

Abstract
Inactivation of tumor-infiltrating lymphocytes (TIL) is one of the mechanisms mitigating anti-tumor immunity during tumor onset and progression. Epigenetic abnormalities are regarded as a major culprit contributing to the dysfunction of TILs within tumor microenvironments. In this study, we used a murine model of melanoma to discover that Tet2 inactivation significantly enhances the anti-tumor activity of TILs with an efficacy comparable to immune checkpoint inhibition imposed by anti-PD-L1 treatment. Single-cell RNA-seq analysis suggested that Tet2-deficient TILs exhibit effector-like features. Transcriptomic and ATAC-seq analysis showed that Tet2 ablation reshapes chromatin accessibility and favors binding of transcription factors geared toward CD8+ T cell activation. Furthermore, the ETS family of transcription factors contributed to augmented CD8+ T cell function following Tet2 depletion. Overall, our study establishes that Tet2 constitutes one of the epigenetic barriers that account for dysfunction of TILs, and that Tet2 inactivation could promote anti-tumor immunity to suppress tumor growth.
Introduction
Dynamic epigenetic alterations have been observed in CD8+ tumor infiltrating lymphocytes (TILs) during tumor progression (1,2). Epigenetic plasticity is one of the critical factors governing the expression of key genes that define the status of TILs during tumor development. Recent studies have shown that DNA methylation plays an essential role in regulating CD8+ T cell immune response during chronic infection and tumorigenesis (3–6). Both effector and inhibitory genes are tightly controlled by DNA methylation during CD8+ expansion, activation and exhaustion (7). For example, the demethylation of Gzmb, Ifng, and Pdcd1 loci are essential for active expression of these genes in CD8+ effector T cells during acute infection (3). In addition, Ghoneim et al reported that blockage of de novo DNA methylation via Dnmt3a deletion resulted in prolonged CD8+ T cell immunity during persistent immune response (4). Clearly, DNA methylation-associated epigenetic reprogramming is critical for the adaptive immune response of CD8+ T cells.
The reversal of DNA methylation, or DNA demethylation, is catalyzed by the Ten-eleven Translocation (TET) protein family (TET1, 2, 3) of dioxygenases (8–10), which successively oxidize 5-methylcytosine (5mC) to yield 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) (8–10). Genetic depletion of Tet could impair chromatin accessibility and perturb the binding of transcription factors (TFs) to their genomic targets in cells of the immune systems, including B cells, T cells and macrophages (11–14). Among all the three Tet proteins, Tet2 is most frequently mutated in the hematopoietic and immune system (15–17). Tet2-deficient hematopoietic stem cells, lymphoid or myeloid cells exhibited aberrant immune activities when responding to external stimulations (18–20). For example, Tet2 ablation in CD8+ T cells promotes memory differentiation and enhances pathogen control (6). In addition, disruption of TET2 in both chimeric antigen receptor (CAR) T-cells and tumor-infiltrating myeloid cells (13,14) improves immunotherapy efficiency, suggesting the beneficial effects of TET2 inactivation during anti-tumor immunotherapy. At the molecular level, we and other groups have shown that Tet-mediated DNA methylation oxidation regulates gene expression by controlling chromatin accessibility and transcription factor binding (TF) during embryonic development (11,21). However, the role of Tet2 in TILs during anti-tumor immunity has not been systematically defined.
In this study, we utilized a murine B16-OVA and Tet2-deficient OT-I mouse model to investigate the function of Tet2 in TILs. We unveiled that Tet2-deficient CD8+ T cells exhibited enhanced cytotoxicity to tumor cells at an early stage of tumorigenesis, which resulted in significantly enhanced anti-tumor efficiency. Tet2-deficient TILs showed similar anti-tumor activity to immune checkpoint inhibitors, e.g., anti-PDL-1. Transcriptomic and epigenomic studies revealed that Tet2 inactivation reshaped the chromatin accessibility at the genomic binding sites of several key transcription factors (e.g., the bZIP and ETS TF families), thereby enhancing the transcription of genes encoding effector molecules (e.g., Tnf and Prf1) to promote the anti-tumor efficiency of TILs and suppress melanoma growth in vivo.
Method and Materials
Mice
C57BL/6J mice (The Jackson Laboratory) and Tet2−/− mice (22) were crossed with OT-1+ mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J, The Jackson Laboratory) bearing transgenic T cell receptors that recognize ovalbumin (OVA257–264) in the context of CD8 co-receptor interaction with MHC class I. Crossed mice in age between 6 and 20 weeks were used for CD8+ T cell isolation. 6–12 week-old CD45.1+ mice (B6.SJL-Ptprca Pepcb/BoyJ, The Jackson Laboratory) were challenged with B16-OVA mouse melanoma tumor cells then used as recipients for adoptive T cell transfer experiments. All mice were maintained in the animal facility at the Institute of Biosciences and Technology, Texas A&M University. All animal studies were approved by the Institutional Animal Care Use Committee (IACUC) of the Texas A&M University Institute of Biosciences and Technology.
Mouse T cell isolation and ex vivo activation.
CD8+ T cells were isolated using a mouse CD8a+ T Cell Isolation Kit (Miltenyi Biotec). Briefly, harvested spleens and lymph nodes from WT-OT1+ and Tet2−/− OT1+ mice were grounded and filtered with 70-μm cell strainers (Falcon) to remove debris. The suspended cells were treated with an ACK (Ammonium-Chloride-Potassium) lysis buffer to lyse red blood cells and residual cells were purified for total CD8+ T cell isolation using the T cell isolation kit according to the manufacturer protocol. Purified CD8+ T cells were activated by anti-CD3 (clone 17A2, InVivoMAb), anti-CD28 (clone 37.51, InVivoMAb) and a high dose of mouse IL-2 (100 unit/ml, eBioscience), and cultured in multi-well plates pre-coated with a goat anti-hamster IgG (H+L, Invitrogen). After 2 days of culture, T cells were removed from the coated plate and seeded with a low dose of mouse IL-2 (20 units/ml, eBioscience) for 24 hrs before injection.
The B16-OVA mouse model of melanoma and adoptive T cell transfer
B16-OVA mouse melanoma cells were obtained from Dr. Anjana Rao’s laboratory in La Jolla Institute (23) and were cultured in complete Dulbecco’s Modification of Eagle’s Medium (DMEM), and passaged at least two times before injection. The cells were tested negative for potential mycoplasma contamination on a monthly basis. Cultured B16-OVA cells were trypsinized and washed with 1X PBS and diluted at 6 million cells / mL. Prepared B16-OVA cells (300,000 cell / mice in 50 μL PBS) were injected intradermally into the dorsal flanks of CD45.1+ recipient mice. Tumor sizes were measured and recorded every day. 10–12 days after B16-OVA inoculation, in vitro cultured and activated WT-OT1+ and Tet2−/− OT1+ CD8+ T cells were retro-orbitally injected into tumor bearing CD45.1+ mice (2 million cells / mice). Mice were sacrificed 3 and 8 days after adoptive T cell transfer. Anti–PD-L1 Ab (clone no. 10F.9G2, BioXCell, 200 μg / mice) or control anti-KLH rat IgG2b (BioXCell) were injected (intraperitoneally, i.p.) in the corresponding groups at Day 3 and Day 6 after adoptive transfer.
CD8+ T cell isolation
Spleens, peripheral blood and tumor mass were dissected at the indicated time points after adoptive T cell transfer. Spleens and peripheral blood undergo red blood cell lysis by using an ACK lysis buffer and used for downstream flow cytometry analysis. Dissected tumors were dissociated using ultra-fine scissors and tweezers, and then incubated in RPMI media with Liberase TL (100 μg / mL, Roche) for 15 min at 37 °C. The same amount of Liberase was added into the sample and incubated for additional 10 min at 37 °C. The dissociated tissues were then passed through 70 μm cell strainers (Falcon) and washed with PBS for three times. Cells were then labeled with a mouse anti-CD63-biotin antibody (Miltenyi Biotec) followed by the negative selection using anti-biotin microbeads (Miltenyi Biotec). The negatively selected cells were then stained with corresponding cell surface markers for flow cytometry analysis or cell sorting.
Cell surface marker staining and in vitro re-stimulation for cytokine staining
In vitro activated CD8+ T cells (before injection, Day 0 control) and purified TILs were stained with corresponding cell surface markers (dilution of 1:100 to 1:200) in FACS buffer (1X PBS with 0.1% FBS and 2mM EDTA) for 20 min. Stained cells were washed twice using FACS buffer. For cytokine staining, same sets of cells were seeded in 48-well plate followed by incubation with T cell media containing 10 nM of Phorbol 12-myristate 13-acetate (PMA, Sigma-aldrich), 500 nM of ionomycin (Thermofisher), and 1 μg/ml of brefeldin (BioLegend) for 4 hrs at 37 °C. Then the cells were stained with corresponding cell surface antibodies (CD45.1, CD45.2 and CD8) followed by fixation and permeabilization with BD Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit (BD biosciences). Fixed and permeabilized cells were then stained with cytokine antibodies and proceed for flow cytometry analysis. All flow cytometry analysis was performed using LSRII (BD Biosciences) and the data was analyzed using FlowJo v.10 (Tree Star, Inc.)
In vitro cytotoxicity assay
B16-OVA cells (250,000 cell / well) were incubated with a Cell Proliferation Dye conjugated with APC (eBioscience) and seeded on 24-well plates for 1 hr at 37 °C. Then, the culture medium was removed and 4X amount of in vitro activated CD8+ T cells with complete T cell media were added into labeled B16-OVA cells for 4 and 8 hrs at 37 °C. Co-cultured cells were then washed twice with 1X PBS and dead cells were stained with the CellEvent™ Caspase-3/7 Green Detection Reagent (Invitrogen) according to the manufacturer instructions. Double stained populations were examined by flow cytometry analysis.
For cell viability, proliferation and cell cycle analysis, CD8+ T cells were co-cultured with pre-stained B16-OVA cells for 4 hrs before analysis. After co-culture, the cell mixture was stained with CD8a antibody (Biolegend). For cell viability analysis, Propidium Iodide (PI, BD Pharmingen, 5 μL) was incubated with the cell mixture for 5 min and the dead cell population was measured by the PI positive population within CD8+ gated cells. For cell proliferation analysis, the cell mixture was fixed and permeabilized using Transcription Factor Buffer Set (BD Pharmingen) followed by incubating with anti-Ki-67 antibody (Biolegend). Actively proliferating cells were detected by the Ki-67 positive population within CD8+ gated cells. For cell cycle analysis, 20 μM of BrdU was incubated with the cultured cell mixture for 2 hrs followed by an additional 2 hrs incubation. Then the cell mixture was stained with anti-CD8a antibody and fixed / permeabilized using Transcription Factor Buffer Set (BD Pharmingen). Fixed cell mixture was then incubated with 20 μg DNAseI (Sigma-aldrich) at 37°C for 1 hr. Then BrdU antibody was used to capture BrdU-pulsed cells and 7-AAD (Tonbo Bioscience) was used to stain DNA.
Pharmacological inhibition treatment
YK-4–279 (Tocris Bioscience, 0.1 μM) and TK216 (MedChemExpress, 0.5 nM) were used in both individual B16-OVA or CD8+ T cell culture, and co-culture experiments.
RNA extraction, reverse transcription and quantitative real-time PCR
Isolated TILs were lysed with RLT buffer (Qiagen) and homogenized using 16G of syringe 5 times. Then total RNA was purified using AllPrep DNA/RNA micro kit (Qiagen) following manufacturer’s instruction. The concentration of RNA was measured by Qubit 4 Fluorometer with Qubit RNA high sensitivity assay kit. Purified RNA (500 ng) was reverse transcribed using ABscript II cDNA First Strand Synthesis kit (ABclonal). Gene expression was quantified on ViiA 7 Real-Time PCR System (Applied Biosystems) using 2X Universal SYBR Green Fast qPCR Mix (ABclonal). Two-step cycling program was used with initial denaturation for 10 min at 95°C, followed by 40 cycles with denaturation for 10 s at 95°C and annealing/elongation at 60°C for 30 s. The total mRNA amount was normalized to endogenous GAPDH mRNA. Primers used for qPCR are listed below:
| GAPDH | For | 5’-GTGTTCCTACCCCCAATGTGT-3’ |
| Rev | 5’-ATTGTCATACCAGGAAATGAGCTT-3’ | |
| Prf1 | For | 5’-AATATCAATAACGACTGGCGTGT-3’ |
| Rev | 5’-CATGTTTGCCTCTGGCCTA-3’ | |
| Icam1 | For | 5’-CAATTTCTCATGCCGCACAG-3’ |
| Rev | 5’-AGCTGGAAGATCGAAAGTCCG-3’ | |
| Itga5 | For | 5’-ACGTTACATAGCATAGTACCTCTTC-3’ |
| Rev | 5’-TACTGATGGTCTAAATTTGAACTGC-3’ |
ATAC-seq library preparation and data analysis
ATAC-seq library preparation was performed as described previously (24). In brief, nuclei were isolated from 50,000 cells in a lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630), then transposition reaction was performed using an Illumina Nextera DNA library preparation kit at 37 °C for 30 min. Tagmented DNA fragments were purified using a MinElute PCR purification kit (Qiagen) and purified DNA were amplified with a KAPA real-time library amplification kit (Roche). Amplified libraries were then purified with AmpureXP beads. The size of DNA libraries was measured by Bioanalyzer using an Agilent High Sensitivity DNA Kit (Agilent). The concentration of DNA Libraries was measured by the Qubit 4 Fluorometer with a Qubit dsDNA high sensitivity assay kit (Thermofisher). Equimolarly combined libraries were sequenced on NextSeq 500 (Illumina) with the NextSeq 500/550 High Output Kit v2.5 (80PE).
Adaptor trimming of raw reads were performed by TrimGalore v0.5.0 with default parameters, and high-quality (Q ≥ 20) reads were uniquely aligned to mm10 reference genome using Bowtie2 with ‘--very-sensitive’ options. Only uniquely mapped reads were finally extracted for downstream analysis. The resulting alignment of each sample (with two biological replicates) was analyzed by Genrich v.0.5 (https://github.com/jsh58/Genrich) with ATAC-seq mode (option: -j, -q 0.05, -d 150). The options to remove PCR duplicates (-r) and to discard alignments to chrM (-e chrM) were used to call chromatin accessible peak regions. Bedtools merge was used to count the reads that fell into non-overlapped peak regions, and the significantly Differential Chromatin Accessible Regions were detected using DESeq2 with normalized peak signals (fold change ≥ 2; FDR < 0.05). Venn diagram was plotted using R package ggplot2 to find group-specific chromatin changed regions. Motif annotation of differential chromatin accessible regions was performed using HOMER software. GREAT (25) analysis was used to perform the functional annotation of differential chromatin accessible regions. The number of mapped reads and mapped ratios were listed in Table S1.
To detect the chromatin accessibility across the transcription factors, published BATF and ETS1 ChIP-seq data were downloaded from GSE54191 and GSE56393, respectively. High-quality reads were mapped to mm10 reference using Bowtie2 with “-very-sensitive” options, and the binding regions were detected using Macs2 with default parameters. The Profile and heatmaps of chromatin accessibility across upstream and downstream of BATF binding loci were generated using the computeMatrix and plotHeatmap scripts from the deepTools package. Bam2wig.py was used to transform the bam file to normalized bigwig files (option: -t 2000000000).
RNA-seq library preparation and data analysis
PolyA tailed mRNA were enriched using NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB). A NEBNext Ultra™ II Directional RNA Library Prep Kit for Illumina (NEB) was used to generate RNA-seq libraries. Then adaptor for Illumine sequencing system was ligated into cDNA followed by final library amplification with illumina TruSeq single index (NEB). Constructed libraries were purified with AmpureXP beads. Size and concentration of generated libraries were verified as described above.
To process the sequencing data, low quality bases and adaptors were trimmed using TrimGalore v.0.5.0 with default parameters (https://github.com/FelixKrueger/TrimGalore). Clean reads of RNA-seq data were aligned to mm10 reference genome using Hisat2 (version, 2.1.0) with default parameters and only uniquely mapped reads were used for downstream analysis. Count matrix for each gene was generated using htseq-count (HTSeq package). DESeq2 was used to identify significantly differentially expressed genes (DEGs) in WT and Tet2KO samples between different time points (fold change ≥ 2; FDR < 0.05). In-house R scripts were used to plot the volcano and scatter plots for DEGs. The Gene Set Enrichment Analysis (GSEA) was performed for the functional enrichment analysis of DEGs. Hierarchical cluster analysis of the union DEGs was used to determine group-specific signature genes. The number of mapped reads and mapped ratios were listed in Table S1.
Single-cell RNA-seq libraries were generated using the Chromium Single-Cell 3′ Reagent V2 Kit (10× Genomics) according to the manufacturer’s instructions. Briefly, single cell GEM was generated and barcoded in a Chromium Controller (10× Genomics). Then RNA transcripts from single cells were reverse transcribed, amplified and fragmented. The library size and concentration were measured using Agilent Bioanalyzer 2100 (Agilent) and Qubit 4 Fluorometer with Qubit dsDNA high sensitivity assay kit as described above. Libraries were subjected to illumina NextSeq 500 system using the NextSeq 500 High Output v2 Kit (Illumina) with a paired end, dual indexing (26/8/0/58-bp) format. The mkfastq in Cellranger (version, 3.0.2) was firstly used to demultiplex the raw sequencing data into fastq files. Cellranger was used to align raw fastq files to mm10 reference genome and perform barcode and UMI counting. The count matrix for each gene of each cell was taken as an input file for R package Seruat (version, 2.3). Reads with the same UMI were combined and then annotated to ensembl genes (mm10). To ensure the data quality, the genes detected in less than 10 cells and the cells with less than 20 genes were filtered out firstly from each dataset. We used the first 8 principle components to perform cell cluster and t-Distributed Stochastic Neighbor Embedding (t-SNE) with resolution = 0.4. To identify the marker genes, differential expression analysis was performed by FindAllMarkers function with Wilcoxon rank sum test. Violin plots were generated using Seurat Vlnplot function. The raw reads of different CD8+ tumor-infiltrating lymphocytes (TILs) cells (bulk RNA-seq dataset) were downloaded from GSE122969, and the data analysis was similar to RNA-seq data of this study. The heatmap of scaled gene expression level across top 20 significant marker genes for each cluster was plotted by R package ComplexHeatmap.
Accession numbers
The sequencing datasets have been deposited into NCBI BioProject under the accession number GSE152496.
Results
Tet2 deletion enhances the anti-tumor activity of CD8+ tumor infiltrated lymphocytes (TILs).
In order to study the function of Tet2 in CD8+ TILs, we crossed Tet2KO mice (22) with OT-I mice (26) to generate a Tet2KO-OT-I mouse model (Figure S1A), which could recognize the chicken ovalbumin (OVA) antigen (27) expressed on the surface of B16 murine melanoma cells (B16-OVA). In this model, engineered B16-OVA melanoma cells could be recognized by adoptively transferred OT-I OVA specific CD8+ T cells during tumor onset and progression (28,29). As shown in Figure 1A, the B16-OVA melanoma cells were intradermally injected in the B6.SJL-Ptprca Pepcb/BoyJ recipient mice (30) expressing CD45.1 isoform in their lymphocytes. In parallel, the CD8+ T cells were purified from wild-type (WT)-OT-I or Tet2KO-OT-I mice with CD45.2 expression in their lymphocytes, and cultured ex vivo for expansion for up to 72 h. Next, the purified CD45.2+CD8+OT-I T cells (WT or Tet2KO; 2 million cells / mice) were adoptively transferred to the tumor-bearing CD45.1 recipient mice at 10–12 days after tumor inoculation via retro-orbital injection. To compare the tumor-suppressive effects between immune checkpoint inhibitors and Tet2 ablation, we further treated the mice with an anti-PD-L1 inhibitor or IgG2b antibody (as control). In mice transferred with Tet2KO TILs, we observed a pronounced reduction of tumor size when compared with the WT group (Figure 1B and S1B, red line). The similar suppressive effect was also noted in mice transferred with WT TILs and treated with anti-PD-L1 antibody (Figure 1B and S1B, green line). Surprisingly, the anti-PD-L1 treatment had minor effects on mice transferred with Tet2KO TILs compared with the control group transferred with Tet2KO TILs (Figure 1B and S1B, blue vs red lines). These data suggested that Tet2 depletion in CD8+ T cells exhibits similar tumor-suppressive effects as anti-PD-L1 treatment does to melanoma. However, the combination of Tet2 loss in CD8+ T cells with anti-PD-L1 treatment does not seem to further benefit the anti-tumor efficacy.
Figure 1. Tet2-deficient TILs exhibit enhanced anti-tumor activity in vivo.
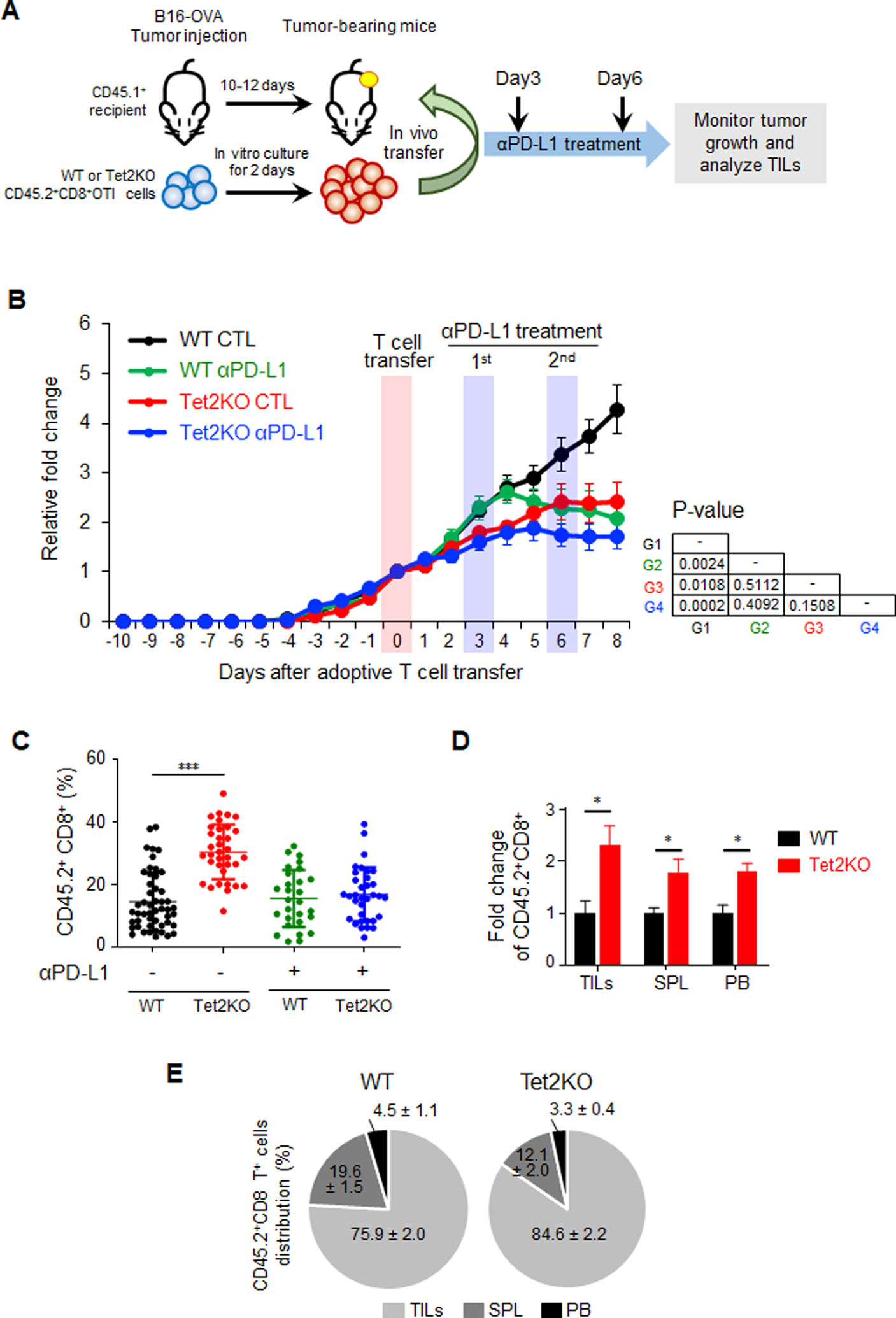
A. Experimental design of adoptive transferring in vitro-generated OT-I CD8+ T cells (WT and Tet2KO; CD45.2+) into recipient mice (CD45.1+) bearing subcutaneous B16-OVA tumors. The mice were then treated with or without an anti-PD-L1 antibody at 3 and 6 days after T cell transfer.
B. Quantification of B16-OVA melanoma tumor size in four experimental groups: WT control (CTL; black), WT with anti-PD-L1 treatment (green), Tet2KO (red), Tet2KO with anti-PD-L1 treatment (blue). Data were shown as mean ± S.D; n=16–27 mice; P values were listed on the right next to the curves (two-tailed Student’s t-test).
C. Quantification of the percentage of CD45.2+CD8+ TILs (WT vs Tet2KO) in CD8+ T cells population with and without anti-PD-L1 treatment at 8 days after adoptive transfer. Data were shown as mean ± S.D; n=12–24 mice, ** P < 0.005 (two-tailed Student’s t-test).
D. Comparison of the relative distribution of adoptively transferred WT (black) and Tet2KO (red) CD45.2+CD8+ cells in tumor (TILs), spleen (SPL) and peripheral blood (PB) at 8 days in the recipient mice. The data were shown as the fold change relative to WT in TILs, spleen and peripheral blood. Data were shown as mean ± S.D; n=6–11 mice, * p < 0.05, by two-tailed Student’s t-test.
E. Percentage of adoptively transferred WT (left) and Tet2KO (right) CD45.2+CD8+ T cells within tumor (TILs), spleen (SPL) and peripheral blood (PB) at 8 days after adoptive transfer. Data were shown as mean ± S.D; n=6–11 mice.
We then moved on to analyze the distribution of donor CD45.2+CD8+ T cells at 8 days after adoptive transfer in the melanoma-bearing CD45.1+ recipient mice (Figure 1C, Figure S1C–D). Within the tumor foci, we observed a significant increase of the fraction of CD45.2+CD8+OT-I TILs in the Tet2KO group when compared with that in the WT group. Interestingly, the expansion of Tet2KO TILs was not evident in the similar recipient mice treated with anti-PD-L1 (Figure 1C), suggesting that anti-PD-L1 treatment might exert inhibitory effects on Tet2KO TILs expansion. To further track the adoptively transferred CD8+ T cells, we measured the CD45.2+CD8+OT-I T cells in the tumor, spleen and peripheral blood (PB) (Figure 1D). We observed a marked increase of CD45.2+CD8+OT-I T cells (~1.5–2.5 fold) in all these tissues for the Tet2KO group, which is consistent with the previous observation that Tet2-deficient lymphocytes exhibit an expansion advantage (6,19). In addition, within all the injected CD45.2+CD8+OT-I T cells, Tet2KO CD8+ T cells tend to be enriched more at tumor foci when compared with the WT group (Figure 1E). Interestingly, in addition to CD45.2+CD8+OT-I T cells, we observed significantly higher endogenous CD45.1+CD8+OT-I T cells within the tumor foci, but not in lymphocytes or peripheral blood (PB) of the recipient mice transferred with Tet2KO CD8+ T cell when compared to the mice transferred with WT CD8+ T cells (Figure S1D–E). These findings suggest that the tumor suppressive effect of Tet2KO TILs might involve both autonomous and non-autonomous cellular effects.
Tet2KO TILs exhibit enhanced immune response and cytotoxicity at the early stage.
To further characterize TILs in the B16-OVA mouse model of melanoma, we isolated the tumors at 3 and 8 days after CD8+ T cell injection, purified CD8+ TILs and performed flow cytometry analysis on the signature marks within both the CD45.2+ (transferred) and CD45.1+ (endogenous) populations. After adoptive transfer in the Tet2KO group, we observed a significant increase in the production of IFN-γ and TNF-α, two signature cytokines reflecting the effector function of CD8+ T cells (Figure 2A), at day 3 but not at day 8, compared with the WT group. A similar trend was noted for the PD-1, Tim-3 positive population (Figure 2B). These data suggested that deletion of Tet2 enhances the anti-tumor efficiency of TILs at an early stage. In parallel, we measured IFN-γ, TNF-α PD-1, and Tim-3 levels in the endogenous CD45.1+CD8+ TILs (Figure S2A–B). No significant difference of IFN-γ or TNF-α production in endogenous CD45.1+CD8+ TILs was detected between the WT and Tet2KO groups (Figure S2A). We observed elevated PD-1 or Tim-3 positive population in mice transferred with Tet2KO CD45.2+CD8+ TILs at 3 days after adoptive transfer (Figure S2B). These results indicate that the production of pro-inflammatory cytokines, e.g., IFN-γ and TNF-α, by Tet2KO CD8+ TILs might be one of the factors that promote the activation of endogenous CD45.1+CD8+ TILs in the recipient mice.
Figure 2. Tet2-deficient TILs show enhanced immune response and cytotoxicity at the early stage of tumorigenesis.
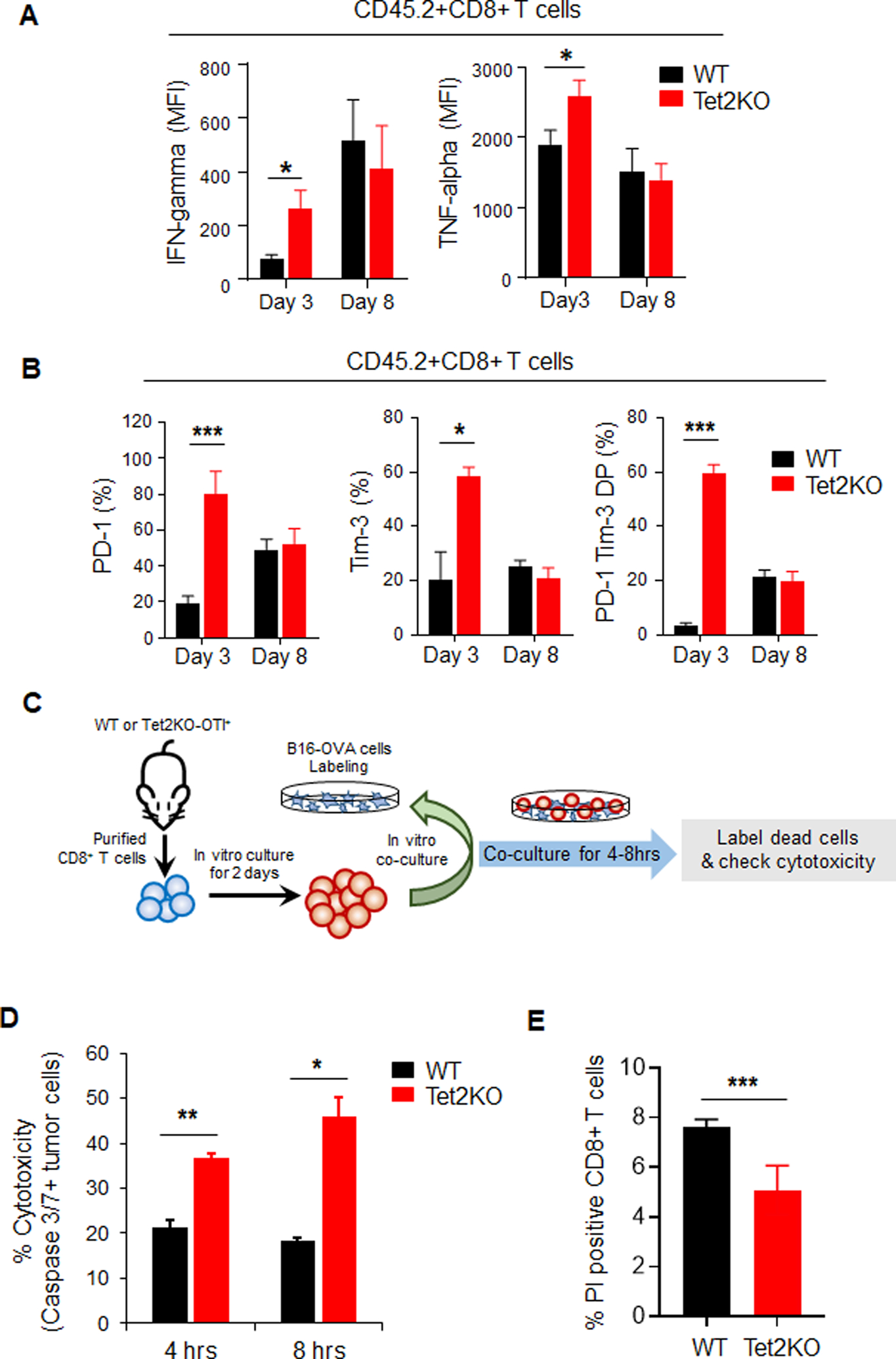
A-B. Quantification of the percentage of IFN-γ or TNF-α (A), or PD-1/Tim-3 (B) positive populations in adoptively transferred WT (black) and Tet2KO (red) CD45.2+CD8+ T cells at 3 and 8 days in the recipient mice. Data were shown as mean ± S.D; n=7–22, * P < 0.05, *** P < 0.0001, by two-tailed Student’s t-test.
C. Experimental design for the in vitro co-culture cytotoxicity assay. In vitro cultured B16-OVA cells were labeled with cell proliferation dye and co-cultured with WT or Tet2KO CD8+ T cells for 4–8 hours. The cytotoxicity of CD8+ T cells were quantified by measuring the caspase 3/7 positive B16-OVA cells using flow cytometry.
D. Quantification of caspase 3/7 positive B16-OVA melanoma cells co-cultured with WT (black) and Tet2KO (red) CD8+ T cells for 4 and 8 hrs in vitro. Data were shown as mean ± S.D; n=3, * P < 0.05, ** P < 0.005, by two-tailed Student’s t-test.
E. The quantification of flow cytometry analysis of Propidium Iodide (PI) staining in WT and Tet2KO CD8+OTI T cells co-cultured with B16-OVA cells for 4 hrs. Data were shown as mean ± S.D; n=3, *** P < 0.0001, by two-tailed Student’s t-test.
To further confirm the cytotoxicity effects of Tet2KO CD8+ T cells, we performed the in vitro co-culture experiment. As shown in Figure 2C, we purified CD8+ T cells from WT-OT-I or Tet2KO-OT-I and cultured them for 2 days. In parallel, we labelled in vitro cultured B16-OVA cells with cell proliferation dyes which can be used to separate them from co-cultured CD8+ T cells in the following experiment (Figure S2C). Then we co-cultured in vitro activated CD8+ T cells and in vitro labelled B16-OVA cells for 4–8 hrs. The cytotoxicity effects were then measured by the caspase3/7 activity in labeled B16-OVA cells (Figure S2C). With this assay, we observed that B16-OVA cells co-cultured with Tet2KO CD8+ T cells displayed significant higher caspase3/7 activity than WT CD8+ T cells at both 4 and 8 hrs after co-culture (Figure 2D). In parallel, we also observed increased cell proliferation (Figure S2D–E) and decreased cell death (Figure 2E) in Tet2KO CD8+ T cells when co-cultured with B16-OVA cells compared with WT group. These data confirmed that Tet2KO CD8+ T cells displayed higher cytotoxic effects than WT CD8+ T cells toward B16-OVA melanoma cells in vitro and this cytotoxic effect is an early event.
Single-cell RNA-seq (scRNA-seq) reveals enhanced activation of Tet2KO TILs.
To further identify the cell populations that might contribute to anti-tumor immunity, we performed scRNA-seq analysis in CD45.2+CD8+ TILs in four experimental groups (WT, Tet2KO, WT + anti-PD-L1, and Tet2KO + anti-PD-L1) (Figure 3A and S3A, Table S1) 8 days after adoptive transfer. We observed three major groups of cells: TILs (CD8+), monocytes (F4/80+), and B16-OVA tumor cells (CD63+) (Figure 3A and S3B). We performed cross-comparison analysis between WT and Tet2KO without anti-PD-L1 treatment, WT with and without anti-PD-L1 treatment, and Tet2KO with and without anti-PD-L1 treatment (Figure 3A). We found that the Tet2KO group exhibited significant reduction in the B16-OVA tumor cell population compared with the WT group (Figure 3A left). Similarly, in the WT group, anti-PD-L1 treatment also led to a notable decrease of B16-OVA tumor cells compared with the untreated group (Figure 3A middle). For the Tet2KO group, anti-PD-L1 treatment did not seem to cause major changes in cell populations (Figure 3A, right). These results echoed the findings shown in Figure 1, implying that Tet2KO TILs indeed suppress B16-OVA cancer cell growth. To further study how Tet2 deletion influences CD8+ TILs, we selected CD8α expressed cells for further cluster analysis (Figure 3B). We identified 5 major clusters in selected CD8+ TILs (Figure 3B, Table S2). The distribution of cell numbers within four experimental groups was different in 5 clusters (Figure 3C). Cluster 0 showed similar cellular distribution among the four groups, while anti-PD-L1 treatment resulted in significant decrease of the cell number in both WT and Tet2KO groups in clusters 1 and 2. Tet2KO resulted in increased cell numbers in clusters 3 and 4; while anti-PD-L1 treatment augmented the cell number only in the WT group for clusters 3 and 4, but not in the Tet2KO group (Figure 3C). To further elucidate the cell identities within these 5 clusters, we mined publicly available RNA-seq data from the purified subsets of CD8+ T cells, including naïve, effector, memory CD8+ T cell, PD1+Tim3+ double positive (DP) CD8+ TILs with and without anti-PD1 treatment (31). Then we selected the top 20 expressed genes within each cluster and examined the expression level of these genes in purified cell types using the published RNA-seq data (31). Based on gene expression patterns, the cells within clusters 0, 1 and 2 had high levels of Pdcd1, Ctla4 and Havcr2 with exhaustion-like features. By contrast, cells in clusters 3 and 4 exhibited effector-like features with high expression of Tnf and NF-κB signaling related genes (Figure 3D–E, Table S2). This analysis demonstrated that, although a subset of Tet2-deficient TILs showed similar exhaustion-like patterns as WT, some Tet2KO TILs displayed enhanced effector-like features and might contribute to the enhanced anti-tumor immunity. Interestingly, we observed that anti-PD-L1 treatment exerted similar effects on WT and Tet2KO TILs bearing exhaustion-like features (cluster 0–2), but had less effects on Tet2KO TILs that showed effector-like features (cluster 3–4), which is consistent with the overall phenotype shown in Figure 1C.
Figure 3. Single-cell RNA-seq (scRNA-seq) reveals enhanced activation of Tet2KO TILs.
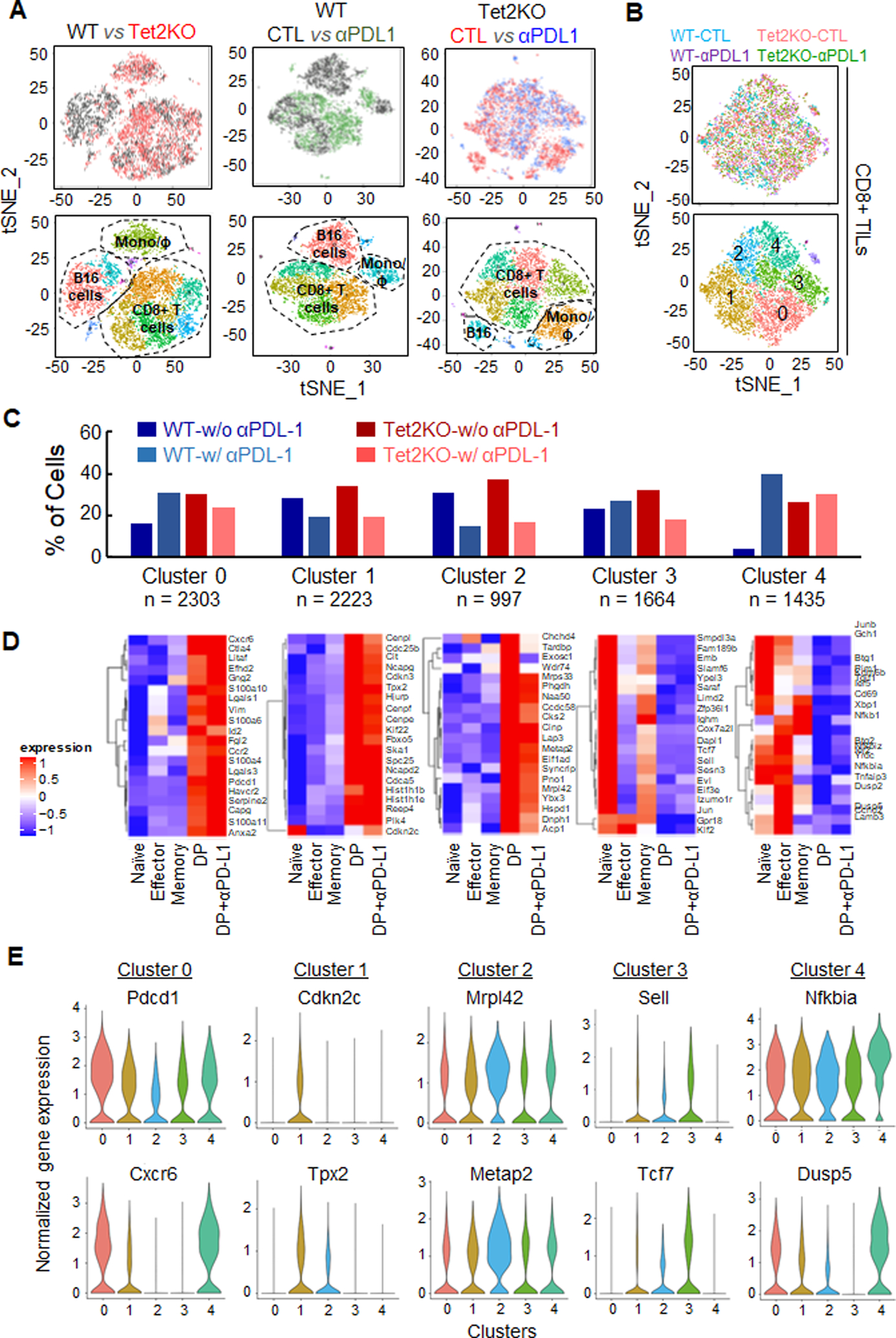
A. tSNE plots of scRNA-seq data obtained from CD45.2+CD8+ TILs (WT or Tet2KO) purified 8 days after adoptive transfer, treated with or without anti-PD-L1. Top: tSNE plot comparison between the indicated experimental groups. Bottom: the matched tSNE plots with identified cell populations.
B. The tSNE plots (top) and clustering analysis (bottom) of CD8α-expressing cells in the indicated TILs purified 8 days after adoptive transfer.
C. The percentage of cells from four experimental groups (WT control, WT anti-PD-L1, Tet2KO control, and Tet2KO anti-PD-L1) within each cluster identified from panel B. The total cell number within each cluster was listed at the bottom.
D. Heatmaps showing the differential expression of signature genes in each cluster identified from Figure 3B in the indicated cell populations. DP: PD1+Tim3+ double positive CD8+ T cells.
E. Violin plots showing the distribution of normalized expression levels of representative cluster specific genes.
Tet2 deletion enhances the transcription of tumor-suppressive genes in TILs.
From scRNA-seq analysis, we noted that the CD45.2+CD8+ selection method still retains B16-OVA tumor cells and monocytes with highly expressed CD63 (Figure 3A, S3B). In addition, we observed significant changes of TNF and IFN-γ production at 3 days after T cell transfer between WT and Tet2KO TILs (Figure 2B). Therefore, we further purified CD45.2+ CD63-CD8+ T cells from tumor tissues at day 0 and 3 after adoptive transfer for further RNA-seq (transcriptome) and ATAC-seq (chromatin accessibility) analysis (Figure 4A, S4A, Table S1, Table S3–4). We observed massive transcriptomic and epigenetic remodeling events from day 0 to day 3 after adoptive transfer in both WT and Tet2KO TILs (Figure S4B). Consistent with previous reports (11,21,32), we observed a strong positive association between transcriptional changes and chromatin accessibility in the analyzed TILs (Figure S4B), suggesting epigenetic remodeling is one of the critical regulators contributing to transcriptional outputs during anti-tumor immunity.
Figure 4. Tet2 deletion enhances the transcription of tumor-suppressive genes in TILs.
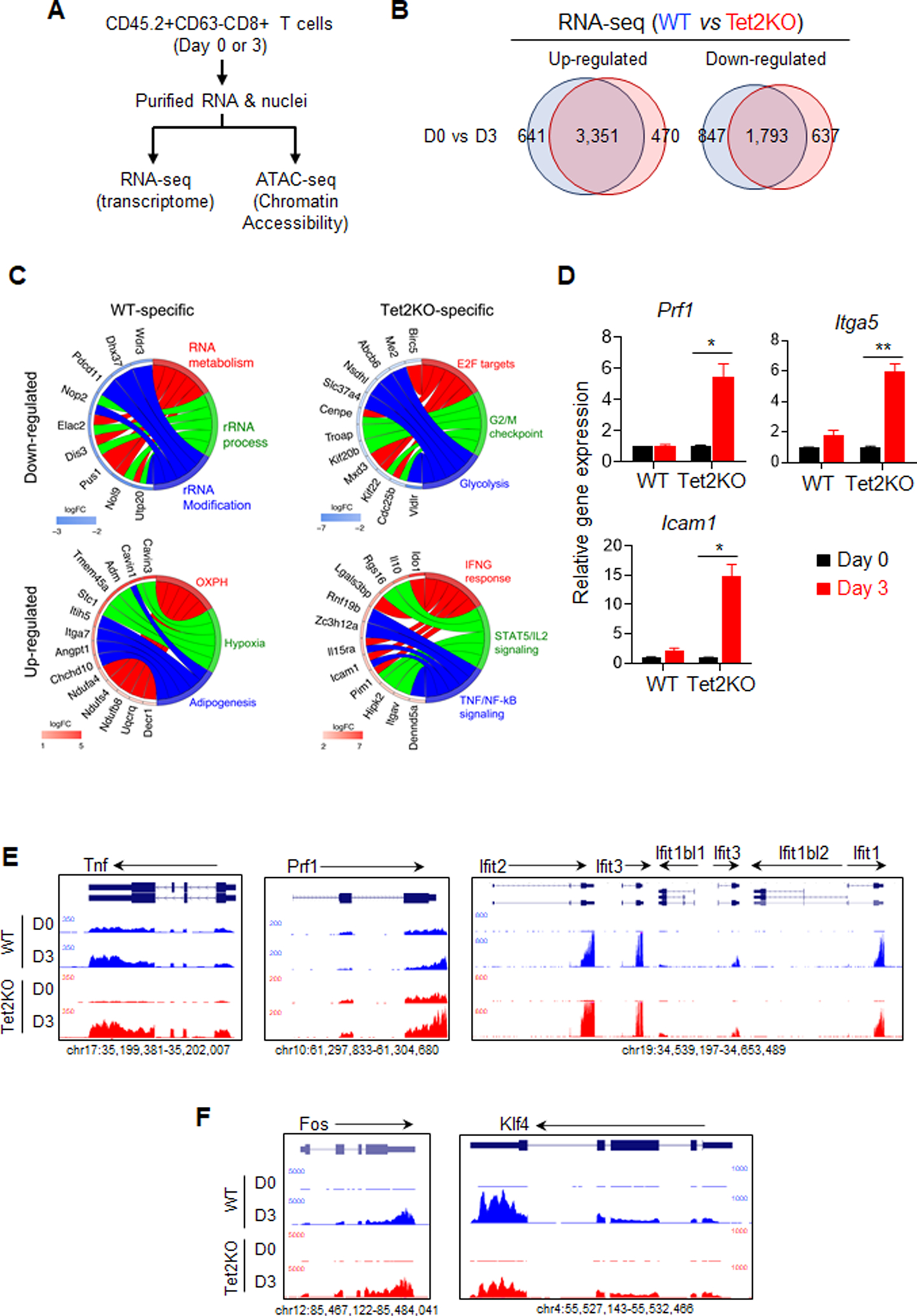
A. The experimental design for comparative RNA-seq and ATAC-seq analyses.
B. Venn diagrams showing WT- and Tet2-specific differentially expressed genes (DEGs) identified between Day 0 and Day 3 adoptively transferred TILs (WT vs Tet2KO).
C. GOplot illustrating the top 5 genes in the top three categories from the GSEA analysis of WT- and Tet2-specific DEGs. The left side of the circle displays the DEGs and color represents the log2 fold change (logFC). Red, upregulated genes; Blue, downregulated genes. The right side of the circle shows the GSEA categories.
D. Real-time quantitative PCR validation of selected DEGs annotated as regulators of immune response.
E-F. The UCSC genome browser view of RNA-seq data for representative genes that are involved in CD8+ T cell immunity and are up-regulated in the Tet2KO group.
To further examine the function of Tet2 during this process, we first compared differentially expressed genes (DEGs) between day 0 and 3 TILs and then compared the DEGs between WT and Tet2KO groups, anticipating to identify WT- or Tet2KO-specific DEGs during anti-tumor immunity at different days (Figure 4B, S4C, Table S3). The Gene Set Enrichment Analysis (GSEA) analysis (33) showed that the genes specifically up-regulated in Tet2KO TILs were enriched with cytokine production and immune signaling pathways (e.g., Interferon-gamma responding genes) (Figure 4C–E, S4D, Table S3). Genes specifically down-regulated in Tet2KO TILs were associated with cell cycle and proliferation (e.g., centromere genes and Cdks) (Figure 4C, S4D, Table S3). In WT TILs, genes that were specifically up- or down-regulated are associated with certain house-keeping functions, such as metabolism (e.g., NADH-Ubiquinone Oxidoreductase), and RNA processing (e.g., WD repeat domains) (Figure 4C, S4D, Table S3). Some of the genes identified from RNA-seq analysis were further validated using real-time qPCR analysis (Figure 4D). For example, we observed over a 5-fold increase of the expression of Prf1 in Tet2KO CD8+ T cells compare with the WT group (Figure 4D–E), which is consistent with the observed enhancement of cytotoxicity (Figure 2D). Interestingly, we also detected several key transcription factors, such as Fos and Klf4, that were differentially expressed in Tet2KO CD8 T cells (Figure 4F), suggesting that Tet2 might regulate key transcription factors in CD8 T cells and indirectly induce the transcriptional alterations during anti-tumor immunity. Collectively, our findings suggest that Tet2 ablation in TILs might lead to increased expression of cytokines and other immunomodulatory genes to enhance anti-tumor immunity.
Tet2 knockout reshapes the chromatin accessibility in TILs.
Next, we performed similar comparative analysis on chromatin accessibility by using ATAC-seq (Figure 5A and S5A–B, Table S4). We first identified differentially enriched regions between day 0 and 3 in WT or Tet2KO TILs. We observed a marked increase in chromatin accessibility from day 0 to day 3 in WT TILs, but to a lesser extent in Tet2KO TILs (Figure 5A, S5A). To further investigate how Tet2 regulates the function of TILs during this process, we identified differential chromatin accessible regions that were specifically altered in WT- or Tet2KO groups (Figure 5A, S5B). Genomic Regions Enrichment of Annotations Tool (GREAT) analysis (25) showed that these regions are close to the genes essential for T cell activation and immune response (Figure S5C). Since chromatin accessible regions are known to be critical for TF binding (34), we analyzed the enrichment of TF motifs within the identified WT- or Tet2-specific differential accessible regions (Figure 5B). We found that bZIP and ETS motifs were most prominently enriched within WT- and Tet2-specific differential accessible regions, respectively (Figure 5B–C). WT-specific open genomic regions were mainly enriched with the motifs for the bZIP TF family, including BATF and AP-1; while Tet2-specific accessible genomic regions were enriched with motifs for the ETS TF family, including ETS1 and ELFs (Figure 5B). To further validate this observation, we compared our own ATAC-seq data with the published BATF (bZIP family) ChIP-seq data collected in lymphocytes (35). We focused on the chromatin accessibility states of the BATF binding regions in both WT and Tet2KO TILs (Figure 5C). At day 0, the chromatin accessibility of the BATF binding regions in Tet2KO CD8+ T cells was significantly higher than of the WT group. At day 3, the chromatin accessibility of the same regions was reduced in the Tet2KO group, but not in the WT group. These findings are consistent with the motif enrichment analysis results, both highlighting that Tet2 deletion reshapes the chromatin accessibility of selected TFs in TILs. Furthermore, genes that showed positive correlations between their expression and chromatin accessibility in CD8+ TILs were found to exhibit differential enrichment of TF motifs (Figure 5D, S5D). For example, BATF was enriched at the Slc14a1 promoters, and we observed relatively higher gene expression and chromatin accessibility of Slac14a1 in WT TILs when compared to Tet2KO TILs (Figure 5E, top). On the other hand, Irf2 gene exhibited both higher expression and chromatin accessibility in Tet2KO TILs 3 days after adoptive transfer than in WT TILs, and we observed ETS1 enrichment, but not BATF at the promoter of Irf2 (Figure 5E, bottom). To further test whether Tet2KO TILs exhibited enhanced transcriptional activity of the ETS family members, we applied two ETS Inhibitors, YK-4–279 and TK-216(36), to both WT and Tet2KO CD8+ OT-I T cells that were co-cultured with B16-OVA tumor cells as shown in Figure 2C, and assessed the degrees of tumor cell apoptosis by using caspase 3/7 staining. We noted that both YK-4–279 and TK-216 treatment led to a significant reduction in B16-OVA tumor cell apoptosis induced by Tet2KO CD8+ OT-I T cells, but had minor effects on B16-OVA tumor cells co-cultured with WT CD8+ OT-I T cells (Figure 5F). As negative control, YK-4–279 and TK-216 treatment per se did not induce major cytotoxicity in B16-OVA cells (Figure S5E). The YK-4–279 and TK-216 mediated suppressive effect on tumor killing might be due to the inhibition of the cytokine production in Tet2KO CD8+ T cells (Figure S5F). Taken together, these data suggest that Tet2 deletion remodels the chromatin accessibility and transcriptional activity of selected TFs to impact gene expression in TILs to affect anti-tumor immunity.
Figure 5. Tet2 knockout reshapes the chromatin accessibility in TILs.
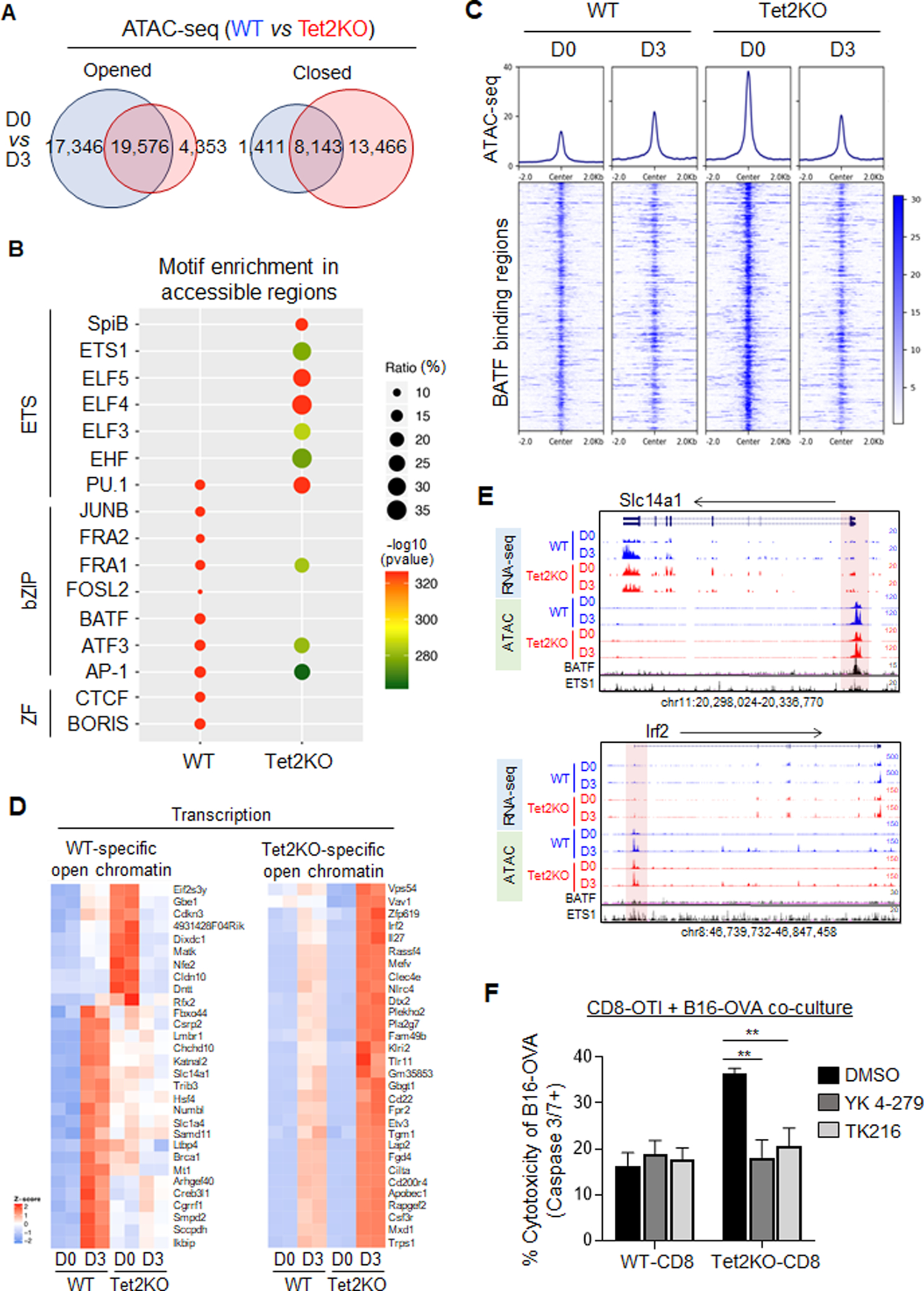
A. Venn diagrams showing WT- and Tet2-specific differential chromatin accessible regions identified in the indicated groups.
B. The top 10 significantly enriched transcription factor binding motifs within the WT- and Tet2-specific differential chromatin accessible regions.
C. The enrichment of chromatin accessibility within BATF binding regions in WT and Tet2KO TILs at day 0 and day 3 after adoptive transfer.
D. Heatmap representation of gene expression for the top 30 selected genes that displayed increased chromatin accessibility in either WT (left) or Tet2KO (right) TILs (day 3 group compared with day 0).
E. The UCSC genome browser view of RNA-seq and ATAC-seq results for representative genes listed in Figure 5D. The published BATF and ETS1 ChIP-seq data were also included. The highlighted regions were promoter regions of the indicated genes.
F. Quantification of caspase 3/7 positive B16-OVA melanoma cells co-cultured with WT or Tet2KO CD8+ T cells for 4 hrs in the presence of DMSO or ETS inhibitors (100 nM YK-4–279 or 500 nM TK-216). Data were shown as mean ± S.D; n=8, ** P < 0.005, by two-tailed Student’s t-test.
Discussion
TET2 is one of the frequently mutated genes in the hematopoietic and immune systems and is a well-known tumor suppressor (15,17). In addition, TET2 mutations are often observed in elderly individuals with clonal hematopoiesis (CH) (16), suggesting that TET2 mutations are also present in the progeny of hematopoietic stem and progenitor cells (HSCPs), such as monocytes and lymphocytes, and subsequently alters their functions during innate and adaptive immune response. Indeed, recent studies have shown that CH-associated Tet2 loss-of-function in HSPCs accelerates atherosclerosis due to the increased inflammasome activity in Tet2-deficient macrophages (37). On the other hand, the deletion of TET2 in chimeric antigen receptor (CAR) T-cells and tumor-associated macrophages (TAM) suppresses the tumor growth and significantly benefits the anti-tumor immunotherapy (13,14). In this study, we investigated the effects of Tet2 LOF in CD8+ TILs during tumor onset and progression. Our study provides additional evidence to support the beneficial role of Tet2 deficiency in the immune system to combat cancer. Mechanistically, we have unveiled that deletion of Tet2 reshapes the chromatin accessibility at genomic binding regions of several key transcription factors, e.g., BATF and ETS1, in CD8+ TILs, thereby enhancing its anti-tumor immunity and suppressing melanoma growth in vivo.
Previous studies using an acute viral infection model have demonstrated that Tet2 loss promotes the early onset of memory CD8+ T cells and enhances their anti-viral activity (6). In our study, we have shown that Tet2-deficient CD8+ TILs display more effective anti-tumor activity than the WT CD8+ TILs. The deletion of Tet2 augments the expansion of CD8+ T cells not only in tumors, but also in spleens and peripheral blood, which is consistent with previous observations made in Tet2 deficient CD4+ T cells and HSCPs (18,19). In addition, we have found that more CD8+ T cells tend to be enriched within the tumor foci in the Tet2KO group than in the WT group. This effect was also observed in CD45.1+CD8+ T cells in the recipient mice transferred with Tet2KO CD8+ T cells, suggesting that Tet2 inactivation could promote the enrichment of CD8+ T cells within tumor foci to enhance the anti-tumor efficiency. In our study, we also observed the up-regulation of Ifn-γ and Tnf-α in Tet2KO TILs, which might mediate the enrichment of CD45.1+ TILs in recipient mice transferred with Tet2KO TILs. Further studies are needed to clarify the underlying mechanism. Nonetheless, our exciting findings establish that Tet2 inactivation not only renders CD8+ TILs more potent in tumor killing, but also leads to a more immunogenic tumor microenvironment to enhance the overall anti-tumor immunity.
Chromatin reprogramming in the tumor microenvironment is one of the critical barriers blocking the anti-tumor function of CD8+ TILs (2). The chromatin reprogramming process is reversible during the early stage of tumor development and gradually becomes irreversible in the late stage (2). In our study, we observed that Tet2 deficiency leads to massive chromatin reprogramming in TILs and enhances their anti-tumor efficiency at an early stage during tumor development. Compared to WT TILs, Tet2-deficient TILs bear higher chromatin accessibility at the ETS (ETS1) family binding regions but show reduced accessibility in the bZIP family binding regions, such as Batf. Pharmacological inhibition of the ETS TF family members reduced the anti-tumor efficiency of Tet2KO CD8+ T cells, but not WT CD8+ T cells, further confirming that Tet2 deficiency reprograms the transcriptional activity of selected TFs. These changes in chromatin accessibility and transcriptional activity might lead to upregulation of genes that are essential for the effector function of TILs, including genes involved in TNF and IFNG pathways. In addition to changes in chromatin accessibility arising from Tet2 depletion in CD8+ TILs, Tet2 loss might abrogate its interaction with negative regulators of gene transcription, such as Sin3a(38) and Hdac2(20), thereby contributing to augmented transcriptional activation in Tet2 deficient cells. Furthermore, the DNA binding of many TFs might be directly influenced by DNA methylation and its oxidative derivatives (5hmC, 5fC and 5caC) generated by the Tet protein family (39–41). Therefore, Tet2 deficiency induced alterations in DNA methylation and its oxidation could add another layer of control over TFs binding to theirs genomic targets in CD8+ TILs. Further studies are needed to explore these possibilities in CD8+ TILs.
In summary, our study highlights the inhibitory role of Tet2 in regulating CD8+ TILs during anti-tumor immunity. Tet2 inactivation significantly enhances the effector-like function of TILs to promote anti-tumor immunity by reshaping the chromatin accessibility in TILs at the early stage during tumor development.
Supplementary Material
Statement of Significance.
This study suggests that ablation of TET2 from TILs could promote their anti-tumor function by reshaping chromatin accessibility for key transcription factors and enhancing the transcription of genes essential for anti-tumor activity.
Acknowledgements
We are grateful for Dr. Jianjun Shen and the MD Anderson Cancer Center next-generation sequencing core at Smithville (CPRIT RP120348 and RP170002), and the Epigenetic core at the Institute of Biosciences and Technology at the Texas A&M University. This work was supported by grants from Cancer Prevention and Research Institute of Texas (RP180131 to D. Sun, RP190581 to M. Moczygemba), National Institute of Health grants (R01HL134780, R21GM138824, and R01HL146852 to Y. Huang; R01GM112003, R21GM132778, and R01CA232017 to Y. Zhou), the Welch Foundation (BE-1913-20190330 to Y. Zhou), the American Cancer Society (RSG-18-043-01-LIB to Y. Huang; RSG-16-215-01-TBE to Y. Zhou), and by an allocation from the Texas A&M University start-up funds (Y. Huang).
Footnotes
Disclosure of Conflicts of Interest: The authors declare no competing interests.
References
- 1.Yang R, Cheng S, Luo N, Gao R, Yu K, Kang B, et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol 2019;21:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017;545:452–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scharer CD, Barwick BG, Youngblood BA, Ahmed R, Boss JM. Global DNA methylation remodeling accompanies CD8 T cell effector function. J Immunol 2013;191:3419–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017;170:142–57 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahn E, Youngblood B, Lee J, Lee J, Sarkar S, Ahmed R. Demethylation of the PD-1 Promoter Is Imprinted during the Effector Phase of CD8 T Cell Exhaustion. J Virol 2016;90:8934–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E, et al. The Loss of TET2 Promotes CD8(+) T Cell Memory Differentiation. J Immunol 2018;200:82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol 2018;18:340–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011;333:1303–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011;333:1300–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lio CW, Zhang J, Gonzalez-Avalos E, Hogan PG, Chang X, Rao A. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsagaratou A, Gonzalez-Avalos E, Rautio S, Scott-Browne JP, Togher S, Pastor WA, et al. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nat Immunol 2017;18:45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018;558:307–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan W, Zhu S, Qu K, Meeth K, Cheng J, He K, et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017;47:284–97 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009;360:2289–301 [DOI] [PubMed] [Google Scholar]
- 16.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012;44:1179–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mullighan CG. TET2 mutations in myelodysplasia and myeloid malignancies. Nat Genet 2009;41:766–7 [DOI] [PubMed] [Google Scholar]
- 18.Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018;23:833–49 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015;42:613–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015;525:389–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang S, Li J, Xiao Y, Lee M, Guo L, Han W, et al. Tet inactivation disrupts YY1 binding and long-range chromatin interactions during embryonic heart development. Nature Communications 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A 2011;108:14566–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, et al. Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U S A 2017;114:E2776–E85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 2015;109:21 9 1–9 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010;28:495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell 1994;76:17–27 [DOI] [PubMed] [Google Scholar]
- 27.Falo LD, Jr., Kovacsovics-Bankowski M, Thompson K, Rock KL. Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. Nat Med 1995;1:649–53 [DOI] [PubMed] [Google Scholar]
- 28.Schuler T, Blankenstein T. Cutting edge: CD8+ effector T cells reject tumors by direct antigen recognition but indirect action on host cells. J Immunol 2003;170:4427–31 [DOI] [PubMed] [Google Scholar]
- 29.Gerner MY, Heltemes-Harris LM, Fife BT, Mescher MF. Cutting edge: IL-12 and type I IFN differentially program CD8 T cells for programmed death 1 re-expression levels and tumor control. J Immunol 2013;191:1011–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janowska-Wieczorek A, Majka M, Kijowski J, Baj-Krzyworzeka M, Reca R, Turner AR, et al. Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood 2001;98:3143–9 [DOI] [PubMed] [Google Scholar]
- 31.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 2019;50:181–94 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Wu X, Zhou Y, Lee M, Guo L, Han W, et al. Decoding the dynamic DNA methylation and hydroxymethylation landscapes in endodermal lineage intermediates during pancreatic differentiation of hESC. Nucleic Acids Res 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 2019;20:207–20 [DOI] [PubMed] [Google Scholar]
- 35.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol 2014;15:373–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spriano F, Chung EYL, Gaudio E, Tarantelli C, Cascione L, Napoli S, et al. The ETS Inhibitors YK-4–279 and TK-216 Are Novel Antilymphoma Agents. Clin Cancer Res 2019;25:5167–76 [DOI] [PubMed] [Google Scholar]
- 37.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017;355:842–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 2011;473:343–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Horton JR, Wang D, Ren R, Li J, Sun D, et al. Structural basis for effects of CpA modifications on C/EBPbeta binding of DNA. Nucleic Acids Res 2019;47:1774–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong S, Wang D, Horton JR, Zhang X, Speck SH, Blumenthal RM, et al. Methyl-dependent and spatial-specific DNA recognition by the orthologous transcription factors human AP-1 and Epstein-Barr virus Zta. Nucleic Acids Res 2017;45:2503–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D, Hashimoto H, Zhang X, Barwick BG, Lonial S, Boise LH, et al. MAX is an epigenetic sensor of 5-carboxylcytosine and is altered in multiple myeloma. Nucleic Acids Res 2017;45:2396–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.