

Abstract
The mutational genetic landscape of colorectal cancer has been extensively characterized, however, the ability of “cooperation response genes” to modulate the function of cancer “driver” genes remains largely unknown. In this study, we investigate the role of aryl hydrocarbon receptor (AhR), a ligand activated transcription factor, in modulating oncogenic cues in the colon. We show that intestinal epithelial cell targeted AhR knockout (KO) promotes the expansion and clonogenic capacity of colonic stem/progenitor cells harboring ApcS580/+; KrasG12D/+ mutations by upregulating Wnt signaling. The loss of AhR in the gut epithelium increased cell proliferation, reduced mouse survival rate, and promoted cecum and colon tumorigenesis in mice. Mechanistically, the antagonism of Wnt signaling induced by Lgr5 haploinsufficiency attenuated the effects of AhR KO on cecum and colon tumorigenesis.
Keywords: aryl hydrocarbon receptor, Lgr5, stem cells, Apc/Kras, colon cancer
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer deaths, in men and women combined, affecting 6% of the U.S. population. A hallmark of this disease is the progressive accumulation of chromosomal and microsatellite instability, and CpG island methylator phenotype, leading to transformation of normal epithelial cells to adenocarcinoma. Approximately, 75% of CRC cases occur sporadically and only 5–10% result from hereditary genetic mutations (1). Generally, in CRC, genetic alterations involve the inactivation of tumor suppressor genes and DNA mismatch repair genes, and/or activation of oncogenes. It is estimated that 2–8 driver gene mutations are required to promote CRC development (2,3), even though the median number of nonsynonymous mutations in sporadic CRC is 66 mutations per tumor (4), in which Adenomatous polyposis coli (Apc) serves as an initiating event, accompanied by mutations of other common genes, such as Kirsten RAS (Kras), Sma- and Mad-related protein 4 (SMAD4) and TP53.
Apc is a tumor suppressor gene encoding a 312 kDa protein that plays an important role in regulating the Wnt signaling pathway, cell migration and adhesion, transcriptional activation, apoptosis and DNA repair (5,6). The multiple functions of Apc are achieved by binding to various protein partners, including β-catenin, Axin, CtBP, Asefs, IQGAP1, EB1 and microtubes (7). The most highly characterized consequence of Apc inactivation in CRC is aberrant activation of the Wnt signaling pathway. In the absence of Wnt ligands, the primary Wnt signaling effector β-catenin is sequestered and targeted for proteasomal degradation in the cytosol by a multiprotein destruction complex, which contains the scaffold components Axin, Apc, GSK3β and CK1α/ϵ kinases. Inactivation of Apc by mutations, deletions, and loss of heterozygocity, leads to an accumulation of β-catenin in the nucleus. Nuclear β-catenin then interacts with Tcf4 (transcription factor) to mediate the transcription of target genes, such as c-myc, cyclin D1, Ascl2 and EphB (8,9). In human CRC, Apc point mutations occur primarily in a mutation cluster region (MCR, codons 1286–1513), generating premature stop codons or frameshift mutations, resulting in the deletion of the C-terminal region of the Apc protein. Approximately 80% of sporadic CRC individuals harbor at least one inactivating Apc mutation (10), which serves as one of the earliest events driving normal-to-adenoma transition. Kras is another commonly mutated driver gene involved in the progression of benign tumors (adenomas) or hyperplastic aberrant crypt focus (ACF). Mutations of this gene occur in approximately 30 to 50% of CRC cases (11). Mutant Kras is also associated with poor prognosis and CRC metastasis (11,12).
With respect to CRC driver genes, the contribution of cooperation response genes, capable of modifying the initiation and/or progression of cancer remains largely unknown. From this perspective, the aryl hydrocarbon receptor (AhR) is increasingly recognized as an important regulator of CRC (13–16). AhR is a ligand activated bHLH transcription factor, capable of promiscuously recognizing small molecules ranging from environmental pollutants as well as dietary and gut microbiota derived tryptophan metabolites, thus acting as an environmental sensor that integrates exogenous environmental stimuli and host response. In the absence of dietary and/or microbial ligands, AhR remains bound to several chaperon proteins in the cytoplasm including heat shock proteins 90, p23 and immunophilin related protein XAP2. Upon ligand binding, cytosolic AhR translocates to the nucleus and forms a heterodimer with the AhR nuclear translocator (ARNT) protein, which then interacts with AhR response elements (AREs) with the core sequence 5’-TNGCGTG-3’ or 5’-CACGCNA-3’ on the promoters of AhR target genes (17). Our lab and others have previously demonstrated that AhR KO promotes colitis-associated colon cancer (14,16). However, the effect of AhR deletion targeted to the epithelium in a genetically susceptible colon tumorigenesis model has not been investigated to date. Therefore, we generated inducible and colon epithelial cell targeted ApcS580/+; KrasG12D/+ with or without AhR. In the context of mutant Apc and Kras, via activation of Wnt signaling to promote colon tumorigenesis, we identified a novel inhibitory role for the AhR and our results support targeting the AhR as a promising strategy for CRC chemoprevention and/or treatment.
Methods
Mice
Animals were housed under conventional conditions, adhering to the guidelines approved by the Institutional Animal Care and Use Committee at Texas A&M University. Lgr5-EGFP-IRES-CreERT2, CDX2P-CreERT2, AhRf/f, KrasLSL G12D/+, and ApcS580/+ mouse strains have all been previously described. Specifically, Lgr5-EGFP-IRES-CreERT2 mice express EGFP and CreERT2 in crypt base columnar cells in the intestine under control of the Lgr5 promoter (18). CDX2P-CreERT2 mice express CreERT2 throughout the entire intestinal epithelium under the control of the CDX2 promoter (19,20). AhRf/f mice carry loxP sites bordering exon 2 of the AhR gene, with recombination resulting in generation of a premature stop codon at codon 29 (21). KrasLSL G12D/+ mice carry a latent point mutant Kras allele and Cre mediated recombination results in the expression of constitutively active KRas (22). In addition, ApcS580/+ mice carry loxP sites bordering exon 14 of the Apc gene, thus recombination results in an inactivating frame shift mutation at codon 580 (23). Mice were intraperitoneally injected with 2.5 mg of tamoxifen (Sigma, T5648) dissolved in corn oil (25 mg/ml) once a day for four consecutive days. Mice were maintained on an AIN-76A semi-purified diet (Research Diets, D12450B), fed ad libitum and housed on a 12 h light-dark cycle. For all experiments, littermate controls were cohoused with the knockout mice, unless specifically indicated.
For genotyping analyses, DNA was extracted from tails using DNeasy Blood and Tissue Kit (Qiagen, 69506). PCR was subsequently performed using the following primer sets: AhR (5’-CAGTGGGAATAAGGCAAGAGTGA-3’ and 5’- GGTACAAGTGCACATGCCTGC-3’), CDX2P-Cre (5’- GGACTTGAGCAGCTAGCTGTGCAACTT-3’ and 5’-TGTCTCGTGCCTGGAATGACCTT-3’), Lgr5-EGFP (5’-CACTGCATTCTAGTTGTGG-3’ and 5’- CGGTGCCCGCAGCGAG-3’), Kras-G12D (5’-TGTCTTTCCCCAGCACAGT-3’, 5’-CTGCATAGTACGCTATACCCTGT-3’, and 5’-GCAGGTCGAGGGACCTAATA-3’), and Apc (5’- GTTCTGTATCATGGAAAGATAGGTGGTC-3’ and 5’-CACTCAAAACGCTTTTGAGGGTTGATTC-3’).
Colonic crypt and single cell isolation and cell sorting
Colonic crypt and single cell isolation were performed as previously described (16). Briefly, colons were isolated, washed with cold PBS without calcium and magnesium (PBS−/−), everted on a disposable blunt end needle (Instech Laboratories), and incubated in 15 mM EDTA in PBS−/− at 37°C for 35 min. Subsequently, following transfer to chilled PBS−/−, crypts were mechanically separated from the connective tissue by rigorous vortexing. Crypt suspensions were then dissociated to individual cells with 0.25% Trypsin-EDTA containing 200 U/ml DNase. Cell suspensions were subsequently filtered through a 40-μm mesh and GFP-expressing cells were collected. GFP positive cells were collected using a MoFlo Astrios Cell Sorter (Beckman Coulter) or Bio-Rad S3e Cell Sorter. Dead cells were excluded by staining with propidium iodide or 7-AAD. Sorted cells were collected in RNA lysis buffer (for RNA isolation) or crypt culture medium (for culturing).
Organoid cultures
Organoid culture from isolated cells was performed as previously described (16). Briefly, ISCs (GFFhigh), progenitor cells (GFPlow) or bulk colonocytes were centrifuged for 3 min at 500xg, resuspended in the appropriate volume of crypt culture medium (100–250 cells μl−1), then seeded (500 cells for ISC or progenitor cells, 1500 cells for bulk colonocytes) onto 30 μl Matrigel containing 1 μM Jagged-1 (Ana-Spec) in a flat bottom 24-well plate. Following Matrigel polymerization, cells were overlaid with 300 μl of crypt culture medium containing Advanced DMEM/F12 (Gibco) supplemented with 2 mM glutamax, penicillin/streptomycin, and 10 mM HEPES (ADF+), 50 ng ml−1 EGF (Life Technologies), 100 ng ml−1 Noggin (Peprotech), 10% R-spondin conditioned medium, 1 μM N-acetyl-l-cysteine (Sigma), 1X N2 (Life Technologies), 1X B27 (Life Technologies) and 50% Wnt conditioned medium as described previously (24) supplemented with 10 μM Y-27632 (Sigma Y0503), 1 μM Jagged-1 and 2.5 μM CHIR99021 (Stemgent, 04–0004). Y-27632, Jagged-1 and CHIR99021 were withdrawn from crypt culture medium 2 d after plating. In select experiments, colonic organoids were cultured in 50% WRN conditioned medium generated from L-WRN cells (ATCC, CRL-3276) and 10% FBS (WRN medium)(25). To measure organoid viability, CellTiter-Blue reagent (Promega) was used according to manufacturer’s instructions. For AhR-related treatments, DMSO or TCDD (10 nM) were added to cultures for 3 d. Fluorescence was measured on a CLARIOstar microplate reader. Cell cycle analysis of organoids was performed as previously described (16).
Gene recombination characterization
For assessment of AhR recombination in vitro, mouse organoids derived from sorted colonic stem cells were incubated with 10 nM TCDD for 1 d. Taqman assays were performed to examine the expression of AhR exon 2, and Cyp1a1 induction, a downstream biomarker of activated AhR. Recombination of ApcS580 and KrasLSL G12D was determined genetically by modifying a previously described multiplex PCR assay (26–28). PCR was performed using Hot-Start-Taq Blue Mastermix (Denville Scientific, CB4040–8) according to the manufacturer’s instructions for 35 cycles using the Apc and Kras primer sets: Apc (5’-GTTCTGTATCATGGAAAGATAGGTGGTC-3′, 5’-CACTCAAAACGCTTTTGAGGGTTGATTC −3′, and 5’-GAGTACGGGGTCTCTGTCTCAGTGAA-3’), and Kras (5’- GTCTTTCCCCAGCACAGTGC-3’, 5’- GCAGCGTTACCTCTATCGTA-3’, and 5’- AGCTAGCCACCATGGCTTGAGTAAGTCTGCA-3′).
Secondary murine organoid assay
For secondary organoid assays, organoids were pretreated with DMSO or 10 nM TCDD for 2 d and then were dissociated for 8 min in 0.25% Trypsin-EDTA at 37°C. Cell suspensions were filtered through a 20-μm mesh, centrifuged for 3 min at 500xg, and resuspended in cold ADF+ medium. Live cell density was assessed and 2500 live cells were seeded into 30 μl Matrigel in a flat bottom 24-well plate. Following Matrigel polymerization, cells were overlaid with 300 μl of crypt culture medium supplemented with 10 μM Y-27632, 1 μM Jagged-1 and 2.5 μM CHIR99021. Y-27632, Jagged-1 and CHIR99021 were withdrawn from crypt culture medium 2 d after plating. The crypt media was changed every 2 d. Organoids were quantified on day 5 of culture, unless otherwise specified.
Organoid conditioned media transfer experiments
HACKG colonic organoids were incubated with crypt culture medium (WREN) containing ADF+, EGF, LDN, R-Spondin, N2, B27, N-acetyl-l-cysteine, and Wnt conditioned medium or crypt culture medium without Wnt3a, R-Spondin1 and EGF (N) for 1 d at 37°C. The concentration of each component is annotated above. Supernatants were then transferred to culture passaged ACKG organoids. Fresh N medium was also used as a control. Organoids were imaged on day 4 of culture.
Tumor study
At 8 to 12 wks of age, male and female mice were administrated 100 mg/kg tamoxifen daily for 4 consecutive days. Mice were terminated 20 wk post last tamoxifen injection. Mice appearing severely moribund or with a body weight decreased by 20% relative to the highest body weight recorded during study were terminated. EdU was injected 2 h prior to termination to examine cell proliferation. On the day of euthanasia, the cecum and colon were harvested, colon lesions were measured, mapped, excised, routinely processed and paraffin embedded. Hematoxylin and eosin (H&E) stained sections were examined using light microscopy by a board-certified veterinary pathologist in a blinded manner. Histological lesions were categorized by predominant change. Specifically, early lesions exhibited mucosal hyperplasia. Mid-stage lesions included foci of adenomatous hyperplasia. Late stage lesions had progressed to adenocarcinoma of the colonic epithelium with invasion into the submucosa. Neoplastic cells were embedded in a proliferative fibrovascular stroma (see Supplemental Figure 3 for details). Tumor volume was calculated as length × width.
RNA isolation and quantitative real-time PCR
RNA isolation and integrity assessment was performed as previously described (16). Briefly, sorted cells or organoids were isolated using the Quick-RNA MicroPrep Kit (Zymo, R1050) and further processed using a DNA removal kit (DNA Free, Ambion, AM1906). RNA integrity was assessed on a Bioanalyzer 2100 (Agilent Technologies), quantified by Nanodrop and stored at −80°C. Real-time PCR was performed on a QuantStudio 3 System (Applied Biosystems) using TaqMan Universal PCR Master Mix (Applied Biosystems). Specific primer/probe mix for each gene was obtained from Applied Biosystems: AhR (Mm00478930_m1), Lgr5 (Mm00438890_m1), Cyp1a1 (Mm00487218_m1), Axin2 (Mm00443610_m1), FoxM1(Mm00514924_m1), Ccnd1 (Mm00432359_m1), and GAPDH (Mm99999915_g1).
Immunohistochemistry
In order to assess cell proliferation in the colon, mice were intraperitoneally injected with EdU 2 h prior to termination as previously described (29). Colonic cell proliferation was measured using the Click-IT EdU kit (Life Technologies, C10340). Antigen retrieval was performed by sub-boiling in 10 mmol/L sodium citrate (pH 6.0) for 20 min. Antibodies used were: mouse polyclonal antibody to β-catenin (BD, 610153) followed by Alexa-568 donkey anti-mouse secondary antibody (Life Technologies, A-10037), rabbit monoclonal antibody to non-phosphorylated β-catenin (Cell Signaling Technology, 8814S) followed by Alexa-555 donkey anti-rabbit secondary antibody (Life Technologies, A31572), FITC conjugated mouse anti-E-catenin (BD, 612130). Prolong Gold antifade with DAPI (Life Technologies, P36935) was used to coverslip the slides. Images of colonic crypts were captured on a Leica DMi8 TCS SPE spectral confocal microscope. Images were processed using ImageJ software (ImageJ 1.51n version). For enumeration of immunohistochemical staining, approximately 40 crypts were assessed from at least three animals per treatment.
Western blotting
Cell lysates from organoids or colon tumor tissues were subjected to standard SDS-PAGE and Western blotting procedures using primary antibodies against β-catenin (1:2000, BD 610154), non-phospho β-catenin (1:2000, 8814S, Cell Signaling Technology), pERK1/2 (1:1000, 4370S, Cell Signaling Technology), ERK1/2 (1:1000, 9107S, Cell Signaling Technology), pAKT (1:1000, 4056S, Cell Signaling Technology), and AKT (1:1000, 2920S, Cell Signaling Technology). Secondary anti-mouse or rabbit conjugated to horseradish peroxidase or secondary StarBright Blue 700 goat anti-mouse IgG (Bio-Rad, #12004159) were used to detect primary antibodies. Signal was imaged with the Bio-Rad Chemidoc System and protein bands quantified using Image Lab 6.0 (Bio-Rad).
RNAseq analysis
Libraries for RNA sequencing were made using 250 ng of total RNA with the QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina kit from Lexogen (Vienna, Austria) following manufacturer’s instructions. The molarity of each library was determined using the NEBNext Library Quant Kit for Illumina (NEB, Ipswich, MA) and the DNA High Sensitivity kit (Agilent, Santa Clara, CA). The libraries were then pooled in an equimolar fashion and sequenced on the NextSeq 500 for 75 bases, single end procedure. Demultiplexed reads from the sequencing runs were then mapped to the mouse genome (version GRCm38.94) using STAR (version 2.6.0) with default settings. The mapped reads were counted using HTSeq-count. Among 54,392 genes, we first filtered the genes that had a count of more than three for both groups in order to assess differential expression (DE). Subsequently, edgeR (software R package) was used to normalize the data and identify the DE genes. Data normalization was performed using the upper-quartile method to adjust for sequencing depth variation between samples. To determine the enrichment of intestinal stem cell signatures, or SANSOM APC Targets gene sets, unbiased gene set enrichment analysis was performed using Gene Set Enrichment Analysis (GSEA v3.0; http://www.broadinstitute.org/gsea) and normalized enrichment scores (NES) with FDRs were generated.
Data availability
The RNAseq data from this publication have been deposited in NCBI Sequence Read Archive with accession number PRJNA682633.
Statistics
Two-tailed Student’s t-tests were used for comparisons between two groups, unless otherwise specified. One-way ANOVA with Tukey’s multiple comparisons test were used for comparisons of more than 2 groups. Log-rank (Mantel-Cox) test was used for comparison of survival curves. All data are presented as mean ± SEM (standard error) and all analyses were conducted using Prism 8 statistical software (GraphPad Software, Inc.).
Results
Loss of AhR perturbs the functionality of colonic stem and progenitor cells.
In a previous study, we showed that AhR signaling regulates normal colonic stem and progenitor cell homeostasis (16), however, it remains to be determined if the effect is phenocopied in colonic stem and progenitor cells harboring ApcS580/+ and KrasG12D/+ mutations. To elucidate the role of AhR on a genetically induced CRC genetic background, we generated compound mutant mice carrying one floxed Apc allele (ApcS580, abbreviated as A) and a floxed, latently oncogenic allele of Kras (KrasLSL-G12D/+, abbreviated as K), with/without two alleles of AhR (AhRf/f, abbreviated as H). By crossing with CDX2P-CreERT2 (abbreviated as C) mice and upon injection of tamoxifen, the entire distal ileum, cecum and colonic epithelium becomes tumorigenic (26). We found that additional AhR deletion promoted organoid forming efficiency and growth of unsorted (bulk) colonocytes from ACK mice 3 weeks post tamoxifen injection (Figure 1A–C). Since crypt stem cells are the cells-of-origin of intestinal cancer (30), we further characterized the effect of AhR KO on the colonic stem and progenitor cell populations with AK mutations. For this purpose, Lgr5-GFP-IRES-CreERT2 (abbreviated as G) reporter mice were crossed with our compound mutant mice. Flow cytometry was used to sort colonic stem cells (defined as GFPhi) and progenitor cells (defined as GFPlow) based on GFP fluorescence intensity. Consistent with the results from unsorted bulk colonocytes, AhR KO promoted enhanced stemness, as measured by organoid forming efficiency and organoid size of both sorted AK colonic stem and progenitor cells (Figure 1D–I). We also validated the genetic mutations from organoids derived from sorted colonic stem or progenitor cells from compound mutant mice (Supplemental Figure 1).
Figure 1.
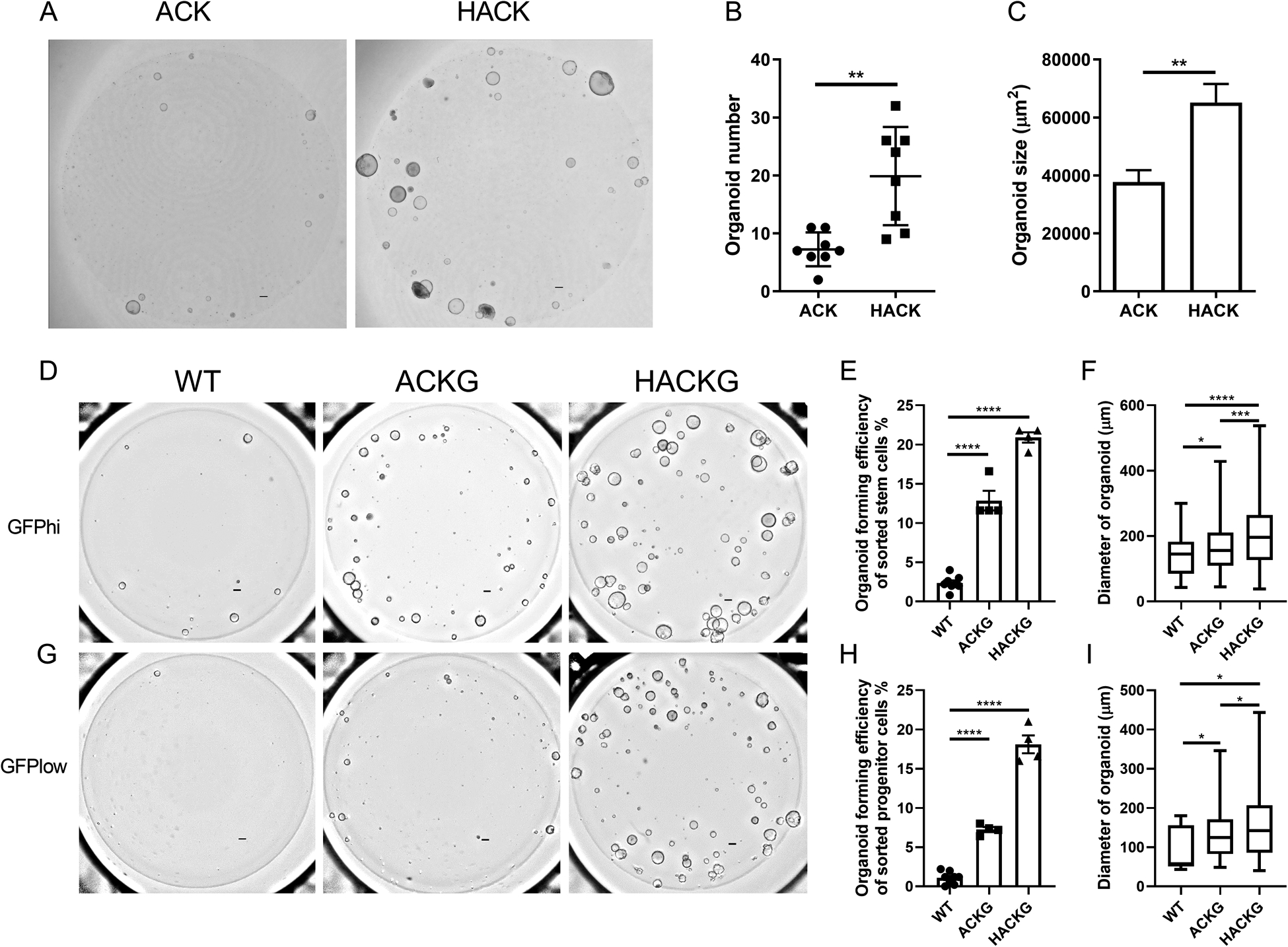
AhR KO promotes organoid growth harboring mutant Apc and Kras. (A) Representative brightfield images of organoids generated from individual bulk colonocytes isolated from 3 wk post tamoxifen treated ACK (ApcS580/+; KrasG12D/+; CDX2P-CreERT2) and HACK mice (AhRf/f; ApcS580/+; KrasG12D/+; CDX2P-CreERT2). Scale bar, 200 μm. (B-C) Quantification of organoid number and size at day 5 after plating, n=8 biological replicates per group from two mice, 2 independent experiments. (D) Representative brightfield images of organoids generated from sorted GFPhi stem cells isolated from 3 wk post tamoxifen treated ACKG (ApcS580/+; KrasG12D/+; CDX2P-CreERT2; Lgr5-GFP- CreERT2) and HACKG mice (AhRf/f; ApcS580/+; KrasG12D/+; CDX2P-CreERT2; Lgr5-GFP- CreERT2). Scale bar, 200 μm. (E-F) Quantification of organoid forming efficiency and size at day 5 after plating, n=4 or 8 biological replicates per group from one or two mice, 3 independent experiments. (G) Representative brightfield images of organoids generated from sorted GFPlow progenitor cells isolated from tamoxifen treated ACKG and HACKG mice. Scale bar, 200 μm. (H-I) Quantification of organoid forming efficiency and size at day 5 after plating, n=4 or 8 biological replicates per group from one or two mice, 3 independent experiments. *p<0.05, **p<0.01, ****p<0.0001.
AhR signaling modulates the percentage of colonic stem cells.
Since deletion of AhR enhanced the clonogenic capacity of organoids derived from Apc and Kras mutant mice, we hypothesized that the increased clonogenic capacity of unsorted colonocytes could be explained by an alteration in the percentage of colonic stem cells. qPCR analysis of colonic stem cell marker genes revealed that AhR KO increased the expression of Lgr5, Ascl2 and Axin2, and AhR activation by TCDD (a high affinity AhR ligand) decreased the expression of Lgr5 and Ascl2 (Figure 2A). In addition, consistent with our previous findings(16), the expression of FoxM1, a pivotal regulator of cell proliferation, was decreased upon ligand activation of the AhR (Figure 2B), and AhR KO increased FoxM1 and CCND1 expression, compared with AhR activation (Figure 2B&C). In addition, we consistently observed that AhR KO organoids expressed an enhanced GFP signal (Figure 2D), indicating that AhR KO promoted the percentage of colonic stem cells, since GFP serves as a proxy biomarker of colonic stem cells. To further confirm this finding, secondary organoid forming efficiency, a proxy for measuring the percentage of colonic stem cells, was determined. We found that TCDD pretreatment decreased secondary organoid forming efficiency in the ACK, but not in the HACK group, compared to DMSO pretreatment (Figure 2E&F). In contrast, AhR KO promoted secondary organoid formation. In addition, TCDD pretreatment had no effect on organoid size in either ACK or HACK group (Figure 2G), suggesting that the AhR activation effect was reversible. Moreover, RNAseq was performed using ACKG and HACKG organoids with or without TCDD treatment. Gene Set Enrichment Analysis (GSEA) revealed that an intestinal stem cell signature was enriched in DMSO treatment compared with TCDD treatment in ACKG organoids, and AhR KO group (HACKG) vs ACKG organoids (Figure 2H), suggesting that AhR signaling modulated the percentage of colonic stem cells. Immunohistochemical analysis of the colonic stem cell marker, GFP, revealed that AhR KO promoted the number of colonic stem cells per crypt (Figure 2I). Collectively, AhR activation by TCDD decreased the percentage of AK colonic stem cells, while AhR KO had the opposite effect.
Figure 2.
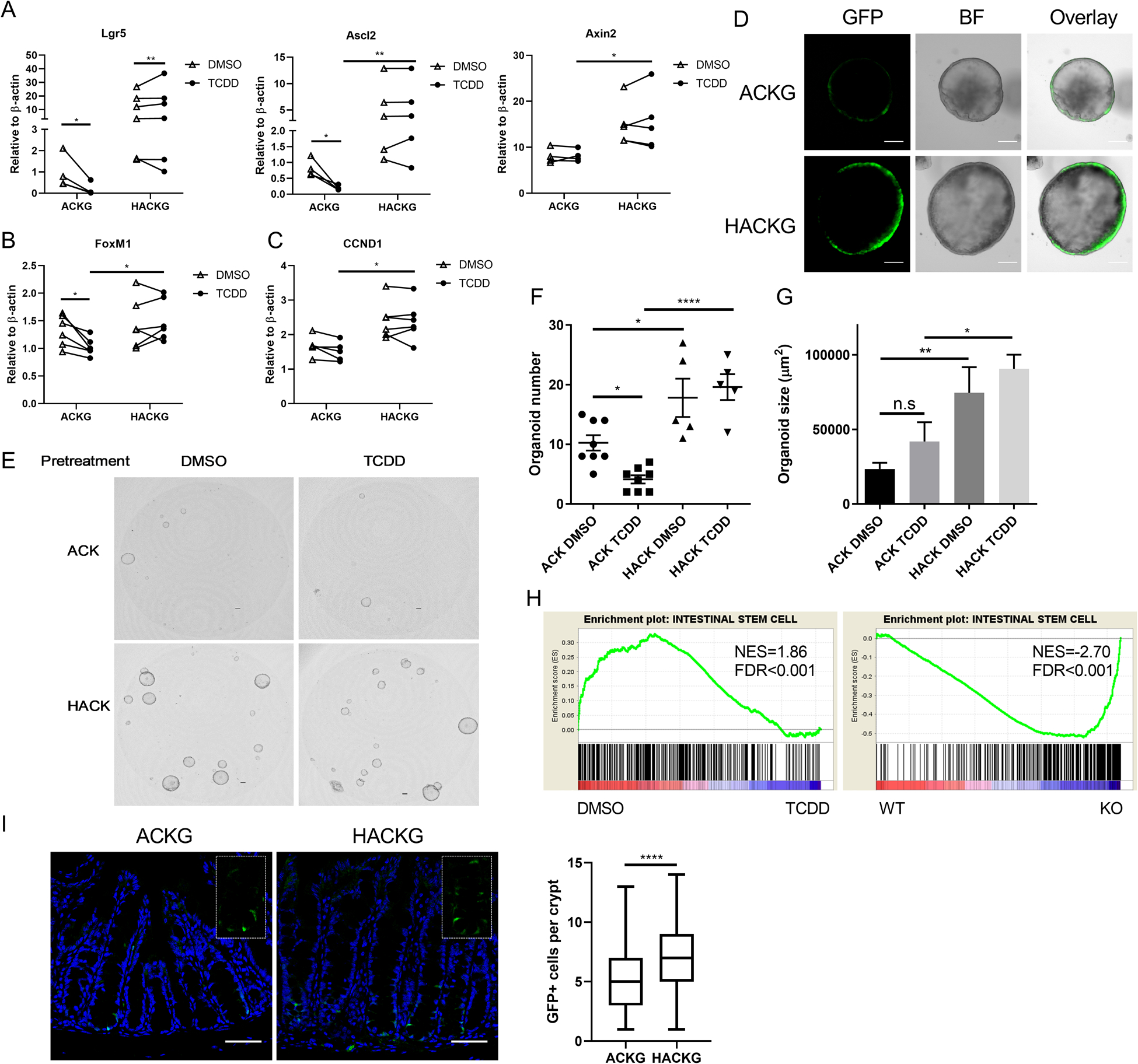
Deletion of AhR promotes colonic stem cell expansion. (A) mRNA expression of mouse colonic stem cell markers Lgr5 and Axin2 following exposure to 10 nM TCDD (AhR agonist) or DMSO (control) in organoids, n=4–6 mice per group. (B-C) mRNA expression of FoxM1 and CCND1 in organoids, n=5–6 mice per group. (D) Representative images of GFP+ organoids from sorted GFPhi stem cells. Scale bar, 50 μm. (E) Secondary organoid growth from ACK and HACK organoids pre-treated with DMSO or 10 nM TCDD. Scale bar, 200 μm. (F-G) Quantification of secondary organoid number and size at day 5 after plating, n=5 or 8 biological replicates per group from two mice. (H) Gene set enrichment plots of INTESTINAL STEM CELL between DMSO and TCDD treatment for 1 d in ACKG organoids or between WT (ACKG) and KO (HACKG) following TCDD treatment for 1 d, n=5 mice per group, n=5 mice per group. (I) Representative immunohistochemistry and box plots of GFP+ stem cells per crypt from 3 wk post tamoxifen treated ACKG and HACKG mice, n=79–94 GFP+ crypts from 5 mice per group. Scale 50 μm. *p<0.05, **p<0.01, ****p<0.0001.
AhR deficiency potentiates Wnt signaling.
Since Apc is a member of the core destruction complex of Wnt/β-catenin signaling, and oncogenic KrasG12D is constitutively activated, we assessed whether AK organoids could grow independently of exogenous Wnt3a, R-spondin 1 and EGF (WRE). By simply removing one or more of these niche factors, it is possible to determine whether intestinal epithelial cells and their growth dynamics are dependent on essential microenvironmental signals (31). Interestingly, wild type (WT, normal mouse with no Apc or Kras mutations) and ACKG organoids were not viable without WRE supplementation, while HACKG organoids exhibited normal growth independent of WRE (Figure 3A). We hypothesized that HACKG organoids may condition the media by producing soluble WRE growth factors in order to sustain their viability. To this end, we performed a conditioned culture medium transfer experiment. Although ACKG organoids exhibited normal growth in whole medium (designated as WREN), neither fresh medium without WRE (designated as N, noggin) nor N supernatant medium derived from HACKG organoids were capable of maintaining ACKG organoids, ruling out the contribution of an autocrine effect in HACKG organoids (Figure 3B). Next, we assessed β-catenin, pERK1/2 and pAKT levels in organoids with and without WRE for 2 d, when ACKG and HACKG organoids exhibited a normal morphology. Consistent with the impact of AhR deletion on organoid growth, withdrawal of WRE decreased non-phosphorylated β-catenin (active β-catenin) and pERK1/2 levels in the ACKG group, while HACKG organoids were still able to maintain the activation status of β-catenin even though pERK1/2 was significantly decreased (Figure 3C&D). The pAKT levels were not affected by withdrawal of WRE either in ACKG or HACKG organoids, and AhR KO significantly increased pAKT levels, but not total AKT (Figure 3C&D). Withdrawal of WRE had no effect on total β-catenin or total ERK1/2 in HACKG organoids (Figure 3E&F). In addition, GSEA analysis of Wnt target genes confirmed that AhR activation suppressed Wnt signaling while AhR KO upregulated Wnt signaling in organoids with ApcS580/+; KrasG12D/+ mutations (Figure 3G). These data indicate that AhR loss potentiates Wnt signaling to enable AK organoids to grow independently of stem cell niche-derived factors.
Figure 3.
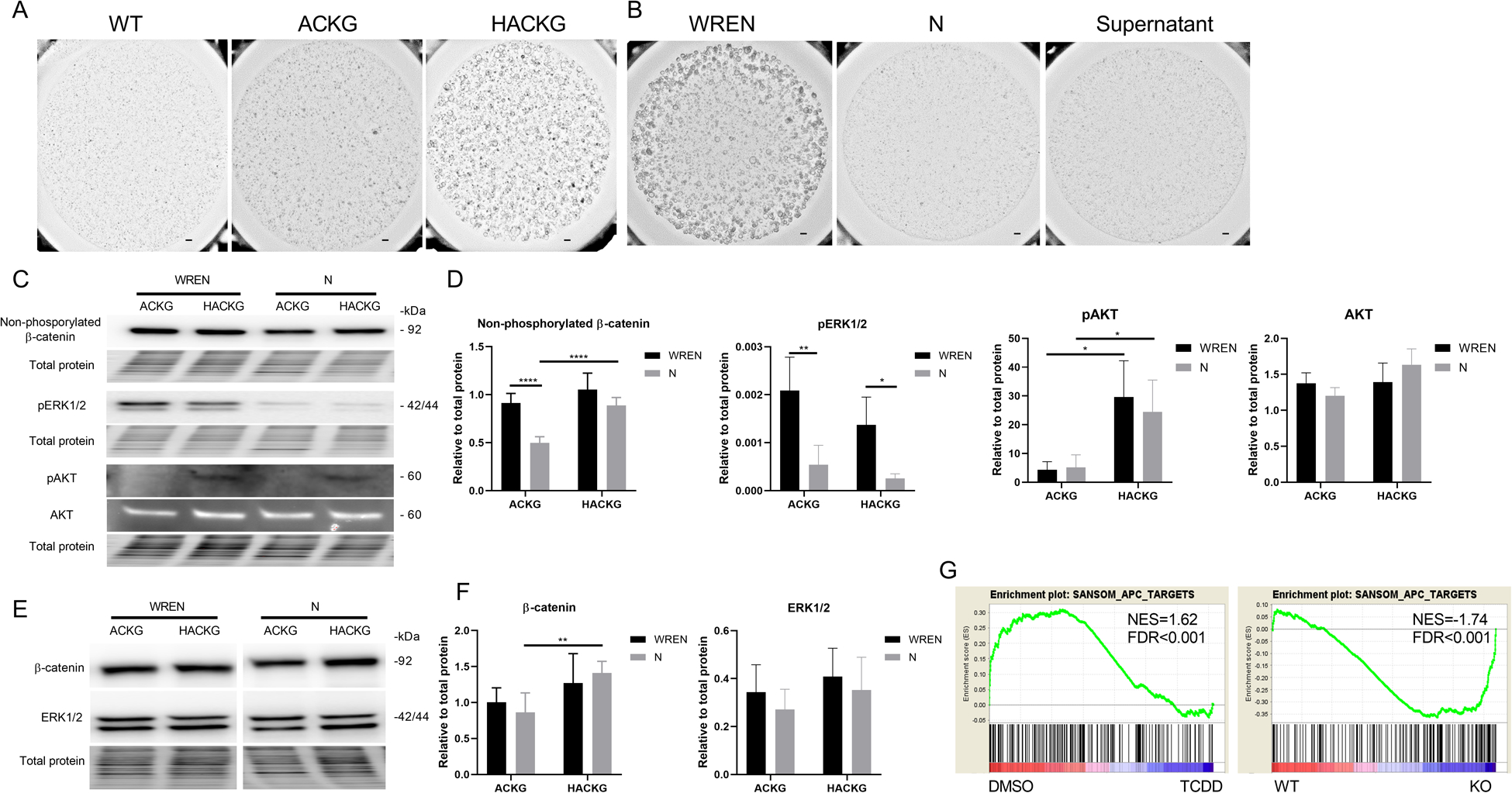
AhR deletion potentiates Wnt signaling. (A) Representative brightfield images of organoids with indicated genotypes grown for 4 d in medium without Wnt3a, R-spondin1, and EGF (N). Scale bar, 200 μm. (B) Representative brightfield images of ACKG organoids grown for 4 d in the indicated media. Scale bar, 200 μm. (C-D) Immunoblot analysis for non-phosphorylated (active) β-catenin, pERK1/2, pAKT, and AKT protein in organoids derived from 3 wk post tamoxifen treated mice, n=3–7 mice per group. (E-F) Immunoblot analysis for total β-catenin and ERK1/2 protein in organoids derived from 3 wk post tamoxifen treated mice, n=3–7 mice per group. (G) Gene set enrichment plots of SANSOM_APC_TARGETS between DMSO and TCDD treatment for 1 d in ACKG organoids or between WT (ACKG) and KO (HACKG) following TCDD treatment for 1 d, n=5 mice per group. *p<0.05, **p<0.01, ****p<0.0001.
AhR deletion promotes cell proliferation in vivo.
Since AhR KO potentiated Wnt signaling in organoids, we sought to determine whether AhR KO could directly modulate cell proliferation in vivo. For this purpose, our compound mutant mice were sacrificed 3 weeks after tamoxifen administration (Figure 4A). Strikingly, HACKG mice developed longer colonic crypts (Figure 4B&C). In addition, cell proliferation (marked by EdU+ cells), total and non-phosphorylated β-catenin levels were elevated following the deletion of AhR (Figure 4D–F, Supplemental Figure 2). To further determine how AhR status affected cell cycle phases, ACKG and HACKG organoids were cultured in WREN medium, and pulsed with EdU 1 h prior to harvest. Flow cytometric cell cycle analysis revealed that AhR KO significantly reduced the percentage of cells in G1 phase and increased the percentage of cells in S and G2/M phases (Figure 4G–H). These data confirm that AhR KO upregulated Wnt signaling to promote cell proliferation in vivo.
Figure 4.
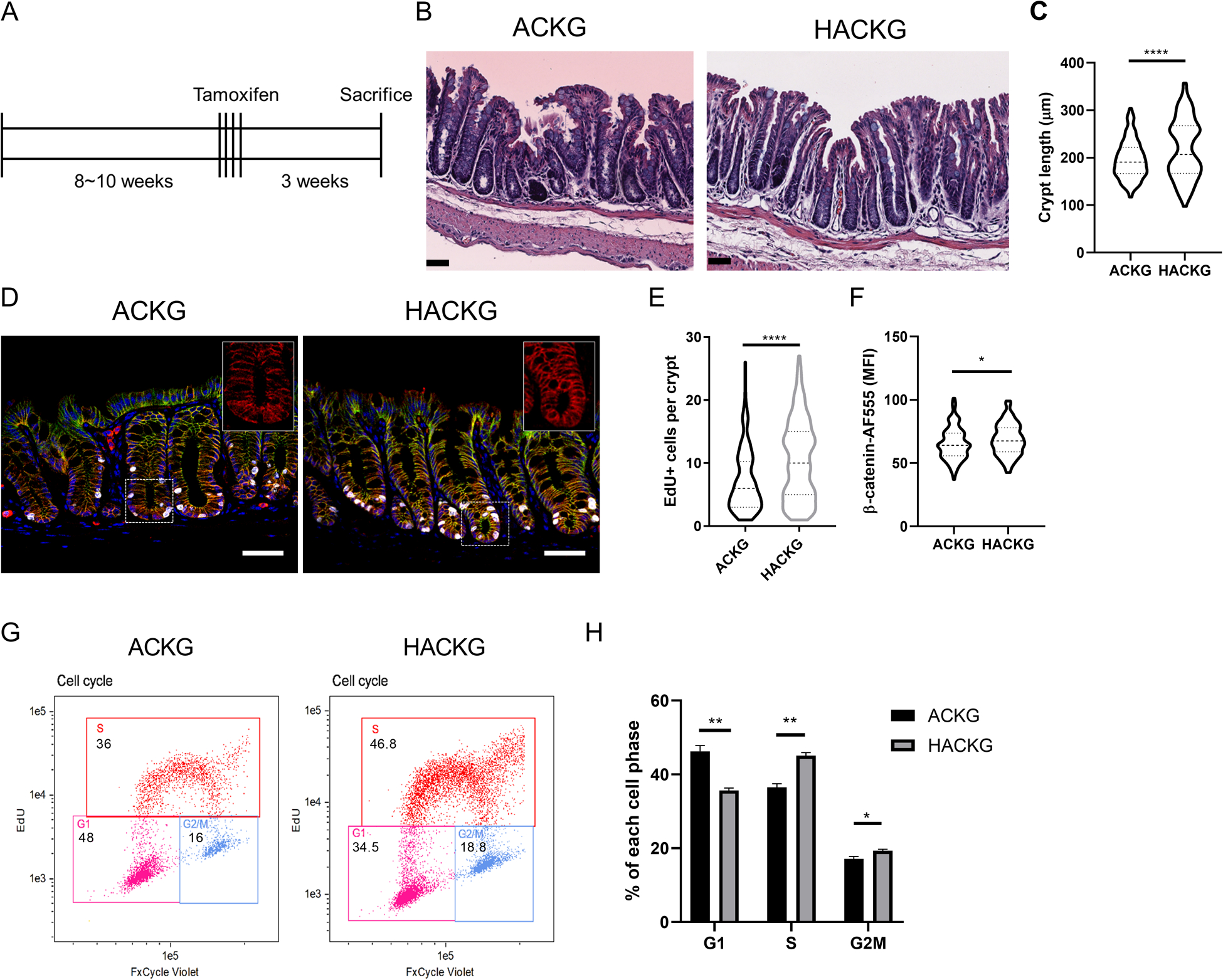
Effect of AhR deficiency on mouse colonic crypt-related phenotypes. (A) Schematic regimen for the 3 wk study period examining cell proliferation. (B) H&E staining of representative colon sections from ACKG and HACKG mice. Scale bar, 50 μm. (C) Quantification of colon crypt length in ACKG and HACKG mice, n=4 mice per group. (D) Representative images of total β-catenin and cell proliferation marked by EdU+ in the distal colon of ACKG and HACKG mice. Blue, DAPI; Green, E-cadherin; Red, β-catenin; and Gray, EdU. Scale bar, 50 μm. (E) Quantification of EdU+ cells per crypt and (F) mean fluorescence intensity (MFI) of β-catenin per crypt, n=4 mice per group. (G-H) Cell cycle analysis comparing ACKG and HACKG organoids, n=3 mice per group. *p<0.05, **p<0.01, ****p<0.0001.
AhR KO promotes colon tumorigenesis in compound mutant Apc and Kras mice.
Considering that AhR KO dysregulated colonic stem cells and promoted Wnt signaling and cell proliferation, we assessed whether AhR modulated colon tumorigenesis in compound mutant Apc and Kras mice. For this purpose, mice were terminated 20 wks after tamoxifen administration (Figure 5A). HACK mice had significantly lower survival rates, compared to ACK mice (Figure 5B). In addition, increased spleen weight was observed in HACK mice (Supplemental Figure 3A), consistent with enhanced tumor-associated inflammation. Similarly, mouse colon length was elongated in compound mutant mice, compared with WT mice and AhR KO further increased colon length (Supplemental Figure 3B), consistent with the increased rates of cell proliferation. Since genetic mutations were targeted to the cecum and colon (CDX2P-CreERT2), we monitored tumor development at these sites. AhR KO enhanced the development of cecum tumors, and significantly increased cecum weights (Figure 5C&D). Pathology analysis of cecal tumors indicated that AhR KO preferentially increased the number of mid-stage cecum tumors and the proportion of tumors in mid-stage (Supplemental Figure 3C–E). Next, we measured the development of colon tumors. The number of colonic tumors exhibited an obvious location-based distribution, with the preponderance of larger tumors occurring in the proximal colon (Figure 5E&F). Specifically, AhR deletion robustly increased the number of tumors in the distal colon, with a tendency to increase tumor volume (Figure 5G&H). Several mice (5/14 in ACK, and 10/17 in HACK) developed very severe tumors in the proximal colon, and it was not feasible to accurately quantify the number of tumors in all of these mice. Instead, we quantified the percentage of involved surface area in the proximal colon occupied by tumors. AhR KO significantly increased the percentage of proximal colon occupied by tumors (Figure 5I). Pathological analysis of colon tumors indicated that AhR KO preferentially increased the number of mid-stage colon tumors and had no effect on the proportion of tumors in each stage (Supplemental Figure 3F&G). Moreover, crypt length in the tumor uninvolved colon was significantly longer in HACK mice, compared with ACK mice at 20 wks post tamoxifen (Figure 5J&K).
Figure 5.
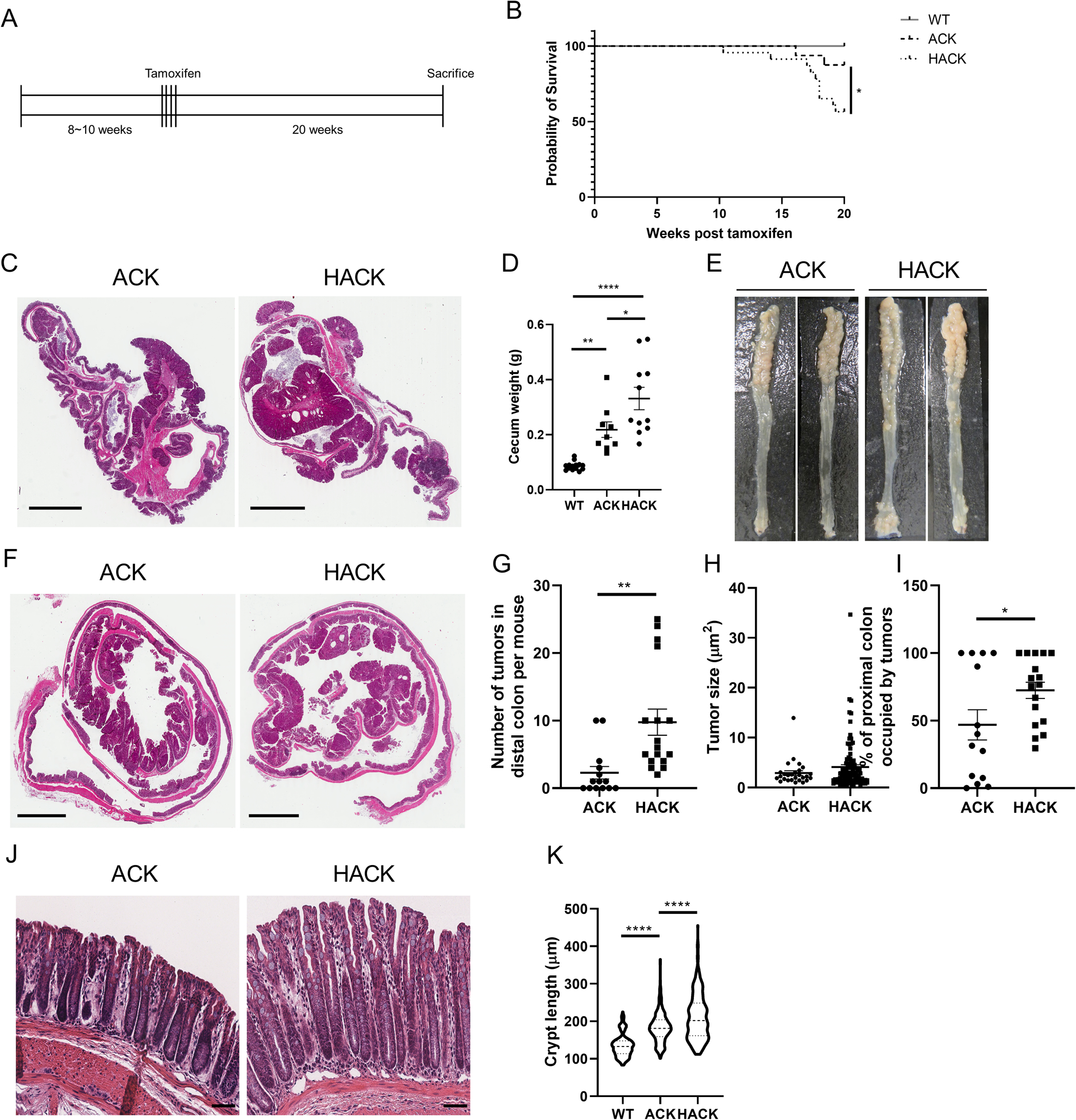
AhR KO promotes colorectal tumor growth in ApcS580/+; KrasG2D/+; CDX2P-CreERT2 (ACK) mice. (A) Schematic regimen for the induction of colon tumorigenesis in a genetically susceptible mouse model. At 8~10 wk of age, inducible ACK (n=16) and HACK (n=23) mice were administrated tamoxifen for 4 consecutive days to induce gene mutations. Mice were terminated 20 wk after the final tamoxifen injection. (B) Kaplan-Meier survival plot indicating percentage survival over a 20 wk period, WT (no mutations, n=29), ACK (n=16), and HACK (n=23). (C-D) Cecum tumor growth in ACK versus HACK mice, n=9–14 mice per group. Scale bar, 4 mm. (E) Representative colons from ACK and HACK mice 20 wk post final tamoxifen injection. (F) H&E staining of representative Swiss roll colon sections. Scale bar, 3 mm. (G) Quantification of the number of tumors and (H) tumor size in the distal colon, n=14–17 mice per group. (I) Quantification of tumor coverage in the proximal colon, n=14–17 mice per group. (J) H&E staining of representative colon uninvolved tissue sections. Scale bar, 50 μm. (K) Quantification of crypt length in the colon, n=4 mice per group. *p<0.05, **p<0.01, ****p<0.0001.
Analysis of β-catenin in tumor bearing mice revealed that non-phosphorylated β-catenin levels were significantly increased in colonic uninvolved regions in HACK mice compared with ACK mice (Figure 6A&B). AhR deletion promoted cell proliferation in uninvolved crypts 20 wks post tamoxifen injection, while the total β-catenin level remained unchanged (Supplemental Figure 4A–C). Moreover, non-phosphorylated β-catenin levels were also increased in tumors from HACK mice, even though total β-catenin levels were unchanged (Figure 6C&D, Supplemental Figure 4D&E). In particular, the number of tumor cells with nuclear β-catenin was much higher in HACK mice compared with ACK mice (Figure 6C and Supplemental Figure 4D). However, total β-catenin and cell proliferation were not altered in tumors upon comparison between ACK and HACK mice (Supplemental Figure 4F). Interestingly, colonic cell proliferation (number of EdU+ cells) was not correlated with total β-catenin levels (Supplemental Figure 4G). We also analyzed the expression of β-catenin and ERK1/2 proteins in proximal colon tumors and observed that non-phosphorylated β-catenin levels were significantly higher in the HACK group while total β-catenin, pERK1/2 or total ERK1/2 remained unaltered (Figure 6E–G). In addition, the growth of organoids derived from HACK proximal tumors was enhanced relative to ACK tumor organoids (Figure 6H). Interestingly, tumor derived organoids grown in medium without WRE supplementation lost the wildtype Apc allele possibly via loss of heterozygosity, although Kras was not affected (Supplemental Figure 4H&I). Collectively, our findings indicate that AhR KO upregulated Wnt signaling to promote colon tumorigenesis in a genetic colon tumor model.
Figure 6.
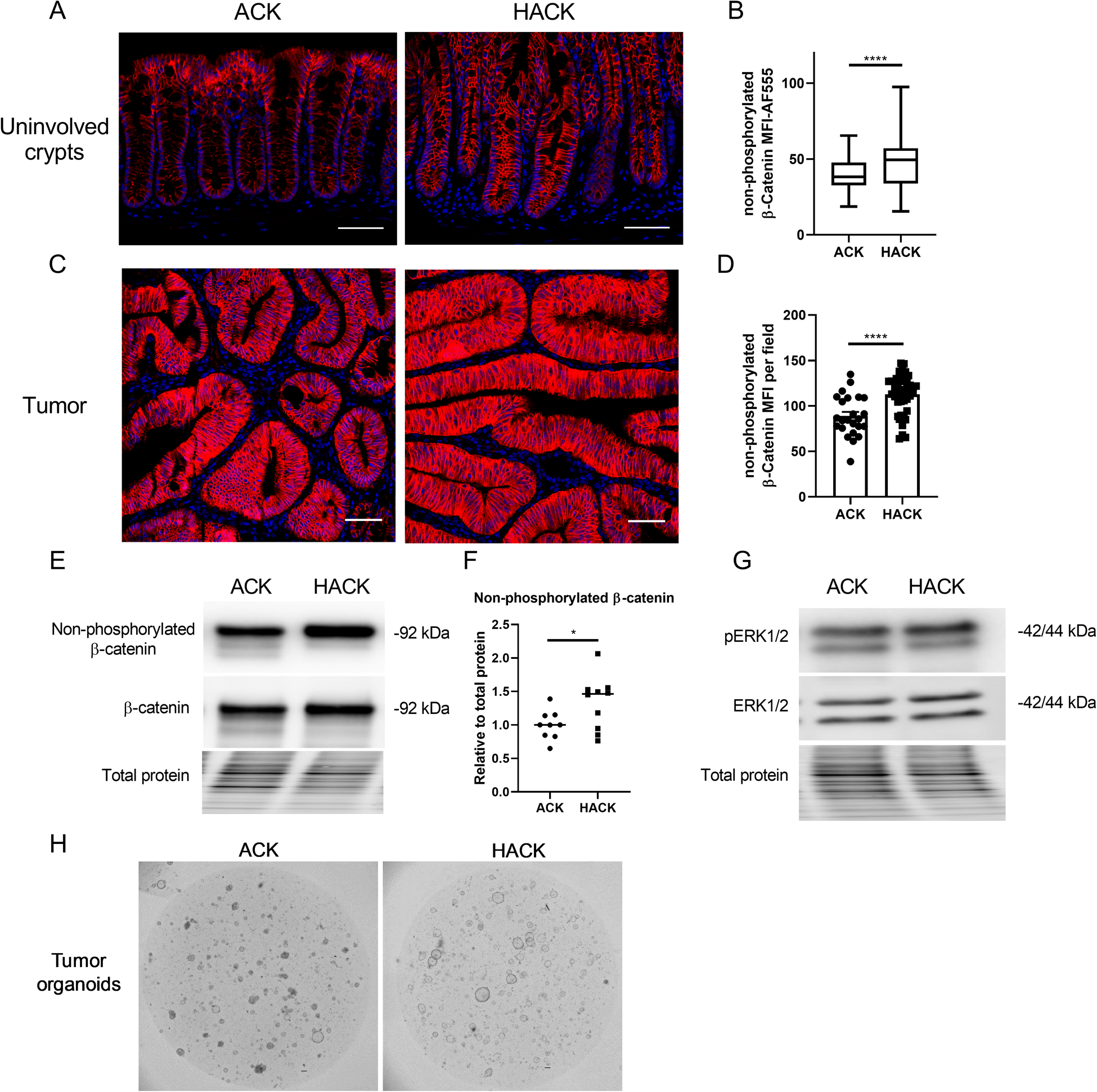
Loss of AhR upregulates Wnt signaling in ACK mice. (A) Representative immunohistochemistry of non-phosphorylated (active) β-catenin in tumor uninvolved tissues. Scale 50 μm. (B) Quantification of MFI of non-phosphorylated β-catenin in lower crypt regions, n=128–180 crypts from 4–6 mice per group. (C) Representative immunohistochemistry of non-phosphorylated (active) β-catenin in colonic tumor tissues. Scale 50 μm. (D) Quantification of MFI of non-phosphorylated β-catenin per field, n=24–45 fields from 3–5 mice per group. (E-F) Representative immunoblots for non-phosphorylated β-catenin (active β-catenin) and total β-catenin in proximal colon tumors from n=9–10 mice. (G) Representative immunoblots for ERK1/2 in proximal colon tumors from n=9–10 mice. (H) Representative brightfield images of organoids generated from proximal colon tumors. Scale bar, 200 μm. *p<0.05, **p<0.01, ****p<0.0001.
Lgr5 haploinsufficiency attenuates AhR KO mediated colon tumorigenesis.
Next, we tested whether upregulated Wnt signaling is required to promote colon tumorigenesis following AhR KO. Lgr5, a leucine-rich repeat-containing G-protein coupled receptor for R-spondins, potentiates Wnt/β-catenin signaling upon R-spondin binding (32). The use of Lgr5-EGFP-IRES-CreERT2 reporter mice enabled us to trace intestinal stem cells. However, in this model one allele of endogenous Lgr5 is disrupted by introducing the knockin of eGFP, which potentially results in Lgr5 haploinsufficiency, affecting Wnt signaling. Therefore, we compared colonic Lgr5 expression in WT and Lgr5-EGFP reporter mice as well as tumorigenic ACK and ACKG mice and found that Lgr5-EGFP knockin significantly decreased the expression of Lgr5 in both comparisons (Figure 7A and Supplemental Figure 5A). The expression of other Wnt downstream targets, Ascl2 and Axin2, was also significantly decreased in ACKG compared with ACK mice (Figure 7A). Consistently, β-catenin levels were significantly decreased in colonic crypts from ACKG compared with ACK mice and abrogated the AhR KO changes in cell proliferation (Figure 7B&C). Therefore, Lgr5-GFP knockin selectively downregulated Wnt/β-catenin signaling in the colon.
Figure 7.
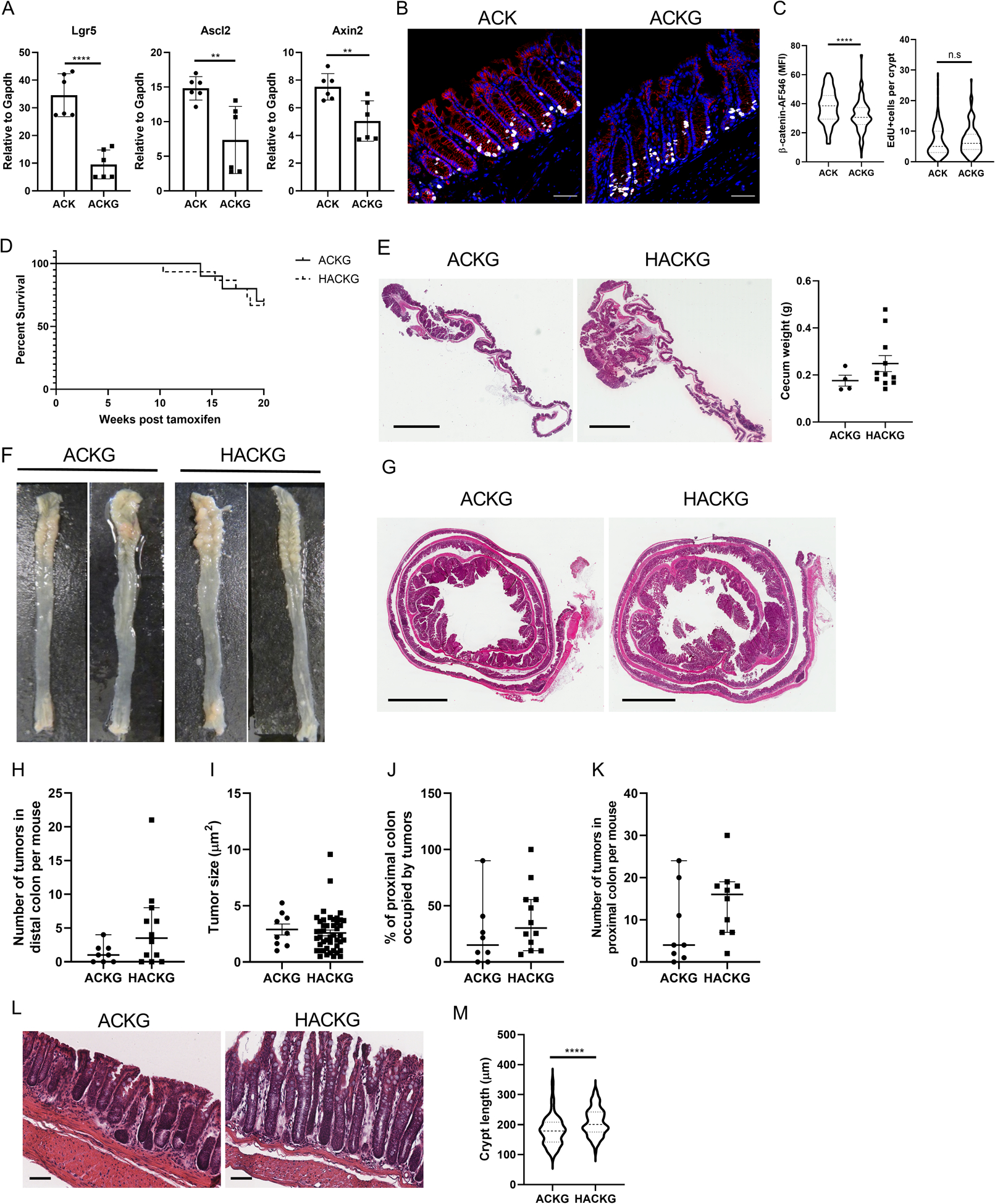
Lgr5 haploinsufficiency attenuates Wnt signaling and colon tumorigenesis in AhR KO mice. (A) mRNA expression of Wnt target genes, Lgr5, Ascl2 and Axin2 in organoids. Lgr5-GFP knock-in generates Lgr5 haploinsufficiency, n=4 from 2 mice per group. (B-C) Lgr5-GFP knock-in reduces total β-catenin levels but has no effect on cell proliferation in colonic crypts. Blue, DAPI; Red, β-catenin; and Gray, EdU. Scale bar, 50 μm. (D) Kaplan-Meier plot of percentage survival over a 20 wk period. At 8~10 wk of age, inducible ACKG (n=10) and HACKG (n=15) mice were administrated tamoxifen for 4 consecutive days to induce gene mutations. Mice were terminated 20 wk after the final tamoxifen injection. (E) Cecum tumor growth from ACKG and HACKG mice, n=4 or 11 mice per group. Scale bar, 3 mm. (F) Representative colon images from ACKG and HACKG mice 20 wk after the final tamoxifen injection. (G) H&E staining of representative entire colon sections. Scale bar, 3 mm. (H) Quantification of the number of tumors and (I) tumor size in the distal colon, n=8–12 mice per group. (J-K) Quantification of tumor growth in the proximal colon, n=8–12 mice per group. (L) H&E staining of representative colon uninvolved tissue sections. Scale bar, 50 μm. (M) Quantification of crypt length in colon, n=4 mice per group. *p<0.05, **p<0.01, ****p<0.0001; n.s, not significant.
We also assessed the effect of Lgr5-haploinsufficiency in ACKG and HACKG mice carrying ApcS580/+; KrasG12D/+ mutations on cecal and colon tumorigenesis. Of note, we found that the knock-in of the Lgr5-GFP reporter remarkably reduced colon tumorigenesis in both ACKG and HACKG mice and abrogated the AhR KO mediated phenotype with respect to mouse survival rate and cecum or colon tumor growth (Figure 7D–K). In addition, there was no significant difference in terms of spleen weight and colon length between ACKG and HACKG mice (Supplemental Figure 5B&C). Similarly, assessment of cecum and/or colon tumors indicated that dampened Wnt/β-catenin signaling abrogated the development of mid-stage cecum and colon tumor growth mediated by AhR KO, even though the proportion of mid-stage cecum tumors was increased in HACKG mice, but not in colon tumors (Supplementary Figure 5D–G). However, decreased Wnt/β-catenin signaling did not affect crypt length, and elongated crypt length was still observed in HACKG mice (Figure 7L&M), which was consistent with the results from the 3 wk study (Figure 4C). IHC analysis for β-catenin and cell proliferation in the colon was also performed. We found that total and non-phosphorylated β-catenin level was comparable between uninvolved crypt regions in ACKG and HACKG mice, while AhR KO enhanced non-phosphorylated but not total β-catenin levels in colon tumors (Supplemental Figure 6A–I). Moreover, AhR KO increased cell proliferation in uninvolved crypt regions but had no effect on colon tumor mass (Supplemental Figure 6G&J). Collectively, these findings show that attenuated Wnt/ β-catenin signaling, due to knockin of the Lgr5-GFP reporter allele, significantly reduced β-catenin stability, and suppressed AhR KO mediated colon tumorigenesis, while having no effect on cell proliferation.
Discussion
To date, the modulatory role of AhR signaling on intestinal tumorigenesis is based on carcinogen-induced or colitis-associated colon tumor models (14,15,33,34). However, the ability of AhR to modulate oncogenic cues in genetically driven colon tumor models has not been well studied to date (13,35). In this study, we provide evidence that the tissue-specific loss of AhR signaling promotes Lgr5+ colonic stem and/or progenitor cell self-renewal, endows features of stemness (i.e., organoid forming capacity, cell proliferation), and enhances cecum and colon tumorigenesis by potentiating Wnt signaling in mice expressing intestinally targeted ApcS580/+ and KrasG12D/+ mutations. This is noteworthy, because Apc and Kras are the two commonly mutated genes associated with the initiation and early progression of human colorectal cancer. Suppression of Wnt signaling by targeting one allele of Lgr5 attenuated AhR KO mediated cecal and colon tumorigenesis.
Kawajiri et al reported that the global deletion of AhR could cooperatively promote cecal and small intestinal carcinogenesis in ApcMin/+ mice, due to upregulated Wnt signaling (13). They noted that AhR acted as a ligand-dependent E3 ubiquitin ligase of β-catenin, and AhR activation by diverse ligands such as 3MC, β-naphthoflavone (βNF) or IAA enhance the ubiquitylation and proteasomal degradation of β-catenin, thus downregulating Wnt signaling and suppressing intestinal tumorigenesis (13). In addition, as a parallel pathway of Wnt signaling regulation, AhR was shown to bind to the promoter of Znrf3 and increase Znrf3 expression (34), which is a negative regulator of the Wnt Frizzled receptor. However, previously, our lab and another group did not find any observable difference in terms of β-catenin expression and its downstream targets between WT and AhR KO colonocytes both in vivo and ex vivo in normal mice with no cancer (14,16). In contrast, in the genetically susceptible colon tumor model (ApcS580/+; KrasG12D/+), increased β-catenin levels were observed in AhR KO (HACK) compared with ACK mice. This unexpected outcome suggests that the regulation of β-catenin by AhR is context dependent, i.e., normal versus malignant transformation conditions. The other possibility is that β-catenin is not a direct target of AhR. This is consistent with a recent study using human colon cancer cell lines, where AhR activation failed to recruit CUL4B or β-catenin to form the complex required for subsequent β-catenin degradation, consistent with the lack of an effect on Tcf4/β-catenin mediated transcriptional activity (36). In addition, following tissue specific AhR KO, we did not observe obvious β-catenin accumulation in the nucleus in normal crypts in the absence of colon cancer (16). It is likely that the AhR affects other signaling pathways that can crosstalk with Wnt signaling in the context of tumorigenesis. Therefore, the association between AhR and β-catenin warrants further experimentation.
Another important observation in our study is that Lgr5-GFP reporter mice significantly attenuated AhR KO mediated colon tumorigenesis, due to the haploinsufficiency of Lgr5 in the Lgr5-GFP reporter background. Lgr5 is an orphan G-protein coupled receptor and constitutes the receptor for R-spondin (37). Upon R-spondin binding, Lgr5 interacts and forms a supercomplex with Rnf43/Znrf3, which antagonizes Wnt signaling by targeting Frizzled receptors for degradation and subsequent co-internalization (38). The Lgr5 expression level was decreased by ~60–70% in Lgr5-GFP knockin mice, compared with control mice. Interestingly, Lgr5 haploinsufficiency did not result in any difference in crypt morphology or cell proliferation, but significantly reduced β-catenin levels and colon tumorigenesis. This observation is consistent with the “just right” theory of Wnt signaling in driving malignant transformation, in which an optimal level of Wnt signaling is required for colon tumorigenesis (39). Extensive studies have shown that mutant Apc and Kras synergistically potentiate Wnt signaling in the intestine (40–43), which is consistent with our studies that ApcS580/+ alone was not enough to induce colon tumorigenesis (data not shown). In addition, it is possible that other cell signaling Lgr5-dependent functions may be involved in the reduced of cecum/colon tumorigenesis in HACKG mice.
We present evidence demonstrating that intestinal targeted ablation of AhR signaling in colonic epithelial cells further stabilized and increased β-catenin levels, thus enhancing tumor burden in mice carrying compound ApcS580/+; KrasG12D/+ mutations. Considering the relatively low tumor burden and the importance of Wnt signaling in tumor development, it is likely that the Wnt signaling levels were just above the threshold required for tumorigenesis in ApcS580/+; KrasG12D/+ mice, and a further increase in Wnt signaling by AhR deletion could therefore functionally promote cecal and colon tumorigenesis. Interestingly, the majority of tumors in ApcS580/+; KrasG12D/+ mice with or without AhR were early or mid-stage, and very few underwent metastasis, which was consistent with a recent study (44), indicating that further transformation of tumors requires additional mutational hits, such as p53 and SMAD4 (45). Interestingly, LOH of wildtype Apc was detected in most large proximal colon tumors (diameter >5mm), which is consistent with a previous study (40). We also examined cecal tumor development in our mouse model. It has been previously reported that the Wnt signaling level is lowest in the cecum, compared with small intestine and colon, and Wnt signaling strength for polyp initiation is relatively low in the cecum (39). Thus, when Wnt signaling becomes a limiting factor to initiate adenoma growth, it should have the greatest impact on tumor burden in the cecum, which is exactly what we observed. Consistent with this finding, we propose that Lgr5 haploinsufficiency titrates Wnt signaling below the threshold required for tumor growth. Hence, dampened Wnt signaling due to Lgr5 haploinsufficiency significantly decreased tumorigenesis in the cecum and colon and attenuated the effect of AhR KO.
Loss of AhR has been shown to promote cell proliferation both at basal homeostasis, following mucosal wounding and during carcinogen-induced tumor initiation (16). We recently demonstrated that the upregulation of FoxM1 signaling was at least partly responsible for the accelerated cell proliferation at basal and DSS-induced injury states, while Wnt signaling was not altered by AhR signaling under steady conditions (16). However, upregulated Wnt signaling and FoxM1 signaling could co-modulate cell proliferation following AhR deletion during the tumor initiation state. This suggests that under different cellular contexts, AhR signaling may differentially impact networks driving cell proliferation.
Currently no loss-of-function related AhR mutations or causal effects of AhR SNPs alone have been linked to CRC risk. Instead, recent studies suggest that AhR ligand availability and its expression level contribute to its signaling strength and duration. The expression of AhR is upregulated in CRC, compared with normal tissue based on The Cancer Genome Atlas (TCGA) database (https://xenabrowser.net/). However, reduced CYP1A1 and CYP1B1 expression levels were detected in colon cancer, compared with normal tissue, implying that AhR signaling is impaired in CRC patients. In addition, depletion of AhR ligand availability promoted carcinogen induced or colitis-associated colon tumorigenesis in mice (15). It is also increasingly appreciated that gut dysbiosis plays an important role in the pathogenesis of colorectal cancer (46). However, it remains to be determined whether gut dysbiosis impairs the production of AhR ligands, and thus AhR activation in CRC individuals. Interestingly, impaired production of AhR ligands has been observed in patients with inflammatory bowel diseases (47–49), which are a risk factor for promoting CRC progression. Thus, it is possible that the increased AhR expression associated with colonic tumors represents a compensatory response to the diminished availability of AhR ligands. In future studies, it will be interesting to determine whether restoration of AhR ligand producing microbiota or dietary intervention can attenuate CRC incidence and progression.
In summary, emerging studies have shown that AhR signaling plays a protective role in carcinogen-induced colon cancer, colitis-associated colon tumorigenesis and ApcMin/+ mouse models. Our findings broaden and redefine the phenotypic features of colonic epithelial cell specific AhR deletion in mice carrying ApcS580/+ and KrasG12D/+ mutations, which are commonly mutated in humans during early phase adenoma development. Findings from the current study suggest that AhR signaling plays an important role in intestinal tumorigenesis, and therefore could be a promising therapeutic target for colon cancer prevention and/or treatment.
Supplementary Material
Implications:
Our findings reveal that AhR signaling plays a protective role in genetically induced colon tumorigenesis at least by suppressing Wnt signaling and provides rationale for the AhR as a therapeutic target for cancer prevention and treatment.
Acknowledgements
We would like to thank Kristina Hielsberg for assistance in tumor studies and Jennifer Goldsby for RNA Seq related analyses. We also thank Hans Clevers and Eric Fearon for providing the Lgr5CreERT2 reporter and CDX2PCreERT2 mice, respectively. Funding was provided by Texas AgriLife Research, the Sid Kyle Chair Endowment, the Allen Endowed Chair in Nutrition & Chronic Disease Prevention, the Cancer Prevention Research Institute of Texas (RP160589), and the National Institutes of Health (R01-ES025713, R01-CA202697, R35-CA197707, P30-ES029067 and T32-CA090301).
Footnotes
Disclosure: The authors declare that there is no conflict of interest.
References
- 1.Yamagishi H, Kuroda H, Imai Y, Hiraishi H. Molecular pathogenesis of sporadic colorectal cancers. Chin J Cancer 2016;35:4 doi 10.1186/s40880-015-0066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carethers JM, Jung BH. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015;149(5):1177–90 e3 doi 10.1053/j.gastro.2015.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proceedings of the National Academy of Sciences of the United States of America 2015;112(1):118–23 doi 10.1073/pnas.1421839112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science 2013;339(6127):1546 doi 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brocardo M, Henderson BR. APC shuttling to the membrane, nucleus and beyond. Trends Cell Biol 2008;18(12):587–96 doi 10.1016/j.tcb.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Jaiswal AS, Narayan S. A novel function of adenomatous polyposis coli (APC) in regulating DNA repair. Cancer letters 2008;271(2):272–80 doi 10.1016/j.canlet.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. Journal of Cell Science 2007;120(19):3327 doi 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- 8.Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 2002;111(2):251–63 doi 10.1016/s0092-8674(02)01015-2. [DOI] [PubMed] [Google Scholar]
- 9.Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Göke B, et al. Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signaling. BMC Genomics 2014;15(1):74 doi 10.1186/1471-2164-15-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joseph R, Little P, Hayes DN, Lee MS. Characterization of the number and site of APC mutations in sporadic colorectal cancer. Journal of Clinical Oncology 2017;35(4_suppl):630– doi 10.1200/JCO.2017.35.4_suppl.630. [DOI] [Google Scholar]
- 11.Xie M-Z, Li J-L, Cai Z-M, Li K-Z, Hu B-L. Impact of primary colorectal Cancer location on the KRAS status and its prognostic value. BMC Gastroenterol 2019;19(1):46– doi 10.1186/s12876-019-0965-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang D, Sun W, Zhou Y, Li P, Chen F, Chen H, et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer metastasis reviews 2018;37(1):173–87 doi 10.1007/s10555-017-9726-5. [DOI] [PubMed] [Google Scholar]
- 13.Kawajiri K, Kobayashi Y, Ohtake F, Ikuta T, Matsushima Y, Mimura J, et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proceedings of the National Academy of Sciences of the United States of America 2009;106(32):13481–6 doi 10.1073/pnas.0902132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz-Diaz CJ, Ronnekleiv-Kelly SM, Nukaya M, Geiger PG, Balbo S, Dator R, et al. The Aryl Hydrocarbon Receptor is a Repressor of Inflammation-associated Colorectal Tumorigenesis in Mouse. Ann Surg 2016. doi 10.1097/SLA.0000000000001874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Metidji A, Omenetti S, Crotta S, Li Y, Nye E, Ross E, et al. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018. doi 10.1016/j.immuni.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han H, Davidson LA, Fan YY, Goldsby JS, Yoon G, Jin UH, et al. Loss of aryl hydrocarbon receptor potentiates FoxM1 signaling to enhance self-renewal of colonic stem and progenitor cells. EMBO J 2020;39(19):e104319 doi 10.15252/embj.2019104319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S, Pei X, Zhang W, Xie HQ, Zhao B. Functional analysis of the dioxin response elements (DREs) of the murine CYP1A1 gene promoter: beyond the core DRE sequence. Int J Mol Sci 2014;15(4):6475–87 doi 10.3390/ijms15046475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449(7165):1003–7 doi 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 19.Hardiman KM, Liu J, Feng Y, Greenson JK, Fearon ER. Rapamycin Inhibition of Polyposis and Progression to Dysplasia in a Mouse Model. PLOS ONE 2014;9(4):e96023 doi 10.1371/journal.pone.0096023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng Y, Sentani K, Wiese A, Sands E, Green M, Bommer GT, et al. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. The American journal of pathology 2013;183(2):493–503 doi 10.1016/j.ajpath.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walisser JA, Glover E, Pande K, Liss AL, Bradfield CA. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proceedings of the National Academy of Sciences of the United States of America 2005;102(49):17858–63 doi 10.1073/pnas.0504757102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001;410(6832):1111–6 doi 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 23.Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 1997;278(5335):120–3. [DOI] [PubMed] [Google Scholar]
- 24.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459(7244):262–5 doi 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 25.Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nature Protocols 2013;8:2471 doi 10.1038/nprot.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res 2007;67(20):9721–30 doi 10.1158/0008-5472.can-07-2735. [DOI] [PubMed] [Google Scholar]
- 27.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2001;15(24):3243–8 doi 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou M, Baitei EY, Al-Rijjal RA, Parhar RS, Al-Mohanna FA, Kimura S, et al. KRASG12D-mediated oncogenic transformation of thyroid follicular cells requires long-term TSH stimulation and is regulated by SPRY1. Laboratory Investigation 2015;95:1269 doi 10.1038/labinvest.2015.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turk HF, Monk JM, Fan YY, Callaway ES, Weeks B, Chapkin RS. Inhibitory effects of omega-3 fatty acids on injury-induced epidermal growth factor receptor transactivation contribute to delayed wound healing. Am J Physiol Cell Physiol 2013;304(9):C905–17 doi 10.1152/ajpcell.00379.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457(7229):608–11 doi 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 31.Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015;521(7550):43–7 doi 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 32.Morgan RG, Mortensson E, Williams AC. Targeting LGR5 in Colorectal Cancer: therapeutic gold or too plastic? Br J Cancer 2018;118(11):1410–8 doi 10.1038/s41416-018-0118-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Villatoro EL, DeLuca JAA, Callaway ES, Allred KF, Davidson LA, Hensel ME, et al. Effects of high-fat diet and intestinal aryl hydrocarbon receptor deletion on colon carcinogenesis. Am J Physiol Gastrointest Liver Physiol 2020;318(3):G451–g63 doi 10.1152/ajpgi.00268.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metidji A, Omenetti S, Crotta S, Li Y, Nye E, Ross E, et al. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018;49(2):353–62.e5 doi 10.1016/j.immuni.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikuta T, Kobayashi Y, Kitazawa M, Shiizaki K, Itano N, Noda T, et al. ASC-associated inflammation promotes cecal tumorigenesis in aryl hydrocarbon receptor-deficient mice. Carcinogenesis 2013;34(7):1620–7 doi 10.1093/carcin/bgt083. [DOI] [PubMed] [Google Scholar]
- 36.Shiizaki K, Kido K, Mizuta Y. Insight into the relationship between aryl-hydrocarbon receptor and β-catenin in human colon cancer cells. PLOS ONE 2019;14(11):e0224613 doi 10.1371/journal.pone.0224613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmon KS, Lin Q, Gong X, Thomas A, Liu Q. LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/beta-catenin signaling. Molecular and cellular biology 2012;32(11):2054–64 doi 10.1128/MCB.00272-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung C, Tan SH, Barker N. Recent Advances in Lgr5(+) Stem Cell Research. Trends Cell Biol 2018;28(5):380–91 doi 10.1016/j.tcb.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 39.Leedham SJ, Rodenas-Cuadrado P, Howarth K, Lewis A, Mallappa S, Segditsas S, et al. A basal gradient of Wnt and stem-cell number influences regional tumour distribution in human and mouse intestinal tracts. Gut 2013;62(1):83–93 doi 10.1136/gutjnl-2011-301601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology 2006;131(4):1096–109 doi 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Hwang J-H, Yoon J, Cho Y-H, Cha P-H, Park J-C, Choi K-Y. A mutant KRAS-induced factor REG4 promotes cancer stem cell properties via Wnt/β-catenin signaling. International journal of cancer 2019;n/a(n/a) doi 10.1002/ijc.32728. [DOI] [PubMed] [Google Scholar]
- 42.Jeong W-J, Ro EJ, Choi K-Y. Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. npj Precision Oncology 2018;2(1):5 doi 10.1038/s41698-018-0049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the Wnt/beta-catenin pathway through LRP6 in colorectal cancer. Oncogene 2015;34(38):4914–27 doi 10.1038/onc.2014.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang J, Feng Y, Kuick R, Green M, Green M, Sakamoto N, et al. Trp53 null and R270H mutant alleles have comparable effects in regulating invasion, metastasis, and gene expression in mouse colon tumorigenesis. Laboratory investigation; a journal of technical methods and pathology 2019;99(10):1454–69 doi 10.1038/s41374-019-0269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017;543(7647):676–80 doi 10.1038/nature21713. [DOI] [PubMed] [Google Scholar]
- 46.Sobhani I, Bergsten E, Couffin S, Amiot A, Nebbad B, Barau C, et al. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proceedings of the National Academy of Sciences 2019;116(48):24285–95 doi 10.1073/pnas.1912129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 2016. doi 10.1038/nm.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunology 2018;11(4):1024–38 doi 10.1038/s41385-018-0019-2. [DOI] [PubMed] [Google Scholar]
- 49.Agus A, Planchais J, Sokol H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018;23(6):716–24 doi 10.1016/j.chom.2018.05.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq data from this publication have been deposited in NCBI Sequence Read Archive with accession number PRJNA682633.