

Abstract
Several master transcription factors (TFs) can activate the epithelial-to-mesenchymal transition (EMT). However, their individual and combinatorial contributions to EMT in breast cancer are not defined. We show that overexpression of EMT-TFs individually in epithelial cells upregulated endogenous SNAI2, ZEB1/2, TCF4 and TWIST1/2 as a result of positive feedback mediated in part by suppression of their negative regulator microRNAs miR200s/203/205. We identified TCF4 as a potential new target of miR200s. Expression of ZEB1/2 strongly correlated with the mesenchymal phenotype in breast cancer cells, with the CD24−/CD44+ stemness profile, and with lower expression of core epithelial genes in human breast tumors. Knockdown of EMT-TFs identified the key role of ZEB1 and its functional cooperation with other EMT-TFs in the maintenance of the mesenchymal state. Inducible ZEB1+2 knockdown in xenograft models inhibited pulmonary metastasis, emphasizing their critical role in dissemination from primary site and in extravasation. However, ZEB1+2 depletion one week post intravenous injection did not inhibit lung colonization, suggesting that ZEB1/2 and EMT are not essential for macrometastatic outgrowth. These results provide strong evidence that EMT is orchestrated by coordinated expression of several EMT-TFs and establish ZEB1 as a key master regulator of EMT and metastasis in breast cancer.
Implications:
The EMT program is orchestrated by coordinated expression of multiple EMT transcription factors, while ZEB1 integrates the EMT master regulatory network and plays the major role in promoting EMT and metastasis.
Introduction
The EMT is an evolutionarily conserved transdifferentiation program that plays an important role in embryonic development and tissue specialization(1). In adult organisms, EMT has been implicated in wound healing, fibrosis, and cancer. Though formally dispensable for tumor initiation, EMT promotes stemness, metastasis and confers increased therapeutic resistance(2).
A handful of master EMT transcription factors have been identified based on their ability to induce EMT when overexpressed individually in epithelial cells, including C2H2-type zinc finger family members SNAI1/2 and ZEB1/2, and bHLH family members TWIST1/2 and TCF3/4(3-5). Though being sufficient for EMT induction, some of them form positive feedback loops leading to activation of other EMT regulators in the same cell. For example, overexpression of SNAI1 (and SNAI2) in many systems activates SNAI2(6, 7), ZEB1/2(8, 9), TWIST1(10, 11) and TCF4(3, 10). ZEB1 and ZEB2 can activate each other(7, 12), while TWIST1-induced EMT is dependent on activation of SNAI2(13). The functional significance of this apparent redundancy and hierarchy between the EMT-TFs have not been thoroughly investigated.
It has been well-established that epithelial-mesenchymal balance in cells is maintained in large part through double negative feedback loops between EMT-TFs and epithelial-specific microRNAs, namely between ZEB1/2 and the miR200 family (miR200c,141;200b,200a,429 – collectively miR200s)(12, 14, 15), miR203(16, 17) and miR205(18, 19); between SNAI2 and the miR200 family(20); between SNAI1/2 and miR203(21, 22) and the miR34 family(2).
Association analyses showed that transcript levels of multiple EMT-TFs in primary breast tumors correlate with worse patient outcome, while expression of the miR200 family microRNAs predict better outcome(23, 24). Similarly, protein expression of multiple EMT-TFs analyzed by IHC predicts poor prognosis in metastatic breast cancer(25).
Although the evidence for a pivotal role of EMT in invasion and metastasis obtained from cell culture and tumor xenograft experiments is compelling, it is rather fragmentary from in vivo studies(26, 27). Recent attempts have begun to address the role of EMT in metastasis in genetically engineered mouse tumor models and lineage-tracing experiments(28). Surprisingly, genetic ablation of the Snai1 or Twist1 genes in a pancreatic mouse tumor model did not affect metastasis(29). However, Zeb1 gene deletion in the same model impaired metastatic dissemination(30), indicating that different EMT-TFs might be required for metastasis in specific contexts. Further, deletion of a single EMT-TF may not be sufficient to prevent EMT due to the redundancy and mutual activation of multiple EMT-TFs during EMT.
Given the significant role EMT plays in tumor metastasis and drug resistance, defining the EMT-TFs crosstalk network will provide critical mechanistic insight allowing the development of novel strategies for the prevention or treatment of metastasis.
Materials and Methods
Reagents and Antibodies.
Doxycycline Hydrochloride (Dox) (Fisher, BP2653). SNAI1/Snail (C15D3) (Cell Signaling, 3879), SNAI2/Slug (C19G7) (Cell Signaling, 9585), ZEB1 (Sigma, HPA027524), ZEB2/SIP1 (BethylLabs, A302-474A), TCF3/E2A (V-18) (Santa Cruz, sc-349), TCF4/ITF-2 (C-1) (Santa Cruz, sc-393255), TWIST1 (Twist2C1a) (Santa Cruz, sc-81417), E-cadherin (BD Biosciences, 610181), Claudin1 (JAY.8) (Invitrogen, 51-9000), Claudin3 (Z23.JM) (Invitrogen, 34-1700), Claudin4 (3E2C1) (Invitrogen, 32-9400), Pan-Cytokeratin (C11) (Santa Cruz, sc-8018), Vimentin (V-9) (Santa Cruz, sc-6260), N-Cadherin (cl.32) (BD Biosciences, 610920), CD44/HCAM (F-4) (Santa Cruz, sc-9960), PDGFRβ (28E1) (Cell Signaling, 3169) CD133 (C24B9) (Cell Signaling, 3663), CD49f/ITGA6 (Cell Signaling, 3570), ALDH1/2 (H-8) (Santa Cruz, sc-166362), MET (C-28) (Santa Cruz, sc-161), GAPDH (Millipore, MAB374), Tubulin α (Sigma, T9026), NEDD9 (2G9) (Santa Cruz, sc-33659), ESRP1/2 (23A7) (Novus, NBP1-77971); secondary horse radish peroxidase (HRP)-conjugated antibodies against mouse and rabbit (Jackson ImmunoResearch).
Cell lines and cell culture.
Primary HMECs were purchased from Lonza and Invitrogen and grown in Mammary epithelial growth medium (MEGM) with BulletKit (Lonza, CC-3150). HMLE cells were described previously(31) and were grown in DMEM/F12 50/50 (ThermoFisher, MT10092CV) supplemented with 5% horse serum (HS; LifeTechnologies, 16050122), 20 ng/ml EGF (Sigma, E4127), 10 μg/ml Insulin (Sigma, I5500), and 0.5 μg/ml Hydrocortisone (Sigma, H4001). MCF10A cells were cultured in the HMLE media plus 100 ng/ml cholera toxin (Sigma, C8052). MDA-MB-231-luc-D3H2LN (MDA231LN) cells were purchased from Caliper Life Science (RRID:CVCL_D257) and grown in Alpha modification of Eagle's medium (AMEM) (ThermoFisher, MT15012CV) supplemented with 10% heat-inactivated fetal bovine serum (HI FBS; HyClone). Parental MDA-MB-231 cells and its three derivative super-metastatic sublines (LM2-4175, BoM-1833, BrM-831) were kindly provided by Dr. Joan Massague (HHMI). The other cell lines were purchased from and authenticated by ATCC and were grown in Dulbecco’s modified eagle medium (DMEM; ThermoFisher, MT10013CV) supplemented with 10% fetal bovine serum (FBS; HyClone). All DMEM/AMEM media were also supplemented with 29.2 mg/ml L-glutamine, 100U/ml penicillin and 100 ug/ml streptomycin (ThermoFisher, MT30009CI). All cells were maintained at 37C in a 5% CO2 incubator. Established cell lines were tested for mycoplasma using MycoAlert® Mycoplasma Detection Kit (Lonza, LT07-318), expanded, frozen and each experiment was started with a new vial from the frozen stock to minimize passaging.
Plasmids.
ORFs for the eight EMT-TFs and genomic fragments for the four microRNA genes/clusters were PCR-amplified using PfuUltraII DNA polymerase and cloned into Dox-inducible pLUT lentiviral expression vector (Suppl.Fig.9) using primers and templates described in Suppl.Table1. All constructs were sequenced to verify absence of mutations. TCF4 and ZEB1 translational reporters: Firefly luciferase reporter vector was prepared by cloning luc2 gene from pGL4.10 into pcDNA3.1-zeo using PCR with #5Luc2-Nhe fwd primer (5’-AGAGGCTAGC-CACCATGGAAGATGC-3’) and #3Luc2-H rev primer (5’-AGAGAAGCTT-ATTACACGGCGATCTTGCC-3’). TCF4 and ZEB1 3’UTRs were then cloned downstream of luc2 in resulting pcDNA3.1-Luc2-zeo. A 1.66-kb cDNA fragment (nt 3589-5260 of accession NM_001243226) representing the partial 3′ UTR of human TCF4 sequence was generated by PCR using #5TCF4 3UTR1-B fwd primer (5’-GAGAGGATCC-TGTGTAGCCGAAGTAGTGT-3’) and #3TCF4 3UTR1-RI rev primer (5’-AGAGAATTC-CTCCACATTTTAGGAACTTATCAACAAATC-3’) on oligo-dT generated total cDNA from HMEC cells. TCF4 reporter with mutations in the miR200c-seed complementary regions was generated by QuickChange PCR mutagenesis using following primer pairs: #5TCF4 200cseed2mt (5’-GAGGAGGGGTAGTGAAATACTTCTCTGGCCAGTATG-3’) and #3TCF4 200cseed2mt (5’-CATACTGGCCAGAGAAGTATTTCACTACCCCTCCTC-3’); #5TCF4 200cseedX1mt (5’-GCCACACAATTCTTGGGCTGTACTAAGAAAAAAAAAAATCCCTGT-3’) and #3TCF4 200cseedX1mt (5’-ACAGGGATTTTTTTTTTTCTTAGTACAGCCCAAGAATTGTGTGGC-3’). A 1.73-kb cDNA fragment (nt 4062-5642 of accession NM_001128128) representing the partial 3′ UTR of human ZEB1 sequence was generated by PCR using #5ZEB1 3UTR1-RI fwd primer (5’-AGAGAATTC-GCCTGAACCTCAGACCTAG-3’) and #3ZEB1 3UTR1-X rev primer (5’-AGACTCGAG-ATGCATTTTATTGTGAGATGGGAG-3’) on pCI-neo-RL-ZEB1 plasmid template (Addgene, 35535)(18).
Luciferase reporter assays.
MDA231 cells on a 12-well plate were reverse transfected with 50 ng of pcDNA3.1-Luc2 reporter vector, with or without TCF4/ZEB1 3’UTR, 20 ng of pRL-TK (Promega) and 25 nM of mirVana™ miRNA Mimic hsa-miR-200c or Negative Control #1. Briefly, lipid-DNA complexes were allowed to form by incubation of TransIT-LT1 (Mirus Bio) and reporter DNA plasmids. In a separate tube, lipid-RNA complexes were allowed to form by incubation of Lipofectamine RNAiMAX transfection reagent (Invitrogen) and microRNA mimics. Lipid-DNA and lipid-RNA complexes were then added to plate wells. Following trypsinization MDA231 cells were resuspended in Opti-MEM and 5 x 104 cells was added to each well bringing the total volume to 0.5 ml. Plates were incubated for 2 h at 37°C in 5% CO2, and then 1 ml of complete growth medium was added to each well. 48 h post-transfection Dual-Luciferase Reporter Assays System (Promega, E1910) was used to measure reporter activity. Signals were normalized to empty vector.
EMT-TFs and microRNAs overexpression and knockdown.
Dox-inducible pLUT-zeo or -puro lentiviral vector (Suppl.Fig.9) was used for overexpression of EMT-TFs ORFs and microRNA genes. Constitutive pGIPZ or Dox-inducible pTRIPZ lentiviral vectors (Dharmacon/Horizon) were used for expression of shRNAs, see Suppl.Table2 for details. Briefly, lentiviral particles were packaged in HEK293T cells (RRID:CVCL_0063) following calcium phosphate cotransfection of transfer vector, psPAX2 (Addgene, 12260), and pCMV-VSV-G (Addgene, 8454). After 2-4 rounds of infection with lentiviral particles, cells were selected in zeocin (50 μg/ml) or puromycin (1 μg/ml) containing media for ten days. Mass culture pools representing hundreds of drug resistant clones were used in all experiments. Double expressing cell lines were established by superinfection with the second shRNA vector. Expression of ORFs or shRNAs was induced by addition of 0.5 μg/ml doxycycline to cell culture media for 7-10 days. pLUT expressing turbo red fluorescent protein (RFP) and pGIPZ/TRIPZ expressing non-targeting shRNA were used as appropriate controls.
Cell proliferation assay.
A total of 2x104 cells were plated in triplicates in 6-well plates, cultured for 2, 4, 6, and 8 days, trypsinized, and counted on Countess (Invitrogen).
RNA isolation and quantitative RT-qPCR.
Total RNA was extracted using mirVana™ miRNA Isolation Kit (Ambion, AM1560) following the manufacturer’s instructions. Two micrograms of total RNA was reverse transcribed using Maxima RT (ThermoFisher, EP0742) and dT20 primer. Primers were designed using RealTime qPCR Assay Tool (IDT). From two to six primer sets were tested for each gene and the best one was chosen for analysis. A water negative control did not produce significant amplification product. qPCR primer sequences are listed in Suppl.Table3. Mature microRNAs were reverse transcribed using TaqMan™ MicroRNA Reverse Transcription Kit (ABI, 4366596) and analyzed using ABI’s microRNA-specific assays described in Suppl.Table4. Quantitative PCR (qPCR) was performed in an ABI-7500 Real-Time PCR Cycler using SYBR Green (Sigma, S4438) or TaqMan™ Universal (ABI, 4440038) master mixes and analyzed using ABI SDS2.06 software. Transcript levels in the breast cancer cell line panel (Suppl.Fig.2A-E) were normalized to the geometrical mean of UBC, PCNA, RPL13A, and tubulin genes; in HMLE sublines expressing ectopic EMT-TFs and MDA231 super-metastatic sublines by the geometrical mean of UBC, PCNA, and B2M; in HMLE CD24+/CD44− and CD24−/CD44+ cells by the geometrical mean of UBC, PCNA, and RPL13A. MicroRNA levels were normalized to U6 small nuclear RNA level. FCs were calculated using the comparative Ct method (ΔΔCt).
Western blotting (WB).
Cells were washed with PBS and lysed in nonreducing Laemmli buffer (50mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol). Lysates were boiled for 5 minutes to shear DNA. Total protein was quantified by bicinchoninic acid (BCA) assay (ThermoFisher), then lysates were supplemented with β-mercaptoethanol (final concentration 200mM) and boiled for 5 minutes. Equal amounts of total protein (typically 30 μg) were separated on 4%-12% NuPAGE Bis-Tris gels (Invitrogen) in MOPS SDS buffer and transferred to a PVDF membrane (Pierce, 88518) using liquid transfer in Tris-Glycine buffer supplemented with 10% methanol. Membranes were blocked in 5% skim milk dissolved in PBS-T (PBS plus 0.05% Tween-20) at room temperature for 1h. All antibody incubations and washes were performed in PBS-T at room temperature. Protein bands were detected using standard chemiluminescence techniques. Blots were stripped in Restore™ PLUS Stripping Buffer (ThermoFisher, 46430) and re-probed with multiple antibodies.
Immunofluorescence (IF).
Cells grown on glass coverslips in 12-well plate for 48 hours were fixed in 4% paraformaldehyde, rinsed once in PBS and twice in PBS-X (PBS plus 0.1% Triton-X100), then permeabilized with methanol for 10 minutes at −20°C, rinsed twice with PBS and blocked with 5% BSA in PBS for 1 hour, incubated with anti-E-Cadherin (BD, 610181) and then anti-mouse AlexaFluor-647 (ThermoFisher) antibodies. Coverslips were mounted on slides using ProLong™ Gold Antifade Mountant with DAPI (ThermoFisher). Images were acquired on Nikon A1R confocal with N-SIM E microscope and 60X Plan Apo objective.
Fluorescence-Activated Cell Sorting and Flow Cytometry.
Cells were prepared according to standard protocols and suspended in DPBS containing 2.5mM EDTA, 25mM HEPES pH=7.0 and 1% FBS on ice. For FACSorting on a BD FACSAria III, CD24-PE (BD, 555428), CD44-FITC (BD, 555478) and FITC Mouse IgG2b κ Isotype Control (BD, 555742) antibodies were used. 7-AAD (BD, 559925) was used to exclude dead cells. For flow cytometry analysis, cocktail of four antibodies (Miltenyi Biotec) CD44-FITC (130-115-867), CD24-APC (130-112-657), CD104/ITGB4-PE-Vio770 (130-101-441), EpCAM-VioBlue (130-113-266) and LIVE/DEAD™ Fixable Near-IR Dead Cell Stain (Invitrogen, L34975) were used. Data were collected and analyzed on BD LSR Fortessa using BD FACSDiva 8.0 Software (BD Biosciences).
Trans-well invasion assay.
A 24-well BD BioCoat™ Tumor Invasion System (BD, 354483) (membrane with 8 μm pores, covered with a layer of growth factor reduced Matrigel) was used according to the manufacturer’s instructions. Briefly, a total of 15,000 cells resuspended in serum-free media were plated in the apical chambers and were allowed to invade towards the basal chambers containing media with 10% fetal bovine serum for 10 hours. Following the incubation, non-invaded cells on the upper surface of the membrane were removed with a cotton swab. The inserts were labeled with 4 μg/mL calcein AM in Hanks’ balanced salt solution (HBSS) for 1 hour. Fluorescently labeled cells were visualized using a Typhoon 9410 imager at 488/526 nm with 25 micron resolution, and signals were quantified using ImageJ software as mean gray values for each well. The experiments were performed at least in triplicates.
Animal studies and bioluminescence imaging.
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were obtained from the Jackson Laboratory. Luciferase-expressing MDA231LN cells were resuspended in PBS and 2x106 cells in 100 μl were injected into the fourth mammary fat pad or 2x105 cells in 250 μl were injected into the tail vein of 7-9-week old NSG mice. Mice were maintained on a control diet or diet supplemented with doxycycline 200 mg/kg (Bio-Serv) to induce shRNA expression. Xenograft tumors were measured with calipers every week and tumor volume was calculated using the formula V=(π/6)(LxW2), where L is the length of the tumor and W its width. Primary tumors and lungs were collected at the endpoint of study, fixed in formalin and analyzed for number of metastases by a pathologist. Portions of primary tumors were lysed for Western blot analysis. For bioluminescent imaging, about 150 mg/kg D-luciferin (Caliper Life Sciences) was injected into the peritoneum. Images were obtained using the IVIS Lumina-II Imaging System and Living Image-4.0 software as described(32).
TCGA data analysis.
Breast TCGA data were downloaded from (http://cancergenome.nih.gov/). Statistical programming software R (version 3.0.1) was used to assemble and process the data as described(32). These data from 105 normal, 354 LumA, 203 LumB, 97 HER2 and 137 basal samples were used to generate boxplots for protein-coding genes in Suppl.Fig.4. Boxplots for microRNA genes in Suppl.Fig.4,5 were generated from 87 normal, 232 LumA, 114 LumB, 46 HER2 and 82 basal samples. The Pearson’s correlations described in Fig.3,Suppl.Fig.6 and Suppl.File2 were carried out using log-transformed values and were generated from 112 normal, 359 LumA, 170 LumB, 60 HER2 and 122 basal samples described in (33). A cutoff Pearson’s R value of ≥ ∣0.25∣ and p-value <0.05 was selected based on the minimal sample size of n=60 in HER2 subtype.
Fig.3. ZEB1/2 and TCF4 strongly correlate with the mesenchymal phenotype in breast cancer cell lines, and with EMT markers in human primary breast tumors.

A, WB analysis of EMT-TFs, EMT and stemness markers in primary HMECs and in the indicated immortalized and breast cancer cell lines of luminal (L), basal (B) and mesenchymal (M) groups. Filled arrow ~78kDa TCF4 isoform, open arrow ~68kDa TCF4 isoform. Tubulin – loading control. * - non-specific band. B, Correlation of expression between each of the EMT-TFs in normal breast tissue and four breast cancer subtypes. Pearson’s R coefficient was computed between an EMT-TF indicated at the top of each column section and the other seven EMT-TFs for each of the five sample groups. R values were plotted as color-coded dots, where each color represents individual EMT-TF according to the legend. See description of dashed boxes in the main text. C, Number of negatively (above 0) and positively (below 0) correlated core epithelial genes with each EMT-TF in the five sample groups indicated at the top. Quantification of data presented in Suppl.Fig.6.
Statistical analysis.
Statistical analyses and graphs were generated with GraphPad Prism5 software using one-way ANOVA with Dunnett’s post-test.
Data availability.
All data supporting the findings in this study are available from the corresponding author upon reasonable request.
Results
Convergence of positive feedback from multiple EMT-TFs to upregulate ZEB1/2, TWIST1/2, SNAI2 and TCF4 in epithelial cells undergoing EMT.
The EMT-TFs bind similar E-box DNA consensus sequences (Suppl.Fig.1) and regulate overlapping sets of genes(9, 10) primarily through direct repression of epithelial targets bearing an E-box(es) in their promoters(4, 14). To gain further insight into their individual functions, we expressed the eight EMT-TFs in a doxycycline (Dox)-inducible manner in immortalized human mammary epithelial HMLE cells. The kinetics of EMT induction was established in a time course experiment with SNAI1. Addition of Dox led to rapid induction of SNAI1 protein within hours, upregulation of mesenchymal markers CD44 and vimentin, and concomitant downregulation of epithelial protein claudin1 within 24 hours (Fig.1A). However, significant suppression of E-cadherin was observed only after five days likely due to slow decay of stable E-cadherin protein (Fig.1A). When expressed for ten days, six of the eight EMT-TFs induced characteristic changes in cell morphology (Fig.1B) and expression of specific marker proteins indicative of EMT, including dramatic suppression of E-cadherin, cytokeratins and claudins, and elevated expression of vimentin and CD44 (Fig.1C). Unexpectedly, high TWIST1/2 overexpression led to suppression of cell growth and limited EMT induction (not shown), suggesting that high TWIST overexpression could be cytotoxic as demonstrated previously(34). Therefore, we excluded TWIST1/2 overexpressing cells from further analysis. Though each of the other six TFs induced EMT, there were distinct differences between the TF families in regard to specific targets. For example, N-cadherin and PDGFR β were induced by ZEB1/2 and TCF3/4 but not by SNAI1/2, NEDD9 was induced by SNAI1/2 only and c-MET was repressed by TCF3/4 only (Fig. 1C). Furthermore, overexpression of ZEB1/2 led to increase in cell number, while ectopic SNAI1/2 and TCF3/4 slowed cell growth down (Fig.1D), indicating that the effects of EMT on cell proliferation are EMT-TF-specific.
Fig.1. Overexpression of exogenous EMT-TFs induces EMT and secondary upregulation of endogenous EMT-TFs in HMLE cells.

HMLE cells were infected with pLUT-EMT-TF lentiviruses and selected with zeocin to establish stable Dox-inducible cell lines. A, WB analysis of EMT markers in HMLE-LUT-SNAI1 cells during time-course induction of SNAI1. Dox was added to culture media and cells were lysed at the indicated time points. B, Phase contrast images, C, WB analysis, and D, cell proliferation assay of the indicated HMLE-LUT-EMT-TFs cell lines. E, Relative mRNA levels of the EMT-TFs shown at the top in the indicated HMLE cell lines shown at the X-axis determined by RT-qPCR. The mean values of the EMT-TFs expression in control HMLE cells were set to 1. The data are mean ± SEM of three biological replicates. D,E, ANOVA with Dunnett’s post-test, * - p<0.05, ** - p<0.01, *** - p<0.001.
Endogenous SNAI2 protein was upregulated by each of the other five EMT-TFs (Fig.1C). ZEB1 and ZEB2 proteins were generally induced by SNAI1/2, ZEB1/2 and TCF3/4, while ZEB1 was most dramatically upregulated by TCF3/4. Interestingly, expression of endogenous TWIST1 protein correlated with the induction of ZEB1. We were unable to detect endogenous levels of TCF3/4 and TWIST2 proteins in HMLE cells and relied on RT-qPCR for their expression analysis. Levels of endogenous TCF4 mRNA were modestly upregulated by SNAI1/2 and ZEB1, while TCF3 mRNA levels were marginally repressed by ZEB1/2 (Fig.1E). These results revealed distinct positive feedback loops orchestrating the expression of multiple EMT-TFs when only one is initially activated.
Suppression of the miR200 family, miR205 and miR203 microRNAs by multiple EMT-TFs correlates with secondary upregulation of endogenous EMT-TFs.
The observed secondary upregulation of multiple EMT-TFs during forced EMT could be due to repression of one or more of their common negative regulator microRNAs. We compared the mRNA sequences of the eight EMT-TFs for the presence of established and predicted binding sites for miR200s/203/205 microRNAs and found that ZEB1/2, TCF4 and to a lesser extent SNAI2 bear the highest number of these sites (Fig.2A,B). To determine if miR200s microRNAs can also directly target TCF4, we subcloned its 3’UTR downstream of a luciferase gene and performed reporter assays in mesenchymal MDA231 cells expressing very low endogenous levels of miR200s. MiR200c mimic repressed expression of the TCF4 3’UTR luciferase reporter as efficiently as of the control ZEB1 3’UTR reporter (Fig.2C). Mutation of the two predicted miR200c binding sites in the TCF4 3’UTR (Fig.2B) abolished the repression (Fig.2C), indicating that TCF4 is a putative target of miR200s.
Fig.2. Reciprocal repression of EMT-TFs and miR200s/203/205 microRNAs.

A, Schematic map of ZEB1/2, TCF4 and SNAI2 transcripts and color-coded location of the microRNA sites. 3’UTR regions of ZEB1 and TCF4 used in luciferase reporter assay in (C) are shown below. (Table) Number of occurrences of predicted and validated sites complementary to seeds of the indicated microRNAs in coding sequences (CDS) and 3’UTRs of the EMT-TFs. B, Conservation of two sites and predicted duplex formation of TCF4 3’UTR from five different species and mature miR200c microRNA. The miR200c 7-nt seed sequence is shown in bold. Nucleotide substitutions of the mutant (Mt) TCF4 3’UTR construct used in (C) are shown in red. C, Repression of the TCF4 and ZEB1 luciferase-3’UTR reporters by miR200c. MDA231 cells were co-transfected with the indicated wild type (WT) or mutant (Mt) constructs and control or miR200c mimics. D, Ectopic EMT-TFs repress endogenous epithelial-specific microRNAs in HMLE cells. Relative levels of four microRNAs in the indicated HMLE-LUT-EMT-TFs cell lines described in Fig.1 determined by RT-qPCR. The mean values of the microRNAs expression in control HMLE cells were set to 1. C,D, The data are mean ± SEM of three replicate transfections or three biological replicates, respectively. ANOVA with Dunnett’s post-test, * - p<0.05, ** - p<0.01, *** - p<0.001. E, Ectopic epithelial-specific microRNAs repress endogenous EMT-TFs in BT549 and HS578T cells. Cells were infected with pLUT lentiviruses encoding indicated microRNAs. WB analysis of EMT-TFs and select EMT markers. Filled arrow ~78kDa TCF4 isoform, open arrow ~68kDa TCF4 isoform. F, Double negative feedback loop model. Red T-ending lines indicate repression by the EMT-TFs of the respective microRNAs and vice versa.
The miR200 family consists of five microRNAs arranged into two gene clusters encoding miR200c/141 and miR200b/200a/429, so we profiled the leading members of each cluster: miR200c and miR200b, which are typically expressed at higher levels. Overexpression of ZEB1 and TCF3/4 led to dramatic suppression of the miR200s and miR205 microRNAs, while SNAI1/2 repressed them only modestly (Fig.2D). Conversely, miR203 was significantly downregulated by SNAI1/2 and TCF3/4, and less efficiently by ZEB1/2. These data indicate that upregulation of endogenous ZEB1/2, TCF4 and SNAI2 by ectopic EMT-TFs strongly correlated with suppression of their common negative regulators miR200s/205/203.
To confirm that the epithelial microRNAs can inhibit multiple EMT-TFs, we overexpressed miR203, miR205, miR200b/a/429 or miR200c/141 clusters in mesenchymal BT549 and HS578T cells that express multiple EMT-TFs. As expected, expression of the miR200s microRNAs led to dramatic downregulation of ZEB1/2 and significant downregulation of TCF4, SNAI2 and TWIST1 proteins, concomitant with reactivation of E-cadherin (Fig.2E). These results indicate that the epithelial microRNAs coordinately suppress multiple EMT-TFs during MET (Fig.2F).
Expression of ZEB1/2 correlates with the mesenchymal phenotype in breast cancer cell lines.
To correlate the expression of each of the eight EMT-TFs with the mesenchymal phenotype, we first profiled a panel of 17 human tumor cell lines representing luminal/ER+ or basal/triple-negative breast cancer subtypes(35, 36), two independent isolates of primary HMECs and two immortalized mammary cell lines, HMLE and MCF10A. The vast majority of cell lines in our panel maintained the epithelial phenotype, i.e. high expression of E-cadherin, cytokeratins, ESRP1/2, claudins, and CD24 (Fig.3A,Suppl.Fig.2E). MDA453 cells carry homozygous nonsense mutation W638* in CDH1 gene (COSMIC database) and do not express E-cadherin protein but maintain overall luminal gene expression profile (Fig.3A and (36)). A subset of basal cell lines in our panel have undergone full EMT, i.e. silenced the epithelial genes and gained high expression of mesenchymal markers vimentin, N-cadherin and CD44 (Fig.3A). Given this clear distinction, we referred to these four cell lines HCC1395, MDA231 (and its derivative MDA231LN), BT549 and HS578T as mesenchymal (M). These cell lines have also been previously shown to be the most invasive (35). In addition, they exhibited the stemness phenotype associated with CD24low/CD44high markers but not with CD133, CD49f or ALDH1/2 (Fig.3A,Suppl.Fig.2E).
Compared to HMECs, expression of SNAI1 protein was higher in most cell lines, especially in basal subtype (Fig.3A,Suppl.Fig.2A), suggesting that SNAI1 is commonly expressed in breast cancer cells. In contrast, SNAI2 protein was undetectable in all luminal and many basal lines. Interestingly, ZEB1/2 were consistently detected at high levels in all mesenchymal cell lines, but not in other subtypes (Fig.3A,Suppl.Fig.2B). High TWIST1/2 levels correlated with high ZEB1/2 levels in mesenchymal cell lines (and ZEB1 in HCC1143), with the exception of MDA231 and MDA231LN, where TWIST1/2 were undetectable (Suppl.Fig.2C). Compared to HMECs, TCF4 levels were low in all cells except three mesenchymal cell lines and basal HCC38, while the levels of TCF3 were either similar or modestly lower (Fig.3A,Suppl.Fig.2D).
These data show that high endogenous expression of an individual EMT-TF does not necessarily elicit full EMT, as is exemplified by SNAI1 (Fig.3A). Highest level of SNAI1 was observed in basal MDA157 cells reaching ~600-fold over the HMECs level (Fig.3A,Suppl.Fig.2A). Though some epithelial markers were decreased in these cells (i.e. cytokeratins), others, such as E-cadherin, CD24 and ESRP1/2 were expressed at levels comparable to many basal and luminal cell lines (Fig.3A), indicating that MDA157 cells have undergone only partial EMT.
In contrast, high ZEB1/2 expression correlated well with full EMT/mesenchymal phenotype (Fig.3A). TCF4, SNAI2 and TWIST1/2 were also expressed in mesenchymal cells, but they were present at similar levels in some epithelial cell lines and HMECs (Fig.3A,Suppl.Fig.2A,C,D), suggesting that individually they were not sufficient to induce full EMT. Interestingly, TCF4 and TWIST1/2 levels were very low in MDA231, indicating that they were not necessary for full EMT in these cells. Together, these results indicate that though many EMT-TFs could be upregulated in breast cancer cells, high expression of ZEB1/2 strongly correlated with full EMT/mesenchymal phenotype. In addition, TCF4, SNAI2 and TWIST1/2 were upregulated in the mesenchymal cell lines, suggesting that full EMT is orchestrated by cooperative action of multiple EMT-TFs. In agreement with the role of miR200s/205/203 as negative regulators of ZEB1/2, TCF4 and SNAI2, endogenous levels of these microRNAs were consistently low in mesenchymal cell lines (Suppl.Fig.3).
Differential expression of EMT-TFs and miR200s/203/205 microRNAs in human breast cancer subtypes.
To correlate our observations in cell lines with patient data, we analyzed expression of the eight EMT-TFs, microRNAs and EMT marker genes in the Breast TCGA RNA-Seq dataset representing ~100 normal breast tissue samples and ~900 primary breast tumors of luminal A, luminal B, HER2 and basal subtypes(33). Compared to normal breast tissue, ZEB1/2, TWIST1/2, TCF4 and SNAI2 were expressed at lower levels in all four intrinsic subtypes and SNAI1 in both luminal subtypes (Suppl.Fig.4). In contrast, TCF3 levels were higher in all subtypes, while SNAI1 levels were elevated in basal tumors. The observed lower expression of the majority of EMT-TFs in tumors is concordant with dramatic suppression of the major inducer of mesenchymal traits, the TGF-β tumor suppressor pathway (via downregulation of TGFBR2 and TGFBR3 receptors) in all subtypes (Suppl.Fig.4), that is commonly observed in primary breast tumors(37, 38). Consistently, compared to normal breast tissue, the tumors expressed higher levels of the majority of epithelial markers and epithelial microRNAs, demonstrating apparent tumor epithelialization (Suppl.Fig.4,5A, Suppl.File1). Arguably, this could potentially be due to reorganization of diseased breast tissue via changes in composition of stromal components and should be investigated in the future. When analyzed only within tumors, ZEB1 and TCF4 levels were lower, while SNAI1 and TCF3 levels were higher in basal compared to other subtypes. In agreement with their antagonistic regulation, highest levels of epithelial-specific miR200s and miR203 in basal subtype correlated with lowest levels of ZEB1 and TCF4 (Suppl.Fig.4). Levels of miR34 family microRNAs did not change significantly across subtypes (Suppl.Fig.5B), indicating that increased SNAI1 level in basal tumors is not due to miR34 change.
ZEB1/2, TCF4 and SNAI2 expression is strongly correlated in breast tumors.
Simultaneous expression of multiple EMT-TFs in the mesenchymal cells lines (Fig.3A) prompted us to analyze correlation of expression among the EMT-TFs in the Breast TCGA dataset (Suppl.File2). SNAI2 expression has been previously shown to be restricted to mammary myoepithelial/basal cells(31, 39). TCF3 was the only EMT-TF which correlated positively with SNAI2 in normal breast tissue (R=0.33), while the other six EMT-TFs correlated negatively with both SNAI2 and TCF3 (Fig.3B, neon green arrow & boxes), suggesting that TCF3 expression is also myoepithelial-specific. The strongest correlations in normal tissue were observed between ZEB1/2 and TCF4 (R~0.95) (Fig.3B, red boxes), which together modestly correlated with TWIST1/2 (R=0.53-0.65). TWIST1/2 most strongly correlated with each other (R=0.81) and with ZEB1/2, TCF4 and SNAI1 (R=0.48-0.65) (Fig.3B, blue boxes), while SNAI1 equally weakly correlated with TWIST1/2, ZEB1/2 and TCF4 (R=0.4-0.51) (Fig.3B, dark green box).
Surprisingly, patterns of SNAI2 correlation with ZEB1/2 and TCF4 flipped from negative in normal tissue to strong positive in breast tumors (Fig.3B, red boxes). Similarly, negative correlation of TCF3 with the six EMT-TFs observed in normal tissue mostly dissipated in tumors. The reversal in SNAI2 (and to some extent in TCF3) correlations with other EMT-TFs could be due to myoepithelial lineage depletion in tumors as discussed below. Strong correlation between ZEB1/2 and TCF4 observed in normal tissue maintained in all subtypes (R=0.68-0.93, red boxes), while patterns of TWIST1/2 and SNAI1 correlations were preserved only in luminal A (Fig.3B, blue and dark green boxes, respectively).
These data indicate that in breast tumors the expression of ZEB1/2, TCF4 and SNAI2 is highly correlated (R=0.6-0.93). In addition, these four EMT-TFs moderately correlate with TWIST1/2 (R=0.26-0.71), suggesting that the EMT program is tightly coordinated by several EMT-TFs.
Correlation of EMT-TFs with epithelial markers in normal breast tissue and primary breast tumors.
To assess potential functional role of the EMT-TFs in regulation of EMT markers, their correlation of expression was computed for normal breast tissue and breast tumors (Suppl.File2). Three published breast EMT signatures(9, 40, 41) were used to select EMT markers. We narrowed our analysis to epithelial genes, assuming that they are specific to tumor cells, while mesenchymal markers could be expressed in partially EMT’ed tumors as well as in mesenchymal stromal components(42, 43). Additionally, most of the EMT-TFs are bona fide direct repressors of epithelial genes(4, 14), while their effect on upregulation of mesenchymal genes could be indirect. To avoid subtype-specific effects we further focused on a 100 core epithelial markers expressed at similar levels across all four intrinsic subtypes (<2-fold difference, p<0.01) (see Methods).
As expected, majority of the epithelial markers correlated negatively with multiple EMT-TFs in normal breast tissue (Suppl.Fig.6). Though, the number of negatively correlated epithelial genes was similar for each of ZEB1/2, TCF4 and TWIST1/2 (66-76 genes), stronger correlations were observed for ZEB1/2 and TCF4 (R ~ −0.9 to −0.55) followed by TWIST1/2 (R ~ −0.7 to −0.3) and SNAI1 (R ~ −0.4 to −0.25) (Fig.3C,Suppl.Fig.6). As discussed above, SNAI2 and TCF3 were specific for myoepithelial lineage and correlated positively with majority of the epithelial markers in normal breast tissue (Fig.3C,Suppl.Fig.6).
Luminal A tumors featured number of pairwise negative gene correlations similar to normal tissue (Fig.3C). However, the relative strengths of these correlations were markedly different (Suppl.Fig.6). Fewer epithelial gene-EMT-TF correlations of weaker strengths were observed in the other subtypes (Fig.3C). Remarkably, SNAI2 and TCF3 switched from generally strong positive correlation with epithelial markers in normal tissue to mostly negative correlation in tumors (Fig.3C,Suppl.Fig.6). This phenomenon is likely a result of depletion of myoepithelial lineage and its gene expression patterns in tumors, which are proposed to originate from luminal progenitors (44). In all tumor subtypes ZEB1/2 and TCF4 negatively correlated with a larger number of epithelial genes compared to other EMT-TFs, and with stronger R values (Fig.3C,Suppl.Fig.6). Interestingly, higher expression of SNAI1 and TCF3 in basal subtype (Suppl.Fig.4) did not result in an increase in number of their negatively correlated core epithelial genes compared to the other subtypes or compared to the other EMT-TFs (Fig.3C,Suppl.Fig.6). This data is in agreement with recent report indicating that SNAI1 appears to exert little effect on EMT program in tumors in vivo(45).
Altogether, these results indicate that among the eight EMT-TFs, ZEB1/2 and TCF4 negatively correlated with more core epithelial genes and with stronger R values in normal breast and in tumors, suggesting that they may play more prominent role in their repression.
ZEB1 plays dominant role in maintenance of the mesenchymal/stem-like phenotype of human mammary epithelial cells.
Low expression of many EMT-TFs in primary breast tumors(46) prompted us to investigate if any of them are enriched in mammary stem cells, which constitute a small cell population in tissue and are considered to be potential precursors of tumor initiating cells(47). Heterogeneous cell populations of primary human mammary epithelial cells (HMECs) and derivative immortalized human mammary epithelial cell line HMLE contain a small proportion of cells (0.5-3%) expressing CD24−/CD44+ (or CD24low/CD44hi) stemness profile(31, 48). When purified by flow cytometry, CD24−/CD44+ cells exhibit hallmarks of stem cells, i.e. greatly increased capacity to form mammospheres in vitro and the ability to reconstitute the mammary gland in mice(31). We purified the CD24−/CD44+ subpopulation from HMLE cells (Suppl.Fig.7) and confirmed that they adopt the mesenchymal state (Fig.4A-C), consistent with previous reports(31, 48). Among the eight EMT-TFs, expression levels of ZEB1 (~500-fold), ZEB2 (~90-fold), TWIST1 (~60-fold) and TWIST2 (~10-fold) were markedly higher in CD24−/CD44+ compared to CD24+/CD44− HMLE cells, while expression of SNAI1/2 and TCF3/4 was not significantly different (Fig.4A,B). Concomitantly, miR200s/203/205 levels were drastically lower in CD24−/CD44+ cells (Fig.4C). These results suggest that though the stemness phenotype could be induced by forced expression of many EMT-TFs, including SNAI1/2 and TCF3/4 (Fig.1C and (31)), the subpopulation of normal mammary stem-like cells naturally overexpresses ZEB1/2 and TWIST1/2.
Fig.4. Normal mammary stem-like CD24−/CD44+ HMLE cells adopt mesenchymal phenotype and express high levels of ZEB1/2 and TWIST1/2.
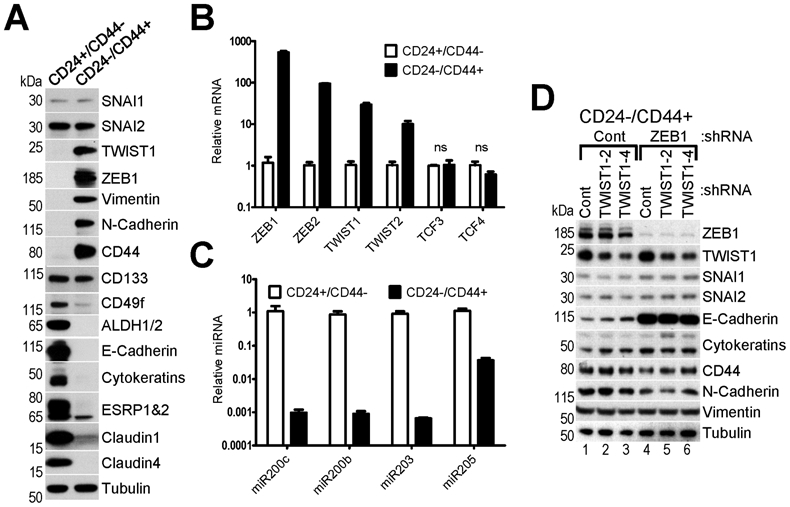
A, WB analysis and B,C RT-qPCR analysis of EMT-TFs, EMT and stemness markers and epithelial-specific microRNAs in two FACS-purified subpopulations of HMLE cells (CD24+/CD44− and CD24−/CD44+). Tubulin – loading control for WB. (B,C) The mean values of expression in CD24+/CD44− cells were set to 1. The data are mean ± SEM of three biological replicates, ns – non-significant. D, Knockdown of ZEB1 but not TWIST1 in CD24−/CD44+ cells leads to partial MET. WB analysis of cells infected with lentiviruses encoding Control shRNA and two shRNAs against TWIST1, which were then reinfected with lentiviruses encoding Control and shRNA against ZEB1.
To determine which of these two families of EMT-TFs plays a major role in the maintenance of the mesenchymal phenotype, we knocked down highly expressed members TWIST1 or ZEB1, or both in CD24−/CD44+ HMLE cells. Depletion of TWIST1 resulted in a modest increase in E-cadherin expression, while knockdown of ZEB1 led to significant upregulation of E-cadherin and downregulation of CD44 and N-cadherin (Fig.4D). Interestingly, depletion of TWIST1 in addition to ZEB1 did not result in further inhibition of EMT (Fig.4D). These results suggest that ZEB1 plays a more dominant role in maintenance of the mesenchymal phenotype of normal mammary stem-like cells.
ZEB1 plays a major role in maintenance of the mesenchymal phenotype of breast cancer cells.
Remarkably, the expression pattern of EMT-TFs in normal mammary stem-like cells resembled the pattern observed in fully EMT’ed breast cancer cell lines (compare Fig.4A and Fig.3A). To establish a functional requirement of individual EMT-TFs for maintenance of the mesenchymal phenotype in tumor cells, we initially focused on the mesenchymal cell line MDA231LN(49). Parental MDA231 is the most commonly used cell line in mouse xenograft models of breast cancer metastasis due to its superior invasiveness(35). Its derivative subline MDA231LN, isolated from a mouse lymph node metastasis, acquired increased metastatic potential(49). Importantly, parental and MDA231LN cells express high levels of SNAI1/2 and ZEB1/2, but low levels of TWIST1/2 and TCF4 (Fig.3A,Suppl.Fig.2A-D), which makes them suitable for interrogation of the individual roles of SNAI1/2 and ZEB1/2 in EMT. Individual knockdowns of each of these four EMT-TFs by two independent shRNAs showed that while depletion of SNAI2 and ZEB2 resulted in a modest upregulation of E-cadherin, indicative of partial MET, suppression of ZEB1 led to dramatic re-expression of epithelial E-cadherin and cytokeratins, and significant downregulation of the mesenchymal marker CD44 (Fig.5A). In agreement with previous report(50), knockdown of SNAI1 was insufficient to significantly upregulate E-cadherin. Interestingly, knockdown of each of the four EMT-TFs in MDA231LN cells did not significantly affect the expression of the others (Fig.5A). This is likely because none of the epithelial-specific microRNAs (miR200s/203/205), which may target multiple EMT-TFs, was reactivated by either SNAI or ZEB knockdowns (not shown).
Fig.5. ZEB1 plays a major role in control of EMT in mesenchymal-like breast cancer cells.
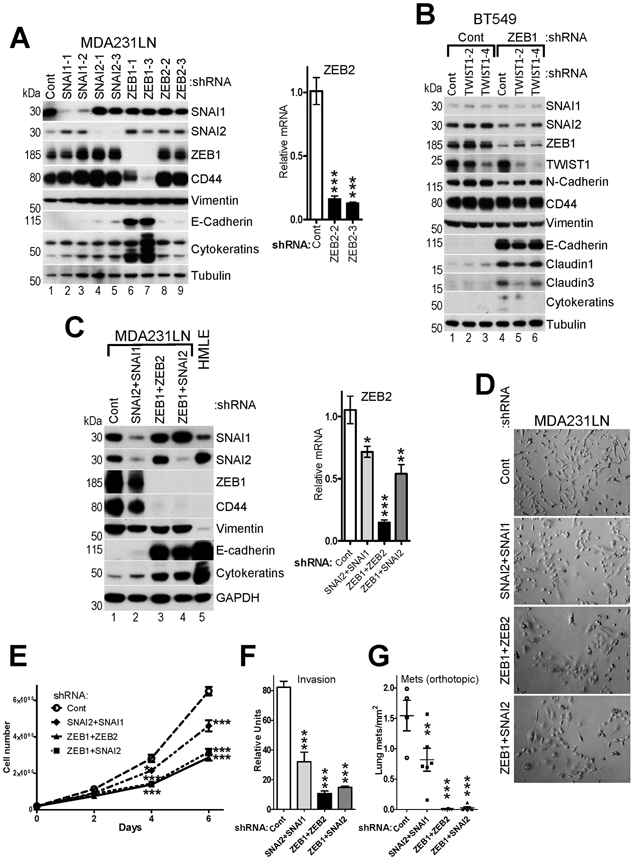
A, Knockdowns of individual EMT-TFs in MDA231LN cells cause partial MET. WB (left panel) and RT-qPCR (right panel) analyses of MDA231LN cells infected with pGIPZ lentiviruses encoding Control and two different shRNAs against each SNAI1/2 and ZEB1/2. B, WB analysis of BT549 cells infected with pTRIPZ lentiviruses encoding Control shRNA and two different shRNAs against TWIST1, which were then reinfected with pTRIPZ lentiviruses encoding Control shRNA and shRNA against ZEB1. C, WB (left panel) and RT-qPCR (right panel) analyses of MDA231LN cells expressing Control, SNAI2-3 or ZEB1-3 shRNAs as shown in (A) reinfected with lentiviruses encoding Control shRNA or shRNA against SNAI1, ZEB2 or SNAI2, respectively. D, Phase contrast images, E, cell proliferation analysis, F, in vitro invasion assay of the indicated MDA231LN double knockdown cell lines described in (C). G, Cell lines described in (C) were injected into mammary fad pad of NSG mice, and the number of metastases was quantified in mouse lungs 5 weeks post-injection (n=4 for control and n=6 per other groups). The data are mean ± SEM of three biological replicates (A,C,E,F). ANOVA with Dunnett’s post-test, ** - p<0.01, *** - p<0.001.
Next, we used mesenchymal BT549 cells, which express high levels of ZEB1/2, TWIST1/2 and TCF4 and low levels of SNAI1/2 (Fig.3A,Suppl.Fig.2A-D). Knockdown of TWIST1 by two different shRNAs showed no significant change in expression of EMT markers, while depletion of ZEB1 led to marked re-expression of epithelial E-cadherin and claudins, and downregulation of the mesenchymal marker N-cadherin (Fig.5B). Depletion of TWIST1 in addition to ZEB1 did not result in further MET.
Together, these results indicate that ZEB1 is one of the most critical regulators of the mesenchymal phenotype in breast cancer cells.
Cooperation of multiple EMT-TFs in control of cell proliferation, invasion and metastasis of MDA231LN cells.
Though ZEB1 played the dominant role, partial MET induced by knockdowns of ZEB2 and SNAI2 in MDA231LN cells indicated that multiple EMT-TFs contribute to the mesenchymal phenotype. To confirm this, we generated MDA231LN cells with double knockdowns of SNAI1+2, ZEB1+2 and ZEB1+SNAI2. Depletion of ZEB2 or SNAI2 in addition to ZEB1 resulted in more profound suppression of EMT and reactivation of E-cadherin to the levels observed in epithelial HMLE cells, while double SNAI1+2 knockdown had only a partial effect (Fig.5C). The knockdowns also led to upregulation of epithelial cytokeratins and downregulation of mesenchymal CD44 and vimentin. Notably, miR200s microRNAs were not reactivated in response to double knockdowns (not shown), indicating that certain EMT-TFs targets may remain stably silenced even in the absence of EMT-TFs expression.
Interestingly, knockdowns of SNAI1+2 and especially ZEB1+2 and ZEB1+SNAI2 led to dramatic change in cell morphology (Fig.5D) and decreased cell proliferation in vitro (Fig.5E). Furthermore, progressive suppression of EMT among the double knockdown lines correlated with decreased invasion in vitro (Fig.5F) and a drastic suppression of spontaneous lung metastasis in a mouse orthotopic xenograft model (Fig.5G).
Together, these results indicate that multiple EMT-TFs expressed in MDA231LN cells contribute to the mesenchymal phenotype characterized as mesenchymal gene expression pattern, increased invasion and metastasis. At the same time, the relative contribution between them varies significantly, with ZEB1 being the most potent.
ZEB1 expression correlates with increased metastatic potential of MDA231 sublines.
Dr. Joan Massague’s group has selected several independent sublines of MDA231 cells with much higher propensity to metastasize and colonize specific distant organs, including lung (MDA231-LM2), bone (MDA231-BoM) and brain (MDA231-BrM)(51). To determine if their super-metastatic potential correlates with EMT, we analyzed expression of EMT-TFs and EMT markers in these sublines. Parental MDA231 cells obtained from Joan Massague’s lab and from ATCC were used as controls. Surprisingly, we found that ZEB1 protein but not mRNA levels were significantly upregulated in all three super-metastatic sublines (Fig.6A,B), indicating that the increase in ZEB1 expression have occurred at the post-transcriptional level. Consistently, miR200s microRNAs were down-regulated in these cells (Fig.6C). In addition, ZEB2 and TCF4 mRNAs were upregulated in BrM cells and in BoM and BrM cells, respectively (Fig.6B). Surprisingly, SNAI1 and to a lesser extent SNAI2 proteins were down-regulated in super-metastatic sublines compared to controls (Fig.6A). Increased ZEB1 expression correlated with progressive EMT of super-metastatic sublines, i.e. elevated levels of vimentin and decreased levels of cytokeratins, claudin4 and epithelial-specific splice isoforms of CD44 (Fig.6A), and increased cell proliferation (Fig.6D).
Fig.6. Expression of ZEB1 correlates with increased metastatic potential of MDA231 cells.
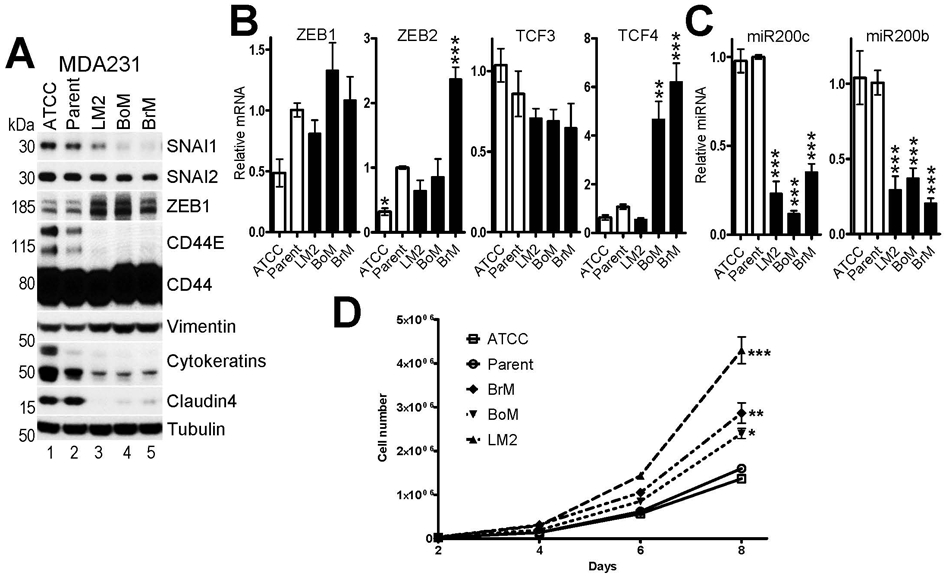
A, WB and B,C RT-qPCR analyses of EMT-TFs, EMT markers and epithelial-specific microRNAs in two parental MDA231 sublines (ATCC and Parent, see main text) and three super-metastatic MDA231 sublines with increased metastatic potential to the lung (LM2), bone (BoM) and brain (BrM). The mean values of expression in Parent MDA231 cells were set to 1. D, cell proliferation assay of the indicated MDA231 sublines. The data are mean ± SEM of three biological replicates. ANOVA with Dunnett’s post-test, * - p<0.05, ** - p<0.01, *** - p<0.001.
These results indicate that increased metastatic potential of the MDA231-LM2, MDA231-BoM and MDA231-BrM sublines is associated with progressive EMT and elevated levels of ZEB1/2 and TCF4.
Suppression of ZEB1+2 and EMT at early stages of tumor growth blocks metastasis in an orthotopic xenograft mouse model.
To determine the effect of ZEB1+2 knockdown and EMT suppression on different stages of metastasis we generated MDA231LN cells with Dox-inducible shRNAs targeting ZEB1+2. Addition of Dox led to dramatic down-regulation of ZEB1 protein within 5 days and concomitant suppression of EMT, loss of spindle-like cell morphology, activation and membranous localization of E-cadherin, and reversal in expression of the CD24−/CD44+ stemness markers (Fig.7A,B,Suppl.Fig.8). Interestingly, wash-out of Dox and culturing cells for 16 days resulted in re-expression of ZEB1 and reactivation of the EMT program, indicating that MET was reversible (Fig.7A, lanes 11-12).
Fig.7. Suppression of ZEB1+2 and EMT inhibits lung metastasis at multiple stages.

A, WB analysis of the indicated EMT markers in MDA231LN cells during time-course induction of Dox-regulated shRNAs against ZEB1+2. Cells were lysed at the indicate time points post Dox addition (lanes 1-10,13). Dox was washed out on day 20 and the cells were lysed 8 and 16 days post Dox withdrawal (lanes 11,12). B, Phase contrast images (top panel) and immunofluorescence for E-Cadherin pseudocolored cyan (bottom panel) of cells shown in (A). C, Timeline for mouse groups M, M>E and E used in xenograft experiments described in (D-J). D-J, MDA231LN/shZEB1+2 cells described in (A&B) were injected into mammary fad pad (D-G), or into tail vein (H-J) of NSG mice. D, WB analysis of the indicated EMT markers in primary tumors of four mice from each of the three groups at endpoint of the orthotopic injection experiment. E, H&E staining of primary tumors (top panel) and lungs (bottom panel) from (D). White arrows point to macrometastases. F-J, Dynamics of tumor growth (F and I) and the number of lung metastases at endpoints (G and J) for orthotopic and intravenous injection experiments, respectively (n=5 mice per group). H, Representative bioluminescent images for the tail vein injection. The data are mean ± SEM. ANOVA with Dunnett’s post-test, * - p<0.05, ** - p<0.01, *** - p<0.001, ns – non-significant.
To test the requirement of EMT for initial stages of metastasis we utilized a mouse orthotopic xenograft model and conditionally inhibited ZEB1+2 expression in MDA231LN cells in a time-resolved manner. The timeline of the experiment is schematically outlined in Fig.7C. The control mouse group never received Dox and cells remained in the mesenchymal state for the whole duration of the experiment (M group). For other two groups Dox (resulting in ZEB1+2 KD) was administered at different time points relative to the injection time (day 0): one week before injection to inhibit EMT before the start of tumor growth (E group) and one week after injection (M>E group) to inhibit EMT after the primary tumor has formed. Five weeks after injection the tumors and lungs were removed and analyzed. Western blot analysis on tumor lysates from the E and M>E groups confirmed equally efficient ZEB1 knockdown and suppression of EMT in vivo, e.g. reactivation of epithelial markers and downregulation of CD44 (Fig.7D). Suppression of EMT in vivo did not significantly affect primary tumor growth (Fig.7F) or histology (Fig.7E, top). However, EMT inhibition led to a >100-fold decrease in the number of lung metastases (Fig.7G,E bottom). Unexpectedly, lung metastasis was also greatly suppressed in the M>E (mesenchymal-turned-epithelial) group (Fig.7G), indicating that EMT suppression during the initial stage of tumor growth is effective in preventing metastasis. These results showed that inhibition of ZEB1+2 and EMT at early stages of tumor growth is sufficient to effectively block metastasis.
Expression of ZEB1+2 and EMT are critical for the extravasation stage of metastasis and are dispensable for outgrowth of seeded macrometastases.
To test a role of ZEB1+2 in outgrowth of already disseminated cells, we injected MDA231LN cells directly into the blood stream and allowed them sufficient time to establish micrometastases(52), i.e. one week before inhibiting EMT (Fig.7C). To monitor the injected tumor cell survival and growth bioluminescence in the lungs was measured on the day of injection (day 0) and then weekly for three weeks (Fig.7H,I). As expected, the control M group produced high metastatic outgrowth (Fig.7H-J). The epithelial E group yielded significantly reduced metastasis, suggesting that the extravasation or/and later stages of metastasis are inhibited by suppression of EMT. Surprisingly, the M>E group, in which mesenchymal cells were switched to the epithelial state one week after injection, produced a similar number of metastases to the control M group (Fig.7I,J), indicating that one week time was sufficient for mesenchymal cells to extravasate and that EMT is not essential for outgrowth of seeded macrometastases. These results indicate that ZEB1+2 expression and EMT are necessary for the transition of circulating cancer cells into the lungs but are not required for the macrometastatic outgrowth.
Discussion
Here we characterized EMT-TF network expression across cancer models and human breast tumors, and used conditional knockdown to demonstrate a critical role of ZEB1/2 as regulators of metastasis. We established ZEB1/2, SNAI2, TCF4 and TWIST1/2 as commonly and cooperatively upregulated during forced EMT of normal mammary epithelial cells. A likely mechanism for the observed secondary upregulation is repression of the common negative regulators of EMT-TFs, the epithelial-specific microRNAs miR200s/203/205 that can target multiple EMT-TFs (9, 20, 21, 53), summarized in Fig.2F. We found that in addition to ZEB1/2 and SNAI2, miR200c could target TCF4 (Fig.2).
We correlated expression of EMT-TFs and EMT markers in multiple breast cancer cells lines. All analyzed mesenchymal cell lines expressed high levels of ZEB1/2, indicating that full EMT is consistently associated with activation of ZEB1/2. In addition to ZEB1/2, mesenchymal cell lines expressed high levels of other EMT-TFs, including in various combinations SNAI2, TCF4, TWIST1/2 and SNAI1, suggesting that a combined action of multiple EMT-TFs may contribute to full EMT.
Furthermore, we identified ZEB1/2, TCF4 and SNAI2 as a group of EMT-TFs with tightly coordinated expression in major breast cancer subtypes (Fig.3B-C). ZEB1/2 and TCF4 negatively correlated with a larger number of core epithelial genes and with stronger R values compared to the other EMT-TFs, suggesting that they may play a more prominent role in EMT.
The most striking difference of the patient data with the cell line panel was the absence of true mesenchymal/full EMT subtype in primary breast tumors, suggesting that the mesenchymal cells are likely present in small subpopulations within a tumor or appear at later stages of tumor progression. Generally low expression of EMT-TFs in the bulk of epithelial tumors(46) suggests that EMT is likely to occur in distinct cell subpopulations that play key roles in tumor progression and metastatic dissemination(27). Since cancer stem cells (CSC) have been proposed as the driving force of tumorigenesis and the source of metastases(47), it is, therefore, logical to consider CSC as preferentially malleable to EMT. In fact, stemness and EMT features often significantly overlap, but the degree to which EMT and stemness are coupled or independent is a topic of current debate(54). Remarkably, activation of the EMT program in non-transformed epithelial cells confers properties of stem cells that are associated with the CD24−/CD44+ phenotype(31, 42). We found that ZEB1/2 and TWIST1/2 are significantly upregulated in CD24−/CD44+ mammary stem-like cells and showed that ZEB1 plays a major role in maintenance of the mesenchymal status of CD24−/CD44+ stem-like cells. Consistent with this, ZEB1/2 were significantly enriched in CD24−/CD44+ cancer stem cell populations isolated from primary human breast cancers(55).
Remarkably, the expression pattern of EMT-TFs in CD24−/CD44+ normal mammary stem-like cells resembles those of the mesenchymal breast cancer cells, suggesting that the latter may represent tumor cells permanently locked in the ultimate stemness phenotype due to compounding genetic alterations. We showed that ZEB1 knockdown led to marked suppression of EMT in mesenchymal breast cancer cells. At the same time, depletion of SNAI1/2, ZEB2 or TWIST1 had only minor effect, indicating that ZEB1 is the major TF required for full EMT. These results are corroborated by earlier genetic studies in mouse embryonic fibroblasts (MEFs), which similarly express Zeb1/2 and Snai1/2. Thus, deletion of Zeb1 in MEFs led to reactivation of E-cadherin and downregulation of vimentin(56), while knockout of Snai1 did not(57). We showed that though knockdown of ZEB1 alone was sufficient to induce MET, simultaneous depletion of ZEB1 with ZEB2 or SNAI2 led to more profound suppression of EMT. Interestingly, knocking out Zeb1 in a mouse pancreatic tumor model resulted in a strong but still incomplete suppression of metastatic ability(30), suggesting that other EMT-TFs may partially substitute for Zeb1 loss.
Importantly, there was a distinct difference in the control of the EMT network between normal mammary epithelial cells and mesenchymal breast cancer cells. While normal epithelial cells induced to undergo EMT maintained positive feedback for coordinated upregulation of multiple endogenous EMT-TFs mediated through down-regulation of their inhibitory microRNAs, mesenchymal cells induced to undergo MET demonstrated impaired or lost co-regulation of EMT-TFs. Similarly, deletion of Zeb1 or Snai1 in MEFs did not significantly affect expression of the other EMT-TFs(56, 57). We did not detect miR200s reactivation in response to ZEB1 knockdown in MDA231LN cells despite overt MET and reactivation of E-cadherin, suggesting that cancer cells could maintain silenced chromatin at specific targets even in the absence of an active repressor. This may have important implications for future therapeutic treatment strategies aimed at reverting EMT. Thus, addition of epigenetic drugs targeting DNA methylation or histone modifications could facilitate reactivation of the epithelial program(53, 58).
MDA231 cells have been widely used as a model human cell line to study invasion and breast cancer metastasis due to its ability to produce spontaneous metastases in immunocompromised mice. Parental MDA231 cells are considered moderately metastatic, and several successful attempts have been made to select super-metastatic variants of MDA231 cells with preferential organ-specific seeding to lung, bone or brain(51). We showed that all three MDA231 super-metastatic sublines express higher levels of ZEB1 and lower levels of miR200s, indicating that their increased metastatic potential is associated with enhanced EMT.
Using Dox-inducible shRNAs, we showed that depletion of ZEB1+2 and suppression of EMT at initial stages of tumor growth blocks spontaneous lung metastasis, suggesting that administration of anti-EMT drugs at early tumor stages could potentially be effective in preventing metastasis. This is consistent with established role of ZEB1 in local invasion and transendothelial migration(59). Next, we showed that EMT is required for later stages of metastasis, after cancer cells enter the circulation. Depletion of ZEB1+2 and suppression of EMT before tail vein injection significantly reduced lung metastasis of MDA231LN. Previously, we have shown that MDA231LN cells injected into the tail vein form micrometastases and resume proliferation in lungs by around three days post-injection(52). Remarkably, silencing of ZEB1+2 and inhibition of EMT seven days after intravenous injection of MDA231LN cells did not reduce metastatic outgrowth in lungs, indicating that EMT may facilitate earlier extravasation and establishment of micrometastases but is dispensable for progression toward macrometastases(27, 60).
Supplementary Material
Acknowledgements:
We thank Sarah McLaughlin, Dr. Katherine Brundage, Fengju Chen, Yuriy Loskutov, Kristina Marinak-Whately and Sila Yanardag for technical assistance, Drs. Steven Frisch and Peter Stoilov for critically reading the manuscript, Drs. Joan Massague, Gregory Goodall and Russ Carstens for providing reagents.
Financial support: This work was supported by grants from Susan G. Komen Foundation (KG110350 to AVI, KG100539 to ENP) American Cancer Society (ACS-IRG-09-061-01 to AVI), NIH/NCRR (P20RR016440 to AVI), NIH/NCI (R01CA148671 to ENP), and from NIH/NCI (P30CA125123 to CJC) and in part from NIH/NIGMS (WV-INBRE P20GM103434). The WVU Flow Cytometry and Animal Models and Imaging Facilities were supported by NIH/NIGMS grants CoBRE P30GM103488, WV-INBRE P20GM103434 and WV-CTSI U54GM104942.
Footnotes
Competing interests: The authors declare no potential conflicts of interest.
References
- 1.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. [DOI] [PubMed] [Google Scholar]
- 2.Mittal V Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol. 2018;13:395–412. [DOI] [PubMed] [Google Scholar]
- 3.Sobrado VR, Moreno-Bueno G, Cubillo E, Holt LJ, Nieto MA, Portillo F, et al. The class I bHLH factors E2-2A and E2-2B regulate EMT. Journal of cell science. 2009;122(Pt 7):1014–24. [DOI] [PubMed] [Google Scholar]
- 4.de Herreros AG, Peiro S, Nassour M, Savagner P. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. Journal of mammary gland biology and neoplasia. 2010;15(2):135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Non-redundant functions of EMT transcription factors. Nature cell biology. 2019;21(1):102–12. [DOI] [PubMed] [Google Scholar]
- 6.Akalay I, Janji B, Hasmim M, Noman MZ, Andre F, De Cremoux P, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer research. 2013;73(8):2418–27. [DOI] [PubMed] [Google Scholar]
- 7.Diaz-Lopez A, Diaz-Martin J, Moreno-Bueno G, Cuevas EP, Santos V, Olmeda D, et al. Zeb1 and Snail1 engage miR-200f transcriptional and epigenetic regulation during EMT. International journal of cancer. 2015;136(4):E62–73. [DOI] [PubMed] [Google Scholar]
- 8.Guaita S, Puig I, Franci C, Garrido M, Dominguez D, Batlle E, et al. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. The Journal of biological chemistry. 2002;277(42):39209–16. [DOI] [PubMed] [Google Scholar]
- 9.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(35):15449–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreno-Bueno G, Cubillo E, Sarrio D, Peinado H, Rodriguez-Pinilla SM, Villa S, et al. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer research. 2006;66(19):9543–56. [DOI] [PubMed] [Google Scholar]
- 11.Smit MA, Geiger TR, Song JY, Gitelman I, Peeper DS. A Twist-Snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Molecular and cellular biology. 2009;29(13):3722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer research. 2008;68(19):7846–54. [DOI] [PubMed] [Google Scholar]
- 13.Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer research. 2011;71(1):245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9(6):582–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. The Journal of biological chemistry. 2008;283(22):14910–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nature cell biology. 2009;11(12):1487–95. [DOI] [PubMed] [Google Scholar]
- 17.Saini S, Majid S, Yamamura S, Tabatabai L, Suh SO, Shahryari V, et al. Regulatory Role of mir-203 in Prostate Cancer Progression and Metastasis. Clin Cancer Res. 2011;17(16):5287–98. [DOI] [PubMed] [Google Scholar]
- 18.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature cell biology. 2008;10(5):593–601. [DOI] [PubMed] [Google Scholar]
- 19.Zhang P, Wang L, Rodriguez-Aguayo C, Yuan Y, Debeb BG, Chen D, et al. miR-205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat Commun. 2014;5:5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu YN, Yin JJ, Abou-Kheir W, Hynes PG, Casey OM, Fang L, et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene. 2013;32(3):296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moes M, Le Bechec A, Crespo I, Laurini C, Halavatyi A, Vetter G, et al. A novel network integrating a miRNA-203/SNAI1 feedback loop which regulates epithelial to mesenchymal transition. PloS one. 2012;7(4):e35440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding X, Park SI, McCauley LK, Wang CY. Signaling between transforming growth factor beta (TGF-beta) and transcription factor SNAI2 represses expression of microRNA miR-203 to promote epithelial-mesenchymal transition and tumor metastasis. The Journal of biological chemistry. 2013;288(15):10241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibbons DL, Creighton CJ. Pan-cancer survey of epithelial-mesenchymal transition markers across the Cancer Genome Atlas. Dev Dyn. 2018;247(3):555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, et al. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun. 2016;7:10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imani S, Hosseinifard H, Cheng J, Wei C, Fu J. Prognostic Value of EMT-inducing Transcription Factors (EMT-TFs) in Metastatic Breast Cancer: A Systematic Review and Meta-analysis. Sci Rep. 2016;6:28587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(33):13820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beerling E, Seinstra D, de Wit E, Kester L, van der Velden D, Maynard C, et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016;14(10):2281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT, MET, Plasticity, and Tumor Metastasis. Trends in cell biology. 2020;30(10):764–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nature cell biology. 2017;19(5):518–29. [DOI] [PubMed] [Google Scholar]
- 31.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Addison JB, Koontz C, Fugett JH, Creighton CJ, Chen D, Farrugia MK, et al. KAP1 promotes proliferation and metastatic progression of breast cancer cells. Cancer research. 2015;75(2):344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt JM, Panzilius E, Bartsch HS, Irmler M, Beckers J, Kari V, et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 2015;10(2):131–9. [DOI] [PubMed] [Google Scholar]
- 35.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer cell. 2006;10(6):515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PloS one. 2009;4(7):e6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paiva CE, Drigo SA, Rosa FE, Moraes Neto FA, Caldeira JR, Soares FA, et al. Absence of transforming growth factor-beta type II receptor is associated with poorer prognosis in HER2-negative breast tumours. Ann Oncol. 2010;21(4):734–40. [DOI] [PubMed] [Google Scholar]
- 38.Dong M, How T, Kirkbride KC, Gordon KJ, Lee JD, Hempel N, et al. The type III TGF-beta receptor suppresses breast cancer progression. The Journal of clinical investigation. 2007;117(1):206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8(2):149–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Creighton CJ, Gibbons DL, Kurie JM. The role of epithelial-mesenchymal transition programming in invasion and metastasis: a clinical perspective. Cancer Manag Res. 2013;5:187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med. 2014;6(10):1279–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem cells (Dayton, Ohio). 2010;28(8):1435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Block CJ, Dyson G, Campeanu IJ, Watza D, Ratnam M, Wu G. A stroma-corrected ZEB1 transcriptional signature is inversely associated with antitumor immune activity in breast cancer. Sci Rep. 2019;9(1):17807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes & development. 2014;28(11):1143–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ni T, Li XY, Lu N, An T, Liu ZP, Fu R, et al. Snail1-dependent p53 repression regulates expansion and activity of tumour-initiating cells in breast cancer. Nature cell biology. 2016;18(11):1221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soini Y, Tuhkanen H, Sironen R, Virtanen I, Kataja V, Auvinen P, et al. Transcription factors zeb1, twist and snai1 in breast carcinoma. BMC cancer. 2011;11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medema JP. Cancer stem cells: the challenges ahead. Nature cell biology. 2013;15(4):338–44. [DOI] [PubMed] [Google Scholar]
- 48.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145(6):926–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jenkins DE, Hornig YS, Oei Y, Dusich J, Purchio T. Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivo detection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res. 2005;7(4):R444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. 2007;26(49):6979–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kozyreva VK, Kiseleva AA, Ice RJ, Jones BC, Loskutov YV, Matalkah F, et al. Combination of Eribulin and Aurora A Inhibitor MLN8237 Prevents Metastatic Colonization and Induces Cytotoxic Autophagy in Breast Cancer. Molecular cancer therapeutics. 2016;15(8):1809–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taube JH, Malouf GG, Lu E, Sphyris N, Vijay V, Ramachandran PP, et al. Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Sci Rep. 2013;3:2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fabregat I, Malfettone A, Soukupova J. New Insights into the Crossroads between EMT and Stemness in the Context of Cancer. J Clin Med. 2016;5(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2014;2(1):78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, El-Naggar S, Darling DS, Higashi Y, Dean DC. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development (Cambridge, England). 2008;135(3):579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rowe RG, Li XY, Hu Y, Saunders TL, Virtanen I, Garcia de Herreros A, et al. Mesenchymal cells reactivate Snail1 expression to drive three-dimensional invasion programs. J Cell Biol. 2009;184(3):399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, Ruh M, et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med. 2015;7(6):831–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Molecular biology of the cell. 2009;20(8):2207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Esposito M, Mondal N, Greco TM, Wei Y, Spadazzi C, Lin SC, et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nature cell biology. 2019;21(5):627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings in this study are available from the corresponding author upon reasonable request.