

Abstract
Ewing sarcoma (ES) is the second most common pediatric bone cancer, with a 5-year survival rate for metastatic disease of only 20%. Recent work indicates that survival is strongly correlated with high levels of tumor infiltrating lymphocytes (TIL), whose abundance is associated with interferon (IFN)-inducible chemokines CXCL10 and CCL5. However, the tumor-intrinsic factors that drive chemokine production and TIL recruitment have not been fully elucidated. We previously showed that ubiquitin-specific protease 6 (USP6) directly deubiquitinates and stabilizes Jak1, thereby inducing an IFN signature in ES cells. Here we show this gene set comprises chemokines associated with immunostimulatory, anti-tumorigenic functions, including CXCL10 and CCL5. USP6 synergistically enhanced chemokine production in response to exogenous IFN by inducing surface upregulation of IFNAR1 and IFNGR1. USP6-expressing ES cells stimulated migration of primary human monocytes and T lymphocytes and triggered activation of natural killer (NK) cells in vitro. USP6 inhibited ES xenograft growth in nude but not NSG mice and was accompanied by increased intra-tumoral chemokine production and infiltration and activation of NK cells, dendritic cells, and macrophages, consistent with a requirement for innate immune cells in mediating the anti-tumorigenic effects of USP6. High USP6 expression in ES patients was associated with chemokine production, immune infiltration, and improved survival. This work reveals a previously unrecognized tumor suppressive function for USP6, which engenders an immunostimulatory microenvironment through pleiotropic effects on multiple immune lineages. This further raises the possibility that USP6 activity may be harnessed to create a "hot" tumor microenvironment in immunotherapy.
Keywords: USP6, TRE17, Ewing Sarcoma, Interferon, CXCL10, CCL5, chemokines, tumor microenvironment
INTRODUCTION
Ewing sarcoma (ES) is the second most common bone cancer, predominantly affecting the pediatric population (1, 2). While patients with localized disease have a five-year survival rate of ~75%, metastatic and recurrent patients have a dismal survival of 20%. This low survival rate reflects the inefficacy of standard therapy, which includes radiation, surgery, and chemotherapy. Patient burden is exacerbated by the lack of markers that can reliably predict recurrence. ES is driven by pathognomonic translocations between the RNA-binding protein EWS and Ets family transcription factors, most commonly FLI1. Agents targeting EWS-FLI1 or its downstream effectors have failed to yield effective treatments in recent clinical trials (3, 4). Furthermore, immunotherapy has had limited success in ES, in part because the immune tumor microenvironment (TME) is poorly characterized (5).
ES, like other sarcomas, has generally been regarded as immunologically “cold,” with few tumor infiltrating T lymphocytes (TILs) (5-7). However, recent studies reveal that TILs actually vary significantly, and that high CD8+ TIL levels are associated with greatly improved overall survival (8, 9). In one study, survival was ~40% at 55 months vs. ~90% at 100 months for patients with low vs. high CD8+ TIL levels, respectively (8). Macrophage infiltration and polarization has also been found to affect ES pathogenesis and progression (7, 10), though the factors contributing to these effects are unknown. Thus, elucidating the mechanisms of TIL recruitment and immunomodulation of the TME represents a window of opportunity for preventing recurrence and improving longterm survival. In the aforementioned study, expression profiling of chemokines revealed that CD8+ TIL abundance correlated with production of the interferon (IFN) γ-inducible factors CXCL9/CXCL10 and CCL5 (8). Receptors for these chemokines (CXCR3 and CCR5, respectively) are highly expressed on CD8+ TILs, other cytotoxic immune cells such as natural killer (NK) cells, and myeloid lineages (11-13). Collectively, these immune features (IFNγ response, CXCL9/10 production, and high CD8+ TIL recruitment) define a "hot" TME, and predict both improved overall survival and response to checkpoint inhibitor immunotherapy across multiple cancers (14, 15). However, the tumor-intrinsic factors that confer this favorable immune phenotype, in ES or any other cancer, are largely unknown.
We recently identified ubiquitin-specific protease 6 (USP6) as a novel regulator of IFN signaling in ES. USP6 functions by directly de-ubiquitinating Jak1, leading to its stabilization and activation, with resultant phosphorylation of the STAT1 and STAT3 transcription factors (16, 17). We found that USP6 is sufficient to trigger an IFN response gene signature, both in ES cells and other neoplastic models (17). USP6 further renders ES cells hypersensitive to exogenous Type I and II IFNs, as reflected by heightened and prolonged STAT1 phosphorylation, and synergistic induction of IFN-stimulated genes (ISGs). However, how USP6 and its activation of IFN signaling impact ES pathogenesis remains unexplored. Indeed, IFNs can exert either anti- or pro-tumorigenic effects depending on the cancer context (18-20).
To date, only pro-tumorigenic functions have been ascribed to USP6. USP6 is the key etiologic agent in several benign mesenchymal neoplasms, including nodular fasciitis (NF) and aneurysmal bone cyst (ABC), where it undergoes promoter-swapping translocations that result in overexpression of wild type USP6 (21). We have developed cellular and animal models of NF and ABC, and shown that overexpression of USP6 in candidate cells of origin (fibroblasts and pre-osteoblasts) is sufficient to induce formation of tumors that recapitulate molecular, histological and clinical features of NF and ABC (22, 23). In these models, activation of STAT3, NF-κB, and β-catenin by USP6 was essential for tumorigenesis, as genetic or pharmacologic inhibition of these pathways significantly attenuated xenograft growth (16, 23, 24). In addition, we recently reported that an IFN signature is triggered by USP6 in NF, both in primary patient samples and in our cellular model (17). How IFN signaling impacts NF pathogenesis is completely unknown. It is of interest to note, however, that NF lesions invariably regress, although the mechanism underlying this spontaneous regression remains unexplored.
In the current study, we investigated the effects of USP6 and IFN signaling on ES tumorigenesis, and uncovered an unexpected tumor inhibitory function for USP6. In ES cells in vitro, USP6 was capable of directly inducing expression of immunostimulatory chemokines, and also mediated their synergistic induction upon treatment with exogenous IFNs. Conditioned medium from USP6-expressing ES cells was competent to enhance migration of multiple immune cell lineages, and co-culturing enhanced activation of NK cells and primary monocytes-derived lineages in vitro. Furthermore, USP6 inhibited ES tumor growth in xenografted nude mice, coincident with increased intra-tumoral chemokine production, and immune cell infiltration and activation. Consistent with this anti-tumorigenic effect in mice, we found that high USP6 mRNA expression was associated with immune cell infiltration and significantly improved event-free and overall survival in ES patients. Together, our studies provide the first demonstration of a tumor suppressive function for USP6, and reveal that it functions to promote an immunogenic tumor microenvironment through enhanced IFN signaling.
MATERIALS AND METHODS
Cell Lines
RD-ES, A673, SK-N-MC, and TC-71 Ewing sarcoma cell lines and their HA-USP6-expressing and Jak1/STAT1 CRISPR derivatives were cultivated as previously described (17). NK92 was from ATCC. Cell line identity was confirmed by STR analysis and EWS-FLI1 expression. All cells were screened for mycoplasma every 2-4 weeks. Primary human monocytes and T cells from peripheral blood were obtained from the Human Immunology Core at the University of Pennsylvania. Monocytes, CD4+ and CD8+ T cells from de-identified healthy adult human donors were purified from apheresis material by negative selection using RosetteSep Human Monocyte, CD4+ or CD8+ Enrichment Cocktails (StemCell Technologies) according to manufacturer instructions.
Reagents
Reagents were obtained from: Doxycycline, ClonTech (#8634-1); Jak Inhibitor I (#420099) and PS-1145 (P6624), Sigma. IFN α and β were from PBL Assay Science (#11410-2 and #11200-1), and IFNγ from PeproTech (#300-02). Lipofectamine 2000 was from Life Technologies. Flow antibodies/reagents were from Biolegend: Zombie UV (#423107), HA (#901526), IFNAR1 (#21370-3), CD11b (#101208), MHCII (I-A/I-E) (#107620), CD86 (#105036), CD11c (#117318), CD45 (#103116 or #103128), F4/80 (#123116), Ly6c (#128033), Ly6g (#127622), mouse Fc Trustain block (#101319), NK1.1 (#108724), anti-mouse CD69 (#104514) and CD25 (#102036). IFNGR (10338-MM05-A) was from Sino Biologic.
Dual Luciferase Assay
CXCL10 promoter reporter constructs were generously provided by Dr. David Proud (University of Calgary). Reporters were co-transfected with constructs encoding renilla luciferase and USP6 into HeLa cells; IFNγ was added, and dual luciferase assays were performed the following day (Promega Dual-Luciferase #E1910). Firefly luciferase/renilla ratio was calculated, then expressed relative to the value for cells expressing the WT promoter with USP6 and treated with IFNγ.
ELISA and Chemokine antibody array
Semi-quantitative chemokine antibody array was purchased from RayBiotech (Array #G5-4). USP6/RD-ES cells were serum starved, with or without dox and IFNγ (50 ng/mL) for 24 hours. The antibody array was incubated with CM overnight at 4C, and processed per manufacturer instructions. Signal intensities were quantified using LI-COR Odyssey.
For the CXCL10 ELISA, USP6/RD-ES or USP6/A673 cells were seeded in growth medium, and treated with or without dox and IFNγ (5ng/ml). CM was collected the following day and subjected to ELISA (Biolegend, #439904) per manufacturer instructions. All samples were analyzed in technical triplicate; data represent n=3 for USP6/RD-ES, and n=2 for USP6/A673.
Migration assays
CM was prepared by starving cells in serum-free medium with or without dox and IFNγ (50ng/ml) for 24 hours. Immune cells (2.5-5.0E5/well in 25ul serum-free medium containing 0.1% bovine serum albumin) were seeded in Transwell chambers (Neuroprobe #101; 5 micron pore size). CM or control medium was dispensed into the distal chamber, and cells were allowed to migrate for 2 hours at 37C. The upper side of the filter was washed three times with PBS to remove non-migrated cells; the plate/filter was then centrifuged at low speed, and cells in the bottom chamber counted either by flow cytometry, or manually using a hemocytometer. All samples were assayed in technical triplicate, for n=3-5 experiments.
NK92 killing and activation assays
HA-USP6/RD-ES (which express GFP) and NK92 were co-cultured at the indicated ratios overnight in the presence of 200 U/mL IL-2 (Biolegend # 589108). To monitor killing, viable HA-USP6/RD-ES cells were quantified by flow cytometry, monitoring GFP+/propidium iodide-excluded cells. To monitor NK92 activation and degranulation, surface levels of CD69 and CD107a were quantified in the CD45+/GFP- population by flow cytometry. RNA was also isolated from the co-cultures and subjected to RT-qPCR.
Mouse xenografts
All procedures were performed under Institutional Animal Care and Use Committee–approved protocols. J:NU mice (4-8 weeks; Jackson labs) were pre-treated with drinking water containing dox (BioWorld #40410005-2; 1 mg/mL) and 2.5% sucrose for 5-7 days prior to xenograft. Mice were subcutaneously injected with indicated cells (2.5E6) resuspended 1:1 in PBS:Matrigel (Corning #356234). Dox water was changed twice weekly for the duration of the experiment. Two protocols were used to determine effects of USP6 on tumor growth. In the first, the primary goal was to document effects on terminal mass at a common timepoint, and mice were sacrificed approximately 3-4 weeks after xenografting. In the second, the goal was to assess the time required to reach a defined volume (2000 mm3). Tumors were measured 2-3 times per week using digital calipers. Tumor volume was calculated [Volume=(π/6)(Length*Width2)], where length is the longest measurement and width the shortest.
Tumor immune infiltration and flow cytometry analysis
Following tumor excision, representative sections of tumor were digested with Miltenyi tumor dissociation kit (#130-096-730). Digested samples were subjected to surface staining for the indicated markers. Cells were resuspended in Cell Staining Buffer (Biolegend #420201) and blocked with either mouse or human Fc Trustain block (Biolegend #101319 or #422302) at RT in the dark. Staining with antibodies was subsequently performed at 4C in the dark for 20 minutes. For experiments monitoring intracellular HA-USP6, after surface staining of the indicated markers, cells were washed fixed in 4% formalin for 20 minutes at RT in the dark. Cells were then permeablized with Biolegend Intracellular Staining Permeabilization Wash Buffer (#421002), followed by staining with anti-HA for 20 minutes at RT in the dark. Samples were washed and resuspended in Cell Stain Buffer. The HA-negative gate was set using HA-USP6 ES cells without dox treatment, which lack expression of HA-USP6. Data was collected using LSR II or Cytoflex flow cytometers, and analyzed with FlowJo.
Statistical and Pathway analysis
All data represent results from at least 3 biological replicates. GSEA and microarray analysis was performed as previously described (17). GO datasets were acquired from https://www.gsea-msigdb.org/gsea/msigdb/index.jsp. For the Kaplan-Meier analysis, only the primary untreated patient samples in GSE17679 were included. To assess immune infiltration in primary ES samples, the CIBERSORT algorithm (29) was adapted by curating the gene lists for each immune lineage, then applying GSEA to compare enrichment of each lineage in USP6high vs USP6low ES patients. Ratio-paired t-test was used to determine significance (p<0.05) for all samples that were comparing the effects of +dox or +IFNγ to +IFNγ+dox. All other comparions were done using paired t-test.
RESULTS
Chemokine production is associated with high USP6 expression in multiple human neoplasms in vivo, and is directly induced by USP6 in vitro
We recently reported that USP6 is associated with an IFN response gene signature in primary patient samples of Ewing sarcoma (ES), nodular fasciitis (NF), and germ cell tumors. Among the genes most consistently associated with high USP6 expression across these neoplasms were chemokines, i.e. chemotactic cytokines. In two independent ES patient datasets, high USP6 expression was strongly associated with multiple genesets for leukocyte chemotaxis and chemokine signaling (Supplemental Figure 1). Across all four genesets, ligands for the chemokine receptors CXCR3 (CXCL9 and CXCL10) and CCR5 (CCL4, CCL5, and CCL8) emerged within the core enrichment set (Supplemental Figure 2). These same genesets and chemokines were also found to be associated with high USP6 expression in germ cell tumors and in USP6-translocated NF (Supplemental Figure 3A), suggesting that USP6 may play a direct role in their induction.
To determine if USP6 was capable of directly triggering expression of these chemokines, studies were performed in ES cell lines inducibly expressing USP6 (17). As previously described, the USP6 locus is highly methylated in all immortalized cell lines examined, resulting in its silencing and necessitating generation of cell lines ectopically expressing USP6 in a doxycyclin (dox)-inducible manner, at physiologic levels (17). In the patient-derived ES cell line RD-ES, upon addition of dox, USP6 triggered the same chemotaxis and chemokine signatures as was observed in the USP6high primary ES tumors (Supplemental Figure 3B). Together, these results demonstrate that high USP6 expression is associated with chemokine production across multiple tumor types in patients, and that USP6 is capable of directly inducing their expression in vitro.
USP6 synergistically enhances chemokine induction upon IFN treatment in vitro
We previously reported that USP6 not only induces an IFN gene signature by itself, but that it also magnifies the response to exogenous Type I and II IFNs (17). Among the genes with the greatest synergistic induction were ligands for CXCR3 and CCR5: USP6 or IFNα individually led to modest increases in levels of these chemokines, whereas expression was increased 2-4 orders of magnitude in USP6-expressing cells treated with IFNα (Supplemental Figure 3B). The chemokines exhibiting the greatest synergistic induction were CXCL9/CXCL10/CXCL11 and CCL4/CCL5/CCL8/CCL20. As mentioned above, CXCL10 and CCL5 production was correlated with CD8+ TIL recruitment and improved survival in ES patients, and CXCL10 has been associated with TIL recruitment and improved prognosis across multiple other malignancies (8, 11, 12, 14). USP6's effect on these chemokines was highly selective, as it did not impact expression of chemokines with pro-tumorigenic, immuno-inhibitory functions (13) (Supplemental Figure 3B).
These RNA-seq results were validated by RT-qPCR in USP6/RD-ES, focusing on CXCL10 and CCL5 as representative ligands for CXCR3 and CCR5, respectively. Induction of USP6, or treatment with either IFNα, IFNβ, or IFNγ individually modestly induced CXCL10 and CCL5 expression, while all three IFNs elicited synergistic induction of these chemokines in the presence of USP6 (Figures 1A/B). To further characterize this IFN-based synergy, we focused on Type II IFNγ since our previous work showed that Type I IFNs (particularly IFNβ) induce apoptosis of USP6-expressing RD-ES through activation of the TRAIL pathway, thus complicating analysis (17). We tested a range of IFNγ doses and found that USP6 amplified chemokine expression at all doses: at a low dose of 0.5 ng/ml, IFNγ had no effect on control RD-ES cells, but induced a 10,000-fold increase in CXCL10 and 100-fold increase in CCL5 expression in USP6/RD-ES (Figure 1B). At a 100-fold higher dose of IFNγ (50 ng/ml), USP6 still enhanced CXCL10 and CCL5 expression more than 2 orders of magnitude (Figure 1B). Similar effects were observed in multiple independent patient-derived ES cell lines, including A673, TC71, and SK-N-MC cells, though the specific chemokines induced and the magnitude of the effect varied between the cell lines (Figure 1C and Supplemental Figure 4A). The extent of induction was also dependent on USP6 dosage as RD-ES cells expressing higher levels of USP6 exhibited greater CXCL10 induction, both basally and in response of IFNγ (Supplemental Figure 4B). Furthermore, screening of conditioned medium (CM) by ELISA (for CXCL10) or a chemokine antibody array (for CCL4/5/8/20) confirmed that synergy of induction could be observed at the protein level (Figure 1D/E and Supplemental Figure 4C). Together, these data suggest that USP6 serves to fine tune and amplify the cellular response to IFNs, and that it could play a pivotal role in modulating the immune TME through production of chemokines.
Figure 1: USP6 elicits synergistic induction of IFN-regulated chemokines in ES cells.
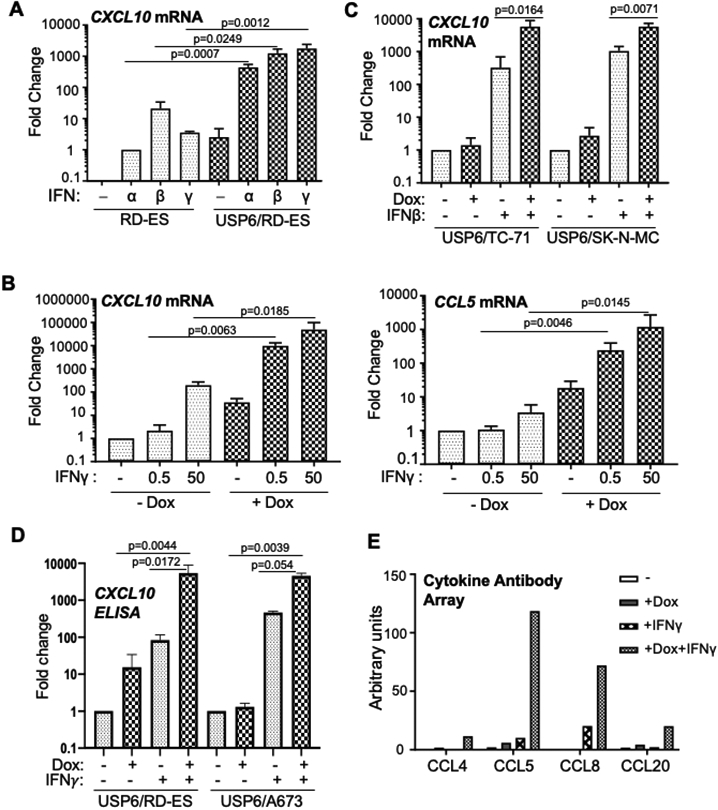
A-C) RT-qPCR was performed for cells treated as indicated. A) Parental RD-ES or USP6(Pool)/RD-ES were treated with dox and the indicated IFN overnight. B) USP6(Pool)/RD-ES were treated with dox and IFNγ overnight. C) ES cell lines TC71 and SK-N-MC were treated as indicated. D) Cells were treated with dox and IFNγ (5 ng/ml) as shown, and CM subjected to CXCL10 ELISA (RD-ES n=3, A673 n=2). See Supplemental Figure 2C for absolute concentrations. E) CM from USP6/RD-ES treated as indicated was used to probe a RayBiotech chemokine antibody array, followed by LI-COR detection. Results represent data from probing a single set of cytokine arrays (n=1).
Synergistic chemokine induction is mediated through Jak1-STAT1
We next explored the mechanisms underlying the synergistic chemokine induction, focusing predominantly on CXCL10, given its strong association with TIL recruitment across cancers. Jak1-STAT1 and NF-κB have previously been shown to regulate expression of CXCL10 and CXCL9 (25). CRISPR deletion of Jak1 or STAT1, but not STAT3, dramatically suppressed the synergistic induction of CXCL10 and CXCL9 by USP6+IFNγ (Figure 2A)(17)). As an alternative means of probing their contributions, pharmacological inhibitors of the pathways were utilized. Induction was almost completely suppressed by a pan-Jak inhibitor, and partially suppressed by the NF-κB inhibitor PS1145 (Figure 2B), indicating that Jak1-STAT1 assumes a dominant role in CXCL10 induction.
Figure 2: USP6-regulated chemokines are induced by Jak-STAT1/NF-κB and can stimulate immune cell migration.

A) USP6/RD-ES with the indicated CRISPR deletions were treated with dox and IFNγ (50 ng/ml), and CXCL9/10 expression was quantified. B) USP6(Pool)/RD-ES were treated with dox and IFNγ overnight, in the presence of PS1145 (15 μM) and Jak inhibitor (1 μM) as indicated, and CXCL9/10 expression quantified. C) Firefly (FF) luciferase driven by the CXCL10 promoter (WT or mutants in the indicated response elements) were co-transfected with renilla luciferase and USP6-encoding vectors. Cells were treated overnight with IFNγ, then subjected to dual luciferase assays. FF/renilla ratios relative to WT (black bar) were calculated; all p values are in comparison to this sample. Transwell migration assays were performed on THP-1 (D), and primary monocytes (E). Migration was monitored after 2 hr in response to serum-free conditioned medium (CM) from USP6/RD-ES cells treated as indicated, control serum-free medium, or recombinant CCL5 (10 ng/ml), CXCL10 (250 ng/ml), or 1% fetal bovine serum (FBS).
To further dissect the contributions of these pathways, we utilized a luciferase reporter construct driven by the proximal CXCL10 promoter comprising 972 base pairs upstream of the transcription start site (26). USP6+IFNγ cooperatively activated this reporter in transiently transfected HeLa cells, although the effects were much less pronounced than the dramatic synergy observed with endogenous CXCL10 (Figure 2C). The reason for this reduced response is unknown, but possibilities are that cis elements outside of the proximal promoter are required, or that the transiently transfected reporter is not properly chromatinized in a manner required for synergy. Irrespective of the cause, we probed the contribution of various response elements within the promoter, which include two STAT1 homo-dimer binding sites, two NF-κB sites, an AP-1 site, and an interferon-sensitive response element (ISRE), which binds to the STAT1-STAT2-IRF9 complex. As shown in Figure 2C, the first STAT1 site, both NF-κB sites, and the ISRE all contributed to activation of the CXCL10 promoter by USP6+IFNγ, whereas the AP-1 site was largely dispensable.
Conditioned medium from USP6 /ES cells induces enhanced immune cell migration
The array of chemokines induced by USP6 have pleiotropic functions, including the ability to stimulate migration of myeloid, NK and T cells (13, 27). To assess their functionality, transwell assays were performed on both primary and immortalized immune cells. CM from RD-ES or A673 cells either expressing USP6 or treated with IFNγ induced a modest increase in migration of THP-1, a monocytic cell line (Figure 2D) and primary human monocytes (Figure 2E). However, CM from USP6-expressing cells treated with IFNγ triggered a cooperative 20-100-fold increase in migration. While it remains to be determined which specific chemokines/factors in this complex cocktail are responsible, it is likely that multiple factors function in concert. Indeed, while recombinant CXCL10 (reCXCL10) alone was a poor chemoattractant for THP-1, when combined with CCL5 a cooperative induction was observed (Figure 2D). Enhanced migration was also induced in primary human CD8+ T cells, although the magnitude of enhancement was not as pronounced (Supplemental Figure 5). We also tested whether CM could stimulate migration of NK cells using the NK92 cell line. However, contrary to prior reports we did not observe significant migration even with reCXCL10, perhaps due to low levels of CXCR3 in our isolates. Nevertheless, in sum these data confirm that USP6 expression in ES cells can stimulate immune cell chemotaxis, both by itself and in the presence of ectopic IFNγ.
We also probed whether hyper-responsiveness of USP6-expressing cells to IFN might arise from effects on IFN receptor (IFNR) levels. As shown in Figure 3A, levels of the IFNGR1 were elevated in the presence of dox, both under basal and IFNγ-stimulated conditions. RT-qPCR confirmed that this effect was post-transcriptional (Figure 3B). Flow cytometry revealed that surface IFNGR1 was significantly increased upon expression of HA-tagged USP6 (Figure 3C/D). Similar effects were seen with the Type I IFN receptor, IFNAR1 (Supplemental Figure 6A-C). Thus, elevated levels of the IFNRs and Jak1 likely both contribute to the hyper-responsiveness of USP6-expressing cells to IFNs.
Figure 3: USP6 increases IFNGR1 surface expression.

A) Top, cells were treated with dox and IFNg as shown, lysed, then subjected to western for IFNGR1 and b-catenin (as a loading control). Bottom, quantification of IFNGR1 protein levels, normalized against b-catenin, using LI-COR. B) RT-qPCR to quantify IFNGR1 mRNA. C) Surface IFNGR1 was quantified by flow cytometry in cells treated with or without dox. USP6/A673 and USP6/RD-ES are pooled cell lines that express heterogenous levels of HA-USP6; in dox-treated samples, IFNGR1 levels were assessed in the HA+ vs. HA− populations. IFNGR1 CRISPR knockout cells were used as an antibody specificity control. D) Quantification of IFNGR1 flow results in C.
USP6 inhibits ES xenograft growth concomitant with enhanced intra-tumoral chemokine production and immune cell infiltration and activation
These results prompted us to investigate USP6's effects on ES tumor growth and the immune TME. Immunocompetent genetically engineered mouse models that accurately recapitulate human ES disease are lacking (28). The field is thus limited to xenografting patient-derived cell lines into immunodeficient strains, such as nude or NSG mice. We first subcutaneously xenografted into nude mice clonal RD-ES cell lines expressing high or intermediate levels of USP6 (USP6(high) and USP6(med), respectively)(17), in the absence or presence of dox in the drinking water. USP6 inhibited growth of RD-ES xenografts in a dose-dependent manner (Figure 4A). Although the effects did not reach statistical significance, there was a clear trend toward suppression of tumor growth by USP6. Indeed, only one out of five animals developed a tumor in the dox-treated USP6(high)/RD-ES cohort. In striking contrast, USP6 had no effect on RD-ES tumor growth when xenografted into NSG mice (Figure 4B). Since the key difference between nude and NSG mice is that the latter lack NK cells and functional macrophages/DCs, this strongly suggests that the immune system plays a significant role in mediating the anti-tumorigenic effect of USP6 in nude mice. Indeed, these results indicate that in the absence of NK cells and macrophages/DCs, USP6 exerts no discernable cell-autonomous inhibitory effect on ES tumor growth in NSG mice.
Figure 4: USP6 inhibits growth of RD-ES xenografts in nude mice coincident with enhanced chemokine production and immune cell infiltration in vivo.

A) Nude mice were subcutaneously injected with clonal RD-ES cell lines expressing high or medium levels of USP6 (USP6(high) and USP6(med), respectively (17)), and maintained on dox or control water. Tumors were harvested after approximately 4 weeks; terminal masses are indicated for each mouse. Though dox treatment yielded a clear inhibitory trend in nude mice, this difference did not reach statistical significance. B) USP6/RD-ES were subcutaneously xenografted into NSG mice and terminal mass determined after approximately 4 weeks; differences in terminal mass were not statistically different. C) RT-qPCR was performed on tumors, normalizing against GAPDH. D) USP6(med) tumors were digested and surface flow cytometry performed using antibodies against: CD45 (pan-leukocyte), CD11b (pan-myeloid), CD11c (dendritic cell), F4/80 (macrophage), Ly6c (monocyte), and Ly6g (granulocyte). The percent of cells positive for the indicated marker relative to the total live cells in the bulk tumor is shown. See Supplemental Figures 8/9 for gating strategy.
RT-qPCR of tumors confirmed that USP6 induced expression of CXCL9, CXCL10, and CCL5 in vivo (Figure 4C). To investigate whether the increased immune infiltrates were associated with inhibition of tumorigenesis, tumors were isolated from the USP6(med)/RD-ES cohorts, dispersed, then analyzed by flow cytometry (tumors in the USP6(high)/RD-ES cohorts were too small to obtain sufficient material for analysis). Strikingly, USP6 significantly increased total infiltrated immune cells, as monitored by the pan-leukocyte marker CD45 (Figure 4D). Analysis of lineage markers revealed that USP6 increased intra-tumoral abundance of macrophages (F4/80+), DCs (CD11c+), and myeloid cells (CD11b+), but not monocytes (Ly6c+) or granulocytes (Ly6g+) (Figure 4D, and Supplemental Figures 7/8 for gating strategy).
USP6's anti-tumorigenic effects were validated using two additional ES cell lines, A673 (Figure 5) and TC71 (Supplemental Figure 9). For the USP6/A673 cells, 10 days after xenografting, palpable tumors were observed in 90% of control mice but only 50% of the dox-treated cohort (Figure 5A). USP6 prolonged the time to reach maximum tumor volume (2000 mm3) (Figure 5B), with two "strong responders" in the +dox cohort failing to reach this endpoint (labeled with asterisks in Figure 5A). While tumor growth rates of the remaining mice in the +dox cohort ostensibly clustered with the -dox cohort, flow cytometric analysis of the tumors revealed that the former had a significantly increased fraction of dead cells (Figure 5C; gray dots correspond to the strong responders; and Supplemental Figure 8). Furthermore, as with the RD-ES xenografts, we observed significant increases in intratumoral levels of total leukocytes (CD45+), macrophages (F4/80+), and DCs (CD11c+), but not CD11b+ cells (Figure 5C and Supplemental Figures 8/9 for gating strategy). Notably, the two strong responder mice had the highest percentages of dead cells and immune cell infiltrates (gray dots in Figure 5C-E). Markers of antigen-presenting cell (APC) activation were also examined: CD86 (a ligand for the co-stimulatory receptor CD28 on T cells), and MHC Class II (MHCII, which mediates antigen presentation). Among the total live population, cells that were positive for CD86 or MHCII were increased upon USP6 induction (Supplemental Figures 10-12). Expression of these markers was examined in classical macrophages (CD11b+ F4/80+) and DCs (CD11b+ CD11c+). In both lineages, USP6 induced a marked increase in the percentage of cells that were single and double positive for CD86 and MHCII (Figure 5F, and Supplemental Figures 10-12).
Figure 5: USP6 stimulates intra-tumoral infiltration and activation of myeloid lineages.

A) Nude mice were subcutaneously injected with USP6/A673, and maintained on dox or control water. Tumors were measured 3 times weekly until a terminal volume of 2000 mm3 was reached. Asterisks, mice whose tumors failed to reach terminal volume ("strong responders"). B) Time to terminal tumor volume was determined (presented as percent of mice whose tumors have yet to reach terminal volume). C-F) USP6/A673 tumors were digested and flow cytometry was performed using the indicated antibodies. Cell viability was assessed by Zombie-UV exclusion (C). Gray dots correspond to the strong responders (i.e. the mice labeled with asterisks in A). The percent of cells positive for the indicated marker relative to the total live cells in the bulk tumor is shown (see Supplemental Figures 8/9 for gating strategy). For p-values in gray, strong responders were included in the calculation; for p-values in black, they were excluded].
We also examined infiltration of NK cells, the main cytolytic effectors present in nude mice. USP6 increased not only the intratumoral abundance of NK cells (Figure 6A and Supplemental Figure 13), but also their activation and proliferative potential (as monitored by CD69 and CD25/IL2Rα, respectively) (Figure 6A and Supplemental Figure 14). Notably, the two strong responder mice exhibited the highest levels of NK cell infiltration, and CD69 and CD25 surface expression (gray dots in Figure 6A). We were unable to detect enhanced IFNγ mRNA production in vivo, likely due to the low absolute numbers of intra-tumoral NK cells, which are the predicted main source of IFNγ.
Figure 6: USP6 promotes NK activation in vivo, and directly induces NK activation in vitro.

A) Digested USP6/A673 tumors were subjected to flow cytometry. The percent of cells positive for NK1.1+ relative to total live cells in the digested tumor was quantified, as was CD69 and CD25 in the NK1.1+/CD45+ live population. See Supplemental Figures 12/13 for gating strategy. B) NK92 (effector, E) cells were co-incubated with USP6/RD-ES (GFP+) (target, T) cells at various effector E:T ratios overnight, in the absence or presence of dox. Percent survival of the USP6/RD-ES (relative to sample without NK92) was quantified by monitoring the GFP+/propidium iodide-excluded population. C/D) USP6/RDES and NK92 were co-cultured overnight with or without dox, then subjected to (C) flow cytometry to monitor surface CD69 and CD107a on the NK92 (CD45+/GFP−) cells, or (D) RT-qPCR to quantify IFNγ, CXCL9, and CXCL10 expression.
USP6 expression in ES stimulates NK cell activation and enhanced paracrine induction of chemokines
The studies above demonstrate that USP6 is sufficient to increase intratumoral levels and activation of NK cells in vivo. To determine whether USP6 could directly induce their activation in vitro, co-culture assays were performed using the human NK cell line, NK92. NK92 cells were co-cultivated at various ratios with USP6/RD-ES (which express GFP), and survival was quantified by monitoring the GFP+/propidium iodide-excluded population. USP6/RD-ES were sensitive to killing by NK92 in the absence of dox, but addition of dox enhanced death at all effector:target (E:T) ratios (Figure 6B). USP6 expression in RD-ES cells dramatically enhanced NK cell activation and degranulation, as measured by surface levels of CD69 and the degranulation marker CD107a, respectively (Figure 6C). Furthermore, we speculated that since NK cells produce IFNγ upon activation, CXCL9/10 induction would be enhanced in dox-treated co-cultures, due to hypersensitivity of USP6-expressing ES cells to IFNγ. This was confirmed in Figure 6D: co-cultivation of USP6/RDE-ES with NK92 induced IFNγ production (independently of dox), and CXCL9/CXCL10 induction was significantly increased upon USP6 expression.
USP6 expression is associated with increased immune cell infiltration in primary ES tumors and improved patient survival
The ability of USP6 to induce immune infiltration and activation in ES xenografts led us to examine its association with immune infiltrates in human ES tumors using an adaptation of the CIBERSORT algorithm that was recently developed to assess the presence of immune lineages in complex tumor samples (29). This algorithm curates approximately 500 immune markers to define 22 distinct immune lineages/activation states. In two independent ES patient datasets, high USP6 expression was found to be associated with enhanced immune infiltration (Supplemental Figure 15). Remarkably, USP6high tumors exhibited increased abundance of multiple anti-tumorigenic lineages, including CD8+ and CD4+ (resting memory) T cells, and activated dendritic, NK and γδT cells. In contrast, levels of pro-tumorigenic lineages such as M2 macrophages were not associated with high USP6 expression ((Supplemental Figure 15).
We next examined whether high USP6 expression was associated with differential prognosis. When patients were stratified by USP6 levels, comparing the top ~1/3 to the bottom ~2/3, those with high USP6 expression exhibited dramatically improved event-free and overall survival (Figure 7A). After five years, patients with high USP6 levels had a nearly 80% survival rate, whereas those with low expression had a ~40% survival rate. It was not possible to confirm USP6 protein levels due to inavailability of antibodies suitable for immunohistochemistry. Nevertheless, taken together these results suggest that in ES patients, as in our xenograft model, USP6 stimulates immune infiltration and suppresses cancer growth.
Figure 7: USP6 Expression is Associated with Increased Immune Cell Infiltration and Prolonged Survival in ES Patients.

A) Primary untreated ES patient samples (GSE17679) (37) were ranked by USP6 expression, and overall and event-free survival was plotted. USP6high, top ~1/3; USP6low, bottom ~2/3; total patients in dataset n=27. B) Model for establishment of an immunostimulatory, anti-tumorigenic TME by USP6 to drive cancer cell clearance. USP6, via Jak1/STAT1, induces low level production of multiple chemokines (red cloud), leading to recruitment of multiple immune lineages (including DCs, macrophages (mφ), NK cells, and CD8+ cytotoxic T lymphocytes) into the TME. USP6 expression activates NK cells, leading to enhanced killing of USP6+ ES cells (green arrow), and IFNγ production. IFNγ feeds back on USP6-expressing ES cells, which synergistically induce production of chemokines (due to upregulation of IFNAR1, IFNGR, and Jak1 in these cells), and amplifying immune cell recruitment and activation. USP6 also induces activation of DCs/macrophages by stimulating their expression of CD86 and MHCII (white arrow), which we speculate would enhance their ability to activate recruited CD8+ CTLs.
DISCUSSION
Hallmark features of a hot TME are induction of IFN response signature, CXCL10 production, and infiltration of CD8+ T lymphocytes (8, 11, 14). In aggregate, these features predict both improved overall survival and response to checkpoint inhibitors, across dozens of cancer types. Yet, remarkably little is known of why a subset of patients spontaneously exhibits this favorable immune phenotype. This study demonstrates that high USP6 expression is associated with all of these hot TME hallmarks in ES patients, and that USP6 is sufficient to drive an IFN response signature and CXCL10 expression in vitro, as well as intra-tumoral immune cell recruitment and activation in vivo.
Our work further provides mechanistic insights into how USP6 functions, and reveals for the first time an anti-tumorigenic role for USP6 in human malignancy. We show that in the context of ES, USP6 functions to amplify IFN signaling, leading to immune infiltration/activation and inhibition of tumor growth. We envisage that USP6 engenders a potent positive feed-forward immunostimulatory loop as follows (model, Figure 7B): USP6 directly induces Jak1 stabilization/STAT1 activation, leading to low level chemokine production, which we speculate stimulates recruitment of myeloid, NK, and T cells; these factors may also enhance immune cell activation and proliferation. USP6 expression in ES can also directly enhance NK cell activation, and we predict that USP6 would also promote activation of T cells in vivo due to their ability to stimulate their migration as well as activate APCs in the TME. Activated NK and T cells produce IFNγ, which could feed back on the USP6-expressing ES cells to synergistically enhance production of CXCL10 and other chemokines (due to their upregulated expression of IFNAR, IFNGR, and Jak1). Immune cell recruitment and activation would thereby be amplified, with resultant tumor cell elimination. We posit that this potent feedforward response underlies the association of high USP6 expression with immune infiltration and dramatically improved survival in ES patients.
While we show that USP6 has pleiotropic effects on multiple immune lineages in vitro and in vivo in nude mice, the full extent of its impact on the immune TME in ES in vivo remains to be enumerated. As mentioned, in vivo studies are restricted to xenografting of human ES cell lines into immunocompromised mice. Nevertheless, despite using a cross-species immunocompromised model, USP6 was able to elicit an anti-tumor immune response. While nude mice have allowed us to probe effects on NK and myeloid lineages, they lack mature T lymphocytes. Thus, validating USP6's effects on acquired immune cells in future in vivo studies will require allografting/xenografting of primary T cells. It is highly likely that USP6 will be able to promote T cell recruitment and activation in vivo, given that USP6 induces robust production of CXCL10, that CM from USP6-expressing ES cells can stimulate migration of primary T lymphocytes, and that ES cells expressing USP6 can induce APC activation in vivo (as monitored by CD86 and MHCII surface upregulation). This notion is further supported by analysis of ES patient samples, which indicates that high USP6 expression is associated with infiltration of CD8+/CD4+ lymphocytes and activated DCs (Supplemental Figure 15). Interestingly, USP6 is also associated with infiltration of γδT lymphocytes in silico, which were recently reported to be the most significant favorable prognostic intra-tumoral immune lineage, across dozens of cancer types, in tens of thousands of samples (30) .
A curiosity of our findings is that the dominant effect of USP6 in ES is tumor suppressive, contrasting its oncogenic activity in NF. We confirmed that this was not a peculiarity of ES, as USP6 also inhibited tumor growth and stimulated immune infiltration in a disparate tumor model, 293T embryonic kidney cells (Supplemental Figure 16A-C). We speculate that whether USP6's pro- or anti-tumorigenic function dominates depends on the oncogenic driver present (model, Supplemental Figure 17). ES is driven by the potent oncogenic driver EWS-FLI1, and in this context the oncogenic functions of USP6 may be superfluous; its anti-tumorigenic activity, mediated through IFN signaling, is therefore its dominant manifestation. In contrast, translocated USP6 is the oncogenic driver in NF (Supplemental Figure 17, bottom). We posit that in the early phase of the disease when the tumor is actively growing, USP6's pro-tumorigenic signaling (via STAT3 and NF-κB) dominates (16, 23, 24). However, USP6 simultaneously triggers IFN signaling (17) and infiltration of anti-tumorigenic immune lineages, which ultimately prevail to drive the spontaneous regression that typifies NF. Indeed, adapted CIBERSORT analysis and IHC of NF confirmed enrichment of CD4+ and CD8+ T lymphocytes, γδT cells, and activated DCs and NK cells, and but not Tregs (Supplemental Figure 18A-C).
On a final note, we conjecture a role for USP6 in cancer immunoediting, the process by which the immune system monitors and modulates the immunogenicity and clearance of developing neoplasms. Cancer immunoediting comprises three stages: elimination, equilibrium, and escape (31). During the elimination phase, nascent neoplastic cells are effectively recognized and cleared by the immune system. Some cells may evade elimination and enter the equilibrium phase, which is considered a state of functional dormancy where there is no detectable increase in tumor burden. Elimination and equilibrium have been extensively dissected mechanistically in mouse models, which reveal a central role for IFN signaling, CXCL9/10 production, and both NK and T cells, although a degree of immunoediting can occur even in the absence of adaptive immunity (31-33). However, confirmation of these mechanisms in humans has been elusive, since elimination/equilibrium lack clinical manifestation and are thus difficult to monitor. Our longstanding studies of NF, a self-limited transient neoplasm, led us to uncover that USP6 can render these defining features of elimination. We thus propose that USP6 may represent the first identified human endogenous elimination factor, in both NF and ES, explaining its association with significantly improved ES patient survival. Our current study demonstrates that USP6 has pleiotropic effects on the immune system, with its potent anti-tumorigenic activity likely mediated through multiple immune lineages. USP6 increases the intra-tumoral abundance of activated macrophages/DCs and NK cells in mice. Recent work has highlighted the importance of these innate immune lineages in combating tumor growth, and suggested that shifting macrophage and NK activation can have a profound impact on the response to existing immunotherapeutics (33-35). Forthcoming studies will dissect how immune subsets are activated by USP6, and assess their contribution to tumor suppression. Furthermore, future work will seek to harness the powerful and pleiotropic immunostimulatory properties of USP6 for therapeutic benefit.
Supplementary Material
SIGNIFICANCE.
This study reveals a novel tumor suppressive function for USP6 by inducing an immunostimulatory microenvironment, suggesting that USP6 activity may be exploited to enhance immunotherapy regimens.
Acknowledgments
We thank Dr. Florin Tuluc and colleagues at the flow cytometry at CHOP, the Human Immunology Core which is co-funded by the Penn Center for AIDS Research (CFAR, P30 AI 045008), and Abramson Cancer Center Support Grant (P30CA016520).
Grant Support: This work was funded by NIH/NCI grants CA168452 and CA178601 (M.M.C.), and T32 GM008076 (I.C.H.); and Kids Beating Cancer, Inc.
Footnotes
Conflict of Interest: This manuscript presents original research that has not been or is in the process of being published elsewhere. The Children’s Hospital of Philadelphia has ownership of PCT/US2019/063594 “Modulation of USP6 Expression as a Cancer Therapeutic” which lists Drs. Henrich and Chou as co-inventors.
REFERENCES
- 1.Lawlor ER, Sorensen PH. Twenty Years On – What Do We Really Know About Ewing Sarcoma And What Is The Path Forward? Crit Rev Oncog 2015;20:155–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grohar PJ, Helman LJ. Prospects and challenges for the development of new therapies for Ewing sarcoma. Pharmacol Ther 2013;137:216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnaldez FI, Helman LJ. New strategies in ewing sarcoma: lost in translation? Clin Cancer Res 2014;20:3050–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pishas KI, Lessnick SL. Recent advances in targeted therapy for Ewing sarcoma. F1000Res 2016;5:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hutzen B, Ghonime M, Lee J, Mardis ER, Wang R, Lee DA, et al. Immunotherapeutic Challenges for Pediatric Cancers. Mol Ther Oncolytics 2019;15:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spurny C, Kailayangiri S, Jamitzky S, Altvater B, Wardelmann E, Dirksen U, et al. Programmed cell death ligand 1 (PD-L1) expression is not a predominant feature in Ewing sarcomas. Pediatr Blood Cancer 2018;65:1–8. [DOI] [PubMed] [Google Scholar]
- 7.Dancsok AR, Gao D, Lee AF, Steigen SE, Blay JY, Thomas DM, et al. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berghuis D, Santos SJ, Baelde HJ, Taminiau AH, Egeler RM, Schilham MW, et al. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J Pathol 2011;223:347–57. [DOI] [PubMed] [Google Scholar]
- 9.Stahl D, Gentles AJ, Thiele R, Gutgemann I. Prognostic profiling of the immune cell microenvironment in Ewing s Sarcoma Family of Tumors. Oncoimmunology 2019;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujiwara T, Fukushi J, Yamamoto S, Matsumoto Y, Setsu N, Oda Y, et al. Macrophage infiltration predicts a poor prognosis for human ewing sarcoma. Am J Pathol 2011;179:1157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review). Oncol Lett 2011;2:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Martin A, Gomez L, Lustgarten J, Mira E, Manes S. Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4(+) and CD8(+) T cells. Cancer Res 2011;71:5455–66. [DOI] [PubMed] [Google Scholar]
- 13.Viola A, Sarukhan A, Bronte V, Molon B. The pros and cons of chemokines in tumor immunology. Trends Immunol 2012;33:496–504. [DOI] [PubMed] [Google Scholar]
- 14.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24:541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quick L, Young R, Henrich IC, Wang X, Asmann YW, Oliveira AM, et al. Jak1-STAT3 Signals Are Essential Effectors of the USP6/TRE17 Oncogene in Tumorigenesis. Cancer Res 2016;76:5337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henrich IC, Young R, Quick L, Oliveira AM, Chou MM. USP6 Confers Sensitivity to IFN-Mediated Apoptosis through Modulation of TRAIL Signaling in Ewing Sarcoma. Mol Cancer Res 2018;16:1834–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Franco S, Turdo A, Todaro M, Stassi G. Role of Type I and II Interferons in Colorectal Cancer and Melanoma. Front Immunol 2017;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol 2015;15:405–14. [DOI] [PubMed] [Google Scholar]
- 21.Oliveira AM, Chou MM. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol 2014;45:1–11. [DOI] [PubMed] [Google Scholar]
- 22.Ye Y, Pringle LM, Lau AW, Riquelme DN, Wang H, Jiang T, et al. TRE17/USP6 oncogene translocated in aneurysmal bone cyst induces matrix metalloproteinase production via activation of NF-kappaB. Oncogene 2010;29:3619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pringle LM, Young R, Quick L, Riquelme DN, Oliveira AM, May MJ, et al. Atypical mechanism of NF-kappaB activation by TRE17/ubiquitin-specific protease 6 (USP6) oncogene and its requirement in tumorigenesis. Oncogene 2012;31:3525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madan B, Walker MP, Young R, Quick L, Orgel KA, Ryan M, et al. USP6 oncogene promotes Wnt signaling by deubiquitylating Frizzleds. Proceedings of the National Academy of Sciences 2016;113:2945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burke SJ, Goff MR, Lu D, Proud D, Karlstad MD, Collier JJ. Synergistic expression of the CXCL10 gene in response to IL-1beta and IFN-gamma involves NF-kappaB, phosphorylation of STAT1 at Tyr701, and acetylation of histones H3 and H4. J Immunol 2013;191:323–36. [DOI] [PubMed] [Google Scholar]
- 26.Hudy MH, Traves SL, Proud D. Transcriptional and epigenetic modulation of human rhinovirus-induced CXCL10 production by cigarette smoke. Am J Respir Cell Mol Biol 2014;50:571–82. [DOI] [PubMed] [Google Scholar]
- 27.Billottet C, Quemener C, Bikfalvi A. CXCR3, a double-edged sword in tumor progression and angiogenesis. Biochim Biophys Acta 2013;1836:287–95. [DOI] [PubMed] [Google Scholar]
- 28.Minas TZ, Surdez D, Javaheri T, Tanaka M, Howarth M, Kang H-J, et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. OncoTarget 2017;8:34141–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015;21:938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smyth MJ, Dunn GP, Schreiber RD. Cancer Immunosurveillance and Immunoediting: The Roles of Immunity in Suppressing Tumor Development and Shaping Tumor Immunogenicity. Advances in Immunology 2006;90:1–50. [DOI] [PubMed] [Google Scholar]
- 32.O'Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med 2012;209:1869–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol 2015;33:445–74. [DOI] [PubMed] [Google Scholar]
- 34.Datta M, Coussens LM, Nishikawa H, Hodi FS, Jain RK. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am Soc Clin Oncol Educ Book 2019;39:165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shevtsov M, Multhoff G. Immunological and Translational Aspects of NK Cell-Based Antitumor Immunotherapies. Front Immunol 2016;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007;11:421–9. [DOI] [PubMed] [Google Scholar]
- 37.Savola S, Klami A, Myllykangas S, Manara C, Scotlandi K, Picci P, et al. High Expression of Complement Component 5 (C5) at Tumor Site Associates with Superior Survival in Ewing's Sarcoma Family of Tumour Patients. ISRN Oncol 2011;2011:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palmer RD, Barbosa-Morais NL, Gooding EL, Muralidhar B, Thornton CM, Pett MR, et al. Pediatric malignant germ cell tumors show characteristic transcriptome profiles. Cancer Res 2008;68:4239–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.