

Abstract
Extremity skeletal muscle injuries result in substantial disability. Current treatments fail to recoup muscle function, and properly designed and implemented tissue engineering and regenerative medicine techniques can overcome this challenge. In this study, we present a nanoengineered, growth factor eluting bioink that utilizes Laponite® nanoclay for the controlled release of vascular endothelial growth factor (VEGF) and a GelMA hydrogel for a supportive and adhesive scaffold that can be crosslinked in vivo. The bioink is delivered with a partially-automated handheld printer for the in vivo formation of an adhesive and three dimensional (3D) scaffold. The effect of the controlled delivery of VEGF alone or paired with adhesive, supportive, and fibrilar architecture has not been studied in volumetric muscle loss (VML) injuries. Upon direct in vivo printing, our constructs were adherent to skeletal muscle and sustained release of VEGF. The in vivo printing of Muscle Ink in a murine model of volumetric muscle loss (VML) injury promotes functional muscle recovery, reduced fibrosis, and increased anabolic response compared to untreated mice. The in vivo construction of a therapeutic eluting 3D scaffold paves the way for the immediate treatment of a variety of soft tissue traumas.
Keywords: In vivo printing, soft tissue injury, handheld printer, volumetric muscle loss, functional recovery, hydrogel scaffolds
Introduction
Skeletal muscle is required to generate motion, maintain glucose homeostasis, maintain body temperature, and provide support and protection of the skeleton and internal organs.[1] Muscle is one of the few organs which maintains a lifelong capacity for regeneration.[2] However, when a large proportion of mass is lost within a given muscle, as the result of a trauma, disease, or surgical ablation, the native regenerative capabilities of muscle become overwhelmed.[3, 4] Instead, skeletal muscle heals with an overactive fibrotic response to volumetric muscle loss (VML) that results in disabling scar tissue.[5] Such injuries are frequent and impose an economic impact on the order of billions of dollars annually.[6]
One current treatment for VML is functional muscle transfer, which is limited by donor site weakness, morbidity, and incomplete functional restoration.[4, 7] Additionally, surgical intervention is further complicated by the technical difficulty of the operation, requiring an experienced surgical team and appropriate treatment facilities.[7] Because of these limitations, regenerative medicine and tissue engineering have been explored for their vast potential to potentially treat VML injuries. However, most current regenerative strategies fail to recover muscle function in VML injuries due to the severity of the injury, and those attempts that have recovered lost muscle function are well below baseline uninjured levels.[8]
The biochemical environment at the injury site has been modified through the delivery of cells, growth factors, and scaffolding materials.[9, 10, 11] The delivery of cells has shown promise to improve muscle regeneration, but it is limited by the complexity of harvesting myogenic cells and guiding the differentiation of non-myogenic stem cells.[10] Further, the ability to economically expand cell sources to a clinically relevant level has yet to be realized.[10] A variety of delivered growth factors have been able to reduce fibrosis, improve vascularization, and enhance myofiber regeneration.[10, 12] It has been shown that the expression of several important growth factors such as insulin-like growth factor-1 (IGF-1) and vascular endothelial growth factor (VEGF) change following VML injury.[3] VEGF, in particular, is a vital growth factor for inducing angiogenesis and vascularization needed for muscle regeneration, but its localized concentration should be sustained, and its activity should be preserved in vivo.[12, 13] Further, VEGF is upregulated after muscle injury, but local delivery of VEGF above native levels can reduce apoptosis and reduce the injured area.[14]
The implantation of scaffolds with or without growth factors or cells can modify the mechanical properties of the wound.[15] VEGF controlled delivery has shown promise to improve reinnervation and flow perfusion in several ischemic models and recently increased muscle action potential in a craniofacial muscle transplantation.[12, 16] Unfortunately, ischemic injuries do not demonstrate the extreme impairment of regenerative ability compared to the loss of muscle in VML because the muscle matrix remains serving as a template for endogenous regeneration.[8] Further, these models have utilized an ex vivo crosslinked alginate hydrogel that is not adequate for VML injuries because it fails to form complex architectures, fails to adhere to remnant muscle, and fails to support mechanical load.[12, 16] No attempt has been made at understanding the effect of the controlled delivery of VEGF on VML injuries where there is no remaining native structural matrix.[8] Recent advancements in biofabrication and biomanufacturing tools such as three dimensional (3D) bioprinters have enabled the fabrication of scaffolds with biomimetic physical, chemical, and architectural properties.[17] However, most implanted scaffolds, whether injected, sutured, or placed into the wound, fail to adhere to the remnant muscle. Unattached scaffolds prevent the proper interaction between the scaffold and host tissue, which fails to provide adequate support for enduring mechanical loading.[15] While some injectable scaffolds adhere to native tissue and have a continuous interface, those that have shown to adhere to the local tissue fail to replicate the fibrillar organization of lost tissue to guide tissue healing.[18] An emerging strategy to overcome this barrier is to directly print a scaffold in vivo. Such strategies have been used for the treatment of skin,[19] cartilage,[20] and muscle injuries.[7] So far, none of these in situ printers have been successful in creating fibrillar scaffolds capable of controlled release of biological factors for the treatment of VML injuries.[7]
To improve the treatment of soft tissue wounds like VML, we hypothesized that the immediate in situ application of an adhesive VEGF eluting bioink would be an important step towards the treatment of VML injuries by modulating the mechanical and biological wound environment. The objective of our research is to develop a rapid portable handheld printer and growth factor releasing bioink for the local delivery of VEGF in situ to synergistically modulate the wound environment to improve functional muscle recovery post-VML injury. The release of VEGF is controlled by incorporation of Laponite®, which is an artificially manufactured 2D nanoclay disks with a positive edge charge, due to protonation of a hydroxyl group, and a negative surface face distribution, due to exposure of oxygen on the surface.[21, 22] The uneven distribution of charges results in unique micro-organization of the Laponite® disks and results in electrostatic binding to proteins.[21] Laponite® has been utilized in many studies for the delivery of therapeutics and proteins because of its sustained release, biocompatibility, and ability to retain protein functionality after binding.[21–23] Unlike several other nanoscale delivery vehicles, Laponite® degrades into nontoxic ionic products.[21] In this study, the VEGF-bound Laponite® is incorporated into a gelatin methacrylate (GelMA) hydrogel that has been shown to be adhesive to wet tissue, biocompatible, mechanically relevant for soft tissue injuries, and photopolymerizable in vivo. Alginate, the most widely used bioink for VEGF delivery in skeletal muscle injuries, is not suitable for in vivo bioprinting because of its difficulty to crosslink in vivo and its lack of adhesion to the surrounding tissues.[12] For simplicity, the printable nanocomposite hydrogel-based ink is called “Muscle Ink.” The manuscript highlights the development of the handheld bioprinter as well as the analysis of protein-Laponite® binding and release kinetics. Finally, the benefit of in vivo printing of Muscle Ink for the treatment of a murine VML injury model is investigated.
Results and Discussions
The partially automated handheld printer was designed using a top-down approach to enable the formation of 3D scaffolds that replicate the geometry of lost tissue (Figure 1A). The device accepted syringes loaded with photopolymerizable nanoengineered bioinks to balance portability with the capability to fill large voids with printed filament scaffolds (Figure 1Ai–ii). The design offered several desirable features including fine-tuned continuous extrusion control, rapid exchange of bioink syringes, thermal insulation of the incorporated syringes, and embedded ultraviolet (UV) crosslinking system (Figure 1B). The extrusion system transferred the rotational motion of an electric motor into a linear motion to compress the plunger of the inserted syringe. The force was transferred through external threads on the motor shaft to internal threads on a bearing-guided push plate. The extrusion rate of the printer was designed to range from 0 to 10 μL/s, which was adequate to form continuous and uniform fibers of printed hydrogel when the nozzle travel was manually controlled (Figure S1). The preference of manual control of the nozzle travel and rate over traditional, automated means were made to optimize portability and simplicity. A trained user of the printer can form scaffolds that adhere to the injured or debrided tissue in a layer-by-layer fashion to form a 3D architecture that resembles the form of the pre-injured muscle (Figure 1C). The ability to print directly into wounds and reconstruct tissue geometry without the need for medical imaging, generation of a stereolithography file, independent scaffold construction, and surgical implantation of the scaffold yields vast potential for immediately available and clinically valuable treatments for soft tissues in a variety of settings.
Figure 1.

In vivo handheld printing of therapeutic eluting scaffolds. a) Conceptual diagram of organized scaffold deposition directly into a wound using a handheld printer. Schematics of the composite bioink (Muscle Ink) (i) printed into filament scaffold and (ii) its components including VEGF bound to Laponite® nanodisks and supported by a GelMA hydrogel network. b) Callouts of the features and components of the handheld printer. The force to compress the plunger of a syringe that is fixed within the printer housing is transferred via the push plate from the rotational shaft of an electric motor. Photopolymerizable materials are crosslinked through the embedded ultraviolet (UV) and blue light. c) The handheld printer is able to reconstruct the geometry of volumetric muscle loss (top) and adhere to local tissue (bottom) through the designed Muscle Ink composite hydrogel.
Muscle Ink was designed to gradually release VEGF into the injury site due to its key role in inducing angiogenesis and regulating muscle regeneration.[24] To ensure a sustained release of VEGF, Laponite® was utilized to electrostatically bind to VEGF. Individual Laponite® nanodisks are 20–50 nm in diameter and 1–2 nm thick (Figure 2A).[21] To effectively bind a protein to Laponite®, the nanodisks must be exfoliated from their packed state to allow for the diffusion of the interstitial sodium ions that bind the faces of two nanodisks.[21, 23] To model the interactions with Laponite® and biological factors such as VEGF (MW 38.2 kDa), bovine serum albumin (BSA, MW 66 kDa) was used as a model protein as shown elsewhere.[21, 25] The proteins were bound to Laponite® nanoclay disks in an aqueous solution under vigorous mixing. The interaction of Laponite® and BSA was first investigated by comparing the zeta potential of plain Laponite® and Laponite® mixed with BSA for one hour. Zeta potential is a measure of the electrostatic potential difference between the surrounding medium and the local charges that surround the suspended particle; it is a useful tool to analyze colloidal stability, sedimentation capability, and surface charge.[26] The plain Laponite® that was suspended in DPBS measured −26.6 ± 1.1 mV (Figure 2B). With the addition of the BSA, the zeta potential was −4.2 ± 0.3 mV and became more positive than the plain Laponite® (p<0.0001) because the negative surface charges became coated with protein, reducing the electrostatic potential between the DPBS and the nanoparticles.
Figure 2.

Protein interaction and release kinetics from Laponite® in physiological buffers and mechanical characterization of Muscle Ink composite hydrogel. a) Diagram of a single Laponite® nanodisk with a diameter ranging from 20–50 nm and a thickness of 1–2 nm. The uneven distribution of negative face charges and positive edge charges enable the electrostatic binding of proteins. Reproduced with permission (pending).[21] b) The zeta potential of Laponite® becomes less negative after binding with BSA, the model protein. c) Binding efficiency of Laponite® was near 100% binding was achieved at 5 mg/mL. d) Cumulative model protein release of VEGF over a period of 22 days. SEM images of e) 7% (w/v) GelMA and f) Muscle Ink at 5,000× (scale bars 10 μm) and at 20,000× (scale bars 5 μm). There is no difference between the pore size of GelMA and Muscle Ink hydrogels. g) Hydrogel disks were compressed (left) to determine the compressive modulus (right). h) Muscle Ink and GelMA were printed onto skeletal muscle to determine ultimate normal adhesion strength.
To further understand the protein and Laponite® interaction, the effect of the Laponite®-to-protein mass ratio on protein binding was investigated. BSA was kept at a constant concentration of 100 μg/mL and as the concentration of Laponite® was increased, the percentage of the protein bound to Laponite® also increased (Figure 2C). A threshold saturation point was reached at around 5 mg/mL and no significant increase in protein between 5 and 10 mg/mL was observed. To ensure that Laponite® adsorbed all added protein during the binding phase, a concentration of 5 mg/mL was chosen for all future tests.
Next, the release kinetics of proteins bound to Laponite® was investigated. First, various concentrations of BSA-bound Laponite® were left under physiological conditions for 8 days. The results suggest an initial burst release, followed by a gradual release for all concentrations (Figure S2). While the release of a higher portion of the therapeutic during the first 24 h has therapeutic implications to induce physiological processes by delivering greater amounts of protein immediately after injury followed by a period of sustained delivery.[13] The release demonstrated that a higher percentage of the model protein was released for a lower concentration of protein-bound Laponite®, but a higher total mass of protein was released with the higher concentration of protein-bound Laponite® due to the higher mass of adsorbed protein. While the inhibitory concentration is up to ten times higher than other nanodelivery systems, Laponite® can have adverse cellular effects at a concentration above 5 mg/mL.[21] Laponite® was to be incorporated into a gel that would undergo slow in vivo degradation that would prevent high concentrations of Laponite® from being exposed to cells. However, the concentration of Laponite® was maintained at 5 mg/mL for the following experiments to negate any adverse effects while maintaining the highest mass of adsorbed protein. A release was then performed using bound VEGF (Figure 4D). A similar release profile is shown for both proteins. It is noteworthy that the initial burst release was much lower than the burst release observed in some other porous drug carriers.[27] As with the 5 mg/mL BSA-bound Laponite®, the VEGF release demonstrated that a portion of the protein was not released into the supernatant during this study and was retained by the Laponite®. The steady release over a period longer than 3 weeks verified that VEGF could be released over long durations.
Figure 4.
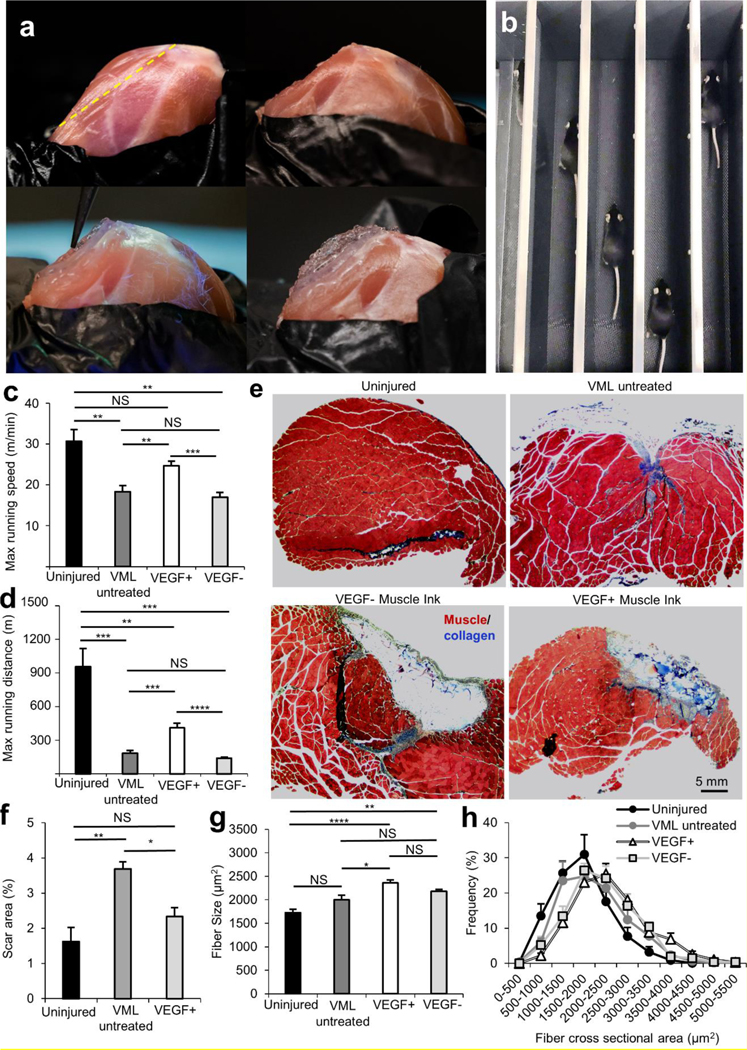
In situ printing and an in vivo murine model demonstrated the feasibility of in vivo printing and functional improvement with the treatment of VEGF+ Muscle Ink. a) In situ printing into an induced VML wound: uninjured quadriceps with a yellow dotted line indicating the location of muscle resection (top left), quadriceps after muscle resection to simulate VML injury (top right), in situ printing of Muscle Ink (bottom left), and reconstructed of Muscle Ink scaffold that replicates the geometry of the native muscle (bottom right). b) The functional performance of the VML animal model after treatment was assessed using a treadmill. Functional treadmill data shows improvement in c) maximum running speed and d) maximum running distance of VEGF+ Muscle Ink treated animals compared to other injured groups. There was no statistical significance in the maximum running speed between VEGF+ Muscle Ink treated group and the uninjured group. e) Trichrome staining was used to investigate fibrosis (blue) formation (scale bar 5 mm). (f) Fibrosis was quantified, and the Muscle Ink VEGF+ treated group showed less fibrosis compared to the VML injured animals. g) Average fiber area (μm2) in quadricep muscles of the uninjured, injured and untreated, injured and treated with printed VEGF- or VEGF+ Muscle Ink scaffolds. h) The distribution of fiber cross-sectional area (μm2) in the tested groups and controls.
The mechanical properties of scaffolds influence the regenerative response of the host during the healing process.[11] The stiffness of an implanted scaffold can affect the migration and differentiation of cells.[28] While stiff materials can limit the tissue function, potentially lead to fibrosis upon their implantation, and stifle myogenic differentiation of migrated cells, the inadequate mechanical strength of scaffolds results in their rupture or detachment. To verify that the mechanical properties of the Muscle Ink provided a suitable mechanical environment, the material and mechanical properties of Muscle Ink were delineated. The implanted constructs must be able to withstand the compressive loads of muscles while maintaining adherence to native tissue. Therefore, a concentration of 7% (w/v) of GelMA was initially chosen for this study based on proper mechanical and biological properties from previous literature.[7]
It has been reported that the incorporation of hard nanoparticles such as Laponite® can change the mechanical properties of hydrogels and polymer networks.[21, 29] Further, Laponite® forms microaggregates in physiological buffers.[21, 30] The effect of Laponite® incorporation on the microstructure of GelMA hydrogels was investigated through SEM imaging. Cross sections of GelMA hydrogels without (Figure 2E) and with the addition of Laponite® (Figure 2F) were imaged at 5,000× and 20,000×. There was no noticeable difference between the pore sizes of either condition at either magnification.
Next, the mechanical properties of the GelMA and Muscle Ink hydrogels were investigated. As the Muscle Ink will be printed in vivo at the relaxed state of skeletal muscle, they will experience compressive forces upon muscle contraction. The compressive modulus and ultimate normal and shear adhesion strengths to skeletal muscle were investigated (Figure 2G–H). The compressive modulus was investigated on hydrogel disks compressed between two plates to ensure that our scaffold could support compressive loads from contractions of remnant muscle while enabling cell migration and differentiation (Figure 2G). The resultant modulus denoted a non-significant increase in the compressive modulus of the Muscle Ink hydrogel (2.3 ± 0.2 kPa) compared to the GelMA hydrogel (1.9 ± 0.4 kPa). None of the constructs failed in compression when tested up to 50% strain and reached up to 4.5 kPa in maximum compressive stress, which is greater than the maximum 40% strain experienced during contraction of most skeletal muscles.[31] Native murine skeletal muscles have an elastic modulus of 12–52 kPa.[32] Our results demonstrate that printed Muscle Ink constructs are less stiff than the skeletal muscle tissue of rodents allowing printed constructs to deform during movement where specific murine muscle forces can exceed 10 kN/m2.[33] Further, the stiffness was within the desired 1–3 kPa range to maximize myogenic differentiation.[28] These results justified the use of 7% GelMA as the hydrogel matrix to support protein-bound Laponite®.
The ultimate adhesion shear stress of the material was measured to determine if the addition of Laponite® to GelMA hydrogels would hamper the ink adhesion to skeletal muscle tissue. GelMA hydrogels alone are capable of crosslinking to tissues once crosslinked directly at the tissue interface.[7] The adhesion shear strength was assessed by printing either GelMA or Muscle Ink between two glass slides and pulling the slides vertically until the material failed in shear (Figure S3). The materials were printed on two different surface conditions to mimic two fundamental situations that would occur in vivo: i) sandwiched between skeletal muscle pieces (SKM-SKM) and ii) attached to skeletal muscle from one side and a 3-(trimethoxysilyl)propyl methacrylate (TMSPMA)-functionalized slide from the other side (SKM-Slide). The SKM-SKM surface conditions replicated the adhesion between the boundaries of the scaffold where the material is surrounded by skeletal muscle tissue, and the SKM-Slide conditions replicated the adhesion between a section of the scaffold where it is only in contact to the tissue from one side. In both conditions, Muscle Ink had lower ultimate adhesion stress (2.1 ± 0.2 kPa for SKM-SKM and 2.9± 0.8 kPa for SKM-Slide) than GelMA alone (3.2 ± 0.6 kPa for SKM-SKM and 3.5 ± 0.6 kPa for SKM-Slide). Even though there are slight decreases in ultimate adhesive shear stress, the differences are subtle. The addition of Laponite® did not hamper adhesion strength to an in vivo VML injury.[7]
The ultimate normal adhesion stress was also analyzed. The ultimate tensile adhesion stress was characterized by printing between two pieces of skeletal muscle that were attached to mounting fixtures (Figure 2H). The Muscle Ink and GelMA hydrogels were pulled until failure or separation occurred. The ultimate normal adhesion stress was lower in Muscle Ink (2.3 ± 0.3 kPa) than in the GelMA hydrogels (3.1 ± 0.3 kPa), but the difference was not statistically significant. The ultimate adhesion stress of GelMA has been shown to be capable of supporting the general loads reported for muscle contraction in a mouse VML injury model, and the addition of Laponite® does not appear to make Muscle Ink statistically less adhesive.[7, 33]
To ensure the lack of cytotoxicity of Muscle Ink, C2C12 myoblasts cultures were interfaced in situ printed scaffolds. The viability of the cells was evaluated using a standard Live/Dead assay on day 1 and day 3 of culture (Figure S4A). Micrographs of the experiment from a fluorescent microscope showed that the majority of cells in both groups (Muscle Ink and tissue culture plate (TCP) control) were alive (green) with few dead cells (red). The images between both groups at each respective time point qualitatively appear similar in morphology as well as the ratio of live to dead cells demonstrating that Muscle Ink did not have an adverse effect on cell growth.
The proliferation rate and viability of cells were quantified using a PrestoBlue™ metabolic assay over 3 days (Figure S4B). The measured fluorescence intensity revealed that the rate of metabolic activity of Muscle Ink exposed cells increased from day 1 to day 3 of culture similar to the rate of the control cells (no significant difference). The results confirmed the Live/Dead assay results and further demonstrated that Muscle Ink did not induce cytotoxicity or any negative impact on cell growth.
To ensure the activity of the encapsulated VEGF within Muscle Ink and its effectiveness in stimulating endothelial cells, two experiments were performed in vitro. A scratch assay was performed to investigate the migration of endothelial cells and a tube formation assay to assess tubulogenesis of endothelial cells.[34, 35] To expose the endothelial cells to Muscle Ink, cell culture inserts were used. The Muscle Ink was crosslinked into the cell culture insert and placed into the cell culture media in both experiments.
A scratch assay was performed to analyze the effect of VEGF release from Muscle Ink on HUVECs migration (Figure 3A). To model the effectiveness of the VEGF release at different exposure times of the VEGF-bound Laponite® to the material, two groups of the Muscle Ink were used. The first Muscle Ink group (fresh) was bound to the Laponite® right before the exposure to the endothelial cells to model the bioink immediately after implantation. The second group (presoaked) was theorized to represent the scaffolds 24 hours after implantation in which VEGF would have time to diffuse from Laponite®, distribute throughout the GelMA hydrogel matrix, and facilitate more VEGF delivery to the cells. All groups showed a partial closure of the scratch over the 4 h period (Figure 3B). The brightfield microscopy images at each time point showed that the relative area change of the Muscle Ink groups was larger than the negative control. At hour 4, the relative scratch closure area revealed that the presoaked group induced more cell migration than the fresh Muscle Ink compared to the negative control (p<0.0001 and p<0.05, respectively) (Figure 3C). The results from the migration of the cells in the scratch assay mirrored the Laponite® long-term protein release.
Figure 3.

VEGF eluting Muscle Ink induced angiogenic potential in HUVECs. a) A schematic representing the scratch assay. (i) A confluent layer of HUVECs was scratched to generate (ii) a gap in the cells. The cells were exposed to (iii) a positive control with VEGF in the media and (iv) VEGF+ Muscle Ink crosslinked into porous cell culture inserts to assess the angiogenic potential of released VEGF. The VEGF+ Muscle Ink was prepared fresh to model scaffolds at the time of implantation or presoaked in DPBS to replicate scaffolds 24 h after implantation. b) Micrographs of the scratch assay to assess the induced migratory effects of VEGF+ Muscle Ink (scale bar 500 μm). c) The relative scratch closure area was quantified over 4 h. d) A tube formation assay was performed to analyze the tubulogenesis of HUVECs that were exposed to VEGF+ Muscle Ink (scale bar 1 mm). e) The number of closed-loop structures shows the networking ability of the tube formation assay. f) Total tube length demonstrates the effect of eluted VEGF on tubulogenesis.
To analyze endothelial activation and tubulogenesis, a process that is vital to the formation of vasculature in vivo, a tube formation assay was performed under similar conditions to the endothelial scratch assay.[35] Similar to the scratch assay experiment, the Muscle Ink was interfaced with the cells using culture inserts in two groups: a presoaked group (presoaked) and a freshly bound group (fresh). An optimized number of HUVECs were seeded in multiwell-plates coated with Matrigel®. The cells were imaged and analyzed over time until the maximum number of tubes were formed in the positive control group. Overall, the HUVECs increased their interaction with each other until they formed tubes in all groups. However, the micrographs from the negative control showed a trend of incomplete and fewer tubes that formed a less extensive network than some of the other groups (Figure 3D). On the other hand, the capillary networks formed in test groups and the positive control were more developed than the negative control group possessing similar tube lengths, densities, and complexity. Furthermore, in some locations, the formed networks for the presoaked group appeared to be more complex.
ImageJ software was used to further quantify and compare the tube formation parameters. The tube networks were analyzed for the number of tubes, average tube length, number of formed loop networks, and total tube length.[36] The total number of tubes was similar among all the groups with the highest number in the presoaked and positive control groups, but the only statistically significant difference was between the positive and negative controls (p<0.01) (Figure S5A). Although the number of tubes was similar between groups, the tubes appeared to be less networked and developed in the negative control. The average tube length showed that the Muscle Ink groups, particularly the fresh group (p<0.01), and the positive control had longer average tube lengths compared to the negative control (Figure S5B). Also, the Muscle Ink fresh and presoaked groups and the positive control all had a significantly higher frequency of closed-loop formations in comparison to the negative control (p<0.05, p<0.05, and p<0.01, respectively) (Figure 3E). To find a balanced quantitative measure between tube number and average length, the total length of all measured tubes was compared (Figure 3F). The presoaked Muscle Ink yielded a higher tube formation length than the fresh Muscle Ink group and had a statistically significant difference with the negative control (p<0.001). The results from the tube formation show that the Muscle Ink is capable of inducing activation and networking of vascular endothelial cells.
To evaluate the practicality of the printing process, a murine model of VML injury was utilized and the handheld printer was used for in vivo and in situ printing of a scaffold directly within the defect site. Animals underwent VML injury to the quadriceps as previously described, controlling for muscle mass resected (Figure 4A).[3, 37] The exposed quadriceps muscle is shown with a yellow line to denote the approximate location of the longitudinal incision. The void from the resected muscle was filled using the handheld pen printer. Individual filaments were printed using the handheld printer and each layer was printed while the UV crosslinking system was activated. The printed 3D scaffold matched the topology of the rough wound interface and adhered to the bulk remnant quadriceps, while the fibrous scaffold architecture mimicking the structure of skeletal muscles was preserved.
The impact of in vivo printing of Muscle Ink with and without VEGF in the treatment of VML injuries was investigated in the same murine model. Animals were divided into four groups: (1) uninjured sham, (2) VML injury with no treatment, (3) VML injury treated with Muscle Ink hydrogels loaded with VEGF (VEGF+), and (4) VML injuries treated with Muscle Ink without VEGF (VEGF-). VML induced mice underwent bilateral resection of their hind limb quadriceps. For the treatment groups, VEGF+ or VEGF- Muscle Ink hydrogels were printed into both of the VML injury defects using the handheld printer to ensure that the same treatment was given to each leg. Bilateral surgery and treatment were completed to enable functional performance assessment. The printed constructs recreated the native volume of lost muscle using longitudinally formed filaments.
Eight weeks after injury, the animals were exercised on a treadmill to assess the function of all groups (Figure 4B). The animals were tested for maximum running speed and maximum running distance (Figure 4C–D). To analyze the function of hind-limb musculature, the animals were placed on a treadmill and the speed was incrementally increased.[38] The maximum speed reached before the animal failed to remain on the track was recorded (Figure 4C). All of the VML induced groups had lower maximum speeds than the uninjured mice, but the difference between the VEGF+ Muscle Ink and the uninjured group was not significant. The nonsignificant difference between the running speed of the VEGF+ treated group and uninjured group is noteworthy because of the difficulty in recovering function in VML models close to baseline values due to the extreme loss of native regenerative components.[8] The VEGF+ Muscle Ink group had the highest maximum speed of all of the injured groups. The increased maximum speed can be attributed to the release of VEGF from the Laponite® within the GelMA hydrogels because the Muscle Ink that was VEGF negative had no significant functional improvement over the VML untreated group.
Similar to the maximum running speed, the maximum distance each animal could reach before exhaustion was recorded as another measure of exercise capacity (Figure 4D). [38] Results revealed that the uninjured animals had the highest maximum running distances in comparison to other groups as expected. While there were significant deficits in all injured animals, the addition of VEGF was shown to have functional improvement. The mice that received VEGF+ Muscle Ink could run about twice as far as either the VML untreated group or VML VEGF- Muscle Ink treated group. There was again no significant difference between VML untreated animals and those that received the VEGF- Muscle Ink. Therefore, it seems that the slow release of VEGF is responsible for the improvement in functional performance. The improvement in muscle function may be due to a hypertrophic response to VML injury and a limit in fibrosis.
After eight weeks, the muscle was harvested for histological analysis. Hematoxylin and eosin (H&E) staining was performed on cross sections of harvested quadriceps to visualize the cellular nuclei (blue) as well as the sarcoplasm and ECM (pink) (Figure S6A). The stable interface between the hydrogel scaffold and the muscle injury showed long-term adhesion of the Muscle Ink to the wound site (Figure S6B). The adhesion after eight weeks confirmed that the biomaterial is suitable to withstand the forces experienced during treadmill exercising. Both of the printed Muscle Inks (VEGF+ and VEGF-) showed cellular infiltration as evidenced by the presence of nuclei in the hydrogel.
To investigate the effect of released VEGF from Muscle Ink on vascularization in the bulk remnant tissue, cross sections of the quadriceps were analyzed through immunohistochemical staining. The cross sections were stained with CD31 (green) and laminin (red) to mark the presence of endothelial cells of the vasculature and the sarcolemma, respectively (Fig. S7A). The staining of the sarcolemma allowed for the identification of individual muscle fibers under fluorescent imaging. The number of CD31 positive capillaries in the bulk remnant muscle was quantified and showed that the average number of blood vessels was higher in all three of the injured groups compared to the uninjured group (Fig. S7B). The increase correlates with the upregulation of VEGF after VML.[14] However, the VEGF+ Muscle Ink group was the only statistically different increase in the number of CD31+ capillaries (p = 0.0013). The increase in capillaries can be attributed to the released VEGF from the scaffold for this increase is not shown in the scaffold only group.
To investigate the effect of Muscle Ink scaffolds and sustained VEGF release on fibrosis, the cross sections were trichrome stained (Figure 4E and S8). In each image, the muscle tissue fibers are shown in red and the collagen, the main component of fibrotic scar tissue, is shown in blue. The wound or scaffold interface is displayed as separate from the bulk remnant muscle by a yellow dotted line (Figure S8). The percentage of scar area was quantified for the uninjured, VML untreated, and VML treated with VEGF+ Muscle Ink groups. The VEGF+ Muscle Ink group displayed less fibrosis than the VML-only group (p = 0.0395). The reduction in fibrosis in the VEGF+ Muscle Ink group demonstrates that the gain in functional performance is not due to functional fibrosis. The decrease in fibrosis compared to VML untreated animals demonstrates the supportive nature of the Muscle Ink scaffold, and a reduction in fibrosis has also been attributed to mechanically adequate scaffolds for the treatment of VML.[15] Further, fibrosis is a sign of wound inflammation that usually results in further muscle degeneration and prevents proper myogenic regeneration.[39] The treatment of VML with in vivo printing of a Muscle Ink scaffold reduced fibrosis without the use of antifibrotic agents, such as Losartan that have been employed by other groups.[39]
Previous studies utilizing our pen printer model, without the use of Muscle Ink, suggested that placement of a scaffold may promote an improved hypertrophic muscle response following VML injury.[7] The muscle fiber size in the native and remnant quadriceps muscle was compared within the four groups: healthy control (no injury), VML injury without treatment (negative control), VML injury treated with an in situ printed Muscle Ink (VEGF-), and VML injury treated with in situ printed Muscle Ink and VEGF (VEGF+) (Figure 4G–H). Following VML injury, remnant muscle fibers undergo hypertrophy, a correlate for muscle strength, to compensate for lost muscle.[7, 40] Because no injury occurred in the uninjured animals, they had the smallest central fiber area (1725 ± 79 μm). The central fiber area was greater in the injured animals compared to the uninjured animals, but the only statistically significant difference between the uninjured animals was the VEGF+ and VEGF- Muscle Ink group treated groups (p < 0.0001 and p = 0.0084, respectively). The VEGF+ Muscle Ink contributed to the greatest anabolic response with an average fiber size 18% larger than (2360 ± 61 μm) and a statistically significant difference (p = 0.022) compared to untreated VML animals (2000 ± 97.7 μm). The VEGF+ Muscle Ink had an average 8% greater fiber cross sectional area than the VEGF- Muscle Ink (2177 ± 47 μm) demonstrating the benefit of the controlled release of VEGF. The anabolic response was greater in the VEGF+ Muscle Ink compared to other treatments in the same VML model such as collagen glycosaminoglycan scaffolds.[37] The improved anabolic response correlates with improved functional performance.
Conclusions
The present study demonstrated a feasible approach for the treatment of VML injuries. In this approach, Muscle Ink made of GelMA and VEGF-eluting Laponite® particles was directly printed into the injury site using a custom-built handheld bioprinter. The slow release of growth factors over days could modulate the biological and mechanical environment of the injury site resulting in improved functional performance and increased CD31+ capillaries, reduced fibrosis, and increased hypertrophy in the remnant muscle. This methodology allows for the immediate application of the therapeutic hydrogel-based bioinks via a handheld printer without the detrimental delay of reconstructive surgery or current regenerative medical approaches.[41] Treatment of injuries with VEGF Muscle Ink scaffold is theorized to improve functional recovery through the suppression of fibrosis and improved anabolic response. While the current research was utilized in a VML injury for the delivery of skeletal muscle, it could be applied for the treatment of other soft tissue wounds. Such a rapid and portable deployment of in situ delivery and reconstruction has serious implications in several settings, specifically military trauma care. The partially automated handheld printer continues to broaden the potential of in vivo delivery of hydrogel scaffolds for clinically relevant treatment.
Experimental Section
GelMA Synthesis
GelMA was synthesized following previously described methods.[42] First, porcine skin gelatin type A (Sigma-Aldrich) was added at 10% (w/v) to Dulbecco’s Phosphate-Buffered Saline (DPBS) (Thermo Fisher Scientific) and dissolved at 50 °C using a magnetic mixer at 200 rpm medium degree of methacrylation was obtained by adding methacrylic anhydride (Sigma-Aldrich) dropwise at 1.25% (v/v). The solution was reacted at 50 °C in the dark for 1 h. The reaction was terminated by a 5x dilution of DPBS at 40 °C. To remove unnecessary salts and excess methacrylic anhydride, the solution was dialyzed against deionized water (diH2O) using a 12 – 14 kDa cutoff dialysis membrane (Thermo Fisher Scientific). The deionized water was maintained at a volume ratio greater than 10:1 to the reacted solution, and the water was exchanged every 12 h for one week. The solution was vacuum filtered using 40 Ashless filter paper (Whatman®, GE Healthcare) and aliquoted. The aliquots were frozen at −20 °C. The frozen GelMA was lyophilized using a FreeZone® benchtop freeze dryer (Labconco®, Kansas City, MO) for one week and stored at 4 °C.
Nanoparticle Preparation
Laponite® was sterilized using a dry autoclave cycle and mixed for 15 min in ethanol on a vortex mixer at 3000 rpm. The mixture was centrifuged at 2000 g for 5 min. The ethanol was aspirated in a biosafety cabinet to leave the settled Laponite®. The Laponite® was washed three times in DPBS by stirring for 5 min, centrifuging, and aspirated as mentioned for the ethanol wash. During use, Laponite® was suspended in DPBS and mixed using a vortex mixer for 1 min at 3000 rpm.
Composite Hydrogel Preparation
Lyophilized GelMA was dissolved in DPBS at 37 °C at 7% (w/v) with LAP photoinitiator (PI) (Allevi, Philadelphia, PA) at 0.06875% (w/v). The GelMA-PI solution was mixed using a vortex mixer. For sterile tests, the PI and GelMA solutions were filtered separately through 0.2 μm syringe filters (Whatman®, GE Healthcare) before their combination. The GelMA-PI was then added to isolated Laponite®. The Laponite®-GelMA solution was mixed via a vortex mixer at 3000 rpm for 1 min to ensure homogeneous mixing.
Protein Binding Studies
Laponite® was diluted from a stock solution of 100 mg/mL into various concentrations (final concentrations of 100, 500, 1,000, 2,000, 5,000, and 10,000 μg/mL with the addition of the protein solution) (n = 4). Dilutions were achieved by vortexing the stock solution to ensure homogeneity, pipetting the desired amount from the middle of the solution, and placing the Laponite® solution into a 2 mL microcentrifuge tube with the appropriate volume of DPBS. Bovine Serum Albumin (BSA) from Pierce™ standard ampules (Thermo Fisher Scientific) was added at 100 μg/mL to each Laponite® solution and bound for 1 h. At the end of the 1 h, the solution was centrifuged for 5 min and the supernatant was collected. The supernatant was analyzed for protein concentration using a Micro BCA™ protein assay kit (Thermo Fisher Scientific) with a Cytation 5 imaging reader (BioTek, Winooski, VT). The concentration of bound protein was determined by taking the difference between initial protein and unbound protein in the supernatant.
Zeta Potential Measurements
Laponite® was mixed into DPBS at 5000 μg/mL. The Laponite® was either left unbound or was bound to BSA protein at 100 μg/mL. The samples were mixed, and the zeta potential was measured using a dynamic light scattering analyzer (Zetasizer Nano ZS90, Malvern Instruments Ltd, UK) (n = 3).
In Vitro Assessment of Protein Release Kinetics
The sustained release of protein from Laponite® was first analyzed using BSA as a model protein. BSA at 100 μg/mL was bound to Laponite® at 5000 μg/mL (n = 4). The protein-bound Laponite® was diluted at various concentrations to assess the effect of nanoclay concentration. For each collection time point, the protein-bound Laponite® was isolated by centrifugation, half of the supernatant was collected, and the collected volume was replaced with fresh DPBS. Samples were incubated at 37 °C between each collection. Supernatants were analyzed for protein concentration by Micro BCA™ protein assay kit with a Cytation 5 imaging reader.
The sustained release of VEGF from Laponite® was similarly assessed. VEGF (68–8784-82, Invitrogen) at 750 ng/mL was bound to Laponite® at 5000 μg/mL (n = 3). The same binding and sample collection methods were completed as described. The concentration of VEGF was identified in the collected sample using a VEGF ELISA kit (Ab222510, Abcam®) as per manufacturer-recommended protocol and the signals were measured using a Cytation 5 imaging reader.
Composite Hydrogel Printing
GelMA-Laponite® composite hydrogels, hereby referred to as Muscle Ink, were loaded into 3 mL syringes. The syringes were stored at 4 °C. The syringes were placed at room temperature for at least 10 min before printing and were left at room temperature for the remainder of the printing process. The syringes were placed into the loading chamber of the handheld printer. The material was extruded out of the syringe through a blunt 22 G taper-tipped needle at a rate controlled by the operator. Printing constructs were created in continuous parallel stacked lines. Each layer of the scaffold was crosslinked for 20 sec; for in situ printing, the UV light was operated during the print and for 20 sec on each successive layer.
Scanning Electron Microscopy (SEM)
GelMA hydrogel and GelMA-composite hydrogel were printed onto glass slides (1 cm × 1 cm). SEM preparation was accomplished as stated elsewhere.[43] The prints were crosslinked for 20 sec and immediately placed in liquid nitrogen. The swollen, frozen hydrogels were then lyophilized for at least 24 h. After lyophilization, the samples were broken to expose their cross sections to enable visualization of internal microstructures. The sections were coated with palladium using a sputter coater (Cressington 106 Auto Sputter 655 Coater, UK). Images of the samples were captured on high vacuum using an SEM (FEI Quanta 200 Environmental, FEI Company, Hillsboro, OR) at 10 kV.
Mechanical Characterization of Hydrogel
Mechanical characterization was performed as previously described.[7] Compression testing of the GelMA hydrogels was performed using molded hydrogels. Polydimethylsiloxane (PDMS) molds were used to cast the bulk GelMA (n = 4) or Muscle Ink hydrogels (n = 5). The molds were filled, topped with a 3-(trimethoxysilyl)propyl methacrylate (TMSPMA)-functionalized slide to allow for adhesion to a supportive substrate to ease removal from the mold, and crosslinked from a distance of 45 mm through the slide for 20 sec. After crosslinking, the molded hydrogel disks that measured 4.5 mm thick and 10 mm in diameter were placed between compression mounting attachments of an Electroforce 3220 (TA Instruments, New Castle, DE) mechanical tester. The samples were compressed at a rate of 0.0166 mm/s. The compressive modulus was calculated by the linear slope of the stress-strain curve for the first 10% of the strain.
Adhesion Strength Assessment
Shear Adhesion Testing.
The ultimate shear adhesion strength of the GelMA-composite and GelMA hydrogels were assessed by modifying the ASTM F2255–05 standard.[44] Two different surface conditions were analyzed: between skeletal muscle tissue and TMSPMA slides (SKM-Slide) or between two pieces of skeletal muscle tissue (SKM-SKM). The TMSPMA slides were cut into 10 mm × 35 mm rectangles. For skeletal tissue, freshly harvested skeletal porcine tissue was cut into rectangular samples (10 mm × 5 mm × 1 mm) and glued to a glass slide. Hydrogel constructs were printed in rectangular samples (10 mm × 5 mm × 2 mm) on the skeletal muscle, the other surface condition was placed in contact with the printed structures, and the hydrogel was crosslinked for 20 sec. The tests were conducted using the Electroforce 3220 mechanical tester at a shear rate of 0.0166 mm/s until the samples detached or ruptured (n = 6).
Normal Adhesion Testing.
The ultimate normal tensile adhesion strength of the GelMA-composite and GelMA hydrogels were assessed by modifying the ASTM F2255–05 standard.[45] Both hydrogels were tested between skeletal muscle. Fresh porcine skeletal muscle was cut into squares (10 mm × 10 mm × 1 mm) and glued to the surface of a 3D printed adhesion interface tool. The hydrogels were printed between two pieces of the skeletal muscle and immediately tested until detachment or rupture on the Electroforce 3220 mechanical tester (n = 6).
In Vitro Biological Characterization
Live/Dead Viability Assay.
Immortalized mouse myoblasts (C2C12) (Sigma-Aldrich) were used to assess material cytotoxicity. The C2C12s were cultured and expanded in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific) supplemented with 10% (v/v) fetal bovine serum (FBS) (Thermo Fisher Scientific) and 1% (v/v) penicillin-streptomycin (Thermo Fisher Scientific). Cells were passaged once they reached 80–85% confluency. For the experiment, cells were detached from the tissue culture plate (TCP) using trypsin-EDTA (0.1%) (Sigma-Aldrich). Cells were counted and resuspended in the growth medium and added to a 24-well TCP at 50,000 cells/ well. The total volume used in each well was 1 mL.
Muscle Ink was prepared as previously mentioned. Enough of the Muscle Ink (200 μL) was added to cover the bottom surface of TCP inserts (Greiner Bio-One, Monroe, NC) with pores of 0.4 μm. The Muscle Ink was crosslinked as previously described. The hydrogel inserts were added to one group of the TCPs immediately after cell seeding. The Laponite®-GelMA hydrogel groups with the cell culture inserts were compared against a control with no treatment.
Cell viability was evaluated using Live/Dead Viability/Cytotoxicity Kit (Invitrogen) on day 1 and day 3. A master mix was mixed using calcein-AM at 0.5 μL/mL and ethidium homodimer at 2 μL/mL in DPBS. The C2C12 cultured plate media was aspirated and the cells were washed with DPBS. The culture plate was then incubated with 180 μL of the master mix per well at 37 °C for 15 min. After the incubation period, the master mix was removed, the cells were washed once with DBPS, and the wells were filled with DPBS. The cells were immediately imaged using a Zeiss Axio Observer fluorescent microscope (Zeiss) where the live cells appear green and the dead cells appear red (n = 3).
Presto Blue Metabolic Assay.
Using the same culturing and seeding methodology for the Live/Dead assay, viability and proliferation were quantified of day 1 and day 3 cells using a Prestoblue™ Cell Viability Assay (Invitrogen) (n = 3). At each time point, the cells were incubated for 1 h at 37 °C with the assay reagent at10% (v/v) in growth medium. After the incubation period, 100 μL of the medium was collected and analyzed for fluorescent intensity at 540 nm excitation and 600 nm emission using a Cytation 5 imaging reader.
Scratch Assay.
A scratch assay was performed to analyze the effect of VEGF release from Muscle Ink. Human umbilical vein endothelial cells (HUVECs)were grown in an EGM™ endothelial cell growth medium (Lonza). Passage 5 HUVECs were detached using trypsin-EDTA (0.1%), resuspended in the growth medium, and seeded at 30,000 cells/well into a 12-well TCP that was coated with a thin layer of Matrigel® (Corning®). The growth medium was changed every 2 days and the cells were allowed to become confluent.
Laponite® at 5000 μg/mL was loaded with human VEGF at 750 ng/mL as previously described. The VEGF-loaded Laponite® was then encapsulated in 7% (w/v) GelMA. The prepolymer hydrogel was pipetted in 200 μL volumes into 0.8 μm pore 12-well TCP inserts (Corning®) and crosslinked for 20 sec.
A scratch was created at hour 0 in the confluent HUVECs by lightly dragging a P200 pipette tip across the bottom of the well plate (n = 4). The media was changed to remove cell fragments and to apply all treatment groups: 1) positive control- growth medium supplemented with 30 ng/mL of human VEGF, 2) negative control- growth medium without supplementation, 3) VEGF-loaded Muscle Ink crosslinked at hour 0, and 4) crosslinked Muscle Ink presoaked in DPBS for 24 h. The growth medium was added into the inserts to prevent evaporation. Four replicates were used per group and were imaged at various time points to compare the scratch closure relative to the initial scratch area. Images were captured utilizing the phase contrast mode on an inverted Zeiss Axio Observer fluorescent microscope. Scratch closure area was analyzed using ImageJ (NIH, Bethesda, MD).
Tube Formation Assay.
HUVECs were cultured as described previously. HUVECs were seeded at 66,666 cells/mL of a 24-well TCP that was coated with Matrigel®. Immediately after the cells were seeded, the same four testing groups as the scratch assay were exposed to the cells. The only difference in preparation was the use of 24-well cell culture inserts with 0.4 μm pores (Griener) and the volumes were scaled to fit the 24-well TCPs. After 12 h, the plates were imaged every hour. At hour 16, the positive control showed the most tubes of any timepoint and was used as the time point for quantification. Images were taken using a Zeiss Axio Observer fluorescent microscope. The number of tubes, average number of junctions, and tube length were quantified at this time point for each test condition (n = 3). The center of the wells was analyzed because this was the location where the surface was the flattest of the Matrigel® coating. The uneven surface at the edges was due to the formation of a meniscus during solidification and affected the formation of tubes.
Animal Study
Animal studies were performed at Brigham and Women’s Hospital in accordance with Protocol 2016N000375 and approved by the Institutional Animal Care and Use Committee (IACUC). Twenty-four 11-week old C57/B16 mice (The Jackson Laboratory, Bar Harbor, ME) were anesthetized using 2–4% isoflurane vapor in an induction chamber. The hair from the hind legs was removed using Nair Hair Removal Lotion (Church & Dwight), and the legs were disinfected using Benzoin Compound Tincture USP (Humco Holding Group). In VML induced animals, both quadriceps were exposed by a longitudinal cut in the skin of the hind legs. Six animals (n = 6) for each treatment were used in four treatment groups: 1) sham (uninjured), 2) VML injury untreated, 3) VML injury with VEGF-loaded Muscle Ink scaffold printed from the handheld printer, 4) VML injury with blank Muscle Ink scaffold printed from the handheld printer. VML injuries were created through muscle resection as elsewhere described.[3, 37] If treated, both legs were treated. Constructs were printed as described earlier. Resected muscle mass was serially performed, if needed, to reach a total resection volume of 80 mg. This represents approximately 50% of the native quadricep volume.
Treadmill Muscle Performance Testing.
Functional assessment was performed using a 6-lane mouse treadmill (model no. CL-4, Omnitech, Columbus, OH), as previously described.[37] Mice were acclimatized for three days a week for seven weeks prior to functional testing by placing the mice on the static treadmill for 10 min with just the shock pad on. On week 7, the running speed was set to 10 m per min. Running time was 10 min on day 1, 20 min on day 3 and 30 min on day 5 of that week. On week 8, mice were placed on the treadmill which was set to a 38° incline and tested for maximum speed. The treadmill was started at 8 m/min and the speed was increased by 2 m/min every min. Maximum speed was defined as the speed at which the mouse was no longer able to run on the treadmill for five consecutive seconds with the shock pad activated. To measure endurance, animal maximal running endurance was evaluated two days later. The treadmill was started at 10 m/min at a constant incline of 10° and the speed was increased by 2 m/min every min. Maximum endurance was defined as the distance traveled at which the mouse was no longer able to run on the treadmill for five consecutive seconds with the shock pad activated. Activity remained ad lib in all mice throughout the time course.
Histological Analyses
Hematoxylin and Eosin (H&E) Staining.
The quadriceps were harvested 8 weeks post-surgery. Tissues were prepared for histological analysis by fixing in a 10% neutral-buffered formalin for up to 40 h. Tissues were then stored in 70% ethanol before sectioning. The tissue was embedded in paraffin, cut into sections, and stained with H&E according to a standard protocol.
Trichrome Staining for Fibrosis.
The preserved sections were also stained by trichrome staining (Ab150686, Abcam) to visualize the presence of fibrosis within the muscle, where the bulk muscle is stained red and collagen blue. A standard protocol was followed. Briefly, de-paraffinized sections were placed in Bouin’s fluid at 60 degrees for 60 min, followed by incubation in Weigert’s iron hematoxylin for 5 min, and then in Biebrich scarlet-acid fuchsin solution for 15 min, with each step followed by rinsing in water. The sections were differentiated in a phosphotungstic acid solution for 10 min and incubated in acetic acid solution (1%) for 5 min. The sections were then dehydrated in 95% alcohol and imaged at 10x and 4x using a Nikon Eclipse E400 light microscope (Nikon Corporation). The percentage of fibrotic scar tissue was quantified using ImageJ (NIH, Bethesda, MD).
Vascularization Characterization.
Preserved sections were stained for cluster of differentiation 31 (CD31) as an endothelial marker and laminin to denote the muscle fiber. After de-paraffinization and heat-induced antigen retrieval in a 10 mM citrate buffer (C2488, Sigma-Aldrich), muscle tissue sections were blocked with 5% BSA and 5% goat serum. The sections were then incubated with rat anti-CD31 (ab56299, Abcam®) and rat anti-laminin (ab11575, Abcam®) antibodies for 2 days at 4 °C. After washing with PBS, the sections were incubated with goat anti-rat IgG Alexa Fluor 488 (A-11006, Thermo Fisher Scientific) and goat anti-rabbit IgG Alexa Fluor 594 (A-11037, Thermo Fisher Scientific) secondary antibodies for 1 h at room temperature. The sections were mounted with Aquamount (194905, Thermo Fisher Scientific) before visualizing with Olympus BX53 microscope (Olympus UCMAD3, T7, Japan). The sections were imaged so that CD31 appeared green and laminin appeared red. The average number of identified blood vessels in the bulk remnant muscle was quantified.
Statistical Analysis
Displayed data are reported as mean ± standard deviation. GraphPad Prism 8.0 software (San Diego, CA) was used to perform statistical analyses. Column analyses were conducted using two-tailed Student’s t-test. Grouped analyses were completed using analyses of variance (ANOVA) testing. Significant ANOVA results underwent Tukey’s multiple comparison post-hoc testing. Values of p<0.05 were considered statistically significant. p<0.05 denoted by *, p<0.01 denoted by **, p<0.001 denoted by ***, and a p<0.0001 denoted by ****.
Supplementary Material
Acknowledgements
Manufacturing and characterization analysis were performed at the NanoEngineering Research Core Facility (NERCF), University of Nebraska-Lincoln. Use of Zetasizer for zeta potential analysis was performed at Pannier Lab, University of Nebraska-Lincoln. Pen printer photography was taken by Jaideep Sahni. The financial support from the National Institutes of Health (GM126831, AR073822) and Stepping Strong Innovator Award are gratefully acknowledged.
Contributor Information
Mr. Jacob P. Quint, Department of Mechanical and Materials Engineering, University of Nebraska, Lincoln, Lincoln, NE, 68588, USA Department of Biomedical Engineering, University of Connecticut, Farmington, CT 06030, USA.
Ms. Azadeh Mostafavi, Department of Mechanical and Materials Engineering, University of Nebraska, Lincoln, Lincoln, NE, 68588, USA
Dr. Yori Endo, Division of Plastic Surgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, 02115, USA
Dr. Adriana Panayi, Division of Plastic Surgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, 02115, USA
Ms. Carina S. Russell, Department of Mechanical and Materials Engineering, University of Nebraska, Lincoln, Lincoln, NE, 68588, USA
Ms. Atousa Nourmahnad, Division of Plastic Surgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, 02115, USA
Mr. Chris Wiseman, Department of Mechanical and Materials Engineering, University of Nebraska, Lincoln, Lincoln, NE, 68588, USA
Ms. Laleh Abbasi, Department of Mechanical and Materials Engineering, University of Nebraska, Lincoln, Lincoln, NE, 68588, USA
Dr. Mohamadmahdi Samandari, Department of Biomedical Engineering, University of Connecticut, Farmington, CT 06030, USA
Prof. Amir Sheikhi, Department of Chemical Engineering, Department of Biomedical Engineering, The Pennsylvania State University, University Park, PA 16802, USA
Dr. Kristo Nuutila, Division of Plastic Surgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, 02115, USA
Prof. Indranil Sinha, Division of Plastic Surgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, 02115, USA.
Prof. Ali Tamayol, Department of Mechanical and Materials Engineering, University of Nebraska, Lincoln, Lincoln, NE, 68588, USA; Department of Biomedical Engineering, University of Connecticut, Farmington, CT 06030, USA.
References
- [1].Cezar CA, Roche ET, Vandenburgh HH, Duda GN, Walsh CJ, Mooney DJ, Proceedings of the National Academy of Sciences 2016, 113, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Almada AE, Wagers AJ, Nat Rev Mol Cell Biol 2016, 17, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nuutila K, Sakthivel D, Kruse C, Tran P, Giatsidis G, Sinha I, Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2017, 25, 408. [DOI] [PubMed] [Google Scholar]
- [4].Grogan BF, Hsu JR, The Journal of the American Academy of Orthopaedic Surgeons 2011, 19 Suppl 1, S35. [DOI] [PubMed] [Google Scholar]
- [5].Garg K, Corona BT, Walters TJ, Front Pharmacol 2015, 6, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Corona BT, Henderson BEP, Ward CL, Greising SM, Physiol Rep 2017, 5, e13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Russell CS, Mostafavi A, Quint JP, Panayi AC, Baldino K, Williams TJ, Daubendiek JG, Hugo Sánchez V, Bonick Z, Trujillo-Miranda M, Shin SR, Pourquie O, Salehi S, Sinha I, Tamayol A, ACS Applied Bio Materials 2020, 3, 1568. [DOI] [PubMed] [Google Scholar]
- [8].Grasman JM, Zayas MJ, Page RL, Pins GD, Acta Biomater 2015, 25, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sicari BM, Rubin JP, Dearth CL, Wolf MT, Ambrosio F, Boninger M, Turner NJ, Weber DJ, Simpson TW, Wyse A, Brown EH, Dziki JL, Fisher LE, Brown S, Badylak SF, Sci Transl Med 2014, 6, 234ra58; [DOI] [PMC free article] [PubMed] [Google Scholar]; Jahromi S. Hamidian, Davies JE, Stem Cells Transl Med 2019, 8, 456; [DOI] [PMC free article] [PubMed] [Google Scholar]; Borselli C, Cezar CA, Shvartsman D, Vandenburgh HH, Mooney DJ, Biomaterials 2011, 32, 8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Qazi TH, Duda GN, Ort MJ, Perka C, Geissler S, Winkler T, J Cachexia Sarcopenia Muscle 2019, 10, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Qazi TH, Mooney DJ, Pumberger M, Geißler S, Duda GN, Biomaterials 2015, 53, 502. [DOI] [PubMed] [Google Scholar]
- [12].Borselli C, Storrie H, Benesch-Lee F, Shvartsman D, Cezar C, Lichtman JW, Vandenburgh HH, Mooney DJ, Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ozawa CR, Banfi A, Glazer NL, Thurston G, Springer ML, Kraft PE, McDonald DM, Blau HM, The Journal of clinical investigation 2004, 113, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Deasy BM, Feduska JM, Payne TR, Li Y, Ambrosio F, Huard J, Molecular therapy : the journal of the American Society of Gene Therapy 2009, 17, 1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kwee BJ, Mooney DJ, Current Opinion in Biotechnology 2017, 47, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Raimondo TM, Li H, Kwee BJ, Kinsley S, Budina E, Anderson EM, Doherty EJ, Talbot SG, Mooney DJ, Biomaterials 2019, 216, 119246. [DOI] [PubMed] [Google Scholar]
- [17].Ostrovidov S, Salehi S, Costantini M, Suthiwanich K, Ebrahimi M, Sadeghian RB, Fujie T, Shi X, Cannata S, Gargioli C, Tamayol A, Dokmeci MR, Orive G, Swieszkowski W, Khademhosseini A, Small 2019, 15, 1805530; [DOI] [PMC free article] [PubMed] [Google Scholar]; Faramarzi N, Yazdi IK, Nabavinia M, Gemma A, Fanelli A, Caizzone A, Ptaszek LM, Sinha I, Khademhosseini A, Ruskin JN, Tamayol A, Advanced Healthcare Materials 2018, 7, 1701347; [DOI] [PMC free article] [PubMed] [Google Scholar]; Fallahi A, Yazdi IK, Serex L, Lesha E, Faramarzi N, Tarlan F, Avci H, Costa-Almeida R, Sharifi F, Rinoldi C, Gomes ME, Shin SR, Khademhosseini A, Akbari M, Tamayol A, ACS Biomaterials Science & Engineering 2020, 6, 1112. [DOI] [PubMed] [Google Scholar]
- [18].Wang L, Cao L, Shansky J, Wang Z, Mooney D, Vandenburgh H, Molecular Therapy 2014, 22, 1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hakimi N, Cheng R, Leng L, Sotoudehfar M, Ba PQ, Bakhtyar N, Amini-Nik S, Jeschke MG, Günther A, Lab on a Chip 2018, 18, 1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].O’Connell CD, Di Bella C, Thompson F, Augustine C, Beirne S, Cornock R, Richards CJ, Chung J, Gambhir S, Yue Z, Bourke J, Zhang B, Taylor A, Quigley A, Kapsa R, Choong P, Wallace GG, Biofabrication 2016, 8, 015019. [DOI] [PubMed] [Google Scholar]
- [21].Gaharwar AK, Cross LM, Peak CW, Gold K, Carrow JK, Brokesh A, Singh KA, Advanced Materials 2019, 31, 1900332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cross LM, Carrow JK, Ding X, Singh KA, Gaharwar AK, ACS applied materials & interfaces 2019, 11, 6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carrow JK, Cross LM, Reese RW, Jaiswal MK, Gregory CA, Kaunas R, Singh I, Gaharwar AK, Proc Natl Acad Sci U S A 2018, 115, E3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Huey KA, Smith SA, Sulaeman A, Breen EC, J Appl Physiol (1985) 2016, 120, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dawson JI, Kanczler JM, Yang XB, Attard GS, Oreffo ROC, Advanced Materials 2011, 23, 3304. [DOI] [PubMed] [Google Scholar]
- [26].Barba AA, Bochicchio S, Dalmoro A, Caccavo D, Cascone S, Lamberti G, in Nanomaterials for Drug Delivery and Therapy, (Ed: Grumezescu AM), William Andrew Publishing, 2019, 267. [Google Scholar]
- [27].Liu JMH, Zhang J, Zhang X, Hlavaty KA, Ricci CF, Leonard JN, Shea LD, Gower RM, Biomaterials 2016, 80, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Costantini M, Testa S, Fornetti E, Barbetta A, Trombetta M, Cannata SM, Gargioli C, Rainer A, Front Bioeng Biotechnol 2017, 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Elkhoury K, Russell CS, Sanchez-Gonzalez L, Mostafavi A, Williams TJ, Kahn C, Peppas NA, Arab-Tehrany E, Tamayol A, Advanced Healthcare Materials 2019, 8, 1900506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ruzicka B, Zaccarelli E, Soft Matter 2011, 7, 1268; [Google Scholar]; Sheikhi A, Afewerki S, Oklu R, Gaharwar AK, Khademhosseini A, Biomaterials Science 2018, 6, 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rassier DE, MacIntosh BR, Herzog W, Journal of Applied Physiology 1999, 86, 1445. [DOI] [PubMed] [Google Scholar]
- [32].Kot BC, Zhang ZJ, Lee AW, Leung VY, Fu SN, PLoS One 2012, 7, e44348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Quarta M, Cromie Lear MJ, Blonigan J, Paine P, Chacon R, Rando TA, NPJ Regen Med 2018, 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Irvin MW, Zijlstra A, Wikswo JP, Pozzi A, Exp Biol Med (Maywood) 2014, 239, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hogan BLM, Kolodziej PA, Nature Reviews Genetics 2002, 3, 513. [DOI] [PubMed] [Google Scholar]
- [36].DeCicco-Skinner KL, Henry GH, Cataisson C, Tabib T, Gwilliam JC, Watson NJ, Bullwinkle EM, Falkenburg L, O’Neill RC, Morin A, Wiest JS, J Vis Exp 2014, e51312; [DOI] [PMC free article] [PubMed] [Google Scholar]; Xie D, Ju D, Speyer C, Gorski D, Kosir MA, J Vis Exp 2016, 54074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Panayi AC, Smit L, Hays N, Udeh K, Endo Y, Li B, Sakthivel D, Tamayol A, Neppl RL, Orgill DP, Nuutila K, Sinha I, Wound Repair and Regeneration 2020, 28, 61. [DOI] [PubMed] [Google Scholar]
- [38].Castro B, Kuang S, Bio Protoc 2017, 7, e2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Greising SM, Rivera JC, Goldman SM, Watts A, Aguilar CA, Corona BT, Scientific Reports 2017, 7, 13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Haun CT, Vann CG, Roberts BM, Vigotsky AD, Schoenfeld BJ, Roberts MD, Front Physiol 2019, 10, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Corona BT, Rivera JC, Greising SM, Clin Transl Sci 2018, 11, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A, Biomaterials 2010, 31, 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shin SR, Bae H, Cha JM, Mun JY, Chen Y-C, Tekin H, Shin H, Farshchi S, Dokmeci MR, Tang S, Khademhosseini A, ACS Nano 2012, 6, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].ASTM Internatational, West Conshohocken, PA 2003.
- [45].ASTM International, West Conshohocken, PA 2005.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.