

Abstract
Mice with a mutation (D244G, DG) in calsequestrin 1 (CASQ1), analogous to a human mutation in CASQ1 associated with a delayed onset human myopathy (vacuolar aggregate myopathy), display a progressive myopathy characterized by decreased activity, decreased ability of fast twitch muscles to generate force and low body weight after one year of age. The DG mutation causes CASQ1 to partially dissociate from the junctional sarcoplasmic reticulum (SR) and accumulate in the endoplasmic reticulum (ER). Decreased junctional CASQ1 reduces SR Ca2+ release. Muscles from older DG mice display ER stress, ER expansion, increased mTOR signaling, inadequate clearance of aggregated proteins by the proteasomes, and elevation of protein aggregates and lysosomes. This study suggests that the myopathy associated with the D244G mutation in CASQ1 is driven by CASQ1 mislocalization, reduced SR Ca2+ release, CASQ1 misfolding/aggregation and ER stress. The subsequent maladaptive increase in protein synthesis and decreased protein aggregate clearance are likely to contribute to disease progression.
Keywords: calsequestrin, EC coupling, ER stress, myopathy, sarcoplasmic reticulum
1 |. INTRODUCTION
Vacuolar aggregate myopathy (VAM, OMIM# 616231) is a rare human muscle disorder, linked to mutations in CASQ1 and presents with elevated serum creatine kinase, fatigue, myalgia, muscle cramps, lower limb hypertrophy, and muscle weakness.1–4 Muscles from VAM patients frequently contain protein aggregates and an increased number of vacuoles.1–4 Mutation of a conserved aspartic acid at position D244 to glycine (D244G, DG) in CASQ1 associated with VAM in humans slows sarcoplasmic reticulum (SR) Ca2+ release and impairs polymerization of CASQ1.1–3,5 Although the age at which VAM is diagnosed is variable, adults in their sixth decade are the most likely to experience muscle weakness and exercise intolerance.1,3 Vacuoles and protein aggregates form mainly in Type II muscle fibers which stain positive for both the mutated protein and several non-mutated proteins,1–4 suggesting the mutated proteins cause misfolding and aggregation of normal proteins.6
The endoplasmic reticulum (ER) and SR of striated muscle are parts of a large, continuous network with distinct subdomains that fulfil the functions of protein and lipid synthesis, protein folding, and translocation (ER) and Ca2+ storage and release (SR). Sub-domains are also formed with the plasma membrane and with other organelles including mitochondria, lysosomes, and the Golgi network.7–12 Canonical ER functions occur in the perinuclear region and throughout the peripheral network of tubules.9,13 ER quality control mechanisms including the unfolded protein response (UPR) and the ubiquitin proteasome system (UPS) function to ensure that misfolded and/or excess proteins do not accumulate and cause long-term activation of stress pathways.14 In contrast, the primary role of the SR is to serve as a reservoir for the Ca2+ needed for excitation-contraction coupling (ECC). CASQ1 is the primary Ca2+ buffer in skeletal muscle SR and modulates SR Ca2+ release through the Ca2+ release channel (type 1 Ryanodine Receptor, RYR1) during ECC. Polymerized CASQ1 is tethered to RYR1 at the junctional SR via two transmembrane proteins, triadin, and junctin.15,16 The role of CASQ1 as a Ca2+ buffer is regulated by its dynamic polymerization and depolymerization as Ca2+ cycles in and out of the SR during ECC.17 As luminal SR Ca2+ increases, CASQ1 polymerizes, increasing its Ca2+ affinity and binding capacity, but, when the luminal Ca2+ concentration drops during ECC, CASQ1 depolymerizes.17–19 Polymerization is initiated by the interaction of CASQ1 monomers to form front-to-front and back-to-back dimers which, in turn, form long, branched polymers, providing additional sites for Ca2+ binding.17,20,21 The location of CASQ1 proximal to RYR1 and the high capacity Ca2+ binding of CASQ1 polymers enables the SR to have a large local pool of Ca2+ available for release to drive muscle contraction, maintain a high total SR Ca2+ level, and avoid excessive depletion during muscle activity.18,22 In addition to regulating its Ca2+ binding properties, CASQ1 polymerization ensures that CASQ1 is correctly targeted to the junctional SR following its synthesis in the ER.23–26
Amino acid D244 in CASQ1 is part of a high affinity Ca2+-binding site5 and the mutation of D244 to a glycine (D244G) increases flexibility of this region of the protein, leading to disorder, decreased Ca2+ affinity and capacity, and alterations in back-to-back polymerization.5 In studies with isolated CASQ1, the mutation causes the formation of misfolded monomers at low Ca2+ concentrations and insoluble aggregates with decreased Ca2+-binding capacity at high Ca2+ concentrations.5,27 The DG mutation in CASQ1 also causes aggregation of STIM127 and SERCA1.1,4 CASQ1 modulates the activity of RYR128 and, consistent with this, the DG mutation slows caffeine-induced SR Ca2+ release.2 However, the current understanding of the functional effects of the DG mutation relies on in vitro systems. The effects of the DG mutation in CASQ1 on SR Ca2+ stores, SR Ca2+ release, force production, and fatiguability of muscle remain to be determined and require an animal model of VAM. An animal model that recapitulates human VAM is also needed for the development of therapeutic interventions. Although mutant CASQ1 is known to cause protein aggregate formation, the effect of these aggregates on muscle function are not known.
Using CRISPR/Cas9 editing, we created heterozygous mice (designated DG mice) with a mutation in Casq1, leading to the D244G mutation in the CASQ1 protein. The DG mice display many of the features of human VAM, including dominant inheritance, late onset of disease, progressive worsening of the symptoms with age, and the appearance of protein aggregates, primarily in Type II muscle fibers. Our studies demonstrate that the DG mice are a good model for studying VAM and, as such, may be useful for the development of therapeutic interventions. We also show that the late onset of the disease is likely due to the age-dependent decrease in the capacity of the proteolytic systems to adequately clear the misfolded proteins, leading to the formation of protein aggregates and persistent and maladaptive ER stress.
2 |. MATERIALS AND METHODS
2.1 |. Materials
The complete list of antibodies used is provided in Table S1. N-benzyl-p-toluene sulfonamide (BTS), thapsigargin, cyclopiazonic acid (CPA), and colchicine were purchased from Tocris Biosciences (Minneapolis, Mn, USA). Proteostat aggresome detection kit (ENZ-51035-K100) and MG132 were purchased from Enzo Life Sciences (Farmingdale, NY, USA). The Ca2+ indicators Mag Fluo-4 AM, Fura-2 AM, Halt protease and phosphatase inhibitor cocktail, ER-Tracker Blue-White DPX, TRIzol reagent and laminin were purchased from Thermo Fisher (Waltham, MA, USA). iScript cDNA Synthesis Kit, iQ SYBR Green Supermix, precast and acrylamide gel kits containing Stain Free reagent, molecular weight markers and Starbright secondary antibodies were purchased from Bio-Rad (Hercules, CA, USA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2 |. Generation of mouse model
The CASQ1D244G mice were generated at the BCM Genetically Engineered Rodent Models Core facility using CRISPR/Cas9-mediated genome engineering technology. Cas9 mRNA, the single guide (sgRNA) targeting D244 region in exon 6 of the CASQ1 gene, and the donor oligonucleotides with D244G mutation were microinjected into zygotes of C57BL/6J mice.
2.3 |. Wire hang test
A wire hang test was used to assess mouse performance.29 Mice were suspended by the forelimbs on a 60 cm wire between two platforms. The number of falls and escapes in a 3-minute test were counted and timed.
2.4 |. Voluntary running
Voluntary mouse activity was assessed using monitored running wheels. For this test, mice were housed individually in cages equipped with in-cage running wheels (STARR life sciences). Average wheel turns per mouse were assessed over a period of 10 days for daytime (6 AM–6 PM) and nighttime (6 PM–6 AM), excluding the initial 3-day acclimation period.
2.5 |. Body composition
Mice were anesthetized with isoflurane and imaged using the UltraFocus ultra-high-resolution dual-energy x-ray absorptiometry (DEXA) system (Faxitron, Tucson, AZ, USA).
2.6 |. Ex vivo force measurement
Intact soleus and extensor digitorum longus (EDL) muscles were dissected and immersed in Krebs’s ringer solution (137 mM NaCl, 5 mM KCl, 1 mM NaH2PO4, 24 mM NaHCO3, 5 mM Glucose, 2 mM CaCl2, and 1 mM MgSO4) oxygenated with a 95/5% mixture of O2/CO2. Muscles were tied with sutures and suspended between a force transducer and stationary anchor and allowed to equilibrate at 34°C. Muscle optimal length was determined via single twitch force generation measurements. Force frequency measurements were obtained by applying 250 ms trains using frequencies from 1 to 300 Hz followed by a 5 minutes fatigue protocol. The specific fatigue protocol for soleus muscle was 30 Hz, 200 ms duration, 1 second intervals and for EDL muscles was 60 Hz, 200 ms duration, 1 second interval. Muscle stimulation in the test chamber was applied using platinum electrodes attached to a Grass S48 stimulator and recorded within Chart5 (version 5.2) software.
2.7 |. Muscle sections
Soleus and EDL muscles were dissected, embedded in OCT compound (Tissue-Tek) and frozen in 2-methylbutane. Frozen muscles were cut into 10 μm sections and stored at −20°C.
2.8 |. Histology
Hematoxylin and eosin (H&E) staining of frozen muscle sections was done in the Pathology and Histology Core at BCM using a Varistain V24-4 automatic slide stainer. NADH-Tetrazolium Reductase (NADH-TR) staining solution was prepared by dissolving ß-NADH (0.04%) and Nitro Blue Tetrazolium (0.1%) in 50 mM Tris Buffer (pH 7.6) and stored at 37°C before use. Frozen muscle sections were immersed in warmed NADH-TR solution and incubated at 37°C for 20 minutes. Sections were rinsed in deionized water for 5 × 5 s and dehydrated in ascending alcohol solutions (1 × 70%, 2 × 95% and 3 × 100%) for 1 minute each. Dehydrated sections were treated with three washes in 100% xylene and mounted with Permount Mounting medium (Fisher Scientific). Modified Gomori trichrome stain was performed by immersing sections in Harris Hematoxylin for 5 minutes. After washing with water, sections were immersed in modified Gomori Trichrome stain (0.6% Chromotrope 2R, 0.3% Fast Green FCF, 0.6% Phosphotungstic acid dissolved in 1% acetic acid, pH 3.4) for 60 minutes. Sections were differentiated in 0.2% acetic acid and dehydrated in ascending alcohol solutions (2 × 95% and 2 × 100%) and cleared using three washes in xylene. Sections were mounted in Permount Mounting medium. All histological stained sections were imaged at 40× magnification on a Nikon Ci-L brightfield microscope.
2.9 |. Fiber typing and cross-sectional area
Sections were fixed in 4% paraformaldehyde and permeabilized in 0.1% TX-100 in phosphate-buffered saline (PBS). After incubation in blocking buffer (4% heat-inactivated goat serum and 0.5% TX-100), sections were incubated in primary antibodies to myosin heavy chain isoforms I (mouse IgG2b), IIa (mouse IgG1) and IIb (mouse IgM) and to laminin (rabbit IgG) for identification of fiber borders. After washing with PBS, sections were incubated with Alexa Fluor 546-conjugated goat anti-rabbit IgG, Alexa Fluor 647-conjugated goat anti-mouse IgG2b, Alexa Fluor 488-conjugated goat anti-mouse IgG1 and Alexa Fluor 594-conjugated goat anti-mouse IgM 594-conjugated goat anti-mouse secondary antibodies diluted 1:200 at for 90 minutes at room temperature. Sections were washed extensively in PBS and in DAPI to stain nuclei. Slides were mounted with Fluoromount mounting media. Images were acquired with a Zeiss LSM 880 confocal microscope. Lambda scanning was done on control slides stained with each secondary antibody to define the emission spectra of each secondary antibody used. Images of muscle sections were acquired with a 10× objective and analyzed using FIJI. Quantitation of fiber type and cross-sectional area was semi-automated using MyoSight.30
2.10 |. Muscle fiber isolation
Muscle fibers were isolated from flexor digitorum brevis (FDB) muscle. FDB muscles were isolated and incubated at 37°C in DMEM supplemented with collagenase and 10% FBS for 75 minutes. Fibers were separated by gentle trituration with a flame polished pipette, plated on laminin coated coverslips and incubated at 37°C. Fibers were always used or fixed within 24 hours of isolation.
2.11 |. Stimulated Ca2+ transients and SR load
To measure stimulated Ca2+ transients, isolated fibers were loaded with 5 μM Fura 2 AM for 30 minutes at room temperature followed by 30 minutes washout with Tyrode’s solution containing: 121 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 500 μM MgCl2, 400 μM NaH2PO4, 100 μM EDTA, 5.5 mM glucose, and 24 mM NaHCO3. pH was regulated with bubbling of CO2/O2 5/95 % and 40 μM BTS was added to inhibit muscle contraction. Electrical stimulation was applied using field stimulation at the following stimulation frequencies: 1 Hz, 15 Hz, 30 Hz, 40 Hz, 60 Hz, 80 Hz, 160 Hz, and 300 Hz (250 ms train duration), separated by a 1 minute rest period. Fibers were imaged using an IonOptix acquisition system (IonOptix Westwood, MA, USA). Analysis of the Ca2+ transient amplitude and kinetics was made using IonWizard software. Store load was assessed in fibers loaded with 5 μM Mag Fluo 4 AM by applying 30 mM caffeine in the presence of EGTA and 0 mM Ca2+. Total Ca2+ was assessed by applying a Ca2+ release cocktail containing 10 μM ionomycin, 30 μM CPA, and 100 μM EGTA (ICE cocktail).31
2.12 |. Ca2+ influx
To determine if sarcolemmal Ca2+ influx was changed, a manganese (Mn2+) quench assay was performed in intact FDB fibers as described previously.32 Mn2+ ions permeate sarcolemmal Ca2+ channels and this assay takes advantage of the ability of Mn2+ ions to quench Fura 2 fluorescence at the dye’s isosbestic point (360 nm). The decay of the Fura 2 fluorescence in a Mn2+-containing, Ca2+ free solution is indicative of the activity of sarcolemmal Ca2+ channels. Fibers from 3-month-old mice were loaded with Fura 2 for 30 minutes at room temperature followed by a 30 minute washout step. Baseline fluorescence was measured in normal Tyrodes (0 Mn2+ 1.8 Ca2+) before the solution was switched to a Ca2+ free solution containing 1.8 mM MnCl2 (0 Ca2+, 1.8 Mn2+). Passive Mn2+ influx was measured for 2 minutes before electrical stimulation was applied (20 × 500 ms trains, 50 Hz, 1 tps). To measure store operated Ca2+ entry (SOCE) a different set of experiments were done where Mn2+ quench was compared in untreated fibers and fibers with depleted SR stores.33 To block SR Ca2+ uptake and deplete SR Ca2+ stores, fibers were incubated with 15 μM CPA and 1 μM thapsigargin during Fura 2 loading and washout (1 hour total) in a nominally Ca2+ free solution containing 145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 0.2 mM EGTA, and pH 7.4 with NaOH. Following 2 minutes of baseline recording the bathing solution was exchanged with one that contained 0.5 mM MnCl2 and the fluorescence decay was monitored for 5 minutes. All solutions contained 40 μM BTS to prevent fiber contraction. Fura 2 excitation (360 nm) and emission (510 nm) were monitored using an IonOptix acquisition system.
2.13 |. Immunofluorescent labeling of FDB fibers
For immunofluorescence studies intact FDB fibers were plated on laminin-coated glass bottom dishes (MatTek, Ashland, MA, USA). Fibers were fixed for 10 minutes in PBS containing 4% formalin and 100 μM EGTA. After washing in PBS, fibers were permeabilized for 30 minutes in 1% TX-100 and incubated blocking buffer (2% goat serum, 0.1% TX-100 and 0.5% BSA) for 1 hour. Fibers were incubated with primary antibodies overnight at 4°C. After washing, fibers were incubated in Alexa Fluor conjugated secondary antibodies for 1 hour at room temperature. Sections were washed in PBS and DAPI and imaged on a Zeiss LSM 880 confocal microscope equipped with FAST Airyscan. Antibody details and dilution factors are detailed in Table S1.
2.14 |. Aggresome measurement
Protein aggregates were detected using the Proteostat Aggresome detection kit as described in the manufacturer’s protocol (Enzo Life Sciences, NY, USA). After isolation and plating on glass bottom dishes, fibers were incubated with either vehicle (DMSO) or 5 μM MG132 for 16 hours. Fibers were then fixed, permeabilized and blocked as described above. Proteostat Aggresome Detection Reagent combined with Hoechst was added to fibers for 2 hours at room temperature. After washing, fibers were imaged using a Zeiss 780 Confocal microscope. Protein aggregates were quantified in FIJI by applying the MaxEntropy threshold on background subtracted images. Particle analysis was used to determine puncta number per fiber.
2.15 |. Live cell ER staining
To visualize the ER and lysosomes, FDB fibers were plated on laminin-coated glass bottomed dishes and incubated with fluorescent markers. Live, intact fibers were incubated for 30 minutes at 37°C with 100 nM ER-tracker Blue-White DPX dissolved in Tyrodes buffer. Fibers were imaged on a Zeiss LSM 880 confocal microscope. ER expansion was quantified in FIJI by measuring the areas of intensified fluorescence identified by applying the MaxEntropy threshold. Particle analysis was used to determine the area of all particles over 0.5 μm in size and the sum of ER puncta area was expressed as a percentage of the total fiber area, obtained by drawing a perimeter around the fiber border.
2.16 |. Western blotting
Muscles were snap frozen in liquid nitrogen and stored at −80°C until use. Muscles were thawed in RIPA buffer supplemented with Halt Protease and phosphatase inhibitor cocktail and homogenized by bead homogenization (Precellys 24 homogenizer, Bertin Instruments, Paris, France). Protein concentrations were determined using a BCA assay. Muscle homogenates were separated by SDS PAGE using the Bio-Rad Stain Free gel system to enable total protein visualization and quantitation. Proteins were transferred to PVDF membrane (Immobilon-FL, EMD Millipore) before blocking and incubation in primary antibodies overnight at 4°C. After washing in PBS containing 0.2% Tween-20, membranes were incubated with fluorophore-tagged Starbright secondary antibodies (Bio-Rad) for 1 hour at room temperature and imaged on a Chemidoc MP system (Bio-Rad). All Antibody details and dilution factors are detailed in Table S1. Immunofluorescence was quantified using Image Lab software (Bio-Rad). Each band was normalized to the total protein for that lane and expressed as a percentage of the average control value on the same blot.
2.17 |. EC coupling gene expression
Gene expression analysis was done as described previously.34 Total RNA was extracted from WT and DG TA muscles using TRIzol reagent and reverse transcribed into cDNA using the iScript cDNA Synthesis Kit. Gene expression assay were done with iQ SYBR Green Supermix (Bio-Rad) using a ViiA 7 RT-PCR System (Applied Biosystems). Technical triplicates were run for all samples, and primer specificity was confirmed by melting curve analysis. The relative expression of target genes was calculated based on the reaction cycle threshold values, calibrated for PCR efficiency35 and normalized to Rn18s levels. Forward and reverse primer sets are shown in Table S2.
2.18 |. Autophagic flux
To analyze autophagic flux in vivo, mice were injected intraperitoneally once per day with either saline or 0.4 mg/kg colchicine in saline on two consecutive days. This colchicine treatment had no effect on body weight. Mice were euthanized and muscles were rapidly dissected and prepared for western blotting, cryosectioning or immunofluorescence. An in vitro assay for autophagic flux was performed by treating FDB fibers with either DMSO or 400 nM bafilomycin A1 for 3 hours at 37°C. Fibers were then immediately fixed, permeabilized, and prepared for immunofluorescence as above.
2.19 |. Electron microscopy
Transmission electron microscopy methods were modified from 36,37. Hindlimbs were dissected and, following removal of skin and other hindlimb muscles, the exposed EDL and soleus were fixed in situ in 2% paraformaldehyde and 2.5% glutaraldehyde overnight at 4°C, removed from the bone, segmented and postfixed in 1% OsO4 and 0.8% potassium ferricyanide in 0.1M cacodylate buffer for 1 hour at 4°C. Segments were then stained en bloc in saturated aqueous uranyl acetate for 1 hour. Following dehydration in ascending ethanol solutions, segments were embedded in Embed 812 resin. Ultrathin muscle sections (50–60 nm) were prepared with a Leica U7 ultramicrotome (Leica Microsystem, Austria) using a Diatome diamond knife (Diatome Ltd. CH-2501 Biel, Switzerland). Sections were stained in saturated aqueous uranyl acetate and lead citrate solutions.
2.20 |. Analysis of TX-100 soluble and insoluble proteins in WT and DG muscle
Triton X-100 soluble and insoluble fractions were prepared as described in 38,39. Briefly, tibialis anterior (TA) muscles from 3- and 8-month-old WT and DG mice were homogenized by bead homogenization in five volumes of sucrose buffer (50 mM Tris-HCl, pH7.4, 1 mM EDTA, and 0.25 M sucrose) with protease inhibitors and phosphatase inhibitors. To remove the tissue debris, muscle homogenates were centrifuged at 500× g for 10 minutes at 4°C and the supernatants were further lysed with lysis buffer (sucrose buffer containing final concentration of 0.5% TX-100) on ice for 30 minutes. To separate the TX-100 soluble supernatant and insoluble pellet, lysates were centrifuged at 13 000× g for 15 minutes. Pellets were resuspended in 2% SDS in PBS and sonicated for full lysis. Thirty μg of pellet or lysate were subject to SDS-PAGE and western blotting using the methods described above.
2.21 |. Statistical analyses
Data were analyzed using GraphPad Prism 7 software. T tests were used to assess the significance of differential effects between two groups with normal distribution. If data failed a normality test, they were subjected to a Mann-Whitney rank sum test. For data where two factors were tested, two-way ANOVA were performed with the relevant post-hoc tests to compare the means of multiple groups. Outliers (max one per group) were identified using Grubbs’ method (alpha = 0.05). Values are presented as mean ± SEM Data where the P-value was less than .05 were considered statistically significant.
3 |. RESULTS
3.1 |. Generation of D244G (DG) mice
We used CRISPR-Cas9-mediated genome editing to generate C57B6/J mice expressing CASQ1 with an autosomal dominant mutation, D244G as described in Figure S1. These mice were backcrossed for at least two generations prior to the onset of functional analyses. The heterozygous and homozygous DG mice were viable with no obvious effects on lifespan. Heterozygous mice (designated DG below) were used in the current study for comparison to human VAM disease, which is inherited in an autosomal dominant fashion. Unless otherwise stated, studies were performed on mice aged 3 months (±2 weeks) and 8 months (±2 weeks).
3.2 |. DG mice as a model of VAM
Muscle function and performance were assessed in 3-month and 8-month-old DG mice compared to their WT littermates. Young DG and WT mice performed equally well in the wire-hang test displaying similar number of falls and number of escapes (Figure 1A,B). In contrast, the 8-month-old DG mice had significantly more falls and fewer escapes than their WT littermates (Figure 1A,B). Voluntary wheel running also significantly declined in older DG mice compared to their WT littermates (Figure 1C). There were no significant differences between WT and DG mice with respect to body weight or body composition until the mice reached ages greater than 1 year. By 18 months of age the DG mice had lower body weights due to decreased lean and fat mass compared to their WT littermates (Figure 1D–H). Comparison of lean weight (Figure 1F) and fat weight (Figure 1H) normalized to total body weight indicate that the loss of fat mass is the greatest contributor to the overall decrease in body weight of older DG mice compared to their WT littermates.
FIGURE 1.
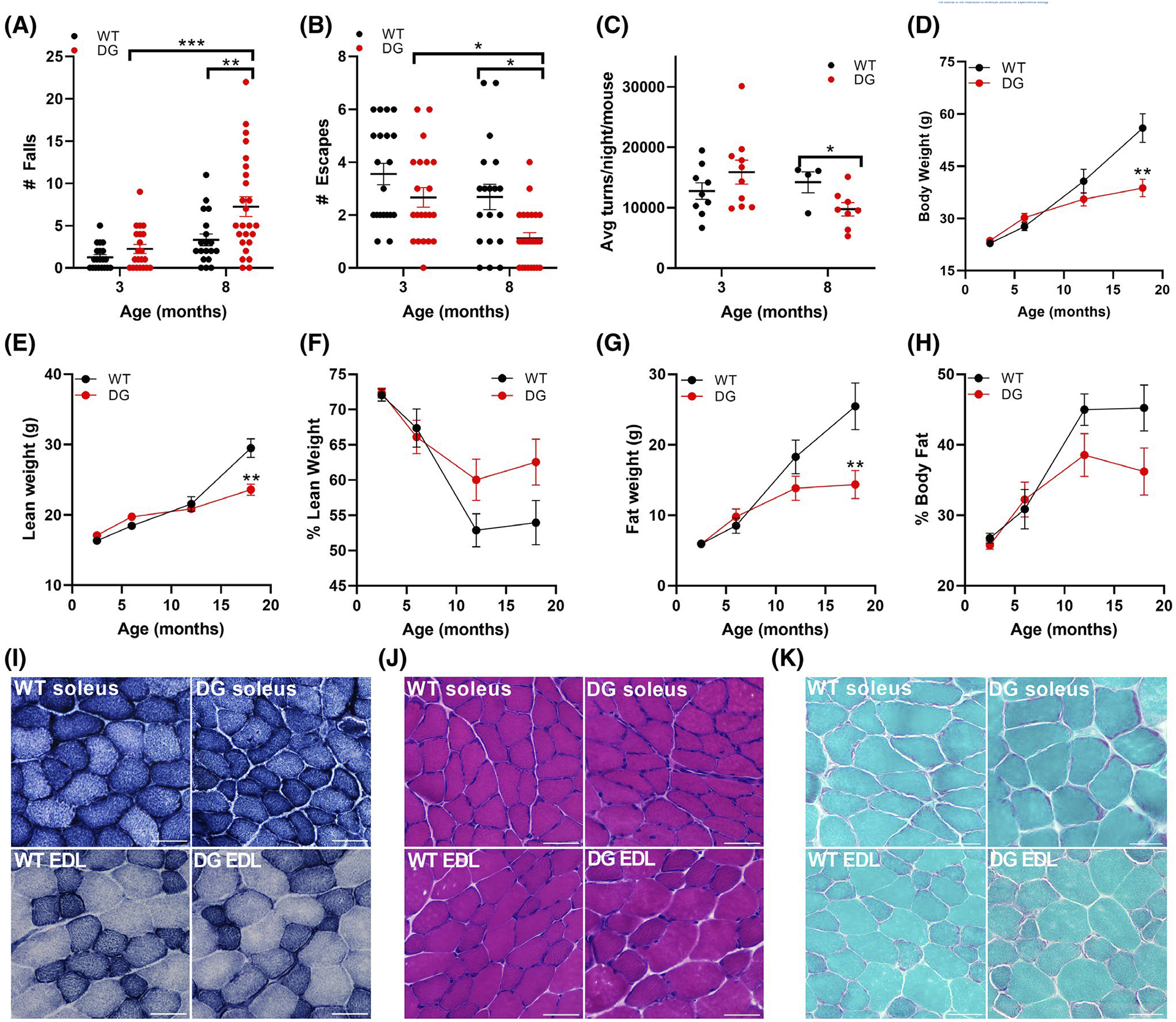
Performance, body composition and histology in WT and DG mice. A,B, Wire-hang test of 3- and 8-month-old WT and DG mice. Number of falls (A) and escapes (B) of mice in 3-minute hang tests. C, Average nightly wheel turns per mouse. D–H, Body composition changes including total body weight (D), lean weight (E), % lean weight (F), fat weight (G), and % body fat (H) between 3 and 18 months of age in WT (n = 44) and DG (n = 52) mice. Error bars in A–H indicate the mean ± SEM. I–K, Histological changes in soleus and EDL sections from 8-month-old mice including (I) NADH-TR (J) H&E and (K) Gomori trichrome. Scale bar is 50 μm. Statistical significance is indicated by *(P < .05), **(P < .005) and ***(P < .001)
Histological examination with NADH-TR, H&E and Gomori trichrome staining of skeletal muscle cross-sections from 8-month-old mice (the age where hang test and running deficiencies were detected) showed no apparent cores (Figure 1I), vacuoles, or other gross abnormalities in fiber morphology (Figure 1J,K). The absence of vacuoles is different from the findings in histological sections from human patients where vacuoles were frequently observed.1–4 While it is possible that vacuoles develop at a later time point in the mouse model, the absence of vacuoles in the mice may reflect the inherent differences between mouse and human muscle.
To assess the effects of the DG mutation in CASQ1 on muscle function, we measured frequency-dependent specific force generation and the rate of onset of muscle fatigue using ex vivo muscles from DG and WT littermate mice. We found no differences in specific force generation at any stimulation frequency in the soleus (Figure S2A) or EDL (Figure S2B) muscles from 3-month-old DG and WT mice. To assess fatiguability of ex vivo muscles, we measured force production with repetitive stimuli over a 5-minute period. No differences in fatigue (the decline in muscle force with repetitive stimulation) were detected in the soleus (Figure S2C) or EDL (Figure S2D) of 3-month-old WT and DG mice. We also did not detect any force deficits in the soleus muscle of 8-month-old DG compared to WT mice (Figure 2A). In contrast, force generation in the EDL muscle of 8-month-old DG mice was significantly decreased across a range of stimulation frequencies (Figure 2B). There were no differences in the rate of onset of fatigue in either the EDL or soleus muscles of 8-month-old DG compared to WT littermates (Figure 2C,D).The difference in the functional consequences of the mutation between the soleus and EDL is similar to the effects of the CASQ1 mutation in humans where the D244G mutation primarily decreases the function of muscles with Type II muscle fiber predominance.3 A greater effect on Type II fibers is also consistent with the higher expression of CASQ1 in Type II compared to Type I fibers.40 To determine if functional changes were attributable to changes in fiber size or distribution, we measured the distribution of individual fiber types and the cross-sectional area (CSA) in muscle cross sections from WT and DG mice. Figure 2E–H shows an immuno-stained cross section that is representative of the images used to quantitate CSA and fiber type distribution. In soleus muscles of DG mice, there were minor shifts to larger Type I and Type IIa fibers, with a more substantial increase in the CSA of Type IIx fibers compared to WT mice (Figure 2I–K). No changes were detected in the fiber type composition of the soleus of DG compared to WT fibers (Figure 2L). In the EDL, the distributions of CSA for Type IIa (Figure 2M), IIx (Figure 2N) and IIb (Figure 2O) fibers displayed a small shift toward larger fibers with no difference in fiber type composition between the EDL muscles of DG and WT mice (Figure 2P). These findings suggest the functional changes in 8-month-old DG EDL muscle are primarily due to a decline in ability of muscle to generate force rather than major changes in muscle CSA or fiber-type distribution.
FIGURE 2.
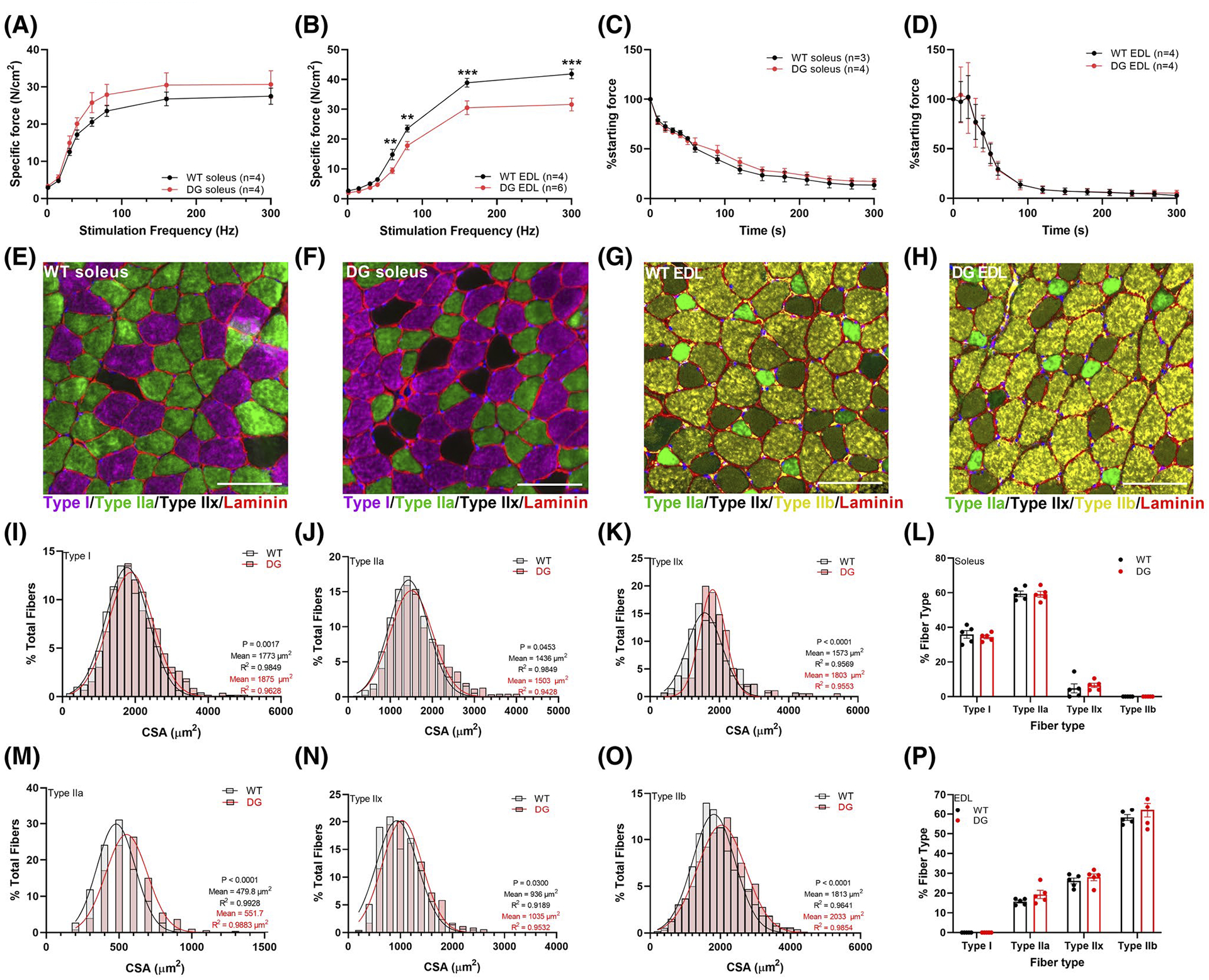
Ex vivo muscle function, muscle fiber typing and CSA in 8-month-old male mice. Muscle force was measured at a range of stimulation frequencies in (A) soleus and (B) EDL muscles. Muscle force was then assessed with repeated stimulation in (C) soleus (1 × 30 Hz train/s) and (D) EDL (1 × 60 Hz train/s) over 5 minutes. Each symbol represents a muscle from one mouse (number of mice indicated in figure legend). E–H, Representative immunofluorescence images of soleus and EDL sections from WT and DG mice. Distribution of CSA measured in (I) Type I (WT n = 888 fibers, DG n = 1011 fibers), (J) Type IIa (WT n = 1457, DG n = 1495) and (K) Type IIx (WT n = 261, DG n = 175) muscle fibers in soleus muscles from five pairs of mice. L, Fiber type distribution in soleus muscles. Distribution of CSA in (M) Type IIa (WT n = 447, DG n = 448), (N) Type IIx (WT n = 740, DG n = 681) and (O) Type IIb (WT n = 1670, DG n = 1518) muscle fibers in EDL muscles from five pairs of mice. P, Fiber type distribution in EDL muscles. Scale bars are 100 μm. Data are presented as the mean ± SEM. Symbols in (L) and (P) represent a single mouse. Statistical significance is indicated by *(P < .05), **(P < .005), and ***(P < .001)
3.3 |. The DG mutation in CASQ1 decreases SR Ca2+ release
To determine if the DG mutation in CASQ1 alters Ca2+ handling in muscle fibers, we measured the Ca2+ transients evoked by electrical stimulation in Fura-2 loaded FDB fibers from 3-month-old DG and WT littermate mice. The amplitude of electrically evoked Ca2+ transients was decreased in DG compared to WT fibers (Figure 3A–D). The half maximal stimulation frequencies for WT and DG fibers (37.1 ± 4.1 Hz and 37.7 ± 4.9 Hz, respectively) were not different (Figure 3E). We did not detect differences in the rate of uptake of twitch Ca2+ (Figure 3F) or in resting cytoplasmic Ca2+ (Figure 3G). CASQ1 is the major SR Ca2+ buffer and the primary determinant of SR storage capacity, but we did not detect any significant differences in SR Ca2+ stores in DG compared to WT fibers using either caffeine or a total SR Ca2+ release cocktail consisting of ionomycin, cyclopiazonic acid (CPA) and EGTA (ICE)31 (Figure 3H,I). It should be noted, however, that these approaches provide a gross assessment of SR Ca2+ levels and are unlikely to reveal changes in microdomain Ca2+ levels arising from decreased polymerization and Ca2+ binding capacity in mutant CASQ1.18
FIGURE 3.
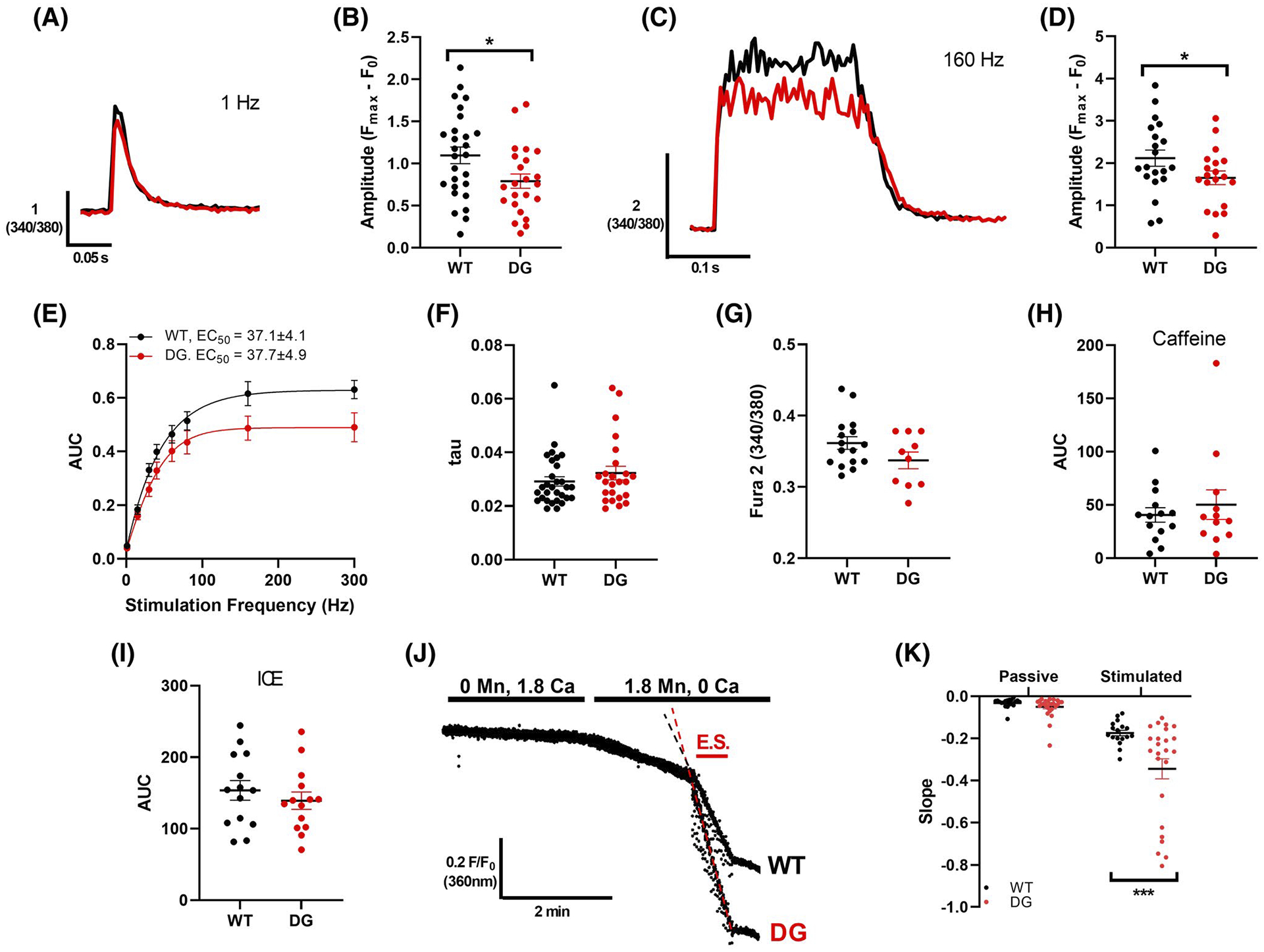
Ca2+ handling in intact FDB fibers. A, Representative Ca2+ transients from Fura-2 loaded WT (black) and DG (red) FDB fibers with 1 Hz stimulation. B, Average amplitude of release with 1 Hz stimulation. C, Representative Ca2+ transients from Fura-2 loaded WT (black) and DG (red) FDB fibers with 160 Hz stimulation. D, Average amplitude of release with 160 Hz stimulation. E, AUC of Ca2+ transients elicited by 300 ms trains at a range of stimulation frequencies in Fura-2 loaded FDB fibers were used to assess the frequency dependence of Ca2+ release. F, Average decay constant (tau) of Ca2+ transient with 1 Hz stimulation. G, Average cytoplasmic Ca2+ levels. SR Ca2+ stores were examined by measuring the area under the curve of Ca2+ release induced with (H) 30 mM caffeine in 0 Ca2+ or (I) 10 μM ionomycin, 30 μM CPA and 100 μM EGTA (ICE) in 0 Ca2+. J, Representative Fura 2 fluorescence in WT and DG fibers during a Mn2+ quench experiment. Influx was measured during the period where fibers were exposed to 1.8 mM Mn and 0 Ca in the absence (passive) or presence (stimulated) or electrical stimulation (E.S.) K, Average slope of decay during passive and stimulated influx are compared in WT and DG fibers. Each symbol indicates a fiber, and fibers were obtained from three to four mice per group. Error bars indicate the mean ± SEM. Statistical significance is indicated by *(P < .05), **(P < .005) and ***(P < .001)
To assess the effects of the DG mutation on sarcolemmal Ca2+ entry, we compared Mn2+ (which enters via Ca2+ entry pathways) quench rates under resting and electrically stimulated conditions. We found no differences in basal Mn2+ influx but did find a significantly larger Mn2+ influx in DG compared to WT fibers during electrical stimulation (Figure 3J,K). Since CASQ1 has been shown to interact with STIM1 and inhibit store-operated Ca2+ entry (SOCE),41 we examined the effects of the DG mutation on SOCE in fibers from the DG and WT mice. To do this, we measured Mn2+ influx in fibers treated with and without a 1-hour pre-incubation with CPA to block SERCA and induce Ca2+ store depletion.33 Very little Mn2+ influx was detected in non-depleted fibers from either DG or WT mice (Figure S3A,B). Pre-treatment with CPA to induce Ca2+ store depletion produced a robust increase in Mn2+ influx in both WT and DG fibers, but SOCE was not increased in DG compared to WT fibers (Figure S3C,D). Hence, increased SOCE in DG fibers is unlikely to explain the increased Ca2+ influx during repetitive stimulation. Another possible explanation of the increased Ca2+influx in the fibers from DG mice is that the mutation alters the interaction of CASQ1 with RYR1 in such a way as to alter the interaction of RYR1 with CaV1.1, leading to an increase Ca2+ influx via CaV1.1 during ECC. The functional significance of small increase in Ca2+ influx in fibers from DG mice is not yet known.
3.4 |. The DG mutation causes CASQ1 mislocalization and aggregation
The D244G mutation in CASQ1 decreases its ability to undergo back-to-back dimerization, a critical step in CASQ1 polymerization and retention at the junctional SR.5 We assessed the localization of CASQ1 in FDB fibers of 3- and 8-month-old WT and DG mice by acquiring images at the mid z-axis level (Figure 4A,B) and at the sub-sarcolemmal region (Figure 4C,D). CASQ1 primarily colocalized with RYR1 at the junctional SR in fibers from 3-month-old DG and WT mice, but some CASQ1 in the DG fibers was also found in the perinuclear ER (Figure 4B,D). CASQ1 was also present at the junctional SR in muscle fibers from the 8-month-old DG mice, but there was increased aggregation of CASQ1 in the intermyofibrillar SR (“streaks”) and accumulation of CASQ1 (some as aggregates) in the perinuclear ER (Figure 4B). These effects were more prominent at the sub-sarcolemmal region of the fiber (Figure 4D) and are consistent with studies showing that the DG-CASQ1 is prone to aggregation.5,27 SERCA1 accumulated in the perinuclear region in both WT and DG fibers of 8-month-old mice (Figure 4E,F) and colocalized with CASQ1 aggregates in the intermyofibrillar SR in fibers from DG mice (Figure 4F). RYR1 was not found in the CASQ1 aggregates in fibers of either 3- or 8-month-old DG mice. Overall, our studies suggest that the DG mutation causes aggregation of CASQ1 in the peri-nuclear ER and in longitudinal ER/SR streaks.
FIGURE 4.
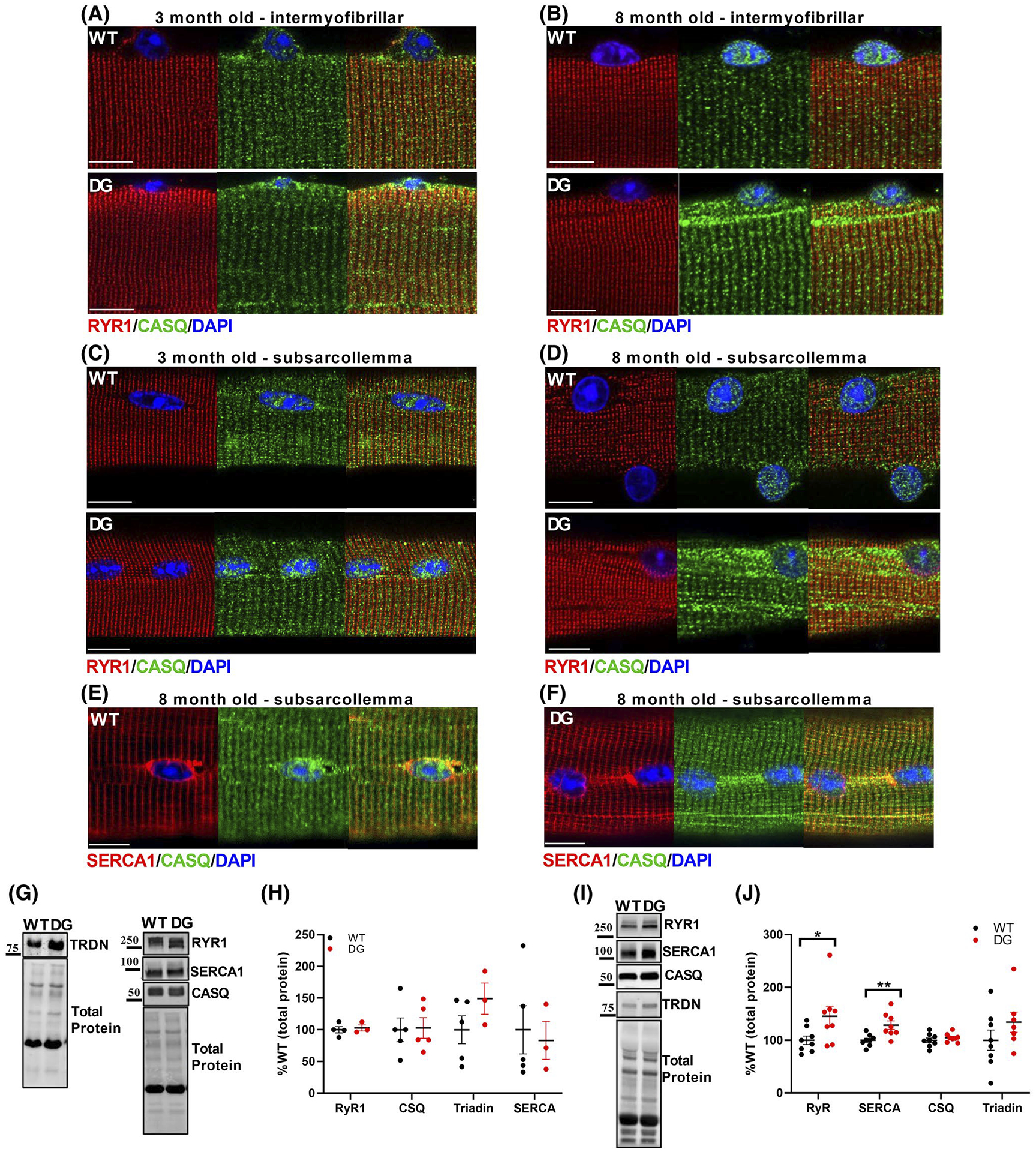
CASQ aggregation and expression of EC Coupling Proteins. Immunofluorescence of CASQ and RyR1 in FDB fibers from 3-month-old (A, C) to 8-month-old (B, D) mice. Images were taken from the mid-z axis of the fiber (A, B) and at the sub-sarcolemmal region (C, D) of fibers. E, F, Immunofluorescence of SERCA1 and CASQ1 in FDB fibers from 8-month-old mice Scale bars are 10 μm. SR protein levels were assessed by western blot in (G, H) 3-month-old and (I, J) 8-month-old mice. G, I, Representative western blots and (H, J) quantitation of SR protein expression in WT and DG EDL muscle homogenates. Protein expression was normalized to the total protein for the lane, detected by Stain-Free imaging. Each symbol in (H) and (J) represents a single muscle from a mouse. Scale bars are 10 μm. Error bars indicate the mean ± SEM. Statistical significance is indicated by *(P < .05) and **(P < .005)
The DG mutation did not alter the expression of major SR proteins in EDL muscle homogenates from 3-month-old WT and DG mice (Figure 4G,H), but RYR1 and SERCA expression was increased in muscle homogenates from the EDL muscle of 8-month-old DG compared to WT mice (Figure 4I,J). We detected no difference in mRNA levels for SERCA and RYR1 in older mice (Figure S4A,B), suggesting that the increase in SERCA and RYR1 expression is likely to reflect a decrease in protein turnover. We found a small but significant decrease in CASQ1 mRNA levels in the muscle of 8-month-old DG compared to WT mice (Figure S4B), suggesting the activation of a compensatory mechanism to decrease misfolded protein load.
3.5 |. The DG mutation in CASQ1 activates UPR/ER stress and causes ER expansion in older mice
Misfolded, aggregated and/or mislocalized proteins trigger the unfolded protein response (UPR) and an ER stress response to reduce the burden of misfolded proteins.42 Although somewhat variable among DG mice, ER stress/UPR markers were not consistently elevated in the muscle of 3-month-old DG mice (Figure S5A–D). Hence our data suggest that the small amount of mislocalized CASQ1 in the 3-month-old mice is not associated with persistent ER stress and that misfolded proteins are adequately cleared in the muscle of these younger mice. However, ER stress markers sXbp1, BiP, and CHOP were significantly elevated in the EDL muscle of 8-month-old DG compared to WT mice (Figure 5A,B). One consequence of ER stress is a decrease in protein synthesis to reduce the misfolded protein load in the ER.43 We assessed the status of mTOR signaling pathways by measuring the phosphorylation status of the mTOR substrates, S6 and 4EBP1. While muscles of 3-month-old DG mice showed no difference in mTOR signaling pathways compared to the muscle of WT mice (Figure S5E–H), there was a significant increase in phosphorylation of the mTOR substrates S6 and 4EBP1 in the EDL muscle of 8-month-old DG mice (Figure 5C,D). An increase in protein synthesis in response to ER stress is maladaptive.44 The soleus muscle from 8-month-old DG mice displayed significant increases in p-EIF2α/EIF2α and CHOP (Figure 5E,F) but no change in mTOR signaling (Figure 5G,H), suggesting that maladaptive protein synthesis was not activated in the soleus.
FIGURE 5.
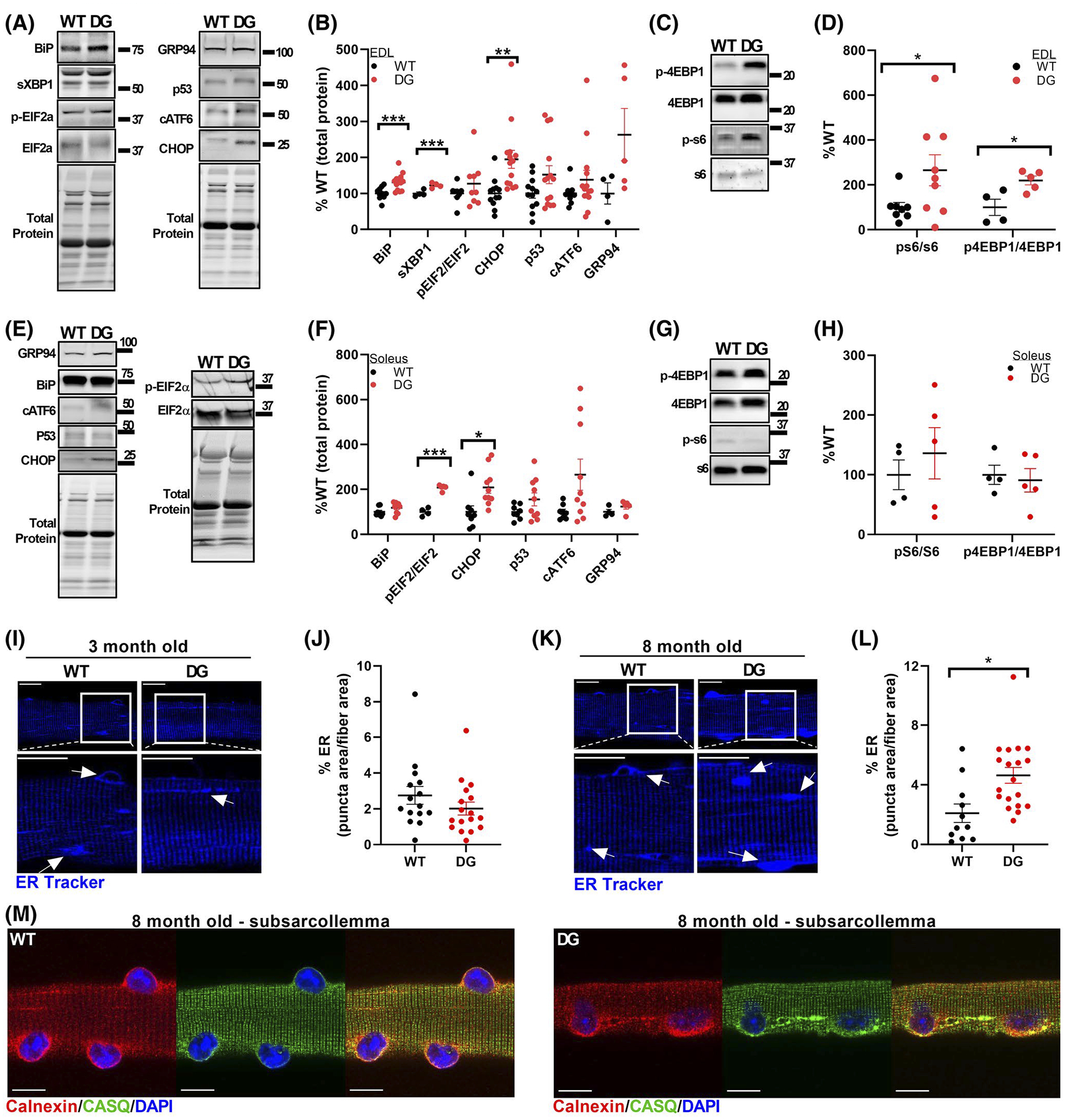
ER stress and remodeling. Representative blots (A) and quantitation (B) of ER stress markers in EDL muscles from 8-month-old mice. Unless otherwise indicated, each ER stress marker was normalized to the total protein for that lane, as detected by Stain-Free imaging. Representative blots (C) and quantitation (D) of total and phospho-S6 and 4EBP1 in EDL homogenates of 8-month-old WT and DG mice. Each symbol in (B) and (D) represents a single muscle from an individual mouse. Representative blots (E) and quantification (F) of ER stress markers in soleus muscles from 8-month-old mice. Unless otherwise indicated, each ER stress marker was normalized to the total protein for that lane, as detected by Stain-Free imaging. Representative blots (G) and quantitation (H) of total and phospho-S6 and 4EBP1 in soleus homogenates of 8-month-old WT and DG mice. Each symbol in (F) and (H) represents a single muscle from an individual mouse. I–L, Representative ER-Tracker Blue-White staining and analysis in intact FDB fibers from WT and DG mice at 3 months (I, J) and 8 months (K, L) of age. The area of puncta that stained more brightly than the background sarcomeric ER regions were quantified and expressed as a percentage of the total fiber area. White boxes indicate regions that are expanded. Arrows indicate examples of features that were quantified. Each symbol in (J) and (L) represents a single fiber, and fibers were obtained from three WT and three DG mice. Scale bars for ER tracker stained fibers are 20 μm. M, Representative immunofluorescence of CASQ1 and calnexin from the subsarcolemmal region of 8-month-old WT and DG FDB fibers. Scale bars are 10 μm. Error bars indicate the mean ± SEM. Statistical significance is indicated by *(P < .05), **(P < .005), and ***(P < .001)
Expansion of the ER, a characteristic of the ER stress response, is a compensatory mechanism to expand protein folding capacity and alleviate ER stress.45,46 We analyzed ER distribution in 3- and 8-month-old mice by staining live FDB fibers with ER-tracker dye (Figure 5I–L). The marker used to track ER morphology, ER-tracker, is unlikely to differentiate between SR and ER and, consistent with this, ER-tracker staining displays both an SR and an ER distribution (dense staining around the nuclei). We found no difference in ER/SR distribution in FDB muscle fibers of 3-month-old DG and WT mice (Figure 5I,J). In contrast, a higher proportion of the total fiber area had a fluorescence intensity above the threshold of normal ER/SR staining in DG versus WT fibers from 8-month old-mice (Figure 5K,L), suggesting expansion of the ER/SR network.47,48 To determine if CASQ1 aggregates colocalized with regions of expanded ER, we examined the distribution of CASQ1 and the canonical ER marker, calnexin. Calnexin and some CASQ1 accumulated in the peri-nuclear region of both WT and DG fibers and colocalized in streaks and clumps close to the nuclei in DG fibers (Figure 5M). Both proteins were also found to be diffusely distributed in the intermyofibrillar region of both WT and DG fibers (Figure 5M).
3.6 |. Aggregates of DG-CASQ1 in the muscle of young mice are adequately cleared by the proteasomes
We evaluated the ability of the ubiquitin-proteasome system (UPS) to clear misfolded protein/protein aggregates in the muscle of DG and WT mice by assessing the formation of aggresomes using the Proteostat aggresome detection reagent. We detected no differences in aggresome levels in fibers of 3-month-old DG and WT mice under basal conditions (Figure 6A,B). However, when proteasome activity was inhibited with 5 μM MG132, there was a significantly larger number of aggresomes formed in muscle fibers of DG compared to WT mice (Figure 6A,B). Inhibition of proteasome function with MG132 also increased CASQ aggregation in muscle fibers from both WT and DG mice. Under basal conditions, DG fibers had a higher number of small CASQ aggregates (0.5–1 μm in size) compared to WT fibers (Figure 6C,D). The number of small aggregates was further increased in WT fibers by treatment with MG132. While MG132 had no effect on the number of small aggregates in DG fibers, it did significantly increase the number of large aggregates >2 μm (Figure 6E). Muscles from WT and DG mice at this age also had similar polyubiquitin levels, as assessed by immunohistochemistry and western blotting (Figure 6F–H). Together these results indicate that the UPS is able to adequately clear CASQ1 aggregates and prevent the accumulation of large aggregates and aggresomes.
FIGURE 6.
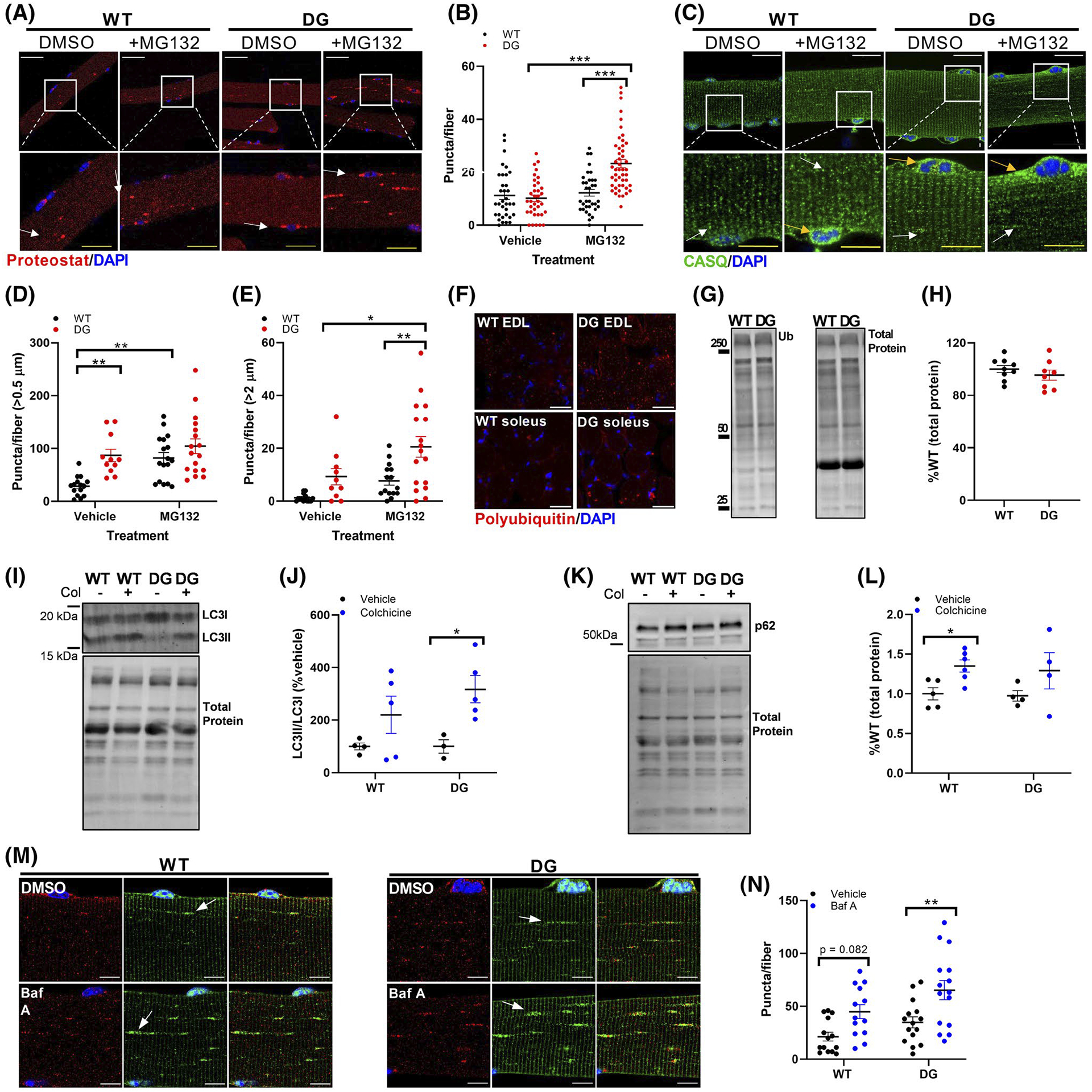
Proteolytic function in 3-month-old mice. A, Proteostat staining of FDB fibers from WT and DG mice. Fibers were treated with either vehicle or 5 μM MG132 overnight. Red puncta indicate aggresome formation and were quantified (B) from multiple fibers from six WT and DG mice. Arrows indicate examples of features that were quantified. Each symbol in (B) represents an individual muscle fiber. White scale bars in (A) are 50 μm, yellow scale bars in expanded images are 20 μm. C, CASQ staining in vehicle and MG132-treated FDB fibers. Aggregates greater than 0.5 μm (white arrows) (D) or 2 μm (yellow arrows) (E) were quantified from multiple fibers from three WT and DG mice. White scale bars in (C) are 50 μm, yellow scale bars in expanded images are 20 μm. White boxes indicate regions that are expanded in the lower panel. F, Polyubiquitin immunofluorescence staining in EDL and soleus muscle sections from WT and DG. Scale bars are 50 μm. Representative western blot (G) and quantitation (H) of ubiquitination in EDL muscle homogenates. I, J, Representative blot and quantitation of LC3II/LC3I ratio and (K, L) p62 expression in vehicle and colchicine treated WT and DG mice. M, Representative LC3 and LAMP2 immunofluorescence in FDB fibers from 8-month-old WT and DG mice. Scale bars are 10 μm. To assess autophagic flux, fibers were treated with either DMSO or 400 nM bafilomycin A for 3 hours. N, Quantitation of LC3 puncta in FDB fibers from three pairs of WT and DG mice. Arrows indicate examples of features that were quantified. Each symbol in (D), (E) and (N) represents an individual muscle fiber. Each symbol in (H), (J) and (L) represents a single muscle from an individual mouse Error bars indicate the mean ± SEM. Statistical significance is indicated by *(P < .05), **(P < .005) and ***(P < .001)
3.7 |. Autophagic flux in the young mice is not altered by the DG mutation in CASQ1
The autophagic/lysosomal degradation pathway works together with the ubiquitin/proteasome system to maintain cellular proteolysis. To assess autophagic flux, 3-month-old WT and DG mice were treated with either vehicle (saline) or colchicine at 0.4 mg/kg/day for 2 days to block autophagosome-lysosome fusion, the final step before degradation of the autolysosome completes the process of autophagic flux. Colchicine treatment induced a robust increase in the LC3II/LC3I ratio in DG muscle, indicating that autophagic flux was intact in the EDL muscle DG mice (Figure 6I,J). p62 is a scaffold protein that isolates substrates for autophagosome sequestration49 and aggresome formation50 prior to their degradation by the lysosome. Inhibition of autophagic flux decreases lysosomal activity and causes accumulation of p62.51,52 We found no difference in the accumulation of p62 in the muscle of 3-month-old DG compared to WT mice (Figure 6K,L), suggesting that autophagic/lysosomal flux was normal in the muscle of young DG mice. Autophagic flux was also assessed in vitro by treating isolated FDB fibers with Bafilomycin A to inhibit autophagosome-lysosome fusion and prevent lysosome acidification. Staining of muscle fibers with LC3 antibodies or expression of fluorescent tagged LC3 produces a sarcomeric staining pattern with LC3 localized at the Z-disc ([53] and Figure 6M). We found dense LC3 puncta, often in close proximity to the lysosome marker LAMP2 (Figure 6M). Bafilomycin A treatment caused a significant increase in LC3 puncta in DG FDB fibers, consistent with the results obtained in vivo with colchicine treatment of WT and DG mice (Figure 6N). The absence of cytoplasmic protein aggregates and the lack of elevated ER stress markers in the muscle of young DG mice suggest that the proteasomal and autophagic proteolysis pathways are adequate for the degradation of misfolded proteins without causing activation of ER stress.
3.8 |. Autophagic flux and aggregate clearance are decreased in muscle of older DG mice
To determine if the increase in CASQ1 and SERCA protein aggregates in the older DG mice was due to age-related decreases in protein degradation,54 we analyzed polyubiquitination, aggresome formation, and autophagy in muscles from 8-month-old DG and WT mice. The number of aggresomes, as detected with Proteostat, were significantly higher in DG compared to WT fibers under baseline conditions and MG132 treatment further increased the aggresome number in both WT and DG muscle fibers (Figure 7A,B). Co-staining of aggresomes and CASQ1 in untreated fibers revealed the presence of large CASQ1 aggregates colocalized or in the vicinity of large aggresomes, which were most visible in the perinuclear regions of the fibers (Figure 7C,D) where aggresomes form before they are degraded by lysosomes.55 These aggresomes were also positive for polyubiquitin (Figure 7E) and p62 (Figure 7F) which tags aggregated proteins for autophagic degradation.49,50 Both p62 and polyubiquitin are common components of aggresomes and other inclusion bodies in a myriad of diseases.55 Elevation of p62 and polyubiquitin in DG muscle was confirmed in EDL muscle cross sections (Figure 7G) and with western blotting of muscle homogenates (Figure 7H,I).
FIGURE 7.

Aggresome formation and composition in 8-month-old mice. To detect protein aggregates, FDB fibers (A) were treated with either vehicle or 5 μM MG132 overnight before being fixed, permeabilized and stained with Proteostat aggresome detection reagent. White boxes indicate regions that were expanded in the lower panel. White scale bars are 50 μm, yellow scale bars in expanded images are 25 μm. Arrows indicate examples of features that were quantified. Red puncta indicate aggresome formation and were quantified (B) in multiple fibers from five WT and five DG mice. Each symbol represents a single fiber. Proteostat composition was assessed by co-staining with Proteostat reagent and antibodies to (C, D) CASQ, (E) polyubiquitin and (F) p62 Scale bars are 10μm. G, Polyubiquitin immunofluorescence staining in EDL muscle sections from WT and DG mice. Scale bars are 50 μm. Representative western blot (H) and quantitation (I) of ubiquitination and p62 in EDL muscle homogenates. Each symbol in (I) represents a single muscle taken from an individual mouse. Statistical significance is indicated by *(P < .05) and **(P < .005)
p62 plays an important role in sequestering proteins for autophagic degradation, and its accumulation is reflective of decreased autophagic flux. Autophagic flux was assessed in vitro by treating isolated FDB fibers with Bafilomycin A. In response to bafilomycin A treatment, WT fibers showed a significant increase in LC3 puncta indicating intact autophagic flux in these fibers. In contrast, bafilomycin A had no effect on LC3 puncta number in DG fibers, suggesting that autophagic flux was inhibited in fibers from 8-month-old DG mice (Figure 8A,B). Consistent with this, we detect increased numbers of lysosomes, autophagosomes, and endocytic multivesicular bodies around the nuclei in electron micrographs of muscle from DG compared to WT mice (Figure 8C,D).
FIGURE 8.
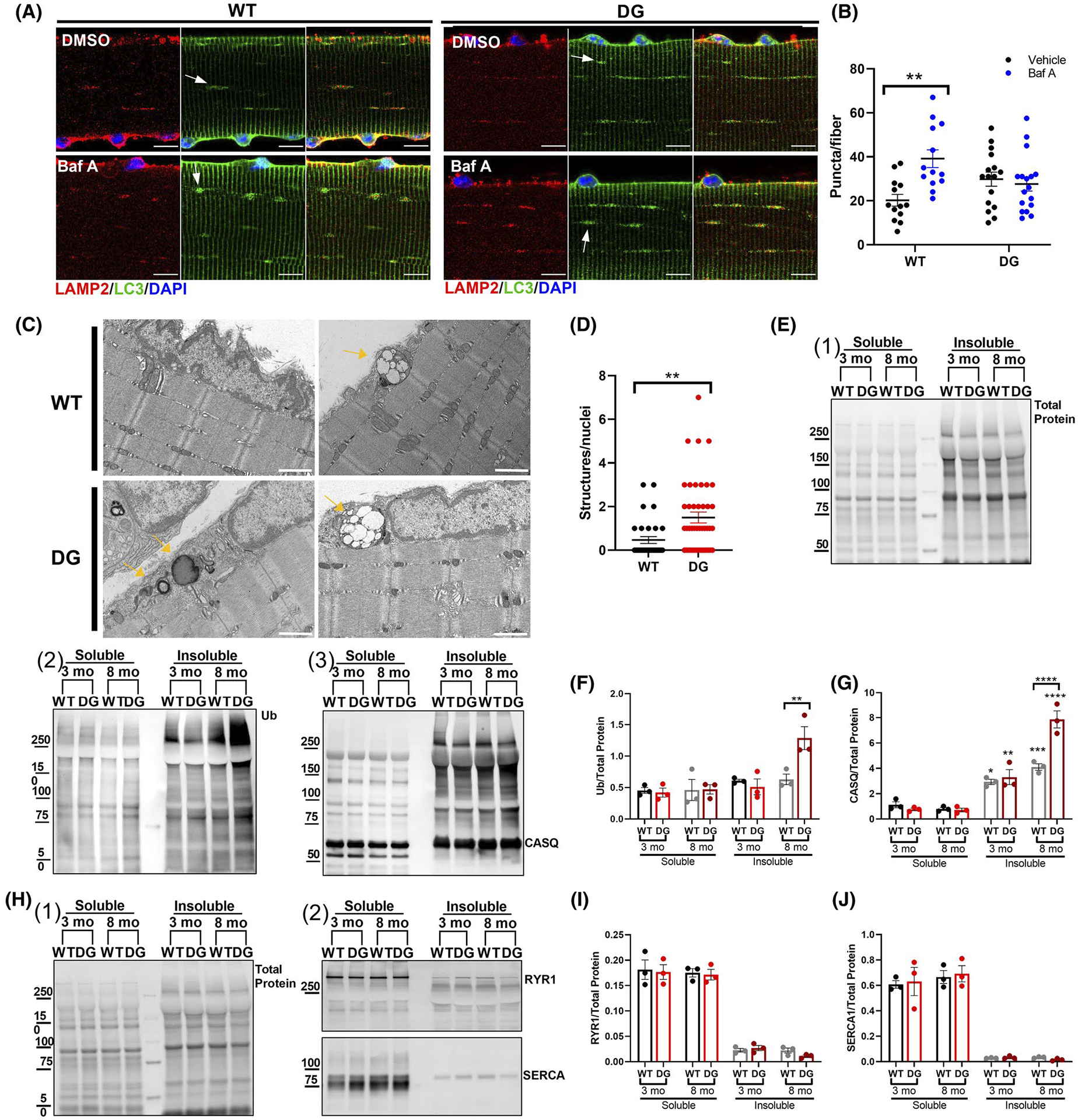
Autophagic flux and muscle fractionation in 8-month-old mice. A, Representative LC3 and LAMP2 immunofluorescence in FDB fibers from 8-month-old WT and DG mice. Scale bars are 10 μm. To assess autophagic flux, fibers were treated with either DMSO or 400 nM bafilomycin A for 3 hours. B, Quantitation of LC3 puncta in FDB fibers from three pairs of WT and DG mice. Arrows indicate examples of features that were quantified. C, Representative transmission electron micrographs from 8-month-old WT and DG EDL muscles. Scale bars are 1 μm. Arrows indicate examples of features that were quantified. D, Quantitation of multivesicular bodies and autophagic structures normalized to number of nuclei in EM images. E, Representative blots of (1) total protein, (2) ubiquitin, and (3) CASQ1 in TX-100 soluble and insoluble fractions from 3- and 8-month-old WT and DG mice. Quantitation of lane (F) ubiquitin and (G) Ub-CASQ1 normalized to the total protein. H, Representative western blots of (1) total protein and (2) RYR1 and SERCA1 in TX-100 soluble and insoluble fractions. Quantitation of (I) RYR1 and (J) SERCA1 western blots normalized to the total protein of the lane. Error bar indicate the mean ± SEM. Statistical significance is indicated by *(P < .05) and **(P < .005)
The accumulation of ubiquitin-enriched aggresomes and decreased autophagic flux causes accumulation of insoluble protein.55–57 To assess this in DG muscle, we fractionated TA muscle homogenates from 3- and 8-month-old mice and separated proteins into TX-100 soluble and insoluble components (Figure 8E–J). The total protein of the fractions are shown in Figure 8E1. Western blots for ubiquitin and CASQ1 are shown in Figure 8E2 and E3, respectively. We found a significant increase in insoluble, ubiquitinated proteins and CASQ1 in muscle from 8-month-old DG mice compared to WT littermate controls (Figure 8F and G, respectively). In both age groups a significant amount of high molecular weight CASQ1 was found in the TX-100 insoluble fraction. Smeared bands characteristic of polyubiquitinated proteins58 above CASQ1 (potentially the poly-Ub-CASQ1) were only found in the insoluble fractions (Figure 8G) and this signal was significantly higher in 8-month-old DG compared to WT (Figure 8F,G). Soluble and insoluble proteins were also analyzed for the presence of RYR1 and SERCA. Figure 8H1 show the total protein staining for the soluble and insoluble proteins and Figure 8H2 shows the western blots for RYR1 and SERCA. RYR1 and SERCA were primarily in the soluble fractions with only small amounts in the insoluble fractions (Figure 8I and J, respectively). While small amounts of SERCA1 and RYR1 were both detected in the insoluble fractions, there were no differences in their distribution to the insoluble fractions from the muscle of DG compared to WT mice, regardless of age (Figure 8H–J). These data suggest that CASQ1 is subject to high turnover by ubiquitin/proteasomes but accumulates in the muscle of DG mice. Collectively, our data suggest that the proteolytic systems are saturated and/or impaired in older DG mice. ER stress/UPR markers and increased aggregates under baseline conditions are likely reflections of this reduced/saturated proteasomal function in 8-month-old mice.
4 |. DISCUSSION
The newly created DG mice undergo an age-related decline in performance and muscle function. Functional changes coincide with an increase of insoluble, ubiquitinated proteins, ER stress and decreased autophagic flux. These results are consistent with earlier studies demonstrating that the DG mutation increases the susceptibility of CASQ1 to misfolding and aggregation.2,5,27 Misfolded CASQ1 that is unable to form linear polymers is trapped in the ER and forms aggregates.26,59 In DG muscle there was accumulation of CASQ1 around the nuclei which colocalized with the ER marker, calnexin. The ER expansion measured in the DG muscle is consistent with ER/SR morphology of muscle biopsies from VAM patients2 and may be an early stage of vacuole formation. The DG mutation in CASQ1 increases aggregation and aggregated CASQ1 is subject to proteasomal and autophagic degradation. However, as the mice age, there is a decline in the ability of the muscle fiber to clear the aggregated CASQ1, leading eventually to the activation of maladaptive protein synthesis, increasing the protein load and the severity of the response. The late onset of disease in DG mice mirrors the disease progression in human patients, where the median age of onset is 60 years old.3 The relatively mild functional impairment in DG mice partially recapitulates the symptoms of patients where the disease often presents with mild myalgia, cramps, muscle weakness, and exercise intolerance.2,3
The DG mutation in CASQ1 decreases SR Ca2+ release in muscle fibers from the DG compared to WT mice. Decreased SR Ca2+ release is an expected finding in this model given the role of polymerized CASQ1 in SR Ca2+ release19,28 and the effects of the DG mutation on CASQ1 polymerization.5,27 The mutation also increases Ca2+ influx during electrical stimulation. We, however, find no evidence of increased SOCE in muscle fibers from DG compared to WT mice. It is possible that the decrease in the interaction of CASQ1 with RYR1 leads to a conformational change in RYR1 that is communicated to CaV1.1. This possibility requires additional study. In addition to reducing SR Ca2+ release, a second direct action of the mutated CASQ1 is to drive an UPR/ER stress response. While ER stress (and SOCE) can be activated by Ca2+ store depletion,60,61 that is not happening in the muscle of the DG mice. SR Ca2+ stores are not significantly depleted and SOCE is not increased in fibers of DG compared to WT mice. UPR/ER stress is also driven by unfolded proteins or excess protein load.62 Since the mutation alters the ability of CASQ1 to polymerize, monomers of CASQ1 would increase. Our data suggest that the mutant CASQ1 accumulates in the perinuclear ER, leading to UPR, ER stress, and eventually accumulation of aggregated proteins. This raises the important question of what is driving the decreased muscle function. It is likely that both changes in Ca2+ release and DG-CASQ1 driven UPR/ER stress/aggregation are contributing to the pathology of the disease but are doing so via relatively independent pathways. Decreased Ca2+ release is probably not directly responsible for the decrease in force generation since force is not altered in young mice. However, the decreased Ca2+ release could lead to decreased activation of Ca2+ regulated pathways, which could impact muscle function. Differentiating between the contributions to the disease process of altered Ca2+ signaling and changes driven by ER stress and uncleared protein aggregates requires further study.
Another important issue is why the protein aggregates accumulate only in the muscle of the older DG mice. Ubiquitination of proteins is an early step in their degradation by the ubiquitin proteasome system, an essential step in ER quality control.63 Numerous protein misfolding diseases are associated with impaired proteasome function56,64–66 due to the toxic accumulation of polyubiquitinated proteins.57,66–68 The increase in insoluble, CASQ1 in older mice suggests that the proteasome is unable to adequately clear the aggregates. An additional factor in DG-CASQ1 aggregation could be the inhibition of autophagic flux. The UPS works in concert with the autophagy-lysosome system to control cellular degradation and it is known that decreased autophagic flux can impair proteasome function.63,69 Further work is needed to determine if the impaired proteasome function in the DG mice is downstream of decreased autophagic flux or occurs via a different mechanism. Our data suggest that a decline in proteasome activity with age contributes to the progression of disease in our mouse model of VAM. Whether there is a decrease in proteolytic function in patients with VAM is not yet known.
The buildup of insoluble, ubiquitinated proteins coincides with the increased expression of ER stress markers and decline in muscle function in the DG EDL muscle. A major difference between EDL and soleus was the phosphorylation status of EIF2α. As an adaptive component of the UPR, enhanced phosphorylation of EIF2α normally suppresses protein translation to decrease the burden of newly translated proteins in the ER.43,70 However, in EDL muscle from older DG mice there was no change in EIF2α phosphorylation, which may account for the EDL muscles having increased mTOR signaling despite activation of ER stress pathways. This maladaptive pathway may partially explain why DG EDL muscles had larger muscle fibers compared to WT. Activation of mTOR is also consistent with the decreased autophagic flux detected in 8-month-old mice.
We previously found that ER stress markers were elevated and CASQ1 was mislocalized in the muscle of mice with a mutation (I4895T in mice, I4898T in humans) in RYR1 associated with a dominantly inherited myopathy.29 That study raised the question of whether misfolded and/or mislocalized CASQ1 could cause ER stress. Our findings here indicate that CASQ1 is subject to ER quality control mechanisms and persistent aggregation of CASQ1 induces ER stress when there is inadequate proteolytic capacity, as may occur with aging. These findings indicate that conditions that induce CASQ1 aggregation, including protein mutations or alterations in SR Ca2+ homeostasis alter the protein homeostasis of the SR/ER compartments in addition to any direct effects the CASQ1 mutation have on EC coupling. Our study complements the findings obtained with CASQ1 knockout (CASQ1-KO) mice. DG mice have a milder phenotype than CASQ1-KO mice. CASQ1-KO mice survive into adulthood71 but are malignant hyperthermia susceptible and males exposed to stress have a shortened lifespan.72 We did not detect either a decrease in lifespan or malignant hyperthermia susceptibility in the DG mice. Unlike the CASQ1-KO mice, the DG mice display only modest changes in releasable Ca2+ with no detectable changes in cytoplasmic Ca2+ or total/ releasable Ca2+ stores. The differences between the CASQ1-KO and DG mice are not surprising since CASQ1-KO is a loss-of-function condition whereas the DG mutation induces a gain-of-function pathology. Both models provide valuable insight into the complex role of CASQ1 in skeletal muscle. A chemical chaperone that promotes protein folding alleviates the ER stress and improves muscle function in our mouse model of a RYR1-linked myopathy29 and this treatment may also be beneficial for the DG mice. This is currently being tested.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Muscular Dystrophy Association Development Grant to A.D. Hanna (548809) and a National Institute of Health (NIH) grant RO1 AR072602 to S.L. Hamilton which included salary support for C.S. Lee and A.D. Hanna. L Babcock was supported by an NIH training grant (T32HL007676, SLH, Program Director). C.S. Lee also receives salary support from NIH AR053349 to S.L. Hamilton. We acknowledge the expertise and resources provided by several Advanced Technology Core facilities at Baylor College of Medicine. The DG mice were generated in the Genetically Engineered Rodent Models Core. Body composition data were acquired in the Mouse Metabolism and Phenotyping Core. Confocal Microscopy was conducted at the Optical Imaging & Vital Microscopy Core. Brightfield Microscopy, Electron Microscopy, and preparation of EM samples was performed by the Integrated Microscopy Core.
Funding information
Muscular Dystrophy Association (MDA), Grant/Award Number: 548809; HHS | NIH | National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), Grant/Award Number: NIH AR072475 and NIH AR053349; HHS | National Institutes of Health (NIH), Grant/Award Number: T32HL007676
Abbreviations:
- CASQ1
human or mouse calsequestrin protein
- Casq1
mouse calsequestrin gene
- CASQ1
human calsequestrin gene
- Cav1.1
L-type voltage dependent Ca2+ channel
- ECC
excitation-contraction coupling
- ER
endoplasmic reticulum
- RYR1
ryanodine receptor 1
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
- SR
sarcoplasmic reticulum
- UPR
unfolded protein response
- UPS
ubiquitin-proteasome system
- VAM
vacuolar aggregate myopathy
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Di Blasi C, Sansanelli S, Ruggieri A, et al. A CASQ1 founder mutation in three Italian families with protein aggregate myopathy and hyperCKaemia. J Med Genet. 2015;52:617–626. [DOI] [PubMed] [Google Scholar]
- 2.Rossi D, Vezzani B, Galli L, et al. A mutation in the CASQ1 gene causes a vacuolar myopathy with accumulation of sarcoplasmic reticulum protein aggregates. Hum Mutat. 2014;35:1163–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Semplicini C, Bertolin C, Bello L, et al. The clinical spectrum of CASQ1-related myopathy. Neurology. 2018;91:e1629–e1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomelleri G, Palmucci L, Tonin P, et al. SERCA1 and calsequestrin storage myopathy: a new surplus protein myopathy. Brain. 2006;129:2085–2092. [DOI] [PubMed] [Google Scholar]
- 5.Lewis KM, Ronish LA, Ríos E, Kang C. Characterization of two human skeletal calsequestrin mutants implicated in malignant hyperthermia and vacuolar aggregate myopathy. J Biol Chem. 2015;290:28665–28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goebel HH, Warlo I. Gene-related protein surplus myopathies. Mol Genet Metab. 2000;71:267–275. [DOI] [PubMed] [Google Scholar]
- 7.Eisner V, Csordás G, Hajnóczky G. Interactions between sarcoendoplasmic reticulum and mitochondria in cardiac and skeletal muscle—pivotal roles in Ca2+ and reactive oxygen species signaling. J Cell Sci. 2013;126:2965–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaakinen M, Papponen H, Metsikkö K. Microdomains of endoplasmic reticulum within the sarcoplasmic reticulum of skeletal myofibers. Exp Cell Res. 2008;314:237–245. [DOI] [PubMed] [Google Scholar]
- 9.Kaisto T, Metsikkö K. Distribution of the endoplasmic reticulum and its relationship with the sarcoplasmic reticulum in skeletal myofibers. Exp Cell Res. 2003;289:47–57. [DOI] [PubMed] [Google Scholar]
- 10.Ralston E, Ploug T, Kalhovde J, Lømo T. Golgi complex, endoplasmic reticulum exit sites, and microtubules in skeletal muscle fibers are organized by patterned activity. J Neurosci. 2001;21:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi D, Barone V, Giacomello E, Cusimano V, Sorrentino V. The sarcoplasmic reticulum: an organized patchwork of specialized domains. Traffic. 2008;9:1044–1049. [DOI] [PubMed] [Google Scholar]
- 12.Volpe P, Villa A, Podini P, et al. The endoplasmic reticulum-sarcoplasmic reticulum connection: distribution of endoplasmic reticulum markers in the sarcoplasmic reticulum of skeletal muscle fibers. Proc Natl Acad Sci. 1992;89:6142–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villa A, Podini P, Nori A, et al. The endoplasmic reticulum-sarcoplasmic reticulum connection. Exp Cell Res. 1993;209:140–148. [DOI] [PubMed] [Google Scholar]
- 14.Samant RS, Livingston CM, Sontag EM, Frydman J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature. 2018;563:407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beard NA, Wei L, Dulhunty AF. Ca2+ signaling in striated muscle: the elusive roles of triadin, junctin, and calsequestrin. Eur Biophys J. 2009;39:27–36. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–23397. [DOI] [PubMed] [Google Scholar]
- 17.Park H, Park IY, Kim E, et al. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J Biol Chem. 2004;279:18026–18033. [DOI] [PubMed] [Google Scholar]
- 18.Launikonis BS, Zhou J, Royer L, Shannon TR, Brum G, Rios E. Depletion “skraps” and dynamic buffering inside the cellular calcium store. Proc Natl Acad Sci U S A. 2006;103:2982–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manno C, Figueroa LC, Gillespie D, et al. Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc Natl Acad Sci. 2017;114:E638–E647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park H, Wu S, Dunker AK, Kang C. Polymerization of calsequestrin: implications for Ca2+ regulation. J Biol Chem. 2003;278:16176–16182. [DOI] [PubMed] [Google Scholar]
- 21.Royer L, Ríos E. Deconstructing calsequestrin. Complex buffering in the calcium store of skeletal muscle. J Physiol. 2009;587:3101–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy RM, Larkins NT, Mollica JP, Beard NA, Lamb GD. Calsequestrin content and SERCA determine normal and maximal Ca2+ storage levels in sarcoplasmic reticulum of fast- and slowtwitch fibres of rat. J Physiol. 2009;587:443–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gatti G, Trifari S, Mesaeli N, Parker JMR, Michalak M, Meldolesi J. Head-to-tail oligomerization of calsequestrin: a novel mechanism for heterogeneous distribution of endoplasmic reticulum luminal proteins. J Cell Biol. 2001;154:525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo A, Cala SE, Song L-S. Calsequestrin accumulation in rough endoplasmic reticulum promotes perinuclear Ca2+ release. J Biol Chem. 2012;287:16670–16680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houle TD, Ram ML, McMurray WJ, Cala SE. Different endoplasmic reticulum trafficking and processing pathways for calsequestrin (CSQ) and epitope-tagged CSQ. Exp Cell Res. 2006;312:4150–4161. [DOI] [PubMed] [Google Scholar]
- 26.McFarland TP, Milstein ML, Cala SE. Rough endoplasmic reticulum to junctional sarcoplasmic reticulum trafficking of calsequestrin in adult cardiomyocytes. J Mol Cell Cardiol. 2010;49:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Böhm J, Lornage X, Chevessier F, et al. CASQ1 mutations impair calsequestrin polymerization and cause tubular aggregate myopathy. Acta Neuropathol. 2018;135:149–151. [DOI] [PubMed] [Google Scholar]
- 28.Wei L, Varsányi M, Dulhunty AF, Beard NA. The conformation of calsequestrin determines its ability to regulate skeletal ryanodine receptors. Biophys J. 2006;91:1288–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee CS, Hanna AD, Wang H, et al. A chemical chaperone improves muscle function in mice with a RyR1 mutation. Nat Commun. 2017;8:14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Babcock LW, Hanna AD, Agha NH, Hamilton SL. MyoSight-semi-automated image analysis of skeletal muscle cross sections. Skelet Muscle. 2020;10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loy RE, Orynbayev M, Xu L, et al. Muscle weakness in Ryr1I4895T/WT knock-in mice as a result of reduced ryanodine receptor Ca2+ ion permeation and release from the sarcoplasmic reticulum. J Gen Physiol. 2011;137:43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee CS, Georgiou DK, Dagnino-Acosta A, et al. Ligands for FKBP12 increase Ca2+ influx and protein synthesis to improve skeletal muscle function. J Biol Chem. 2014;289:25556–25570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michelucci A, Boncompagni S, Pietrangelo L, Takano T, Protasi F, Dirksen RT. Pre-assembled Ca2+ entry units and constitutively active Ca2+ entry in skeletal muscle of calsequestrin-1 knockout mice. J Gen Physiol. 2020;152:e202012617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang HJ, Lee CS, Yee RSZ, et al. Adaptive thermogenesis enhances the life-threatening response to heat in mice with an Ryr1 mutation. Nat Commun. 2020;11:5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boncompagni S, Loy RE, Dirksen RT, Franzini-Armstrong C. The I4895T mutation in the type 1 ryanodine receptor induces fiber-type specific alterations in skeletal muscle that mimic premature aging. Aging Cell. 2010;9:958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lavorato M, Franzini-Armstrong C. Practical solutions to frequent problems encountered in thin sections electron microscopy. Microscopy Today. 2017;25:40–45. [Google Scholar]
- 38.Fuqua JD, Mere CP, Kronemberger A, et al. ULK2 is essential for degradation of ubiquitinated protein aggregates and homeostasis in skeletal muscle. FASEB J. 2019;33:11735–12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- 40.Lamboley CR, Murphy RM, McKenna MJ, Lamb GD. Endogenous and maximal sarcoplasmic reticulum calcium content and calsequestrin expression in type I and type II human skeletal muscle fibres. J Physiol. 2013;591:6053–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Zhang L, Li S, et al. Retrograde regulation of STIM1-Orai1 interaction and store-operated Ca2+ entry by calsequestrin. Sci Rep. 2015;5:11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. [DOI] [PubMed] [Google Scholar]
- 44.Zito E Targeting ER stress/ER stress response in myopathies. Redox Biol. 2019;26:101232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. 2009;187:525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. 2016;73:79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.An H, Ordureau A, Paulo JA, Shoemaker CJ, Denic V, Harper JW. TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol Cell. 2019;74:891–908.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith MD, Harley ME, Kemp AJ, et al. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-Phagy and pancreatic ER proteostasis. Dev Cell. 2018;44:217–232.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. [DOI] [PubMed] [Google Scholar]
- 50.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao Y, Ma C, Yi J, et al. Suppressed autophagy flux in skeletal muscle of an amyotrophic lateral sclerosis mouse model during disease progression. Physiol Rep. 2015;3:e12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strucksberg K-H, Tangavelou K, Schröder R, Clemen CS. Proteasomal activity in skeletal muscle: a matter of assay design, muscle type, and age. Anal Biochem. 2010;399:225–229. [DOI] [PubMed] [Google Scholar]
- 55.Garcia-Mata R, Gao Y-S, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic. 2002;3:388–396. [DOI] [PubMed] [Google Scholar]
- 56.Fratta P, Engel WK, McFerrin J, Davies KJA, Lin SW, Askanas V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-β precursor protein-overexpressing cultured human muscle fibers. Am J Pathol. 2005;167:517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol. 2004;36:2519–2530. [DOI] [PubMed] [Google Scholar]
- 58.Newton K, Matsumoto ML, Wertz IE, et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. [DOI] [PubMed] [Google Scholar]
- 59.Sanchez EJ, Lewis KM, Munske GR, Nissen MS, Kang C. Glycosylation of skeletal calsequestrin: implications for its function. J Biol Chem. 2012;287:3042–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol. 2011;3(6):a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshida I, Monji A, Tashiro K-I, Nakamura K-I, Inoue R, Kanba S. Depletion of intracellular Ca2+ store itself may be a major factor in thapsigargin-induced ER stress and apoptosis in PC12 cells. Neurochem Int. 2006;48:696–702. [DOI] [PubMed] [Google Scholar]
- 62.Hwang J, Qi L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci. 2018;43:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–1398. [DOI] [PubMed] [Google Scholar]
- 64.Askanas V, Engel WK, Nogalska A. Inclusion body myositis: a degenerative muscle disease associated with intra-muscle fiber multi-protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol. 2009;19:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75:436–439. [DOI] [PubMed] [Google Scholar]
- 66.McNaught KSP, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci Lett. 2001;297: 191–194. [DOI] [PubMed] [Google Scholar]
- 67.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. [DOI] [PubMed] [Google Scholar]
- 68.Gregori L, Fuchs C, Figueiredo-Pereira ME, Van Nostrand WE, Goldgaber D. Amyloid β-protein inhibits ubiquitin-dependent protein degradation in vitro. J Biol Chem. 1995;270:19702–19708. [DOI] [PubMed] [Google Scholar]
- 69.Cohen-Kaplan V, Livneh I, Avni N, Cohen-Rosenzweig C, Ciechanover A. The ubiquitin-proteasome system and autophagy: Coordinated and independent activities. Int J Biochem Cell Biol. 2016;79:403–418. [DOI] [PubMed] [Google Scholar]
- 70.Han J, Back SH, Hur J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paolini C, Quarta M, Nori A, et al. Reorganized stores and impaired calcium handling in skeletal muscle of mice lacking calsequestrin-1. J Physiol. 2007;583:767–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dainese M, Quarta M, Lyfenko AD, et al. Anesthetic- and heat-induced sudden death in calsequestrin-1-knockout mice. FASEB J. 2009;23:1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.