

Abstract
Neutrophils are the most abundant circulating leucocytes and are essential for innate immunity. In cancer, pro- or anti-tumor properties have been attributed to tumor-associated neutrophils (TAN). Here, focusing on TAN accumulation within lung tumors, we identify Glut1 as an essential glucose transporter for their tumor supportive behavior. Compared to normal neutrophils, Glut1 and glucose metabolism increased in TANs from a mouse model of lung adenocarcinoma. To elucidate the impact of glucose uptake on TANs, we used a strategy with two recombinases, dissociating tumor initiation from neutrophil-specific Glut1 deletion. Loss of Glut1 accelerated neutrophil turnover in tumors and reduced a subset of TANs expressing SiglecF. In the absence of Glut1 expression by TANs, tumor growth was diminished and the efficacy of radiotherapy was augmented. Our results demonstrate the importance of Glut1 in TANs, which may affect their pro- versus anti-tumor behavior. These results also suggest targeting metabolic vulnerabilities to favor anti-tumor neutrophils.
Keywords: Neutrophils, lung adenocarcinoma, Glut1, tumor microenvironment, tumor-associated neutrophils, genetically-engineered mouse model, radiotherapy, innate immunity
Introduction
Neutrophils are critical for the innate immune response (1). The expected short half-life of these cells might have contributed to underestimating their importance in tumor progression in past research. However, neutrophil functions in tumors have begun to be revealed in different types of cancer including breast and lung cancers (2). Although they have now been considered in several cancer models, only a few studies used autochthonous mouse models closely recapitulating the characteristics of the human disease (3). Neutrophils with pro-tumor properties have been extensively described in the context of breast cancer metastasis (4,5), and have also been linked to immune-suppression and pro-angiogenesis (6,7). Immune signature analyses of lung adenocarcinoma (LUAD) from the KrasLox-STOP-Lox-G12D/WT; Tp53Flox/Flox (KP) mouse model (8) demonstrated that neutrophils are the most prevalent immune cells within the tumor mass (9) and may possess tumor-supportive properties (10). Specifically, a TAN subset defined by high SiglecF membrane protein expression showed a tumor-associated transcriptional profile and an increased ROS production in a KP cell-derived transplantation model in vivo. Although this subset remains to be fully characterized, SiglecFhigh TANs have tumor-promoting functions compared to SiglecFlow cells (11). This suggests that gaining fundamental knowledge about neutrophil heterogeneity in cancer will enable selective targeting of pro-tumor neutrophils without affecting other neutrophils that are essential for the host defense.
Although multiple metabolic alterations of tumor cells are known to help them survive and proliferate in harsh environmental conditions (12,13), much less is known about the rewired metabolism of non-tumor cells from the tumor microenvironment. Because neutrophils are exposed to the same hypoxic environment with possibly reduced access to nutrients, we hypothesized that their glucose metabolism changes within solid tumors and impacts tumor development. To test this, we explored the effect of the lung tumor microenvironment on TAN metabolism, survival and turnover as well as the consequences of glucose transporter Glut1-deficient (Glut1KO) TANs on disease progression.
Materials and Methods
Key resources
A Key Resources Table is provided as Table S1.
Key Resources Table.
| Reagent type (species) or resource | Source or reference | Identifiers | Additional information |
|---|---|---|---|
| Antibodies | |||
| Anti-mouse PD-1 (clone 29F.1A12, rat, monoclonal) | Bio X Cell | Cat# BE0273 | Immunotherapy treatment |
| RRID:AB_2687796 | 200 μg/mouse twice a week for 2 weeks | ||
| Anti-mouse G-CSF (clone 67604, rat, monoclonal) | R&D Systems | Cat# MAB414 | Anti G-CSF treatment |
| RRID:AB_2085954 | 10 μg every day for 14 days | ||
| Anti-GLUT1 (rabbit, polyclonal) | Millipore | Cat# 07-1401 | WB |
| RRID:AB_1587074 | IF (1:5’000) | ||
| Anti-GLUT1 (rabbit, monoclonal) | Abcam | Cat# ab115730 | IHC (1:650) |
| RRID:AB_10903230 | |||
| Anti-Histone H3 (rabbit, polyclonal) | Abcam | Cat# ab1791 | WB |
| RRID:AB_302613 | |||
| Anti-pHH3 (rabbit, polyclonal) | Cell Signaling Technology | Cat# 9701S | IHC (1:100) |
| RRID:AB_331535 | |||
| Anti-pErk (rabbit, monoclonal) | Cell Signaling Technology | Cat# 4370S | IHC (1:400) |
| RRID:AB_2315112 | |||
| Anti-S100A9 (rabbit, polyclonal) | Novus biologicals | Cat# NB110-89726 | IHC (1:5’000) |
| RRID:AB_1217846 | IF (1:20’000) | ||
| Anti-CD4 (clone SP35, rabbit, monoclonal) | Roche Diagnostics | Cat# 05552737001 | IF |
| Anti-CD8 (clone SP57, rabbit, monoclonal) | Roche Diagnostics | Cat# 05937248001 | IF |
| Anti-mouse-CD45-BUV661 (clone 30-F11, rat, monoclonal) | BD Biosciences | Cat# 565079 | Flow cytometry |
| RRID:AB_2739057 | |||
| Anti-mouse/human-CD11b-BV711 (clone M1/70, rat, monoclonal) | BioLegend | Cat# 101242 | Flow cytometry |
| RRID:AB_2563310 | |||
| Anti-mouse-Ly6G-PE (clone 1A8, rat, monoclonal) | BioLegend | Cat# 127607 | Flow cytometry |
| RRID:AB_1186104 | |||
| Anti-mouse-Siglec-F-PE-Vio 615 (clone REA798, human, monoclonal) | Miltenyi Biotec | Cat# 130-112-172 | Flow cytometry |
| RRID:AB_2653444 | |||
| Anti-BrdU-APC (clone Bu20a, mouse, monoclonal) | BioLegend | Cat# 339808 | Flow cytometry |
| RRID:AB_10895898 | |||
| Anti-PD-L1-BV-785 (clone 10F.9G2, rat, monoclonal) | BioLegend | Cat# 124331 | Flow cytometry |
| RRID:AB_2629659 | |||
| Recombinant proteins | |||
| Recombinant murine G-CSF | PeproTech | Cat# 250-05 | G-CSF treatment |
| 10 μg, daily injection | |||
| Recombinant murine TNF | PeproTech | Cat# 315-01A | TNF stimulation |
| 10 ng/ml, 4 hours | |||
| Viral vector | |||
| Ad5CMVFlpo (adenoviral vector) | University of Iowa Viral Vector Core Facility | ||
| Sequence-based reagents | |||
| Rpl30 (Mus musculus) | Thermo Fisher Scientific | Mm01611464_g1 | TaqMan probe |
| (Housekeeping gene) | |||
| Slc2a1 (Glut1) (Mus musculus) | Thermo Fisher Scientific | Mm00441480_m1 | TaqMan probe |
| Slc2a3 (Glut3) (Mus musculus) | Thermo Fisher Scientific | Mm00441483_m1 | TaqMan probe |
| Arg1 (Mus musculus) | Thermo Fisher Scientific | Mm00475988_m1 | TaqMan probe |
| Hk2 (Mus musculus) | Thermo Fisher Scientific | Mm00443385_m1 | TaqMan probe |
| Siglecf (Siglec5) (Mus musculus) | Thermo Fisher Scientific | Mm00523987_m1 | TaqMan probe |
| Ikbke (Mus musculus) | Thermo Fisher Scientific | Mm00444862_m1 | TaqMan probe |
| Apoe (Mus musculus) | Thermo Fisher Scientific | Mm01307193_g1 | TaqMan probe |
| Pfkp (Mus musculus) | Thermo Fisher Scientific | Mm00444792_m1 | TaqMan probe |
| Cxcl3 (Mus musculus) | Thermo Fisher Scientific | Mm01701838_m1 | TaqMan probe |
| Grx1 (Mus musculus) | Thermo Fisher Scientific | Mm01352826_g1 | TaqMan probe |
| Il23a (Mus musculus) | Thermo Fisher Scientific | Mm00518984_m1 | TaqMan probe |
| Padi4 (Mus musculus) | Thermo Fisher Scientific | Mm01341658_m1 | TaqMan probe |
| Csf3 (G-CSF) (Mus musculus) | Thermo Fisher Scientific | Mm00438334_m1 | TaqMan probe |
| Chemicals | |||
| Isoflurane | Piramal | Cat# 56.761.002 | Anaesthetic |
| BrdU | Merck | Cat# 10280879001 | BrdU assay |
| 2 mg of freshly prepared BrdU into 100 μl of PBS 1X | |||
| Glucose | VWR | Cat# 1.08337.0250 | Seahorse |
| 10 mM glucose | |||
| 2-deoxyglucose (2-DG) | Merck | Cat# D8375-5G | Seahorse |
| 50 mM 2-DG | |||
| Crystal violet | Merck | Cat# C3886 | |
| Experimental Models: Cell lines | |||
| B16-F1 (Mus musculus) | RRID:CVCL_0158 | Melanoma lung metastases | |
| SV2 (Mus musculus) | Developed in our laboratory | ||
| Experimental Models: Organism/Strains | |||
| KrasLox-STOP-Lox-G12D/WT (K) | The Jackson Laboratory | RRID:IMSR_JAX:008179 | KrasLoxSTOPLox-G12D/WT |
| K and P were interbred at EPFL | |||
| Tp53Flox/Flox (P) | The Jackson Laboratory | RRID:IMSR_JAX:008462 | Trp53Flox/Flox |
| K and P were interbred at EPFL | |||
| KrasFrt-STOP-Frt-G12D/WT (KFrt) | The Jackson Laboratory | RRID:IMSR_JAX:008653 | KPFrt were provided by D.G. Kirsch, Duke University Medical Center in a mixed 129-C57BL/6 background |
| Tp53Frt/Frt (PFrt) | The Jackson Laboratory | RRID:IMSR_JAX:017767 | KPFrt were provided by D.G. Kirsch, Duke University Medical Center in a mixed 129-C57BL/6 background |
| Glut1Flox/Flox (G1) | The Jackson Laboratory | RRID:IMSR_JAX:031871 | Glut1Flox/Flox were provided by E. Dale Abel, University of Iowa in a C57BL/6 background |
| Glut3Flox/Flox (G3) | Duke University Transgenic Facility | ||
| Ly6gCre (Catchup) | Ly6gCre (Catchup) were provided by M. Gunzer, University Duisburg-Essen, in a C57BL/6 background | ||
| KrasFrt-STOP-Frt-G12D/WT; Tp53Frt/Frt; Ly6gCre/WT; Glut1Flox/Flox | G1, Ly6gCre and KPFrt mice were interbred at EPFL | ||
| KrasFrt-STOP-Frt-G12D/WT; Tp53Frt/Frt; Ly6gWT/WT; Glut1Flox/Flox | G1, Ly6gCre and KPFrt mice were interbred at EPFL | ||
| KrasFrt-STOP-Frt-G12D/WT; Tp53Frt/Frt; Ly6gCre/WT; Glut1WT/WT | G1, Ly6gCre and KPFrt mice were interbred at EPFL | ||
| Commercial assays and kits | |||
| Anti-mouse CD45 MicroBeads (clone 30F11.1, monoclonal) | Miltenyi Biotec | Cat# 130-052-301 | CD45 magnetic cell sorting |
| RRID:AB_2877061) | |||
| Anti-mouse Ly-6G MicroBead (monoclonal) | Miltenyi Biotec | Cat# 130-092-332 | Ly-6G magnetic cell sorting |
| Anti-mouse Ly-6G MicroBeads UltraPure (clone REA526, monoclonal) | Miltenyi Biotec | Cat# 130-120-337 | Ly-6G magnetic cell sorting |
| Centricon Plus-70 Centrifugal Filter (3 kDa cutoff) | Merck | Cat# UFC700308 | Size exclusion separation of proteins |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat# 4368814 | Reverse transcription |
| BCA Protein Assay Kit | Merck | Cat# 71285-3 | Protein quantification |
| CountBright™ Absolute Counting Beads | Thermo Fisher Scientific | Cat# C36950 | Flow cytometry |
| Anti-rabbit Immpress HRP (horse) | Vector Laboratories | Cat# MP-7401 | Peroxidase Polymer Detection Kit |
| RRID:AB_2336529 | |||
| Rhodamine-6G | Roche Diagnostics | Cat# 07988168001 | TSA kit |
| DCC | Roche Diagnostics | Cat# 07988192001 | TSA kit |
| Red 610 | Roche Diagnostics | Cat# 07988176001 | TSA kit |
| Cyanine 5 | Roche Diagnostics | Cat# 07551215001 | TSA kit |
| Glucose-uptake cell-based assay kit | Cayman Chemical | Cat# CAY-600470-1 | Glucose Uptake Cell-Based Assay Kit |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7570 | ATP production |
| Software and Algorithms | |||
| Analyze 12.0 | PerkinElmer | Tumor volume analysis | |
| OsiriX MD | Pixmeo | RRID:SCR_013618 | Tumor volume analysis |
| QuPath | RRID:SCR_018257 | ||
| Fiji | RRID:SCR_002285 | ||
| FlowJo | RRID:SCR_008520 | Flow cytometry | |
| GraphPad Prism | RRID:SCR_002798 | ||
| (version 6) | |||
| Instruments | |||
| X-Ray microtomography | PerkinElmer | Quantum FX microCT | |
| 20 mm2 collimator | Radiotherapy treatment | ||
| 13 mA, 3 mm Cu filter, 225 keV | |||
| 11.6 Gy with a single dose in 256 seconds | |||
| Automated Ventana Discovery ULTRA | Roche Diagnostics | Human sample multiplexing | |
| Olympus slide scanner | Olympus | VS120-L100 | Slide scanner |
| XF96 Extracellular Flux analyser | Seahorse Bioscience | XF96 | Seahorse |
| LSRII SORP | Becton Dickinson | Flow cytometry | |
Animal studies
KrasLox-STOP-Lox-G12D/WT; Tp53Flox/Flox (KP) (RRID:IMSR_JAX:008179, RRID:IMSR_JAX:008462) mice were described before (10) and were used in this study for the experiments comparing wild-type TANs to wild-type HLNs, represented in Figure 2. KrasFrt-STOP-Frt-G12D/WT (KFrt) (RRID:IMSR_JAX:008653) mice, previously generated by T. Jacks (14), Massachusetts Institute of Technology, were bred with Tp53Frt/Frt (PFrt) (RRID:IMSR_JAX:017767) mice (15). The resulting animals, KrasFrt-STOP-Frt-G12D/WT; Tp53Frt/Frt (KPFrt) mice, were kindly provided by D.G. Kirsch, Duke University Medical Center in a mixed 129-C57BL/6 background. The generation of the Glut1Flox/Flox (G1) (RRID:IMSR_JAX:031871) mice was described previously (16), and the mice in a C57BL/6 background were provided by E. Dale Abel, University of Iowa. Glut3Flox/Flox (G3) mice were produced at the Duke University Transgenic Facility upon blastocyst microinjection of targeted ES cells obtained from KOMP Repository (project CSD48048). The resulting mice were then crossed to Flp transgenic mice to remove the neomycin resistance cassette and generate mice with a Glut3Flox allele, in which exon 6 is flanked by LoxP sites. Ly6gCre, also called Catchup mice (17) were obtained from M. Gunzer, University Duisburg-Essen, in a C57BL/6 background. G1, and Ly6gCre mice were bred with KPFrt mice to obtain the following genotypes: KrasFrt-STOP-Frt-G12D/WT; Tp53Frt/Frt; Ly6gCre/WT; Glut1Flox/Flox, KrasFrt-STOP-Frt-G12D/WT; Tp53Frt/Frt; Ly6gWT/WT; Glut1Flox/Flox, or KrasFrt-STOP-Frt-G12D/WT; Tp53Frt/Frt; Ly6gCre/WT; Glut1WT/WT, the latter two being used as control (no Cre, or Glut1WT/WT). Tumors were initiated upon intratracheal instillation of 107 PFU per mouse of a commercially available adenoviral CMV-Flp vector (Ad5CMVFlpo, University of Iowa Viral Vector Core Facility) to activate oncogenic KrasG12D and delete Tp53. To follow tumor development, mice were anesthetized using isoflurane (Piramal, 56.761.002) and maintained under anesthesia during the scanning procedure. Lungs were imaged with an X-Ray microtomography (Quantum FX; PerkinElmer) at a 50 μm voxel size, with retrospective respiratory gating. Individual tumor volumes were measured and calculated using Analyze 12.0 (PerkinElmer) or Osirix MD (Pixmeo, RRID:SCR_013618). For the BrdU assay, 2 mg of freshly prepared BrdU (Merck, 10280879001) was injected intraperitoneally into 100 μl of PBS 1x. Immunotherapy treatment was performed with an intraperitoneal injection of InVivoMAb anti-mouse PD-1 (clone 29F.1A12, Bio X Cell, BE0273, RRID:AB_2687796) at a dose of 200 μg/mouse twice a week for 2 weeks. Anti G-CSF treatment was performed with an intraperitoneal injection of 10 μg anti-mouse G-CSF antibody (clone 67604, R&D Systems, MAB414, RRID:AB_2085954) every day for 14 days. G-CSF treatment was performed with daily intraperitoneal injection of 10 μg of recombinant mouse G-CSF (PeproTech, 250–05). Tumor-bearing mice were subjected to radiotherapy with a 20 mm2 collimator (13 mA, 3 mm Cu filter, 225 keV) 11.6 Gy with a single dose delivered in 256 seconds as previously described (9). Experimental melanoma lung metastases were obtained by injecting 3 × 105 B16-F1 cells (RRID:CVCL_0158) into the tail vein of female syngeneic recipient mice, either control (Ly6gWT/WT; Glut1Flox/Flox or Ly6gCre/WT; Glut1WT/WT) or Glut1KO (Ly6gCre/WT; Glut1Flox/Flox). All lungs were collected, fixed and paraffin-embeded 21 days after the injection. Hematoxylin and Eosin staining was performed on sections to detect the experimental metastases, which were counted blindly.
Figure 2. Tumor-associated neutrophils are more glycolytic than neutrophils from healthy lung.
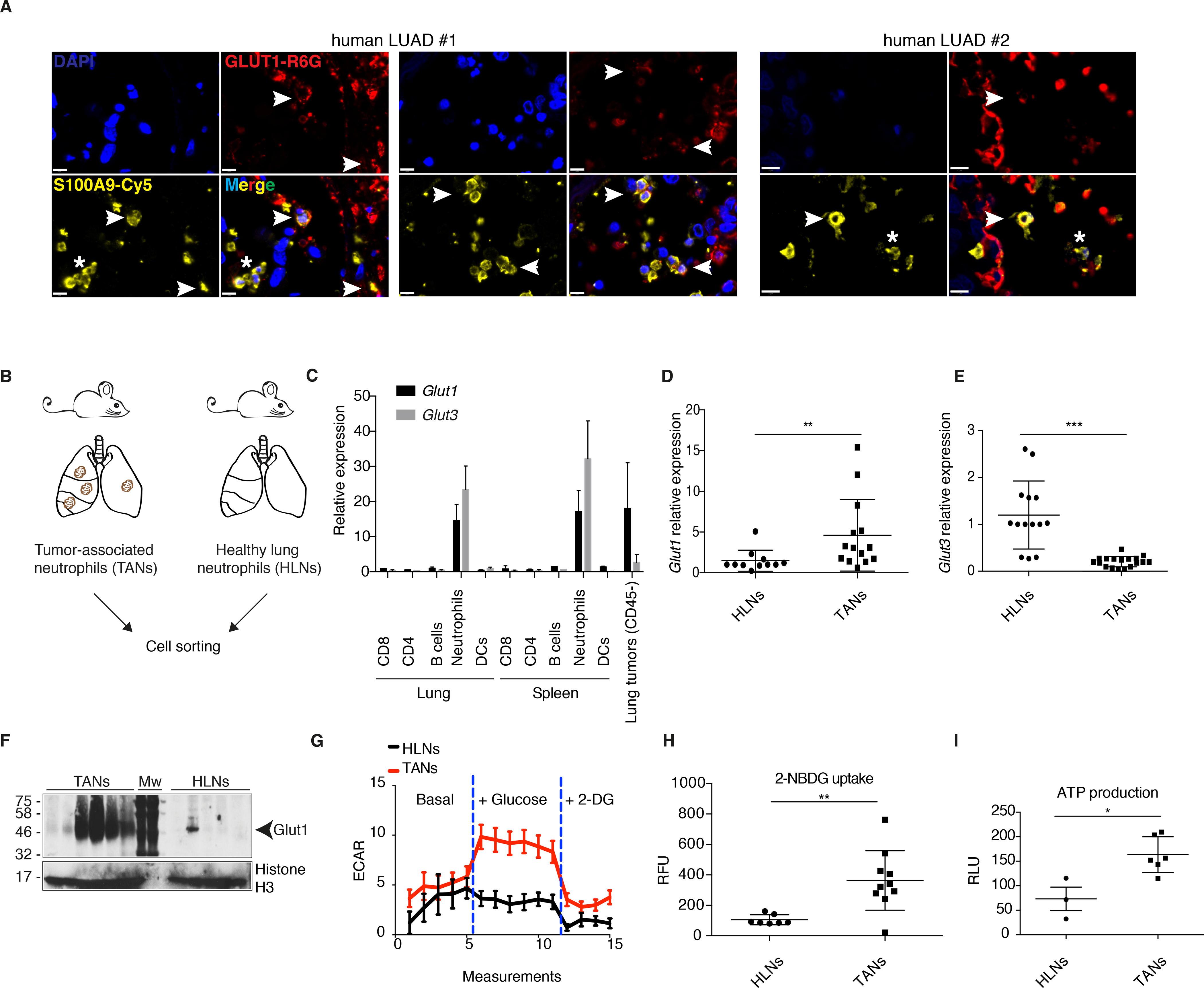
A) Two human LUAD samples (with two different regions for the first one) showing TANs expressing (white arrowheads) or not (white asterisks) GLUT1. Staining was done with DAPI (dark blue), S100A9 (yellow) and GLUT1 (red). Scale bars: 10 μm. B) Schematic representation of the isolation of TANs and healthy lung neutrophils (HLNs). C) Glut1 and Glut3 expression (mean ± s.d.) in different cell-sorted populations from healthy lung or spleen, or from the CD45- fraction of lung tumors. D-E) Real-time PCR analyses of Glut1 and Glut3 expression (mean ± s.d.) in HLNs compared to TANs. F) Western blot of Glut1 from HLNs and TANs. Mw, molecular weight marker. G) Seahorse analysis to measure ECAR in response to glucose addition to the medium in HLNs compared to TANs (mean ± s.e.m.). H) Measurements of 2-NBDG uptake (mean ± s.d.) in HLNs compared to TANs. RFU, relative fluorescence units. I) ATP production in HLNs compared to TANs (mean ± s.d.). RLU, relative light units.
Study approval
All mouse experiments were performed with the permission of the Veterinary Authority of Canton de Vaud, Switzerland (license number: VD2391).
Tumor and tissue-derived supernatant (SN) production
KPFrt tumors and healthy tissues (lung and spleen) were isolated and dissociated as previously described (10). The cell suspension was then cultured in complete DMEM (Thermo Fisher Scientific, 41965062) supplemented with 10% Fetal Bovine Serum (FBS) (Thermo Fisher Scientific, 10270106) for 24 hours at a concentration of 107 cells per ml. The day after, supernatant (SN) was collected, filtered (0.22 μm), aliquoted after centrifugation (5 minutes, 2000 rpm) and stored at −80°C or used immediately. For CD45+ and CD45- fraction-derived SN, CD45 positive cells were sorted using CD45 MicroBeads (clone 30F11.1, Miltenyi Biotec, 130–052-301, RRID:AB_2877061), using the manufacturer’s instructions. Both positive and negative fractions were cultured for 24 hours hours at a concentration of 107 cells per ml.
Mouse neutrophil isolation
Tumor-associated neutrophils (TANs), healthy lung neutrophils (HLNs), and bone marrow neutrophils were isolated using anti-Ly6G MicroBead Kit (Miltenyi Biotec, 130–092-332) and anti-Ly6G MicroBeads UltraPure (clone REA526, Miltenyi Biotec, 130–120-337) according to the manufacturer’s instructions. Where indicated, bone marrow neutrophils were stimulated with TNF (PeproTech, 315–01A, 10 ng/ml, 4 hours) before RNA isolation.
Neutrophil in vitro survival assay
105 freshly-isolated neutrophils were cultured for 20 or 48 hours in 200 μl of complete DMEM (Thermo Fisher Scientific, 41965062, 10270106) or complete DMEM supplemented with 16% of tumor-derived SN. For size exclusion separation of proteins from the tumor-derived SN, the Centricon Plus-70 Centrifugal Filter (Merck, UFC700308) was used according to the manufacturer’s instructions.
Cell lines
The SV2 cell line was generated in our laboratory in 2013 from a lung tumor of a KP mouse; it was passaged > 20 times before use. Around the time of the experiments, SV2 cells were not tested for Mycoplasma. The B16-F1 cells were obtained in 2020 from D. Constam (ISREC, EPFL) and were Mycoplasma negative.
Neutrophil-tumor cell co-culture to measure tumor cell growth
100, 200 or 400 KP-derived cells (SV2) were cultured alone or in presence of 104 control or Glut1KO TANs. After 10 days in culture, cells were fixed for 10 minutes at room temperature in 3.6% formaldehyde (VWR Chemicals, 20909.290) and colored with crystal violet (Merck, C3886) for 5 minutes at room temperature. The percentage of covered area was then measured and quantified using Fiji (RRID:SCR_002285).
CD45 negative cell enrichment
Individual tumors were isolated and dissected and CD45 positive cells were depleted using CD45 MicroBeads (clone 30F11.1, Miltenyi Biotec, 130–052-301, RRID:AB_2877061), using the manufacturer’s instructions.
RNA isolation and real-time PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen, 15596018) according to the manufacturer’s instructions. cDNAs were synthesized from 1 μg total RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, 4368814). Real-time PCR was performed using 5 ng of cDNA. The gene quantification was performed using the following commercially available probes from Thermo Fisher Scientific: Rpl30 (Mm01611464_g1, used for normalization), Glut1 (Slc2a1) (Mm00441480_m1), Glut3 (Slc2a3) (Mm00441483_m1), Arg1 (Mm00475988_m1), Hk2 (Mm00443385_m1), Siglecf (Siglec5) (Mm00523987_m1), Ikbke (Mm00444862_m1), Apoe (Mm01307193_g1), Pfkp (Mm00444792_m1), Cxcl3 (Mm01701838_m1), Grx1 (Mm01352826_g1), Il23a (Mm00518984_m1), Padi4 (Mm01341658_m1) and G-CSF (Csf3) (Mm00438334_m1). Ct values were analyzed according to the comparative Ct method.
RNA sequencing
RNA from the Ly6g positive or CD45 negative fractions was extracted using TRIzol. Multiplexed libraries for mRNA-seq were prepared following SMARTer Stranded Total RNA-seq Pico v2 prep, starting from 1 ng total RNA from the Ly6g positive fractions; and following TruSeq stranded mRNA LT prep, starting from 375 ng of RNA from CD45 negative fractions. Sequencing was subsequently performed on a NextSeq 500 instrument (Illumina) on a high-output flow cell, yielding single-end reads of 75 nucleotides. Adapter sequences and low-quality ends were removed with cutadapt (v1.14) as needed. Reads were aligned to mouse genome build mm10 using HISAT2 aligner (v2.10). Genes with low expression were filtered out (average transcripts per kilobase million < 6 or average raw counts < 50). Counts were normalized for library size using TMM method from EdgeR (v3.24.3) and voom from limma. Differential expression was computed with limma between TANs and HLNs (significance cutoff LFC > 1 or < -1 and adjusted P-value < 0.05), Glut1KO and control TANs, Glut1KO and control non-immune fraction (significance cutoff LFC > 1 or < -1 and P-value < 0.05).
We investigated over-representation of pathways as defined in the Hallmark collection of MSigDB (v6.0) among the top 200 genes up-regulated in TANs versus HLNs or down-regulated in Glut1KO versus control TANs.
Western blot
Western blots were performed on protein extracts from freshly isolated neutrophils. Cells were lysed in RIPA buffer (20 mM Tris pH8, 50 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM Na3VO4, protease inhibitor cocktail (complete, Roche)) at 4°C for 30 minutes. BCA Protein Assay Kit (Merck, 71285–3) was used for protein quantification. Anti-Glut1 (Millipore, 07–1401, RRID:AB_1587074) and anti-Histone H3 antibodies (Abcam, ab1791, RRID:AB_302613) were used.
Flow cytometry
Flow cytometry analysis of tumors was performed as previously described (10). Antibodies and fluorochromes used were the following: anti-CD45-BUV661 (clone 30-F11, BD Biosciences, 565079, RRID:AB_2739057), anti-CD11b-BV711 (clone M1/70, BioLegend, 101242, RRID:AB_2563310), anti-Ly6G-PE (clone 1A8, BioLegend, 127607, RRID:AB_1186104), anti-Siglec-F-PE-Vio 615 (clone REA798, Miltenyi Biotec, Cat# 130–112-172, RRID:AB_2653444), anti-BrdU-APC (clone Bu20a, BioLegend, 339808, RRID:AB_10895898), anti-PD-L1-BV-785 (clone 10F.9G2, BioLegend, 124331, RRID:AB_2629659). Samples were acquired using the LSRII SORP (Becton Dickinson), a 5-laser and 18-detector analyser at the EPFL Flow Cytometry Core Facility. CountBright™ Absolute Counting Beads (Thermo Fisher Scientific, C36950) were used for Trucount experiments using the manufacturer’s instructions. Data analyses were performed using FlowJo (FlowJo LLC ©, RRID:SCR_008520).
Immunohistochemistry and Immunofluorescence
Preparation of mouse lung sections was explained before (10). Antibodies used for immunohistochemistry from mouse tissue sections are anti-GLUT1 (Abcam, ab115730, RRID:AB_10903230, 1:650), anti-pHH3 (Cell Signaling Technology, 9701S, RRID:AB_331535, 1:100), anti-pErk (Cell Signaling Technology, 4370S, RRID:AB_2315112, 1:400) and anti-S100A9 (Novus Biologicals, NB110–89726, RRID:AB_1217846, 1:5’000). For human sample multiplexing, the 4plex immunofluorescence was performed using the fully automated Ventana Discovery ULTRA (Roche Diagnostics, Rotkreuz, Switzerland). All steps were performed on the instrument with Ventana solutions following their recommendations. Briefly, dewaxed and rehydrated paraffin sections were pretreated with heat using standard conditions for 40 minutes in CC1 solution. Primary antibodies were rabbit anti-GLUT1 (Millipore, 07–1401, RRID:AB_1587074, 1:5’000), rabbit anti-CD4 (clone SP35, Roche Diagnostics, 05552737001), rabbit anti-CD8 (clone SP57, Roche Diagnostics, 05937248001), rabbit anti-S100A9 (Novus Biologicals, NB110–89726, RRID:AB_1217846, 1:20’000). They were applied sequentially and incubated during 1 hour (Glut1 and S100A9) or 20 minutes (CD4 and CD8) at 37°C (the data obtained with CD4 and CD8 staining were not used in this study). After incubation with an anti-rabbit Immpress HRP (ready to use, Vector Laboratories, MP-7401, RRID:AB_2336529), they were sequentially revealed with the Rhodamine-6G (Roche Diagnostics, 07988168001), DCC (Roche Diagnostics, 07988192001), Red 610 (Roche Diagnostics, 07988176001) and Cyanine 5 (Roche Diagnostics, 07551215001) TSA kits. Heat denaturation was applied after each TSA revelation. Slides were then acquired with an Olympus slide scanner (Olympus, VS120-L100) and analyzed using QuPath (RRID:SCR_018257). Permission to use the human material was granted by the local ethics committee (KEK: 200/2014).
Seahorse
Extracellular acidification rate (ECAR) measurements were performed using XF96 Extracellular Flux analyser (Seahorse Bioscience, North Billerica, MA). Briefly, 100,000 freshly isolated neutrophils from healthy lungs or KP tumors were plated into a XF96 V3 polystyrene cell culture plate (Seahorse Bioscience). Cells were incubated for 3 hours in a humidified 37°C incubator with 5% CO2 in RPMI with no glucose (Thermo Fisher Scientific, 11879020). All experiments were performed at 37°C. Each measurement cycle consisted of a mixing time of 3 minutes and a data acquisition period of 2 minutes. For glycolysis assay, 10 mM glucose (VWR, 1.08337.0250) was added during the seahorse experiment followed by 50 mM 2-deoxyglucose (2-DG) (Merck, D8375–5G). At the end of the experiment, proteins were extracted using RIPA buffer (20 mM Tris pH8, 50 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM Na3VO4, protease inhibitor cocktail (complete, Roche)) to determine protein concentration in each well. ECAR was calculated as normalized against protein concentration.
Glucose uptake
2-NBDG glucose uptake experiments were performed on freshly isolated neutrophils using a glucose-uptake cell-based assay kit (Cayman Chemical, CAY-600470–1) according to the manufacturer’s instructions. Data were normalized against protein concentration.
ATP production assay
Production of ATP in freshly isolated neutrophils was measured using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, G7570) according to the manufacturer’s instructions. Data were normalized against protein concentration.
Statistical analysis
Mice were age-matched and randomized according to tumor progression and gender. For metabolic and survival assays, μCT follow-up, flow cytometry, IHC and real-time PCR, the samples were processed and analyzed blindly. All results are represented as mean ± s.d. unless stated otherwise. Each point represents an independent tumor (for in vivo experiments) or an independent well (for ex vivo experiments). Comparisons between groups were made as indicated in the Figure Legends. Statistical significance is indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, or ns (not significant), using two-sided Mann-Whitney tests unless specified otherwise. Statistical analysis was performed using GraphPad Prism (version 6, RRID:SCR_002798).
Data Availability
The RNA sequencing data have been deposited to the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and assigned the identifier: GSE140159.
Results
Glucose transporter Glut1 mediates neutrophil survival
To address how the tumor microenvironment may alter neutrophils, we firstly compared the fate of freshly-isolated bone marrow-derived neutrophils, cultured either in complete medium or in complete medium supplemented with a KP lung tumor-derived supernatant (SN) (Figure S1A). After 20 or 48 hours in culture, the medium supplemented with tumor-derived SN increased neutrophil survival significantly (Figure S1B-C). Size exclusion separation of proteins revealed that only the protein fraction of >3 kDa increases neutrophil survival (Figure S1D), excluding a pro-survival effect mediated by pH or small metabolites. We next generated tumor supernatants from CD45+ (immune) or CD45- (non-immune) cells. The CD45- derived SN was sufficient to increase neutrophil survival, whereas the CD45+ SN had no effect (Figure S1E). Thus, this experimental system, which enables to extend neutrophil lifespan ex vivo, could be used to identify mechanisms altering their survival in tumors.
To determine if neutrophil survival is enabled by an increased metabolic activity, we next monitored the expression of two high affinity glucose transporters, Glut1 and Glut3, in response to tumor-derived SN. Surprisingly, the two transporters were regulated in an opposite manner, with Glut1 being up- and Glut3 down-regulated upon incubation with tumor-derived SN (Figure 1A).
Figure 1. Glut1 expression is necessary for prolonged neutrophil survival.
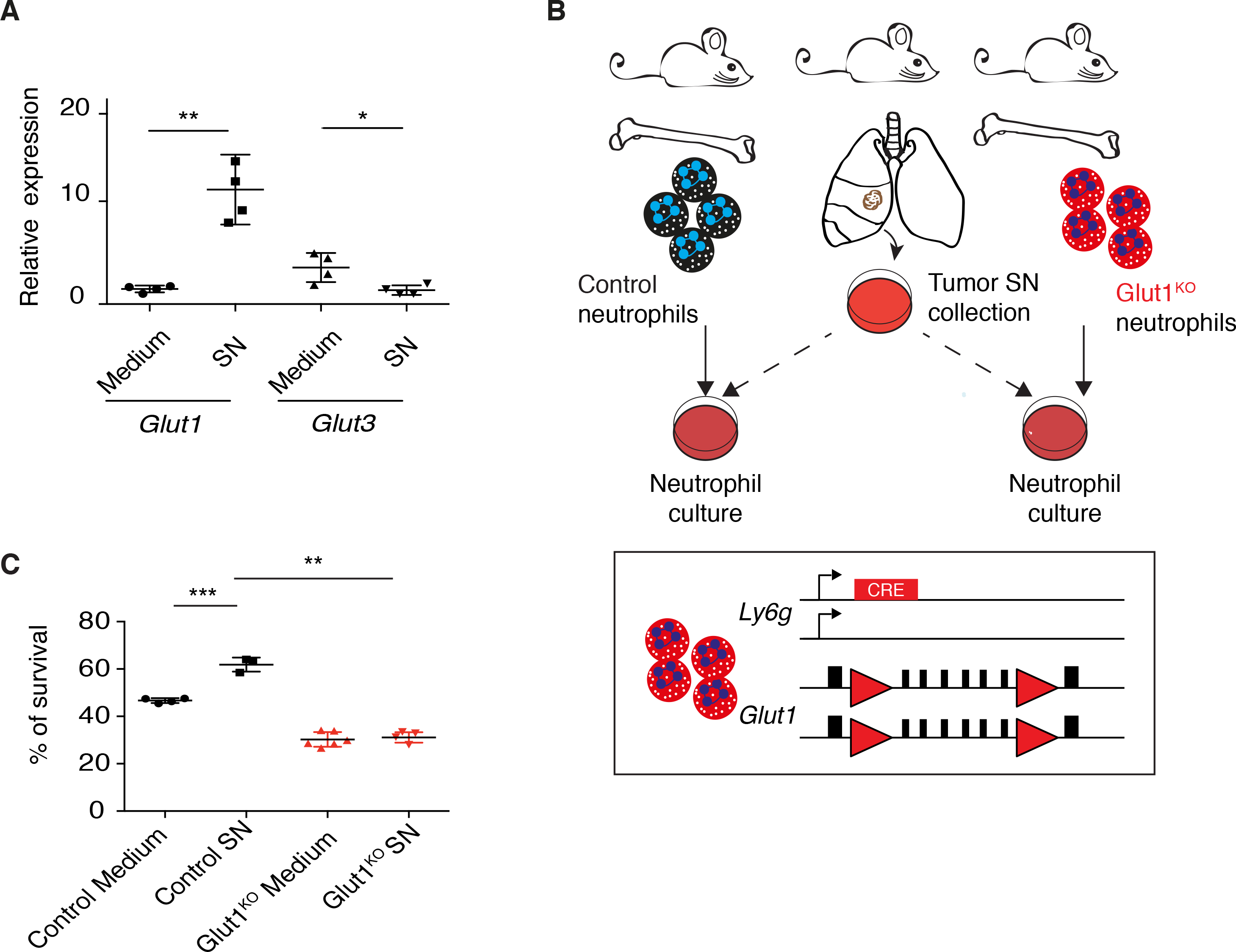
A) Relative Glut1 and Glut3 expression by real-time PCR (mean ± s.d.) in response to supernatant (SN) or control medium. B) Schematic representation of the Glut1KO neutrophil model and survival assay. Red triangles, LoxP sites. C) WT or Glut1KO neutrophil survival (mean ± s.d.) after 20 hours in control medium or in presence of tumor-derived SN.
To directly test the importance of Glut1 for neutrophil survival, we crossed Ly6GCre mice, where the Cre recombinase gene is knocked into the Ly6g allele (17), to Glut1Flox/Flox mice (16), generating mice with neutrophil-specific Glut1 deletion (Figure 1B). In the blood of healthy mice, both control and Glut1KO neutrophils represent between 8 and 10 percent of the immune cell population (Figure S2A). We isolated Glut1KO and control neutrophils, and monitored a partial-to-complete loss of survival benefit upon tumor-derived SN incubation in the Glut1KO cells (Figure 1B–C and S2B). Altogether, our ex vivo data indicate that soluble factors produced in lung tumors increase neutrophil survival through Glut1 up-regulation, a response that may favor an extended neutrophil lifespan within the tumor microenvironment.
Glut1 and glucose metabolism are enhanced in tumor-associated neutrophils
To determine if GLUT1 is expressed in TANs from human LUAD, we used immunofluoresecence and stained tumor sections with anti-S100A9 to mark neutrophils, and with anti-GLUT1. We identified both GLUT1 positive and GLUT1 negative human TANs, which suggests a metabolic heterogeneity for intra-tumoral neutrophils (Figure 2A).
To better understand the metabolic alterations of TANs in vivo, we isolated neutrophils from age-matched healthy lung (HLNs) and from KP tumors (TANs) (Figure 2B). RNA sequencing analyses identified 1470 up-regulated genes and 487 down-regulated genes in TANs compared to HLNs (Table S2). Pathway analysis revealed glycolysis as the most significantly up-regulated pathway in TANs (Figure S3A, S3B and Table S3). By real-time PCR analysis, we validated the TAN signature with multiple genes from the lists of up- and down-regulated genes (Figure S3C). The mRNA expression of each of Glut1 and Glut3 was stronger in neutrophils compared to other immune cell populations isolated from healthy lung or spleen (Figure 2C) and, similar to the tumor SN experiments (see Figure 1A), Glut1 was higher and Glut3 lower in TANs compared to HLNs (Figure 2D–E). The strong Glut1 expression in TANs was particularly evident at protein level (Figure 2F). Furthermore, the expression of the glycolytic gene, hexokinase 2 (Hk2), was significantly more elevated in TANs than in HLNs (Figure S3C). Congruent with the up-regulated glycolysis gene signature, including stronger Hk2 and Glut1 expression, we demonstrated that TANs undergo a metabolic switch toward increased glycolysis, since they exhibit an elevated extracellular acidification rate in response to glucose (Figure 2G), absorb glucose more efficiently (Figure 2H) and produce more ATP (Figure 2I) compared to HLNs. Thus, when compared to healthy lung neutrophils, lung TANs are characterized by a shift toward increased glucose usage and metabolism.
An accelerated turnover defines Glut1-deficient tumor-associated neutrophils
To directly interrogate the importance of Glut1 in TANs, we crossed a second-generation KrasFrt-STOP-Frt-G12D/WT; Tp53Frt/Frt (referred to as KPFrt thereafter) tumor mouse model (15) to Ly6gCre knockin and Glut1Flox/Flox (16,17) conditional knockout mice (Figure 3A). In this experimental configuration, intratracheal instillation of adenoviral-Flp activates oncogenic Kras (G12D) and deletes Tp53, resulting in tumor development in a context where neutrophils and TANs lack Glut1.
Figure 3. Neutrophil-specific Glut1 deletion reduces the proportion of SiglecFhigh TANs.
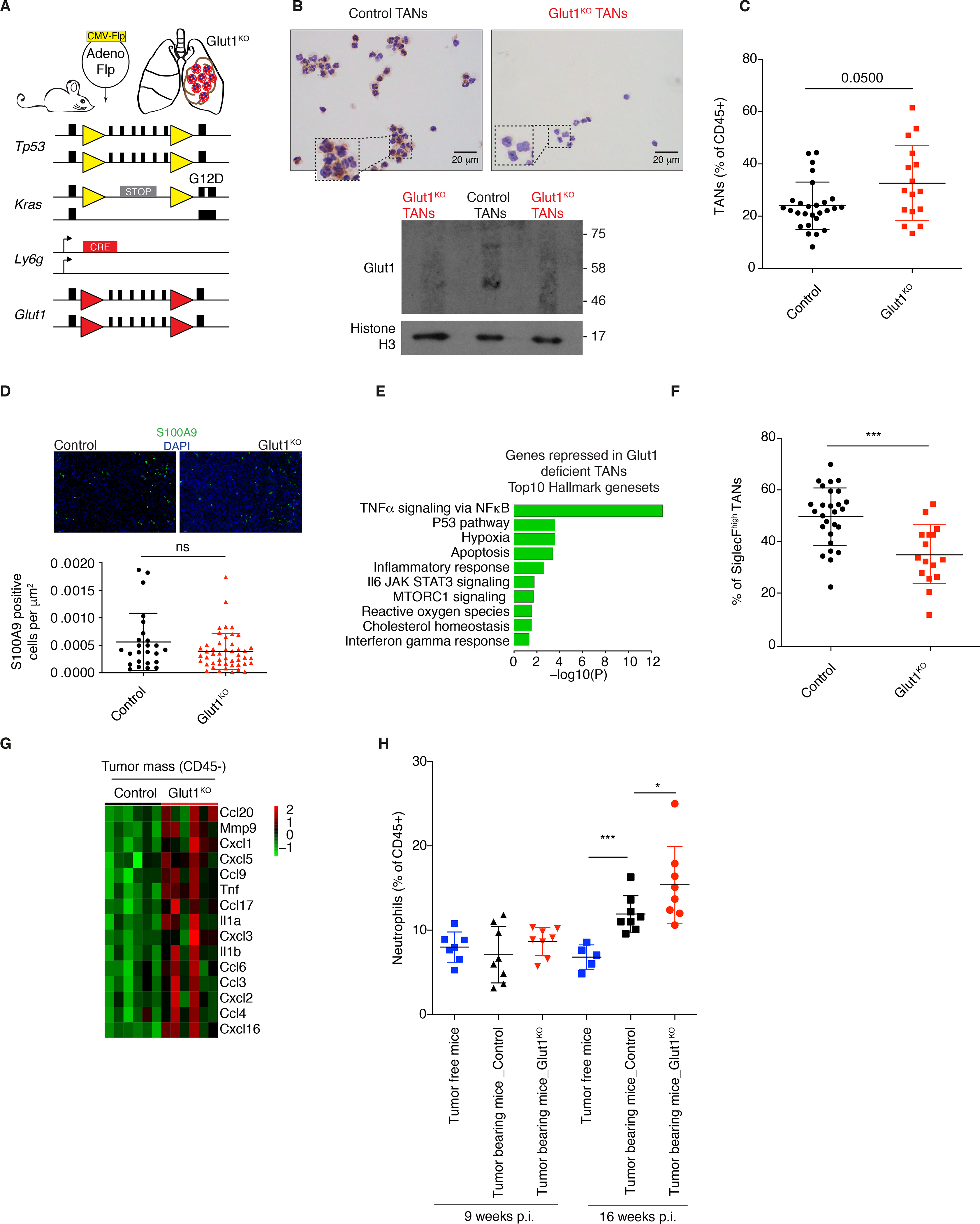
A) Schematic representation of the mouse model used. Yellow triangles, Frt sites; Red triangles, LoxP sites. B) Glut1 protein expression levels in isolated Glut1KO TANs or control TANs by immunocytochemistry. C) Neutrophil prevalence (mean ± s.d.) in KP tumors in control and Glut1KO conditions. D) Representative staining and quantification of S100A9 immunofluorescence from n= 25 control and n= 47 Glut1KO tumors. E) Top 10 pathways from Hallmark over-represented among the top 200 genes most repressed in Glut1KO TANs compared to WT TANs. P-value was computed with Fisher exact test. F) SiglecFhigh cells among control and Glut1KO TANs (mean ± s.d.). G) Heatmap of chemoattractant genes overexpressed in CD45- cells in Glut1KO compared to control neutrophil conditions. H) Neutrophil proportions in the blood of healthy and tumor-bearing mice 9 or 16 weeks post-tumor initiation (p.i.) (mean ± s.d.).
First, we used immunocytochemistry and Western blot to confirm the loss of Glut1 in TANs at protein level (Figure 3B). Contrary to our expectations, there was a higher prevalence of Glut1KO neutrophils in the tumor mass compared to control neutrophils (Figure 3C and S3D); however, their absolute numbers based on the quantification of S100A9 staining by immunofluorescence were not increased (Figure 3D). Next, we monitored the molecular changes consequent to Glut1 loss in TANs using RNA sequencing (Table S4). The downregulation of the “TNFα signaling via NF-κB” gene signature in Glut1KO TANs (Figure 3E and Table S5), which was higher in wild-type TANs compared to HLNs (see Figure S3B, left panel), might indicate that TNF and NF-κB signaling in TANs is linked to Glut1-dependent survival (18,19). This possibility was illustrated by an induction of Glut1 in response to TNF stimulation of bone marrow-derived neutrophils (Figure S3B, right panel).
A SiglecFhigh neutrophil subset was recently reported to have an increased lifespan in lung adenocarcinoma (20) and in myocardial infarction (21). In Glut1KO TANs, the SiglecFhigh subset was very significantly decreased (Figure 3F). To further explore this subset, we analyzed data from different neutrophil subpopulations recently identified by single cell RNA sequencing in another KP-derived model, where 6 subsets of neutrophils were identified, in blood (N1 and N2) and in tumors (N3-N6) (22). We observed that the SiglecFhigh cells were predominant in the N4, tumor-specific subpopulation. In addition, N4 was defined by high Glut1, Hk2 and Pd-l1, and by low Glut3 expression (Figure S4A), highlighting the relevance of Glut1 for this TAN subset. From autochtonous KP tumors, we demonstrated that Glut3 but not Glut1 expression was significantly reduced in the SiglecFhigh compared to the SiglecFlow neutrophil subpopulation, leaving Glut1 as the predominant glucose transporter of SiglecFhigh TANs (Figure S4B).
To investigate the mechanisms by which neutrophil turnover might be enhanced upon Glut1 deletion, we performed an RNA sequencing of the non-immune fraction (CD45-) of the tumor mass. Among the highest up-regulated genes in tumors having Glut1KO neutrophils, we identified several genes involved in neutrophil recruitment (Figure 3G and Table S6). Additionally, while there is no significant difference in neutrophil percentage between the genotypes in healthy mice (see Figure S2A), we measured a higher proportion of blood-circulating neutrophils in control tumor-bearing compared to healthy mice, which was further increased in KPFrt; Ly6gCre/WT; Glut1Flox/Flox tumor-bearing mice (Figure 3H). These results suggest an elevated neutrophil turnover during cancer growth with Glut1KO TANs. Neutrophil turnover was measured from different anatomical sites using an in vivo BrdU tracking experiment, in healthy and tumor-bearing mice. Specifically, bone marrow, blood, spleen, lung and tumors were collected 2.5 and 6.5 days after a single BrdU injection (Figure 4A). In all healthy tissues, the BrdU kinetics was similar, with a peak (colonization phase) at 2.5 days reaching nearly 50% BrdU positive neutrophils (Figure 4A–C). After 6.5 days, the majority of neutrophils were BrdU negative, showing that the entire pool had been renewed (extinction phase) (Figure 4A–C). As exception, in lungs a small fraction (10%) of neutrophils was still BrdU positive 6.5 days after injection. In contrast, in lung tumors the kinetics was altered, with only 10% of BrdU positive neutrophils after 2.5 days, rising up to 30% after 6.5 days, thus still being in the colonization phase. These data demonstrate a reduced neutrophil turnover specifically in the tumor mass, leading to an aberrantly large proportion of old (6.5 days) neutrophils (Figure 4A–C). In tumors, at 2.5 days all the young BrdU positive neutrophils (< 2.5 days-old) were SiglecFlow while, at 6.5 days, the old BrdU positive TANs were mainly SiglecFhigh (Figure 4B), consistent with a time-dependent acquisition of this membrane protein in lung tumors.
Figure 4. Neutrophil-specific Glut1 deletion accelerates their turnover.
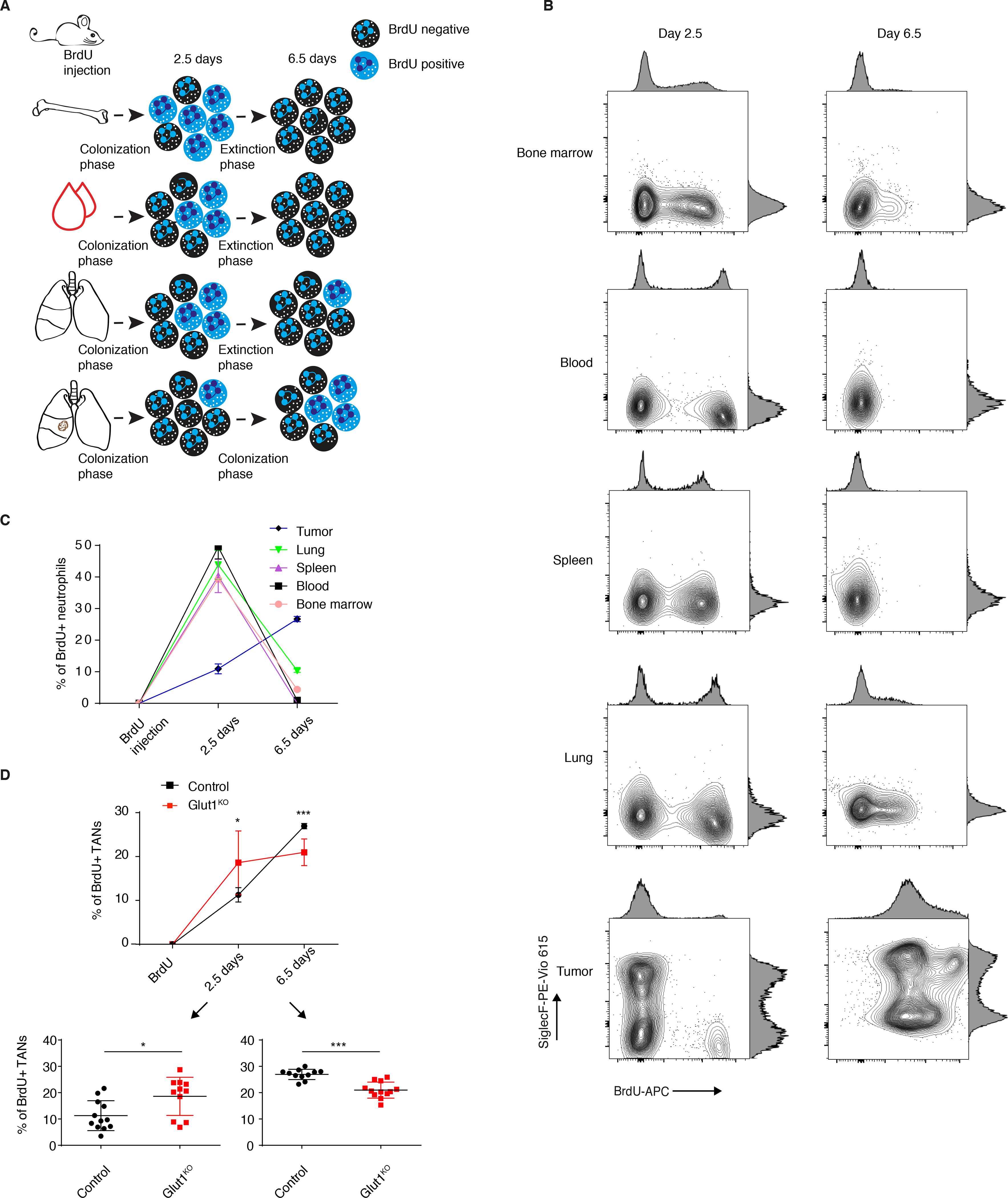
A) Schematic representation of the BrdU experiments. B) Representative BrdU and SiglecF staining in neutrophils from the bone marrow, blood, healthy spleen, healthy lung and tumors 2.5 and 6.5 days after a single BrdU injection. C) BrdU positive neutrophil percentage (mean ± s.d.) among neutrophils in the bone marrow, blood, healthy spleen, healthy lung and tumors 2.5 and 6.5 days after a single BrdU injection. D) Neutrophil turnover kinetics experiment, with detailed measurements at 2.5 (lower left) and 6.5 days (lower right) after BrdU injection in tumors in control and Glut1KO conditions (mean ± s.d.).
When comparing tumors with Glut1KO neutrophils to controls, we identified that Glut1KO TANs have an accelerated turnover with more BrdU positive cells at 2.5 days post-BrdU injection and a reduced old neutrophil fraction 6.5 days after (Figure 4D). Thus, in tumors with Glut1KO neutrophils, reduced neutrophil survival and increased neutrophil recruitment together contribute to a higher neutrophil turnover during cancer progression. These results also position the old, SiglecFhigh and Glut1high TAN subset as an inhibitor of young neutrophil recruitment, possibly acting by a communication with the tumor epithelial cells.
Glut1KO but not Glut3KO neutrophils negatively impact tumor growth
Because the pro- and anti-tumor properties of neutrophils require a better understanding (23), we decided first to explore the consequences of Glut1KO TANs on tumor progression (Figure 5A). Tumors with Glut1KO TANs had a reduced cell proliferation as measured by phospho-Histone H3 (pHH3) and a reduced phospho-ERK1/2 (pERK) staining, a marker of tumor progression (Figure 5B–C). Moreover, tumor growth rates monitored by longitudinal micro-computed tomography (μCT) were significantly diminished in mice with Glut1KO TANs (Figure 5D). Next, to determine if TANs affect lung tumor cells in vitro, we measured their behavior using a KP-derived cell line (SV2) cultured alone, with control TANs or with Glut1KO TANs. A stronger negative impact on tumor cell growth was observed in Glut1KO TANs, as revealed by a reduced spreading of co-cultured SV2 cells (Figure 5E). This suggests that Glut1KO TANs have a reduced tumor-supportive capacity or are endowed with anti-tumor properties.
Figure 5. Glut1 deletion in neutrophils reduces tumor growth.
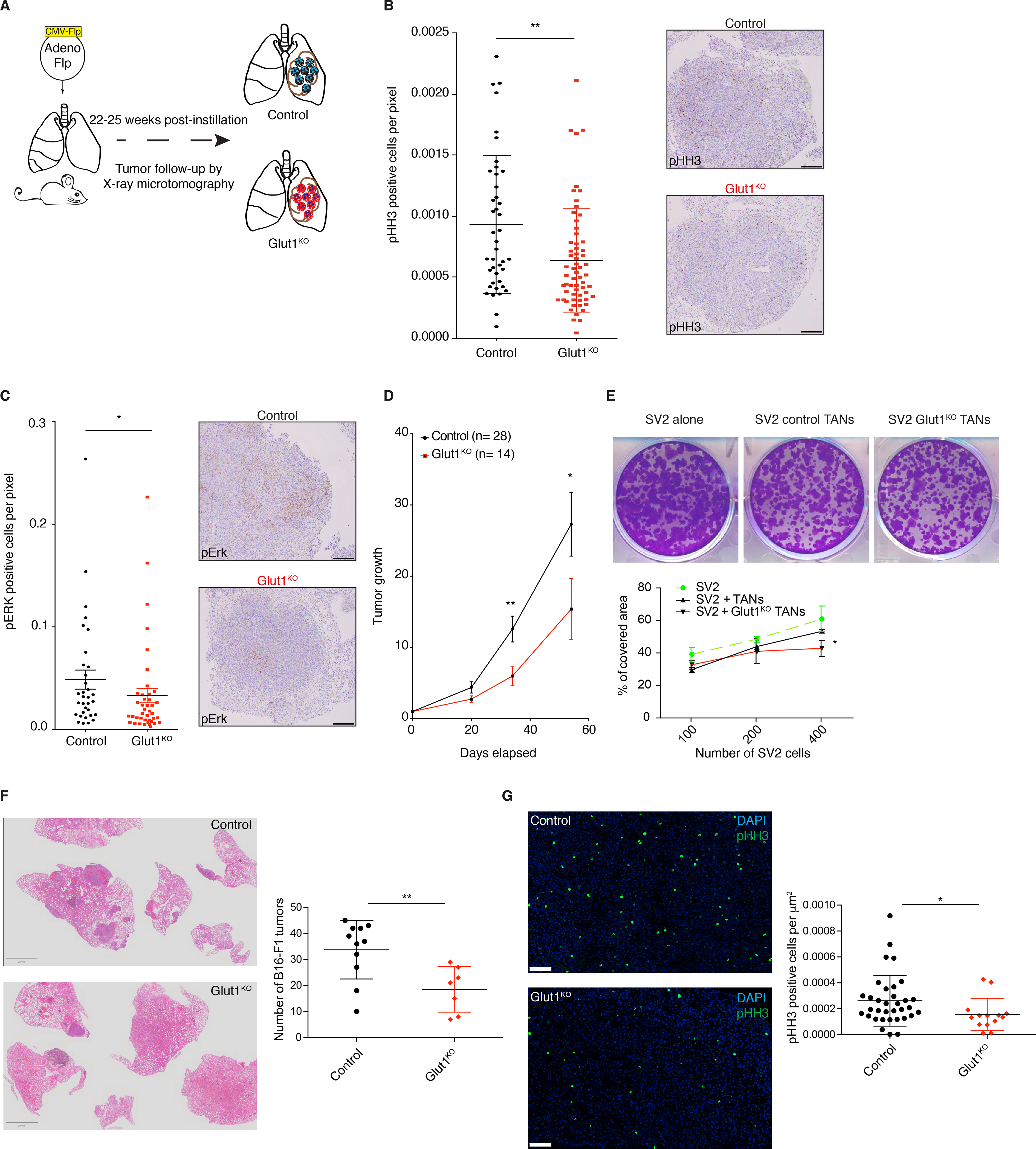
A) Schematic representation of the experiments. B) (left) Quantification of phospho-Histone H3 (pHH3) staining in Glut1KO or control tumors (mean ± s.d.). (right) Representative staining of pHH3 in tumors. Scale bars: 200 μm. C) (left) Quantification of phospho-ERK (pERK) staining in tumors (mean ± s.d.). (right) Representative staining of pERK in tumors. Scale bars: 200 μm. D) Long-term μCT analysis of tumor-bearing mice. Data represent tumor volumes (mean ± s.e.m.) normalized to the first volume (set to 1) of the same tumors in control or GLUT1KO. Control n= 28 tumors; Glut1KO n= 14 tumors. E) Representative images (upper panel) of the SV2 cell line spreading assay, cultured alone or in presence of control or Glut1KO TANs. (lower panel), cell-covered area quantification of SV2 cell lines cultured alone or in presence of control or Glut1KO TANs. F) Representative Hematoxylin and Eosin (H&E) staining of experimental lung metastases from B16-F1 melanoma cells (left panel, scale bars: 2 mm). (right panel) Tumor quantification. G) Representative pHH3 imunofluorescence staining (left panel, scale bars: 100 μm) and quantification (right panel).
To test if the reduced tumor-supportive function of Glut1KO TANs can be extended to other models, we injected B16-F1 melanoma cells via the tail vein of control or Glut1KO syngeneic recipient mice, and counted the number of lung lesions three weeks later. In this experimental metastasis model, there were fewer lesions in Glut1KO conditions (Figure 5F). Furthermore, tumor cell proliferation was reduced in these tumors compared to control tumors, as indicated by a reduced proportion of pHH3 positive cells (Figure 5G). Thus, TAN-mediated tumor support relies on Glut1 in different cancer types.
Intrigued by the opposite regulation of Glut1 and Glut3 in response to tumor-derived SN, in HLNs versus TANs, and by their different expression patterns in the neutrophil subsets (see Figures 1A, 2D–E and S4A), we decided to interrogate directly the consequences of Glut3 gene deletion in neutrophils (Figure S5A–B). In contrast to our data obtained with Glut1KO, we monitored a reduced proportion of Glut3KO neutrophils in the blood of healthy mice (Figure S5C), no tumor growth delay in mice with Glut3KO neutrophils compared to controls (Figure S5D–F), and no change in the proportion of the SiglecFhigh TAN subset compared to controls (Figure S5G). Thus, Glut1 but not Glut3 deficiency in neutrophils diminishes tumor growth.
To understand the mechanisms by which the loss of Glut1 in TANs reduces tumor progression, we hypothesized that the turnover of neutrophils may be critical to regulate the neutrophil subset balance. Specifically, considering that G-CSF affects neutrophil mobilization and survival (24), we decided to explore its role in this balance using loss- and gain-of-function approaches. Daily injections of G-CSF neutralizing antibodies in tumor-bearing mice significantly reduced the number of TANs (Figure 6A–B). This decrease could be explained by a reduced mobilization or by a recruitment defect at the tumor site, as illustrated by the very significant loss of young BrdU+ neutrophils (2.5 days) without affecting the acquisition of SiglecF (Figure 6C–E). This decreased number of the entire TAN pool led to a reduced tumor growth, highlighting the pro-tumoral properties of these innate immune cells (Figure 6F). Accordingly, G-CSF treatment reduced the proportion of SiglecFhigh TANs (Figure S6A–C), but only in Glut1KO conditions and increased the abundance of young (BrdU+ at 2.5 days) neutrophils in tumors with control and Glut1KO neutrophils (Figure S6D–E). Although G-CSF decreased the growth rate of control tumors, its impact was more pronounced in Glut1KO tumors (Figure S6F). Thus, the anti-tumor properties of young neutrophils seem to be amplified upon Glut1 loss. Altogether, our data suggest the existence of a functional link between neutrophil turnover and tumor growth (Figure 6G).
Figure 6. G-CSF neutralization reduces TAN numbers and tumor growth.
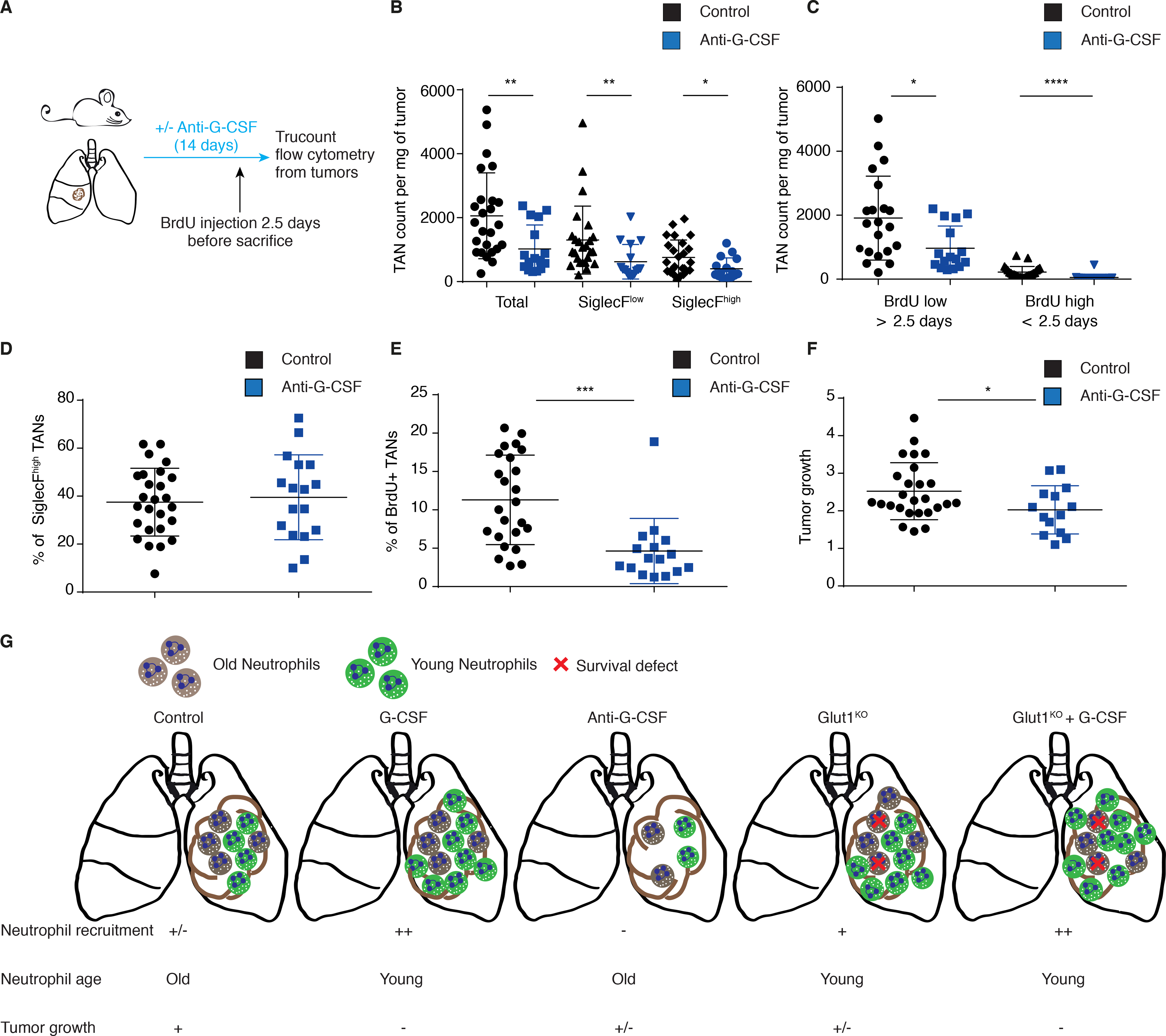
A) Schematic representation of the experiment. B) Trucount flow cytometry of total, SiglecFlow or SiglecFhigh TANs in control or anti-G-CSF conditions (mean ± s.d.). C) Trucount flow cytometry of BrdU positive and negative neutrophils in control or anti-G-CSF conditions (mean ± s.d.). D) Percentage of SiglecFhigh TANs in control or anti G-CSF conditions (mean ± s.d.). E) Percentage of young TANs (< 2.5 days) in control or anti G-CSF conditions (mean ± s.d.). F) μCT analysis of tumor growth in control or anti G-CSF conditions (mean ± s.d.). G) Schematic representation to summarize the conclusions linking neutrophil lifespan and tumor progression.
Glut1KO neutrophils sensitize lung tumors to radiotherapy
Because neutrophils have been previously linked to immuno- and radiotherapy responses (10,25), we next wondered if Glut1 deficiency in these cells could potentiate such treatments. Despite their reduced expression of PD-L1, Glut1KO neutrophils failed to sensitize KP tumors to anti-PD1 (Figure S7A–B). In contrast, the anti-tumor response to radiotherapy was markedly enhanced, with multiple (62.5%) regressing tumors monitored two weeks after irradiation, and a long-term (at least 28 days) loss of tumor growth specific to Glut1KO TAN-irradiated mice (Figure 7A). Thus, Glut1 deletion in neutrophils increases KP lung tumor sensitivity to radiotherapy, leading to a durable growth impairment.
Figure 7. Glut1 deletion in neutrophils increases radiotherapy efficacy.
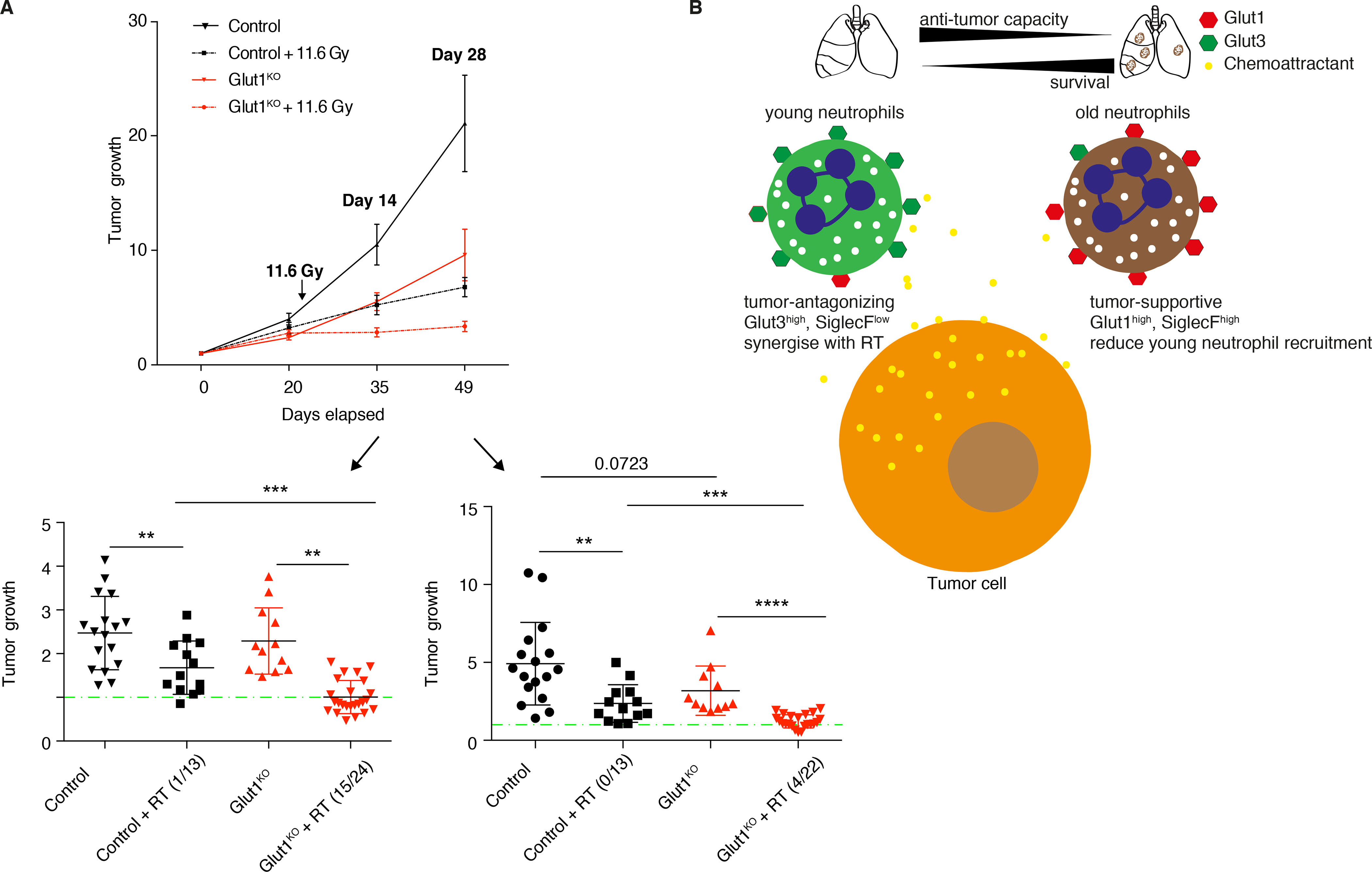
A) μCT analysis of control or Glut1KO tumors before and after 11.6 Gy radiotherapy (RT) or untreated (mean ± s.e.m.), normalized to the first volume (set to 1) of the same tumors. The tumors’ progression or regression are detailed at 14 days (lower left, n= 17, 13, 12 and 24) and 28 days (lower right, n=17, 13, 11 and 22) after radiotherapy (mean ± s.e.m.). The green line indicates a growth of 1 and numbers indicate the number of tumors with a reduced volume. B) Model illustrating the findings from this study. In the tumor, two neutrophil subpopulations exist. The young Glut3high anti-tumor neutrophils are recruited by chemoattractants secreted by the non-immune fraction of the tumor, presumably tumor cells. Within the tumor, neutrophils become dependent on Glut1 to survive and to support on-site aging and differentiation. This extensive survival forms a second subpopulation of neutrophils expressing SiglecF and exhibiting tumor-supportive properties. These old neutrophils limit chemoattractant production by tumor cells, thus inhibiting the recruitment of young neutrophils.
Discussion
In this study, we demonstrated that tumor-derived secreted factors increase neutrophil survival, which is enabled by glucose transporter Glut1. Using a mouse model of lung adenocarcinoma, we identified a reduced neutrophil turnover specifically in the tumor compartment. We found that an important proportion of TANs can survive at least 6.5 days, providing an abnormally high level of old neutrophils when compared to other compartments including the healthy lung.
Within lung tumors, the aberrantly long neutrophil lifespan provides sufficient time to acquire tumor-associated markers such as SiglecF and PD-L1. Whether the acquisition of tumor-supportive properties is passive and linked to the reduced TAN turnover, or active and triggered by specific factors remains to be elucidated. Moreover, in vivo G-CSF treatment or neutralization failed to demonstrate a direct link with the acquisition of SiglecF, whereas its inhibition strongly impacted neutrophil infiltration in the tumor. On the contrary, G-CSF treatment led to a massive recruitment of young neutrophils, accompanied by a reduced tumor development. The impact of this cytokine on young neutrophil recruitment and tumor growth was exacerbated when neutrophils did not express Glut1, reducing the proportion and number of old, SiglecFhigh neutrophils. These results suggest the existence of functionally diverse and competing neutrophils in tumors. A facet of this putative competition was observed when the old SiglecFhigh subpopulation decreased as a consequence of Glut1 loss. When TANs lack Glut1, neutrophil chemoattractants are induced in tumors, which may contribute to an enhanced recruitment of young neutrophils, a response that is accompanied by a reduction of tumor growth. Such a systemic response could resemble one described for TNF-related apoptosis-inducing ligand receptor (TRAIL-R)-mediated apoptosis in myeloid-derived suppressor cells (26); alternatively an increased homing mediated by local chemoattractants might occur upon Glut1 deficiency. The precise mechanisms and the cells responsible in this increased recruitment remain to be elucidated.
Metabolic alterations in the tumor immune compartment have been described in multiple cell types (27). Whereas here we focused on glucose transporters in TANs, another study using a 4T1 mammary tumor cell line transplantation model reported a switch toward oxidative metabolism (28) in cKit positive, immature TANs. Additionally, bone marrow neutrophils from mice with early-stage cancer were characterized by enhanced spontaneous migration compared to neutrophils from tumor-free mice (29). These cells had a higher rate of glycolysis and ATP production compared to control neutrophils, suggesting that some neutrophils are prone to rewire their glucose metabolism directly at the bone marrow stage. Undoubtedly, more work will be needed to characterize and compare how neutrophil subsets vary metabolically and functionally in different tumor types and during tumor evolution.
The accelerated turnover of neutrophils in Glut1KO or under G-CSF treatment was sufficient to reduce tumor growth significantly. However, the alterations of tumor progression were visible only after a few weeks of monitoring, and we did not observe tumor regression. In contrast, the loss of Glut1 augments radiotherapy efficacy, leading to a tumor regression never observed previously with different radiotherapy protocols (this study and (25,30)). Upon radiotherapy, proliferating cancer cells accumulate double-strand breaks and die. The anti-tumor role of neutrophils seems to rely on direct cancer cell killing (31), which might be stimulated after radiotherapy treatment where cells are stressed and exposed to a strong local inflammation. The link between inflammation, anti-tumor neutrophils and radiotherapy treatment needs to be better understood to envisage possible future neutrophil-based strategies against cancer.
In conclusion, our work highlights the importance of enhanced glucose uptake and metabolism for tumor-associated neutrophils, enabling tumor support. Although more experiments will be needed to test this directly, our data favor the hypothesis that the presence of old and possibly pro-tumor neutrophils competes with young anti-tumor neutrophils. We suggest that pro- or anti-tumor properties of neutrophils are a function of their turnover, glucose transporter expression, and differentiation status (Figure 7B), a model that may reconcile earlier studies describing opposite roles for neutrophils in cancer. In such a scenario, therapeutic agents reducing neutrophil survival or increasing young neutrophil recruitment to the tumor may counteract tumor growth when used alone or in combination with conventional anti-cancer treatment modalities such as radiotherapy. However, a limitation of our work is that the data linking TAN survival to tumor support are mainly indirect, and firm proof for this link should be obtained from future investigations.
Finally, our study positions glucose metabolism and particularly Glut1/3-mediated glucose uptake as a crucial node for TAN turnover that may control the equilibrium between pro- and anti-tumor neutrophils in lung cancer.
Supplementary Material
Significance.
Lung tumor support and radiotherapy resistance depend on Glut1-mediated glucose uptake in tumor-associated neutrophils, indicating that metabolic vulnerabilities should be considered to target both tumor cells as well as innate immune cells.
Acknowledgements
PBA, SS, JP, JYP, SP and EM were supported by a grant from the Swiss Cancer Research Foundation (KFS-4555–08-2018). PBA, CC, GB and EM were supported by a grant from the Swiss National Science Foundation (310030_179324). CC, NZ and EM were supported by the Emma Muschamp Foundation. DGK was supported by R35CA197616 from the National Cancer Institute in the United States.
We thank the EPFL SV Flow Cytometry, Gene Expression Core and Histology Core Facilities for help with flow cytometry, RNA sequencing, tissue preparation and multiplexing, respectively. We thank S. Berezowska, I. Zlobec and the Translational Research Unit of the Institute of Pathology (University of Bern, Switzerland) for generously providing the human LUAD samples. We thank D. Constam for providing the B16-F1 cells and G. Diaceri for injecting them (both from EPFL, Switzerland). We thank M. Pittet (MGH, USA) for critically reading the manuscript.
Conflicts of Interest Statement
DGK is a co-founder of Xrad Therapeutics, which is developing radiosensitizers, and serves on the Scientific Advisory Board of Lumicell, which is commercializing intraoperative imaging technology. DGK also receives funding for a clinical trial from a Stand Up To Cancer (SU2C) Catalyst Research Grant with support from Merck. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript. The laboratory of DGK currently receives funding or reagents from Xrad Therapeutics, Merck, Amgen, Bristol-Myers Squibb, Varian Medical Systems, and Calithera, but this did not support the research described in this manuscript. JCR has held stock equity in Sitryx and within the past two years has received unrelated research support, travel, and honorarium from Sitryx, Caribou, Kadmon, Calithera, Tempest, Merck, Mitobridge, and Pfizer. SP has received education grants, provided consultation, attended advisory boards, and/or provided lectures for: Abbvie, Amgen, AstraZeneca, Bayer, Biocartis, Bioinvent, Blueprint Medicines, Boehringer-Ingelheim, Bristol-Myers Squibb, Clovis, Daiichi Sankyo, Debiopharm, Eli Lilly, F. Hoffmann-La Roche, Foundation Medicine, Illumina, Incyte, Janssen, Merck Sharp and Dohme, Merck Serono, Merrimack, Novartis, Pharma Mar, Pfizer, Regeneron, Sanofi, Seattle Genetics and Takeda, from whom she has received honoraria (all fees to institution). The other authors report no conflict of interest.
References
- 1.Rungelrath V, Kobayashi SD, DeLeo FR. Neutrophils in innate immunity and systems biology-level approaches. Wiley Interdiscip Rev Syst Biol Med. 2019;e1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ocana A, Nieto-Jiménez C, Pandiella A, Templeton AJ. Neutrophils in cancer: prognostic role and therapeutic strategies. Mol Cancer. 2017;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eruslanov EB, Singhal S, Albelda SM. Mouse versus human neutrophils in cancer–a major knowledge gap. Trends Cancer. 2017;3:149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, et al. IL17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil (TAN) Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaul ME, Levy L, Sun J, Mishalian I, Singhal S, Kapoor V, et al. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boivin G, Kalambaden P, Faget J, Rusakiewicz S, Montay-Gruel P, Meylan E, et al. Cellular Composition and Contribution of Tertiary Lymphoid Structures to Tumor Immune Infiltration and Modulation by Radiation Therapy. Front Oncol. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faget J, Groeneveld S, Boivin G, Sankar M, Zangger N, Garcia M, et al. Neutrophils and Snail Orchestrate the Establishment of a Pro-tumor Microenvironment in Lung Cancer. Cell Rep. 2017;21:3190–204. [DOI] [PubMed] [Google Scholar]
- 11.Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science. 2017;358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 13.Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front Immunol. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young NP, Crowley D, Jacks T. Uncoupling cancer mutations reveals critical timing of p53 loss in sarcomagenesis. Cancer Res. 2011;71:4040–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee C-L, Moding EJ, Huang X, Li Y, Woodlief LZ, Rodrigues RC, et al. Generation of primary tumors with Flp recombinase in FRT-flanked p53 mice. Dis Model Mech. 2012;5:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, et al. Modulation of Glucose Transporter 1 (GLUT1) Expression Levels Alters Mouse Mammary Tumor Cell Growth In Vitro and In Vivo. PLOS ONE. 2011;6:e23205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasenberg A, Hasenberg M, Männ L, Neumann F, Borkenstein L, Stecher M, et al. Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat Methods. 2015;12:445–52. [DOI] [PubMed] [Google Scholar]
- 18.Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pires BRB, Silva RCMC, Ferreira GM, Abdelhay E. NF-kappaB: Two Sides of the Same Coin. Genes. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfirschke C, Engblom C, Gungabeesoon J, Lin Y, Rickelt S, Zilionis R, et al. Tumor-Promoting Ly-6G+ SiglecFhigh Cells Are Mature and Long-Lived Neutrophils. Cell Rep. 2020;32:108164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vafadarnejad E, Rizzo G, Krampert L, Arampatzi P, Nugroho VA, Schulz D, et al. Time-resolved single-cell transcriptomics uncovers dynamics of cardiac neutrophil diversity in murine myocardial infarction. bioRxiv. 2019;738005. [DOI] [PubMed] [Google Scholar]
- 22.Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity. 2019;50:1317–1334.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu L, Saxena S, Awaji M, Singh RK. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basu S, Hodgson G, Katz M, Dunn AR. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood. 2002;100:854–61. [DOI] [PubMed] [Google Scholar]
- 25.Wisdom AJ, Hong CS, Lin AJ, Xiang Y, Cooper DE, Zhang J, et al. Neutrophils promote tumor resistance to radiation therapy. Proc Natl Acad Sci U S A. 2019;116:18584–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Condamine T, Kumar V, Ramachandran IR, Youn J-I, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R–mediated apoptosis. J Clin Invest. American Society for Clinical Investigation; 2014;124:2626–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassim S, Pouyssegur J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int J Mol Sci. 2019;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun. 2018;9:5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel S, Fu S, Mastio J, Dominguez G, Purohit A, Kossenkov A, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol. 2018;19:1236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torok JA, Oh P, Castle KD, Reinsvold M, Ma Y, Luo L, et al. Deletion of ATM in tumor but not endothelial cells improves radiation response in a primary mouse model of lung adenocarcinoma. Cancer Res. 2018;canres.3103.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masucci MT, Minopoli M, Carriero MV. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front Oncol. 2019;9:1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data have been deposited to the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and assigned the identifier: GSE140159.