwent high-resolution MRI at the following time points:+0, +14, + 21, +28, +35, +44, +54, and +61 days after treatment. Animals were anaesthetized with 1.5 – 2% isoflurane (Aerrane, Baxter, United States) vapourized in a mixture of medical air and oxygen (60 : 40 vol:vol, flow rate 0.8 L/min) via a nose cone. Exhaled gas was actively vacuumed away via a built-in vacuum line. To detect the depth of anesthesia and the animal health condition during the study, the respiratory rate was monitored by a pneumatic sensor. The animals’ temperature was kept at 36.5 ± 0.5°C by means of a warm-water circuit integrated into the animal holder. Mice were positioned on an animal bed equipped with a nose cone for gas anesthesia and a three point-fixation system (tooth-bar and ear-plugs). Mice underwent high-resolution MRI with the following protocol: a T2-weighted Rapid Acquisition with Reduced Echoes (RARE) sequence (TR = 3360 ms, TE = 35 ms, in plane resolution = 100 × 100 um2, slice thickness = 400 um, 4 averages, total acquisition time of 5 min 36 sec) and two T1-weighted RARE sequences (TR = 510 ms, TE = 8 ms, in plane resolution = 78 × 78 um2, slice thickness = 400 um, 6 averages, total acquisition time of 9 min 47 sec) acquired before and after intraperitoneal administration of 100 microlitersof Gadoliunium-based contrast medium (Gadovist 1.0 mmol/mL, Bayer, Germany). All imaging sequences were acquired along the same coronal geometry (400 um thick continuous slices), with slice package posterior to olfactory bulb and anterior to cerebellum. T1-weighted images were visually inspected and every slice was evaluated in terms of contrast agent-induced signal enhancement, which was interpreted as being due to a blood brain barrier lesion.
Statistical analysis
Data were summarized as the mean±SD. Student t-test or two-way ANOVA were used to determine statistically significant differences between treatment groups, with Bonferroni’s correction for multiple comparisons when appropriate (Prism 6: GraphPad Software). Survival analysis was performed using the Kaplan-Meier method and the Mantel-Cox log rank test was applied (Prism 6: GraphPad Software). All P values less than 0.05 were considered statistically significant.
Results
Modifications of the scFv abrogate CAR tonic signaling
We have previously reported on the generation of a CAR targeting the chondroitin sulphate proteoglycan 4 (CSPG4)(12;27). This CAR was obtained using the scFv 763.74(A) from the 763.74 murine monoclonal antibody. T cells expressing CSPG4-specific CAR show antitumor effects in multiple tumors expressing CSPG4 (12;27). However, T cells expressing the scFv 763.74(A) CAR encoding either CD28 or 4–1BB costimulatory endodomains showed release of IFNγ in the absence of antigen stimulation (Fig. 1A–B), a phenomenon defined as CAR tonic signaling (6). Spontaneous IFNγ release by T cells expressing the scFv 763.74(A) CARs was strictly dependent on CAR signaling because mutations of the tyrosine of the immunoreceptor tyrosine-based activation motifs (ITAMs) of the CAR-CD3ζ chain (Fig. 1C), that prevent tyrosine phosphorylation, completely abrogated the spontaneous IFNγ release (Fig. 1D–E). To study the distribution of CAR molecules on the cell surface of T cells, we generated scFv 763.74(A) CARs in which the CD3ζ chain of the CAR was fused at COOH terminal with GFP. Using confocal microscopy imaging, we found that scFv 763.74(A) CARs form membrane clusters in the absence of CAR crosslinking, likely indicating self-aggregation of CAR molecules (Fig. 1F). The sequence of the scFv 763.74(A) was obtained from an early passage of the hybridoma 763.74 secreting the murine IgG1 monoclonal antibody (mAb) that recognizes a peptide epitope of human CSPG4 (28). It is well-established that culture passages of hybridomas affect their growth rate and the yield of the secreted antibody (29). It has also been described that upon culture passage, amino acid substitutions may occur in both CDRs and FWRs in subclones derived from the hybridoma (30). In light of this possibility, we sequenced the VL and VH domains of a late passage of the 763.74 hybridoma. We obtained two VL and VH sequences in which amino acid substitutions were identified in the FWRs (FWR1 and FWR3) of both VL and VH (Supplementary Fig. S1A). We assembled a new scFv called scFv 763.74(B), generated new scFv 763.74(B) CARs, and compared them side-by-side with scFv 763.74(A) CARs for evidence of tonic signaling. All CARs were equally expressed in T cells (Fig. 2A, Supplementary Fig. S1B), and CAR-T cells equally expanded in vitro (Supplementary Fig. S1C). However, T cells expressing the scFv 763.74(B) CAR encoding either CD28 or 4–1BB costimulatory endodomains did not show spontaneous release of IFNγ (Fig. 2B). We also observed a more homogeneous distribution of the scFv 763.74(B) CARs on the membrane of T cells using confocal microscopy of GFP-tagged CARs (Fig. 2C). We quantified the relative variability of GFP-tagged CAR expression by calculating the Coefficient of Variation. The distribution of scFv 763.74(A) was significantly more variable than scFv 763.74(B) (P<0.0001), regardless from the type of costimulatory molecule (Fig. 2D). Cross-linking of CARs expressed in T cells mediated by an anti-idyotipic mAb (MK2–23 mAb) caused significant cluster formation of CAR molecules, regardless the type of scFv expressed, further indicating that clusters identified by confocal microscopy reflected the formation of CAR aggregates (Supplementary Fig. S1D). Phenotypic analysis of T cells expressing scFv 763.74(A) CARs or scFv 763.74(B) CARs did not show differences in the expression of memory and exhaustion markers (Supplementary Fig. S1E–F), suggesting that tonic signaling may not induce an exhaustion phenotype identified by phenotypic markers during the 10–14 days of culture usually required to manufacture CAR-T cells for clinical use (31;32). Overall, these data indicated that amino acid substitutions within the FWRs of a scFv are sufficient in causing self-aggregation of the scFv in the CAR format and tonic signaling in T cells.
Figure 1. Tonic signaling of CAR-T cells expressing the CAR encoding scFv 763.74(A).
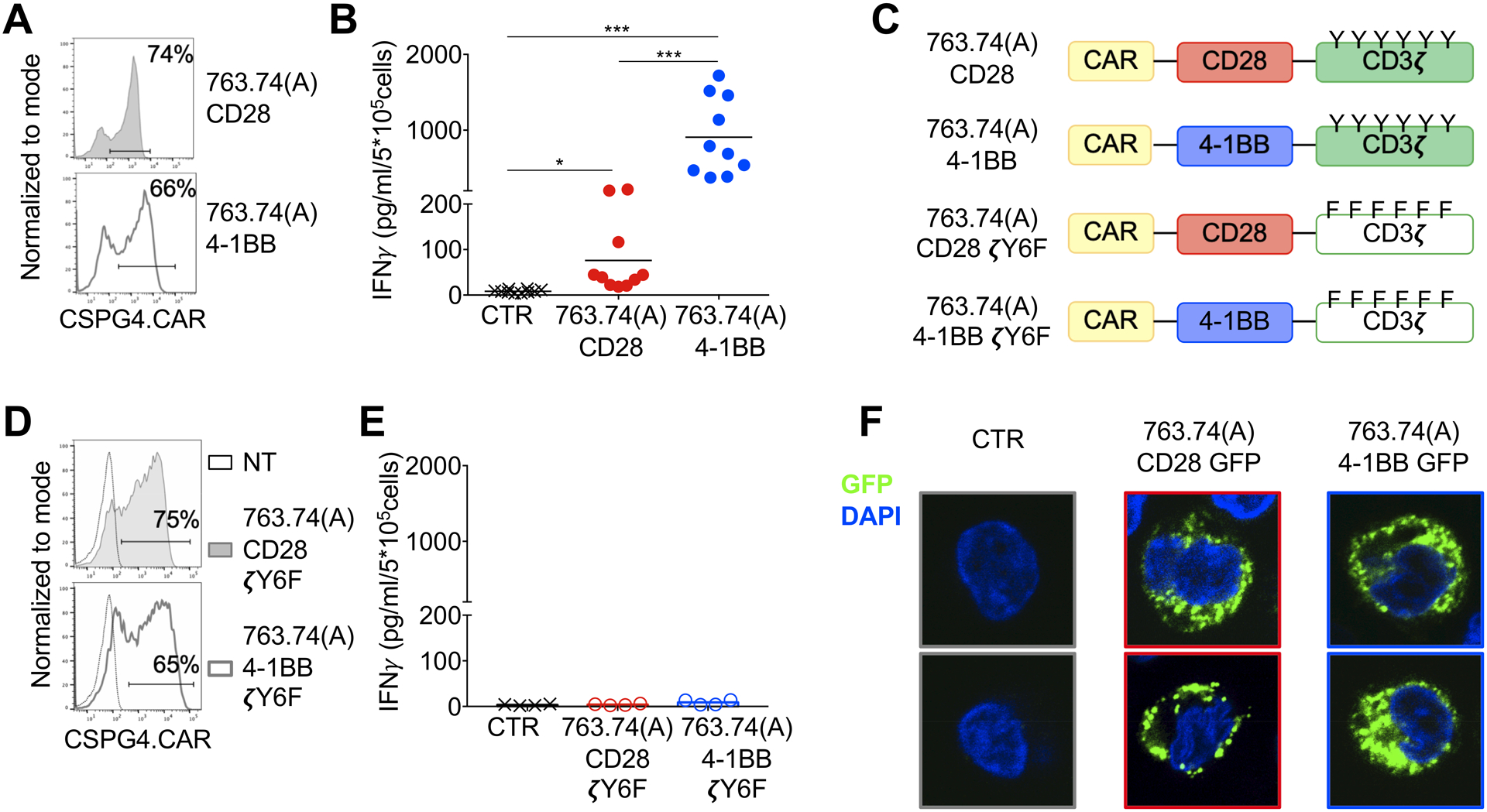
A. Representative flow cytometry plots showing CAR expression in T cells assessed at day 8 of culture. 763.74(A) CD28 and 763.74(A) 4–1BB indicate the specific CAR expressed in T cells. CAR expression was assessed using an anti-idyotipic antibody followed by the staining with a secondary rat anti-mouse antibody. B. Quantification of IFNγ released into supernatants by T cells expressing control CAR (CTR), 763.74(A) CD28, or 763.74(A) 4–1BB CAR without CAR-specific activation after 24 hours (n=10, mean shown). *P=0.0184; ***P=0.0003 (CTR vs. 763.74(A) CD28); ***P=0.0004 (763.74(A) CD28 vs. 763.74(A) 4–1BB), paired t-test. C. Schema of the 763.74(A) CAR constructs encoding either CD3ζ wild-type or CD3ζ in which all tyrosine amino acids of ITAMs have been mutated. D. Representative flow cytometry plots showing CAR expression in T cells assessed at day 8 of culture. 763.74(A) CD28 ζY6F and 763.74(A)4–1BB ζY6F indicate the specific CAR expressed in T cells. CAR expression was assessed using an anti-idyotipic antibody followed by the staining with a secondary rat anti-mouse antibody. Non-transduced (NT) T cells were a negative control. E. Quantification of IFNγ released into supernatants by T cells expressing control CAR (CTR), 763.74(A) CD28ζY6F or 763.74(A) 4–1BBζY6F CAR without CAR-specific activation after 24 hours (n=4, mean shown). F. Representative confocal microscopy imaging showing GFP aggregation in T cells expressing GFP-tagged CARs (green) in which the CARs are obtained using the scFv 763.74(A) and either CD28 or 4–1BB endodomains. CTR indicates control T cells. Blue staining indicates the DAPI. Shown are representative cell of a single field (Magnification 63X).
Modifications of FWRs of the scFv cause protein destabilization
To study if differences in amino acids between the scFv 763.74(A) and scFv 763.74(B) cause destabilization of the scFv, we generated the 3D conformation of the scFv 763.74(B) through homology modeling and optimized the structure for in silico mutagenesis (Fig. 3A). We employed the Eris tool to delineate the effect of FWR mutations, L3K, T5S, A9S, E83Q, I123V, Q124K, V126K, Q127E, and L230V on the structure of the scFv 763.74(B). Eris estimated the ΔΔGmut for the above-mentioned mutations as 2.41, 1.26, 2.29, 0.47, 0.79, 1.87, 4.58, 2.70, and 0.67 kcal/mol, respectively (Fig. 3B). Specifically, the mutations destabilized the scFv 763.74(A) structure (ΔΔGmut>0) and subsequently affected the spontaneous CAR aggregation. To cross-validate the structural conformation of scFv 763.74(B), we performed Eris analysis to identify stabilizing mutations (ΔΔGmut<0) at the FWR-mutated sites. Our analysis indicated that mutations, such as E83L, E83I, T5M, and Q124M, resulted in negative ΔΔGmut, notifying their potential stabilizing capability (Supplementary Fig. S2). We also identified that the residues, such as I123 and Q127, were critical for the stability of the scFv 763.74(B) structure. The substitution of any of the other 19 amino acids at I123 or Q127 positions destabilized the scFv 763.74(B) structure (ΔΔG>0) and thereby affected the protein aggregation (Supplementary Fig. S2). Overall, these data indicated that amino acid substitutions within the FRWs of a scFv destabilize the protein and cause self-aggregation of the scFv.
Figure 3. Amino acid substitutions in the FWRs of the scFv 763.74(A) destabilize the scFv.
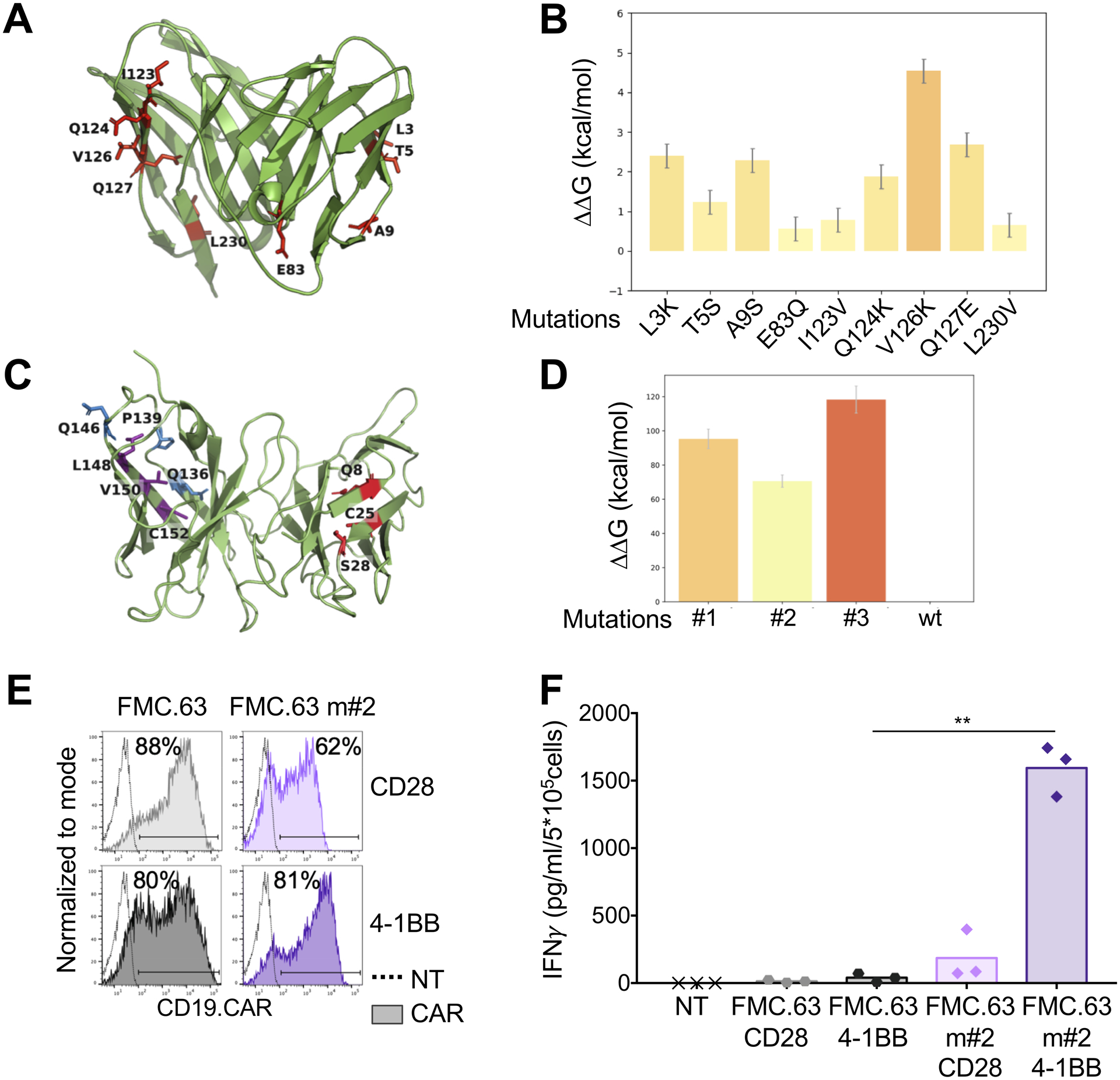
A. Structural conformation of the scFv 763.74(B) generated through computational modeling. Protein is shown in cartoon representation and FWR mutations in stick representation. B. Chosen amino acid mutations evaluated for their influence on scFv 763.74(B) stability (n=50, mean and SD shown). C. Structural conformation of the scFv FMC.63 generated through computational modeling. Protein is shown in cartoon representation and FWR mutations in stick representation. D. Chosen amino acid mutations (Q8W|C25R|S28P, Q136W|P139L|Q146P, and L148Y|V150W|C152W) evaluated for their influence on the scFv FMC.63 stability (n=50, mean and SD shown). E. Representative flow cytometry plots showing CAR expression in T cells engineered with the scFv FMC.63 CARs and the destabilized scFv FMC.63 m#2 CARs encoding either CD28 or 4–1BB endodomains. CAR expression was assessed using an anti-idyotipic antibody followed by the staining with a secondary rat anti-mouse antibody. F. Quantification of IFNγ released into supernatants by T cells expressing the different CARs without CAR-specific activation after 24 hours (n=3, mean shown). **P=0.0047, paired t test.
To validate the computational model, we selected the murine scFv FMC.63 (recognizes the human CD19), which is known to lack tonic signaling in the CD19-specific CAR. We generated the 3D-conformation of the scFv FMC.63 through homology modeling and optimized the structure for in silico mutagenesis (Fig. 3C). We then employed the Eris tool to predict destabilizing mutations of this scFv and identified three sets of mutations (Q8W|C25R|S28P, Q136W|P139L|Q146P, and L148Y|V150W|C152W) within the FWRs that destabilized the FMC.63 scFv (Fig. 3D, Supplementary Fig. S3A). Eris estimated the ΔΔGmut for the three mutations as 95.3, 70.6, and 118.3 kcal/mol, respectively. We generated CARs containing these three predicted destabilized scFvs encoding either CD28 or 4–1BB (FMC.63(m#1), FMC.63(m#2), FMC.63(m#3)). Upon transduction of T cells, only the FMC.63(m#2) CAR was detectable by flow cytometry (Fig. 3E, Supplementary Fig. S3B–C). We compared T cells expressing the FMC.63(m#2) CARs side-by-side with T cells expressing the FMC.63 CARs for evidence of tonic signaling. The destabilized FMC.63(m#2) CAR showed spontaneous production of IFNγ, especially when encoding 4–1BB (Fig. 3F). Overall these data validated the computational model’s ability to predict scFv stability, supporting our finding that destabilizing mutations of the FWRs are sufficient in causing self-aggregation of the scFv in the CAR format and tonic signaling in T cells.
Modifications of the FWRs of the scFv enhance function of CAR-T cells
To determine if the amino acid substitutions within the FWRs of the scFv affected the anti-tumor activity of CAR-T cells, we used the melanoma cell lines WM115 (CSPG4+) and M14 (CSPG4−)(Supplementary Fig. S4A). Paralleling the spontaneous release of IFNγ, T cells expressing the 763.74(A) CAR with 4–1BB showed compromised capacity to eliminate tumor cells in vitro (residual tumor cells 43.6% ± 26.0%) after a 4-day coculture of CAR-T cells and tumor cells at the ratio 1:5 (Fig. 4A–B). In contrast, CD28 costimulation seemed to allow complete tumor elimination for both 763.74(A) and 763.74(B) CARs (residual tumor cells 2.2% ± 2.7% and 2.9% ± 2.6%, respectively). T cells expressing the 763.74(B) CAR with 4–1BB costimulation showed improved antitumor effects compared to T cells expressing the 763.74(A) CAR with 4–1BB, but did not completely eliminate the tumor cells (residual tumor cells 14.1% ± 8.0% and 43.6% ± 26.0%, respectively)(Fig. 4A–B). CAR-T cells did not eliminate the melanoma cell line M14 (lacks CSPG4 expression), indicating that antigen specificity was not affected by amino acid substitutions within the FWRs. Only T cells expressing the 763.74(B) CARs consistently released detectable amounts of IFNγ in the culture supernatant with WM115 tumor cells (Supplementary Fig. S4B). The superior antitumor effects of 763.74(B) CAR-T cells was more evident in vivo using the eGFP-FFLuc WM115 xenogeneic NSG mouse model (Fig. 4C). T cells expressing the 763.74(B) CAR with CD28 exhibited the most prominent antitumor effects measured as both tumor bioluminescence (Fig. 4D, Supplementary Fig. S4C) and tumor size (Fig. 4E). Enhanced functions of CAR-T cells expressing the 763.74(B) CAR were confirmed in our previously described glioblastoma (GBM) tumor model in which nude mice were engrafted in the brain with primary GBM-NS cells and treated via intratumor inoculation of CAR-T cells (12)(Fig. 5A). Tumor engraftment and progression were monitored by MRI. As previously described (12), we observed rapid tumor progression in mice treated with control T cells or T cells expressing the 763.74(A) CAR encoding CD28, and in these animals, tumor masses occupied whole hemispheres and infiltrated the contralateral one (Fig. 5B–C). Mice treated with T cells expressing the 763.74(B) CAR encoding CD28 showed the most evident antitumor effects, indicated by smaller and more circumscribed lesions (Fig. 5D), but tumor control was also observed in mice treated with T cells expressing either 763.74(A) or 763.74(B) CAR encoding 4–1BB, although antitumor effects were less dramatic (Fig. 5E–F). T cells expressing either 763.74(A) or 763.74(B) CARs encoding 4–1BB prolonged survival compared to mice treated with control T cells (p<0.0001). However, T cells expressing the 763.74(B) CAR encoding CD28 were the most effective in prolonging survival (p<0.0001 vs. CTR; p=0.04 vs. 763.74(A) with 4–1BB, p=0.01 vs. 763.74(B) with 4–1BB). (12)(Fig. 5G). To further characterize the antitumor effects of T cells expressing the 763.74(B) CAR encoding CD28 in the GBM model, we investigated the activation status of T cells immediately after intracranial infusion. We explanted the tumor masses 2, 4, 6, 12, 24, and 48 hours after CAR-T cell infusion and observed that T cells expressing the 763.74(B) CAR with CD28 upregulated CD69 within 2 hours after inoculation (42.5 ± 2.1%) and maintained high CD69 expression for 24 hours (22.5 ± 1.5% CD69+ T cells), which is consistent with previous reports indicating the fast activity of CAR-T cells encoding the CD28 endodomain (33;34)(Fig. 5H). Additional experiments in vitro further demonstrated the rapid antitumor effects of T cells expressing the 763.74(B) CAR with CD28 (Supplementary Fig. S5). Overall these data indicated that the correction of the tonic signaling caused by the self-dimerization of the scFv improves the antitumor effects of CAR-T cells.
Figure 4. T cells expressing the 763.74(B) CAR with CD28 are superior in mediating tumor elimination in a melanoma tumor model.
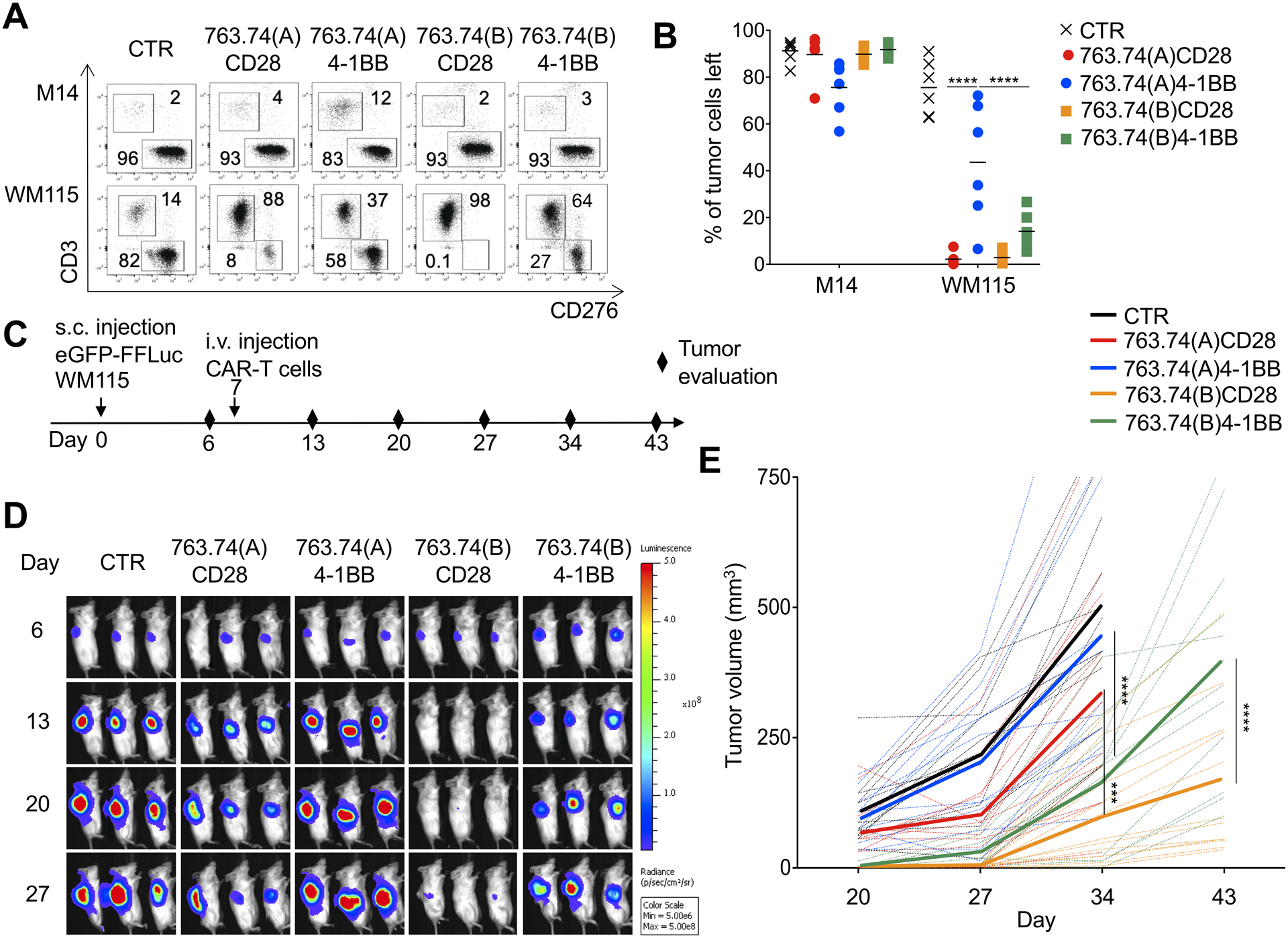
A Representative flow plots and B. summary of the quantification of residual tumor cells of experiments in which control T cells (CTR) and T cells engineered with the scFv 763.74(A) and scFv 763.74(B) CARs encoding either CD28 or 4–1BB endodomains. CAR-T cells were cocultured with melanoma cell lines (E:T=1:5) for 5 days and then collected and stained with anti-CD3 and anti-CD276 (B7-H3) to identify T cells and melanoma cells, respectively, by flow cytometry. Data are presented as mean±SD, n=6. ****P<0.0001, two-way ANOVA with Bonferroni’s correction. C. Experimental schema of the melanoma xenograft model. eGFP-FFLuc WM115 (5 × 105 cells) were injected s.c., and 7 days later, mice were injected i.v. with control T cells (CTR) or T cells engineered with the scFv 763.74(A) and scFv 763.74(B) CARs encoding either CD28 or 4–1BB endodomains (5×106 cells). D. Representative tumor bioluminescence (BLI)(color scale: min=5 × 106; max=5 × 108) in mice treated as illustrated in (C). E. Tumor volume in mice engrafted in (D). Dotted lines represent individual mice, and bolded solid lines represent the mean for the group. Summary of 4 independent experiments (n=12 for each condition). ***P=0.00012; ****P<0.0001, two-way ANOVA with Bonferroni’s correction.
Figure 5. T cells expressing the 763.74(B) CAR with CD28 have rapid antitumor activity in a glioblastoma tumor model.
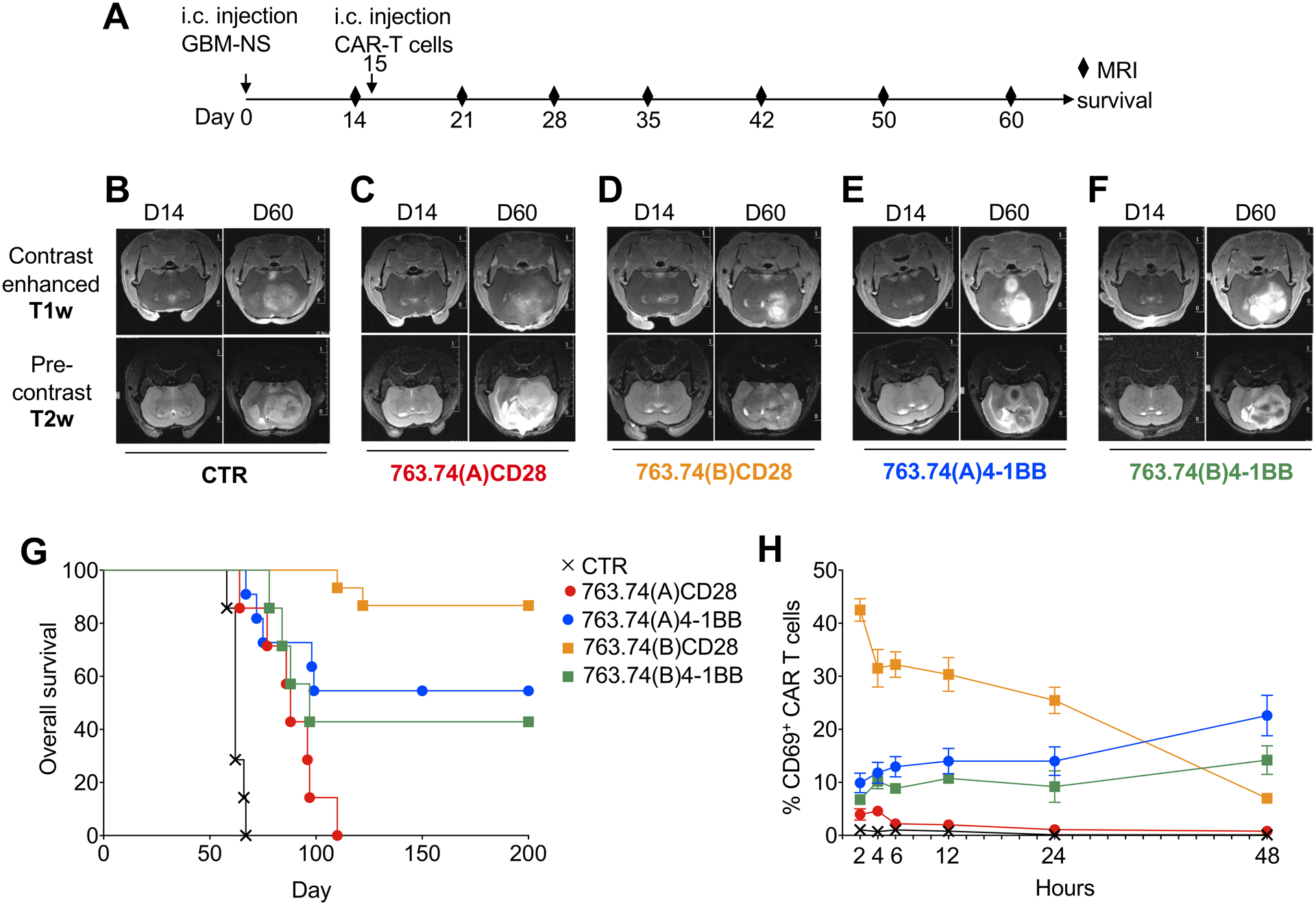
A. Experimental schema of the GBM xenograft model. GBM-NS (1×105 cells) were injected into the caudate nucleus, and 15 days later, mice were injected intratumorally with control T cells (CTR) or T cells engineered with the scFv 763.74(A) and scFv 763.74(B) CARs encoding either CD28 or 4–1BB endodomains (2×106 cells). Tumor growth was monitored with MRI. B-F. Representative MRI performed with T1-weighted images (T1-wi) with contrast medium injection and T2-weighted images (T2-wi) showing the pattern of tumor progression and infiltration in mice treated as in (A). G. Kaplan-Meier survival curves of mice treated as in (A). n=7 mice/group for CTR, 763.74(A)CD28, 763.74(B)4–1BB. n=15 mice/group for 763.74(B) CD28 and 763.74(A) 4–1BB. Overall survival statistical analysis was performed using the Mantel-Cox log rank test. H. Time course of CD69 expression in CAR-T cells isolated from the tumor at the indicated time points after intratumor delivery of T cells. Data are presented as mean±SD, n=2.
Humanization of the FWRs of the scFv abrogates CAR tonic signaling
Humanization of murine derived scFvs is a strategy proposed to prevent humoral and T-cell responses to the CAR (35;36). We asked the question whether substituting the murine FWRs of the scFv with human FWRs could be used to abrogate the CAR tonic signaling. The sequence of 763.74(A) and the stable human framework rFW1.4 were aligned, and the critical amino acids were identified (37). One CDR graft with no mutations in the rFW1.4 sequence and seven variants with up to 24 mutations in the critical regions were designed in the first round of engineering (Supplementary Fig. S6A). Humanized scFv variants alone (i.e., only extracellular domain of CARs) and the wild-type murine scFv were expressed in E.coli, refolded, and purified by size-exclusion chromatography (SEC). The wild-type murine 763.74(A) scFv variant was not refoldable due to aggregation. Therefore, we were unable to purify it in a soluble form. This result further suggested that the murine 763.74(A) scFv was unstable. The humanized variant with no mutations in the rFW1.4 was expressed but did not bind CSPG4. Nearly all other humanized scFv variants were successfully expressed and able to bind CSPG4+ cells. In the next engineering rounds, humanized variants with the minimal number of 763.74(A) murine FW residues were further subjected to chain shuffling of the VH and VL. A total of 26 humanized scFvs were produced (Supplementary Table S1). Four humanized scFvs (h763.74 #2, h763.74 #3, h763.74 #4 and h763.74 #5) with the minimal number of murine FWR residues and retained CSPG4-binding activity were selected for further studies (Supplementary Fig. S6B). The scFvs were characterized for binding affinity (EC50) to CSPG4-expressing cells by flow cytometry. The humanized scFvs h763.74 #2, h763.74 #3, h763.74 #4 and h763.74 #5 bound CSPG4 with comparable affinities (Supplementary Fig. S6C). In a separate experiment, we compared the scFvs h763.74 #5 and 763.74(B). The latter bound CSPG4 with a higher binding affinity than the scFv h763.74 #5 (p<0.0001 and p=0.0007 at 500 and 1000 ng/mL, respectively) (Supplementary Fig. S6D). We generated CARs with all four humanized scFv 763.74 with CD28 endodomain and performed in vitro coculture experiments with tumor cells. T cells expressing h763.74.CAR #2 and h763.74.CAR #5 showed a trend for better antitumor activity and higher production of IFNγ and IL2 in vitro and were selected for further studies (Supplementary Fig. S6E–G). To further characterize the two selected humanized scFvs h763.74.CAR #2 and h763.74.CAR #5 (Supplementary Fig. S7A), we performed storage stability studies with purified soluble scFvs. Proteins were prepared at 1 mg/mL and stored for 48 hours at 4°C and 37°C. After incubation, samples were analyzed by SEC to estimate the percentage of monomeric proteins. Under tested conditions, no detectable protein loss was observed, and the percentage of monomers remained above 91%−97% (Supplementary Fig. S7B). This method demonstrated that humanized scFvs were monomeric under the tested conditions and did not dimerize or aggregate upon storage at 4°C and 37°C. We also measured the thermal unfolding temperature of h763.74 #5 scFv and the murine 763.74(B) scFv with nanoDSF and observed that both proteins remain folded up to 60°C (Supplementary Fig. S7C). We also employed DMD (19–21) to perform a computational molecular dynamics simulation to compare the stability. The free energy (763.74(B) < h763.74 #2 < h763.74 #5) and the average RMSF (763.74(B) < h763.74 #2 < h763.74 #5) both suggested that 763.74(B) was more stable than h763.74 #2 and h763.74 #5 (Supplementary Fig. S7D). The h763.74.CAR #2 and h763.74.CAR #5 were equally expressed in T cells (Fig. 6A, Supplementary Fig. S8A), and T cells did not show spontaneous release of IFNγ (Fig. 6B). T cells expressing the h763.74.CAR #2 and h763.74.CAR #5 successfully controlled the CSPG4+ WM115 melanoma cell growth in vitro (residual tumor cells 11% ± 15% and 10% ± 17% respectively), whereas they did not target the CSPG4− M14 melanoma cell line, indicating that antigen specificity was maintained (Fig. 6C, Supplementary Fig. S8B). The anti-tumor activity of T cells expressing h763.74.CAR #2 and h763.74.CAR #5 was corroborated by specific production of IFNγ and IL2 (Supplementary Fig. S8C). Finally, we compared T cells expressing the h763.74.CAR #2 and h763.74.CAR #5 with T cells expressing the 763.74(B) CAR encoding the CD28 endodomain in the xenogeneic WM115 melanoma mouse model (Fig. 6D). T cells expressing the h763.74.CAR #2 and h763.74.CAR #5 showed potent antitumor activity (Fig. 6E, Supplementary Fig. S8D). Furthermore, T cells were detectable in the peripheral blood of treated mice at different time points (Supplementary Fig. S8E) and in the liver and spleen at the time of euthanasia (Fig. 6F), and T cells retained CAR expression (Fig. 6G). T cells expressing h763.74.CAR #2 and h763.74.CAR #5 did not show increased expression of PD-1 compared to T cells with the 763.74(B) CAR with CD28 (Supplementary Fig. S8F–G). Overall, these data showed that the humanization of a scFv can be used to eliminate tonic signaling of CAR molecules, maintaining specific antitumor effects.
Figure 6. Humanization of the FWRs of the scFv 763.74(A) abrogates CAR tonic signaling without affecting the anti-tumor activity.
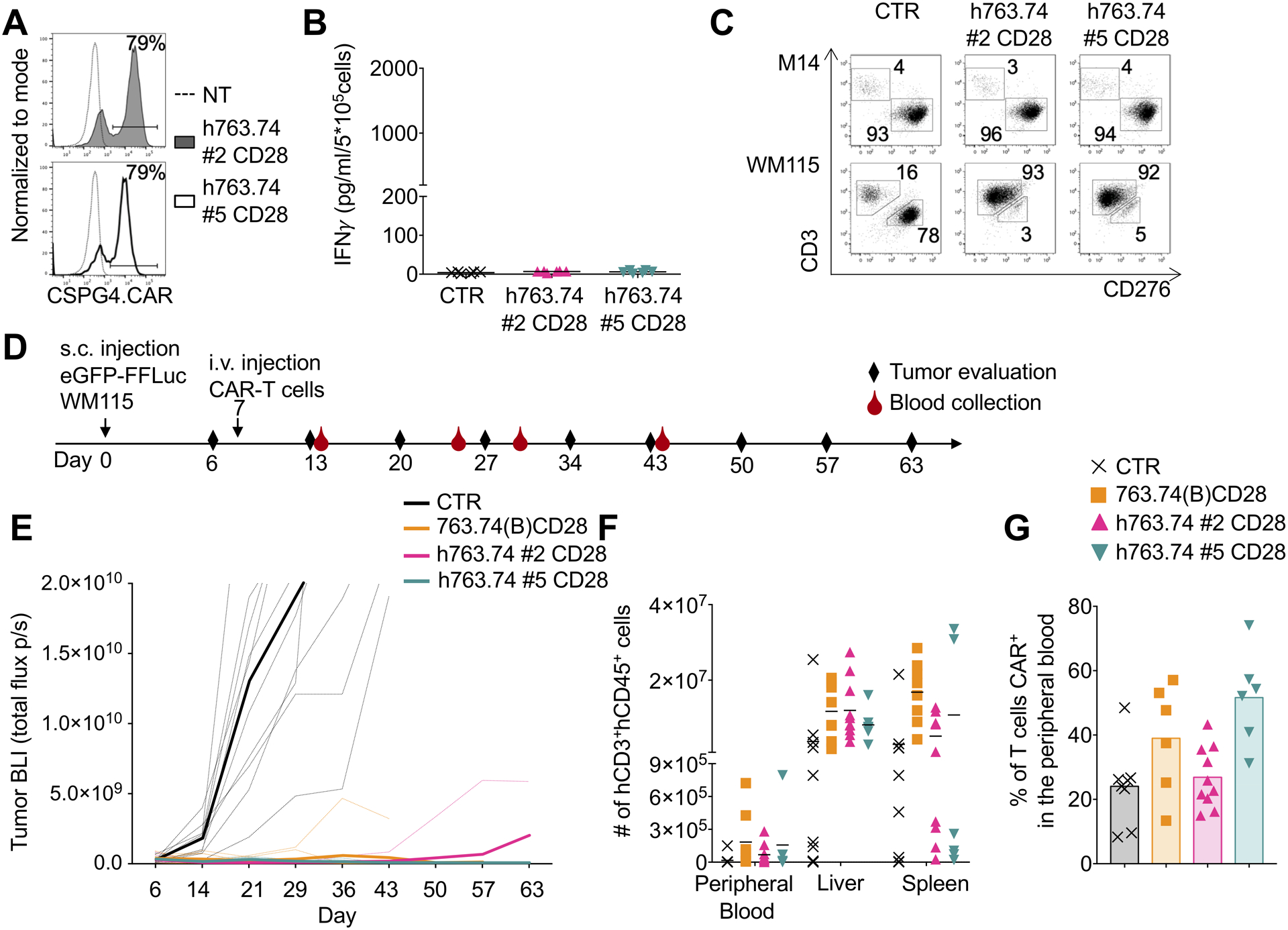
A. Representative flow cytometry plots showing CAR expression in T cells engineered with h763.74 #2 and h763.74 #5 CARs encoding CD28 (h763.74 #2 CD28 and h763.74 #5 CD28) as assessed at day 8 of culture. CAR expression was assessed using a biotinylated antibody recognizing the human FWRs followed by the staining with streptavidin protein. Non-transduced (NT) T cells were a negative control. B. Quantification of IFNγ in supernatants of control (CTR), h763.74 #2 CD28, and h763.74 #5 CD28 CAR-T cells collected after 24 hours. Data are presented as mean±SD, n=6. C. Representative flow plots of coculture experiments in which control (CTR) or T cells expressing either h763.74 #2 CD28 or h763.74 #5 CD28 CARs were plated with melanoma cell lines (E:T=1:5) for 5 days. Cells were then collected and stained with the anti-CD3 and anti-CD276 to identify T cells and melanoma cells, respectively, by flow cytometry. D. Experimental schema of the melanoma xenograft model. eGFP-FFLuc WM115 (5×105 cells) were injected s.c., and 7 days later, mice were injected i.v. with control T cells (CTR) or T cells engineered with 763.74(B) CD28 CAR, h763.74 #2 CD28, or h763.74 #5 CD28 CARs (5×106 cells). E. Tumor BLI kinetics of mice treated according to scheme (D). Dotted lines represent individual mice, and bolded solid lines represent the mean for the group. Summary of 2 independent experiments (n=10 for each group). F. Quantification of human CD3+CD45+ cells in the peripheral blood, liver, and spleen at the time of euthanasia of tumor-bearing mice treated as in (D). Data are presented as mean±SD, n=6. G. Percentage of CAR-T cells in the peripheral blood, gated on human CD3+CD45+ cells, at sacrifice in tumor-bearing mice treated as in (D). Data are presented as mean±SD, n=6.
Discussion
Unstable murine scFvs cause self-aggregation, and murine sequences can theoretically induce immune responses in human subjects. Here, we demonstrated that the instability of the scFv was critical in causing tonic signaling when the scFv was assembled into the CAR format, and the CAR was expressed in T cells. We demonstrated that tonic signaling can be corrected either by the substitution of the amino acids, causing instability within the murine FWRs of the scFv, or humanization of the FWRs. Correction of the tonic signaling enhanced the antitumor effects of the CAR-T cells.
TCR-mediated tonic signaling is a well-characterized homeostatic property of naïve T cells and plays a critical role in promoting their long-term persistence (38). However, TCR-mediated tonic signaling requires TCR engagement with self-peptides presented either in Class I or II, and is strictly confined to T cells located in lymphoid organs (39). In sharp contrast, CAR-mediated tonic signaling in T cells, in its strictest sense, refers to CAR signaling that is independent from any specific CAR engagement and defined as spontaneous release of cytokines, such as IFNγ (6). The event triggering CAR-mediated tonic signaling has been identified as the spontaneous aggregation of a sufficient number of CAR molecules, which leads to initiation of signaling (6). Here, we confirmed that tonic signaling was due to self-aggregation of CAR molecules, and further demonstrated that the CAR-CD3ζ chain was exclusively responsible of the spontaneous cytokine release because loss of function of the CAR-CD3ζ chain completely abrogated the spontaneous release of IFNγ. Spontaneous cytokine release, rather than detection of cell surface markers associated with T-cell exhaustion, was the most robust and reliable assay to define the presence of CAR tonic signaling and reduced functionality.
The instability of synthetic scFvs has long been recognized to cause spontaneous aggregation, hindering the generation of soluble reagents (7;9). Here, we demonstrated that an unstable scFv caused self-aggregation of CAR molecules, leading to CAR tonic signaling in T cells. The CSPG4-specific CAR we generated used a murine scFv from the 763.74 monoclonal antibody. This scFv expressed in E.coli showed a tendency to aggregate, and the scFv was not producible in a soluble form. We have previously demonstrated that structural modeling and mutagenesis driven by computational protein design can be used to restore specificity of scFvs distorted by fusing VL and VH domains (40). Here, we showed that the instability of the scFv was the exclusive cause of self-aggregation and tonic signaling of the CAR. In general, the thermodynamic stability of a protein (ΔG) was crucial for its biological functionality. Amino acid mutations in proteins can disrupt important residue interactions, alter protein active sites, and protein stability. These unstable mutant protein conformations are the underlying sources of numerous human disorders (41). Hence, quantifying the effect of mutations on protein structure is important to estimate the protein stability and their functionality. Here, we employed computational mutagenesis, facilitated by the Eris tool, to delineate the effect of FWR mutations on the structure of the scFv 763.74(A). Using a stable scFv like the scFv FMC.63, we validated the computational modeling, demonstrating how specific amino acid substitutions within the FWRs affected the stability of the scFvs which caused tonic signaling. Finally, we demonstrated that substituting the murine FWRs with a stable human FWRs, such as the framework rFW1.4, also corrected CAR tonic signaling without modifying the antigen specificity (13). The stable human framework rFW1.4 can accommodate CDRs of different origin and allows analyses of soluble scFv proteins to assess their physicochemical stability. This approach thus can be used to select scFvs with optimal physical and binding properties to design CARs. Taken together, the data suggested that stable and monomeric scFvs are applicable to generate CARs that avoid antigen-independent tonic signaling. ScFvs engineered with the human framework rFW1.4 may also have a low likelihood for immunogenicity and could potentially be repeatedly infused to the patients with full efficacy.
Conflicting data have been reported on the role of the costimulatory CD28 and 4–1BB endodomains in exacerbating or attenuating the CAR tonic signaling in T cells using CARs containing different hinge/spacer regions or different vectors/promoters (6;42–44). Despite these conflicting data, it is possible to conclude that an intrinsic instability of the scFv cannot be corrected by the type of costimulation used or by modifying other components of the CAR structure. Indeed, CAR tonic signaling is frequently considered a good reason to abandon a specific scFv from further CAR development, despite the original antibody having excellent antigen specificity and affinity. Our data indicated that using CARs with the intracytoplasmic tail of the costimulatory endodomain CD28 and 4–1BB can cause tonic signaling. Stabilization of the scFv abrogated tonic signaling regardless of the costimulation used, indicating that an unstable scFv can be rescued by either mutations or humanization of the FWRs and that both CD28 and 4–1BB costimulation can be used.
CAR tonic signaling attributes negative effects in T cells and in particular causes poor antitumor effects due to rapid exhaustion (6). Our data in vitro and in vivo support the notion that CAR tonic signaling causes impaired antitumor activity. In our xenograft models, correction of the stability of the scFv and abrogation of the CAR tonic signaling significantly enhanced the antitumor effects of CAR-T cells encoding the CD28 endodomain. This observation is in line with previous work suggesting that 4–1BB slows antitumor effects and can be mechanistically related to the 4–1BB-mediated recruitment of phosphatases within the CAR synapse (33;34). We propose that tonic signaling, defined as the spontaneous release of IFNγ, by CAR-T cells due to self-aggregation of the scFv should be considered as a distinct phenomenon from the spontaneous proliferation of CAR-T cells (43). The latter may be a positive attribute of the CAR-T cells that persist in the absence of an immediate antigen stimulation, especially in patients with solid tumors. Spontaneous proliferation and enhanced survival of CAR-T cells encoding 4–1BB may be due to sustained NF-κB signaling, rather than proximal CAR signaling, and this phenomenon requires further studies to be fully mechanistically defined (42;45;46).
Overall, we demonstrated that tonic signaling of a CAR was due to self-aggregation of an unstable scFv and that it could be abrogated by stabilization of the FWRs of scFv obtained by amino acid substitutions or humanization. Correction of the scFv stability and elimination of CAR tonic signaling enhanced the antitumor effects of CAR-T cells.
Supplementary Material
Acknowledgments
The Microscopy Services Laboratory, Department of Pathology and Laboratory Medicine, is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. This work was supported by R01 CA193140 (GD), 1R35 GM134864 (NVD), UL1 TR002014 (NVD), R35 GM131923 (BK), RO1DE028172 (SF), RO3CA239193 (SF), and RO3CA216114 (SF) from National Institutes for Health, Passan Foundation (NVD), Department of Defense W81XWH-16-1-0500 (SF) and Il Fondo di Gio Onlus (SP). GD also received support from Cell Medica. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Sequences of the 763.74 monoclonal antibody and its humanized form have been submitted as provisional patent.
Footnotes
Disclosure of Potential Conflicts of Interest
Drs Dotti, Savoldo and Ferrone hold patents in the field of T cell engineering and have sponsor research agreements with Bluebird Bio and Bellicum Pharmaceutical. Dr. Dotti serves in the scientific advisory board of Bellicum Pharmaceutical and Catamaran.
Reference List
- 1.Eshhar Z, Waks T, Gross G, and Schindler DG 1993. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. U. S. A 90:720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finney HM, Lawson AD, Bebbington CR, and Weir AN 1998. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol 161:2791–2797. [PubMed] [Google Scholar]
- 3.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, and Campana D 2004. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18:676–684. [DOI] [PubMed] [Google Scholar]
- 4.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al. 2014. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med 371:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dotti G, Gottschalk S, Savoldo B, and Brenner MK 2014. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev 257:107–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR et al. 2015. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med 21:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worn A, and Pluckthun A 2001. Stability engineering of antibody single-chain Fv fragments. J Mol. Biol 305:989–1010. [DOI] [PubMed] [Google Scholar]
- 8.Young NM, MacKenzie CR, Narang SA, Oomen RP, and Baenziger JE 1995. Thermal stabilization of a single-chain Fv antibody fragment by introduction of a disulphide bond. FEBS Lett. 377:135–139. [DOI] [PubMed] [Google Scholar]
- 9.Miller BR, Demarest SJ, Lugovskoy A, Huang F, Wu X, Snyder WB, Croner LJ, Wang N, Amatucci A, Michaelson JS et al. 2010. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng Des Sel 23:549–557. [DOI] [PubMed] [Google Scholar]
- 10.Kugler M, Stein C, Schwenkert M, Saul D, Vockentanz L, Huber T, Wetzel SK, Scholz O, Pluckthun A, Honegger A et al. 2009. Stabilization and humanization of a single-chain Fv antibody fragment specific for human lymphocyte antigen CD19 by designed point mutations and CDR-grafting onto a human framework. Protein Eng Des Sel 22:135–147. [DOI] [PubMed] [Google Scholar]
- 11.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, and Dotti G 2010. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 24:1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pellegatta S, Savoldo B, Di IN, Corbetta C, Chen Y, Patane M, Sun C, Pollo B, Ferrone S, DiMeco F et al. 2018. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borras L, Gunde T, Tietz J, Bauer U, Hulmann-Cottier V, Grimshaw JP, and Urech DM 2010. Generic approach for the generation of stable humanized single-chain Fv fragments from rabbit monoclonal antibodies. J Biol. Chem 285:9054–9066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, and Savoldo B 2017. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther 25:580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landoni E, Smith CC, Fuca G, Chen Y, Sun C, Vincent BG, Metelitsa LS, Dotti G, and Savoldo B 2020. A High-Avidity T-cell Receptor Redirects Natural Killer T-cell Specificity and Outcompetes the Endogenous Invariant T-cell Receptor. Cancer Immunol. Res 8:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rezacova P, Lescar J, Brynda J, Fabry M, Horejsi M, Sedlacek J, and Bentley GA 2001. Structural basis of HIV-1 and HIV-2 protease inhibition by a monoclonal antibody. Structure. 9:887–895. [DOI] [PubMed] [Google Scholar]
- 17.Webb B, and Sali A 2016. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci 86:2. [DOI] [PubMed] [Google Scholar]
- 18.Kota P, Ding F, Ramachandran S, and Dokholyan NV 2011. Gaia: automated quality assessment of protein structure models. Bioinformatics. 27:2209–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding F, Tsao D, Nie H, and Dokholyan NV 2008. Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure. 16:1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shirvanyants D, Ding F, Tsao D, Ramachandran S, and Dokholyan NV 2012. Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. J Phys. Chem. B 116:8375–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dokholyan NV, Buldyrev SV, Stanley HE, and Shakhnovich EI 1998. Discrete molecular dynamics studies of the folding of a protein-like model. Fold. Des 3:577–587. [DOI] [PubMed] [Google Scholar]
- 22.Yin S, Ding F, and Dokholyan NV 2007. Eris: an automated estimator of protein stability. Nat. Methods 4:466–467. [DOI] [PubMed] [Google Scholar]
- 23.Yin S, Biedermannova L, Vondrasek J, and Dokholyan NV 2008. MedusaScore: an accurate force field-based scoring function for virtual drug screening. J Chem. Inf. Model 48:1656–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu C, Dukhovlinova E, Council O, Ping L, Faison EM, Prabhu SS, Potter EL, Upton SL, Yin G, Fay JM et al. 2019. Rationally designed carbohydrate-occluded epitopes elicit HIV-1 Env-specific antibodies. Nat. Commun 10:948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dagliyan O, Tarnawski M, Chu PH, Shirvanyants D, Schlichting I, Dokholyan NV, and Hahn KM 2016. Engineering extrinsic disorder to control protein activity in living cells. Science 354:1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dagliyan O, Shirvanyants D, Karginov AV, Ding F, Fee L, Chandrasekaran SN, Freisinger CM, Smolen GA, Huttenlocher A, Hahn KM et al. 2013. Rational design of a ligand-controlled protein conformational switch. Proc. Natl. Acad. Sci. U. S. A 110:6800–6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geldres C, Savoldo B, Hoyos V, Caruana I, Zhang M, Yvon E, Del VM, Creighton CJ, Ittmann M, Ferrone S et al. 2014. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin. Cancer Res 20:962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reinhold U, Liu L, Ludtke-Handjery HC, Heuser C, Hombach A, Wang X, Tilgen W, Ferrone S, and Abken H 1999. Specific lysis of melanoma cells by receptor grafted T cells is enhanced by anti-idiotypic monoclonal antibodies directed to the scFv domain of the receptor. J Invest Dermatol. 112:744–750. [DOI] [PubMed] [Google Scholar]
- 29.Correa AL, Senna JP, and de Sousa AP 2016. Effects of passage number on growth and productivity of hybridoma secreting MRSA anti-PBP2a monoclonal antibodies. Cytotechnology 68:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xin H, and Cutler JE 2006. Hybridoma passage in vitro may result in reduced ability of antimannan antibody to protect against disseminated candidiasis. Infect. Immun 74:4310–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, Liu E, Carrum G, Kamble RT, Gee AP et al. 2016. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J. Clin. Invest 126:2588–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, Bilgi M, Wu MF, Liu H, Grilley B et al. 2017. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin. Invest 127:3462–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, Plotkin J, and Sadelain M 2015. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun C, Shou P, Du H, Hirabayashi K, Chen Y, Herring LE, Ahn S, Xu Y, Suzuki K, Li G et al. 2020. THEMIS-SHP1 Recruitment by 4–1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 37:216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun M, Shi H, Liu C, Liu J, Liu X, and Sun Y 2014. Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res. 16:R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A et al. 2015. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med 7:275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ewert S, Honegger A, and Pluckthun A 2004. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods 34:184–199. [DOI] [PubMed] [Google Scholar]
- 38.Myers DR, Zikherman J, and Roose JP 2017. Tonic Signals: Why Do Lymphocytes Bother? Trends Immunol. 38:844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hochweller K, Wabnitz GH, Samstag Y, Suffner J, Hammerling GJ, and Garbi N 2010. Dendritic cells control T cell tonic signaling required for responsiveness to foreign antigen. Proc. Natl. Acad. Sci. U. S. A 107:5931–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krokhotin A, Du H, Hirabayashi K, Popov K, Kurokawa T, Wan X, Ferrone S, Dotti G, and Dokholyan NV 2019. Computationally Guided Design of Single-Chain Variable Fragment Improves Specificity of Chimeric Antigen Receptors. Mol. Ther. Oncolytics 15:30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Redler RL, Das J, Diaz JR, and Dokholyan NV 2016. Protein Destabilization as a Common Factor in Diverse Inherited Disorders. J Mol. Evol 82:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, Cabral JMS, Rooney CM, Orange JS, Brenner MK et al. 2017. Tonic 4–1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 21:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD Jr. et al. 2015. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res 3:356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, Brenner MK, Leen AM, and Vera JF 2016. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. 5:e1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, Wang X, Guan L, Beatty N, Abate-Daga D et al. 2018. 4–1BB enhancement of CAR T function requires NF-kappaB and TRAFs. JCI. Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Philipson BI, O’Connor RS, May MJ, June CH, Albelda SM, and Milone MC 2020. 4–1BB costimulation promotes CAR T cell survival through noncanonical NF-kappaB signaling. Sci. Signal 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.