

Summary:
Cardiac Pacemaker Cells (CPCs) rhythmically initiate the electrical impulses that drive heart contraction. CPCs display the highest rate of spontaneous depolarization in the heart despite being subjected to inhibitory electrochemical conditions that should theoretically suppress their activity. While several models have been proposed to explain this apparent paradox, the actual molecular mechanisms that allow CPCs to overcome electrogenic barriers to their function remain poorly understood. Here we have traced CPC development at single cell resolution and uncovered a series of cytoarchitectural patterning events that are critical for proper pacemaking. Specifically, our data reveal that CPCs dynamically modulate adherens junction (AJ) engagement to control characteristics including surface area, volume, and gap junctional coupling. This allows CPCs to adopt a structural configuration that supports their overall excitability. Thus, our data have identified a direct role for local cellular mechanics in patterning critical morphological features that are necessary for CPC electrical activity.
Keywords: Sinoatrial Node, Cardiac Pacemaker Cell, Adherens Junction, Electrophysiology, Gap Junction, Cytoarchitecture, Cardiac Development
eTOC Blurb
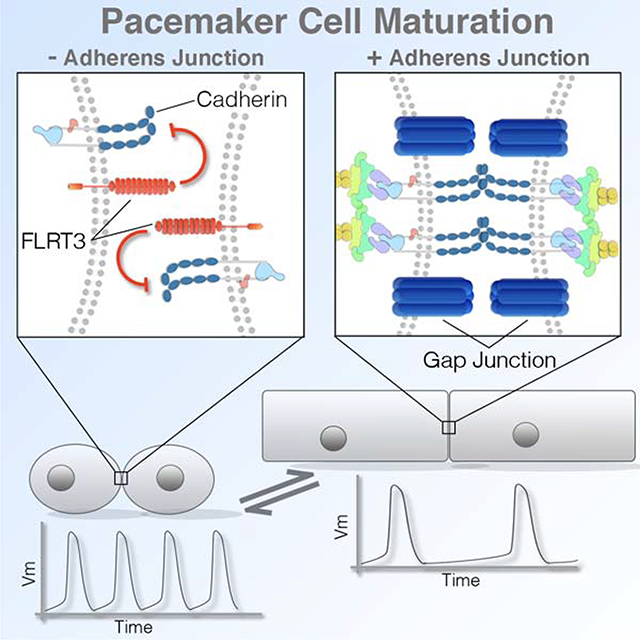
Thomas et al. identify a link between cytoarchitectural features that influence cardiac pacemaker cell excitability and the unique modulation of the cardiac adherens junction (AJ) that occurs during sinoatrial node morphogenesis. Pacemaker cells specifically diminish AJ formation during cardiac morphogenesis resulting in small cell size and limited gap junctional coupling.
Graphical Abstract
Introduction:
A network of cardiac pacemaker cells (CPCs) located in the sinoatrial node (SAN) autonomously initiate the electrical impulses that trigger rhythmic cardiac contraction. CPCs represent only a fraction of the total cellular volume of the heart (Joyner and van Capelle, 1986; Kapoor et al., 2012; Unudurthi et al., 2014), yet they must generate sufficient ionic current to activate the entire working myocardium (WM). It has long been recognized that the low proportion of CPCs relative to the WM represents a significant obstacle to rhythmic impulse generation and propagation (Grijalva et al., 2019; Joyner and van Capelle, 1986; Unudurthi et al., 2014). CPCs need to depolarize while in electrical communication with a large volume of cells that are actively maintaining a negative membrane potential. In theory, this ionic configuration would be predicted to elicit subthreshold current flows from CPCs to WM, delaying or preventing the ability of CPCs to build up enough local charge to initiate an action potential (Fast and Kléber, 1995; Joyner and van Capelle, 1986; Kirchhof et al., 1987; Kleber and Rudy, 2004; Nikolaidou et al., 2012; Rohr et al., 1997; Rohr et al., 1998; Unudurthi et al., 2014). The ability of CPCs to escape such inhibitory forces is a topic of fundamental interest to our overall understanding of cardiac electrophysiology, however, the mechanisms that dictate how CPCs regulate the extent of their electrogenic interaction with the WM remain poorly understood. Previously, we identified that CPCs differentiate relatively late during cardiac morphogenesis and integrate into an already electrically active embryonic heart (Bressan et al., 2013). Thus, we hypothesized that the mechanisms required for CPCs to overcome WM-mediated electrical suppression should rapidly manifest shortly after they are added to the heart.
In this study we have now identified a critical relationship between two parameters that significantly impact CPC electrical excitability during cardiogenesis: cell size regulation and cell-cell coupling. Importantly, our data indicate that these features are related to one another, both in terms of their predicted influence on CPC function and in regard to their molecular regulation. Furthermore, we show that the cardiac adherens junction (AJ) plays a direct role as a molecular regulator of CPC structural patterning. Collectively, these findings have uncovered a cellular mechanics-based model for CPC developmental maturation that defines specific cytoarchitectural features that support CPC function and identifies how these characteristics are patterned and maintained in the forming heart.
Results:
Computational modeling of the embryonic CPC/atrial interface predicts that an interplay between membrane capacitance and intercellular conductance governs successful pacemaking activity.
To explore the cellular parameters that may influence CPC activity and/or excitability, we generated a computational model of the CPC/WM electrical interface based on the geometry of the embryonic heart and probed for critical features that lead to SAN failure (Figures 1 and S1). Herein, 300um thick sections of the embryonic SAN/atria were imaged, digitally segmented, and reconstructed in silico (Figures 1A–1D). CPCs and atrial ionic currents were modeled based on Fabbri et al. (Fabbri et al., 2017) and Courtemanche et al.(Courtemanche et al., 1998) respectively. Modeled CPCs were assigned to the region of the myocardium that our voltage imaging demonstrated displayed pacemaking activity and expressed the CPC functional marker, Hcn4 (Bressan et al 2018) (Figures 1A–1D). Voltage imaging from live cardiac slices were used to qualitatively assess fundamental features of the modeled data including action potential kinetics, conduction velocities, and activation patterns to ensure that computational simulations replicated basic features of the embryonic SAN/Atrial interface (Figure 1E, see Methods).
Figure 1. Mathematical simulation of electrical interactions in the embryonic heart cellular parameters critical for successful pacemaking.
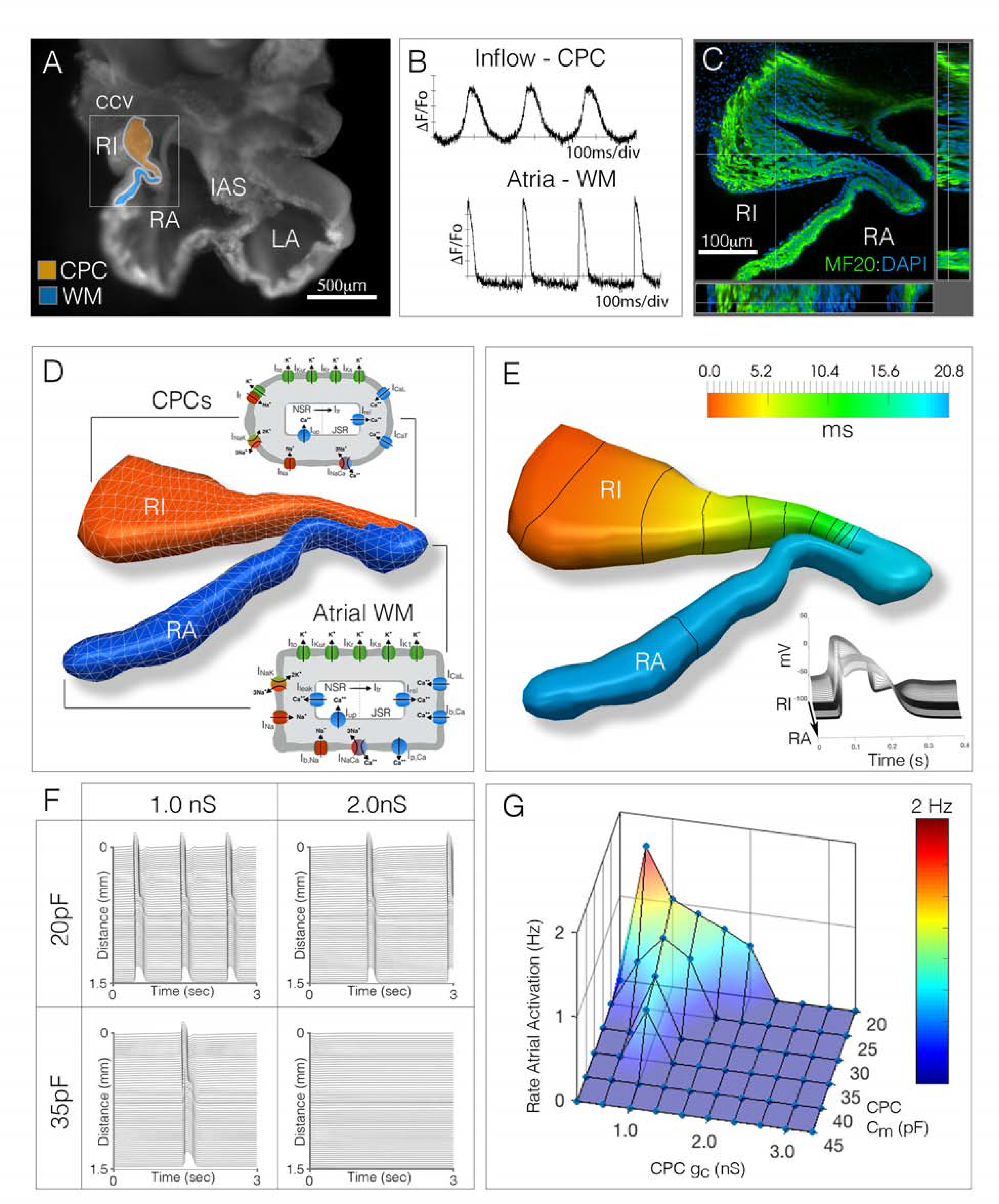
A) Cardiac slice preparation for live-imaging of electrical activity in the embryonic chick heart. Orange region denotes the right inflow (RI) myocardium which displays cardiac pacemaker cell (CPC)-like electrical behavior, blue region denotes adjacent right atrial (RA) muscle with working myocardium (WM)-like physiological features. IAS – interatrial septum, ccv – common cardinal vein. Scale bare – 500μm. B) Electrical activity recorded from inflow (orange region from “A”) and right atria (blue region from “A”). C) 300μm thick section through the CPC/WM interface, tissue was stained with the muscle marker MF20 (green) and DAPI (blue). Scale bar - 100 μm D) Tissue geometry from “C” was segmented and the region of the tissue that displayed CPC physiological activity (from “A,B”) was simulated as containing cells utilizing CPC ionic behavior (orange), while tissue regions that displayed atrial-like physiological features (from “A,B”) were simulated as containing atrial WM ionic features (blue region). E) Simulation of impulse generation and propagation through the segmented geometry from “D.” Isochronal map indicates initial activation in the right inflow (red) and propagation into the atrial myocardium (blue). Lines denote propagation at 5 ms/div. Inset shows action potential shapes as the impulse propagates from right inflow to right atria. F) Changes to CPC cellular parameters, such as cell size (equivalent to cell capacitance, pF) or coupling conductance (nS), directly impact rate of activity. G) Sensitivity analysis comparing variations in CPC cell size and conductance (Cm [pF] vs gc (nS)) on rate of activity.
While constructing the computational model, physiological and structural features such as cellular capacitance (Cm), membrane surface to tissue volume ratio (χ), current densities, and the tissue conductivity (σ) were modified to investigate the resultant effects on CPC activity (see Methods). In agreement with previous modeling studies, our data indicated that coupling conductance (gc) plays a critical role in maintaining rate of activity and 1:1 CPC/WM entrainment (Figures 1F and S1E), with gc needing to be held at low values to prevent WM-mediated suppression of CPC function (Glynn et al., 2014; Inada et al., 2014; Joyner and van Capelle, 1986; Zhang et al., 2000). In addition to gc, our data also highlighted that membrane capacitance (Cm) dramatically influenced behavior in the simulations. Increasing CPC Cm decreased the rate of CPC oscillation and blocked the ability of CPCs to drive the atria (Figures 1F and S1D). Given the predicted influence that both gc and Cm displayed in our computational modeling, we simultaneously altered the ranges of both parameters. In general, lowering Cm increased the ranges of gc that could be tolerated before pacemaking failure with a reciprocal effect in which lowering gc expanded the values of Cm that allowed for effective pacing (Figures 1G, S1E). Importantly, these data demonstrate that both gc and Cm may influence pacemaking activity in the embryonic heart and indicate that changes in one may affect the range of values that are possible in the other.
Data from the above computer simulations are in agreement with the known cellular architecture of mature CPCs. Total membrane capacitance is proportional to cell size (Satoh et al., 1996), and adult CPCs are substantially smaller and have lower Cm than their WM counterparts (Honjo et al., 1996; Jones et al., 2008; Lyashkov et al., 2007; Masson-Pevet et al., 1979; Miyamoto et al., 2002; Shimada et al., 2004). Additionally, gc is believed to be determined by gap junction-mediated coupling and mature CPCs have few gap junctions (GJs)(Boyett et al., 2000; Forbes and Sperelakis, 1985; Honjo et al., 2002; Miyamoto et al., 2002; Shimada et al., 2004). Therefore, our simulations identified known cellular features of the adult heart as potentially important for embryonic CPC electrical activity and highlighted that developmental events upstream of cell size regulation and GJ coupling may be critical for proper cardiac pacemaking.
CPCs structurally diverge from the WM after becoming electrically active.
Motivated by the computational model predictions, we conducted a developmental survey of cellular architecture to determine whether CPCs regulate their morphology in a manner consistent with our simulations.
Initially, we confirmed the location of CPCs at various stages of heart development by monitoring electrical activity using both voltage imaging and micro-electrode array recordings (MEA). Consistent with previous work (Bressan et al., 2018; Bressan et al., 2013), our data indicated that CPCs were present in the right inflow myocardium from embryonic day 3 (E3) through E9 of avian cardiac morphogenesis (Figure 2A–D). To evaluate regional patterning of cell size, we generated mosaic hearts using in vivo transfection of a transposable expression construct encoding a palmitoylated membrane RFP reporter and GFP nuclear reporter (Figure S2A) (Goudy et al., 2019) and three-dimensional renderings of individual myocytes were generated to quantify cellular geometry (Figures S2B–2E). At E3, no statistical differences in cell volume or surface area were noted between CPCs and atrial WM (Figures 2E–2G). However, following three additional days of incubation, statistically significant differences in cell structure began to emerge (Figures 2E–2G). At E6, CPCs retained the structural characteristics present in E3 myocytes, while atrial WM had increased in both surface area and volume. By the completion of cardiac morphogenesis at E9, CPCs had still not changed in their basic structural features compared to their E3 counterparts. In contrast, atrial WM had increased both their surface area and volumes by factors of 2.1 and 2.4-fold respectively (Figures 2E–2G). These data demonstrate that CPCs and atrial WM initially share similar cytoarchitectural features, however, as cardiac morphogenesis proceeds, WM increase in size while CPCs do not.
Figure 2. Cell size is differentially regulated between the CPC and WM lineages during cardiac morphogenesis.
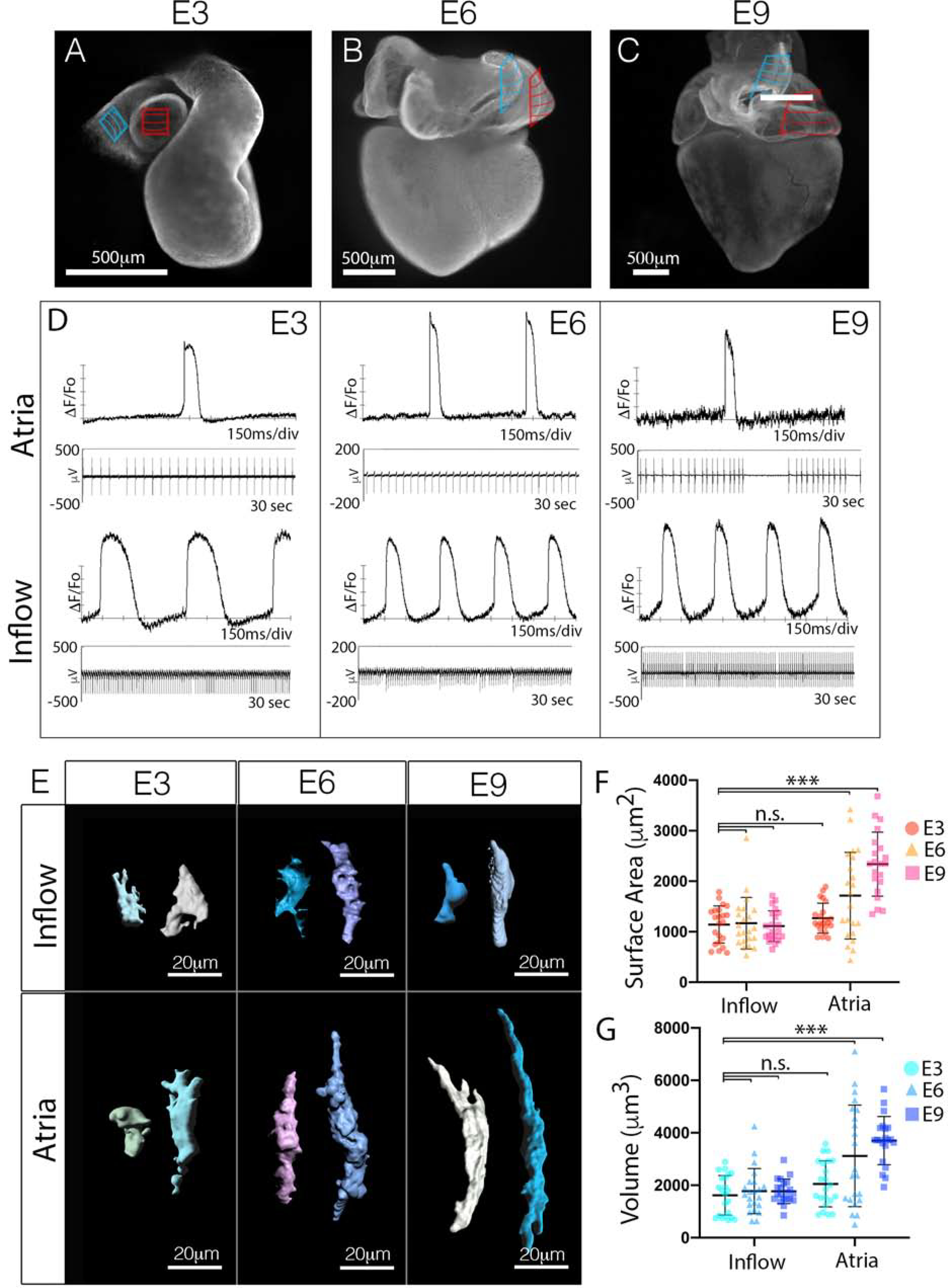
A-C) Whole mount staining for the muscle marker MF20 at embryonic day 3 (A), embryonic day 6 (B), and embryonic day 9 (C). The location of the CPCs within the inflow myocardium is indicated by the blue hatched area, the atrial WM used for size analysis is indicated by the red hatched area. D) Voltage sensitive dye-based (top) and micro-electrode array (MEA) (bottom) recordings of electrical activity from isolated segments of tissues (blue and red regions from (A-C)). Scale bar – 500μm. E) Volumetric reconstruction of CPCs from the inflow and atrial WM at different stages of cardiac morphogenesis (See also Figure S1). F) Quantification of surface area of CPCs from the inflow and atrial WM at different stages of cardiac morphogenesis (n=129). G) Quantification of cell volume of CPCs from the inflow and atrial WM at different stages of cardiac morphogenesis (n=129). Mean +/− SD are indicated by line and bars for F and G.
CPCs lose the ability to form AJ as they diverge from the WM.
Having determined a developmental window during which CPCs and WM structurally diverge, we sought to identify mechanisms regulating cell size. Transmission electron microscopy (TEM) was used to view the ultrastructure of CPCs vs WM at stages when differences in cell structure first became apparent (E6). Consistent with previous data from adult vertebrates (Lu et al., 1993; Masson-Pevet et al., 1984; Opthof, 1988), CPCs located in the forming SAN (Figure 2A) (Bressan et al., 2018; Bressan et al., 2013) contained fewer myofibrils than the atrial WM (Figures 3A–3F). Within the atrial WM, large electron dense structures were noted spanning opposing plasma membranes (Figure 3A–3C). These structures averaged ~600μm in length and were tethered to collections of actin filaments (Figures 3A–3C, 3G), indicative of classic AJs (Gutstein et al., 2003; Merkel et al., 2019; Yonemura et al., 1995). Of note, CPCs displayed long stretches of membrane between adjacent cells with few or no AJ-like structures. When potential AJs were observed, they appeared shorter than in the WM and had few actin fibers associated with them (Figures 3D–3F). These clear differences in cell-cell adhesion were largely restricted to AJs, as our TEM imaging noted that desmosomes were detected at similar abundance among CPCs and atrial WM (Figure 3C and 3F) which is in agreement with studies from the adult SAN (Mavroidis et al., 2020; Mezzano et al., 2016).
Figure 3. CPCs uniquely lose the capacity to form adherens junctions as they structurally diverge from the working myocardium.
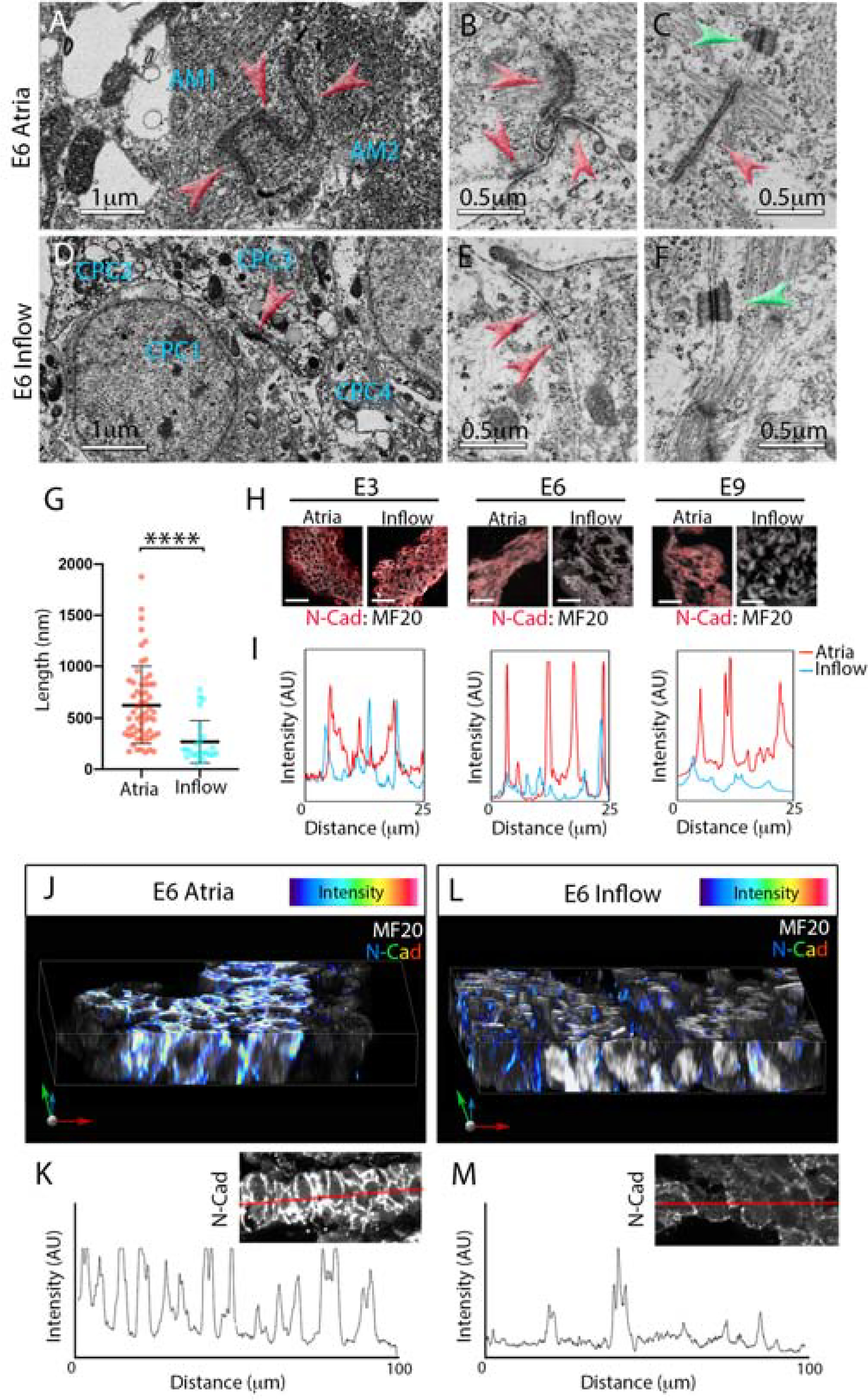
A) Transmission electron microscope (TEM) image of two atrial myocytes (AM1 and AM2). Red arrowheads indicate a series of adherens junctions (AJs) between the two myocytes. B) Higher magnification image of a series of AJs between three atrial myocytes (red arrowheads), these structures are tethered to large bundles of actin filaments. C) Within the atrial WM, AJs are often associated with nearby desmosomes (green arrowhead). D) TEM image of four CPCs (CPC1–4) located within the inflow myocardium. These cells have low sarcomere density consistent with adult CPCs. Note: only one junction is present between the four cells (red arrowhead). E) Higher magnification image of membrane interface between two inflow cells, small electron dense structures that are not tethered to actin are present (red arrowheads). F) Typical desmosome present between inflow myocytes (green arrowhead). G) Quantification of measured AJs length based on TEM images (n=89). Mean +/− SD are indicated by line and bars. H) Immunolocalization of the AJ protein, N-cadherin (N-Cad, red), within E3, E6, and E9 atrial and inflow myocardium. Sections are counterstained with MF20 (white). Scale bar – 25μm. I) Line scan profile comparing intensity of atrial (red trace) and inflow (blue trace) N-Cad staining at E3, E6, and E9. J) Reconstruction of a 30μm Z-stack of atrial WM taken from a 200μm thick vibratome section. N-Cad intensity is color coded. K) Line scan of N-Cad staining taken from a 200μm section of atrial tissue. Inset shows N-Cad staining, red line indicates position of the line scan. L, M) As in (K,L) for inflow myocardium.
The lack of AJs among CPCs was of immediate interest given the known role for these structures in conveying the biomechanical signals that induce hypertrophic growth in a variety of cell types including myocytes (Chopra et al., 2011; Janssens et al., 2001; Li et al., 2012; Luo, 2003; Luo et al., 2001; Vite et al., 2018; Wickline et al., 2016). Thus, we verified our TEM studies by examining the localization of proteins associated with formation of the cardiac AJ via immunostaining for N-cadherin (N-Cad), Beta-Catenin (β-cat), and alphaT-Catenin (αT-cat) at E3, E6, and E9. Interestingly at E3, when CPCs and atrial WM display similar structural characteristics (Figure 2E–2G), no regional differences in N-Cad, β-cat, or αT-cat protein localization were noted (Figures 3H,3I,S3A, and S3B). However, by E6 and continuing through E9, AJ proteins were largely absent from the inflow cells, while they were clearly present at cell borders in the atrial WM (Figures 3H,3I,S3A, and S3B). At stages when N-cad protein levels were diminished in CPCs, mRNA transcript levels were similar with those detected in the WM (Figures S3C and S3D). To confirm these data in a mammalian embryo, we examined sections through an E14.5 embryonic mouse heart. Consistent with our data from the chick, HCN4-positive CPCs in the mouse embryonic SAN showed diminished staining for N-Cad and αT-cat when compared with the atrial WM (Figures S3E,S3F). To confirm that diminished immunoreactivity was not due to variations in section plane we cut 200μm thick sections through the inflow and atria of E6 hearts and examined N-Cad distribution (Figures 3J–3M). Line scans through these sections confirmed that N-Cad was largely absent among CPCs.
Inhibition of the cardiac AJ converts atrial WM to a CPC-like morphology.
To test whether diminished AJ formation alone could generate myocytes with CPC-like structural characteristics, we blocked AJ formation in the WM. We developed two independent shRNAs against N-Cad(Goudy et al., 2019) and generated a mutated N-Cad expression construct lacking the extracellular domain required for intercellular binding (N-cadΔ112–624) (Ong et al., 1998). When transfected into primary embryonic WM, both shRNAs and the N-cadΔ112–624 construct diminished cell-cell contacts and AJ-formation in cultured primary embryonic WM (Figure S4).The N-cadΔ112–624 construct was slightly more effective at disrupting AJ formation in vitro (Figures S4A and S4C), therefore, we used this construct to evaluate the role of AJ formation in cell size regulation in vivo. Following transfection at E2.5 and incubation to E9 (Figure 4A), atrial WM expressing N-cadΔ112–624 displayed a 29.1% decrease in surface area and a 41.0% decrease in volume when compared to control transfected cells (Figures 4C–4H), resulting in WM with CPC-like morphologies in vivo.
Figure 4. Inhibition of the cardiac AJ converts atrial WM to a CPC-like morphology.
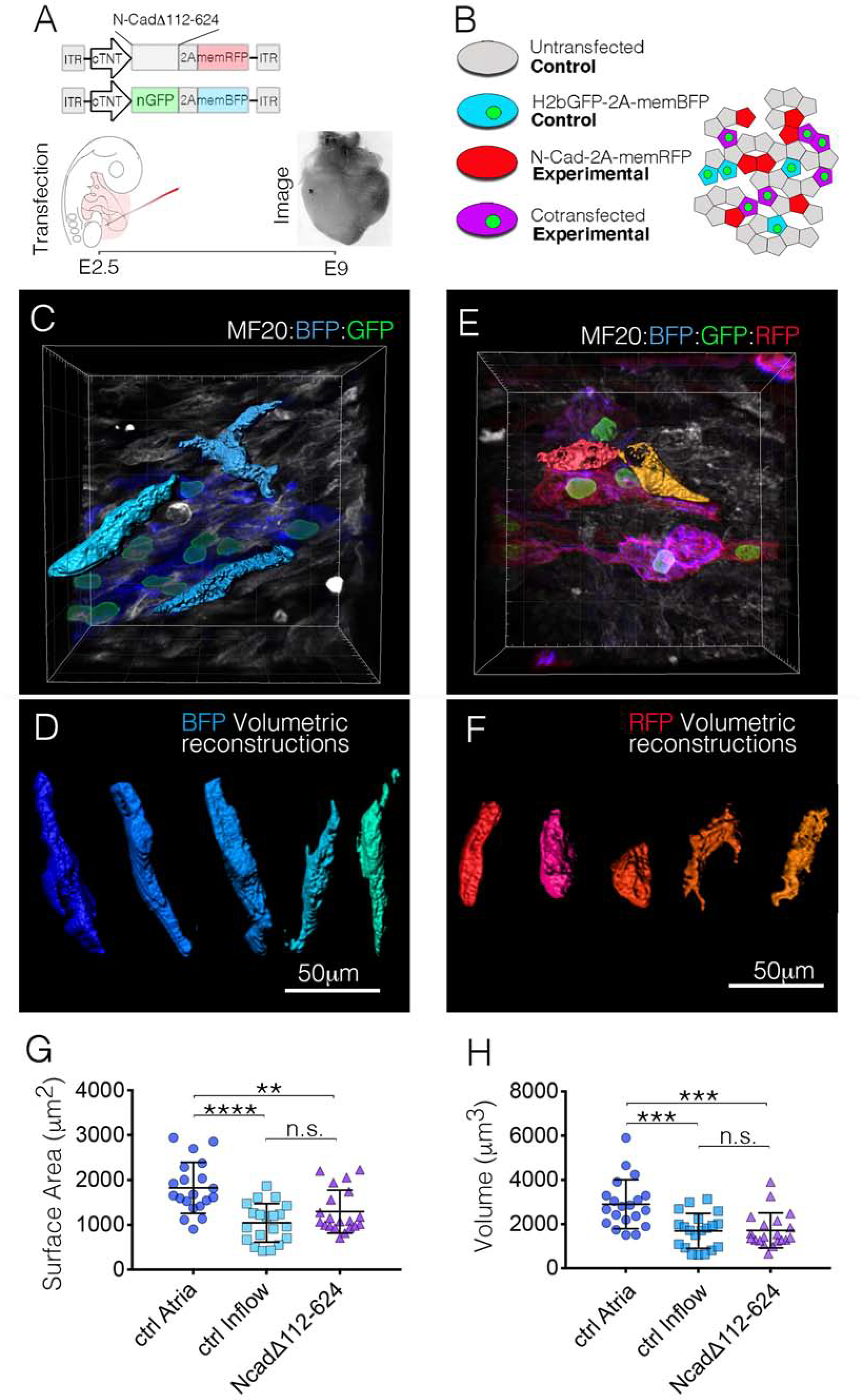
A) Diagram of the experimental procedure for in vivo mosaic inhibition of AJ formation. B) Following manipulation, several cell populations are present in each heart. Control cells (nuclear GFP – Membrane BFP) were compared with experimental cells (nuclear GFP – Membrane BFP / N-CadΔ112–624 – membrane RFP). C) in vivo imaging of control (nuclear GFP – Membrane BFP) atrial WM. Three of these cells were digitally isolated for volumetric reconstruction. D) Examples of in vivo volumetric reconstructions from control atrial WM. Scale Bar – 50μm E) in vivo imaging of experimental (nuclear GFP – Membrane BFP/N-CadΔ112–624 – membrane RFP) atrial WM. F) Typical examples of volumetric reconstructions from experimental cells. G) Quantification comparing surface area between control atrial WM (n=20), control inflow CPCs (n=20), and N-CadΔ112–624 expressing atrial WM (n=20). H) Quantification comparing cellular volume between control atrial WM (n=20), control Inflow CPCs (n=20), and N-CadΔ112–624 expressing atrial WM (n=20). Mean +/− SD are indicated by line and bars for G and H.
Force-mediated AJ engagement induces a WM-like phenotype in CPCs.
We next tested whether forcing AJ engagement would alter the phenotype of embryonic CPCs. At E2.5, hearts were transfected with a construct expressing a membrane localized RFP (palmTagRFP). Inflow and atrial WM were then isolated at E6. Fluorescent in situ hybridization (RNAscope) against the CPC marker gene Hcn4 was used to confirm isolated inflow cells showed molecular characteristics of CPCs (Figures S5A–S5C). Dissociated inflow and atrial cells were then cultured for 72 hours on polyacrylamide gels of varying stiffness (4kPa-60kPa) coated with recombinant N-Cad. In agreement with previous studies (Chopra et al., 2011), atrial WM displayed N-Cad-mediated, stiffness-dependent, regulation of morphology. On relatively compliant gels (4kPa) atrial cells were small and round (Figure 5A). However, atrial WM cultured on stiffer gels (60kPa) exhibited increases in both surface area and volume (Figures 5A–5C). Importantly, CPCs plated on N-Cad coated polyacrylamide gels displayed morphological changes similar to the atrial WM. On 4kPa gels, CPCs had an average surface area of 1697 +/− 931.2 μm2 and an average volume of ~2217.5 +/− 1384.7 μm3. However, when CPCs were plated on 60kPa gels coated with N-Cad both surface area and volume increased by 1.48-fold and 1.84-fold respectively(Figures 5A–5C). These data indicate that WM and CPCs displayed similar size characteristics when presented with similar levels of force-dependent, AJ-engagement. Importantly, size changes among CPCs and atrial myocytes converged towards common characteristics in these studies and did not simply scale based on their initial properties at the time of isolation (CPCs were smaller than atrial cells at the time of isolation), indicating that cell size regulation in both the embryonic WM and CPC lineages is plastic and responsive to local biomechanical cues.
Figure 5. Force-mediated AJ engagement induces a WM-like phenotype in CPCs.
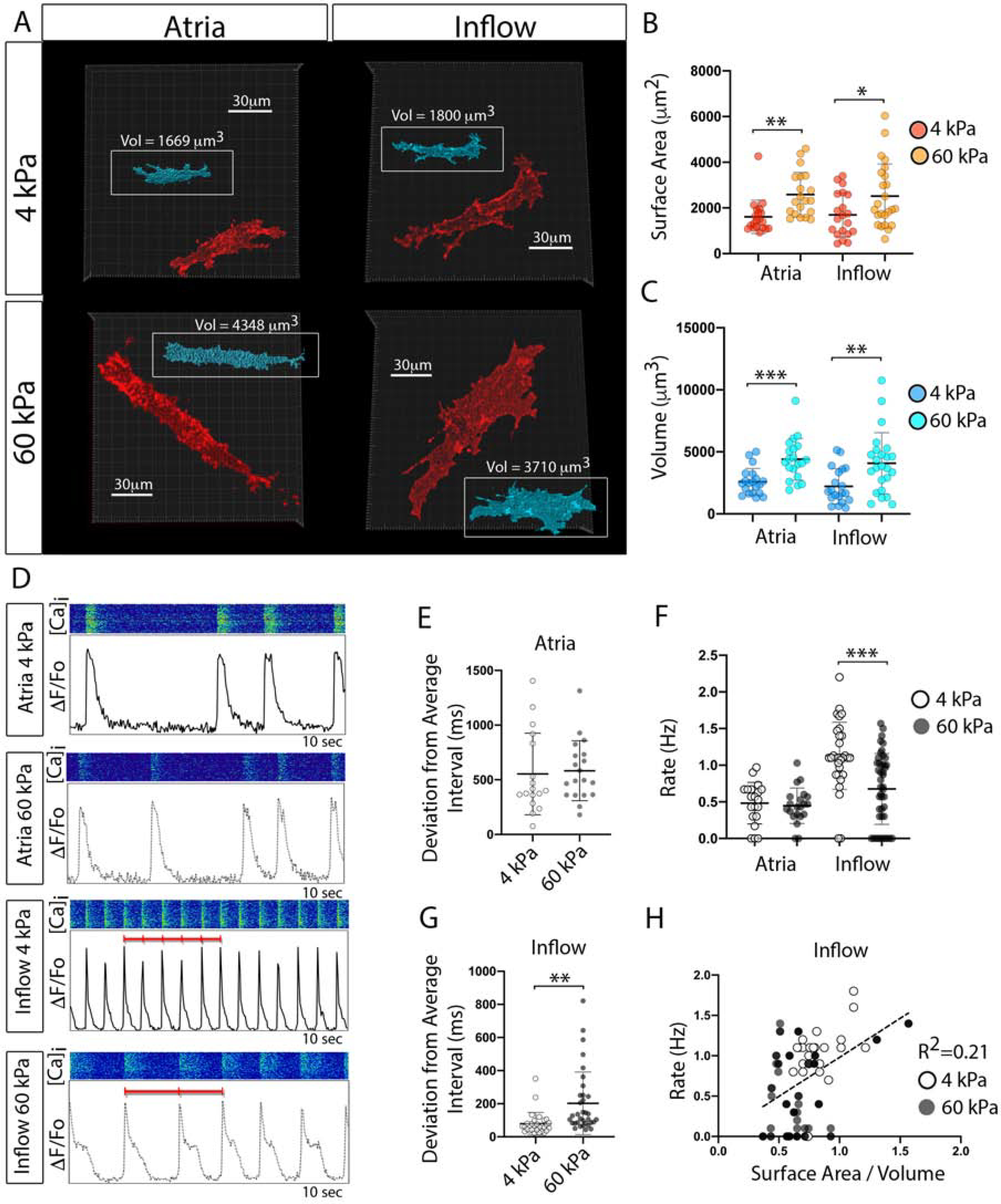
A) Maximum projection images of membrane RFP positive atrial WM and inflow cells cultured on 4kPa and 60kPa N-Cad cross-linked, polyacrylamide gels. Insets (Blue) show volumetric reconstructs of each cell. Scale Bar - 30 μm. B) Quantification of surface area of cells cultured as described for “A” (n=40 for atria, n=44 for inflow). C) Quantification of cellular volume for cells cultured as described in “A” D) Examples of typical physiological behavior of cells from “A-C.” Intracellular calcium transients were recorded from cells cultured on different stiffnesses of N-Cad coated gels. E) Quantification of variation in transient interval on different gel stiffness for atrial cells (n=40). F) Quantification of rate of activation (n=40 for atria, n=44 for inflow). G) Quantification of variation in transient interval on different gel stiffness for inflow cells (n=44). H) Linear regression between inflow cells surface area/volume ratio and rate of activity. Mean +/− SD are indicated by line and bars for B,C,E,F, and G.
To test whether AJ formation altered CPC function, cells from the above analysis were live-imaged and calcium transient activity was monitored to assay the rate and rhythmicity of electrical activity. Regardless of N-Cad substrate stiffness, atrial WM displayed similar functional characteristics, with impulse generation being relatively arrhythmic and spontaneous depolarization occurring at an average rate of 0.46 Hz (Figure 5D–5F). CPCs cultured on soft N-Cad gels were 5.0-fold more rhythmic than WM and were 2.5-fold faster in their rate of activity (Figures 5E–5F). However, the rate and rhythmicity of CPCs significantly dropped when the cells were subjected to stiffer N-Cad coated substrates (Figure 5F,5G), with a weak positive correlation between surface area/volume ratio and rate of activity apparent across both conditions(R2=0.21, Figure 5H).
The above studies are in agreement with our computational modeling that suggested that increasing surface area should negatively impact CPC activity. However, these studies could not rule out that the dominant effect on our cultured cells was a response to substrate stiffness as opposed to changes in cell size. To address this, we plated cells on intermediate 14kPa gels coated with fibronectin (FN) or N-Cad and monitored cell size and activity at 24 and 72 hrs. of culture. On both FN and N-Cad, CPCs were relatively small at 24 hrs. but increased by 4.9 and 2.8-fold respectively by 72hrs (Figures S5F,S5G). The larger CPCs at 72 hrs. of culture decreased rate of activity on both FN and N-Cad despite the fact that substrate stiffness was held constant throughout the analysis (Figure S5G) In addition, we plated CPCs on 50kPa gels with micropatterned wells to constrain their size on a stiff substrate. CPCs on micropatterned PA gels displayed only minor increases in size between 24 and 72hrs of culture (31%) and showed no statistical differences in rate of activity (Figure S5F, S5G) demonstrating that maintaining small cell size preserved function despite the high stiffness of the substrate. Furthermore, when comparing the area vs rate of all of the conditions above, we noted that while small CPCs show a range of possible frequencies (between 0.5–3.5 Hz), larger CPCs lost the capacity for higher rate activity (Figure S5H). Collectively, these data indicate that maintenance of small cell size is beneficial for preserving the potential for high rate, rhythmic, activity in CPCs.
AJ formation regulates CPC gap junction formation.
In agreement with previous studies (Glynn et al., 2014; Inada et al., 2014; Joyner and van Capelle, 1986; Zhang et al., 2000), our computer modeling highlighted gc as an important feature for successful CPC function. Interestingly, several AJ-proteins are known to impact the stability of the gap junction (GJ) proteins that determine gc in the heart (Kostetskii et al., 2005; Li, 2005; Li et al., 2012; Shaw et al., 2007; Smyth et al., 2010; Swope et al., 2012). Therefore, we tested whether the regulation of AJ engagement might contribute to titration of CPC GJ coupling by examining AJ and GJ protein trafficking in primary embryonic CPCs. The GJ protein Connexin 40 (Cx40) was evaluated as our previous RNAseq data indicating it was the most abundant GJ transcript in all regions of the avian heart during early morphogenesis despite being more highly expressed in the WM versus CPCs (Bressan et al., 2018). CPCs and atrial WM were isolated and plated at low density and the distribution of N-Cad and Cx40 was tracked. When either atrial WM or inflow cells were found in isolation (no cell-cell contacts), small puncta of Cx40 and N-Cad were detectable throughout the cells (Figure 6A). However, in regions of the cultures where neighboring cells were in contact, intense N-Cad and Cx40 staining was detected along cell-cell interfaces (Figure 6A).
Figure 6. AJ and gap junction proteins colocalize within embryonic CPCs and atrial WM.
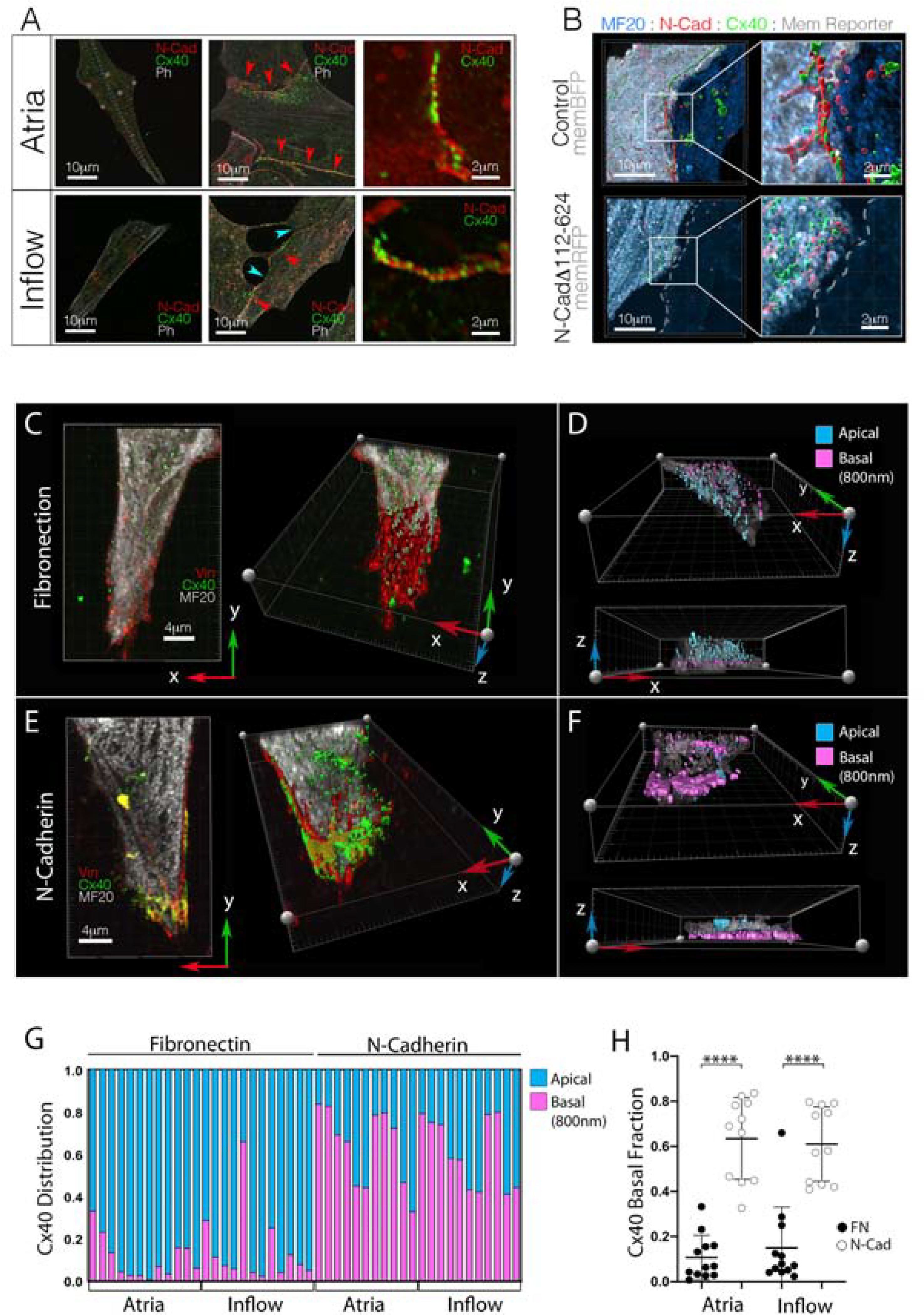
A) Cellular distribution of the AJ protein, N-Cad, and GJ protein, Cx40. Regardless of source tissue, isolated cells (no cell-cell contacts) show little N-Cad or Cx40 immunoreactivity. However, when cell junctions are present, both N-Cad and Cx40 traffic to the cell-cell interface (red arrowheads). Note: inflow cells form junctions in culture, but they tend to show disrupted morphology (blue arrowheads). Scale bars - as indicated in panels. B) Representative images of N-Cad (red) and Cx40 (green) distribution in control vs N-CadΔ112–624 overexpressing atrial WM cell pairs. Scale bars - as indicated in panels. C) Distribution of Vinculin (red) and Cx40 (green) in isolated inflow cell plated on Fibronectin. Cell is viewed from two angles: from above and a rotated view exposing the basal surface of the cell in contact with the fibronectin coating. Scale bars - 4μm D) Cell from “C,” color coded to indicate distance of Cx40 positive staining from the basal surface of the cell. E) as in “C,” for an inflow cell plated on a coverslip coated with N-Cadherin.. F) As in “D,” for the cell plated on N-Cad in “E.” G) Quantification of Cx40 distribution in cells cultured either on Fibronectin or N-Cadherin. Each column represents an individual cell and the proportion of Cx40 immunoreactivity detected within 800nm of the coverslip is indicated in pink. H) Change in Cx40 distribution based on substrate composition. Mean +/− SD are indicated by line and bars.
Upon examination of over 250 junctions, we noted that no Cx40 positive junction was detected independent from an AJ. To test whether the AJ directed GJ formation, we overexpressed the AJ blocking construct (N-cadΔ112–624) in atrial WM and examined Cx40 localization in culture. Across 12 control cell pairs we measured 369.4 μm of total opposing cell membrane (distance between two cells of less than 2μm). Of this, 346 μm (93.7%) was composed of mixed N-cad/Cx40 positive junctions. Among N-cadΔ112–624 expressing cells we measured 270.8 μm of opposing membrane, of which only 65.4 μm (24.1%) was composed of mixed junctions. Indeed, only 2 of 17 N-cadΔ112–624 expressing atrial cells contained Cx40 positive junctions in contrast to 11 of 12 control cell pairs.
To test whether the AJ directly controlled Cx40 trafficking in CPCs, E6 atrial and inflow cells were isolated and cultured on cover glass coated with either FN or N-Cad. Following 72 hours of culture, cells were stained with vinculin, which localizes to both focal adhesions and AJs (Zemljic-Harpf et al., 2009), and Cx40 distribution was scored. Cx40 was rarely detected along a free edge or at the basal surface of atrial cells cultured on FN (Figure S6B). However, among atrial cells cultured on N-Cad, Cx40 dramatically re-localized toward the cover glass (Figure S6C). Similarly, inflow cells cultured on FN displayed large areas of vinculin staining along their basal surfaces, but these regions were largely devoid of Cx40 (Figures 6C,6D). Conversely, when cultured on N-Cad, both vinculin and Cx40 colocalized to the membrane in contact with the cover glass (Figures 6E,6F). To quantify this effect, we determined the proportion of Cx40 immunoreactivity proximal to the cover glass (basal 800nm) relative to the total volume of Cx40 immunoreactivity per cell. Independent of whether a cell originated from the inflow or atria, the presence of N-Cad along the cells basal surface caused a significant redistribution of the total Cx40 protein to the basal cell surface (Figures 6G,6H. Collectively, these data demonstrate that disruption of the AJ blocks GJ formation and N-Cad engagement alone is sufficient to direct Cx40 trafficking to the cell surface in embryonic atrial WM and CPCs.
CPC AJ engagement is regulated by regional enrichment of the transmembrane protein FLRT3.
As described above, no statistical difference for N-Cad transcript levels were noted between WM and CPCs (Figures S3C,S3D). We therefore focused on identifying factors that might post-transcriptionally regulate AJ formation. We compared publicly available RNAseq data obtained from the SAN of E3 avian embryos(Bressan et al., 2018), E14.5 mouse embryos(Vedantham et al., 2015), and E17.5 mouse embryos(van Eif et al., 2019) screening for genes that displayed at least two-fold enrichment over atrial WM in all three datasets (Figure 7A). This yielded a list of 53 commonly enriched genes. We then cross referenced this list with genes in the Gene Ontology (GO) term cell adhesion (GO:0007155), which further refined our candidate list to 12 factors. Fibronectin Leucine Rich Transmembrane Protein 3 (FLRT3) was identified among the common cell adhesion regulators through this approach. FLRT3 has previously been identified as a negative regulator of classical cadherin function (Chen et al., 2009; Karaulanov et al., 2009; Ogata et al., 2007) and as a factor capable of promoting homotypic cell sorting(Egea et al., 2008), but its function in heart development is not known(Maretto et al., 2008; Müller et al., 2011). Therefore, we examined transcript and protein levels of FLRT3 during cardiogenesis. In agreement with the published RNAseq datasets, FLRT3 displayed highly specific enrichment in the inflow myocardium during stages when CPCs are structurally diverging from the WM (Figures 7B and S7B–S7D). Importantly, FLRT3 protein was found to localize on the membrane of CPCs and was particularly enriched at cell contacts (Figures 7C and S7F).
Figure 7. Negative Regulator of Cadherin Function, FLRT3, Disrupts CPC AJ Engagement.
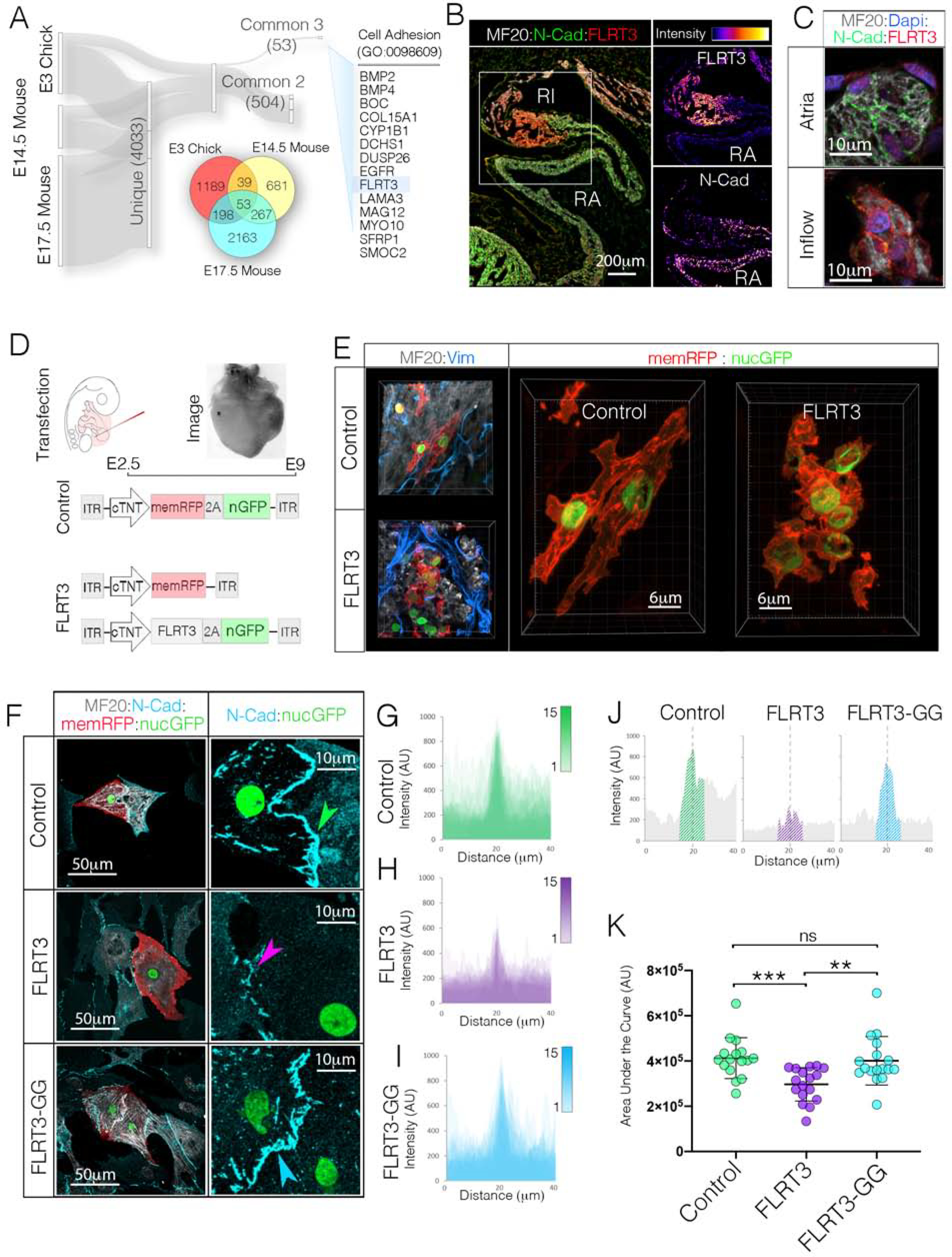
A) RNAseq data sets from E3 Chick (GSE112894 (Bressan et al., 2018)), E14.5 Mouse (GSE65658 (Vedantham, 2015)), and E17.5 Mouse (GSE125932 (van Eif et al., 2019)) sinoatrial nodes were screened for common genes that displayed greater than 2-fold enrichment over atrial WM. The 53 genes common to all three data sets were then assessed for known biological functions related to cell adhesion. This analysis identified FLRT3 as a potential candidate regulator of CPC AJ formation. B) Immunohistochemical localization of FLRT3 and N-Cad. Single channel intensity plots demonstrate that the FLRT3 positive Inflow myocardium shows little immunoreactivity for N-Cad. Scale bar - 200μm C) Higher magnification imaging of FLRT3 and N-Cad immunostaining (see also Figures S7E, S7F). Scale bar – 10μm. D) Diagram for FLRT3 in vivo over expression approach. E) Immunostaining for MF20 (white) and Vimentin (blue) in Control and FLRT3 overexpressing hearts. Scale bar - 6 μm F) Comparison of control, FLRT3, and FLRT3-GG expression on AJ formation in vitro. Scale bars - as indicated in panels. G) Overlay of intensity profiles of N-Cad staining across 15 control cell pairs. Position of cell-cell contact (ex. green arrowhead in “F”) for each cell pair is superimposed at dotted line. H) As in “G,” for cell pairs FLRT3 (n=15). Position of cell-cell contact (ex. purple arrowhead in “F”) are superimposed at dotted line. I) As in “G” and “H” for cells expressing FRLT3-GG (n=15). J) Intensity profile of a single N-cad junctions from control, FLRT3, and FLRT3-GG cells from “F”. Hatched region indicates “area under the curve” quantified in “K.” K) Quantification of N-Cad staining at cell contacts. Mean +/− SD are indicated by line and bars.
To test the function of FLRT3, we overexpressed FLRT3 in the WM outside the forming SAN (Figure 7D). Strikingly, mosaic overexpression of FLRT3 resulted in significant in vivo remodeling of myocardial tissue architecture (Figure 7E and S7G). FLRT3 overexpressing myocytes did not display the elongated aspect ratio present in most WM, but instead appeared small, rounded, and tightly clustered (Figure 7E). Due to the tight clustering of FLRT3 positive cells, it was difficult to determine the size of individual cells in vivo. Therefore, we dissociated FLTR3 overexpressing cells and plated them in dense, multi layered cultures (1×106 cells per cm2). In these cultures the majority of cells are not interacting with the substrate. Therefore, we imaged cells in the upper layers of the cultures and quantified cell area. Compared to control cells, FLRT3 overexpression resulted in a 2.3-fold decrease in area (Figure S7I). Additionally, we overexpressed an shRNA against FLRT3 in the SAN and atria (Tomás et al., 2011). While shFLRT3 did not affect atrial myocardial surface area or volume in vivo, it caused a statistically significant increases in surface area and volume in CPCs (Figures S7J, S7K).
The alteration in cellular morphology induced by FLRT3 lead us to examine whether it directly influenced cardiomyocyte AJ formation. To test this, hearts were transfected with control constructs, FLRT3 expression constructs, or an FLRT3 variant containing a two amino acid substitutions (FLRT3-GG) predicted to disrupt the ability of this protein to influence classical cadherin localization (Karaulanov et al., 2009; Ogata et al., 2007). In control or FLRT3-GG expressing atrial cell pairs, long “ribbon-like” N-Cad positive junction were present (Figures 7F). However, N-Cad positive junctions were less frequent among FLRT3 expressing cells. Moreover, when junctions were detected, both N-Cad intensity and distribution around the junction were statistically diminished (Figure 7G–7K). Collectively, these data demonstrate that FLRT3 is highly enriched in the SAN, modulates the size of cardiomyocytes in a cell autonomous manner, and disrupts cardiac cell AJ engagement.
Discussion:
How the small number of CPCs found in the vertebrate heart are capable of maintaining their excitability while electrically interacting with large numbers of hyperpolarized WM has been a point of significant interest for the past 30 years(Joyner and van Capelle, 1986). Several elegant models have emerged based on the architecture of the adult sinoatrial node (Glynn et al., 2014; Inada et al., 2014; Joyner and van Capelle, 1986; Zhang et al., 2000). The current paradigm holds that reduced coupling conductance serves to insulate CPCs (Fast and Kléber, 1995; Glynn et al., 2014; Inada et al., 2014; Joyner and van Capelle, 1986; Kleber and Rudy, 2004; Rohr et al., 1997; Rohr et al., 1998; Shaw and Rudy, 1997; Zhang et al., 2000). The molecular mechanisms that impart the unique structure/function relationships in the hearts pacemaking complex, however, have yet to be determined. We examined this question under the assumption that aspects of CPC patterning that are required for their function should emerge during embryonic stages when ionic source-sink relationships between CPCs and WM would become untenable to impulse generation. Indeed our mathematical modeling indicated that cell size and coupling would be predicted to influence CPC activity as cardiac morphogenesis proceeds, which was supported by our data that cells within the forming sinoatrial node remain relatively small throughout heart development and loss the ability to mechanically couple with one another via loss of AJs.
The AJ is a major mediator of mechanotransduction in a variety of cell types (Barry et al., 2014; Huveneers and de Rooij, 2013; Liu et al., 2010; Nagafuchi et al., 1994; Yonemura et al., 2010). Furthermore, the cardiac AJ plays a direct role in maintaining the localization and stability of GJ proteins that electrically couple the WM in the adult heart (Kostetskii et al., 2005; Li, 2005; Li et al., 2012; Shaw et al., 2007; Smyth et al., 2010; Swope et al., 2012). As such, the lack of AJ formation among developing CPCs immediately led us to the hypothesis that disruption of cell junctions would serve to blunt the biomechanical inputs that stimulate hypertrophic growth in the WM, while simultaneously eliminating the interfaces required for stable GJ formation. Indeed, our data demonstrate that blocking AJ formation is sufficient to generate CPC-like morphological features outside of the forming SAN (Figure 4) and that presenting CPCs with a surface coated with N-Cad induces Cx40 trafficking to the plasma membrane(Figures 6E–6H).
Previous work has indicated that CPCs express transcription factors that suppress GJ expression (Espinoza-Lewis et al., 2009; Hoogaars et al., 2007; Mommersteeg et al., 2007; Wiese et al., 2009), which has led to a model in which transcriptional regulation of connexins determines CPC intracellular conductivity. Our data indicate that, in parallel to inhibition of mRNA production, embryonic CPCs also control extent of coupling by limiting connexin protein trafficking.
Somewhat surprisingly, both our simulations and cellular studies indicated that increasing CPC size negatively impacted function (Figures 1 and 5). How CPC size influences activity has remained unclear as previous reports have come to differing conclusions. Specifically, when the native heterogeneity of CPCs size present in the adult SAN has been compared to function, either no correlation was observed (Lyashkov et al., 2007; Monfredi et al., 2018), or larger cells isolated from the adult SAN were actually reported to have higher rates of activity (Honjo et al., 1996). Conversely, increased CPC size due to aging is associated with decreased activity (Larson et al., 2013), and transient swelling of CPCs also lowers their firing rate(Lei and Kohl, 1998). We see a dramatic impact on CPC function when these cells are placed under conditions that increase their size (Figures 5D, 5F–5G, and S5F–S5H). Interestingly, among relatively small CPCs in our studies we do see a broad range of possible rates (0.5–3.5hz) which would agree with studies from adult CPCs (Lyashkov et al., 2007; Monfredi et al., 2018). However, CPCs that undergo induced hypertrophy lost the capacity for the higher rates of activity (Figures 2E–2G, 5D–5G, S5F–S5H). Thus, from a developmental standpoint our data indicate that CPCs are capable of acquiring WM-like size characteristics and that events that prevent the hypertrophic growth that occurs in the WM may be necessary to preserve CPC function during cardiac morphogenesis. We would emphasize that our data do not implicate either the AJ or FLRT3 as regulators of cell fate decisions. Our data indicate that equal levels of N-Cad are present among CPCs and atrial WM at early developmental stages when CPCs have already functionally and molecularly diverged from the WM (Figures 2A and 3H–3I)(Bressan et al., 2013). As such, it appears that AJ engagement modulates characteristics such as cell size at time points well after lineage decisions have been made and serve as critical regulator of cellular features that impact CPC excitability.
In summary, our current study highlights that during embryonic cardiac morphogenesis CPCs structurally diverge from the WM, attaining cytoarchitectural features that would be predicted advantageous for overall excitability. Through this process, CPCs are held back from undergoing the hypertrophic growth detected in the WM. Furthermore, CPCs uniquely lose AJ engagement during these stages, which mechanically isolates them while also lowering their capacity to electrically couple to neighboring cells. Final our data highlight the sinoatrial node enriched factor, FLRT3, as a cell autonomous modulator of myocyte AJ engagement. These findings indicate that regulation of AJ formation is a critical step in maintaining structure/function relationships during developmental CPC maturation and that AJ dynamics may need to be considered as rational design approaches for generating cellular-based “biological pacemakers” continue to evolve for clinical uses.
Limitations of the Study:
We would highlight that increasing CPC size would not only be predicted to increase membrane capacitance but would also be predicted to affect the densities of the ionic currents required for electrical automaticity. In this report we do not address how individual channels change expression and/or localization in response to increasing cell size. As such our current data only indicate that increasing embryonic CPC size should negatively impact rate of activity and that the cells are not capable of acutely modulating current densities to compensate during the stages when they are structurally diverging from the WM. Similarly, we have used a culture system to induce hypertrophic growth in CPCs. We chose this system as it was not possible to examine functional differences among CPCs of differing sizes in vivo due to the syncytial electrical behavior of cardiomyocytes. As such, it is possible that mechanisms that allow CPCs to compensate for altered structure may exist in the developing heart and that such mechanisms are absent in our culture system. Finally, our data support a model by which electrical insulation can emerge as a product of decreased gap junctional density following AJ inhibition. Thus, mechanical insulation and electrical insulation would by intimately related. In the current study, however, we do not directly measure intercellular resistance between ether control or AJ manipulated CPCs. Therefore, we cannot rule out that an atypical form of electrical coupling that is below our resolution to detect can form independent of an AJ.
STAR Methods Text:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Michael Bressan (Michael_Bressan@med.unc.edu).
Materials availability
All reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
The three-dimensional monodomain equations were used to simulate the conduction of the electrical propagation through embryonic cardiac tissue. The monodomain model can be derived from a homogenized description of the excitation propagation that neglects the specific details of the tissue microarchitecture(Keener and Sneyd, 1998). In this model, the current density flux J is modeled using Ohm’s law, such that J = −σ∇V, where σ is the tissue conductivity tensor and V is the transmembrane potential difference. In general, the conductivity tensor σ includes information about the alignment of the cells, such that σ = σtI + (σl − σt)f ⊗ f, in which f is a vector parallel to the average cellular orientation, I is the identity tensor, and σt and σl are the conductivity coefficients in the transverse and longitudinal cell directions, respectively. Because the transverse propagation is negligible in the simulations, we simplify the conductivity tensor to be isotropic, such that σ = σI, with σ = σl = σt. The monodomain model reads:
in which χ is the ratio of membrane surface to tissue volume, Csp is the specific membrane capacitance, Iion is the sum of the transmembrane currents per unit area which depend on the gating variables w and on the voltage V. In the following, l and w denote the length and width of the cell respectively, α the volume fraction of the extracellular space, σc the cytosol conductance per unit length, and gc the gap junctional conductance. Following the approach of Hand et al.(Hand et al., 2009), assuming a structured network of cells with uniform (averaged) electrical properties, we can link the macroscopic conductivity of the tissue to the cell dimensions via:
Assuming the membrane surface area A and the cell capacitance C are related by the equation Cm = CspA, we can derive the cell dimensions l and w and the volume fraction of the extracellular space α. More specifically, we assume 1:10 ratio between the l and w, such that
in which is the cell volume, VCPC is the volume of the region containing the CPCs and nCPC is the number of CPCs contained in VCPC. Finally, the value of the membrane surface to tissue volume ratio χ is computed as nCPCA ∕ VCPC.
We extract the values of nCPC and VCPC from imaging data (see Figures 1C and S1). We derive the specific capacitance Csp in the CPCs region assuming CPCs are cylindrical and their capacitance changes with cell size between 20pF and 60pF as reported in the literature(Inada et al., 2014). We use the modified Courtmanche-Ramirez-Nattel ionic model(Courtemanche et al., 1998; McDougal et al., 2017) for the human atrial action potential to describe the ionic current density Iion in the working myocardium. We use the CPC model of Fabbri et al. 2017 to represent the action potential in the SAN region. The monodomain model is solved using a finite linear element method based on piecewise linear finite elements along with a semi-implicit time-stepping scheme(Rossi and Griffith, 2017).
Embryonic tissue geometry was segmented using Imaris (Bitplane). The three-dimensional geometry was prepared for simulations using MeshMixer (Autdodesk), QuadRemesher (Exoside) and Gmsh(Geuzaine and Remacle, 2009). Simulations were run using the reconstructed tissue geometry and features including CPC slow diastolic depolarization, action potential duration, propagation pattern, and conduction velocities within and at the periphery of the forming sinoatrial node were compared back to recorded data from tissue slices. For sensitivity analysis (Cm vs gc) slope of diastolic depolarization, action potential duration, and conduction velocities were allowed to diverge from recorded data and rate, rhythmicity, and the ability of the SAN to initiate an action potential and/or drive the atria was scored to determine parameters ranges that allowed for functional pacemaking (Figure S1).
The code developed in this work, BeatIt (available at github.com/rossisimone/beatit), relies on the parallel C++ finite element library libMesh(Kirk et al., 2006) and on PETSc linear solvers(Balay et al., 2017; Balay et al., 1997). The visualization of the results and their analysis have been carried out using Paraview(Ahrens, 2005) and MATLAB (The Mathworks, Inc.).
| Csp[μF/cm] | Cm [pF] | χ [1/cm] | σ [mS/cm] | σc [mS/cm] | gc [nS] | VCPC [cm3] | nCPC | |
|---|---|---|---|---|---|---|---|---|
| CPCs | 3 | 15 – 60 | 1800 – 5640 | 0.0045 – 0.1318 | 6.7 | 0.3 – 3.3 | 3.496e-6 | 986 |
| WM | 1 | – | 3400 | 1.0 | – | – | – | – |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Fertilized chicken eggs were obtained from Pilgrim’s Farms (Sanford, NC, USA) and Allen Harim Hatchery (Liberty, NC, USA), and placed at 38°C in a humidified incubator. Eggs were incubated to desired developmental stages according to Hamburger-Hamilton(Hamburger and Hamilton, 1992). Wildtype mice embryos were kindly provided by Joan Taylor. For immunofluorescence studies mice embryos were harvested at E13.5 for adherens junction protein expression analysis. Animal procedures and protocols were authorized by the University of North Carolina’s American Association for Accreditation of Laboratory animal Care committee.
METHOD DETAILS
Transfection
Transfections were carried out as previously described(Goudy et al., 2019). In brief, chicken embryos were incubated at 38°C until Hamilton-Hamburger stage 16. Eggs were windowed and a DNA solution (Lipofectamine 3000 reagent (Invitrogen) mixed with plasmid DNA diluted in Opti-MEM (Gibco)) was micro-injected into the pericardial space surrounding the heart of the embryo. Eggs were sealed and placed in an incubator at 38°C until the desired developmental stage. Several plasmids were described and validated previously including: U6-NCad shRNA 1/2, ITR-αMHC-HyPBase-CAG-palmTagRFPII-2A-h2bEGFP-ITR, and ITR-αMHC-HyPBase-CAG-TagBFP-2A-h2bEGFP-ITR (see table 1 for references). The ITR-αMHC-Hybase-CAG-palmTagBFP-2A-h2bEGFP-ITR was made by inserting palmTagBFP into the palmTagRFPII position of the palmTagRFPII-2A-h2bEGFP-ITR construct using Gibson Assembly cloning (GeneArt). The ITR-αMHC-Hybase-cNCadΔ112–624-2A-palmTagRFPII-ITR construct was generated by subcloning a fragment of the chick N-cadherin cDNA lacking ectodomains 1–5 (Ong et al., 1998) into the ITR-αMHC-HybaseCAG-palmTagRFPII-2A-h2bEGFP-ITR construct. The ITR-CMV-HyBase-CAG-cFLRT3–2A-h2bEGFP-ITR and ITR-CMV-HyBase-CAG-cFLRT3[GG]-2A-h2bEGFP-ITR constructs were generated by cloning chick FLRT3 or a mutated chick FLRT3 in which amino acids 579–580 were replaced with Glycines into the palmTagRFPII position of the previously described ITR-CMV-HyBase-CAG-palmTagRFPII-2A-h2bEGFP-ITR (Goudy et al., 2019) using Gibson assembly (NEBuilder HiFi DNA Assembly Cloning Kit, New England Biolabs, Ipswich, MA). The ITR-U6-FLRT3 shRNA-hPGK-EGFP-ITR plasmid containing the previously described shRNA AATTTCAGGCTACTGCTGCGATT (Tomas et al., 2011), was synthesized by VectorBuilder Inc. (Chicago, IL).
Optical Mapping and Calcium Imaging
Optical mapping (voltage Imaging) analysis was done using previously described methods(Bressan et al., 2018; Bressan et al., 2013; Bressan et al., 2014a; Bressan et al., 2014b). Briefly, inflow and atrial tissue were explanted and placed at 37°C in fresh 1X HBSS (1.26 mM CaCl2 anhydrous, 0.49 mM MgCl2–6H2O, 0.41 mM MgSO4–7H2O, 5.33 mM KCl, 0.44 mM KH2PO4, 4.17 mM NaHCO3, 137.93 NaCl, 0.33 mM Na2HPO4 anhydrous, 5.56 mM D-Glucose) solution for 10 minutes to recover. Explants were then place into a staining solution of 10μM Di-4 ANEPPS (Invitrogen) voltage sensitive dye and 10μM (−)Blebbistatin (Millipore Sigma) in 1X HBSS for 10 minutes at 37°C with 95% O2, and 5% CO2 incubation. After incubation, explants’ action potentials were recorded in 1X HBSS solution perfused with 95% O2, and 5% CO2 on a temperature-controlled imaging chamber (36 +/−0.5°C).
Calcium transient activity of cultured cells was imaged by staining cells with Cal-520, AM (AAT Bioquest, Sunnyvale, CA); 5uM in in DMEM/F12 containing 15% FBS. Cultures were allowed to recover for 30 minutes at 37°C with 5% CO2. Following recovery individual cells were imaged for calcium activity in a Tokai Hit Stage Top Incubator at 37°C and 5% CO2. Analysis and quantification methods are described below.
Cytopainter – Cell Plasma Membrane Staining Kit – Orange Fluorescence (Abcam, Cambridge, MA) was used for a subset of cells that did not contain a genetic membrane (Figures S5F–S5K). Prior to plating on micropatterned polyacrylamide gels, dissociated cell pellets were stained with assay buffer containing 1X staining solution. The pellets were incubated at 37°C with 5% CO2 for 20 minutes. After 3–5 washes in complete media (15% FBS in DMEM), cells were resuspended and plated on PA gels.
Micro-electrode Array Recordings
At the desired developmental stage, heart tissue was isolated using surgical forceps, clipper neuro scissors (Fine Science Tools), and curved tenotomy scissors and placed in pre-warmed 1X HBSS solution. Explants in 1X HBSS were then placed at 37°C for 25 minutes to recover. Explants were then transferred to a 6-well MEA recording chamber (Multichannel systems) precoated with fibronectin (Corning) at 2 μg/cm and pre-warmed to 37°C. Explants were cultured in Dulbecco’s Modified Eagle’s Medium (Sigma Aldrich), 15% fetal bovine serum (Gibco), and 1% Penicillin-Streptomycin (Gibco) for 16 hrs to allow for adherence to the MEA. Three sets of field potential recordings were taken per explant (60 seconds per recording) using three technical replicates per group. Data were recorded and analyzed using MC_Rack V.4.6.2 and MEA_select V.1.3.0 software (Multichannel systems).
Immunofluorescence
Whole mount –
Whole hearts were fixed in 4% PFA at 4°C, rocking for 1 hour, then washed three times with 1X DPBS (2.67 mM KCl, 1.47 mM KH2PO4, 137.93 mM NaCl, 8.06 mM Na2HPO4–7H2O) with 0.1% Tween-20 for 20 minutes. Tissue was blocked at room temperature in 1x DPBS,1% BSA, 0.1% Tween-20 for 2 hours. Simultaneously, the primary antibody MF20 (Invitrogen), was blocked at 1:500. Primary antibody was applied at 4°C overnight followed by six 30 minute washes at room temperature in 1X DPBS. Secondary antibody (1:500) and DAPI (1:2000) were applied overnight at 4°C in 1X DPBT. The following day tissue was washed six times at room temperature in 1X DPBS for 30 minutes in the dark.
Thick section –
Whole hearts were fixed in 4% PFA at 4°C, rocking for 20 minutes, then washed three times with 1X DPBS with 0.1% Tween-20 (1X DPBT) for 15 minutes. Tissue was blocked at room temperature in 1X DPBS, 1% BSA, 0.1% Tween-20 for 1 hour. Simultaneously, the primary antibodies MF20 (Invitrogen) and N-Cadherin (Millipore Sigma), were blocked at 1:500 and 1:500 dilutions, respectively. Primary antibodies were applied at 4°C overnight followed by four 15 minute washes at room temperature in 1X DPBS. Secondary antibodies (1:500) were applied at room temperature in the dark for 2 hours in 1X DPBS. Tissue was washed 4 times with 1X DPBT for 15 minutes followed by application of DAPI (1:1000) for 30 minutes at room temperature in the dark. Tissues were washed three times with 1X DPBT for 15 minutes at room temperature in the dark.
Thin section –
Paraffin sections and cryosections were prepared using standard techniques. Paraffin sections were heated to 55°C for 15 minutes and were deparaffinized using xylene washes for 60 minutes. Sections were then rehydrated through a graded ethanol series. Antigen retrieval was performed for 30 minutes in citrate buffer (10 mM Tri-sodium citrate,0.05% Tween-20, pH 6.0 with HCl). Slides were allowed to cool and then washed with 1X DPBS for 5 minutes and blocked (1X DPBS, 1% BSA, 0.1% Tween-20) for 1 hour concurrent with antibody blocking. Primary antibodies MF20, N-cadherin, β-catenin (ThermoFisher Scientific), and αT-catenin (CTNNA - Abcam) were blocked at 1:500 dilution. Slides were incubated at 4°C overnight in primary antibody. Slides were washed three times with 1X DPBS for 10 minutes. Secondary antibodies were applied at 1:500 dilutions in 1X DPBS for 1 hour in the dark and were washed three times in 1X DPBS for 10 minutes. Cryosections were washed three times with 1X DPBS with 0.1% Tween-20 (1X DPBT) for 5 minutes and blocked (1X DPBS, 1% BSA, 0.1% Tween-20) for 1 hour concurrent with antibody blocking. Primary antibodies, MF20, N-cadherin, β-catenin, and αT-catenin, were blocked at 1:500 dilution. Slides were incubated at 4°C overnight in primary antibody. Slides were washed three times with 1X DPBS for 10 minutes. Secondary antibodies were applied at 1:500 dilutions in 1X DPBS for 1 hour in the dark and washed off using three 10 minutes washes in 1X DPBS. Slides were mounted with a 24×55 microscope cover glass (Fisher Scientific) and Flouroshield mounting media with DAPI (Abcam) or ProLong Gold (Invitrogen).
Cell Dissociation
Dissociations were carried out using previously described protocols(Goudy et al., 2019). Transfected isolated cardiac tissue was placed in a 0.17% trypsin (Sigma-Aldrich, St. Lois, MO) solution for 30 minutes and centrifuged at 3000 rpm for 5 minutes to obtain a pellet of cells. After 3–5 washes in complete media (15% FBS in DMEM), cells were plated on human fibronectin coated 8-well culture slides (Corning) or 35/10 mm glass bottom culture dishes (Greinerbio one). Cultures were fixed at 4°C for 30 minutes in 4% PFA. Immunohistochemistry was carried out using N-cadherin, MF20, Connexin-40 (Life Span Life Sciences), GFP (Invitrogen), and Alexa Fluor 647 Phalloidin (Invitrogen) primary antibodies.
RNAscope In Situ Hybridization
Fluorescent RNA staining was performed using the RNAScope Multiplex Fluorescent Assay (ACDBio, Newark, CA) following manufacturer’s protocol for cultured adherent cells. Cultures were fixed in 4% PFA at 4°C for 30 minutes, permeabilized by washing three times with 1X DPBT for 10 minutes and dehydrated in a graded ethanol series at 4°C. Cells were then rehydrated at 4°C and treated with Protease III (1:150 in 1X DPBT) for 10 minutes at room temperature and washed with 1X DPBT. Hybridization with the target probes (Hcn4, Gja5, Flrt3) was carried out at 40°C for 2 hours. Cultures were then immersed in an alternating series of wash buffers at room temperature and fluorescent amplification buffers at 40°C before being counterstained with DAPI and mounted with ProLong Gold (Invitrogen). Cultures were stored at 4°C until imaging.
Polyacrylamide gels
Polyacrylamide (PA) gels were prepared using previously described methods(Chopra et al., 2011). Commercially available solutions of 40% acrylamide and 2% bisacrylamide (Sigma-Aldrich, St. Louis, MO) were added to final concentrations of 7.5% and 0.05% respectively or 10% and 0.5% respectively to produce gels of 4 kPa and 60 kPa sheer moduli. Beads of 2 μm diameter (Thermo Fisher Scientific, Waltham, MA) were added to the acrylamide solutions as 3D volumetric standards for downstream analysis. To polymerize, 2 μL tetramethylene diamine (Sigma-Aldrich, St. Louis, MO) and 200 μL of 1% ammonium persulfate (Sigma-Aldrich, St. Louis, MO) were added to the solutions to a total volume of 2 mL in 1X DPBS. A 400 μL aliquot of the polyacrylamide was added to a 35/10 mm glass bottom dish (Greiner Bio One, Monroe, NC). Dishes were plasma activated (Harrick Plasma, Ithaca, NY) before being treated with 3-aminopropyltriethanolamine (Sigma-Aldrich, St. Louis, MO) and glutaraldehyde (Sigma-Aldrich, St. Louis, MO). Gels were covered with a 1 inch diameter by 1/16 inch thick quartz coverslip (Thermo Fisher Scientific, Waltham, MA) pretreated with Sigmacote (Sigma-Aldrich, St. Louis, MO). After a 30-minute polymerization step, the coverslip was removed and a solution of 50 mM HEPES buffer (pH 8) containing the cross-linker sulfo-N-sulfosuccinimidyl-6-(4′-azido-2′-nitrophenylamino) hexanoate (Sigma-Aldrich, St. Louis, MO) at a concentration of 0.5 mg/mL was added to the dishes, submerging the gels. The dishes were then placed under ultraviolet light (Jetlight Company, Inc., Irvine, California) and irradiated for 7 minutes. Anti-Human Fc antibody (Thermo Fisher Scientific, Waltham, MA) was added at a concentration of 30 μg/cm2 and incubated overnight at 4°C with rocking. After washes in 50 mM HEPES buffer, recombinant Human N-cadherin Fc chimera (R&D Systems, Minneapolis, MN) was added at a concentration of 10 μg/cm2 and incubated overnight at room temperature with rocking. PA gels were then incubated with 1.5% BSA in 50 mM HEPES at room temperature for 1 hour and washed prior to cell plating. Micropatterned gels were custom fabricated from 50 kPa PA (4Dcell). Single cells seeded on 20um diameter micropatterns were selected for imaging.
Transmission Electron Microscopy (TEM)
E6 chicken embryos were fixed overnight in 4% PFA at 4°C and washed three times in 1X DPBS for 20 minutes. Embryos were embedded in 3% low melting temperature agarose (LMA, Apex) in 1X DPBS. Embedded tissue was cut on a Vibratome into 200 μm thick sections. Sections were immersion-fixed in 4% paraformaldehyde/1% glutaraldehyde/0.1M sodium phosphate, pH 7.4, and stored for several days at 4°C. After post-fixation for 1 hour in potassium ferrocyanide-reduced osmium (1% osmium tetroxide/1.25% potassium ferrocyanide/0.1M sodium phosphate buffer), samples were dehydrated through a graded series of ethanol (30%, 50%, 75%, 90%, 100%, 100%) and propylene oxide, then infiltrated and embedded in PolyBed 812 epoxy resin (Polysciences, Warrington, PA). Using a diamond knife, 1 μm semi-thin sections were cut, mounted on slides, and stained with 1% toluidine blue to examine by light microscopy and isolate the region of interest. Ultrathin sections (70–80 nm) were cut with a diamond knife and mounted on 200 mesh copper grids followed by staining with 4% aqueous uranyl acetate for 12 minutes and lead citrate for 8 minutes.
Stained sections were imaged with a ThermoFisher (FEI) Tecnai 12 G2 transmission electron microscope (Hillsboro, OR) operated at 80 kV using a 1k x 1k CCD camera (Model 794, Gatan, Pleasanton, CA) equipped with Digital Montage software or at 120kV using a 4k x 4k CMOS camera (Model Rio16, Gatan, Pleasanton, CA). CPCs were defined as the cell population located in the right inflow myocardium overlapping with the action potential initiation site (Figure 1A)((Bressan et al., 2018; Bressan et al., 2013)). This region was also confirmed to be enriched for Hcn4, Shox2, Bmp2, Smoc2, and depressed in Nkx2.5 and Gja5 expression as previously reported (Christoffels et al., 2006; Espinoza-Lewis et al., 2009; van Eif et al., 2019; Ye et al., 2015).
RT-qPCR
Cardiac tissue was isolated and placed in TRIzol (Invitrogen) and RNA was extracted using manufacturer’s protocol for PureLink RNA Mini Kit (Invitrogen). Approximately 20 ng of RNA was obtained from each sample and cDNA was generated using Superscript IV according to the manufacturer’s guidelines (Invitrogen). RT-qPCR was performed on a Quantastudio 6 Flex Real-Time PCR System (Invitrogen) using iTaq 2X SYBR Green Supermix (Bio-Rad) with 3 biological replicates, run in triplicate. HCN4 (Forward primer: GCGACTTCAGGTTTTATTGGG Reverse primer: GAGGACAAGGTCGATGAGGA), N-cadherin (Forward primer: ATGGCAAATGAAGGTGAAGC Reverse primer: CTTCAGATGGCTGCTGTCCT), αT-catenin (Forward primer: TTTGTGCGGTATATTGCTCAGAAG Reverse primer: CTTGGAGATACTGGCTTTAAGATGC), FLRT3 (Forward Primer: GAACAGCCACAGTGAAAGCA Reverse Primer: TCGATGCATTCAGGTGTCTC) and gamma-tubulin (Forward primer: CAACTTCGATGAGCTGGACA Reverse primer: CCTCTTGTCCCCATGTCACT), were amplified and expression levels were normalized to Gapdh (Forward primer: CCCCCAATGTCTCTGTTGTT Reverse primer: CATCCAAGGTGGAGGAATGG). Calculated fold changes are relative to the atria.
QUANTIFICATION AND STATISTICAL ANALYSIS
Microscopy
Optical mapping samples were recorded using a CMOS Camera (MiCAM Ultima, SciMedia) and vertical THT Microscope (SciMedia). The data was processed using BV-Ana software (SciMedia). TEM samples were observed using a JEOL JEM-1230 transmission electron microscope operating at 80kV (JEOL USA, Inc., Peabody, MA) and images were acquired with a Gatan Orius SC1000 CCD Digital Camera and Gatan Microscopy Suite 3.0 software (Gatan, Inc., Pleasanton, CA). Whole mount and thick section images were acquired using the LSM 800 confocal microscope (Zeiss) and 25x liquid immersion lens (NA 1.4). Thin sections were imaged using the Olympus IX-81 widefield inverted microscope and Metamorph software. Dissociated cell cultures were imaged on a LSM 880 laser scanning confocal microscope with an Airyscan super resolution sensor. Post processing was performed using ZEN Blue Imaging software (Zeiss). Cells on polyacrylamide gels were imaged using an Olympus FV3000RS confocal microscope with a live cell Tokai Hit stage-top incubator. 3D reconstruction was carried out by batch deconvolution of confocal images in AutoQuantX3 (Media Cybernetics). The deconvolution files were then opened in IMARIS 9.3 software (Bitplane) to render individual cell surfaces for quantification of cell features such as surface area and volume. Calcium transient data was processed using ImageJ V2.0.0 (NIH). RNAScope data was analyzed using ImageJ V2.0.0 (NIH). All statistical analysis of data was assessed using a two-tailed unpaired Student t-test in Microsoft Excel Software. Significance was determined based on p-values as follows: one star if p-value < 0.05, two stars for p-values < 0.01, three stars to indicate p-values < 0.001, four stars indicate p-values < .0001. All experimental numbers (n) are documented in figure legends.
Quantification Area, Surface area, and Volume
Deconvolved image files were quantified in IMARIS. For in vivo imaging, each visible cell was manually selected based on the membrane reporter signal. Briefly, the construct surface and mask-selection features in IMARIS were used to select a 3D space around each cell. The membrane reporter signal within the manually created surface was then used to render the surface structure the cell and the statistical analysis feature in IMARIS was used to export surface area and volume data. In vitro cell size information was generated using the same workflow, however, 2μm diameter beads embedded in the PA gels were used to standardize surface area and volume data. For a select set of studies (micropatterned PA gels), beads could not be embedded to serve as size standards. For these studies (X,Y) area is reported for the size of the cells. Correlation between (X,Y) area and 3D surface area and volume are provided in Figures S5D and S5E.
Quantification of Cell Contacts
Confocal images of genetically modified cells in vitro, were assessed for the number of cell contacts based on N-Cad IHC. Cells were plated at a density of 1×105 cells per cm2 to avoid multilayered cultures. Transfected cells were randomly selected based on fluorescent reporter signals and z-stacks were taken for the field of cells in the immediate area (n=20 images per condition). For each genetically modified cell, the number of N-Cad positive interfaces (puncta or ribbons) were manually counted to determine the number of cell contacts each cell made.
Quantification of Calcium Transients
Three 10 second acquisitions were recorded for each cell at 30 fps on an Olympus FV3000RS confocal microscope. Using ImageJ V2.0.0 (NIH) an intensity profile of each cell per frame was plotted to generate a line graph. Each of the three series were analyzed for the number of calcium transient peaks and averaged to determine a mean rate (Hz) per cell over the three series. A custom macro was written in excel to calculate time between calcium transient peaks for each imaging series and the deviation from the average interval per cell was used to calculate rhythmicity. Linear Regression (surface are/volume ration vs rate) was performed using (GraphPad Prism Software).
Quantification of N-Cadherin Junctional Intensity
Maximum intensity z-projections were generated for each cell pair using ImageJ V2.0.0 (NIH). A 5μm wide strip was then drawn through each junction (perpendicular to the plane of cell-cell contact) and the average intensity across the strip was calculated using the plot profile function in ImageJ. Intensity profiles were then exported to excel where regions +/− 10μm from the cell contact were quantified and the area under the curve was calculated.
Supplementary Material
Highlights.
Adherens junction (AJ) engagement impacts cardiac pacemaker cell (CPC) excitability
Blocking AJ formation is sufficient to generate CPC-like morphological features
AJ formation induces gap junction trafficking to the plasma membrane in CPCs
FLRT3 is a cell autonomous regulator of CPC cytoarchitecture
Acknowledgements:
Mouse tissue was a generous gift from Joan Taylor’s laboratory. This work was supported by National Institutes of Health grants T32HL06976 (NHLBI) to K.T., UL1TR002489 (NCATS) to W.J.P, R01HL146626 (NHLBI) to M.B., and American Heart Association grants 19POST34380213 to R.S. and 19CDA34760248 to M.B. Imaging was performed in the Hooker Imaging Core Facility and Neuroscience Microscopy Core at the University of North Carolina (supported NIH-NINDS P30 NS045892 and NIH-NICHD U54 HD079124).
Footnotes
Declaration of Interests:
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Ahrens J, Geveci B, and Law C (2005). Paraview: An End-User Tool for Large Data Visualization. In Visualization Handbook, pp. 717–731. [Google Scholar]
- Balay S, Abhyankar S, Adams M, Brown J, Brune P, Buschelman K, Dalcin LD, Eijkhout V, Gropp W, Kaushik D, et al. (2017). PETSc Users Manual Revision 3.8 (; Argonne National Lab. (ANL), Argonne, IL (United States)), pp. Medium: ED; Size: 264 p. [Google Scholar]
- Balay S, Gropp WD, McInnes LC, and Smith BF (1997). Efficient Management of Parallelism in Object-Oriented Numerical Software Libraries. In Modern Software Tools for Scientific Computing, Arge E, Bruaset AM, and Langtangen HP, eds. (Boston, MA: Birkhäuser Boston; ), pp. 163–202. [Google Scholar]
- Barry AK, Tabdili H, Muhamed I, Wu J, Shashikanth N, Gomez GA, Yap AS, Gottardi CJ, de Rooij J, Wang N, et al. (2014). α-catenin cytomechanics--role in cadherin-dependent adhesion and mechanotransduction. Journal of Cell Science 127, 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyett MR, Honjo H, research, I.K.C., and 2000 (2000). The sinoatrial node, a heterogeneous pacemaker structure. academicoupcom. [DOI] [PubMed] [Google Scholar]
- Bressan M, Henley T, Louie JD, Liu G, Christodoulou D, Bai X, Taylor J, Seidman CE, Seidman JG, and Mikawa T (2018). Dynamic Cellular Integration Drives Functional Assembly of the Heart's Pacemaker Complex. CELREP 23, 2283–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan M, Liu G, and Mikawa T (2013). Early mesodermal cues assign avian cardiac pacemaker fate potential in a tertiary heart field. Science 340, 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan M, Yang PB, Louie JD, Navetta AM, Garriock RJ, and Mikawa T (2014a). Reciprocal myocardial-endocardial interactions pattern the delay in atrioventricular junction conduction. 141, 4149–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan MC, Louie JD, and Mikawa T (2014b). Hemodynamic Forces Regulate Developmental Patterning of Atrial Conduction. PLoS ONE 9, e115207–115220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Koh E, Yoder M, and Gumbiner BM (2009). A Protocadherin-Cadherin-FLRT3 Complex Controls Cell Adhesion and Morphogenesis. PLoS ONE 4, e8411–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra A, Tabdanov E, Patel H, Janmey PA, and Kresh JY (2011). Cardiac myocyte remodeling mediated by N-cadherin-dependent mechanosensing. American journal of physiology Heart and circulatory physiology 300, H1252–H1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels VM, Mommersteeg MT, Trowe MO, Prall OW, de Gier-de Vries C, Soufan AT, Bussen M, Schuster-Gossler K, Harvey RP, Moorman AF, et al. (2006). Formation of the venous pole of the heart from an Nkx2–5-negative precursor population requires Tbx18. Circ Res 98, 1555–1563. [DOI] [PubMed] [Google Scholar]
- Courtemanche M, Ramirez RJ, and Nattel S (1998). Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am J Physiol 275, H301–321. [DOI] [PubMed] [Google Scholar]
- Egea J, Erlacher C, Montanez E, Burtscher I, YAMAGISHI S, Hess M, Hampel F, Sanchez R, Rodriguez-Manzaneque MT, Bosl MR, et al. (2008). Genetic ablation of FLRT3 reveals a novel morphogenetic function for the anterior visceral endoderm in suppressing mesoderm differentiation. Genes & Development 22, 3349–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza-Lewis RA, Yu L, He F, Liu H, Tang R, Shi J, Sun X, Martin JF, Wang D, Yang J, et al. (2009). Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2–5. Developmental Biology 327, 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri A, Fantini M, Wilders R, and Severi S (2017). Computational analysis of the human sinus node action potential: model development and effects of mutations. J Physiol 595, 2365–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fast VG, and Kléber AG (1995). Block of impulse propagation at an abrupt tissue expansion: evaluation of the critical strand diameter in 2- and 3-dimensional computer models. Cardiovascular Research 30, 449–459. [PubMed] [Google Scholar]
- Forbes MS, and Sperelakis N (1985). Intercalated discs of mammalian heart: A review of structure and function. Tissue and Cell 17, 605–648. [DOI] [PubMed] [Google Scholar]
- Geuzaine C, and Remacle J-F (2009). Gmsh: A 3-D finite element mesh generator with built-in pre- and post-processing facilities. International Journal for Numerical Methods in Engineering 79, 1309–1331. [Google Scholar]
- Glynn P, Onal B, and Hund TJ (2014). Cycle length restitution in sinoatrial node cells: a theory for understanding spontaneous action potential dynamics. PLoS One 9, e89049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudy J, Henley T, Méndez HG, and Bressan M (2019). Simplified platform for mosaic in vivo analysis of cellular maturation in the developing heart. Scientific Reports 9, 10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grijalva SI, Gu JM, Li J, Fernandez N, Fan J, Sung JH, Lee SY, Herndon C, Buckley EM, Park SJ, et al. (2019). Engineered Cardiac Pacemaker Nodes Created by TBX18 Gene Transfer Overcome Source-Sink Mismatch. Adv Sci (Weinh) 6, 1901099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutstein DE, Liu FY, Meyers MB, Choo A, and Fishman GI (2003). The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J Cell Sci 116, 875–885. [DOI] [PubMed] [Google Scholar]
- Hamburger V, and Hamilton HL (1992). A series of normal stages in the development of the chick embryo. 1951. Dev Dyn 195, 231–272. [DOI] [PubMed] [Google Scholar]
- Hand PE, Griffith BE, and Peskin CS (2009). Deriving macroscopic myocardial conductivities by homogenization of microscopic models. Bull Math Biol 71, 1707–1726. [DOI] [PubMed] [Google Scholar]
- Honjo H, Boyett MR, Coppen SR, Takagishi Y, Opthof T, Severs NJ, and Kodama I (2002). Heterogeneous expression of connexins in rabbit sinoatrial node cells: correlation between connexin isotype and cell size. Cardiovascular Research 53, 89–96. [DOI] [PubMed] [Google Scholar]
- Honjo H, Boyett MR, Kodama I, and Toyama J (1996). Correlation between electrical activity and the size of rabbit sino-atrial node cells. J Physiol 496 ( Pt 3), 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogaars WM, Engel A, Brons JF, Verkerk AO, de Lange FJ, Wong LY, Bakker ML, Clout DE, Wakker V, Barnett P, et al. (2007). Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev 21, 1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huveneers S, and de Rooij J (2013). Mechanosensitive systems at the cadherin-F-actin interface. J Cell Sci 126, 403–413. [DOI] [PubMed] [Google Scholar]
- Inada S, Zhang H, Tellez JO, Shibata N, Nakazawa K, Kamiya K, Kodama I, Mitsui K, Dobrzynski H, Boyett MR, et al. (2014). Importance of gradients in membrane properties and electrical coupling in sinoatrial node pacing. PLoS One 9, e94565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens B, Goossens S, Staes K, Gilbert B, van Hengel J, Colpaert C, Bruyneel E, Mareel M, and Van Roy F (2001). αT-Catenin: a novel tissue-specific β-catenin-binding protein mediating strong cell-cell adhesion. Journal of Cell Science 114, 3177–3188. [DOI] [PubMed] [Google Scholar]
- Jones SA, Yamamoto M, Tellez JO, Billeter R, Boyett MR, Honjo H, and Lancaster MK (2008). Distinguishing Properties of Cells From the Myocardial Sleeves of the Pulmonary Veins: A Comparison of Normal and Abnormal Pacemakers. Circulation: Arrhythmia and Electrophysiology 1, 39–48. [DOI] [PubMed] [Google Scholar]
- Joyner RW, and van Capelle FJ (1986). Propagation through electrically coupled cells. How a small SA node drives a large atrium. Biophys J 50, 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor N, Liang W., n, E.M.a., and Cho HC (2012). Direct conversion of quiescent cardiomyocytes to pacemaker cells by expression of Tbx18. Nature Biotechnology 31, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaulanov E, Böttcher RT, Stannek P, Wu W, Rau M, Ogata S, Cho KWY, and Niehrs C (2009). Unc5B interacts with FLRT3 and Rnd1 to modulate cell adhesion in Xenopus embryos. PLoS ONE 4, e5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keener JP, and Sneyd J (1998). Mathematical physiology (New York: Springer; ). [Google Scholar]
- Kirchhof CJ, Bonke FI, Allessie MA, and Lammers WJ (1987). The influence of the atrial myocardium on impulse formation in the rabbit sinus node. Pflugers Arch 410, 198–203. [DOI] [PubMed] [Google Scholar]
- Kirk BS, Peterson JW, Stogner RH, and Carey GF (2006). libMesh: a C++ library for parallel adaptive mesh refinement/coarsening simulations. Engineering with Computers 22, 237–254. [Google Scholar]
- Kleber AG, and Rudy Y (2004). Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 84, 431–488. [DOI] [PubMed] [Google Scholar]
- Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, and Radice GL (2005). Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. 96, 346–354. [DOI] [PubMed] [Google Scholar]
- Larson ED, Clair J, the, W.S.P.o., and 2013 (2013). Depressed pacemaker activity of sinoatrial node myocytes contributes to the age-dependent decline in maximum heart rate. National Acad Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, and Kohl P (1998). Swelling-induced decrease in spontaneous pacemaker activity of rabbit isolated sino-atrial node cells. Acta Physiol Scand 164, 1–12. [DOI] [PubMed] [Google Scholar]
- Li J (2005). Cardiac-Specific Loss of N-Cadherin Leads to Alteration in Connexins With Conduction Slowing and Arrhythmogenesis. 97, 474–481. [DOI] [PubMed] [Google Scholar]
- Li J, Goossens S, van Hengel J, Gao E, Cheng L, Tyberghein K, Shang X, De Rycke R, Van Roy F, and Radice GL (2012). Loss of αT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. Journal of Cell Science 125, 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linscheid N, Logantha SJRJ, Poulsen PC, Zhang S, Schrölkamp M, Egerod KL, Thompson JJ, Kitmitto A, Galli G, Humphries MJ, et al. (2019). Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nature Communications, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, and Chen CS (2010). Mechanical tugging force regulates the size of cell-cell junctions. Proceedings of the National Academy of Sciences of the United States of America 107, 9944–9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, James TN, Yamamoto S, and Terasaki F (1993). Cardiac conduction system in the chicken: Gross anatomy plus light and electron microscopy. The Anatomical Record 236, 493–510. [DOI] [PubMed] [Google Scholar]
- Luo Y (2003). Cadherin-mediated adhesion is essential for myofibril continuity across the plasma membrane but not for assembly of the contractile apparatus. Journal of Cell Science 116, 1471–1479. [DOI] [PubMed] [Google Scholar]
- Luo Y, Ferreira-Cornwell M, Baldwin H, Kostetskii I, Lenox J, Lieberman M, and Radice G (2001). Rescuing the N-cadherin knockout by cardiac-specific expression of N- or E-cadherin. Development 128, 459–469. [DOI] [PubMed] [Google Scholar]
- Lyashkov AE, Juhaszova M, Dobrzynski H, Vinogradova TM, Maltsev VA, Juhasz O, Spurgeon HA, Sollott SJ, and Lakatta EG (2007). Calcium Cycling Protein Density and Functional Importance to Automaticity of Isolated Sinoatrial Nodal Cells Are Independent of Cell Size. Circulation Research 100, 1723–1731. [DOI] [PubMed] [Google Scholar]
- Maretto S, Müller P-S, Aricescu AR, Cho KWY, Bikoff EK, and Robertson EJ (2008). Ventral closure, headfold fusion and definitive endoderm migration defects in mouse embryos lacking the fibronectin leucine-rich transmembrane protein FLRT3. Developmental Biology 318, 184–193. [DOI] [PubMed] [Google Scholar]
- Masson-Pevet M, Bleeker WK, and Gros D (1979). The plasma membrane of leading pacemaker cells in the rabbit sinus node. A qualitative and quantitative ultrastructural analysis. Circ Res 45, 621–629. [DOI] [PubMed] [Google Scholar]
- Masson-Pevet MA, Bleeker WK, Besselsen E, Treytel BW, Jongsma HJ, and Bouman LN (1984). Pacemaker cell types in the rabbit sinus node: a correlative ultrastructural and electrophysiological study. J Mol Cell Cardiol 16, 53–63. [DOI] [PubMed] [Google Scholar]
- Mavroidis M, Athanasiadis NC, Rigas P, Kostavasili I, Kloukina I, Te Rijdt WP, Kavantzas N, Chaniotis D, van Tintelen JP, Skaliora I, et al. (2020). Desmin is essential for the structure and function of the sinoatrial node: implications for increased arrhythmogenesis. Am J Physiol Heart Circ Physiol 319, H557–H570. [DOI] [PubMed] [Google Scholar]
- McDougal RA, Morse TM, Carnevale T, Marenco L, Wang R, Migliore M, Miller PL, Shepherd GM, and Hines ML (2017). Twenty years of ModelDB and beyond: building essential modeling tools for the future of neuroscience. J Comput Neurosci 42, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel CD, Li Y, Raza Q, Stolz DB, and Kwiatkowski AV (2019). Vinculin anchors contractile actin to the cardiomyocyte adherens junction. Mol Biol Cell 30, 2639–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzano V, Liang Y, Wright AT, Lyon RC, Pfeiffer E, Song MY, Gu Y, Dalton ND, Scheinman M, Peterson KL, et al. (2016). Desmosomal junctions are necessary for adult sinus node function. Cardiovasc Res 111, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Zhang L, Sekiguchi A, Hadama T, and Shimada T (2002). Structural differences in the cytoarchitecture and intercalated discs between the working myocardium and conduction system in the human heart. Heart and Vessels 16, 232–240. [DOI] [PubMed] [Google Scholar]
- Mommersteeg MT, Hoogaars WM, Prall OW, de Gier-de Vries C, Wiese C, Clout DE, Papaioannou VE, Brown NA, Harvey RP, Moorman AF, et al. (2007). Molecular pathway for the localized formation of the sinoatrial node. Circ Res 100, 354–362. [DOI] [PubMed] [Google Scholar]
- Monfredi O, Tsutsui K, Ziman B, Stern MD, Lakatta EG, and Maltsev VA (2018). Electrophysiological heterogeneity of pacemaker cells in the rabbit intercaval region, including the SA node: insights from recording multiple ion currents in each cell. Am J Physiol Heart Circ Physiol 314, H403–H414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller P-S, Schulz R, Maretto S, Costello I, Srinivas S, Bikoff E, and Robertson E (2011). The fibronectin leucine-rich repeat transmembrane protein Flrt2 is required in the epicardium to promote heart morphogenesis. 138, 1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafuchi A, Ishihara S, and Tsukita S (1994). The roles of catenins in the cadherin-mediated cell adhesion: functional analysis of E-cadherin-alpha catenin fusion molecules. The Journal of Cell Biology 127, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaidou T, Aslanidi OV, Zhang H, and Efimov IR (2012). Structure-function relationship in the sinus and atrioventricular nodes. Pediatr Cardiol 33, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata S, Morokuma J, Hayata T, Kolle G, Niehrs C, Ueno N, and Cho KWY (2007). TGF-beta signaling-mediated morphogenesis: modulation of cell adhesion via cadherin endocytosis. Genes & Development 21, 1817–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong LL, Kim N, Mima T, Cohen-Gould L, and Mikawa T (1998). Trabecular myocytes of the embryonic heart require N-cadherin for migratory unit identity. Dev Biol 193, 1–9. [DOI] [PubMed] [Google Scholar]
- Opthof T (1988). The mammalian sinoatrial node. Cardiovascular Drugs and Therapy 1, 573–597. [DOI] [PubMed] [Google Scholar]
- Rohr S, Kucera JP, Fast VG, and Kléber AG (1997). Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science 275, 841–844. [DOI] [PubMed] [Google Scholar]
- Rohr S, Kucera JP, and KLÉBER AG (1998). Slow Conduction in Cardiac Tissue, I: Effects of a Reduction of Excitability Versus a Reduction of Electrical Coupling on Microconduction. Circulation Research 83, 781–794. [DOI] [PubMed] [Google Scholar]
- Rossi S, and Griffith BE (2017). Incorporating inductances in tissue-scale models of cardiac electrophysiology. Chaos 27, 093926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Delbridge LM, Blatter LA, and Bers DM (1996). Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophys J 70, 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, and Jan LY (2007). Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RM, and Rudy Y (1997). Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circulation Research 81, 727–741. [DOI] [PubMed] [Google Scholar]
- Shimada T, Kawazato H, Yasuda A, Ono N, and Sueda K (2004). Cytoarchitecture and intercalated disks of the working myocardium and the conduction system in the mammalian heart. Anat Rec A Discov Mol Cell Evol Biol 280, 940–951. [DOI] [PubMed] [Google Scholar]
- Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC, and Shaw RM (2010). Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J Clin Invest 120, 266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope D, Cheng L, Gao E, Li J, and Radice GL (2012). Loss of cadherin-binding proteins beta-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol Cell Biol 32, 1056–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomás AR, Certal AC, and Rodríguez-León J (2011). FLRT3 as a key player on chick limb development. Developmental Biology 355, 324–333. [DOI] [PubMed] [Google Scholar]
- Unudurthi SD, Wolf RM, physiology, T.H.F.i., and 2014 (2014). Role of sinoatrial node architecture in maintaining a balanced source-sink relationship and synchronous cardiac pacemaking. frontiersinorg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eif VWW, Stefanovic S, van Duijvenboden K, Bakker M, Wakker V, de Gier-de Vries C, Zaffran S, Verkerk AO, Boukens BJ, and Christoffels VM (2019). Transcriptome analysis of mouse and human sinoatrial node cells reveals a conserved genetic program. Development (Cambridge, England) 146, dev173161–173129. [DOI] [PubMed] [Google Scholar]
- Vedantham V (2015). New Approaches to Biological Pacemakers: Links to Sinoatrial Node Development. Trends in Molecular Medicine 21, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantham V, Galang G, and Evangelista M (2015). RNA sequencing of mouse sinoatrial node reveals an upstream regulatory role for Islet-1 in cardiac pacemaker cells. Circulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vite A, Zhang C, Yi R, Emms S, and Radice GL (2018). α-Catenin-dependent cytoskeletal tension controls Yap activity in the heart. Development (Cambridge, England) 145, dev149823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickline ED, Dale IW, Merkel CD, Heier JA, Stolz DB, and Kwiatkowski AV (2016). αT-Catenin Is a Constitutive Actin-binding α-Catenin That Directly Couples the Cadherin·Catenin Complex to Actin Filaments. The Journal of biological chemistry 291, 15687–15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Grieskamp T, Airik R, Mommersteeg MT, Gardiwal A, de Gier-de Vries C, Schuster-Gossler K, Moorman AF, Kispert A, and Christoffels VM (2009). Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ Res 104, 388–397. [DOI] [PubMed] [Google Scholar]
- Ye W, Wang J, Song Y, Yu D, Sun C, Liu C, Chen F, Zhang Y, Wang F, Harvey RP, et al. (2015). A common Shox2-Nkx2–5 antagonistic mechanism primes the pacemaker cell fate in the pulmonary vein myocardium and sinoatrial node. Development 142, 2521–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Itoh M, Nagafuchi A, and Tsukita S (1995). Cell-to-cell adherens junction formation and actin filament organization: similarities and differences between non-polarized fibroblasts and polarized epithelial cells. J Cell Sci 108 ( Pt 1), 127–142. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Wada Y, Watanabe T, Nagafuchi A, and Shibata M (2010). Alpha-Catenin as a tension transducer that induces adherens junction development. Nature Cell Biology 12, 533–542. [DOI] [PubMed] [Google Scholar]
- Zemljic-Harpf A, Manso AM, and Ross RS (2009). Vinculin and talin: focus on the myocardium. J Investig Med 57, 849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Holden AV, Kodama I, Honjo H, Lei M, Varghese T, and Boyett MR (2000). Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am J Physiol Heart Circ Physiol 279, H397–421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The three-dimensional monodomain equations were used to simulate the conduction of the electrical propagation through embryonic cardiac tissue. The monodomain model can be derived from a homogenized description of the excitation propagation that neglects the specific details of the tissue microarchitecture(Keener and Sneyd, 1998). In this model, the current density flux J is modeled using Ohm’s law, such that J = −σ∇V, where σ is the tissue conductivity tensor and V is the transmembrane potential difference. In general, the conductivity tensor σ includes information about the alignment of the cells, such that σ = σtI + (σl − σt)f ⊗ f, in which f is a vector parallel to the average cellular orientation, I is the identity tensor, and σt and σl are the conductivity coefficients in the transverse and longitudinal cell directions, respectively. Because the transverse propagation is negligible in the simulations, we simplify the conductivity tensor to be isotropic, such that σ = σI, with σ = σl = σt. The monodomain model reads:
in which χ is the ratio of membrane surface to tissue volume, Csp is the specific membrane capacitance, Iion is the sum of the transmembrane currents per unit area which depend on the gating variables w and on the voltage V. In the following, l and w denote the length and width of the cell respectively, α the volume fraction of the extracellular space, σc the cytosol conductance per unit length, and gc the gap junctional conductance. Following the approach of Hand et al.(Hand et al., 2009), assuming a structured network of cells with uniform (averaged) electrical properties, we can link the macroscopic conductivity of the tissue to the cell dimensions via:
Assuming the membrane surface area A and the cell capacitance C are related by the equation Cm = CspA, we can derive the cell dimensions l and w and the volume fraction of the extracellular space α. More specifically, we assume 1:10 ratio between the l and w, such that
in which is the cell volume, VCPC is the volume of the region containing the CPCs and nCPC is the number of CPCs contained in VCPC. Finally, the value of the membrane surface to tissue volume ratio χ is computed as nCPCA ∕ VCPC.
We extract the values of nCPC and VCPC from imaging data (see Figures 1C and S1). We derive the specific capacitance Csp in the CPCs region assuming CPCs are cylindrical and their capacitance changes with cell size between 20pF and 60pF as reported in the literature(Inada et al., 2014). We use the modified Courtmanche-Ramirez-Nattel ionic model(Courtemanche et al., 1998; McDougal et al., 2017) for the human atrial action potential to describe the ionic current density Iion in the working myocardium. We use the CPC model of Fabbri et al. 2017 to represent the action potential in the SAN region. The monodomain model is solved using a finite linear element method based on piecewise linear finite elements along with a semi-implicit time-stepping scheme(Rossi and Griffith, 2017).
Embryonic tissue geometry was segmented using Imaris (Bitplane). The three-dimensional geometry was prepared for simulations using MeshMixer (Autdodesk), QuadRemesher (Exoside) and Gmsh(Geuzaine and Remacle, 2009). Simulations were run using the reconstructed tissue geometry and features including CPC slow diastolic depolarization, action potential duration, propagation pattern, and conduction velocities within and at the periphery of the forming sinoatrial node were compared back to recorded data from tissue slices. For sensitivity analysis (Cm vs gc) slope of diastolic depolarization, action potential duration, and conduction velocities were allowed to diverge from recorded data and rate, rhythmicity, and the ability of the SAN to initiate an action potential and/or drive the atria was scored to determine parameters ranges that allowed for functional pacemaking (Figure S1).
The code developed in this work, BeatIt (available at github.com/rossisimone/beatit), relies on the parallel C++ finite element library libMesh(Kirk et al., 2006) and on PETSc linear solvers(Balay et al., 2017; Balay et al., 1997). The visualization of the results and their analysis have been carried out using Paraview(Ahrens, 2005) and MATLAB (The Mathworks, Inc.).
| Csp[μF/cm] | Cm [pF] | χ [1/cm] | σ [mS/cm] | σc [mS/cm] | gc [nS] | VCPC [cm3] | nCPC | |
|---|---|---|---|---|---|---|---|---|
| CPCs | 3 | 15 – 60 | 1800 – 5640 | 0.0045 – 0.1318 | 6.7 | 0.3 – 3.3 | 3.496e-6 | 986 |
| WM | 1 | – | 3400 | 1.0 | – | – | – | – |