

Abstract
Covalent crosslinking and mass spectrometry techniques hold great potential in the study of multiprotein complexes, but a major challenge is the inability to differentiate intra- and inter-protein crosslinks in homomeric complexes. In the current study we use CYP102A1, a well-characterized homodimeric P450, to examine a subtractive method that utilizes limited crosslinking with disuccinimidyl dibutyric urea (DSBU) and isolation of the monomer, in addition to the crosslinked dimer, to identify inter-monomer crosslinks. The utility of this approach was examined with the use of MS-cleavable crosslinker DSBU and recently published cryo-EM based structures of the CYP102A1 homodimer. Of the 31 unique crosslinks found, 26 could be fit to the reported structures whereas 5 exceeded the spatial constraints. Not only did these crosslinks validate the cryo-EM structure, they point to new conformations of CYP102A1 that bring the flavins in closer proximity to the heme.
Keywords: crosslinking, mass spectrometry, CXL-MS, CYP102A1, BM3, protein-protein interactions
INTRODUCTION
Covalent crosslinking and mass spectrometry (CXL-MS) is a widely used method that employs bifunctional chemical crosslinking reagents and facile peptide sequencing by high resolution mass spectrometry to identify specific amino acids that were crosslinked on the protein. This technique can be used to identify protein-protein interactions, to define interaction sites in protein complexes, as well as characterize the structure of proteins based on the crosslinker’s known length. One particular challenge of this method is the inability to distinguish between inter- and intra- protein crosslinks in homomultimeric protein complexes. Lima et al. [1] has noted the prevalence and importance of homodimeric and homomultimeric proteins in biological processes and has developed a technique that utilizes stable-isotope labeling of one of the monomers in a homodimer to directly address this issue. This rigorous method to differentiate the inter- and intra- monomeric crosslinks requires the ability to express isotopically enriched protein, to purify the protein, and to reconstitute the labeled monomer with an unlabeled monomer to form a functional dimer. Unfortunately, the ability to reconstitute the dimer is dependent on the protein of interest. Another approach, albeit less rigorous, is to carefully limit the crosslinking reaction such that both crosslinked dimers and monomers can be separated by denaturing SDS-PAGE, so that the monomer, which contains only intra-monomer crosslinks, can be compared to the dimer, which contains both intra- and inter- monomer crosslinks [2–7]. In this way, the intra-monomer crosslinks can be identified with certainty and essentially be subtracted from the crosslinks found in the dimeric sample, leaving a set of crosslinks for further analysis by orthologous methods. There have been only a handful of studies to employ this method [2–7].
In our study, we have utilized this subtractive CXL-MS method in the course of our structural studies on the homodimeric CYP102A1 enzyme. In particular, we wished to examine how well CXL-MS data would compare to a recently reported cryo-EM-based structural model of the full-length enzyme [8] and to explore how well the subtractive CXL-MS method performed when applied to this well characterized homodimeric P450 enzyme. CYP102A1 is a self-sufficient cytochrome P450 enzyme from Bacillus megaterium that catalyzes the hydroxylation of fatty acids and other molecules. The catalysis involves the transfer of electrons derived from NADPH to the FMN/FAD containing reductase domain of CYP102A1 to the heme in the oxygenase domain. This allows for the sequential one-electron transfer of electrons from the FMN to the heme, a requisite one-electron acceptor, to enable dioxygen activation and insertion of one the oxygen atom into the substrate. The exact mechanism of how these electron transfer reactions occur is a topic of intense interest, especially in light of interest in utilizing CYP102A1 as a biocatalyst.
In the current study disuccinimidyl dibutyric urea (DSBU), a MS-cleavable crosslinker that allows high-confidence identification of crosslinked residues by high resolution mass spectrometry [9,10], was used. The subtractive method successfully identified inter-monomeric crosslinks with residues originating in the oxygenase domain that fit in the cryo-EM derived structures of full length CYP102A1. However, the subtractive method falsely identified crosslinks spanning residues in the reductase domain as inter-monomeric crosslinks, as all crosslinks identified in this domain fit better to the cryo-EM derived structure as intra-monomeric crosslinks. Thus, the success of the subtractive method is likely dependent on the nature of the structural domains and how they are impacted as crosslinking proceeds. Moreover, the crosslinks are consistent with the dynamic movements of the FMN domain of one monomer to the heme domain of another monomer that was proposed for CYP102A1 from the cryo-EM derived structures in open and closed conformations [8]. Thus, the crosslinking studies provide important information in support of the cryo-EM derived structures as well as new evidence for a conformation that brings the FMN closer to the heme, allowing for facile electron transfer and enabling efficient catalysis.
MATERIALS AND METHODS
Materials.
Disuccinimidyl dibutyric urea (DSBU) Lot UG281415 was purchased from Thermo Scientific. Sodium chloride Lot 16620 was from Fisher BioReagents and 1X phosphate-buffered saline pH 7.4 Lot 2052719 was from Gibco. Anhydrous DMSO Lot SHBK9388, 2-mercaptoethanol Lot SHBG9616V, and ammonium bicarbonate Lot 116K0130 were from Sigma. Bio-Safe Coomassie stain Lot BR190408, 2x Laemmli Lot 64224909, and 4–15% gradient SDS-PAGE gels Lot 64294610 were purchased from Bio-Rad.
Protein expression, purification, and characterization.
The full-length A82F variant of CYP102A1 with an N-terminal His6 tag, referred to as full-length CYP102A1, was expressed and purified as previously described [8]. This mutant is the same protein used in elucidating the cryo-EM-derived structural model of the CYP102A1 [8]. The SEC-MALS analysis of full-length CYP102A1 shows a single peak with a mass of 238 ± 8.8 kDa, which is consistent with a homodimer. The full-length preparation displays NADPH consumption at ~1,220 min−1 in the presence of omeprazole [8].
Chemical crosslinking and SDS-PAGE analysis.
Purified full-length CYP102A1was diluted to a final concentration of 10 μM in 10 μl of PBS pH 7.4 supplemented with salt to give a final concentration of 255 mM NaCl. The mixture was treated with the desired amount of DSBU, which was dissolved in anhydrous DMSO, for 5 or 15 minutes at room temperature with rotation. The DMSO was always below 10% (v/v) of total reaction volume. Reactions were quenched with 2 μl 0.5 M ammonium bicarbonate and mixed for 5 minutes at 4°C, then diluted with an equal volume of 2x Laemmli Sample Buffer containing 10% (v/v) 2-mercapotethanol. Samples were boiled for 5 minutes and aliquots (3 μg of protein) were submitted for SDS-PAGE on 4–15% gradient gels typically run for 75 minutes at 50 mAmps/gel. Gels were stained for 1 hour with ProteinSafe Coomassie Stain and destained in MilliQ water. Gels were imaged with a LI-COR Odyssey Fc and bands corresponding to monomer and dimer were quantified by densitometric analysis with the use of ImageStudio software (version 5.2). A standard curve of bovine serum albumin and full-length CYP102A1 showed a linear range from 0 to 6 μg protein per lane.
Mass spectrometry and peptide assignment.
Cross-linked protein samples were separated by SDS-PAGE. Protein bands corresponding to monomeric or dimeric CYP102A1 were submitted for in-gel trypsinolysis and subsequent analysis of the tryptic peptides on a Thermo Scientific Q Exactive HF Orbitrap MS at the University of Michigan Mass Spectrometry-Based Proteomics Resource Facility. Peptide assignments were performed using MeroX (version 2.0) to specifically search for peptides containing the signature doublet that DSBU produces upon fragmentation. MeroX software compares the experimental secondary MS to a library of all theoretically possible DSBU-crosslinked peptides and scores the results based on how well each MS/MS spectrum matches its theoretical counterpart [9,10]. MS datasets were analyzed with primary and secondary fragment mass deviations of 10 and 50 ppm, respectively, with mass limits of 600–6000 Da. Score cut-offs calculated for a False Discovery Rate (FDR) < 0.01% were applied [11,12]. The MS/MS spectra were also manually checked, as another layer of quality control, using MeroX and XCalibur (version 3.0).
Mapping of crosslinks onto three-dimensional models of CYP102A1.
Crosslinks were mapped to recently published structural models of CYP102A1 using the Xlink Analyzer Plugin [13] in UCSF Chimera [14]. These models were derived from cryo-EM data of the same full-length A82F variant CYP102A1 used in our current study [8]. The models are the first full-length structures of the CYP102A1 and utilized the EM density as well as rigid-body fitting of the crystal structures of individual heme, FMN, and FAD domains [4KEW, 1BVY, 4DQK] [15–17]. The structures represent homodimers of CYP102A1 with both heme and FAD domains in contact with each other. At least three major conformations of full-length CYP102A1 were detected representing one closed state where the FAD and FMN are in close contact and two open conformations where the FMN domain is rotated away from the FAD and is closer to the adjacent heme domain of the opposing monomer possibly favoring a trans electron transfer. Input files containing crosslinks were manually generated and 27 Å Cα-Cα Euclidean distance cutoffs for the DSBU linker arm were applied in Xlink Analyzer.
RESULTS
Crosslinking of CYP102A1 with DSBU leads to formation of a covalently linked dimer.
In this study we used an A82F variant of full-length CYP102A1, which is identical to that used in a recently published cryo-EM derived full-length structure of the P450 enzyme [8]. This full-length CYP102A1 is highly similar to the wild-type enzyme with a molecular weight determined by MALS of 238 ± 8.8 kDa, consistent with a homodimer, and is fully functional with NADPH consumption of ~1220 min−1 in the presence of omeprazole [8]. As shown in Fig. 1A, analysis of the full-length CYP102A1 by denaturing SDS-PAGE and Coomassie staining gives rise to a visible band migrating slightly above the 100 kDa marker, corresponding to each monomer (lane 1, M). Treatment of full-length CYP102A1 with 50-fold molar excess of DSBU gave a time-dependent increase in formation of one major crosslinked product, which is visible above the 250 kDa molecular weight marker (lanes 2–8, D). Although it migrates much higher in mass than expected for the native dimeric band, SEC analysis of the reaction mixture shows that the crosslinked product elutes with a highly similar retention time as the native dimer of untreated CYP102A1. Thus, we conclude that the SDS-PAGE does not provide the accurate molecular mass. Furthermore, analysis by a linear sucrose gradient shows that the crosslinked full-length CYP102A1 elutes in the same fractions as the native dimer (data not shown). Thus, we denote this species as the crosslinked dimer (D). As shown in Fig. 1B, the quantification of the monomer and dimer bands by densitometric analysis reveals a clear loss of the monomeric band over time (closed squares) and a concomitant increase in the dimeric band (closed circles). The sum of the densities of the monomer and dimer bands equals the total density of the starting untreated CYP102A1 band indicating that we have accounted for the major products of the crosslinking reaction. The formation of the crosslinked dimer of CYP102A1 is dependent on the concentration of DSBU (Fig. 1C, lanes 1–6). The quantification of the bands once again shows that monomer is converted stoichiometrically to the crosslinked dimer product (Fig. 1D).
Figure 1. Formation of covalently crosslinked CYP102A1 dimer after treatment with DSBU.
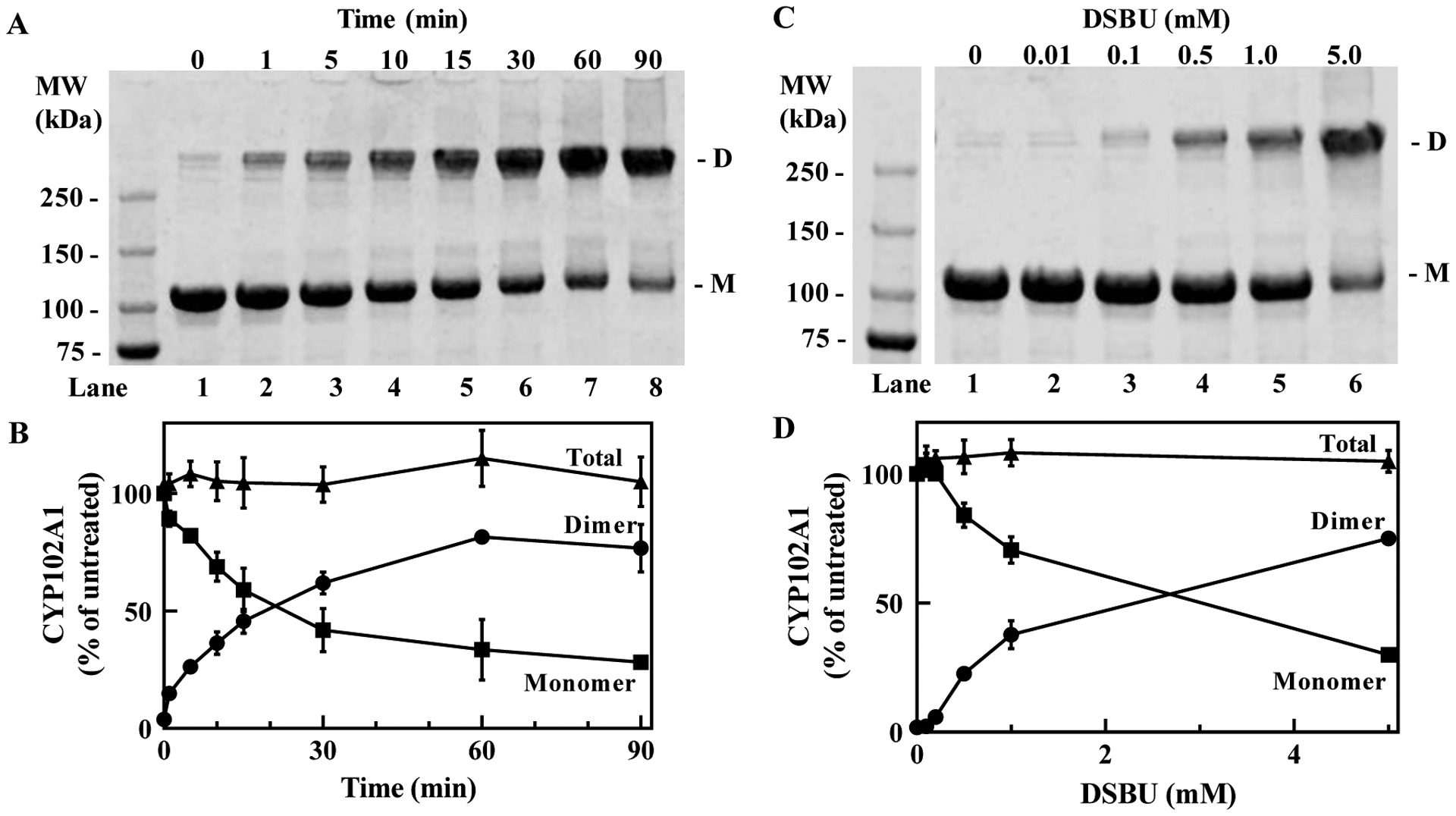
(A), Time-dependent formation of crosslinked CYP102A1 after treatment with DSBU. Full-length CYP102A1 (10 μM) was treated with 0.5 mM DSBU for the indicated duration and aliquots (3 μg) of the reaction mixtures were submitted to SDS-PAGE and stained with Coomassie Blue as described in Methods. D, crosslinked dimeric CYP102A1; M, monomer of CYP102A1. (B), Quantification of bands seen in A. Bands corresponding to the crosslinked dimer (closed circles) and monomer (closed squares) were quantified by densitometric analysis. The sum of the dimer and monomer was also calculated (closed triangle). Mean ± SD derived from three independent reaction mixtures (n=3). (C), Formation of the crosslinked CYP102A1 is dependent on the concentration of DSBU. CYP102A1 was treated with the indicated concentrations of DSBU for 5 min and the reaction mixture analyzed as in A. (D), Quantification of bands seen in C. The amount of dimeric CYP102A1 (closed circles), monomeric CYP102A1 (closed squares), and the sum total (closed triangles) was quantified as in B. Mean ± SD (n=3). Densities determined for all bands are within the linear range of detection.
High-resolution tandem mass spectrometric analysis of the monomer band on SDS-PAGE identifies intra-monomer crosslinked residues in CYP102A1.
Since native CYP102A1 exists as a non-covalently associated homodimer, crosslinking can give rise to either intra-monomer (within one monomer) or inter-monomer (across monomers) covalent crosslinks. We first examined the monomer band from SDS-PAGE gels after DSBU crosslinking as they can only contain intra-monomer crosslinks. We examined samples of full-length CYP102A1 after crosslinking for 5 min or 15 minutes with DSBU at 0.5 mM final concentration. Although we cannot visualize the extent of monomer crosslinking by SDS-PAGE, we do know that approximately 26% and 46% of the starting CYP102A1 was crosslinked to the dimer band after 5 min and 15 min, respectively. Bands were excised in duplicate and submitted for MS analysis, and sites of crosslinks were identified as described in Methods. As shown in Table 1, we found that crosslinking for a short duration gives rise to five major intra-monomer crosslinks with three of the crosslinks starting at residue K77 in the oxygenase domain. Interestingly, K77 exists on the B’-helix located above the substrate binding site of the heme binding pocket of CYP102A1 [18]. As shown in Fig. 2A, when the intra-monomer crosslinks are mapped onto a linear representation of the CYP102A1 monomer, we can readily visualize that four of the crosslinks are within the oxygenase domain with one crosslink in the linker region. At longer durations of crosslinking, we see three of the same crosslinks as in the early time sample but also crosslinks within the FAD domain concentrated around the FAD cofactor binding site (Table 2 and Fig. 2B). We will explore how these crosslinks map to known structures of these regions in the next subsection.
Table 1.
Crosslinked tryptic peptides identified after analysis of the monomer band derived from CYP102A1 treated with DSBU for 5 min
| Site 1 | Site 2 | Peptide 1 | Peptide 2 | m/z | Score |
|---|---|---|---|---|---|
| K10 | K42 | EMPQPKTFGELK | IADELGEIFKFEAPGR | 848.691 | 169 |
| K77 | K188 | NLSQALKFVR | ALDEAMNKLQR | 665.615 | 181 |
| K77 | K203 | NLSQALKFVR | ANPDDPAYDENKR | 959.150 | 87 |
| K77 | K350 | NLSQALKFVR | EDTVLGGEYPLEKGDELMVLIPQLHR | 1081.327 | 138 |
| K469 | K474 | KIPLGGIPSPSTEQSAKK | KAEDAHDTPLLVLYGSNMGTAEGTAR | 954.100 | 21 |
Score values are calculated as indicated in methods.
Figure 2. Schematic representation of the intra-monomer crosslinks found in the monomer band derived from CYP102A1 treated with DSBU for 5 min (A) or 15 min (B).
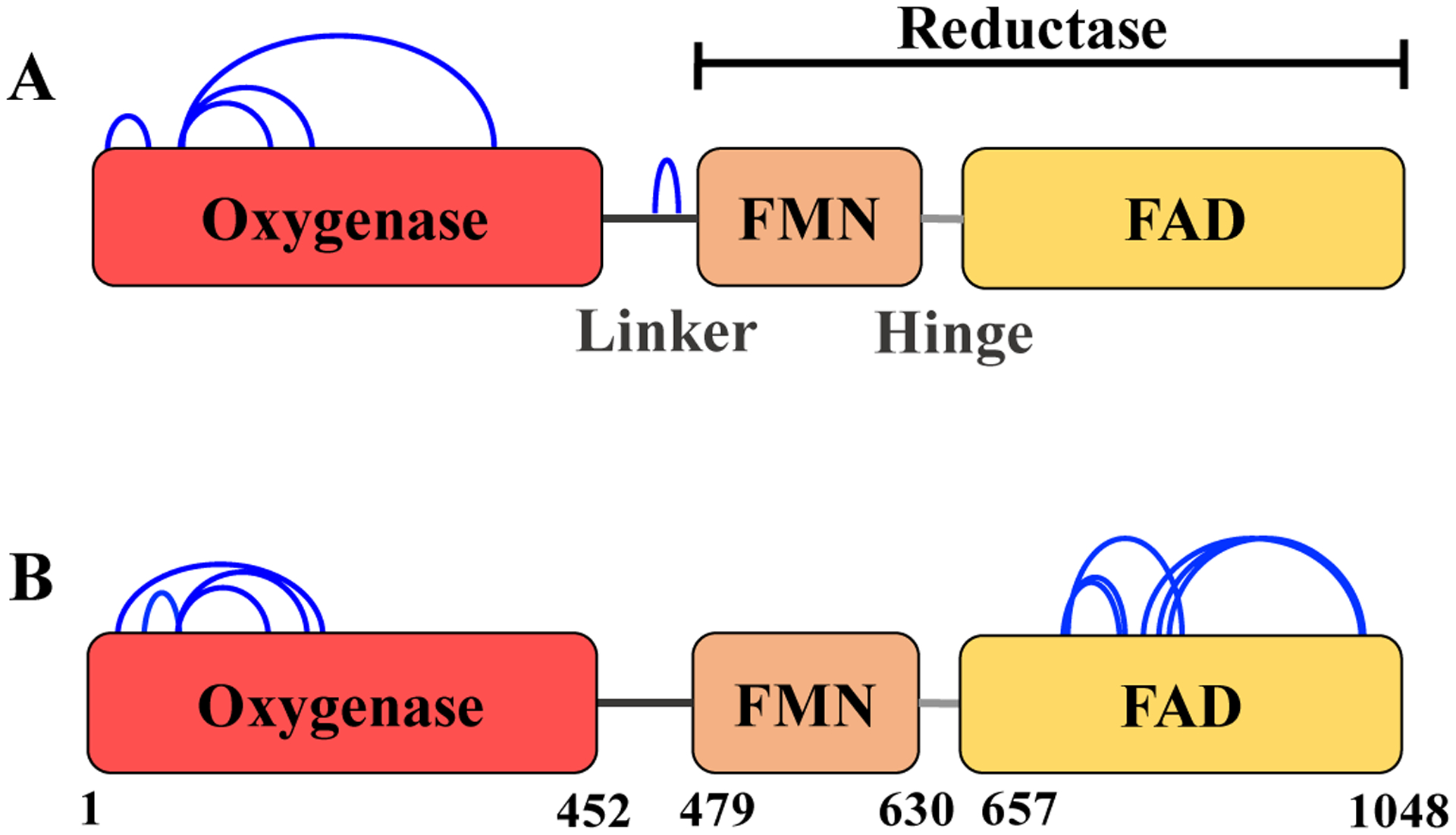
Blue arcs, crosslinks; red, oxygenase domain; orange, FMN domain; yellow, FAD domain. The FMN and FAD domains together represent the reductase domain.
Table 2.
Crosslinked tryptic peptides identified after analysis of the monomer band derived from CYP102A1 treated with DSBU for 15 min
| Site 1 | Site 2 | Peptide 1 | Peptide 2 | m/z | Score |
|---|---|---|---|---|---|
| K25 | K365 | NLPLLNTDKPVQALMKIADELGEIFK | DKTIWGDDVEEFRPER | 1020.331 | 70 |
| K77 | K188 | NLSQALKFVR | ALDEAMNKLQR | 665.615 | 160 |
| K77 | K203 | NLSQALKFVR | ANPDDPAYDENKR | 959.484 | 36 |
| K77 | K350 | NLSQALKFVR | EDTVLGGEYPLEKGDELMVLIPQLHR | 1081.327 | 119 |
| K735 | K771 | LEAEEEKLAHLPLAK | AMAAKTVCPPHK | 640.144 | 183 |
| K735 | T772 | LEAEEEKLAHLPLAK | AMAAKTVCPPHK | 640.143 | 160 |
| K735 | K814 | LEAEEEKLAHLPLAK | YPACEMKFSEFIALLPSIRPR | 883.071 | 45 |
| K787 | K1039 | VELEALLEKQAYK | LWLQQLEEKGR | 1043.572 | 263 |
| K791 | K1039 | QAYKEQVLAK | LWLQQLEEKGR | 693.883 | 171 |
| K797 | K1039 | EQVLAKR | LWLQQLEEKGR | 610.344 | 135 |
Score values are calculated as indicated in methods.
Intra-monomer crosslinks mapped to a Cryo-EM-derived structural model of full-length CYP102A1.
With the exception of one crosslink in the linker region, the intra-monomer crosslinks identified in the previous section (Fig. 2) spanned residues within the oxygenase domain (5 crosslinks) or the FAD domain (6 crosslinks) where high resolution crystal structures exist [15,17].
In Fig. 3, we show the 3D structural model that was constructed by fitting the structure of the oxygenase and reductase domains as rigid bodies into the density map generated from cryo-EM studies [8]. As shown in the upper half of the figure, the five crosslinks identified in the oxygenase domain were mapped onto a crystal structure of the CYP102A1 oxygenase domain as determined by Butler et al. [15] All the crosslinks mapped within the Cα-Cα Euclidean distance of the DSBU linker arm of 27 Å and thus appear in close agreement with the crystal structure. The available crystal structure of the FAD domain determined by Joyce et al. [17] contains residues for only three of the crosslinked adducts we identified. As shown in Fig. 3 lower half, the three crosslinks involving residue K1039 were mapped (red bars) onto structure of the FAD domain and found to have Cα-Cα distances greater than 27 Å, ranging from 39.86 Å (K791-K1039) to 43.24 Å (K787-K1039). Thus, these crosslinks do not fit well to the crystal structure of the FAD domain. The remaining three FAD crosslinks involving K735, which is located on an unresolved loop on the proximal side of the cofactor binding site, as well as the crosslink on the linker region could not be mapped onto the crystal structure and we rely on cryo-EM based modeling of the regions where the crystal structures do not exist [8]. Although the CYP102A1 was found in several different conformations in the cryo-EM studies, there is only one structure where the linker region was resolved. Thus, we used this conformation to examine the remaining intra-monomeric crosslinks (Fig. 3). The cryo-EM derived density maps allowed the modeling of the previously unresolved loop in the FAD domain and we were able to map the three remaining FAD crosslinks from residue K735 within the DSBU distant restraint. Moreover, we also mapped the K469-K474 crosslink within the linker region to well within the DSBU distance restraint. It is noteworthy that with the exception of three crosslinks involving K1039 within the FAD domain, all the intra-monomeric crosslinks are consistent with the full-length cryo-EM derived model of the CYP102A1 structure as well as the available crystal structures of the subdomains.
Figure 3. Structure of the CYP102A1 monomer derived from the cyro-EM model constructed with the use of the crystal structure of the oxygenase and reductase domain, which contains the FMN and FAD binding domains [8].
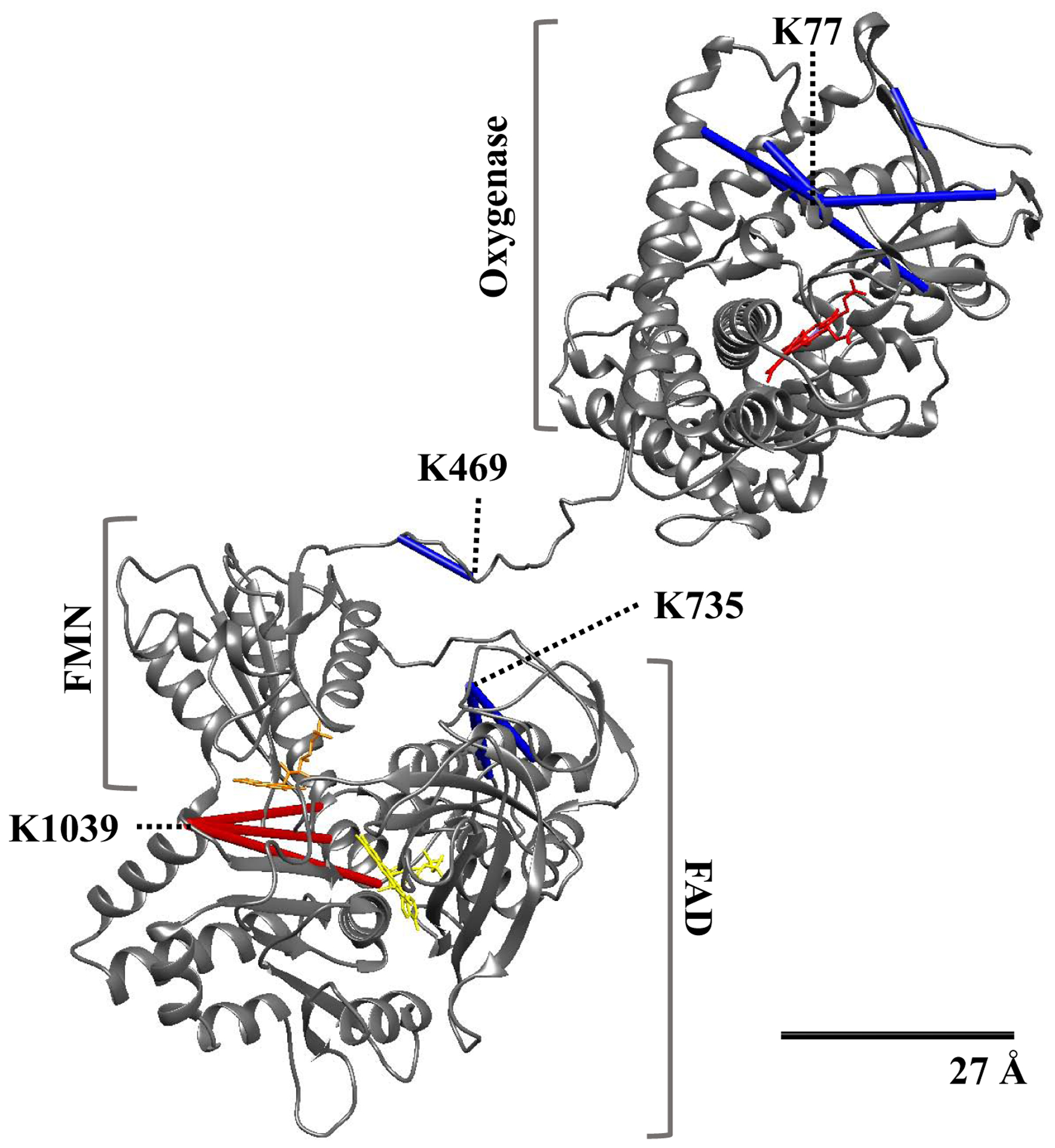
Backbone ribbon model of CYP102A1 is shown in grey and the 27Å scale bar indicates the accepted Cα-Cα distance restraint for the DSBU crosslinker. Blue bars, crosslinks that map within 27 Å; red bars, crosslinks that exceed 27 Å; red, heme prosthetic moiety; orange, FMN; yellow, FAD.
High-resolution tandem mass spectrometric analysis of the dimer band identifies both inter- and intra- monomer crosslinked residues in CYP102A1.
We now focus on the mass spectrometry data derived from the dimer band generated after crosslinking with DSBU. The crosslinks from the dimeric band comprise a mixture of intra- and inter- monomeric crosslinks with those formed at 5 min and 15 min treatments with DSBU shown in Tables 3 and 4, respectively. As a starting point for our analysis, we used an approach used by others that subtracts the crosslinks found in the monomer band, which are intra-monomer crosslinks, from the crosslinks found from the dimer band to give a data set of putative inter-monomer crosslinks [2–7]. Thus, we have greyed out from Tables 3 and 4 the intra-monomer crosslinks identified from the monomer band. We have also verified by use of the cryo-EM derived dimer structure that these intra-monomer crosslinks indeed are better mapped as intra- and not inter- monomer crosslinks (data not shown). Although some studies have utilized the subtractive method to assign the remaining crosslinks as inter-monomer crosslinks, we more carefully examine this assumption by mapping the remaining crosslinks to the cryo-EM derived structures of the CYP102A1 dimer.
Table 3.
Crosslinked tryptic peptides identified after analysis of the dimer band derived from CYP102A1 treated with DSBU for 5 min
| Site 1 | Site 2 | Peptide 1 | Peptide 2 | m/z | Score |
|---|---|---|---|---|---|
| S66 | K1039 | LIKEABDESR | LWLQQLEEKGR | 704.617 | 165 |
| K77 | K188 | NLSQALKFVRR | ALDEAMNKLQR | 665.615 | 175 |
| K290 | K691 | NPHVLQKAAEEAAR | HLEIELPKEASYQEGDHLGVIPR | 872.657 | 44 |
| T577 | K1039 | NWATTYEKVPAFIDETLAAK | LWLQQLEEKGR | 966.515 | 83 |
Table 4.
Crosslinked tryptic peptides identified after analysis of the dimer band derived from CYP102A1 treated with DSBU for 15 min
| Site 1 | Site 2 | Peptide 1 | Peptide 2 | m/z | Score |
|---|---|---|---|---|---|
| K10 | K42 | EMPQPKTFGELK | IADELGEIFKFEAPGR | 1131.249 | 221 |
| K25 | K365 | NLPLLNTDKPVQALMKIADELGEIFK | DKTIWGDDVEEFRPER | 1023.531 | 119 |
| K60 | K561 | LIKEACDESR | QFVDWLDQASADEVKGVR | 1160.237 | 39 |
| S66 | K1039 | LIKEACDESR | LWLQQLEEKGR | 704.616 | 233 |
| K70 | K561 | FDKNLSQALK | QFVDWLDQASADEVKGVR | 856.189 | 136 |
| K77 | K188 | NLSQALKFVR | ALDEAMNKLQR | 665.614 | 159 |
| K77 | K203 | NLSQALKFVR | ANPDDPAYDENKR | 959.151 | 110 |
| K77 | K350 | NLSQALKFVR | EDTVLGGEYPLEKGDELMVLIPQLHR | 1081.32 | 118 |
| K219 | K448 | VMNDLVDKIIADRK | ETLTLKPEGFVVKAK | 697.795 | 169 |
| K310 | K691 | QVKQLK | HLEIELPKEASYQEGDHLGVIPR | 892.985 | 115 |
| K310 | K735 | QVKQLK | LEAEEEKLAHLPLAK | 658.128 | 71 |
| K313 | Y695 | QLKYVGMVLNEALR | HLEIELPKEASYQEGDHLGVIPR | 892.677 | 122 |
| S507 | K791 | DLADIAMSKGFAPQVATLDSHAGNLPR | QAYKEQVLAK | 1042.793 | 30 |
| K508 | K735 | DLADIAMSKGFAPEVATLDSHAGDLPR | LEAEEEKLAHLPLAK | 940.691 | 46 |
| K508 | K778 | DLADIAMSKGFAPEVATLDSHAGDLPR | TVCPPHKVELEALLEK | 974.905 | 25 |
| T516 | K735 | DLADIAMSKGFAPQVATLDSHAGNLPR | LEAEEEKLAHLPLAK | 1171.113 | 144 |
| K561 | K787 | QFVDWLDQASADEVKGVR | VELEALLEKQAYK | 1264.654 | 204 |
| K573 | Y790 | YSVFGCGDKNWATTYQK | VELEALLEKQAYK | 939.219 | 192 |
| K573 | K791 | YSVFGCGDKNWATTYQK | QAYKEQVLAK | 850.169 | 131 |
| K573 | K1039 | YSVFGCGDKNWATTYQK | LWLQQLEEKGR | 905.697 | 171 |
| K691 | K841 | HLEIELPKEASYQEGDHLGVIPR | VDEKQASITVSVVSGEAWSGYGEYK | 1379.439 | 170 |
| K735 | K771 | LEAEEEKLAHLPLAK | AMAAKTVCPPHK | 640.144 | 162 |
| K735 | T772 | LEAEEEKLAHLPLAK | AMAAKTVCPPHK | 640.143 | 175 |
| K791 | K1039 | QAYKEQVLAK | LWLQQLEEKGR | 693.883 | 169 |
| K797 | K1039 | EQVLAKR | LWLQQLEEKGR | 610.344 | 140 |
Analysis of crosslinks obtained from the dimer band with the use of the cryo-EM structural models of the CYP102A1 homodimer.
Our overall approach was to map dimer-specific crosslinks as either intra- or inter- monomer crosslinks onto the three published cryo-EM derived structural models [8] to determine the Cα-Cα Euclidean distance of each crosslink scenario. As shown in Table 5, the crosslinks that were not greyed out from Tables 3 and 4 are listed along with the location of the crosslinks with respect to the domains. A structure of the closed conformation, which was utilized above to map the intra-monomer crosslinks, was used (Closed) in addition to two open conformations (Open I and Open II) representing structures where the FMN domain appears to rotate away from the FAD domain in varying degrees, resulting in its closer proximity to the heme. A simplified model of the CYP102A1 homodimer in these three conformations is shown in Fig. 4. For each crosslink, the Cα-Cα Euclidean distance for each structure mapped as the inter-monomer or intra-monomer crosslink was determined. Since the homodimers are not symmetrical in these conformations, each crosslink can have two inter-monomeric possibilities arbitrarily denoted as α-β and β-α, as well as two intra-monomeric possibilities denoted as α-α and β-β. The distance is depicted in bold type in those cases where the distance is equal to or less than the 27 Å Cα-Cα linker distance.
Table 5. Distances of the crosslinked residues mapped as inter- and intra- monomer crosslinks onto the three available conformations of the full-length CYP102A1 structure.
All crosslinks from Table 3 and 4 that were not greyed out were used. Since the dimer in these conformations is not symmetrical, we arbitrarily assigned one monomer α and the other β. Thus, there are four possible distances for each crosslink in each conformation. Crosslink configurations that mapped to distances within the 27 Å distance restraint for the DSBU linker arm are shown in bold.
| Crosslink Identity | Structural Conformation | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Closed | Open I | Open II | ||||||||||||
| ⍺-β | β-⍺ | ⍺-⍺ | β-β | ⍺-β | β-⍺ | ⍺-⍺ | β-β | ⍺-β | β-⍺ | ⍺-⍺ | β-β | |||
| # | Residues | Domains | C⍺-C⍺ Distance (Å) | |||||||||||
| 1 | K60-K561 | Oxy-FMN | 19.3 | 19.3 | 96.7 | 96.7 | 27.9 | 41.2 | 97.6 | 93.8 | 33.0 | 41.9 | 101.9 | 95.2 |
| 2 | S66-K1039 | Oxy-FAD | 51.8 | 51.3 | 105.1 | 105.1 | 47.4 | 47.1 | 101.8 | 109.4 | 72.3 | 35.5 | 99.8 | 112.4 |
| 3 | K70-K561 | Oxy-FMN | 22.8 | 22.5 | 92.8 | 92.8 | 25.6 | 42.1 | 96.6 | 91.9 | 37.1 | 44.1 | 100.8 | 93.2 |
| 4 | K219-K448 | Oxy-Oxy | 17.9 | 17.9 | 36.5 | 36.5 | 14.3 | 16.4 | 36.5 | 36.5 | 14.0 | 15.4 | 36.5 | 36.5 |
| 5 | K290-K691 | Oxy-FAD | 26.8 | 26.4 | 57.7 | 57.7 | 32.3 | 36.1 | 61.4 | 62.6 | 52.6 | 28.0 | 61.6 | 66.0 |
| 6 | K310-K691 | Oxy-FAD | 31.4 | 31.1 | 49.1 | 49.1 | 32.6 | 37.1 | 52.2 | 54.9 | 50.5 | 31.8 | 53.3 | 57.8 |
| 7 | K310-K735 | Oxy-FAD | 23.3 | 21.4 | 46.8 | 45.1 | 29.0 | 25.8 | 39.0 | 45.8 | 34.2 | 28.0 | 46.8 | 42.4 |
| 8 | K313-Y695 | Oxy-FAD | 28.4 | 28.0 | 64.1 | 64.1 | 31.7 | 33.2 | 64.3 | 68.1 | 52.1 | 25.7 | 65.5 | 69.1 |
| 9 | S507-K791 | FMN-FAD | 79.9 | 79.8 | 27.9 | 27.9 | 79.2 | 76.5 | 22.9 | 27.8 | 78.1 | 75.2 | 22.9 | 27.8 |
| 10 | K508-K735 | FMN-FAD | 58.4 | 59.3 | 20.2 | 20.9 | 59.3 | 53.0 | 23.0 | 21.2 | 57.8 | 51.6 | 23.0 | 21.2 |
| 11 | K508-K778 | FMN-FAD | 78.2 | 78.1 | 31.3 | 31.3 | 80.7 | 74.8 | 24.5 | 31.2 | 79.2 | 73.3 | 24.5 | 31.2 |
| 12 | T516-K735 | FMN-FAD | 57.6 | 59.3 | 25.0 | 27.7 | 69.0 | 56.9 | 36.5 | 30.0 | 67.8 | 56.0 | 36.5 | 30.0 |
| 13 | K561-K787 | FMN-FAD | 97.4 | 97.2 | 35.3 | 35.3 | 96.7 | 93.3 | 16.1 | 34.5 | 95.3 | 92.2 | 16.1 | 34.5 |
| 14 | K573-Y790 | FMN-FAD | 78.3 | 78.3 | 22.3 | 22.3 | 79.3 | 82.8 | 42.4 | 21.8 | 78.0 | 81.7 | 42.4 | 21.8 |
| 15 | K573-K791 | FMN-FAD | 80.5 | 80.5 | 19.0 | 19.0 | 81.8 | 85.0 | 40.8 | 18.4 | 80.7 | 83.9 | 40.8 | 18.4 |
| 16 | K573-K1039 | FMN-FAD | 95.6 | 95.7 | 20.9 | 20.9 | 94.1 | 96.0 | 24.1 | 22.6 | 94.5 | 95.2 | 24.1 | 22.6 |
| 17 | T577-K787 | FMN-FAD | 88.2 | 88.2 | 26.9 | 26.9 | 87.6 | 90.7 | 37.2 | 25.5 | 86.4 | 89.7 | 37.2 | 25.5 |
| 18 | K577-K1039 | FMN-FAD | 100.7 | 100.8 | 17.7 | 17.7 | 99.6 | 100.2 | 20.6 | 21.8 | 99.8 | 99.6 | 20.6 | 21.8 |
| 19 | K691-K841 | FAD-FAD | 60.6 | 60.5 | 7.3 | 7.3 | 59.0 | 59.0 | 7.3 | 7.3 | 59.1 | 59.2 | 7.3 | 7.3 |
Figure 4. Three conformations of the CYP102A1 homodimer identified in Cryo-EM structural model.
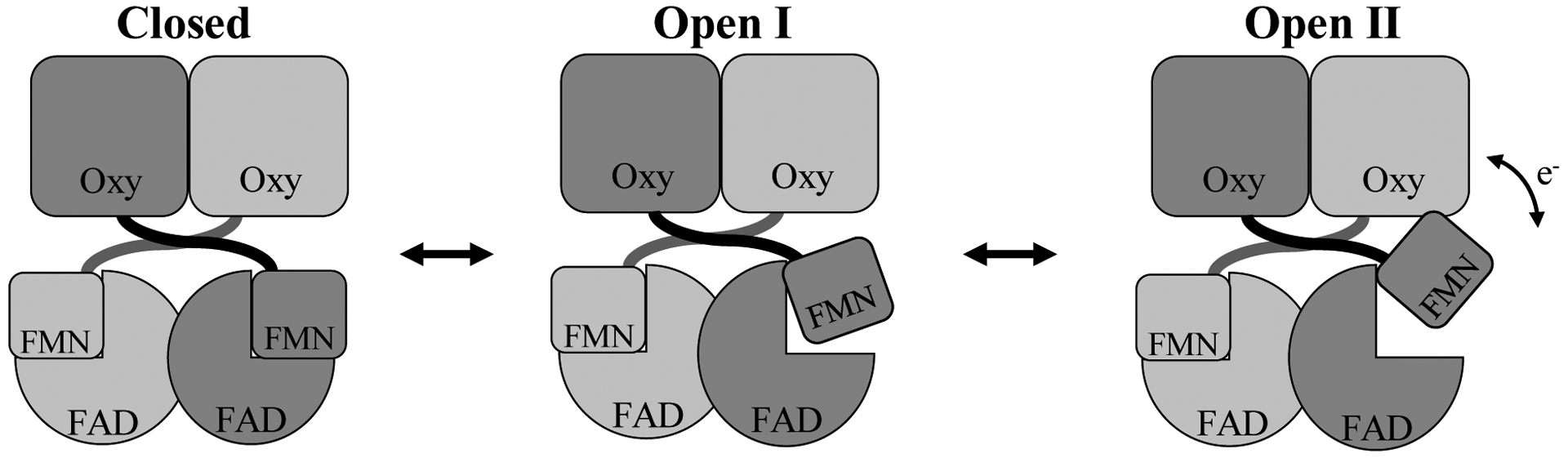
Simplified models of the homodimer in its Closed, Open I, and Open II conformational states are shown that represent conformations captured in recent cryo-EM studies of the enzyme [8].
Oxygenase domain crosslinks (#1–8) –
Six of the eight crosslinks originating in the oxygenase domain (#1,3–5,7,8) could be mapped within the linker distance of 27 Å as inter-monomer crosslinks to at least one of the three conformations, with the closed conformation fulfilling five of the crosslinks. While all conformations could map crosslink #4, interestingly crosslink #8 was best only mapped to the Open II conformation. Two of these oxygenase domain crosslinks (#2,6) did not map to any of the conformations within the linker distance of 27 Å; however, the shortest distances were clearly mapped as the inter-monomer. In fact, all the crosslinks originating in the oxygenase domain were better fit as inter-monomer. Thus, for the mapping of the oxygenase domain contacts, the subtraction method utilizing the crosslinks found in the monomer band was fully validated. It appears that the Closed and Open II conformations captured the extremes of the crosslinks while the Open I conformation was intermediate between these extremes with regard to the crosslinks.
Reductase domain crosslinks (#9–19) –
In sharp contrast to that found for the crosslinks originating from the oxygenase domain, crosslinks entirely within the reductase domain (containing the FMN and FAD subdomains) fit within a linker distance of 27 Å as intra-monomer crosslinks and not inter-monomer crosslinks. Moreover, all the crosslinks found could be fit to at least one conformation. Interestingly, seven of the eleven crosslinks could be fit on what we designated as the β-monomer of all three conformations whereas the α-monomer fit the four remaining crosslinks better.
To better understand the crosslinks in relation to the dynamic nature of the CYP102A1 structure as well as define what monomers were depicted as α and β, we have mapped all of the above crosslinks within the 27 Å linker distance (bolded values from Table 5) onto the Closed and Open II structures since they appear to be the extremes of the conformations with respect to the mapping of the crosslink distances (Fig. 5). Figure 5A shows the inter-monomeric crosslinks on the structure of CYP102A1 in the Closed conformation in all 10 possible combinations between the α-monomer (light grey) and β-monomer (dark grey). There is a tight association of the oxygenase domains containing the heme prosthetic group (red) to form the dimeric structure as well as seemingly looser association with the FMN (orange) and FAD (yellow) containing reductase domains of the other monomer forming a trans-type configuration of the dimer. There are eight crosslinks bridging oxygenase and reductase domains, as well as two crosslinks across the oxygenase domains. As shown in Fig. 5B, the Open II conformation of the CYP102A1 reveals the same two crosslinks that are preserved between the oxygenase domains. However, the open conformation reflects a large conformational change in the reductase domain of α-monomer with the FMN (orange) moving in closer proximity to the heme (red) of the β-monomer. This leads to a loss of the eight crosslinks found in the Closed conformation but a new crosslink between residue Y695 of the FAD domain and residue K313 of the oxygenase domain is able to fit the structure. There are crosslinks between these same residues that are only 28 Å in the Closed conformation so this is likely not specific to the open conformation. However, there is a crosslink between S66-K1039 (Table 5, #2) that is 35.5 Å in distance in the Open II conformation (Fig 5B, red bar) but is much longer (51 Å) in the Closed conformation (not shown). It is possible that crosslink sampled a conformation where these residues are much closer than they appear in the Open II conformation and likely reflects a conformation where the heme and FMN are much closer than captured by the Open II structure. We will examine the crosslinks that could not be assigned within the distance constraint as a group in the Discussion. We also mapped the intra-monomer crosslinks (Table 5, #9–19) to the structure of the closed and open conformations of CYP102A1 in a similar manner. As shown in Fig. 5C, the Closed conformation maps eight of the eleven intra-monomer crosslinks on each monomer. On the β-monomer (dark grey), we can see three main groups of crosslinks at residue K508, centered around residue K573, and one short crosslink at residue K691. Although the reductase domains in the dimer are not symmetrical, we can still observe that these crosslinks are essentially mirrored on the α-monomer (light grey). There is an additional crosslink at residue 516 on the α-monomer and the β-monomer has an analogous crosslink that failed to meet our distance cutoff by only 0.7 Å. In Fig. 5D, the intra-monomer crosslinks were mapped on the Open II conformation. The β-monomer (dark grey) is highly similar to that found for the β-monomer of the Closed conformation and the three sets of crosslinks are present. In contrast, the α-monomer undergoes large conformational changes involving the movement of the FMN prosthetic group (orange) with a notably different pattern of crosslinks present. In fact, all three of the remaining crosslinks at residues S507, K508, and K561 that were not accounted for by the closed conformation are compatible with the open conformations.
Figure 5. Crosslinks mapped to cryo-EM structural model of the CYP102A1 homodimer in Closed (A and C) and Open II (B and C) conformations.
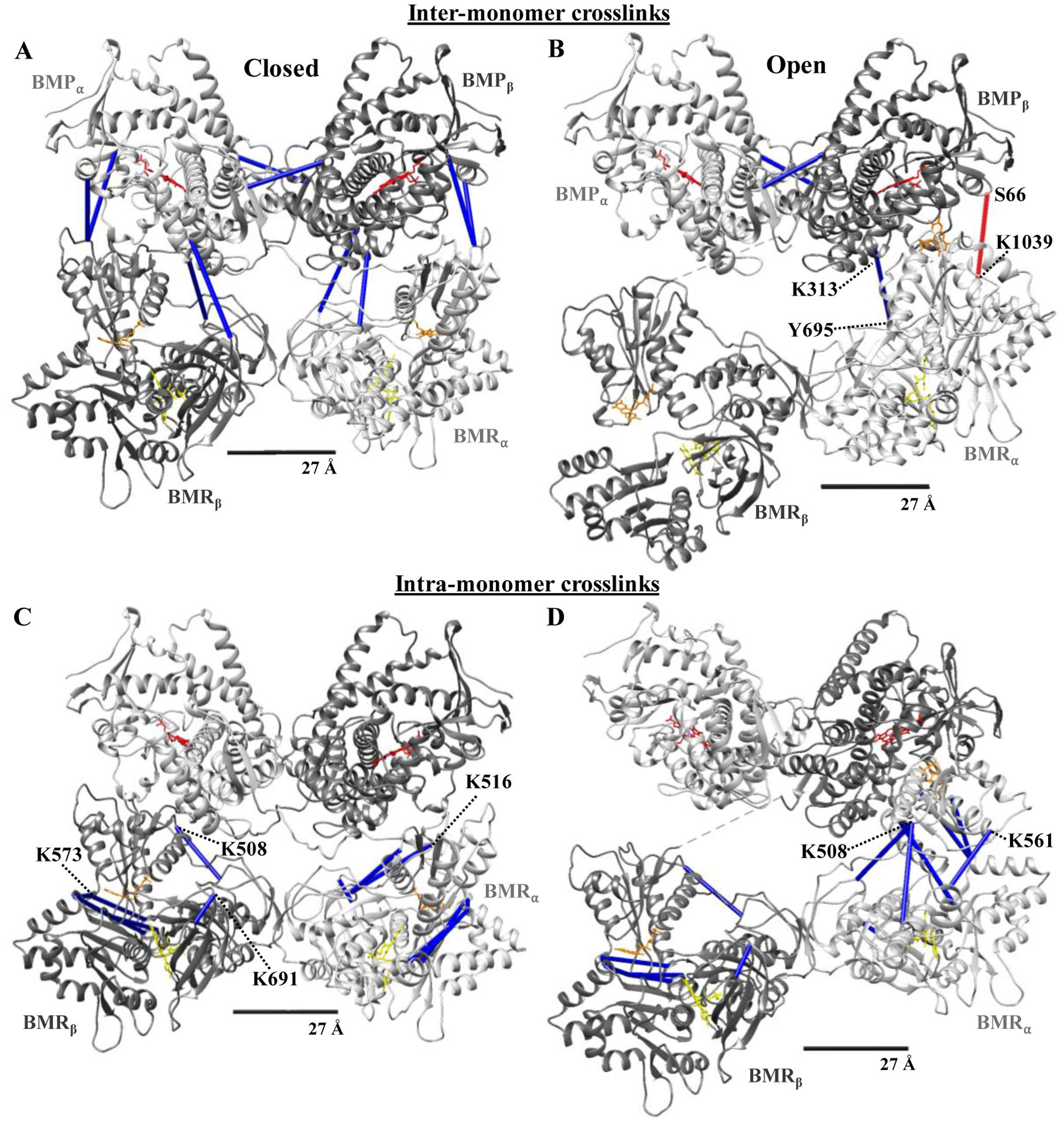
Since the dimer is not symmetrical, we arbitrary labeled one monomer α (light grey) and the other β (dark grey). The heme-containing oxygenase (BMP) and flavin-containing reductase (BMR) domains of each monomer are labeled accordingly. Cofactors and crosslinks are colored as in Figure 3. 27Å scale bar indicates the accepted Cα-Cα distance restraint for the DSBU crosslinker. Crosslinks #1–8 with the exception of #6 from Table 5 are shown in A and B. Crosslinks #9–19 from Table 5 are shown in C and D.
DISCUSSION
In the current study, DSBU was used in CXL-MS experiments to map CYP102A1 residues that are close enough to be spanned by the crosslinker when the CYP102A1 homodimer is in its native state. Although modern mass spectrometry methods can readily identify the crosslinked residues, it is more difficult to determine if the residues are the result of inter- or intra- monomer crosslinks as both monomers have identical amino acid sequences. In lieu of technological challenges of isotopically labeling one monomer, we chose to follow a subtractive method used previously in several CXL-MS studies of homomeric proteins [2–7]. Specifically, we carefully controlled the reaction to obtain a mixture of crosslinked dimers and monomers, which could be separated by SDS-PAGE. The crosslinked monomers were utilized to study intra-monomeric crosslinks, which mapped nicely to known structures of the protein. The crosslinked residues found in the dimer sample comprise inter-monomer as well as intra-monomer crosslinks. This subtractive method worked well for the crosslinks involving at least one residue in the heme-containing oxygenase domain of CYP102A1, as evident by mapping to a recently reported cryo-EM derived structural model of the full-length dimeric protein [8]. The remaining crosslinks, which bridged residues entirely in the reductase domain, could not be mapped as inter-monomeric crosslinks. Even though these crosslinks were not found in the monomer sample data set, they appear to fit more consistently as intra-monomer crosslinks in the cryo-EM structures. This may reflect the inherent conformational flexibility of the reductase domain and its ability to sample different conformations more often after inter-monomer crosslinks are formed that lock the monomers together. Alternatively, perhaps once certain intra-monomeric crosslinks are formed, the CYP102A1 reductase domain can no longer stay in the dimeric state. In either case, we are left with a monomer band that does not give rise to the same intra-monomer crosslinks as the dimer band. Thus, this subtractive method has its limitations and is certainly not as rigorous as labeling one monomer with a stable isotope [1].
Of the 31 total unique crosslinks identified, we successfully mapped 26 to the cryo-EM structure, suggesting a high degree of correspondence between these two methods. However, we could not map five crosslinks within the 27 Å distance restraint of the DSBU linker arm. As shown in Fig. 6, we have mapped these five crosslinks to the residues representing the shortest distance in the Open II conformation of the CYP102A1. Four of these crosslinks involved K1039 crosslinked to either a residue in the oxygenase domain (S66) or to three residues closely clustered on the reductase domain (K787, K791, K797) near the FAD. The distances between these residues vary between 31.8 Å to 43.2 Å in the cryo-EM derived structures. Although the low resolution of the cryo-EM structure precludes definitive statements, it’s possible that conformational flexibility in these regions is reflected by the crosslinking but not apparent in the structural studies. This is noteworthy as electrons donated from NADPH must shuttle from the FAD to FMN to heme for catalytic activity [19–23]. More specifically, a further extension of the Open II conformation of the CYP102A1 homodimer would bring the residue pairs S66-K1039 and K310-K691 closer to each other giving rise to a conformation where perhaps the FAD moves closer to the prosthetic heme in solution. Interestingly, a crystal structure of a truncated CYP102A1 with the oxygenase and part of the reductase domain showed the FMN domain directly in contact with the opposing oxygenase domain [16,24]. The direct interaction of reductase and opposing oxygenase domain is further supported by recent hydrogen-deuterium exchange studies [25]. More recently, a computational modeling study of the interaction of CYP1A1 with cytochrome P450 reductase suggests that transient interactions between heme and FAD domain are likely [26]. Thus, perhaps both FMN and FAD can be closer to the heme during catalysis than is apparent from the current cryo-EM derived structures.
Figure 6. All five crosslinks that exceeded the distance restraint of the DSBU linker arm were mapped to shortest distances in the open II conformation of the CYP102A1 homodimer.
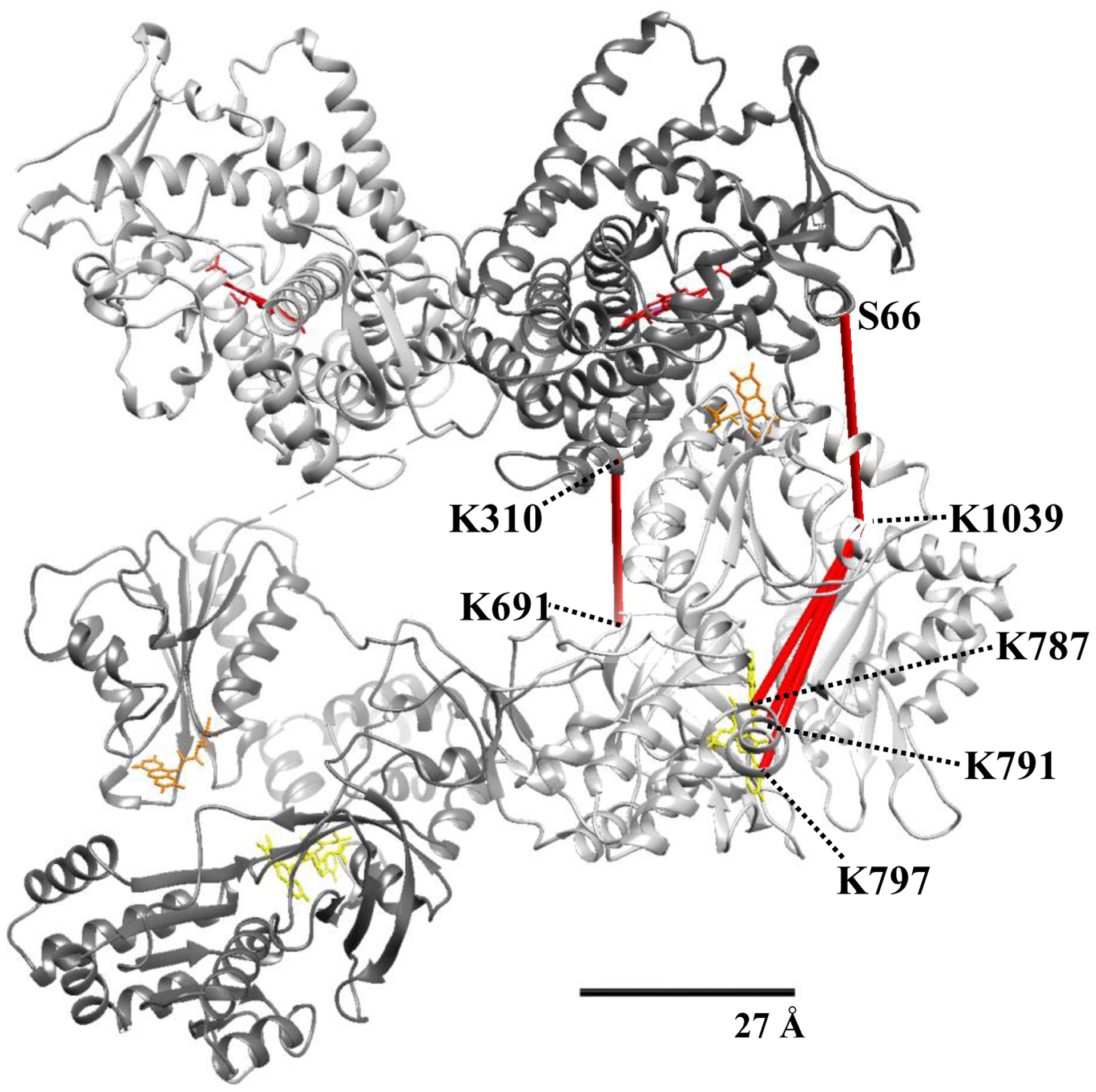
Monomers and cofactors are colored as in Figure 4. 27Å scale bar indicates the accepted Cα-Cα distance restraint for the DSBU crosslinker.
Highlights.
Crosslinking and mass spectrometry complements Cryo-EM structural information
Mapping of protein-protein interaction sites in CYP102A1 by crosslinking and MS
Insights into the application of CXL-MS to homodimeric multiprotein complexes
Acknowledgements
Proteomics Resource Facility, University of Michigan was used to perform mass spectrometry analysis of samples.
Funding
This work was supported in part by National Institutes of Health grants ES007062 (to DF), GM077430, and NS055746, as well as from the University of Michigan’s Protein Folding Disease Initiative.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Lima DB; Melchior JT; Morris J; Barbosa VC; Chamot-Rooke J; Fioramonte M; Souza TACB; Fischer JSG; Gozzo FC; Carvalho PC; Davidson WS Characterization of Homodimer Interfaces with Cross-Linking Mass Spectrometry and Isotopically Labeled Proteins. Nature Protocols 2018, 13 (3), 431–458. 10.1038/nprot.2017.113. [DOI] [PubMed] [Google Scholar]
- 2.Bennett KL; Kussmann M; Björk P; Godzwon M; Mikkelsen M; Sørensen P; Roepstorff P Chemical Cross-Linking with Thiol-Cleavable Reagents Combined with Differential Mass Spectrometric Peptide Mapping--a Novel Approach to Assess Intermolecular Protein Contacts. Protein Science 2000, 9 (8), 1503–1518. 10.1110/ps.9.8.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu B; Peisley A; Richards C; Yao H; Zeng X; Lin C; Chu F; Walz T; Hur S Structural Basis for DsRNA Recognition, Filament Formation, and Antiviral Signal Activation by MDA5. Cell 2013, 152 (1–2), 276–289. 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 4.Zeng-Elmore X; Gao X-Z; Pellarin R; Schneidman-Duhovny D; Zhang X-J; Kozacka KA; Tang Y; Sali A; Chalkley RJ; Cote RH; Chu F Molecular Architecture of Photoreceptor Phosphodiesterase Elucidated by Chemical Cross-Linking and Integrative Modeling. Journal of Molecular Biology 2014, 426 (22), 3713–3728. 10.1016/j.jmb.2014.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karagöz GE; Acosta-Alvear D; Nguyen HT; Lee CP; Chu F; Walter P An Unfolded Protein-Induced Conformational Switch Activates Mammalian IRE1. eLife 6. 10.7554/eLife.30700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Z; Szarecka A; Yonkunas M; Speranskiy K; Kurnikova M; Cascio M Crosslinking Constraints and Computational Models as Complementary Tools in Modeling the Extracellular Domain of the Glycine Receptor. PLoS One 2014, 9 (7). 10.1371/journal.pone.0102571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu F; Thornton DT; Nguyen HT Chemical Cross-Linking in the Structural Analysis of Protein Assemblies. Methods 2018, 144, 53–63. 10.1016/j.ymeth.2018.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su M; Chakraborty S; Osawa Y; Zhang H Cryo-EM Reveals the Architecture of the Dimeric Cytochrome P450 CYP102A1 Enzyme and Conformational Changes Required for Redox Partner Recognition. Journal of Biological Chemistry 2020, 295 (6), 1637–1645. 10.1074/jbc.RA119.011305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Götze M; Pettelkau J; Fritzsche R; Ihling CH; Schäfer M; Sinz A Automated Assignment of MS/MS Cleavable Cross-Links in Protein 3D-Structure Analysis. Journal of the American Society for Mass Spectrometry 2015, 26 (1), 83–97. 10.1007/s13361-014-1001-1. [DOI] [PubMed] [Google Scholar]
- 10.Iacobucci C; Götze M; Ihling CH; Piotrowski C; Arlt C; Schäfer M; Hage C; Schmidt R; Sinz A A Cross-Linking/Mass Spectrometry Workflow Based on MS-Cleavable Cross-Linkers and the MeroX Software for Studying Protein Structures and Protein–Protein Interactions. Nature Protocols 2018, 13 (12), 2864–2889. 10.1038/s41596-018-0068-8. [DOI] [PubMed] [Google Scholar]
- 11.Iacobucci C; Sinz A To Be or Not to Be? Five Guidelines to Avoid Misassignments in Cross-Linking/Mass Spectrometry. Analytical Chemistry 2017, 89 (15), 7832–7835. 10.1021/acs.analchem.7b02316. [DOI] [PubMed] [Google Scholar]
- 12.Iacobucci C; Piotrowski C; Aebersold R; Amaral BC; Andrews P; Bernfur K; Borchers C; Brodie NI; Bruce JE; Cao Y; Chaignepain S; Chavez JD; Claverol S; Cox J; Davis T; Degliesposti G; Dong M-Q; Edinger N; Emanuelsson C; Gay M; Götze M; Gomes-Neto F; Gozzo FC; Gutierrez C; Haupt C; Heck AJR; Herzog F; Huang L; Hoopmann MR; Kalisman N; Klykov O; Kukačka Z; Liu F; MacCoss MJ; Mechtler K; Mesika R; Moritz RL; Nagaraj N; Nesati V; Neves-Ferreira AGC; Ninnis R; Novák P; O’Reilly FJ; Pelzing M; Petrotchenko E; Piersimoni L; Plasencia M; Pukala T; Rand KD; Rappsilber J; Reichmann D; Sailer C; Sarnowski CP; Scheltema RA; Schmidt C; Schriemer DC; Shi Y; Skehel JM; Slavin M; Sobott F; Solis-Mezarino V; Stephanowitz H; Stengel F; Stieger CE; Trabjerg E; Trnka M; Vilaseca M; Viner R; Xiang Y; Yilmaz S; Zelter A; Ziemianowicz D; Leitner A; Sinz A First Community-Wide, Comparative Cross-Linking Mass Spectrometry Study. Analytical Chemistry 2019, 91 (11), 6953–6961. 10.1021/acs.analchem.9b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kosinski J; von Appen A; Ori A; Karius K; Müller CW; Beck M Xlink Analyzer: Software for Analysis and Visualization of Cross-Linking Data in the Context of Three-Dimensional Structures. Journal of Structural Biology 2015, 189 (3), 177–183. 10.1016/j.jsb.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera—A Visualization System for Exploratory Research and Analysis. Journal of Computational Chemistry 2004, 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 15.Butler CF; Peet C; Mason AE; Voice MW; Leys D; Munro AW Key Mutations Alter the Cytochrome P450 BM3 Conformational Landscape and Remove Inherent Substrate Bias. Journal of Biological Chemistry 2013, 288 (35), 25387–25399. 10.1074/jbc.M113.479717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sevrioukova IF; Li H; Zhang H; Peterson JA; Poulos TL Structure of a Cytochrome P450-Redox Partner Electron-Transfer Complex. Proceedings of the National Academy of Sciences 1999, 96 (5), 1863–1868. 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joyce MG; Ekanem IS; Roitel O; Dunford AJ; Neeli R; Girvan HM; Baker GJ; Curtis RA; Munro AW; Leys D The Crystal Structure of the FAD/NADPH-Binding Domain of Flavocytochrome P450 BM3. The FEBS Journal 2012, 279 (9), 1694–1706. 10.1111/j.1742-4658.2012.08544.x. [DOI] [PubMed] [Google Scholar]
- 18.Whitehouse CJC; Bell SG; Wong L-L P450 BM3 (CYP102A1): Connecting the Dots. Chemical Society Reviews 2012, 41 (3), 1218–1260. 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]
- 19.Munro AW; Lindsay JG; Coggins JR; Kelly SM; Price NC Structural and Enzymological Analysis of the Interaction of Isolated Domains of Cytochrome P −450 BM3. FEBS Letters 1994, 343 (1), 70–74. 10.1016/0014-5793(94)80609-8. [DOI] [PubMed] [Google Scholar]
- 20.Munro AW; Daff S; Coggins JR; Lindsay JG; Chapman SK Probing Electron Transfer in Flavocytochrome P-450 BM3 and Its Component Domains. European Journal of Biochemistry 1996, 239 (2), 403–409. 10.1111/j.1432-1033.1996.0403u.x. [DOI] [PubMed] [Google Scholar]
- 21.Munro AW; Leys DG; McLean KJ; Marshall KR; Ost TWB; Daff S; Miles CS; Chapman SK; Lysek DA; Moser CC; Page CC; Dutton PL P450 BM3: The Very Model of a Modern Flavocytochrome. Trends in Biochemical Sciences 2002, 27 (5), 250–257. 10.1016/S0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- 22.Neeli R; Girvan HM; Lawrence A; Warren MJ; Leys D; Scrutton NS; Munro AW The Dimeric Form of Flavocytochrome P450 BM3 Is Catalytically Functional as a Fatty Acid Hydroxylase. FEBS Letters 2005, 579 (25), 5582–5588. 10.1016/j.febslet.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 23.Girvan HM; Dunford AJ; Neeli R; Ekanem IS; Waltham TN; Joyce MG; Leys D; Curtis RA; Williams P; Fisher K; Voice MW; Munro AW Flavocytochrome P450 BM3 Mutant W1046A Is a NADH-Dependent Fatty Acid Hydroxylase: Implications for the Mechanism of Electron Transfer in the P450 BM3 Dimer. Archives of Biochemistry and Biophysics 2011, 507 (1), 75–85. 10.1016/j.abb.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Sevrioukova IF; Hazzard JT; Tollin G; Poulos TL The FMN to Heme Electron Transfer in Cytochrome P450BM-3: Effect of Chemical Modification of Cysteines Engineered at the FMN-Heme Domain Interaction Site. Journal of Biological Chemistry 1999, 274 (51), 36097–36106. 10.1074/jbc.274.51.36097. [DOI] [PubMed] [Google Scholar]
- 25.Jeffreys LN; Pacholarz KJ; Johannissen LO; Girvan HM; Barran PE; Voice MW; Munro AW Characterization of the Structure and Interactions of P450 BM3 Using Hybrid Mass Spectrometry Approaches. Journal of Biological Chemistry 2020, 295 (22), 7595–7607. 10.1074/jbc.RA119.011630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee G; Nandekar PP; Wade RC An Electron Transfer Competent Structural Ensemble of Membrane-Bound Cytochrome P450 1A1 and Cytochrome P450 Oxidoreductase. Communications Biology 2021, 4 (1), 55. 10.1038/s42003-020-01568-y. [DOI] [PMC free article] [PubMed] [Google Scholar]