

Abstract
Mobile genetic elements (MGEs) sequester and mobilize antibiotic resistance genes across bacterial genomes. Efficient and reliable identification of such elements is necessary to follow resistance spreading. However, automated tools for MGE identification are missing. Tyrosine recombinase (YR) proteins drive MGE mobilization and could provide markers for MGE detection, but they constitute a diverse family also involved in housekeeping functions. Here, we conducted a comprehensive survey of YRs from bacterial, archaeal, and phage genomes and developed a sequence‐based classification system that dissects the characteristics of MGE‐borne YRs. We revealed that MGE‐related YRs evolved from non‐mobile YRs by acquisition of a regulatory arm‐binding domain that is essential for their mobility function. Based on these results, we further identified numerous unknown MGEs. This work provides a resource for comparative analysis and functional annotation of YRs and aids the development of computational tools for MGE annotation. Additionally, we reveal how YRs adapted to drive gene transfer across species and provide a tool to better characterize antibiotic resistance dissemination.
Keywords: antibiotic resistance, evolution, horizontal gene transfer, mobile genetic elements, tyrosine recombinases
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Ecology; Microbiology, Virology & Host Pathogen Interaction
A systematic resource for tyrosine recombinase annotation is presented. Comparative sequence analysis of the protein family enables the functional classification of these enzymes and the identification of mobile genetic elements in bacterial genomes.
Introduction
Tyrosine recombinases (YRs) form a large family of proteins that perform site‐specific DNA recombination in a wide variety of biological processes (Grindley et al, 2006; Jayaram et al, 2015). They promote post‐replicative segregation of plasmids and circular chromosomes upon cell division, thereby protecting genome integrity in bacteria, archaea, and phages. For example, the highly conserved Xer proteins (e.g., XerC and XerD in E. coli) resolve chromosome multimers formed after DNA replication in prokaryotes (reviewed in (Midonet & Barre, 2014)), and the Cre recombinase separates dimers of the P1 phage genome (reviewed in Van Duyne, 2015). Other YRs act as genetic switches, triggering phenotype variation within bacterial populations via DNA inversion or deletion (Klemm, 1986; Manso et al, 2014; Li et al, 2016).
In addition, YRs can drive the movement of mobile genetic elements (MGEs), including phages and transposons. Some MGEs hijack host‐encoded Xer proteins (Huber & Waldor, 2002; Das et al, 2013), while others encode specific YRs to promote their own integration and transfer in bacterial genomes (Van Houdt et al, 2012; Wang et al, 2018). Prominent examples of YR‐carrying MGEs include the integrative and conjugative elements (ICEs), also referred to as conjugative transposons. These elements combine features of phages and plasmids, because they actively integrate into genomes and disseminate by conjugative transfer (Burrus & Waldor, 2004). ICEs and related mobilizable elements are abundant in bacterial genomes and provide efficient vehicles for horizontal transfer of genetic traits such as virulence and antibiotic resistance (Franke & Clewell, 1981; Harbottle et al, 2006; Stokes & Gillings, 2011; Bellanger et al, 2014; Guédon et al, 2017). Furthermore, YRs are present in antibiotic resistance‐carrying non‐conjugative transposons (Vanhooff et al, 2006; Siguier et al, 2014), and they mediate the acquisition and activation of antibiotic resistance genes in complex functional units in integrons (reviewed in (Mazel, 2006)).
Therefore, YRs contribute to the emergence and spread of bacterial virulence and antibiotic resistance in several ways and characterizing their distribution and functions is instrumental to understanding horizontal gene transfer and adaptation in bacteria. Yet, functional annotation of these proteins in genome sequence data has been hindered by their large diversity, leaving the vast majority of YRs without a reliable functional prediction and a comprehensive classification. This limits our understanding of the diversity and phylogenetic relationships of different YR groups and hampers automated identification and functional annotation of YRs and YR‐carrying MGE.
Here, we assembled an extended set of prokaryotic YR sequences. By sequence analysis and classification, we showed that YRs of ICEs and phages emerged from a Xer‐like ancestor by acquisition of an additional DNA‐binding domain. In turn, short “simple” YRs function in chromosome dimer resolution, integron recombination, or mobilization of non‐conjugative elements. YRs within phylogenetic groups share specific characteristics, such as distinctive structural features and preferred host taxonomy. With this YR classification system, we further developed a tool to identify MGEs in genome sequence data and used it to detect and characterize numerous novel elements in diverse bacteria. These results illuminate YR diversity, distribution, and evolution, provide a resource for mapping YR function, and help to characterize the role of these proteins in shaping bacterial genomes and the spread of antibiotic resistance.
Results
Identification and analysis of tyrosine recombinases
Previous structural and sequence analyses indicated that YRs generally have two main functional domains: The core‐binding (CB) domain binds the recombination DNA site, and the catalytic (CAT) domain catalyzes all DNA cleavage and joining reactions required for recombination (Esposito & Scocca, 1997; Nunes‐Düby et al, 1998; Swalla et al, 2003). Some YRs have an additional N‐terminal arm‐binding (AB) domain that recognizes accessory DNA sequences, so‐called arm sites, near the recombination sites. Crystal structures showed that the CAT domain has a similar fold in diverse YRs (Guo et al, 1997; Subramanya et al, 1997; Tirumalai et al, 1997; Skaar et al, 2015) and comparative sequence analyses revealed two highly conserved regions (referred to as boxes) and three patches with less significant conservation (Esposito & Scocca, 1997; Nunes‐Düby et al, 1998). Conserved regions include the catalytic residues, i.e., the tyrosine nucleophile and the catalytic pentad RKHRH (Jayaram et al, 2015), as well as the hydrophobic protein core. The CB domain is much less conserved on the sequence level, but its structural architecture is also preserved (Swalla et al, 2003). In turn, the AB domain is highly variable with substantial structural and sequence diversity between YR family members (Clubb et al, 1999; Fadeev et al, 2009; Szwagierczak et al, 2009).
To analyze the diversity of YRs, we employed the following strategy. First, we performed an iterative jackhmmer search against the UniProt reference proteomes database using the prototypical XerD protein from Escherichia coli as an initial query. In every cycle of this search, the hit sequences were aligned and a profile hidden Markov model (profile HMM) was built. Profile HMM is a probabilistic model used to describe characteristic sequence features of the alignment. This profile HMM was then used as a new query in the next search cycle. This iterative procedure allows identification of distantly related homologues of the original query (Johnson et al, 2010; Potter et al, 2018). The resulting sequences were then clustered, and the representatives of the clusters were aligned. The alignment was truncated to contain only the CB and CAT regions, which are ubiquitously present in all YR proteins. This resulting alignment was then used to reconstruct the phylogenetic tree with the PhyML package (Fig 1A and Appendix Fig S1). The tree topology was supported by parametric aBayes and non‐parametric SH‐LRT tests (Anisimova et al, 2011). Based on phylogeny, we then divided YRs into subgroups with significant branch supports (over 0.98 and 0.85 for aBayes and SH‐LRT, respectively; Appendix Table S1). For each subgroup, we created a distinctive profile HMM, which we then used to find all YR homologues in the UniProt reference proteomes collection. For the resulting sequences, we created sequence logos to visualize conserved regions within subgroups (Appendix Figs S2–S4) and analyzed the specific differences between subgroups (Fig 2). We mapped all YR proteins to their genomes of origin and tracked the taxonomic distribution of each subgroup (Fig 1B, Dataset EV1). Finally, we extracted the fifty most abundant YR proteins and characterized their distribution, classification, and putative function (Fig 1C, Dataset EV2).
Figure 1. Diversity and distribution of tyrosine recombinases (YRs).
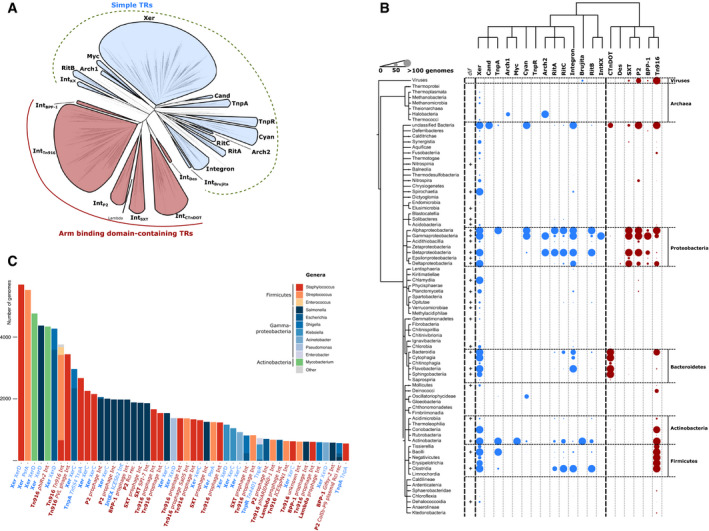
- Maximum‐likelihood phylogenetic tree of YRs. Two major groups of YRs, simple and arm‐binding (AB) domain‐containing YRs, are highlighted in blue and red, respectively. YR subgroups are shown as leaves in the tree. Statistical support for branching was evaluated by aBayes, and for all of the subgroups, its value is more than 0.98.
- Taxonomic distribution of YRs. On the top, a schematic tree of the YR phylogeny corresponding to panel (A) is shown (only nodes with statistical support of more than 0.98 are shown). Phylogeny of the bacterial taxa is shown on the left. The abundance of YRs from a specific subgroup in a particular taxon is indicated by different size dots in the plot (colored as in (A)). The exact numbers of genomes are provided in Dataset EV1.
- The fifty most abundant YR proteins found in the genomic sequences available from NCBI. The bars indicate YR abundance in different bacterial taxa with distinct colors. The YRs are named by the subgroup name (in bold) and functional classification. The names of simple and AB domain‐containing YRs are colored like in (A). NCBI GI numbers for all the sequences are available in Dataset EV2.
Source data are available online for this figure.
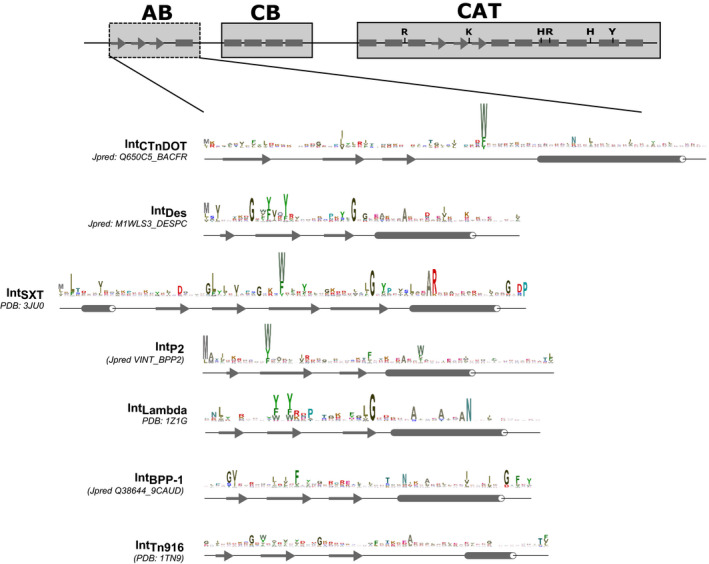
Figure 2. Conservation analysis of tyrosine recombinase (YR) subgroups.

For each of the subgroups, secondary structures of a representative family member were predicted using Jpred or retrieved from corresponding Protein Data Bank (PDB) entries. Helices and strands are shown as rectangles and arrows, respectively. The tyrosine nucleophile and the catalytic RKHRH pentad are marked. Characteristic structural variations of YRs that are conserved within distinct subgroups are highlighted in red. AB—arm‐binding domain; CB—core‐binding domain; CAT—catalytic domain; DUF3701—domain of unknown function (Pfam accession number—PF12482).
This analysis showed that all YRs can be classified into two major phylogenetic groups: simple YRs, which consist of a CB and a CAT domain, and complex YRs, which contain an additional AB domain (Figs 1A and 2). Within these main groups, smaller subgroups were identified, which share a generally conserved domain architecture, but vary in specific structural and sequence features (Appendix Fig S1). Notably, YRs within subgroups have a characteristic taxonomic distribution and share similar predicted functions. In the following sections, we summarize the key sequence features and functional characteristics of all major groups and subgroups.
Simple YRs
The first major YR group revealed in our study includes simple YRs. Members of this group usually comprise only CB and CAT domains and can be further classified into fourteen subgroups (Figs 1A and 2, Appendix Fig S1).
The largest subgroup, Xer, mainly contains recombinases that are responsible for chromosome dimer resolution in bacteria and archaea, such as XerC/D, XerH, XerS, and XerA (Carnoy & Roten, 2009; Cortez et al, 2010; Nolivos et al, 2010; Debowski et al, 2012). Sequence comparisons revealed that proteins in this subgroup are highly conserved, with numerous residues conserved also outside of the active site pocket and the hydrophobic core (Appendix Figs S2–S4). The subgroup is widely distributed, and its members are present in almost all analyzed bacterial and archaeal classes (Fig 1B, Dataset EV1), which is consistent with the essential role of these proteins. In the remaining taxa, other class‐specific simple YRs may compensate for Xer function. For example, in Halobacteria we found a specific type of simple YRs, named Arch1, which resemble Xer but contain short distinct sequence insertion (Fig 2 and Appendix Fig S3). Similarly, Oscillatoriophycideae lack a Xer protein and instead contain members of the separate Cyan subgroup (named after Cyanobacteria, a phylum of the class). Furthermore, the Cand subgroup unites Xer‐related YRs from unclassified “Candidate” phyla, a “microbial dark matter” (Rinke et al, 2013).
The remaining subgroups of simple YRs do not seem to mediate essential cellular functions, but are linked to MGEs. Members of the RitA, RitB, and RitC subgroups are found on “recombinases in trio” transposable elements (Ricker et al, 2013) and contain added structural features compared with the Xer proteins. RitA YRs have two sequential CB domains, and RitC YRs contain an additional conserved C‐terminal helix, both of which are absent in all other YRs (Fig 2 and Appendix Fig S5). The TnpA subgroup unites YRs related to the transposase of the Tn554 transposon (Bastos & Murphy, 1988), which have two beta‐strands inserted on their N‐terminus and an alpha helix insertion between CB and CAT domains (Fig 2 and Appendix Fig S5). The integron‐like subgroup contains the integrase proteins of integrons, which act to reshuffle gene cassettes in integron recombination platforms to promote bacterial adaptation (Collis & Hall, 1992). These proteins contain a specific insertion within the CAT domain fold, which is not fully conserved in the subgroup, but is essential for integron function (Fig 2 and Appendix Fig S3) (MacDonald et al, 2006). The TnpR subgroup includes the TnpR protein from the carbapenemase‐carrying Tn4401 transposon (Naas et al, 2008) and related proteins found in beta‐ and gammaproteobacteria. Notably, TnpR is one of the most abundant and widely distributed YRs in bacterial genomes, which suggests a prominent role for Tn4401 in propagating carbapenem resistance (Fig 1C). Proteins in the TnpR subgroup have a helical N‐terminal DUF3701 domain in addition to the canonical CB and CAT domains (Fig 2 and Appendix Fig S1). The function of this domain is not known, but it contains a helix–hairpin–helix motif, suggesting a role in DNA binding. The IntBrujita subgroup is formed by YRs of mycobacterial phages. Its best‐characterized member is the Brujita phage integrase, which was shown to mediate insertion of the phage genome in target bacteria (Lunt & Hatfull, 2016). Arch2 subgroup includes proteins related to the integrase of the temperate pleolipovirus SNJ2 from halobacteria (Wang et al, 2018). These YRs have a specific short sequence insertion in the CAT domain (Fig 2 and Appendix Fig S3). Finally, the IntKX subgroup (defined in (Roche et al, 2010)) comprises YRs found in known pathogenicity islands in human pathogenic gammaproteobacteria (such as PAPI‐1, ICESb2, and ICEEc2; Dataset EV3) (Collyn et al, 2004; Mohd‐Zain et al, 2004; Harrison et al, 2010; Roche et al, 2010; Seth‐Smith et al, 2012; Juhas et al, 2013). Taken together, these analysis results revealed that most of the simple Xer‐related YR proteins act in chromosome maintenance, with some distinct, smaller subgroups specifically linked to MGEs. Notably, the latter subgroups are characterized by an increased structural complexity compared to canonical Xer proteins.
Arm‐binding domain‐containing tyrosine recombinases
The second large YR group unites proteins that contain, an AB domain in addition to the CB and CAT domains (Appendix Fig S1). The best‐characterized members of this group act as integrases of phages or ICEs. This AB domain‐containing YR group consists of six major subgroups that are discussed in detail in the following sections.
IntTn916 subgroup
The largest subgroup of AB domain‐containing YRs is the IntTn916 subgroup. It is the most diverse among the AB domain‐containing YRs and contains integrases of numerous well‐documented ICEs and phages. Its members are most highly represented in gram‐positive bacteria, but we also found some examples in other taxa, such as Fusobacteria, Synergista, and Chlamydia (Fig 1B). This subgroup contains some of the most abundant AB domain‐containing YRs, such as the mycobacterial phiRV2 prophage integrase (Cole et al, 1998) and the integrase of the tetracycline resistance‐carrying Tn916 transposon (Franke & Clewell, 1981), each found in the genomes of about 4,000 bacterial strains (Fig 1C).
Generally, members of the subgroup contain an AB domain on their N‐terminus, which features three beta‐strands and one alpha helix (Figs 2 and 3), as seen in the NMR structure of the Tn916 integrase AB domain (Wojciak et al, 1999). In some cases, the AB domain was not directly predicted by Pfam (Appendix Fig S1), but our subsequent sequence analysis revealed that the AB domain is preserved throughout the subgroup (Fig 3). Another characteristic feature of the IntTn916 subgroup is a conserved beta‐stranded insertion between the second and third beta‐strands in the CAT domain (Fig 2 and Appendix Fig S3). Recent structural and biochemical work on the Tn1549 integrase showed that this protein segment is important for shaping the DNA substrate for recombination (Rubio‐Cosials et al, 2018).
Figure 3. Sequence conservation of the arm‐binding domains of tyrosine recombinases (YRs).
For each subgroup, web logos were produced after HMM search against the UniProt reference proteomes database and secondary structures were predicted using Jpred or retrieved from corresponding PDB entries (shown below the logos). The logos are colored by residue type, and the typical YR domain composition is shown above the logos as in Fig 2.
Notably, the phage‐ and ICE‐related members of this subgroup do not form separate clusters; instead, most clusters contain integrases from both ICEs and phages (Appendix Fig S6). For example, many actinomycete ICE integrases cluster together with the integrases from actinobacterial phages (see cluster pSAM2 in Appendix Fig S6). Interestingly, many YRs within the clusters integrate their respective MGEs at specific genomic sites, with a reoccurring preference for the conserved flanks of essential genes, such as tRNA encoding genes (Appendix Fig S6). A notable exception is the specific cluster that includes the Tn916 and Tn1549 integrases, which insert into AT‐rich regions without a strict sequence specificity (Trieu‐Cuot et al, 1993; Scott et al, 1994; Wang et al, 2000; Lambertsen et al, 2018). This feature might have contributed to the success of the respective MGEs in spreading to a broad range of bacteria.
IntBPP‐1 subgroup
The IntBPP‐1 is a smaller AB domain‐containing YR subgroup, which is closely related to IntTn916. Its members are found in gammaproteobacteria, betaproteobacteria, and phages (Fig 1B). Examples of this subgroup include putative integrases of the Bordetella BPP‐1 phage, the Stx2a phage, and the Salmonella Gifsy‐2 phage (McClelland et al, 2001; Liu et al, 2004; Ogura et al, 2015), the latter being one of the most abundant proteins in this subgroup (Fig 1C). IntBPP‐1 YRs feature an AB domain that is annotated as DUF3596 in Pfam (PF12167; Appendix Fig S1) and exhibits a canonical three beta‐strand/one helix structure (Fig 3). Similar to IntTn916 members, the IntBPP‐1 subgroup features a beta‐stranded insertion between the second and third beta‐strands in the CAT domain fold (Fig 2 and Appendix Fig S3). Members of the family also have weaker conservation of the first histidine in the catalytic RKHRH pentad (Appendix Fig S4).
IntCTnDOT subgroup
The second largest AB domain‐containing YR subgroup is IntCTnDOT. It includes proteins from Bacteroidetes (Fig 1B), such as integrases of the ICE CTnDOT and mobilizable element NBU1 (Shoemaker et al, 1996; Whittle et al, 2002), as well as YRs from the Salmonella genomic island 1 (SG1) (Doublet et al, 2005; Douard et al, 2010) (Dataset EV3). Initial Pfam annotation suggested that YRs in this subgroup contain only CB and CAT domains, with a substantially larger predicted CB domain than the one found in simple YRs. However, secondary structure predictions previously proposed that the integrase of a prototype CTnDOT element from Bacteroides comprises a canonical AB domain (Kim et al, 2010) (Fig 3) and subsequent biochemical experiments confirmed its interaction with subterminal arm DNA sites in the transposon (DiChiara et al, 2007; Wood et al, 2010). In agreement, our comparative analysis revealed that the N‐terminal segment of all IntCTnDOT members consists of two conserved domains: a canonical CB domain and an upstream AB domain (Fig 3 and Appendix Fig S1). Accordingly, we have updated the corresponding Pfam annotation, which is now available in the new version (Pfam 32.0).
Analyzing sequence logos, we further noted that YRs of the IntCTnDOT subgroup show a weaker conservation of the first arginine residue in the otherwise strictly preserved catalytic RKHRH pentad (Box I in Appendix Fig S2) in the CAT domain. Arginine is present in this position in NBU1, NBU2, and Tn4555 integrases, but it is absent in the integrases of CTnDOT, ERL (S), and Tn5520 elements (Cheng et al, 2000). Previous biochemical experiments showed that in the CTnDOT integrase, this residue is functionally substituted by another arginine located further downstream in the protein sequence (Kim et al, 2010). Consistently, we found that this alternative arginine is conserved in many integrases in the IntCTnDOT subgroup (see conserved R in IntCTnDOT logo in Appendix Fig S3). Thus, YRs of this subgroup carry the catalytic arginine in one of two alternative locations, resulting in a weaker overall conservation.
IntSXT subgroup
The next large subgroup of AB domain‐containing YRs is IntSXT, which comprises integrases of several ICEs, genomic islands, and phages. A characteristic feature of this subgroup is the presence of an N‐terminal DUF4102 domain (Appendix Fig S1). This was previously annotated as an AB domain of genomic island integrases (Szwagierczak et al, 2009) and contains an additional beta‐strand and an alpha helix compared with AB domains of other YRs (Figs 2 and 3). Phylogenetic analysis revealed that two out of six clusters within the IntSXT subgroup contain integrases from both ICEs and phages (Appendix Fig S7). Members of major clusters share distinct genomic insertion profiles, integrating their MGEs near essential genes. For example, integrases of the P4 and Sf6 phages cluster together with various ICE YRs, all of which insert downstream of tRNA genes (P4 cluster, Appendix Fig S7) (Boyd et al, 2009; Van Houdt et al, 2012). Similarly, integrases of the epsilon15 phage, the CMGI‐3 element, and related elements form a separate cluster, and all target the 3′ flank of the guaA gene involved in GMP biosynthesis (Kropinski et al, 2007; Bi et al, 2012) (epsilon15 cluster, Appendix Fig S7). The same pattern is seen for integrases of the Enterobacterial cdt1 phage, the SXT element, and closely related ICEs, all of which insert next to the prfC gene encoding a factor involved in termination of translation (Hochhut & Waldor, 1999; Asakura et al, 2007) (SXT cluster; Appendix Fig S7). Thus, members of each IntSXT cluster seem to drive their diverse MGEs into specific locations, perhaps owing to characteristic features in the integrase sequences. Their preference for the flanks of conserved genes might help promote their dissemination between species and explain their characteristic taxonomic distribution. In addition, the mixed distribution of ICE and phage integrases suggests that these elements frequently exchange their integrases. This is also supported by previous observations that ICEs with different conjugation machineries have closely related integrases (Cury et al, 2017).
IntP2 subgroup
The IntP2 subgroup of AB domain‐containing YRs contains integrases from proteobacterial phages, such as HP1 and P2. Another interesting member of this subgroup is the plasmid‐borne Rci recombinase, which regulates R64 plasmid conjugation by reshuffling distinct gene segments to generate diverse pili proteins (Komano et al, 1987; Gyohda & Komano, 2000; Roche et al, 2010). The CAT domains of YRs in this subgroup are highly similar to those of simple YRs, as also seen with previously determined crystal structures (Hickman et al, 1997; Skaar et al, 2015). Most YRs in this subgroup contain an AB domain with a classical fold (Fig 3), except the Rci recombinases that lack the AB domain. In agreement with previous sequence analyses (Boyd et al, 2009), our phylogenetic reconstructions suggest that IntP2 YRs are related to the lambda phage integrase; however, this clustering is not well supported by statistical analysis (Fig 1A and Appendix Fig S1). Although the well‐studied lambda phage integrase is often used as a prototype for the tyrosine recombinase superfamily (Landy, 2015), our analysis revealed that it is quite different from other YRs. It contains substantial alterations even in the CAT domain, including an insertion of two beta‐strands after the third beta‐strand of the canonical fold, and the replacement of the C‐terminal alpha helix with a beta‐strand (Fig 2, Appendix Figs S3 and S4).
IntDes subgroup
Finally, IntDes is a small subgroup of AB domain‐carrying YRs. Its members are found only in the genus Desulfovibrio of Deltaproteobacteria (Fig 1B). This subgroup features specific sequence perturbations in the catalytic core: Namely the first arginine residue of the RKHRH pentad is shifted in comparison with other YRs and the first histidine is substituted with a tyrosine (Appendix Figs S2 and S4). The biological function of these YRs has remained unknown to date.
Identification and classification of integrative and conjugative elements
The vast majority of the YRs that we analyzed remain unannotated in genomic databases. This particularly hinders identification and characterization of YR‐carrying MGEs. To test whether our classification system can help predict YR function, we next checked whether the unannotated YRs found in ICE‐related subgroups are indeed integrases of ICEs. For this, we examined the YRs’ genomic neighborhood to identify known conjugative machinery proteins (as in Guglielmini et al, 2014; Abby et al, 2016). If an integrase was found in proximity (± 100 kb) to known conjugation machinery proteins, then the corresponding region was considered to be a putative ICE (Fig 4A). ICEs retrieved from the ICEberg database were used for benchmarking. This analysis revealed a total of 59 previously unannotated ICEs (Appendix Fig S8, Dataset EV4). The putative ICEs were then further validated by manual identification of their terminal repeat sequences. We confidently identified terminal repeats in 50 out of 59 predicted ICEs. For 49 of these, the conjugation machinery was found within the predicted borders of the element, further confirming their identity. In one predicted element, the conjugation machinery was located outside of the borders (Dataset EV4), suggesting a coincidental co‐occurrence of YR and conjugation genes in this instance.
Figure 4. Tyrosine recombinase‐based ICE identification and characterization.
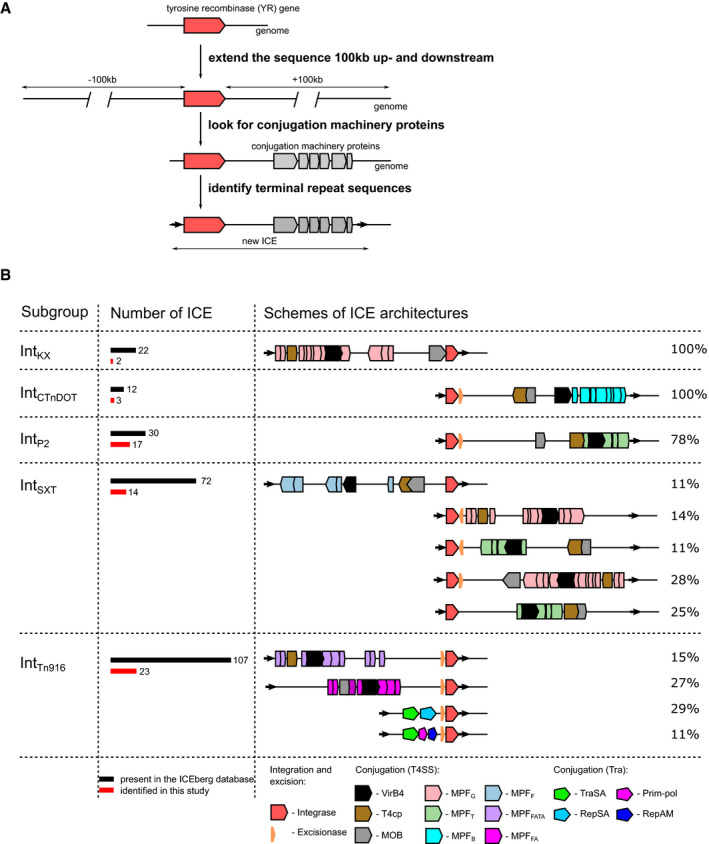
- Overview of the computational pipeline for ICE identification. The genomic regions of the tyrosine recombinase (YR) genes were expanded 100 kb upstream and downstream and analyzed for the presence of conjugation‐related genes and repeat sequences.
- Structural diversity of YR‐carrying ICEs. All ICEs clustered into five subgroups based on their YR classification (left). The numbers of ICEs in each of the subgroups are displayed as bars with numbers (middle). Schematic representations of ICE architectures are shown, aligned by their integrase genes (red symbol, right). Protein open reading frames of various types of conjugation machineries are depicted with different colors as indicated at the bottom of the figure.
To further characterize the detected ICEs, we aimed to reconstruct the naive insertion site (i.e., the bacterial genomic sequence prior to integration) of the identified ICEs and look for such undisrupted sites in closely related genomes. As functional ICEs can move to new genomic sites, successful identification of naive sites can provide ultimate confirmation of their identity and mobile nature. However, identification of such naive sites requires recent mobility of the ICE and may also be challenged by a limited availability of complete genome sequence data for related species in public databases. Nevertheless, we found naive sites for 18 out of the 49 ICEs, which further validates these elements and indicates their recent activity (Dataset EV4, Appendix Fig S9).
YRs in the new ICEs belonged to five YR subgroups (Fig 4B, Dataset EV4), with most examples found in the IntTn916 (23), IntP2 (17) and IntSXT (14) subgroups. To further analyze the detected ICEs, we next reconstructed the phylogeny of their YRs and plotted the genetic structure of their respective conjugation machineries (Fig 4B and Appendix Fig S8). ICEs with closely related YRs were generally associated with closely related conjugation systems, but ICE groups with somewhat more distantly related YR proteins often contained unrelated types of conjugation modules (Fig 4B and Appendix Fig S8). For instance, ICE groups that carry YRs from the diverse IntTn916 and IntSXT subgroups revealed various conjugation systems. In turn, some clusters of the IntSXT YRs and the distinct IntKX YRs associated with the same conjugation system, called MPFG (Fig 4B and Appendix Fig S8), located on different sides of the YR. Altogether, this suggests recurrent exchange of conjugation modules between distantly related ICEs, in accordance with previous reports (Cury et al, 2017).
Furthermore, to complete the characterization of the ICEs' mobilization machinery we looked for excisionase (Xis) genes within newly identified and previously reported ICEs (Fig 4B and Appendix Fig S8). Xis regulates the directionality of the recombination reaction in some of the known YR‐containing systems (Connolly et al, 2002; Wood & Gardner, 2015). We found that only AB‐containing YRs are associated with Xis proteins, which may suggest potential cooperation between the AB domain and Xis. Consistent with this idea, a physical interaction was recently proposed for the integrase and Xis of the lambda phage (Cho et al, 2002; Laxmikanthan et al, 2016). We could not detect Xis in any of the 15 ICEs with simple YRs from the IntKX subgroup.
Taken together, successful identification of new ICEs confirms the predictive value of our classification system for automated annotation of YR function and demonstrates its utility to improve characterization of the bacterial mobilome.
Discussion
Xer tyrosine recombinases are conserved and ancient
In the present study, we devise a classification system for bacterial YRs that are related to Xer recombinases. Based on phylogenetic reconstruction, we divided all YR proteins into two groups and twenty subgroups. Of these, the most abundant and widely distributed is the Xer subgroup of simple YRs, which includes close homologues of the chromosome dimer resolution proteins XerC/D from 16 bacterial phyla (Fig 1 and Appendix Fig S1). Notably, recent reports described an identical taxonomic distribution of conserved dif DNA sequences (Kono et al, 2011), which serve as Xer recombination sites on bacterial chromosomes, thus implying a widely conserved functional role for these proteins. The phylogeny of XerC/D recombinases in proteobacteria was further found to be correlated with their host taxonomy, suggesting that their evolution follows a strictly vertical trajectory (Carnoy & Roten, 2009). This wide distribution and vertical inheritance of Xer‐dif systems indicate that Xer proteins are the most ancient type of bacterial YRs, which may have served as the evolutionary source for other more complex YRs.
Simple tyrosine recombinases can drive the movement of mobile genetic elements
Besides the chromosomal Xer proteins, we identified several simple YRs encoded on MGEs, such as phages or ICEs. Most of these YRs belonged to two subgroups, the IntBrujita subgroup found in mycophages and the IntKX subgroup from gammaproteobacterial ICEs; a few isolated examples fell into the Xer and RitB subgroups (Datasets EV1 and EV3). Notably, several of these proteins have been shown to be required for mobilization of their respective elements (Qiu et al, 2006; Fischer et al, 2010; Flannery et al, 2011; Lunt & Hatfull, 2016). For example, the simple YR of the Brujita phage actively drives its excision and integration (Lunt & Hatfull, 2016), and XerT from Helicobacter pathogenicity islands is needed for their horizontal transfer (Fischer et al, 2010). The close relationship of these MGE‐borne YRs with the Xer subgroup and their significance for MGE function suggest that phages and ICEs have repeatedly sequestered Xer genes from their host genomes and functionally repurposed these to drive their transfer. Alternatively, MGE integrases may have been domesticated by the host for dimer resolution functions, as proposed recently (Koonin et al, 2020). However, this contradicts the observed vertical evolution and wide taxonomical distribution of Xer genes.
Acquisition of Arm‐binding domain drove the evolution of mobile genetic element integrases
While some phages and ICEs carry simple YRs, most of them possess more complex AB domain‐containing YRs. In fact, our analysis detected 27 phages with simple YRs versus 410 with AB domain‐carrying YRs (Dataset EV1). Similarly, we found 12 ICEs that carry only simple YRs, whereas 211 had AB domain‐containing YRs (Dataset EV3). These complex YRs cluster into a monophyletic group with six distinct subgroups, where all subgroups contain an N‐terminal AB domain with similar secondary structure (Fig 3), suggesting a common evolutionary origin. Although the AB domain is less conserved than the CAT domain, sequence similarity can be detected between different YR subgroups. Pfam AB domains from all subgroups belong to the same clan (CL0081), and sequence logos show significant conservation within and across subgroups (Fig 3). In agreement, previous structural studies also revealed a similar AB fold in the lambda phage integrase and in IntTn916 and IntSXT family YRs (Clubb et al, 1999; Wojciak et al, 2002; Szwagierczak et al, 2009).
The functional role of the AB domain may be inferred from its function in the lambda phage integrase, one of the best‐characterized members of this YR group. Here, the AB domain binds to internal arm DNA sequences within the phage genome and plays an essential role in guiding DNA recombination toward excision or integration in different stages of the phage lifecycle (Tirumalai et al, 1996; Biswas et al, 2005; Radman‐Livaja et al, 2006). The presence of an AB domain in the vast majority of ICE and phage integrases indicates that these elements generally benefit from the function of this domain in regulating their integration and excision. MGEs that carry simple YRs may represent an earlier step in evolution with less intricate regulatory features (Lunt & Hatfull, 2016). Interestingly, large serine recombinases that perform integration and excision of discrete phages also require a separate DNA‐binding domain for precise regulation (Rutherford & Van Duyne, 2014), indicating that acquisition of accessory DNA‐binding domains may be a common strategy in MGE evolution.
Tyrosine recombinase classification aids the annotation of mobile genetic elements in bacterial genomes
The recent increase in bacterial genome sequence data highlighted the impact of MGEs and motivated the development of automated sequence‐based MGE mining tools. For insertion sequences, the simplest MGEs that typically contain an RNaseH‐like DDE transposase flanked by short inverted repeats, existing pipelines provide confident annotation through homology‐based prediction (Xie & Tang, 2017). The recently communicated TIGER pipeline also maps various integrative genomic elements by using Pfam‐based annotations (Mageeney et al, 2020). However, for YR‐carrying elements the close relationship of YR family transposases/integrases to essential bacterial genes has greatly hampered functional annotation. In particular, the TIGER software tackles this by discarding Xer and Integron‐related sequences, assuming that all other YRs are MGE integrases, which results in false‐positive hits (Mageeney et al, 2020). Through comprehensive YR classification, we found that the presence of an AB domain in a YR is a strong predictor of its function in phage or ICE mobility. In fact, acquisition of this AB domain by YRs and their cooperation with Xis proteins may have driven evolutionary adaptation of YRs to a “mobile lifestyle”, helping to promote and regulate MGE movement in and between bacterial cells. Sequence features in all YR domains further support functional annotation of MGE‐borne proteins and can even enable identification of simple YRs with a specific mobility function. These direct sequence‐to‐function relationships open up new opportunities for functional annotation and provide clear rules for predicting the mobile nature of YRs and the genomic regions they reside in.
By using our classification system, we identified new ICEs in diverse genomes with mobile YRs as markers. We found 59 previously unannotated ICEs, substantially expanding the repertoire of known elements and illustrating the power of our approach for automated MGE detection. Going forward, application of our pipeline will help explore the abundance and diversity of MGEs in bacterial genomes in diverse environments. Comprehensive studies of MGE content, distribution, and dynamics are necessary to track MGE transfer and to assess their impact on genetic exchange and bacterial adaptation. These studies are important to better understand the dynamics of microbial communities and to follow the spread of genetic traits, such as antibiotic resistance. Our work prepares the stage for analyzing the full repertoire of MGEs and their cargo in bacterial genomes, thus offering new opportunities to characterize gene flow in bacterial communities.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Software | ||
| PhyML | Guindon et al, 2010 | 3.0 |
| Jackhmmer | https://www.ebi.ac.uk/Tools/hmmer/search/jackhmmer | 2016_01 |
| Hmmer | http://hmmer.org/ | 3.1b2 |
| Skylign | http://skylign.org/ | – |
| CD‐HIT | http://weizhongli‐lab.org/cdhit_suite/cgi‐bin/index.cgi | – |
| MAFFT | Katoh & Standley, 2013 | Version 7 |
| iTol | https://itol.embl.de/ | V2 |
| Mauve | Darling et al, 2010 | Snapshot_2015‐02‐25 |
Methods and Protocols
Identification and phylogenetic analysis of tyrosine recombinases from bacterial genomes
As the first step, we used the XerD amino acid sequence (GenBank accession number CDO12527) as a query in a jackhmmer iterative search against the rp15 (v.2019_08) database with different e‐values (from 1e‐05 to 1e‐40). Our aim was to include as many remote Xer homologues as possible, which is possible through the jackhmmer procedure (Park et al, 1998), but at the same time avoid inclusion of distantly related sequences, such as eukaryotic YRs from DIRS retrotransposons, that hamper reconstruction of the phylogenetic tree. We chose to use e‐value of 1e‐30 as this threshold discards 96% of eukaryotic YRs (from those included in Pfam family) and retains more than 80% of bacterial YRs (Appendix Table S2) (Goodwin & Poulter, 2001; Van Houdt et al, 2012). We then repeated the same analysis with this e‐value but now against the full UniProt reference proteomes database (converged after 37 iterations; accessed 13.01.2016, http://hmmer.janelia.org/search/jackhmmer currently moved to https://www.ebi.ac.uk/Tools/hmmer/search/jackhmmer; (Finn et al, 2011)). Other well‐known YRs, including diverse bacterial or phage YRs for which crystal structures, are available (XerD, XerH, integrases of phages λ, HP1 and P2 and integron integrase) and most ICE‐encoded YRs from the ICEberg database passed this threshold and therefore were included in the study. 187 out of 191 (~98%) YRs of single YR‐containing ICEs in ICEberg also passed the threshold and were annotated in our study (Dataset EV3). At the next step, all the sequences that passed the threshold (altogether 9,909 proteins) were then clustered with a 40% identity threshold using CD‐HIT (Fu et al, 2012) and the representatives of clusters with more than one sequence were used for further analysis. This was implemented to exclude singletons and rare sequences with truncations. Then, altogether 866 sequences were aligned using the E‐INS‐I method from the MAFFT software, recommended for sequences where several conserved motifs are embedded in long unalignable regions (Katoh & Standley, 2013). In the alignment, columns containing more than 80% of gaps and the N‐terminal region of the alignment (corresponding to AB) were removed. The final alignment is available in Source Data. The phylogenetic tree was constructed using the PhyML package (Guindon et al, 2010) with LG+I + G model of protein evolution and evaluated by ProtTest (Abascal et al, 2005). In the course of the reconstruction, we built 1,000 trees using both NNI and SPR moves as topology search and random tree as starting trees. A tree with the highest maximum‐likelihood value was used as the reconstruction of the YR phylogeny (Fig 1A). The branch support was evaluated with aBayes and non‐parametric SH‐aLRT (Shimodaira & Hasegawa, 1999; Anisimova et al, 2011). We further defined subgroups as clades that (i) include more than four sequences, (ii) have aBayes and SH‐aLRT branch support values of more than 0.98 and 0.85, respectively, and (iii) exhibit similar domain composition of its members (i.e., presence/absence of distinct Pfam domains (Finn et al, 2016)). The tree with the domain composition retrieved for each of the sequences is available as Appendix Fig S1. The same tree with all branch supports is available at http://itol.embl.de/shared/gera for interactive inspection.
The phylogenetic trees for IntTn916 and IntSXT subgroups (Appendix Figs S6 and S7, respectively), as well as for ICE YRs from Appendix Fig S8, were produced in the same way as for the general tyrosine recombinases tree and their branch support was evaluated by aBayes. The sequences were mined from the ICEberg database (Bi et al, 2012) and the UniProtKB phage section, using profile HMMs specific for the subgroups (see section below). For Appendix Fig S8, the sequences of the newly identified ICEs and ICEs from ICEberg database were used for the reconstruction (Datasets EV3 and EV4).
Annotation of tyrosine recombinase putative function, domain organization, conservation, and host organism
In order to annotate all YRs from the UniProt reference proteomes, we created profile HMMs for each of the subgroups. For that, we retrieved the complete sequences of the proteins forming the group and reran the MAFFT alignment of the proteins with the same settings as described above. Then, we built a profile HMM for this alignment using HMMER3 (Eddy, 1998) and set the gathering threshold for the profile as the lowest score that was produced by the sequences used to build the group. The collection of the 20 profile HMMs specific to each subgroup is available in Source Data.
To analyze YR distribution, we mapped all proteins that were retrieved in the UniProt reference proteomes search to their genomic locations and built a plot of the distribution of YRs from each subgroup in bacteria, archaea, and viruses (Fig 1B, Dataset EV1), visualized using the iTOL webserver (Letunic & Bork, 2011). In addition, to shed some more light on the abundance of the YRs in bacterial genomes we extracted fifty of the most abundant YRs found in the sequenced genomes and annotated their distribution in bacteria, classification, and putative function (Fig 1C, Dataset EV2).
To visualize the similarities and differences between YRs from the different groups and subgroups, we created logos representing the sequences from each of the subgroups (Appendix Figs S2–S5). For that we used the protein alignments, containing all hits produced by the profile HMM‐based search against the UniProt reference proteomes database in the previous step, and visualize them using the Skylign webserver (Wheeler et al, 2014). The logo for the Lambda integrase‐related YRs, which failed to form a subgroup as they have only two members of the clade in the tree (Appendix Fig S1), was created with only two sequences used for the corresponding profile HMM reconstruction. Secondary structure predictions were done using Jpred 4 (Drozdetskiy et al, 2015).
To retrieve functional annotation of the proteins, we run the profile HMM against databases of the proteins with known functions. These included Protein Data Bank (PDB, www.rcsb.org; (Berman et al, 2000)), ICEberg (Dataset EV3; http://db‐mml.sjtu.edu.cn/cgi‐bin/ICEberg/; (Bi et al, 2012)), and ACLAME (http://aclame.ulb.ac.be; (Leplae et al, 2010)). In addition, the homologues of the identified YRs were detected using BLAST searches available from ISFinder (database on the insertion sequences, www‐is.biotoul.fr; (Siguier et al, 2012)), INTEGRALL (integron database, http://integrall.bio.ua.pt, (Moura et al, 2009)), and NCBI CDD (Marchler‐Bauer et al, 2017).
Annotation of new putative ICEs
To predict new ICEs, we created a Perl script NeighborsScan, which is available from GitHub repository (https://github.com/smyshlya/NeighborsScan). This script uses protein accession numbers as an input, retrieves their genomic position, expands this position 100 Kb usptreams and downstream, downloads all proteins from this region, and runs hmmsearch against these proteins using user provided collection of profile HMMs (such as conjugation machinery protein profile HMMs). The results of this run can be directly visualized using iTol web server (Letunic & Bork, 2011). Although some ICEs may be up to 600 Kb in length, we chose to cover only ± 100 kb region as a good compromise between the sensitivity of the search and the computational time. The conjugation machinery proteins were identified using hmmscan search against profile HMMs available from CONJdb with the default e‐value of 1e‐3 (Guglielmini et al, 2014; Abby et al, 2016). For excisionase protein identification, we used Pfam profile HMMs HTH_17, HTH_31, Phage_AlpA, MerR, and DUF2384 with corresponding gathering thresholds. Genomic regions that contain integrase and conjugation machinery proteins were predicted as ICEs. All of the full‐length ICEs present in the ICEberg database were confirmed using this approach, providing a benchmark for the method.
To further validate the identification of new ICEs, we used three levels of computational verification as follows: First, we predicted the borders of the identified ICEs. In known ICEs, terminal repeats define the ends of the element, which are recognized by the integrase enzyme to execute ICE excision and integration. Typically, the integrase gene is located adjacent to one of these terminal repeats. Thus, identification of such terminal repeats and verification of their location relative to the integrase gene will verify the identity of our newly annotated ICEs. These repeats are reported in the Dataset EV4.
Second, we tested whether a complete conjugation system is encoded within the predicted borders of the candidate ICEs and shows sequence conservation in closely related elements. Active ICEs generally include conjugation machinery genes, which “travel” with the ICEs and are required for their intercellular transfer. The requirement for these modules implies a certain level of conservation within ICE families. Our analysis revealed that closely related ICEs have closely related conjugation systems (Fig 4 and Appendix Fig S8).
Third, we attempted to identify naive (ICE‐free) insertion sites for confirmed ICEs in closely related genomes (Appendix Fig S9, visualized with Mauve software Darling et al, 2010). Successful identification of such naive sites provides direct confirmation of ICE movement but may be compromised by the limited availability of related genomes in public databases.
All ICEs identified in the study are presented in Dataset EV4 and were submitted to the ICEberg database.
Author contributions
GS performed the study. GS, AB, and OB designed the study and wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Appendix
Review Process File
Acknowledgements
The authors thank Michael Chandler and Cecilia Zuliani for reading and commenting on the manuscript, and members of the Bateman and the Barabas labs for helpful discussions. This work was supported by the EMBL, the Federal Ministry of Education and Research of Germany (BMBF) under JPIAMR 2016 (01KI1706, JumpAR) and the EMBL Interdisciplinary Postdoc (EIPOD) program under Marie‐Curie COFUND (291772 G.S.). Open Access funding enabled and organized by Projekt DEAL.
Mol Syst Biol. (2021) 17: e9880
Data availability
The computer code produced in this study is available in the following GitHub repository: https://github.com/smyshlya/NeighborsScan.
References
- Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best‐fit models of protein evolution. Bioinformatics 21: 2104–2105 [DOI] [PubMed] [Google Scholar]
- Abby SS, Cury J, Guglielmini J, Neron B, Touchon M, Rocha EP (2016) Identification of protein secretion systems in bacterial genomes. Sci Rep 6: 23080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Gil M, Dufayard J‐F, Dessimoz C, Gascuel O (2011) Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood‐based approximation schemes. Syst Biol 60: 685–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura M, Hinenoya A, Alam MS, Shima K, Zahid SH, Shi L, Sugimoto N, Ghosh AN, Ramamurthy T, Faruque SM et al (2007) An inducible lambdoid prophage encoding cytolethal distending toxin (Cdt‐I) and a type III effector protein in enteropathogenic Escherichia coli . Proc Natl Acad Sci USA 104: 14483–14488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos MC, Murphy E (1988) Transposon Tn554 encodes three products required for transposition. EMBO J 7: 2935–2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanger X, Payot S, Leblond‐Bourget N, Guédon G (2014) Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol Rev 38: 720–760 [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi D, Xu Z, Harrison EM, Tai C, Wei Y, He X, Jia S, Deng Z, Rajakumar K, Ou H‐Y (2012) ICEberg: a web‐based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res 40: D621–D626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas T, Aihara H, Radman‐Livaja M, Filman D, Landy A, Ellenberger T (2005) A structural basis for allosteric control of DNA recombination by lambda integrase. Nature 435: 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd EF, Almagro‐Moreno S, Parent MA (2009) Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol 17: 47–53 [DOI] [PubMed] [Google Scholar]
- Burrus V, Waldor MK (2004) Shaping bacterial genomes with integrative and conjugative elements. Res Microbiol 155: 376–386 [DOI] [PubMed] [Google Scholar]
- Carnoy C, Roten C‐A (2009) The dif/Xer recombination systems in proteobacteria. PLoS One 4: e6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Paszkiet BJ, Shoemaker NB, Gardner JF, Salyers AA (2000) Integration and excision of a bacteroides conjugative transposon, CTnDOT. J Bacteriol 182: 4035–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EH, Gumport RI, Gardner JF (2002) Interactions between integrase and excisionase in the phage lambda excisive nucleoprotein complex. J Bacteriol 184: 5200–5203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clubb RT, Wojciak JM, Connolly KM (1999) NMR structure of the Tn916 integrase‐DNA complex. Nat Struct Biol 6: 366–373 [DOI] [PubMed] [Google Scholar]
- Cole St, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon Sv, Eiglmeier K, Gas S, Barry Ce et al (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544 [DOI] [PubMed] [Google Scholar]
- Collis CM, Hall RM (1992) Site‐specific deletion and rearrangement of integron insert genes catalyzed by the integron DNA integrase. J Bacteriol 174: 1574–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collyn F, Billault A, Mullet C, Simonet M, Marceau M (2004) YAPI, a new Yersinia pseudotuberculosis pathogenicity island. Infect Immun 72: 4784–4790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly KM, Iwahara M, Clubb RT (2002) Xis protein binding to the left arm stimulates excision of conjugative transposon Tn916. J Bacteriol 184: 2088–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Quevillon‐Cheruel S, Gribaldo S, Desnoues N, Sezonov G, Forterre P, Serre M‐C (2010) Evidence for a Xer/dif system for chromosome resolution in archaea. PLoS Genet 6: e1001166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cury J, Touchon M, Rocha EPC (2017) Integrative and conjugative elements and their hosts: composition, distribution and organization. Nucleic Acids Res 46: 202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT (2010) Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5: e11147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Martínez E, Midonet C, Barre F‐X (2013) Integrative mobile elements exploiting Xer recombination. Trends Microbiol 21: 23–30 [DOI] [PubMed] [Google Scholar]
- Debowski AW, Carnoy C, Verbrugghe P, Nilsson H‐O, Gauntlett JC, Fulurija A, Camilleri T, Berg DE, Marshall BJ, Benghezal M (2012) Xer recombinase and genome integrity in Helicobacter pylori, a pathogen without topoisomerase IV. PLoS One 7: e33310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiChiara JM, Mattis AN, Gardner JF (2007) IntDOT interactions with core‐ and arm‐type sites of the conjugative transposon CTnDOT. J Bacteriol 189: 2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douard G, Praud K, Cloeckaert A, Doublet B (2010) The salmonella genomic island 1 is specifically mobilized in trans by the IncA/C multidrug resistance plasmid family. PLoS One 5: e15302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublet B, Boyd D, Mulvey MR, Cloeckaert A (2005) The Salmonella genomic island 1 is an integrative mobilizable element. Mol Microbiol 55: 1911–1924 [DOI] [PubMed] [Google Scholar]
- Drozdetskiy A, Cole C, Procter J, Barton GJ (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43: W389–W394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14: 755–763 [DOI] [PubMed] [Google Scholar]
- Esposito D, Scocca JJ (1997) The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res 25: 3605–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadeev EA, Sam MD, Clubb RT (2009) NMR structure of the amino‐terminal domain of the lambda integrase protein in complex with DNA: immobilization of a flexible tail facilitates beta‐sheet recognition of the major groove. J Mol Biol 388: 682–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador‐Vegas A et al (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44: D279–D285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Windhager L, Rohrer S, Zeiller M, Karnholz A, Hoffmann R, Zimmer R, Haas R (2010) Strain‐specific genes of Helicobacter pylori: genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res 38: 6089–6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery EL, Antczak SM, Mobley HLT (2011) Self‐transmissibility of the integrative and conjugative element ICEPm1 between clinical isolates requires a functional integrase, relaxase, and type IV secretion system. J Bacteriol 193: 4104–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke AE, Clewell DB (1981) Evidence for a chromosome‐borne resistance transposon (Tn916) in Streptococcus faecalis that is capable of ‘conjugal’ transfer in the absence of a conjugative plasmid. J Bacteriol 145: 494–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD‐HIT: accelerated for clustering the next‐generation sequencing data. Bioinformatics 28: 3150–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin TJ, Poulter RT (2001) The DIRS1 group of retrotransposons. Mol Biol Evol 18: 2067–2082 [DOI] [PubMed] [Google Scholar]
- Grindley NDF, Whiteson KL, Rice PA (2006) Mechanisms of site‐specific recombination. Annu Rev Biochem 75: 567–605 [DOI] [PubMed] [Google Scholar]
- Guédon G, Libante V, Coluzzi C, Payot S, Leblond‐Bourget N (2017) The obscure world of integrative and mobilizable elements, highly widespread elements that pirate bacterial conjugative systems. Genes 8: 337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmini J, Néron B, Abby SS, Garcillán‐Barcia MP, de la Cruz F, Rocha EPC (2014) Key components of the eight classes of type IV secretion systems involved in bacterial conjugation or protein secretion. Nucleic Acids Res 42: 5715–5727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard J‐F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307–321 [DOI] [PubMed] [Google Scholar]
- Guo F, Gopaul DN, Van Duyne GD (1997) Structure of Cre recombinase complexed with DNA in a site‐specific recombination synapse. Nature 389: 40–46 [DOI] [PubMed] [Google Scholar]
- Gyohda A, Komano T (2000) Purification and characterization of the R64 shufflon‐specific recombinase. J Bacteriol 182: 2787–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbottle H, Thakur S, Zhao S, White DG (2006) Genetics of antimicrobial resistance. Anim Biotechnol 17: 111–124 [DOI] [PubMed] [Google Scholar]
- Harrison EM, Carter MEK, Luck S, Ou H‐Y, He X, Deng Z, O’Callaghan C, Kadioglu A, Rajakumar K (2010) Pathogenicity islands PAPI‐1 and PAPI‐2 contribute individually and synergistically to the virulence of Pseudomonas aeruginosa strain PA14. Infect Immun 78: 1437–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman AB, Waninger S, Scocca JJ, Dyda F (1997) Molecular organization in site‐specific recombination: the catalytic domain of bacteriophage HP1 integrase at 2.7 A resolution. Cell 89: 227–237 [DOI] [PubMed] [Google Scholar]
- Hochhut B, Waldor MK (1999) Site‐specific integration of the conjugal Vibrio cholerae SXT element into prfC. Mol Microbiol 32: 99–110 [DOI] [PubMed] [Google Scholar]
- Huber KE, Waldor MK (2002) Filamentous phage integration requires the host recombinases XerC and XerD. Nature 417: 656–659 [DOI] [PubMed] [Google Scholar]
- Jayaram M, Rowley PA, Fan H‐F, Kachroo AH, Voziyanov Y, Guga P, Ma C‐H (2015) An overview of tyrosine site‐specific recombination: from an Flp perspective. Microbiol Spectr 3: MDNA3‐0021‐2014 [DOI] [PubMed] [Google Scholar]
- Johnson LS, Eddy SR, Portugaly E (2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11: 431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhas M, Dimopoulou I, Robinson E, Elamin A, Harding R, Hood D, Crook D (2013) Identification of another module involved in the horizontal transfer of the Haemophilus genomic island ICEHin1056. Plasmid 70: 277–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Swalla BM, Gardner JF (2010) Structure‐function analysis of IntDOT. J Bacteriol 192: 575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm P (1986) Two regulatory fim genes, fimB and fimE, control the phase variation of type 1 fimbriae in Escherichia coli . EMBO J 5: 1389–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komano T, Kubo A, Nisioka T (1987) Shufflon: multi‐inversion of four contiguous DNA segments of plasmid R64 creates seven different open reading frames. Nucleic Acids Res 15: 1165–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono N, Arakawa K, Tomita M (2011) Comprehensive prediction of chromosome dimer resolution sites in bacterial genomes. BMC Genom 12: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Wolf YI, Krupovic M (2020) Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat Rev Genet 21: 119–131 [DOI] [PubMed] [Google Scholar]
- Kropinski AM, Kovalyova IV, Billington SJ, Patrick AN, Butts BD, Guichard JA, Pitcher TJ, Guthrie CC, Sydlaske AD, Barnhill LM et al (2007) The genome of epsilon15, a serotype‐converting, Group E1 Salmonella enterica‐specific bacteriophage. Virology 369: 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen L, Rubio‐Cosials A, Patil KR, Barabas O (2018) Conjugative transposition of the vancomycin resistance carrying Tn1549: enzymatic requirements and target site preferences. Mol Microbiol 107: 639–658 [DOI] [PubMed] [Google Scholar]
- Landy A (2015) The λ integrase site‐specific recombination pathway. Microbiol Spectr 3: MDNA3‐0051‐2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxmikanthan G, Xu C, Brilot AF, Warren D, Steele L, Seah N, Tong W, Grigorieff N, Landy A, Van Duyne GD (2016) Structure of a Holliday junction complex reveals mechanisms governing a highly regulated DNA transaction. Elife 5: e14313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplae R, Lima‐Mendez G, Toussaint A (2010) ACLAME: a CLAssification of mobile genetic elements, update 2010. Nucleic Acids Res 38: D57–D61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P (2011) Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39: W475–W478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Li J‐W, Feng Z, Wang J, An H, Liu Y, Wang Y, Wang K, Zhang X, Miao Z et al (2016) Epigenetic switch driven by DNA inversions dictates phase variation in Streptococcus pneumoniae . PLoS Pathog 12: e1005762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Gingery M, Doulatov SR, Liu Y, Hodes A, Baker S, Davis P, Simmonds M, Churcher C, Mungall K et al (2004) Genomic and genetic analysis of Bordetella bacteriophages encoding reverse transcriptase‐mediated tropism‐switching cassettes. J Bacteriol 186: 1503–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt BL, Hatfull GF (2016) Brujita integrase: a simple, arm‐less, directionless, and promiscuous tyrosine integrase system. J Mol Biol 428: 2289–2306 [DOI] [PubMed] [Google Scholar]
- MacDonald D, Demarre G, Bouvier M, Mazel D, Gopaul DN (2006) Structural basis for broad DNA‐specificity in integron recombination. Nature 440: 1157–1162 [DOI] [PubMed] [Google Scholar]
- Mageeney CM, Lau BY, Wagner JM, Hudson CM, Schoeniger JS, Krishnakumar R, Williams KP (2020) New candidates for regulated gene integrity revealed through precise mapping of integrative genetic elements. Nucleic Acids Res 48: 4052–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manso AS, Chai MH, Atack JM, Furi L, De Ste Croix M, Haigh R, Trappetti C, Ogunniyi AD, Shewell LK, Boitano M et al (2014) A random six‐phase switch regulates pneumococcal virulence via global epigenetic changes. Nat Commun 5: 5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler‐Bauer A, Bo Yu, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR et al (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45: D200–D203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazel D (2006) Integrons: agents of bacterial evolution. Nat Rev Microbiol 4: 608–620 [DOI] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F et al (2001) Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413: 852–856 [DOI] [PubMed] [Google Scholar]
- Midonet C, Barre F‐X (2014) Xer site‐specific recombination: promoting vertical and horizontal transmission of genetic information. Microbiol Spectr 2: MDNA3‐0056‐2014 [DOI] [PubMed] [Google Scholar]
- Mohd‐Zain Z, Turner SL, Cerdeño‐Tárraga AM, Lilley AK, Inzana TJ, Duncan AJ, Harding RM, Hood DW, Peto TE, Crook DW (2004) Transferable antibiotic resistance elements in Haemophilus influenzae share a common evolutionary origin with a diverse family of syntenic genomic islands. J Bacteriol 186: 8114–8122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura A, Soares M, Pereira C, Leitao N, Henriques I, Correia A (2009) INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics 25: 1096–1098 [DOI] [PubMed] [Google Scholar]
- Naas T, Cuzon G, Villegas M‐V, Lartigue M‐F, Quinn JP, Nordmann P (2008) Genetic structures at the origin of acquisition of the β‐Lactamase blaKPC gene. Antimicrob Agents Chemother 52: 1257–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolivos S, Pages C, Rousseau P, Le Bourgeois P, Cornet F (2010) Are two better than one? Analysis of an FtsK/Xer recombination system that uses a single recombinase. Nucleic Acids Res 38: 6477–6489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes‐Düby SE, Kwon HJ, Tirumalai RS, Ellenberger T, Landy A (1998) Similarities and differences among 105 members of the Int family of site‐specific recombinases. Nucleic Acids Res 26: 391–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Mondal SI, Islam MR, Mako T, Arisawa K, Katsura K, Ooka T, Gotoh Y, Murase K, Ohnishi M et al (2015) The Shiga toxin 2 production level in enterohemorrhagic Escherichia coli O157:H7 is correlated with the subtypes of toxin‐encoding phage. Sci Rep 5: 16663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Karplus K, Barrett C, Hughey R, Haussler D, Hubbard T, Chothia C (1998) Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J Mol Biol 284: 1201–1210 [DOI] [PubMed] [Google Scholar]
- Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD (2018) HMMER web server: 2018 update. Nucleic Acids Res 46: W200–W204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Gurkar AU, Lory S (2006) Interstrain transfer of the large pathogenicity island (PAPI‐1) of Pseudomonas aeruginosa . Proc Natl Acad Sci USA 103: 19830–19835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman‐Livaja M, Biswas T, Ellenberger T, Landy A, Aihara H (2006) DNA arms do the legwork to ensure the directionality of lambda site‐specific recombination. Curr Opin Struct Biol 16: 42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricker N, Qian H, Fulthorpe RR (2013) Phylogeny and organization of recombinase in trio (RIT) elements. Plasmid 70: 226–239 [DOI] [PubMed] [Google Scholar]
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J‐F, Darling A, Malfatti S, Swan BK, Gies EA et al (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437 [DOI] [PubMed] [Google Scholar]
- Roche D, Fléchard M, Lallier N, Répérant M, Brée A, Pascal G, Schouler C, Germon P (2010) ICEEc2, a new integrative and conjugative element belonging to the pKLC102/PAGI‐2 family, identified in Escherichia coli strain BEN374. J Bacteriol 192: 5026–5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio‐Cosials A, Schulz EC, Lambertsen L, Smyshlyaev G, Rojas‐Cordova C, Forslund K, Karaca E, Bebel A, Bork P, Barabas O (2018) Transposase‐DNA complex structures reveal mechanisms for conjugative transposition of antibiotic resistance. Cell 173: 208–220.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, Van Duyne GD (2014) The ins and outs of serine integrase site‐specific recombination. Curr Opin Struct Biol 24: 125–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JR, Bringel F, Marra D, Van Alstine G, Rudy CK (1994) Conjugative transposition of Tn916: preferred targets and evidence for conjugative transfer of a single strand and for a double‐stranded circular intermediate. Mol Microbiol 11: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Seth‐Smith H, Fookes Mc, Okoro Ck, Baker S, Harris Sr, Scott P, Pickard D, Quail Ma, Churcher C, Sanders M et al (2012) Structure, diversity, and mobility of the Salmonella pathogenicity island 7 family of integrative and conjugative elements within Enterobacteriaceae. J Bacteriol 194: 1494–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M (1999) Multiple comparisons of log‐likelihoods with applications to phylogenetic inference. Mol Biol Evol 16: 1114–1116 [Google Scholar]
- Shoemaker NB, Wang GR, Salyers AA (1996) The Bacteroides mobilizable insertion element, NBU1, integrates into the 3’ end of a Leu‐tRNA gene and has an integrase that is a member of the lambda integrase family. J Bacteriol 178: 3594–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P, Gourbeyre E, Chandler M (2014) Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol Rev 38: 865–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P, Varani A, Perochon J, Chandler M (2012) Exploring bacterial insertion sequences with ISfinder: objectives, uses, and future developments. Methods Mol Biol 859: 91–103 [DOI] [PubMed] [Google Scholar]
- Skaar K, Claesson M, Odegrip R, Högbom M, Haggård‐Ljungquist E, Stenmark P (2015) Crystal structure of the bacteriophage P2 integrase catalytic domain. FEBS Lett 589: 3556–3563 [DOI] [PubMed] [Google Scholar]
- Stokes HW, Gillings MR (2011) Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram‐negative pathogens. FEMS Microbiol Rev 35: 790–819 [DOI] [PubMed] [Google Scholar]
- Subramanya HS, Arciszewska LK, Baker RA, Bird LE, Sherratt DJ, Wigley DB (1997) Crystal structure of the site‐specific recombinase, XerD. EMBO J 16: 5178–5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swalla BM, Gumport RI, Gardner JF (2003) Conservation of structure and function among tyrosine recombinases: homology‐based modeling of the lambda integrase core‐binding domain. Nucleic Acids Res 31: 805–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szwagierczak A, Antonenka U, Popowicz GM, Sitar T, Holak TA, Rakin A (2009) Structures of the arm‐type binding domains of HPI and HAI7 integrases. J Biol Chem 284: 31664–31671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumalai RS, Healey E, Landy A (1997) The catalytic domain of λ site‐specific recombinase. Proc Natl Acad Sci USA 94: 6104–6109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumalai RS, Pargellis CA, Landy A (1996) Identification and characterization of the N‐ethylmaleimide‐sensitive site in lambda‐integrase. J Biol Chem 271: 29599–29604 [DOI] [PubMed] [Google Scholar]
- Trieu‐Cuot P, Poyart‐Salmeron C, Carlier C, Courvalin P (1993) Sequence requirements for target activity in site‐specific recombination mediated by the Int protein of transposon Tn 1545. Mol Microbiol 8: 179–185 [DOI] [PubMed] [Google Scholar]
- Van Duyne GD (2015) Cre recombinase. Microbiol Spectr 3: MDNA3‐0014‐2014 [DOI] [PubMed] [Google Scholar]
- Van Houdt R, Leplae R, Lima‐Mendez G, Mergeay M, Toussaint A (2012) Towards a more accurate annotation of tyrosine‐based site‐specific recombinases in bacterial genomes. Mob DNA 3: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhooff V, Galloy C, Agaisse H, Lereclus D, Revet B, Hallet B (2006) Self‐control in DNA site‐specific recombination mediated by the tyrosine recombinase TnpI. Mol Microbiol 60: 617–629 [DOI] [PubMed] [Google Scholar]
- Wang H, Roberts AP, Mullany P (2000) DNA sequence of the insertional hot spot of Tn916 in the Clostridium difficile genome and discovery of a Tn916‐like element in an environmental isolate integrated in the same hot spot. FEMS Microbiol Lett 192: 15–20 [DOI] [PubMed] [Google Scholar]
- Wang J, Liu Y, Liu Y, Du K, Xu S, Wang Y, Krupovic M, Chen X (2018) A novel family of tyrosine integrases encoded by the temperate pleolipovirus SNJ2. Nucleic Acids Res 46: 2521–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TJ, Clements J, Finn RD (2014) Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics 15: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle G, Shoemaker NB, Salyers AA (2002) The role of Bacteroides conjugative transposons in the dissemination of antibiotic resistance genes. Cell Mol Life Sci 59: 2044–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak JM, Connolly KM, Clubb RT (1999) NMR structure of the Tn916 integrase‐DNA complex. Nat Struct Biol 6: 366–373 [DOI] [PubMed] [Google Scholar]
- Wojciak JM, Sarkar D, Landy A, Clubb RT (2002) Arm‐site binding by lambda ‐integrase: solution structure and functional characterization of its amino‐terminal domain. Proc Natl Acad Sci USA 99: 3434–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MM, Dichiara JM, Yoneji S, Gardner JF (2010) CTnDOT integrase interactions with attachment site DNA and control of directionality of the recombination reaction. J Bacteriol 192: 3934–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MM, Gardner JF (2015) The integration and excision of CTnDOT. Microbiol Spectr 3: MDNA3‐0020‐2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Tang H (2017) ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 33: 3340–3347 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Appendix
Review Process File
Data Availability Statement
The computer code produced in this study is available in the following GitHub repository: https://github.com/smyshlya/NeighborsScan.