

Abstract
There is a strong correlation between myeloid derived suppressor cells (MDSCs) and resistance to immune checkpoint blockade (ICB), but the detailed mechanisms underlying this correlation are largely unknown. Using single-cell RNA-seq analysis in a bilateral tumor model, we found that immunosuppressive myeloid cells with characteristics of fatty acid oxidative metabolism dominate the immune-cell landscape in ICB-resistant subjects. In addition, we uncovered a previously underappreciated role of a serine/threonine kinase, PIM1, in regulating lipid oxidative metabolism via PPARγ-mediated activities. Enforced PPARγ expression sufficiently rescued metabolic and functional defects of Pim1−/− MDSCs. Consistent with this, pharmacological inhibition of PIM kinase by AZD1208 treatment significantly disrupted the myeloid cell–mediated immunosuppressive microenvironment and unleashed CD8+ T cell–mediated antitumor immunity, which enhanced PD-L1 blockade in preclinical cancer models. PIM kinase inhibition also sensitized non-responders to PD-L1 blockade by selectively targeting suppressive myeloid cells. Overall, we have identified PIM1 as a metabolic modulator in MDSCs that is associated with ICB resistance and can be therapeutically targeted to overcome ICB resistance.
Keywords: PIM1, Metabolism, anti-PD-L1 therapy, MDSC, Immunotherapy Resistance
Introduction
Immune checkpoint blockade (ICB) has generated remarkable responses in treating cancer (1, 2). However, this success has been limited to a small proportion of patients due to multiple resistance mechanisms, including T-cell exhaustion and an immunosuppressive tumor microenvironment (TME) (3–5). Among these mechanisms, myeloid-derived suppressor cells (MDSCs), which constitute the largest population of suppressor cells in the TME, are considered a major barrier to successful ICB therapy (4, 6). MDSCs adapt to the glucose-limited and lipid-rich TME by changing their energy consumption toward fatty acid oxidative (FAO) metabolism (7, 8). Although it is known that MDSCs in the TME increase expression of key FAO enzymes, mitochondrial mass and function, and oxygen consumption rate (OCR), which are all associated with elevated suppressive activity (7), the detailed mechanisms by which oxidative metabolism is regulated in MDSCs remain unclear.
To identify the cellular and molecular mechanisms that cause ICB resistance, we performed single-cell RNA sequencing (scRNA-seq) on immune cells from responder and non-responder tumors following PD-1 blockade. This led us to discover that the expression of Pim1 strongly correlated with increased FAO and PPARγ-driven lipid metabolism in MDSCs. PIM1 is a member of a serine/threonine protein kinase family that functions downstream of the JAK-STAT pathway (9–11). PIM kinases are constitutively active when expressed in cells, and their activity is directly correlated with their expression levels (9, 10). Previous research has shown that PIM kinases, predominately PIM1, regulate various aspects of cellular metabolism in myocardiocytes, adipocytes, and T cells (10, 12–14). However, the function of PIM1 in myeloid cells remains largely unknown. Furthermore, it has been shown that PIM kinases function as an emergency backup of the AKT-mTOR pathway in effector T cells, and thereby are dispensable for normal T-cell activation, expansion, and function (10, 13). Taken together, these data suggest that targeting PIM-dependent metabolic pathways in immunosuppressive myeloid cells would have minimal effects on T-cell antitumor activity.
In this study, we have demonstrated that PIM1 was highly expressed in MDSCs with enhanced FAO metabolism, and this was strongly associated with ICB resistance in both mice and humans. Genetic deletion of Pim1 in MDSCs impaired fatty acid uptake and their immunosuppressive function by reducing the expression of PPARγ, a key regulator of lipid uptake and oxidative metabolism. Furthermore, pharmacological inhibition of PIM kinases selectively diminished numbers of MDSCs in the TME, which in turn promoted the antitumor function of cytotoxic T cells and tumor control. Last, PIM kinase inhibition not only improved the efficacy of PD-L1 blockade but also overcame ICB resistance in non-responders.
Materials and Methods
Mice
Six- to eight-week-old male and female mice were used for tumor studies. C57BL/6 and C57BL/6-CD45.1 mice were purchased from Charles River. Pim1−/− mice were originally obtained from Dr. Robert Woodland (University of Massachusetts) and were backcrossed to C57BL/6 background for at least 12 generations in our animal facility prior to use. All mice were bred and maintained under the guideline of IACUC of the Medical College of Wisconsin.
Tumor inoculation and treatments
MC38, B16-GM-CSF (Gvax), and B16-Bl6 tumor cell lines were originally purchased from ATCC and kindly provided by Dr. Lily Wang (Cleveland Clinic) in 2016. These cell lines have not been further authenticated or tested for Mycoplasma since. All tumor cell lines were cultured in DMEM media (Cat# 12-64-F, Lonza) supplemented with 10% FCS (Cat# SH3007103, Hyclone), 2mmol/L glutamine (Cat# 25-005-CI, Corning) and 100U/mL penicillin/streptomycin (Cat# 30-002-CI, Corning) and passaged fewer than 8 times prior to inoculation. To establish the bilateral tumor model, 1 × 106 MC38 cells were subcutaneously injected into the left and right flanks of mice. Once the size of the tumors reached 200 mm3, mice were randomized and received the following treatments: 1) vehicle control, 2) oral administration of AZD1208 (Selleckchem) for 10 days, 3) three doses of intraperitoneal (i.p.) injection with 200 μg anti-PD-L1 (clone# 10F.9G2, BioXcell) every other day, or 4) combination of AZD1208 and α-PD-L1 mAb. For the scRNA-seq study, tumors on the left side were surgical removed one day after the completion of anti-PD-L1 treatments to isolate immune cells, whereas tumors from the right side were kept intact to determine responder status.
For the melanoma model, 5 × 104 B16-BL6 cells were injected intradermally in the left flank. The tumor-bearing mice were treated on days 3, 6, and 9, with the following treatments: 1) Injection of 106 irradiated (16,000 rads) B16-GM-CSF cells (GVax) on the right flank, 2) Gvax treatment and oral administration of AZD1208, 3) Gvax treatment and i.p. injection with 200 μg and-PD-L1, or 4) combination of Gvax, AZD1208, and anti-PD-L1. Tumor volumes were measured three times a week and calculated as [longest dimension × (perpendicular dimension) 2]/2. Mice were euthanized when the tumor was greater than 2000 mm3. Mice were considered cured when tumor size was lower than 10 mm3.
Immune-cell isolation
Tumors were harvested from mice and minced into small pieces, followed by a one-hour incubation at 37°C with RPMI supplemented with 1% FBS, 2 mg/mL Collagenase Type I (Cat# LS004196, Worthington Biochemical Corporation) and Collagenase Type XI (Cat# LS004188, Worthington Biochemical Corporation), and 30 mg/mL DNase (Cat# 11284932001, Sigma-Aldrich). After incubation, tumor samples were mashed against a 70μm cell strainer to harvest immune cells, which were subsequently enriched by Lymphocyte Cell Separation Medium (Cat# 25–072-CV, Corning) and lysed in ACK lysis buffer (Cat# BW10548E, Lonza). Single-cell suspensions were then used for flow cytometry staining and fluorescence-activated cell sorting (FACS).
Flow cytometry
Single-cell suspensions were blocked with CD16/32 Fc blocking antibody (Cat# 101320, Biolegend) for 15 min and incubated with antibodies against surface markers for 30 min at 4°C, followed by 3 washes in FACS buffer. These samples were then run on an LSR II Green flow cytometer (BD Biosciences) and analyzed by FlowJo software (BD Biosciences).
Intracellular transcription factor staining was performed after surface staining. The cells were fixed with buffer from the True-Nuclear Transcription Factor Buffer Set (Cat# 424401, BioLegend) for one hour according to the manufacturer’s protocol. Cells were then washed with permeabilization buffer and stained with antibodies against transcription factors in permeabilization buffer.
For intracellular cytokine staining, single-cell suspensions were stimulated with anti-CD3/CD28 (Cat# 100340 and 102116, respectively, Biolegend) in the presence of brefeldin A (Cat# 420601, BioLegend) and 10 ng/mL IL
2 (Cat# 212–12, Peprotech) for 6 hours at 37°C. Subsequently, cells were collected for staining of surface markers and fixed by fixation buffer. Cells were then washed with permeabilization buffer and stained for cytokine-specific antibodies in permeabilization buffer.
All antibodies used for flow cytometry are listed in Supplemental Table 1.
Single-Cell RNA Sequencing
Cell sorting for scRNA-seq
Immune cells were isolated from tumors and stained as previously described. Then, CD45+ (Cat# 103106, Biolegend) and live (7AAD−, Cat# 420404, Biolegend) cells were sorted using a BD FACSMelody cell sorter.
scRNA-seq library generation
About 1 × 104 sorted cells for each sample were loaded onto the 10x Chromium Controller (10x Genomics). The scRNA-seq libraries were generated by Chromium Single Cell 3’ v2 Reagent Kit (Cat# PN-120267, 10x Genomics) and sequenced using a NextSeq 500/550 High Output Kit v2 (150 cycles) (Illumina) according to the manufacturer’s protocol. The single-cell suspensions of live CD45+ cells were diluted in nuclease-free water and then loaded to Chromium Controller. RNA transcripts were uniquely barcoded and reverse-transcribed within nanoliter-scale droplets. 10x barcoded full-length cDNA were then pooled and amplified via PCR. Then, the amplified cDNA went through an end repair process, A-tailing, adaptor ligation, and sample index PCR. The libraries were sequenced using a NextSeq 500/550 High Output Kit v2 (150 cycles) (Illumina, 20024904).
scRNA-seq analysis
Raw sequencing data were demultiplexed and converted to gene-barcode matrices using the Cell Ranger (version 2.2.0) mkfastq and count functions, respectively (10x Genomics). The mouse reference genome mm10 was used for alignment. Data were further analyzed in R (version 3.6.1) using Seurat (version 3.1.0) (15). A total of 3,214 cells from the non-responder and 1,864 cells from the responder mice were recovered and merged into one Seurat object. The number of genes detected per cell, number of UMIs, and percentage of transcripts derived from the mitochondria were plotted; cells that expressed less than 200 or more than 2,500 genes and cells with percent mitochondrial genes over 5% were removed to filter out doublets and cells with low read quality. Differences in cell library sizes (number of UMIs) and percentage of reads derived from the mitochondria were regressed out to prevent these technical variables from influencing cell clustering. Raw UMI counts were normalized and log-transformed. Principal component analysis was performed using variable genes, and the top 10 most statistically significant principal components were used for t-Distributed Stochastic Neighbor Embedding (t-SNE) analysis. These first 10 principle components were used to cluster the cells with Seurat’s implementation of a shared nearest neighbor (SNN) modularity optimization based clustering algorithm (Louvain’s original algorithm described in 10.1140/epjb/e2013-40829-0). To identify marker genes, the FindAllMarkers function was used with wilcoxon test test for single-cell gene expression. For each cluster, only genes that were expressed in more than 25% of cells with at least 0.25-fold difference were considered. To characterize cell types, we performed annotation in SingleR for each single cell independently based on the ImmGen, GeneQuery, and Enrichr databases. Normalized data were used in feature plots or violin plots. Mean expression of markers inside each cluster was used to perform gene set enrichment analysis (GSEA) using the fgsea R package (16). The volcano plot was generated by R package EnhancedVolcano.
Bulk RNA Sequencing
For each biological replicate, 0.5 × 106 live (7AAD−) bone marrow–derived MDSCs (BM-MDSCs) were FACS-sorted. Total RNA was extracted with the RNeasy Plus Micro kit per the manufacturer’s protocol (Cat# 74034, QIAGEN). Library preparation was performed according to the Smart-seq2 protocol (17). Sequencing was performed on a NextSeq 500/550 High Output Kit v2 (75 cycles) (Illumina) in a 37 bp paired-end mode. Sequenced reads were mapped to the mouse reference genome sequence (mm10) using TopHat v2.1.1 in combination with Bowtie2 v2.2.8 and Samtools v1.3. Fragments per kilobase of exon per million mapped fragments (FPKMs) were calculated and differential expression analysis was performed using Cufflinks v2.2.2. Heatmaps were generated with the R package pheatmap. GSEA was performed using the fgsea R package. Ingenuity Pathway Analysis (Qiagen Bioinformatics) was used for bioinformative analysis of the data.
Data availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request. RNA-seq data were deposited in the NCBI GEO database under accession codes GSE166308 and GSE166309.
Clinical data analysis
To further test if myeloid cells with gene signatures of high FAO metabolism correlated with ICB resistance, we analyzed scRNA-seq data from nivolumab-treated melanoma patients (accession: GSE120575). Data from all 10 ICB responder (R) and all 19 ICB non-responder (NR) patients were used for analysis. Cells were clustered using Seurat in a manner similar to the murine data above. One cluster was identified as myeloid cells based on high expression of ITGAM (encodes CD11b) and CD33. GSEA was performed on these cells after separating the cells by patient status (R vs. NR) using the clusterProfiler package (v 3.16.1) in R and gene sets from the Kyoto Encyclopedia of Genes and Genomes (KEGG).
To correlate the expression of PIM1 and survival, the web-based tool Gene Expression Profiling Interactive Analysis (GEPIA) was used to analyze datasets from The Cancer Genome Atlas (TCGA). Overall survival (OS) analysis was performed for colon adenocarcinoma (n=270), skin cutaneous melanoma (n=458) and combined 27 types of solid tumors (n=3,834) datasets, and the significance was determined using Log-rank test in GEPIA.
Cleavage Under Targets and Tagmentation (CUT&Tag) chromatin profiling
100,000 cells from Control and AZD groups were used for library construction using the CUT&Tag ChIP-Seq approach (version 2) (18) (stepwise protocol can be found at https://www.protocols.io/view/bench-top-cut-amp-tag-z6hf9b6). Thirty-seven cycles of paired-end sequencing were performed on an Illumina Nextseq 500 and 5 to 10 million reads were generated for each sample. Each dataset was downsampled to equal read depths. For standardization between experiments, Escherichia coli DNA derived from transposase protein production was used to normalize sample read counts based on the recommendation of CUT&Tag protocol. Reads were aligned to the Mus musculus mm10 genome and E. coli (strain K12) using Bowtie2 (version 2.2.5) (19) with options: --local --very- sensitive-local --no-unal --no-mixed --no-discordant --phred33 -I 10 -X 700. Peaks were called using SEACR (version1.1) (20), with options: 0.01 non stringent. Peaks were visualized by IGV (version 2.8.2).
Generation of BM-MDSCs and human MDSCs
Femurs were collected from C57BL/6 or Pim1−/− mice and flushed with 10% FBS RPMI medium to harvest bone marrow precursors. These cells were cultured at 37°C, 5% CO2 in the presence of GM-CSF (40 ng/mL, Cat# 200-15, Shenandoah) and IL6 (40 ng/mL, 200-02, Shenandoah) for 7 days to generate BM-MDSCs. To generate human MDSCs, the mononuclear cell suspension was prepared from peripheral blood by differential density gradient separation (Ficoll-Hypaque; Cat# 10771, Sigma-Aldrich). These cells were cultured in complete-RPMI-1640 media with 30% v/v tumor supernatants, 10% FBS, 1% Pen/Strep, GM-CSF (Cat# 100-08, Shenandoah), IL6 (Cat# 100-10, Shenandoah) and G-CSF (Cat# 100-72, Shenandoah) for 4 days.
In vitro suppression assay
For the in vitro suppression assay, activated CD8+ T cells were co-cultured with BM-MDSCs. The CD8+ T cells were isolated from naïve C57BL/6 mice (Cat# 19853, Stemcell) and labeled with CellTrace Violet (CTV, Cat# C34557, Thermo Fisher) followed by activation with anti-CD3/28 mAb. Then BM-MDSCs were added to the culture at different ratios. The proliferation of CTV-labelled CD8+ T cells was analyzed by flow cytometry.
Adoptive transfer of BM-MDSCs
A total of 2 × 106 BM-MDSCs were adoptively transferred into each C57BL/6 CD45.1+ congenic mouse bearing an MC38 tumor. Two weeks later, tumors from recipient mice were harvested and processed for flow cytometry analysis as described above.
Fatty acid uptake assay
BM-MDSCs were incubated with BODIPY FL C16 (1 μg/mL, Cat# D382, Invitrogen) in glucose-free RPMI 1640 medium (Cat#11879020, Thermofisher) for 30 min at 37°C and flow cytometry was used to measure its uptake.
Seahorse assay
The Seahorse XF Cell Mito Stress Test Kit (Cat# 103015-100, Seahorse Bioscience) was used ccording to the manufacturer’s protocol to determine the mitochondrial function of BM-MDSCs by measuring the OCR with an XF96 analyzer (Seahorse Bioscience). About 2 × 105 cells were seeded per well in a XF96 cell culture microplate and compounds were injected during the assay at the following final concentrations: 1.0 μM oligomycin, 1.5 μM FCCP, 0.5 μM rotenone, 0.5 μM antimycin A, and 100 μM etomoxir. The XF-Palmitate-BSA FAO substrate (Cat# 102720-100, Seahorse Bioscience) was used to evaluate the oxidation of exogenously added fatty acids and the proportion of respiration supported by exogenous fatty acids according to the manufacturer’s protocol (Seahorse Bioscience, #102720-100). The BM-MDSCs were inoculated with either palmitate-BSA (200 µM palmitate conjugated with 34 µM BSA) or BSA (34 µM) (Seahorse Bioscience) and the OCR was analyzed as described above.
Retroviral transduction
The coding sequences of mouse Pparg were obtained from Addgene (Cat# 17442) and cloned into MSCV-IRES-Thy1.1 (MIT). To produce retroviral supernatant to express Pparg, 293T cells were transfected with either MIT empty, or MIT-PPARg vector along with the pcLEco ecotropic packaging plasmid. After 48 hours, the retrovirus was collected from the culture supernatant and added to Pim1−/− BM-MDSCs followed by spinning transduction. After an additional 3 days of culturing in GM-CSF and IL6, the positively transduced cells, defined by expression Thy1.1, were sorted for transfer on a FACS Aria III flow cytometer (BD Biosciences).
Statistical analysis
All experiments were performed using randomly assigned mice without investigator blinding. All data points and p-values reflect biological replicates from at least three independent experiments. Statistical analysis was performed using GraphPad PRISM 7. Unpaired, two-tailed Student’s t-tests and one-way ANOVA tests with post hoc Tukey−Kramer corrections were used to assess statistical significance. For Figure 2G, Fisher’s exact test was used to evaluate the statistical significance of the ratios of two groups, each with two conditions (e.g., ratio of PIM1+: PIM1− cells in responders vs. non-responders). p-values < 0.05 were considered to indicate a significant difference. The classification and regression trees (CART) is a supervised machine learning method that builds a tree-like predictive model from data. The tree is built based on splitting the samples into two sub-groups. The splitting rule is to search for a best variable that achieves either the best separation in different groups (classification) or achieves the minimum regression error (regression). The tree will grow as this process is repeated in each sub-group recursively, and will stop when further split does not make meaningful improvement.
Figure 2. Heightened FAO metabolism in myeloid cells correlates with ICB resistance.
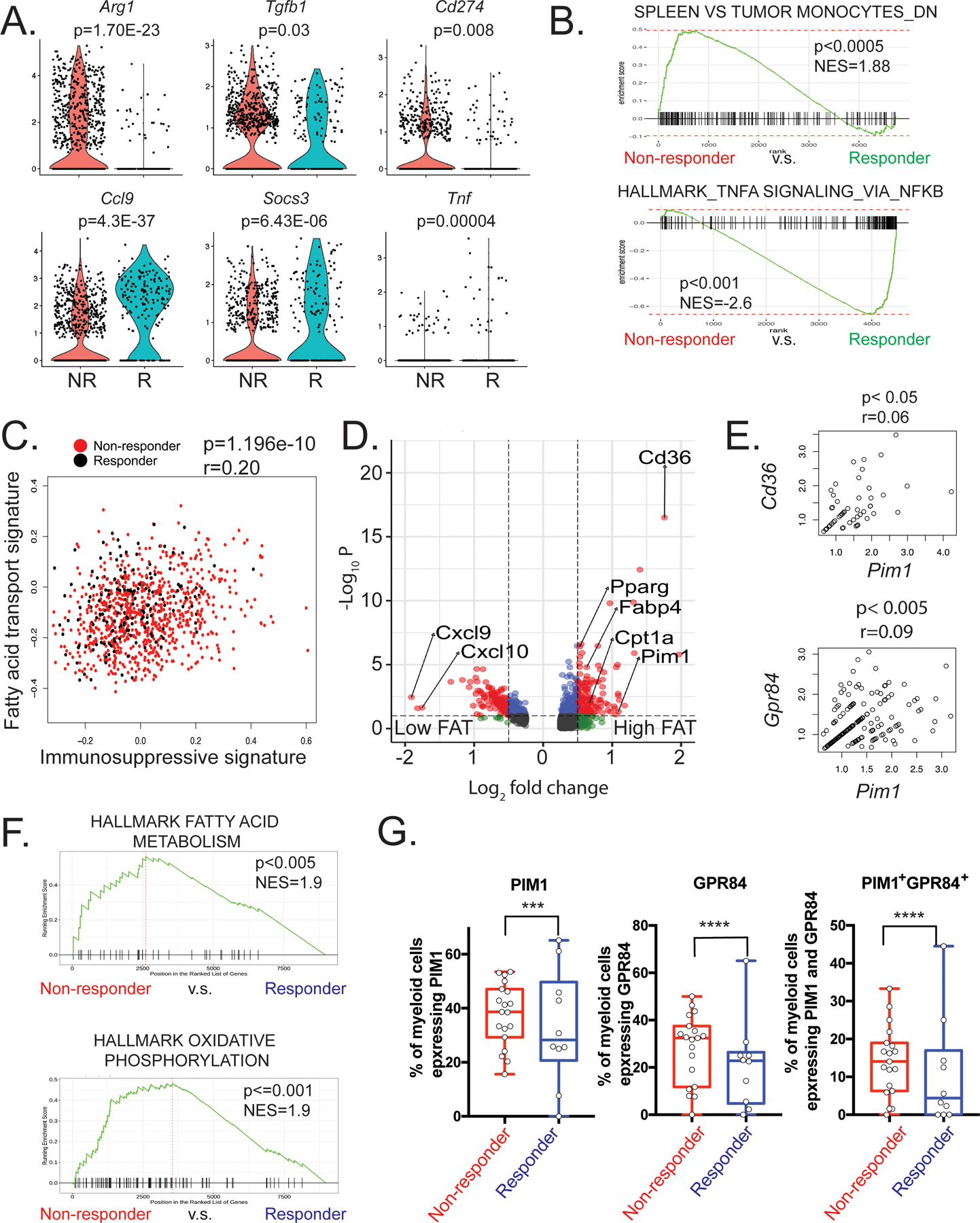
(A) Violin plots showing differentially expressed genes in myeloid cells between responder and non-responder samples. (B) GSEA plots evaluating expression in non-responders and responders of published datasets of genes downregulated in spleen versus tumor monocytes (top) and genes involved in TNFα signaling (bottom). (C) Scatter plot showing the relationship between immunosuppressive activity and fatty acid transport (FAT) activity. The y-axis represents relative expression of immunosuppressive gene signature and the x-axis represents the relative expression of FAT-related gene signature. Each dot represents a single cell from the myeloid populations pooled from responder (black) and non-responder (red). (D) Volcano plot showing differentially expressed genes between myeloid cells with high or low FAT activity. Each dot denotes one gene. (E) The correlation of Pim1 expression and genes related to FAT. (F) GSEA analysis of CD33+ ITGAM+ (encodes CD11b) myeloid cells from scRNA-seq data derived from melanoma patients treated with checkpoint inhibitor therapy reveals the enrichment of fatty acid metabolism and oxidative phosphorylation gene signatures in non-responder myeloid cells. (G) Box plots showing the frequencies of myeloid cells expressing PIM1, GPR84, and PIM1 and GRP84 from non-responders (n=19) and responders (n=10). Statistical significance was determined using Fisher’s exact test with a confidence level of 95%. *** p < 0.001, **** p < 0.0001.
Results
Single-cell transcriptomics reveal an association between the immune landscape and ICB resistance
To dissect the transcriptional and metabolic mechanisms of ICB resistance, we employed scRNA-seq to analyze the tumor-infiltrating immune cells in a murine MC38 colorectal cancer model. Consistent with previous work, we found that ~50–60% of MC38-bearing mice treated with anti-PD-L1 experienced complete tumor regression (21). To obtain immune cells for scRNA-seq analysis before tumors disappeared in responders, we established a bilateral tumor model by inoculating MC38 on both flanks of the mice and demonstrated the response to PD-L1 blockade in a symmetric manner, as previously published (22) (Supplemental Figure 1A). This model enabled us to examine the immune cells from the left flank tumor via surgical removal after the last dose of anti-PD-L1 administration, when the treatment outcomes were still indistinguishable, yet continuously monitor tumor regression or progression on the right flank to determine responder or non-responder status, respectively (Figure 1A).
Figure 1. Single-cell transcriptomics reveal the distinct immune cell landscapes from an ICB responder and non-responder.
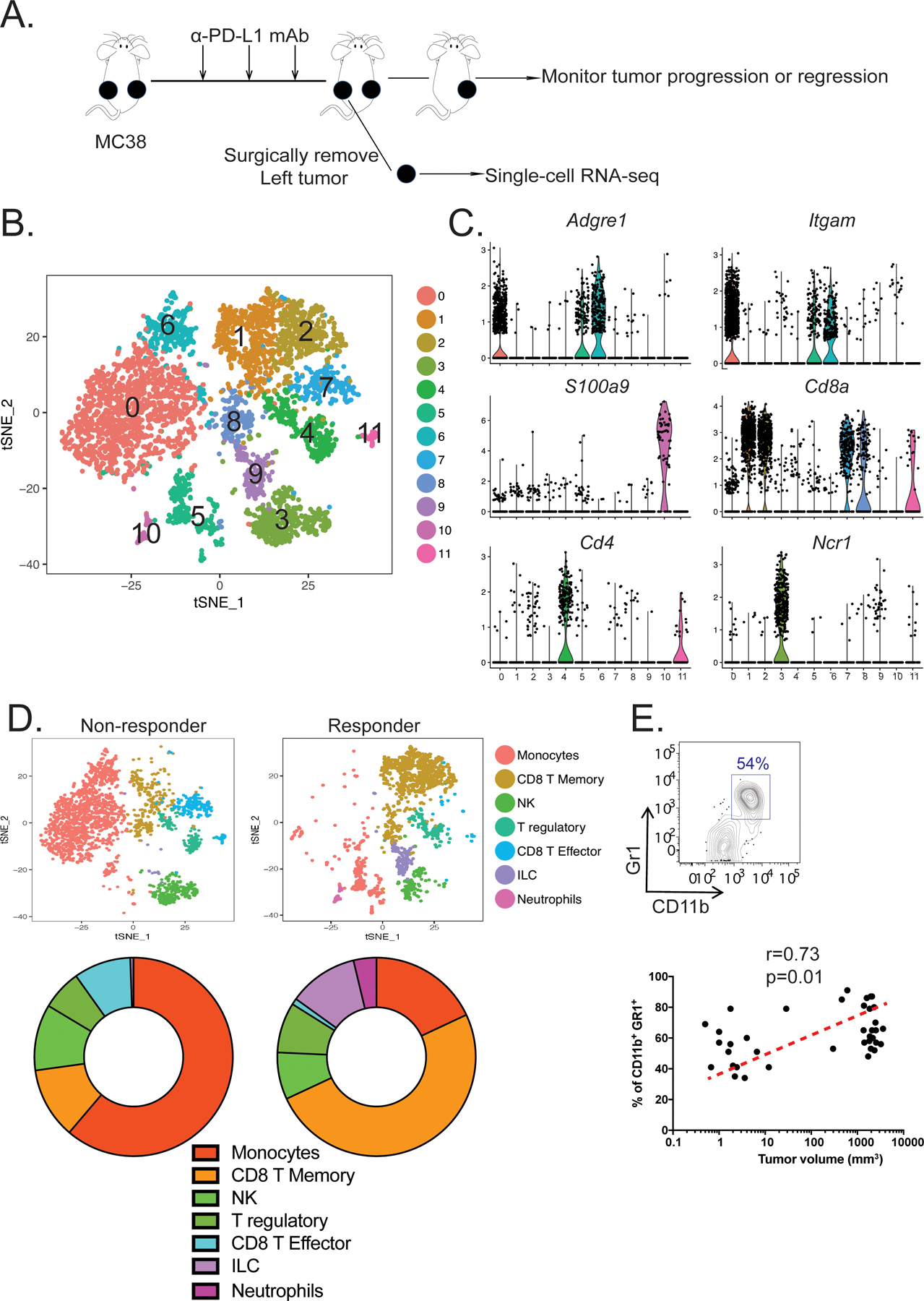
(A) Experimental design for establishing a bilateral tumor model to study the immune cell landscape associated with resistance to ICB immunotherapy in the MC38 colorectal cancer model. (B) t-distributed scholastic neighbor embedding (t-SNE) plot showing 12 immune cell clusters pooled from a responder and a non-responder. (C) Violin plots showing the gene expression distribution of various cell markers in all immune cell clusters. (D) t-SNE plots (top) demonstrating the different tumor-infiltrating immune cells between the non-responder (left) and responder (right). Proportions of immune cell-subsets found in the tumors are shown using pie charts (bottom). (E) The frequencies of intratumoral CD11b+Gr1+ MDSCs detected by flow cytometry (top) were plotted against the tumor volumes (bottom) for correlation (n=38, spearman’s correlation test, p < 0.05, r = 0.34).
After confirming that the bilateral tumors shared similar immune cell compositions and phenotypes (Supplemental Figure 1B–C), we next examined how the immune landscape differed between responders and non-responders. Two days following the end of anti-PD-L1 administration, single-cell transcriptomic datasets from CD45+ immune cells from a responder and a non-responder were successfully obtained. We recovered 1,864 cells from the responder and 3,214 cells from the non-responder with an average recovery of ~1,500 genes/cell. These separated into 12 clusters using unbiased dimensionality reduction and clustering algorithms in the Seurat R package (Figure 1B) (23, 24). The expression profile of various myeloid (Adgre1, which encodes F4/80, Itgam, which encodes CD11b, and S100a9) and lymphoid (Cd8a, Cd4, and Ncr1) lineage markers allowed us to assign biological identities to each cluster (Figure 1C). We identified four distinct CD8+ T-cell clusters (clusters 1, 2, 7, and 8), two distinct CD4+ T-cell clusters (clusters 4 and 11), three myeloid-cell clusters (clusters 0, 5, and 6), one neutrophil cluster (cluster 10), and two NK-cell clusters (clusters 3 and 9) (Figure 1B).
To delineate the relationship between the immune landscape and responsiveness to PD-L1 blockade, we performed comparative analysis between the responder and non-responder (Figure 1D). We observed that myeloid cells dominated in the non-responder tumor (61.22%), whereas the percent of CD8+ T cells (19.26%) was low (Figure 1D). In contrast, the responder’s tumor was predominantly infiltrated by CD8+ T cells (50.75%), whereas myeloid cells comprised only 18.03% of all CD45+ immune cells (Figure 1D). We also found that tumor progression following PD-L1 blockade positively correlated with the frequency of CD11b+Gr1+ tumor-infiltrating myeloid cells (Figure 1E).
Given that ICB resistance is associated with tumor-reactive CD8+ T-cell dysfunction (25), we compared the differentially expressed genes in CD8+ T cells from the responder and non-responder (Supplemental Figure 1D–F). This gene set enrichment analysis (GSEA) revealed that CD8+ T cells from the responder expressed higher levels of genes associated with T-cell co-stimulation and effector function, such as Cd28, Tnfsf4, Icos, Gzmb, Fasl, and Prf1 (Supplemental Figure 1E) and had an enriched transcriptional signature of effector CD8+ T cells (Supplemental Figure 1F). In contrast, CD8+ T cells from the non-responder expressed higher levels of genes associated with exhaustion such as Pdcd1 (Supplemental Figure 1E) and positively correlated with an exhausted T-cell signature (Supplemental Figure 1F).
Myeloid cells with high fatty acid oxidative metabolism correlate with ICB resistance
To gain a more in-depth understanding of how tumor-infiltrating myeloid cells are metabolically regulated in the context of ICB resistance, we further analyzed the differentially expressed genes in the myeloid clusters between responder and non-responder tumors. Tumor-infiltrating myeloid cells from the non-responder exhibited higher expression of immunosuppressive molecules such as Arg1, Tgfb1, and Cd274 (Figure 2A). Furthermore, GSEA revealed that transcriptional signatures related to intratumoral MDSCs were significantly enriched in the non-responder (Figure 2B). In contrast, many pro-inflammatory genes, such as Ccl9, Socs3, and Tnf, were upregulated in the responder along with enriched TNFα signaling, consistent with potential antitumor function.
To better understand mechanisms by which tumor-infiltrating myeloid cells adapt to the nutrient-imbalanced tumor microenvironment, we sought to identify which metabolic processes support their survival and immunosuppressive function. GSEA showed that the signature of oxidative phosphorylation (OXPHOS) was significantly enriched in myeloid cells from the non-responder (Supplemental Figure 2A). As OXPHOS in MDSCs is mainly fueled by exogenous fatty acids (26), we explored the association between the fatty acid transport (FAT) pathway and immunosuppressive function using the R package Gene Set Variation Analysis (GSVA) to quantify the activity of these biological pathways at the single-cell level (27). The activity score of immunosuppressive activity was calculated for each cell based on the expression level of the genes encoding inhibitory modules, and fatty acid metabolic activity was determined by the level of genes in the Gene Ontology (GO) fatty acid transport signature. As expected, we found a significant positive correlation between these two pathways (Figure 2C), which corroborated previous findings showing an essential role for lipid metabolism in the inhibitory functions of MDSCs (28).
By comparing the differentially expressed genes between total myeloid cells with high and low FAT activity, we found that Cd36 and Pim1 were highly upregulated in the high FAT group (Figure 2D). Previous research has shown that PIM kinases, predominantly PIM1, regulate various aspects of cellular metabolism in myocardiocytes, adipocytes, and T cells (10, 12–14). Thus, we hypothesized that PIM1 regulates FAT activity in tumor-infiltrating myeloid cells for their metabolic adaptation and function in the TME. In support of this idea, Pim1 expression significantly correlated with the expression of FAT-related genes such as Cd36 and Gpr84, which encodes GPR84, a G protein-coupled receptor for medium- and long-chain fatty acids (29) (30) (Figure 2E). Furthermore, we stratified myeloid cells into Pim1high and Pim1low cells based on their Pim1 expression and identified differentially regulated genes between the two. GSEA demonstrated that Pim1high cells were highly enriched for fatty acid transport genes and IL6/STAT3 signaling pathway genes (Supplemental Figure 2B). Taken together with the fact that PIM1 is a well-known downstream target of STAT3 signaling (31, 32) and that IL6 is abundant in the TME (33), we hypothesize that the IL6/STAT3 pathway is a major driver of PIM1 upregulation in myeloid cells in the TME.
To further test if myeloid cells with gene signatures of high FAO metabolism correlated with ICB resistance, we analyzed scRNA-seq data from nivolumab-treated melanoma patients (34) (Accession# GSE120575). Similar to our murine colorectal carcinoma data, tumor-infiltrating myeloid cells in non-responder patients exhibited significant enrichment of fatty acid metabolism and oxidative phosphorylation genes (Figure 2F). In addition, greater frequencies of PIM1- and/or GPR84-expressing myeloid cells in patients significantly and positively correlated with ICB resistance (Figure 2G). Expression of PIM1 and GPR84 significantly and positively correlated with each other (Supplemental Figure 2C), suggesting that these could be used as biomarkers either alone or in combination to predict the outcome of ICB therapy. To further evaluate the correlation between PIM1 expression and patient survival, we used the Gene Expression Profiling Interactive Analysis (GEPIA) web server to analyze RNA sequencing expression data from tumor samples from the TCGA and the GTEx projects (35). In the 27 types of solid tumors cohorts analyzed, high levels of PIM1 were associated with reduced disease-free survival rates, whereas lower expression of PIM1 was associated with significantly longer survival (p = 0.002 by log-rank test, Supplemental Figure 2D). Collectively, our results suggest that PIM1 expression and associated metabolic reprogramming in tumor-infiltrating myeloid cells might be responsible for ICB resistance.
PIM1 is required for the immunosuppressive properties of myeloid cells
To test the necessity of PIM1 for the function of immunosuppressive myeloid cells, we analyzed BM-MDSCs (36). In this in vitro model, expression of PIM1 increased with the gradual enrichment of MDSCs by day 7 (Supplemental Figure 3A). Lack of PIM1 led to a reduced formation of total CD11b+Gr1+ MDSCs and frequency of Ly6G+CD11b+Gr1+ polymorphonuclear MDSCs (PMN-MDSCs), but it did not affect Ly6C+CD11b+Gr1+ monocytic MDSCs (M-MDSCs) (Supplemental Figure 3A). To further assess the role of PIM1 in MDSCs, we sorted CD11b+Gr1+ cells and performed bulk RNA sequencing (RNA-seq) analysis to compare gene expression profiles between wild type (WT) and Pim1−/− BM-derived MDSCs. The expression of genes encoding inhibitory molecules, such as indolamine-2, 3-dioxygenase (Ido1), arginase 1 (Arg1), TGFβ1 (Tgfb1), and PD-L1 (Cd274), were substantially reduced in the absence of PIM1 (Figure 3A). In agreement with these observations, flow cytometry analysis revealed that protein levels of Arg1 were significantly decreased in Pim1−/− BM-MDSCs (Supplemental Figure 3B). In addition, the ability of Pim1−/− MDSCs to inhibit T-cell proliferation in vitro was significantly compromised (Figure 3B). Likewise, pharmacologic inhibition of PIM kinases by AZD1208, a potent and selective pan-PIM kinase inhibitor (37, 38), also led to similar functional defects of BM-MDSCs (Supplemental Figure 3C–D). Together, these results uncover an essential role of PIM1 in regulating the suppressive function of MDSCs in vitro.
Figure 3. PIM1 is required for the immunosuppressive phenotype of MDSCs.
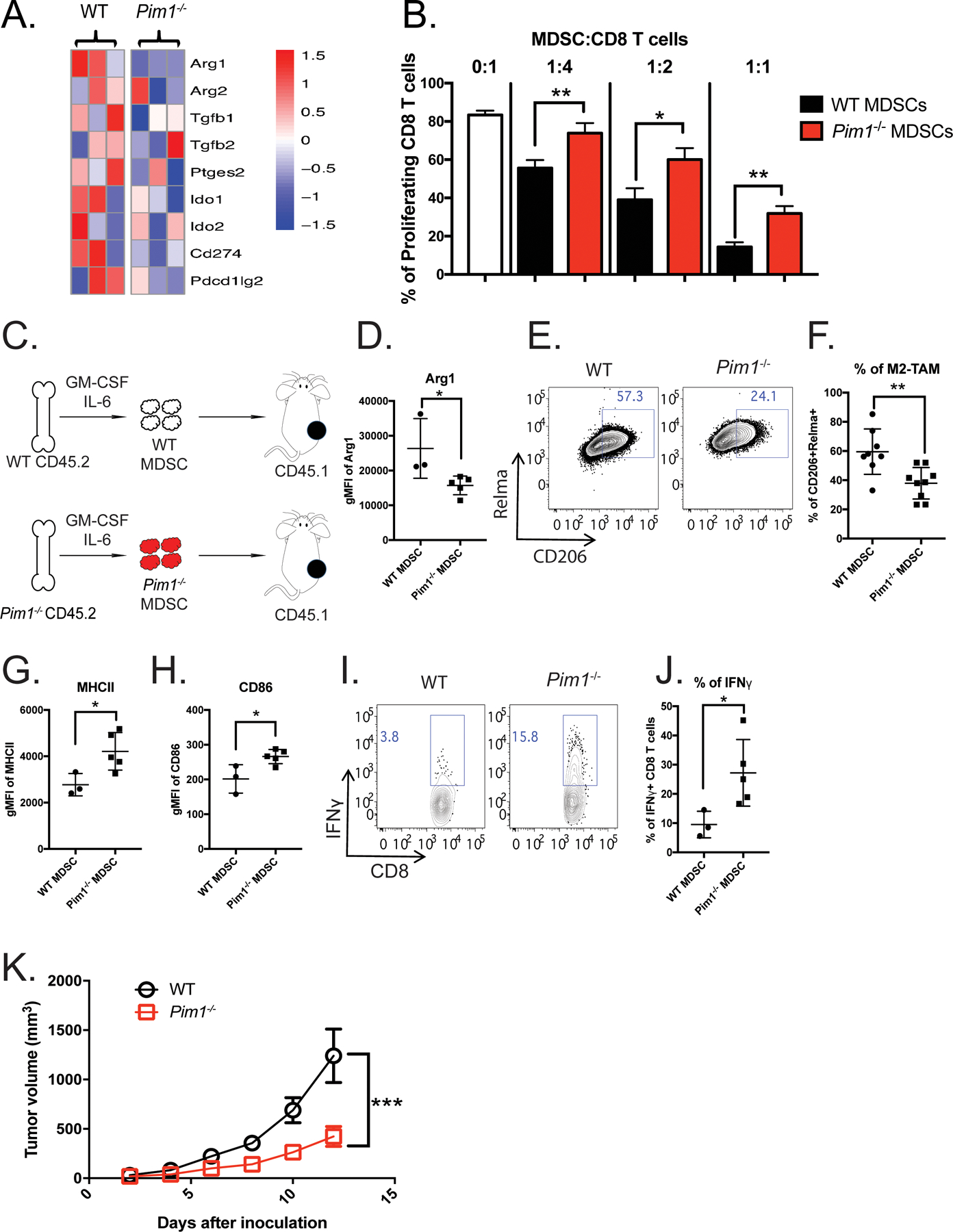
(A) Heatmap showing expression of genes associated with immunosuppressive functions in BM-MDSCs generated from WT (n=3) and Pim1−/− (n=3) mice. (B) WT (n=3) and Pim1−/− (n=3) MDSC suppressive activities were evaluated by the ability to inhibit CD8+ T-cell proliferation. Bar graphs were plotted from three independent experiments. (C-K) WT (n=3) and Pim1−/− (n=5) BM-MDSCs were purified and transferred into CD45.1+ congenic recipient mice bearing an MC38 tumor. (C) Experimental design. (D) Levels of Arg1 protein expression in intratumoral myeloid cells derived from WT and Pim1−/− donor cells. (E) Representative plot showing the frequencies of M2 macrophages evaluated by CD206 and Relmα expression from WT and Pim1−/− donor cells. (F) Quantification of E. (G-H) Protein expression levels of MHCII and CD86. (I-J) Intratumoral CD8 T cells from WT and Pim1−/− MDSC recipients were stimulated and evaluated for IFNγ production by flow cytometry as shown in representative (I) and quantitative (J) plots. (K) Tumor growth was measured using calipers for two weeks and plotted. Data are expressed as mean ± SEM from two experiments with 3 mice per group and significance was determined by t-test. * p < 0.05, ** p < 0.01, *** p < 0.001.
Next, we evaluated whether the loss of PIM1 in myeloid cells would affect the immunosuppressive TME in vivo. The necessary role of PIM1 in hematopoietic stem cell homing and engraftment precludes the possibility of generating bone marrow chimeric mice with specific deletion of PIM1 in myeloid cells (39) (40). To overcome this challenge, we adoptively transferred WT or Pim1−/− (CD45.2+) BM-MDSCs into congenic (CD45.1+) WT mice (Figure 3C). We then inoculated these two groups of recipient mice with MC38 tumor cells. Two weeks later, we found comparable frequencies of CD11b+ cells between adoptively transferred CD45.2+ WT and Pim1−/− cells in tumors (Supplemental Figure 3E). The majority of donor cells isolated from tumors had differentiated into F4/80+CD11b+ tumor-associated macrophages (TAMs) (Supplemental Figure 3F–G). We then assessed whether PIM1 deficiency affected the ability of transferred myeloid cells to inhibit antitumor immunity. First, we found that protein levels of Arg1 were significantly reduced in CD11b+ intratumoral myeloid cells from Pim1−/− compared to WT donor (Figure 3D). Furthermore, Pim1−/− CD11b+ cells exhibited significantly lower expression of the M2 macrophage markers Relmα and CD206 (Figure 3E–F), but expressed significantly higher levels of CD86 and MHC II than their WT counterparts (Figure 3G–H). This is consistent with our in vitro observations that Pim1−/− BM-derived macrophages (BMDM) exhibited higher expression of M1 markers than WT BMDM (Supplemental Figure 3H). Furthermore, another important M1 gene signature, antigen presentation, was also enriched in Pim1−/− BMDMs (Supplemental Figure 3I). These data suggest that PIM1-deficient myeloid cells fail to maintain their immunosuppressive phenotype and instead differentiate toward a more immune-stimulating state. Supporting this notion, we observed that tumor-infiltrating CD8+ T cells from mice that received Pim1−/− MDSCs produced more effector cytokines, such as interferon-γ (IFNγ), than those from mice that received WT MDSCs (Figure 3I–J). As a result, tumor progression in mice that received Pim1−/− MDSCs was significantly slower than that in mice that received WT MDSCs (Figure 3K). Collectively, these data revealed a previously unappreciated role of PIM1 in regulating immunosuppressive properties of MDSCs in vivo.
PIM1 regulates FAO metabolism in MDSCs
To evaluate if PIM1-mediated immunosuppression relates to cellular metabolism of MDSCs, we compared the metabolic gene expression profiles of WT and Pim1−/− BM-MDSCs. Genetic ablation of Pim1 resulted in reduced gene expression related to multiple aspects of fatty acid metabolism, including fatty acid degradation (e.g. Acsl1, Acadm, Acca2, and Hadh), fatty acid transportation (e.g. Cpt1a, Cpt1c, and Cpt2) and mitochondrial beta-oxidation (e.g. Acox1, Acads, and Acat1) (Figure 4A). GSEA confirmed that fatty acid metabolism and oxidative phosphorylation signatures were suppressed in Pim1−/− BM-MDSCs (Figure 4B). To determine whether these transcriptional alternations led to any metabolic changes, we first measured fatty acid uptake upon the addition of fluorescently-labeled palmitate (Bodipy FL C16) and observed that Pim1−/− MDSCs exhibited diminished fatty acid uptake (Figure 4C–D). We then measured OCR using a Seahorse extracellular flux analyzer and found that WT MDSCs had significantly higher basal respiratory capacity (BRC) and nonsignificantly higher spare respiratory capacity (SRC) than Pim1−/− MDSCs (Figure 4E–F). Exposure to exogenous bovine serum albumin (BSA)-conjugated XF-palmitate substrate resulted in a significant increase in the basal OCR in WT MDSCs, whereas such an increase was largely diminished in PIM1-deficient MDSCs (Figure 4G–H). Conversely, glycolytic pathways, as measured by ECAR and glucose uptake, were not affected by PIM1 deficiency (Supplemental Figure 4A–C). Consistent with these results, pharmacologic inhibition of PIM1 by AZD1208 in BM-derived MDSCs resulted in similar metabolic phenotypes (Supplemental Figure 4D–G). Overall, these data suggest that PIM1 regulates FAO metabolism in MDSCs, at least in part, by regulating fatty acid uptake.
Figure 4. Pim1−/− BM-MDSCs exhibit impaired FAO metabolism.
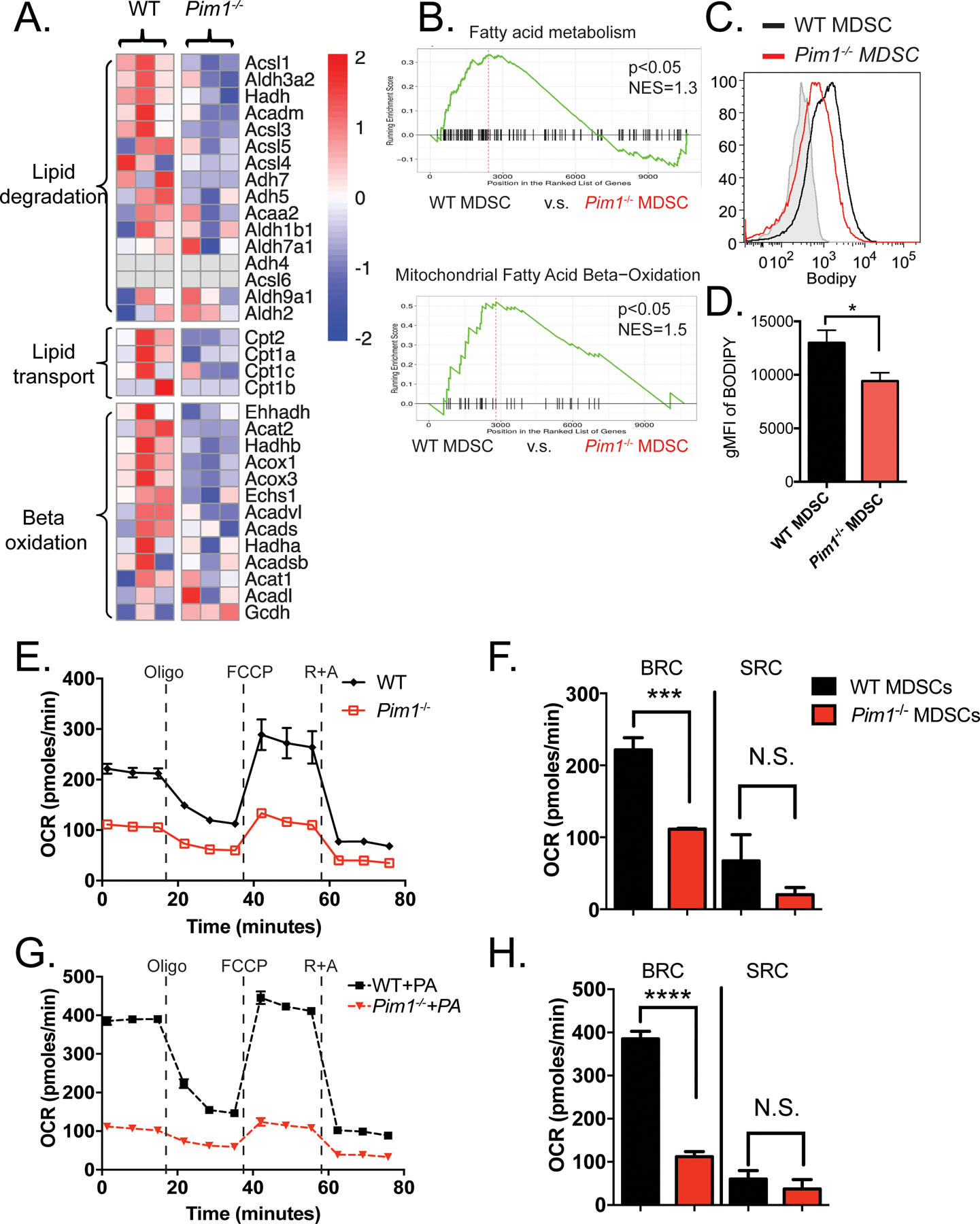
(A) Heatmap depicting genes related to fatty acid metabolism differentially expressed between WT (n=3) and Pim1−/− (n=3) BM-MDSCs. (B) GSEA analysis demonstrating that Pim1−/− BM-MDSCs negatively correlate with gene signatures related to FAO and OXPHOS metabolism. (C) Histograms showing representative fatty acid uptake from WT (n=3) and Pim1−/− (n=3) BM-MDSCs as assessed by flow cytometry. (D) Quantification of C. (E) Line graphs depicting the oxygen consumption rate (OCR) of WT and Pim1−/− BM-MDSCs in response to the Mito Stress assay. (F) Bar graphs quantifying basal respiratory capacity (BRC) and spare respiratory capacity (SRC) in WT and Pim1−/− BM-MDSCs. (G-H) Graphs depicting OCR from WT and Pim1−/− BM- MDSCs following exposure to exogenous XF Palmitate-BSA. Data are expressed as mean ± SEM from two experiments and significance was determined by t-test. * p < 0.05, *** p < 0.001, **** p < 0.0001.
The PIM1–PPARγ axis regulates FAO metabolism in MDSCs
To explore the possible molecular mechanisms by which PIM1 affects the metabolism and function of MDSCs, we performed Ingenuity Pathway Analysis (IPA) using differentially expressed genes between WT and Pim1−/− BM-derived MDSCs. This analysis revealed that peroxisome proliferator-activated receptor (PPAR) signaling was significantly reduced by the ablation of PIM1 (Figure 5A). This was further confirmed by GSEA analysis (Figure 5B). The expression of PPARγ targets, which include genes that encode transcriptional factors related to lipid metabolism, FAO enzymes, and lipid transporters, were substantially downregulated in Pim1−/− MDSCs (Figure 5C). Furthermore, protein levels of PPARγ and one of its targets, CD36, were significantly downregulated in Pim1−/− MDSCs (Figure 5D–G). Together, these results suggest that PPARγ may function as a major downstream target of PIM1 to regulate fatty acid metabolism in MDSCs.
Figure 5. The PIM1-PPARγ signaling axis regulates cellular metabolism and function of MDSCs.

(A-B) Ingenuity Pathway Analysis (IPA) (A) and GSEA (B) on bulk RNA sequencing data from WT (n=3) and Pim1−/− (n=3) MDSCs reveal that PPARγ signaling is significantly disturbed in the absence of Pim1. (C) Heatmap showing gene expression of Pparg and its targets in WT and Pim1−/− cells. (D-G) Flow cytometry analyses of CD36 (D-E) and PPARγ (F-G) expression in WT and Pim1−/− cells. (H-L) Metabolic and functional analysis of Pim1-deficient MDSCs following ectopic expression of Pparg. Expression levels of CD36 (H-I), fatty acid uptake (J-K), and in vitro suppression activity (L) were measured by flow cytometry in Pim1−/− BM-MDSCs expressing empty vector (MIT) (n=3) or Pparg (n=3). Data are expressed as mean ± SEM from two experiments and significance was determined by t-test. * p < 0.05, *** p < 0.001.
To determine whether reduced PPARγ expression was responsible for the metabolic and functional abnormalities in the PIM1-deficient MDSCs, we attempted to rescue these defects by ectopically expressing PPARγ in Pim1−/− BM-derived MDSCs (Supplemental Figure 5A). We found that forced expression of PPARγ bypassed the need for PIM1, restored the expression of CD36 (Figure 5H–I), and subsequently led to increased fatty acid uptake (Figure 5J–K). PPARγ overexpression also significantly increased Arg1 protein expression (Supplemental Figure 5B) and improved the ability of Pim1−/− MDSCs to inhibit T-cell proliferation (Figure 5L). Taken together, these findings support a mechanism by which PIM1 promotes FAO metabolism and immunosuppressive function in MDSCs via the induction of PPARγ expression.
To determine the mechanisms by which PIM1 regulates the transcription of Pparg, which is known to contain STAT3 binding sites (41), we postulated that PIM1 could regulate STAT3 phosphorylation. In support of this idea, AZD1208 treatment inhibited phosphorylation of the serine 727 (S727) site without affecting phosphorylation of tyrosine 705 (Y705) or total STAT3 protein expression levels (Supplemental Figure 5C). To further test whether this altered phosphorylation would affect the binding of STAT3 to the cis-regulatory elements of the Pparg locus, Cleavage Under Targets and Tagmentation (CUT&Tag) chromatin profiling was performed on MDSCs treated with either PBS or AZD1208. Consistent with published work (41), we observed similar binding patterns of STAT3 to the enhancer regions of Pparg and Csf2ra in the PBS treated control group (Supplemental Figure 5D). In contrast, AZD1208 treatment significantly diminished STAT3 binding to the enhancer regions of Pparg without affecting binding to the Csf2ra locus (Supplemental Figure 5D). These observations collectively suggest that PIM1-mediated phosphorylation of STAT3 at S727 enhances STAT3 transcriptional activity and results in heightened PPARγ expression in MDSCs.
PIM kinase inhibition improves the antitumor efficacy of PD-L1 blockade
Previous research has demonstrated that PIM kinases function as an emergency backup of the AKT-mTOR pathway in effector T cells and are therefore dispensable for normal T-cell activation, expansion, and function (10, 13). Thus, we reasoned we could selectively target PIM-dependent metabolic pathways in immunosuppressive myeloid cells while sparing the antitumor activity of T cells within the tumor. To test this idea, we first verified that pharmacological inhibition of PIM kinase by AZD1208 did not have direct toxicity against MC38 tumor cells in vitro compared with AZD1208-sensitive AML-AF9 leukemia cells (Supplementary Figure 6A). Next, we treated MC38 tumor bearing mice with AZD1208 for 9 days, which sufficiently induced complete tumor regression in 30% of treated mice, while anti-PD-L1 treatment alone led to a complete response in 50% of treated mice (Figure 6A). Co-administration of AZD1208 and anti-PD-L1 induced robust tumor eradication in 70% of mice, greater than either single-agent treatment alone (Figure 6A). These findings suggest that pharmacological inhibition of PIM kinases leads to an effective antitumor immune response and enhances ICB response.
Figure 6. PIM kinase inhibition enhances PD-L1 blockade for cancer immunotherapy.
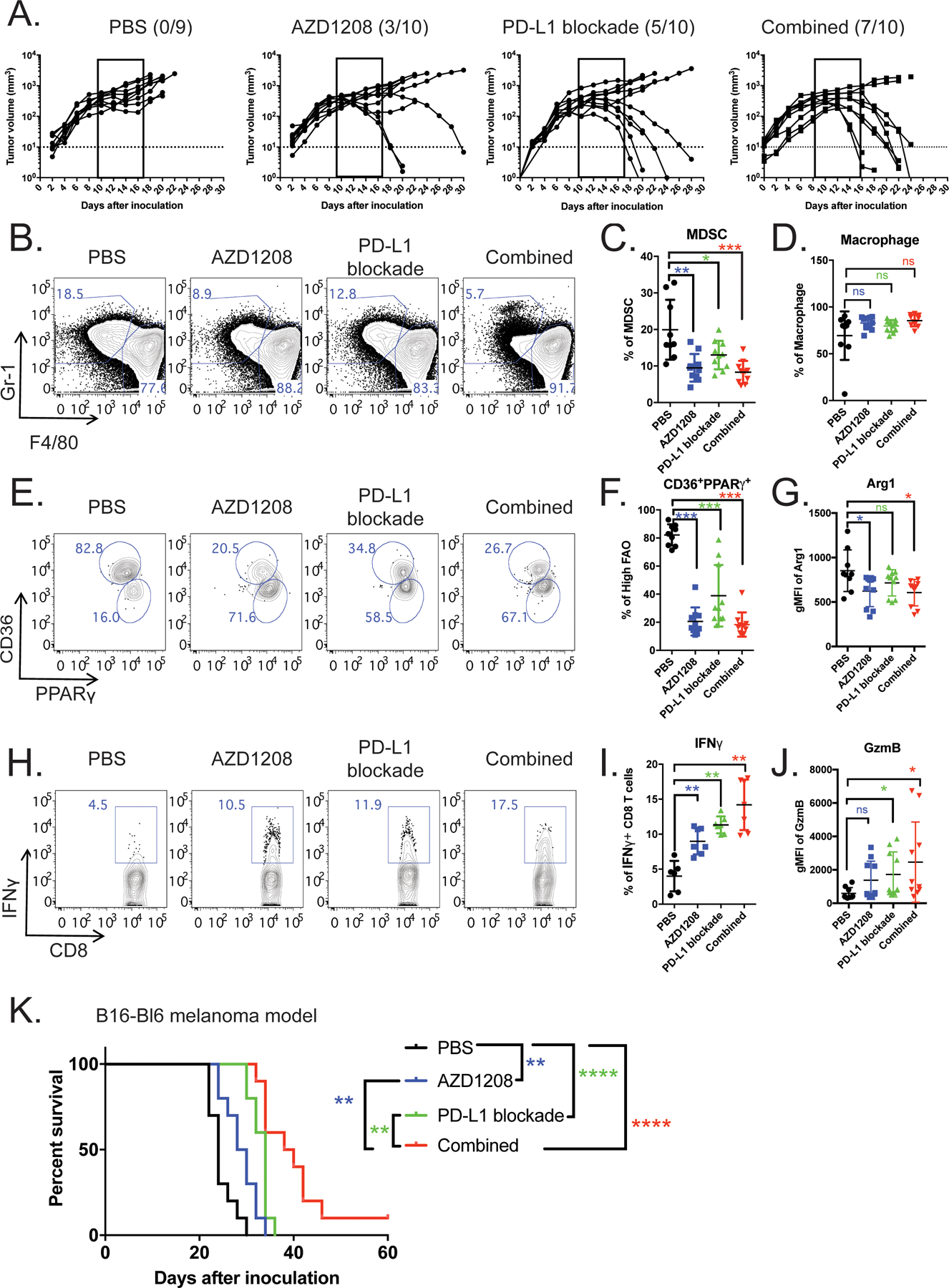
(A) Bilateral MC38 tumor-bearing mice received the following treatments: vehicle control (n=9), AZD1208 (n=10), anti-PD-L1 (n=10), or a combination of AZD1208 and anti-PD-L1 (n=10). Individual tumor growth curves are shown. Dotted lines indicate the level of detection. (B-I) Tumors on the left flank were surgically removed for immune cell analysis. Representative flow plots (B) and quantification of CD11b+Gr1+F4/80low MDSCs (C) and CD11b+Gr1−F4/80high macrophages (D) are shown. (E-F) Within the MDSCs, frequencies of CD36highPPARγhigh cells are shown in representative flow plots (E) and quantitative scatter plots (F). (G) Scatter plot depicting Arg1 levels as determined by flow cytometry. (H-J) Representative flow plots and quantitative scatter plots of IFNγ and granzyme B expression in tumor-infiltrating CD8+ T cells from four treatment groups. (K) Kaplan-Meier survival curves show B16-Bl6 melanoma mice treated with vehicle control, anti-PD-L1, AZD1208, or a combination of AZD1208 and anti-PD-L1. Data represent cumulative results from two independent experiments with n = 9 for vehicle control and combination groups and n = 10 for AZD1208 and α-PD-L1 mAb groups. Cellular significance was determined by t-test. Survival curve was analyzed by the log-rank (Mantel–Cox) test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
To delineate the cellular mechanisms underlying the observed effects of PIM kinase inhibition, we analyzed the immune cells in the tumors. AZD1208 alone or AZD1208 + anti-PD-L1 treatment resulted in a significant reduction in the abundance of CD11b+Gr1+ MDSCs (Figure 6B–C) and M-MDSCs (Supplemental Figure 6B–C) and mildly increased PMN-MDSCs (Supplemental Figure 6B–C) and CD11b+F4/80+ macrophages (Figure 6D). To further evaluate the effects of PIM kinase inhibition on myeloid cells, protein levels of CD36 and PPARγ were measured to quantify FAO metabolic activity. Consistent with our in vitro data, CD36highPPARγhigh myeloid cells expressed higher levels of PIM1 (Supplemental Figure 6D) and were significantly decreased in abundance in AZD1208-treated groups, both alone and in combination with anti-PD-L1 (Figure 6E–F). We also observed down-regulation of Arg1 in these two groups (Figure 6G) and reduced ability to suppress CD8+ T cells (Supplemental Figure 6E). Next, we characterized the phenotypes of TAMs. Althiugh PIM inhibition has minimal impact on the frequency of CD11b+F4/80high TAMs, their expression of immunosuppressive M2 macrophage markers Relmα and CD206 was significantly reduced (Supplemental Figure 6F–G). To evaluate potential off-target effects of AZD1208, Pim1−/− BM-MDSCs were transferred into MC38 tumor-bearing mice and treated with either vehicle or AZD1208. Pharmacological PIM inhibition did not further retard tumor progression compared with vehicle control in Pim1−/− MDSCs (Supplemental Figure 6H). Collectively, these findings suggest that AZD1208 treatment subverts the immunosuppressive phenotype of myeloid cells in the TME.
When disrupting the suppressive phenotype of myeloid cells, we also observed significant improvement in CD8+ T-cell function. AZD1208 treatment alone induced a 2-fold expansion of IFNγ-producing CD8+ T cells in comparison with the PBS control group, and PD-1 blockade alone led to a 2.8-fold increase compared with PBS control group (Figure 6H–I). Combination therapy with AZD1208 + anti-PD-L1 further augmented the frequency of IFN-γ+ CD8+ T cells to 14%, a 3.5-fold increase over the control group (Figure 6I). A similar additive effect was observed with the expression of another effector molecule, granzyme B (Gzmb) (Figure 6J). Motivated by these results, we further evaluated the therapeutic effect of this combination therapy on a syngeneic B16-BL6 melanoma model. Co-administration of AZD1208 and anti-PD-L1 promoted strong antitumor activity and enhanced the survival of tumor-bearing mice when compared to either treatment alone (Figure 6K and Supplemental Figure 7A–E). Taken together, our data suggest that pharmacological inhibition of PIM kinase diminishes the function of immunosuppressive myeloid cells in the TME, which in turn releases the antitumor CD8+ T cell response from inhibition. This treatment further augments the therapeutic efficacy of PD-L1 blockade.
PIM kinase inhibition overcomes ICB resistance
The scRNA-seq analyses shown in Figure 2 revealed a strong correlation between FAT signature and ICB resistance, which prompted us to ask whether the presence of myeloid cells with FAT phenotypes could be used to predict tumor progression after treatment. We thus employed PPARγ and CD36 as indicators of FAT and identified a subset of MDSCs co-expressing both these markers in the circulating blood of MC38 tumor-bearing mice (Figure 7A). Notably, the frequencies of CD36highPPARγhigh myeloid cells positively correlated with tumor volumes following various treatments\ (Figure 7B). The Classification and Regression Trees (CART) supervised machine learning method estimated that mice with low levels (<50%) of PPARγhighCD36high circulating MDSCs were more likely to experience tumor shrinkage compared to mice with high levels (>50%) of PPARγhighCD36high MDSCs (Supplemental Figure 8A).
Figure 7. PIM kinase inhibition overcomes PD-L1 blockade resistance.
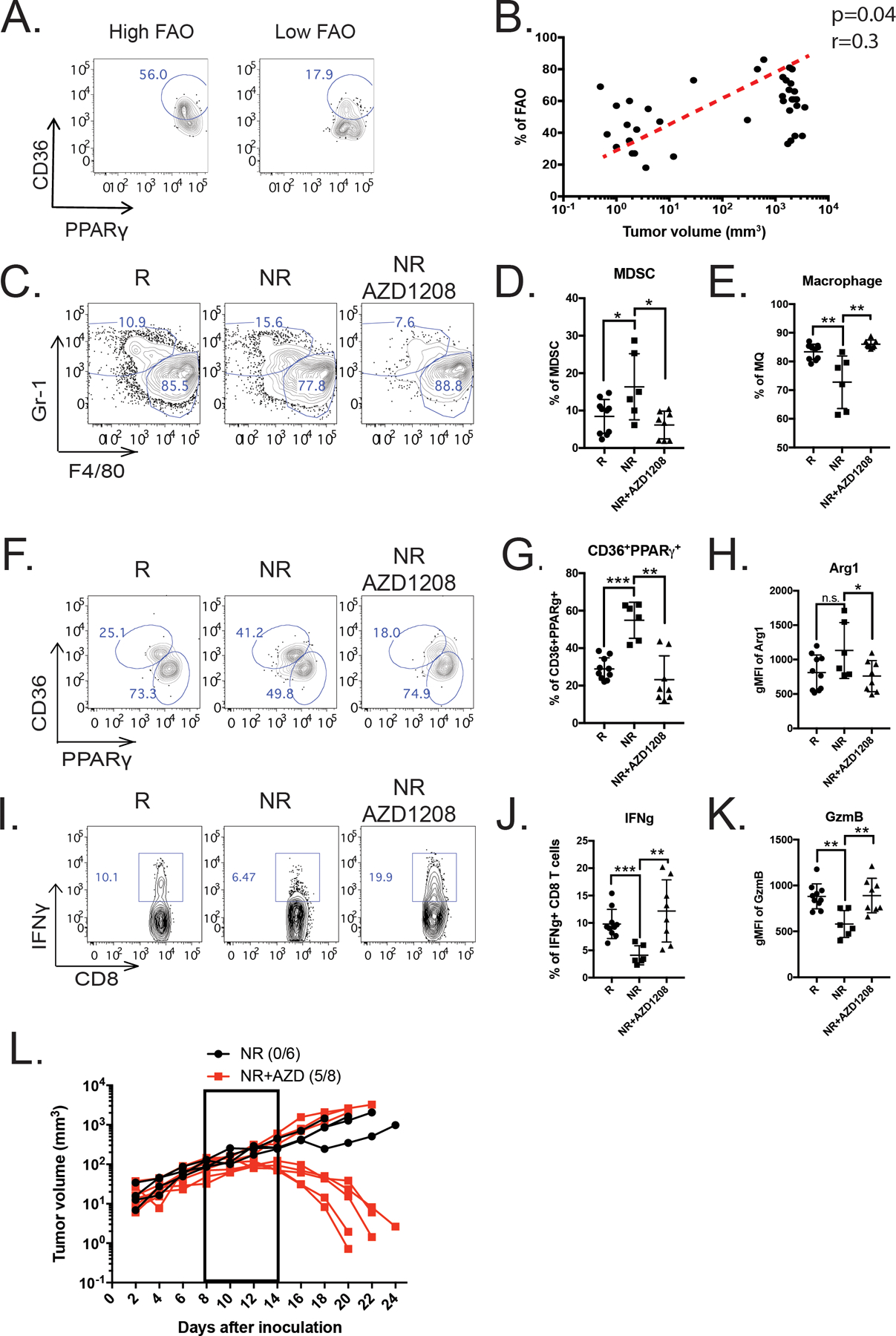
(A) Proportions of CD36highPPARγhigh myeloid cells are shown in representative flow plots. (B) Scatter plot depicting the correlation between the frequencies of CD36highPPARγhigh myeloid cells and tumor progression (n=38, spearman’s correlation test, p = 0.04, r = 0.3). (C-L) Bilateral MC38 tumor-bearing mice were treated with two doses of anti-PD-L1, and then were stratified into responders (R) and non-responders (NR) based on frequencies of CD36highPPARγhigh cells. All mice received one more dose of anti-PD-L1, while NRs received additional daily treatments of either vehicle control or AZD1208 for 10 days (see Supplemental Figure 7B). Upon completion of the full therapeutic regimen, left-sided tumors were harvested for flow cytometry analysis. (C-G) CD11b+Gr1−F4/80low MDSCs (C-D), CD11b+Gr1−F4/80high macrophages (C and E), and CD36highPPARγhigh MDSCs (F-G) are shown in representative contour plots and quantitative scatter plots. (H) Scatter plot shows the levels of Arg1 expression in MDSCs as measured by flow cytometry. (I-J) Intratumoral CD8+ T cell IFNγ secretion in response to anti-CD3/CD28 stimulation is shown in representative contour plots (I) and quantitative scatter plots (J). (K) Quantification of granzyme B expression in MDSCs as measured by flow cytometry (R=10, NR=6 and NR+AZD1208=8). (L) Individual tumor growth curves of non-responder mice treated with vehicle control (black dotted dash line) or AZD1208 (red solid line). Curves represent pooled data from three independent experiments with n = 6 for NR + vehicle and n = 8 for NR + AZD1208. Significance was determined by t-test, * p < 0.05, ** p < 0.01, *** p < 0.001.
Next, we sought to determine whether PIM kinase inhibition can reverse tumor progression in non-responders. To test this idea, mice bearing bilateral MC38 tumors were treated with two doses of anti-PD-L1 and the non-responder group received daily administration of either vehicle control or AZD1208 for 10 days (Supplementary Figure 8B). Compared to the responder group, we observed a significant accumulation of Gr-1+CD11b+ MDSCs in non-responders (Figure 7C–D). This enrichment was subverted by AZD1208 treatment, with mild effects on F4/80+CD11b+ TAMs (Figure 7E). With AZD1208 treatment, the frequencies of CD36highPPARγhigh myeloid cells from the non-responder group were significantly reduced to a level similar to the responder group (Figure 7F–G). More interestingly, this reduced FAT activity correlated with decreased expression of M2 markers (CD206 and Relmα) in TAMs (Supplemental Figure 8C–D) and Arg1 in MDSCs (Figure 7H). Consistent with these data, the effector function of CD8+ T cells in AZD1208-treated non-responders was significantly increased, as evidenced by their enhanced production of IFNγ and granzyme B (Figure 7I–K). Subsequently, 4 out of 7 AZD1208-treated mice originally deemed non-responders achieved complete tumor regression, while none of the vehicle-treated mice originally deemed non-responders eradicated their tumors (Figure 7L). In addition, we found that PIM inhibition induced similar functional and metabolic changes in human MDSCs via reduction of PPARγ (Supplementary Figure 8E–G). In conclusion, these findings suggest that PIM kinase inhibition can selectively target immunosuppressive myeloid cells with high fatty acid oxidative metabolism and in turn restore sensitivity to ICB in non-responders.
Discussion
To dissect the cellular and molecular mechanisms of ICB resistance, we employed scRNA-seq and identified that distinct features of the immune landscape in the TME, especially enhanced infiltration of immunosuppressive myeloid cells, correlated with non-responsiveness to ICB. In-depth computational analysis further unveiled a previously unappreciated role of PIM1 in regulating FAO metabolism in myeloid cells. Mechanistically, PIM1 regulates lipid metabolism by inducing PPARg and its downstream targets, such as CD36, which are responsible for lipid uptake in tumor-infiltrating myeloid cells. Consistent with these data, PIM1-deficient myeloid cells exhibited an impaired ability to inhibit the proliferation and antitumor function of effector CD8+ T cells. Targeting this pathway in MDSCs by pharmacologic inhibition of PIM kinases significantly increased the antitumor immune response and sensitized hosts to anti-PD-L1 therapy, overcoming ICB resistance.
A large body of clinical observations reveals that patients with elevated levels of MDSCs have impaired T-cell function and respond poorly to ICB (42) (43). Supporting this notion, selective elimination of MDSCs in various preclinical and clinical studies shows beneficial effects when combined with immunotherapy for treating solid tumors (44) (45) (46). As the importance of MDSCs in promoting resistance to immunotherapy is becoming increasingly recognized, understanding the fundamental biology of MDSCs has garnered an incredible amount of interest (46). In recent years, it has become well appreciated that metabolic pathways in these myeloid cells are not only required for fulfilling their energetic demands but are also critical for their immunosuppressive functions (47) (48). It is increasingly clear that lipid uptake for fueling mitochondrial OXPHOS is enhanced in M2 macrophages (49) (50) (51) and inhibition of lipid uptake significantly diminishes FAO metabolism, thereby impeding the immunosuppressive activity of MDSCs and enhancing tumor control in a T cell–dependent manner (52) (53). Consistent with this, we demonstrated that an increased FAO signature in tumor-infiltrating myeloid cells positively correlates with tumor progression and ICB resistance. More importantly, our scRNA-seq analyses provided some mechanistic insights into how the distinct metabolic features of MDSCs are regulated. To this end, a previously unappreciated role of PIM1 was identified: that of a key inducible kinase that regulates fatty acid uptake and oxidative metabolism in MDSCs to facilitate their survival and function in the TME. Our data have revealed significant deficiencies in fatty acid metabolism and suppressive function of MDSCs following genetic ablation or pharmacologic inhibition of PIM1. However, the impact of PIM1 on other metabolic pathways, such as glutaminolysis, is still unclear. Future investigations in identifying the alternative energy source or sources employed by Pim1−/− MDSCs are warranted to better ameliorate the suppressive activity of these cells.
The role of PIM1 in energy metabolism has been implicated in various types of cells, such as adipocytes, cardiomyocytes, colorectal cancer cells, and T cells (14) (54) (55). Nevertheless, little is known about how PIM1 regulates multiple aspects of cellular metabolism (56). Genetdeletion of Pim1 enabled us to identify possible metabolic pathways downstream of PIM kinases in MDSCs. To this end, our transcriptomic data revealed that expression levels of PPARγ, a key transcription factor that regulates lipid metabolism, are critically regulated by PIM1 activity. Although the detailed molecular mechanisms by which PIM1 regulates PPARγ expression remain unknown, our data have shown that overexpression of PPARγ in Pim1−/− MDSCs can sufficiently rescue their metabolic and functional defects and promote immunosuppression. Mechanistically, the downregulation of PPARγ in the absence of Pim1 is likely a consequence of decreased PIM1-mediated STAT3 S727 phosphorylation, which is integral for the maximal transcription activity (52) (53). In response to ligand stimulation such as IL6, activated JAK proteins phosphorylate the Tyr705 residue of STAT3 to induce its homodimerization and nuclear translocation (54). Another regulatory residue on STAT3 is Ser727, whose phosphorylation takes place in the C-terminal transactivation domain to maximize transcriptional activity (55) (56). Our results showed that pharmacological inhibition of PIM kinases specifically decreases STAT3Ser727, but not STAT3Tyr705, phosphorylation. More importantly, this altered STAT3 phosphorylation impaired STAT3 binding and transcription of Pparg. Taken together, our findings support a novel mechanism by which PIM1 enhances phosphorylation of STAT3 at the Ser727 site to maximize Pparg expression, which in turn promotes fatty acid metabolism and immunosuppressive activity in MDSCs.
Metabolic reprogramming of immune cells, such as CD8+ T cells, has emerged as a promising approach to enhance the effectiveness of immunotherapies (57, 58). However, few strategies have been developed to metabolically target MDSCs. Given that PIM1 is highly expressed in the myeloid lineage (Supplemental Figure 2E) but only serves as a backup for the AKT-mTOR pathway in T cells (13) (59), PIM kinase inhibition should preferentially disrupt the metabolism and function of MDSCs and have little effect on tumor-reactive T cells. Supporting this idea, we observed improved antitumor effector function in CD8+ T cells when the pan-PIM kinase inhibitor AZD1208 and anti-PD-L1 were co-administered. This is consistent with previous observations (13) (59) and suggests that PIM kinase inhibition-promoted function of CD8+ T cells is likely due to their release from MDSC-mediated suppression. Interestingly, a recent report has shown that PIM kinase inhibition improves the efficacy of immunotherapy by promoting memory phenotypes of T cells (60). Nonetheless, whether these phenotypic changes are intrinsic to tumor reactive T cells remain unclear.
PIM kinases are broadly expressed in both hematopoietic and non-hematopoietic cells (39) (40). Thus, it is possible that PIM kinase inhibition could cause off-target effects. For example, given that PIM kinases are expressed in cardiomyocytes and exert potent cardiac protective function by protecting mitochondrial integrity (61), it is possible that AZD1208 treatment may lead to cardiovascular-related adverse effects. Further work on selectively targeting PIM kinases specifically in MDSCs would be ideal for future clinical applications.
Taken together, our findings demonstrate that the PIM1-PPARγ axis regulates FAO metabolism in suppressive myeloid cells and promotes ICB resistance. This provides new insights into developing targeted approaches to abolish immunosuppression and in turn unleash the T-cell response to control tumors.
Supplementary Material
Synopsis:
Myeloid-derived suppressor cells (MDSC) are a barrier to successful anti-PD-L1 therapy. The authors show that PIM1 facilitates CD36-mediated fatty acid uptake via the PPARγ pathway, promoting MDSC immunosuppressive activity and that PIM1 inhibition overcomes resistance to anti-PD-L1 therapy.
Funding Information:
This work is supported by NIH grants AI125741 (W.C.) and AI148403 (W.C.), American Cancer Society Research Scholar Grant (W.C.), and A Healthier Wisconsin (AHW) Grant (W.C). G.X. is supported by The Elizabeth Elser Doolittle Postdoctoral Fellowship and PIIO-OSUCCC startup funding (46050 061052). W.C. is supported by the Medical College of Wisconsin Cancer Center and The Bartlog Endowment Fund.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Zielonka J, Sikora A, Joseph J, Kalyanaraman B. Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide: direct reaction with boronate-based fluorescent probe. J Biol Chem. 2010;285(19):14210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8(9):1069–86. [DOI] [PubMed] [Google Scholar]
- 3.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175(4):998–1013 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell. 2018;175(4):984–97 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111(32):11774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hossain F, Al-Khami Amir A., Wyczechowska Dorota, Hernandez Claudia, Zheng Liqin, Reiss Krzysztof, Valle Luis Del et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer immunology research 2015;canimm-0036. [DOI] [PMC free article] [PubMed]
- 8.Hammami I, Chen Jingkui, Murschel Frederic, Bronte Vincenzo, Gregory De Crescenzo, and Mario Jolicoeur. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC cell biology. 2012;13(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014;34(1):136–59. [DOI] [PubMed] [Google Scholar]
- 10.Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest. 2005;115(10):2618–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JL, Limnander A, Rothman PB. Pim-1 and Pim-2 kinases are required for efficient pre-B-cell transformation by v-Abl oncogene. Blood. 2008;111(3):1677–85. [DOI] [PubMed] [Google Scholar]
- 12.Din S, Mason Matthew, Völkers Mirko, Johnson Bevan, Cottage Christopher T., Wang Zeping, Joyo Anya Y. et al. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proceedings of the National Academy of Sciences. 2013;201213294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox CJ, Hammerman Peter S., and Thompson Craig B. The Pim kinases control rapamycin-resistant T cell survival and activation. Journal of Experimental Medicine. 2005;201(2):259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park YK, Obiang-Obounou Brice Wilfried, Lee Kyung-Bok, Choi Jong-Soon, and Jang Byeong-Churl. AZD1208, a pan-Pim kinase inhibitor, inhibits adipogenesis and induces lipolysis in 3T3-L1 adipocytes. Journal of cellular and molecular medicine 2018;22(4):2488–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nature biotechnology. 2015;33(5):495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sergushichev AA. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. BioRxiv. 2016;060012.
- 17.Picelli S, Faridani Omid R., Björklund Åsa K., Winberg Gösta, Sagasser Sven, and Sandberg Rickard. Full-length RNA-seq from single cells using Smart-seq2. Nature protocols. 2014;9(1):171–81. [DOI] [PubMed] [Google Scholar]
- 18.Kaya-Okur HS, Wu Steven J., Codomo Christine A., Pledger Erica S., Bryson Terri D., Henikoff Jorja G., Ahmad Kami, and Henikoff Steven. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature communications. 2019;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langmead B, and Steven L. Salzberg. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9(4):357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meers MP, Dan Tenenbaum, and Steven Henikoff. Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics & chromatin. 2019;12:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau J, Cheung Jeanne, Navarro Armando, Lianoglou Steve, Haley Benjamin, Totpal Klara, Sanders Laura et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nature communications. 2017;8:14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zemek RM, Emma De Jong, Wee Loong Chin, Schuster Iona S., Fear Vanessa S., Casey Thomas H., Forbes Cath et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Science translational medicine 2019;11(501):eaav7816. [DOI] [PubMed] [Google Scholar]
- 23.Stuart T, Butler Andrew, Hoffman Paul, Hafemeister Christoph, Papalexi Efthymia, Mauck William M. III, Hao Yuhan, Stoeckius Marlon, Smibert Peter, and Satija Rahul. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler A, Hoffman Paul, Smibert Peter, Papalexi Efthymia, and Satija Rahul. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology. 2018;36(5):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang P, Gu Shengqing, Pan Deng, Fu Jingxin, Sahu Avinash, Hu Xihao, Li Ziyi et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nature medicine. 2018;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan D, Adeshakin Adeleye O., Xu Meichen, Afolabi Lukman O., Zhang Guizhong, Chen Youhai H., and Wan Xiaochun. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Frontiers in immunology. 2019;10:1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hänzelmann S, Robert Castelo, and Justin Guinney. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics. 2013;14(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Khami AA, Zheng Liqin, Valle Luis Del, Hossain Fokhrul, Wyczechowska Dorota, Zabaleta Jovanny, Sanchez Maria D., Dean Matthew J., Rodriguez Paulo C., and Ochoa Augusto C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. 2017;6(10):e1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montgomery MK, Osborne Brenna, Brandon Amanda E., O’Reilly Liam, Fiveash Corrine E., Brown Simon HJ, Wilkins Brendan P. et al. Regulation of mitochondrial metabolism in murine skeletal muscle by the medium-chain fatty acid receptor Gpr84. The FASEB Journal. 2019;33(11): 12264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Recio C, Lucy Daniel, Purvis Gareth, Iveson Poppy, Zeboudj Lynda, Iqbal Asif Jilani, Lin Da et al. “. Activation of the Immune-Metabolic Receptor GPR84 Enhances Inflammation and Phagocytosis in Macrophages. Frontiers in immunology. 2018;9:1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Block KM, Hanke Neale T., Maine Erin A., and Baker Amanda F. IL-6 stimulates STAT3 and Pim-1 kinase in pancreatic cancer cell lines. Pancreas. 2012;41(5):773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao X, Liu Xiangping, Lu Yangyong, Wang Yu, Cao Weihong, Liu Xiaoyi, Hu Haiyan, and Wang Haibo. PIM1 is responsible for IL-6-induced breast cancer cell EMT and stemness via c-myc activation. Breast Cancer. 2019;26(5):663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones SA, and Jenkins Brendan J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nature Reviews Immunology. 2018;18(12):773–89. [DOI] [PubMed] [Google Scholar]
- 34.Tirosh I, Izar Benjamin, Prakadan Sanjay M., Wadsworth Marc H., Treacy Daniel, Trombetta John J., Rotem Asaf et al. “. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang Z, Li Chenwei, Kang Boxi, Gao Ge, Li Cheng, and Zhang Zemin. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research. 2017;45(W1):W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marigo I, Bosio Erika, Solito Samantha, Mesa Circe, Fernandez Audry, Dolcetti Luigi, Ugel Stefano et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity. 2010;32(6):790–802. [DOI] [PubMed] [Google Scholar]
- 37.Cortes J, Tamura Kenji, DeAngelo Daniel J., de Bono Johann, Lorente David, Minden Mark, Uy Geoffrey L. et al. Phase I studies of AZD1208, a proviral integration Moloney virus kinase inhibitor in solid and haematological cancers. British journal of cancer. 2018;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keeton EK, Kristen McEachern, Dillman Keith S., Palakurthi Sangeetha, Cao Yichen, Grondine Michael R., Kaur Surinder et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2013. [DOI] [PMC free article] [PubMed]
- 39.Białopiotrowicz E, Patryk Górniak, Noyszewska-Kania Monika, Bartosz Puła, Hanna Makuch-Łasica, Nowak Grażyna, Bluszcz Aleksandra et al. Microenvironment-induced PIM kinases promote CXCR 4-triggered mTOR pathway required for chronic lymphocytic leukaemia cell migration. Journal of cellular and molecular medicine. 2018;22(7):3548–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Decker S, Finter Johannes, Forde Aaron James, Kissel Sandra, Schwaller Juerg, Mack Thomas Sebastian, Kuhn Anabel et al. PIM kinases are essential for chronic lymphocytic leukemia cell survival (PIM2/3) and CXCR4-mediated microenvironmental interactions (PIM1). Molecular cancer therapeutics. 2014;13(5): 1231–45. [DOI] [PubMed] [Google Scholar]
- 41.Hutchins AP, Stéphane Poulain, and Diego Miranda-Saavedra. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119(13):e110–e9. [DOI] [PubMed] [Google Scholar]
- 42.Gide TN, Quek, Menzies Alexander M., Tasker Annie T., Ping Shang, Jeff Holst, Jason Madore et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer cell 2019;35(2):238–55. [DOI] [PubMed] [Google Scholar]
- 43.Weber R, Fleming Viktor, Hu Xiaoying, Nagibin Vasyl, Groth Christopher, Altevogt Peter, Utikal Jochen, and Umansky Viktor. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Frontiers in immunology. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veglia F, Tyurin Vladimir A., Blasi Maria, De Leo Alessandra, Kossenkov Andrew V., Donthireddy Laxminarasimha, Jerrick To Tsun Ki et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019;569(7754):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar V, Cheng Pingyan, Condamine Thomas, Mony Sridevi, Languino Lucia R., McCaffrey Judith C., Neil Hockstein et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity. 2016;44(2):303–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Veglia F, Michela Perego, and Dmitry Gabrilovich. Myeloid-derived suppressor cells coming of age. Nature immunology. 2018;19(2):108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van den Bossche J, O’Neill Luke A., and Deepthi Menon. Macrophage immunometabolism: where are we (going)? Trends in immunology 38, no 6 (2017): . 2017;38(6):395–406. [DOI] [PubMed] [Google Scholar]
- 48.Diskin C, and Pålsson-McDermott Eva M. Metabolic modulation in macrophage effector function. Frontiers in immunology. 2018;9:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang SC-C, Everts Bart, Ivanova Yulia, O’sullivan David, Nascimento Marcia, Smith Amber M., Beatty Wandy et al. Cell-intrinsic lysosomal lipolysis is essential for macrophage alternative activation. Nature immunology. 2014;15(9):846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vats D, Mukundan Lata, Odegaard Justin I., Zhang Lina, Smith Kristi L., Morel Christine R., Greaves David R., Murray Peter J., and Chawla Ajay. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell metabolism. 2006;4(1): 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’neill LA, and Pearce Edward J. Immunometabolism governs dendritic cell and macrophage function. Journal of Experimental Medicine. 2016;213(1):15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hossain F, Al-Khami Amir A., Wyczechowska Dorota, Hernandez Claudia, Zheng Liqin, Reiss Krzystoff, Valle Luis Del et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer immunology research. 2015;3(11):1236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Al-Khami AA, Zheng Liqin, Valle Luis Del, Hossain Fokhrul, Wyczechowska Dorota, Zabaleta Jovanny, Sanchez Maria D., Dean Matthew J., Rodriguez Paulo C., and Ochoa Augusto C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. 2017;6(10):e1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borillo GA, Mason Matt, Quijada Pearl, Völkers Mirko, Cottage Christopher, McGregor Michael, Din Shabana et al. Pim-1 kinase protects mitochondrial integrity in cardiomyocytes. Circulation research. 2010;106(no. 7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang M, Liu Tingting, Sun Hui, Weng Weiwei, Zhang Qiongyan, Liu Chenchen, Han Yang, and Sheng Weiqi. Pim1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect. Cancer science 2018;109(no. 5): 1468–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahmadian M, Suh Jae Myoung, Hah Nasun, Liddle Christopher, Atkins Annette R., Downes Michael, and Evans Ronald M. PPARγ signaling and metabolism: the good, the bad and the future. Nature medicine 2013;19(5):557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Sullivan D, Sanin David E., Pearce Edward J., and Pearce Erika L. Metabolic interventions in the immune response to cancer. Nature Reviews Immunology. 2019;19(5):324–35. [DOI] [PubMed] [Google Scholar]
- 58.Chatterjee S, Daenthanasanmak Anusara, Chakraborty Paramita, Wyatt Megan W., Dhar Payal, Panneer Selvam Shanmugam, Fu Jianing et al. CD38-NAD+ axis regulates immunotherapeutic anti-tumor T cell response. Cell metabolism. 2018;27(1):85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hammerman PS, Fox Casey J., Birnbaum Morris J., and Thompson Craig B. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood. 2005;105(11): 4477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chatterjee S, Chakraborty Paramita, Daenthanasanmak Anusara, Iamsawat Supinya, Andrejeva Gabriela, Luevano Libia A., Wolf Melissa et al. Targeting PIM Kinase with PD1 Inhibition Improves Immunotherapeutic Antitumor T-cell Response. Clinical Cancer Research. 2019;25(3):1036–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Borillo GA, Mason Matt, Quijada Pearl, Völkers Mirko, Cottage Christopher, McGregor Michael, Din Shabana et al. Pim-1 kinase protects mitochondrial integrity in cardiomyocytes. Circulation research. 2010;106(7):1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. RNA-seq data were deposited in the NCBI GEO database under accession codes GSE166308 and GSE166309.