

Abstract
Background
Neuroinflammation plays a pathogenic role in Parkinson's disease (PD). Immunotherapies that restore brain homeostasis can mitigate neurodegeneration by transforming T cell phenotypes. Sargramostim has gained considerable attention as an immune transformer through laboratory bench to bedside clinical studies. However, its therapeutic use has been offset by dose-dependent adverse events. Therefore, we performed a reduced drug dose regimen to evaluate safety and to uncover novel disease-linked biomarkers during 5 days/week sargramostim treatments for one year.
Methods
Five PD subjects were enrolled in a Phase 1b, unblinded, open-label study to assess safety and tolerability of 3 μg/kg/day sargramostim. Complete blood counts and chemistry profiles, physical examinations, adverse events (AEs), immune profiling, Movement Disorder Society-Sponsored Revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS) scores, T cell phenotypes/function, DNA methylation, and gene and protein patterns were evaluated.
Findings
Sargramostim administered at 3 μg/kg/day significantly reduced numbers and severity of AEs/subject/month compared to 6 μg/kg/day treatment. While MDS-UPDRS Part III score reductions were recorded, peripheral blood immunoregulatory phenotypes and function were elevated. Hypomethylation of upstream FOXP3 DNA elements was also increased.
Interpretation
Long-term sargramostim treatment at 3 μg/kg/day is well-tolerated and effective in restoring immune homeostasis. There were decreased numbers and severity of AEs and restored peripheral immune function coordinate with increased numbers and function of Treg. MDS-UPDRS Part III scores did not worsen. Larger patient numbers need be evaluated to assess conclusive drug efficacy (ClinicalTrials.gov NCT03790670).
Funding
The research was supported by community funds to the University of Nebraska Foundation and federal research support from 5 R01NS034239-25.
Keywords: Parkinson's disease, Granulocyte macrophage-colony stimulating factor, Unified Parkinson's Disease Rating Scale, Regulatory T cells, Neuroprotection, Sargramostim
Research in context.
Evidence before this study
Sargramostim is a known immunomodulator as supported by extensive pre-clinical and clinical studies. Its potential as a long-term therapeutic in neurodegenerative disorders and Parkinson's disease in particular have recently been recorded. Immune-basded biomarkers were previously recorded and demonstrate that sargramostim induces Treg neuroprotective activities. Moderate adverse events were observed that could require drug dose reductions.
Added value of this study
Reductions in adverse events followed dose reductions from 6 to 3 μg/kg/day. MDS-UPDRS Part III scores did not worsen. Peripheral blood Treg numbers, function, and hypomethylation of upstream FOXP3 DNA elements were increased in the lowered dose regimen providing a biomarker signature for sargramostim therapy.
Implications of all the available evidence
Increases in Treg numbers and function support a neuroprotective biomarker phenotype. Sargramostim can be administered safely for one year.
Alt-text: Unlabelled box
1. Introduction
Parkinson's disease (PD) is the most common neurodegenerative motor disorder heralded by reductions in striatal dopamine and numbers of dopaminergic neurons in the substantia nigra pars compacta [1]. While palliative therapies abound, clinical trials designed for efficacy of disease-modifying strategies have largely failed, suggesting that either hypotheses are limited or limitations are inherent in study design, implementation, or clinical outcome assessment [2]. The linkages between clinical and disease biology may parallel the heterogeneity of diverse PD pathobiology. Of the suspected etiologies of PD, neuroinflammation and peripheral immune dysfunction stand concordant [1,3,4]. Targeting immune response components can potentially mitigate disease. This is achieved by balancing numbers and function of regulatory and effector T cells (Tregs and Teffs) in the periphery and along the nigrostriatal axis [4]. We, along with others, demonstrated that Tregs attenuate neuroinflammation and protect dopaminergic neurons from injury and loss [4], [5], [6], [7]. PD patient Tregs are impaired in their immunosuppressive activities, and Teff subsets with neurotoxic potential are present during disease [8], [9], [10], [11]. Studies from our group indicate that increased Teff phenotypes are associated with worsening of UPDRS Part III scores, while others suggest that α-synuclein reactive T cells are elevated in early disease but wane over time [8,10]. Nonetheless, the presence of autoreactive T cells and elevated proinflammatory responses is confirmed, but the exact association and time-course with respect to disease progression is still being explored. Potentiation of Treg function and modulation of Teff responses restores adaptive immune regulation and represents a means to harness neurodegeneration [4]. Our own works demonstrated that granulocyte-macrophage colony-stimulating factor (GM-CSF, sargramostim, Leukine) increases Treg numbers and function and protects dopaminergic neurons [5,12,13]. Translation into humans validated GM-CSF activities in a double-blind, placebo-controlled Phase 1 PD clinical trial [13]. Daily administration of high-dose sargramostim (6 μg/kg/day) increased Treg numbers and function with improved UPDRS-scored motor function and magnetoencephalography assessed neurophysiological activities. Although treatment was generally well-tolerated, sargramostim led to select adverse events including injection site reactions, bone pain, and immune reactions including urticaria and vasculitis. Therefore, we lowered the dosing regimen and extended the time of the study evaluation. Safety and tolerability of a year-long reduced dose sargramostim treatment regimen was evaluated in PD.
2. Methods
2.1. Study design and subject enrollment
The study is an unblinded, open-label, single-center phase 1 clinical trial performed at the University of Nebraska Medical Center (UNMC), Omaha, NE, USA designed to test safety, tolerability, and biomarker discovery utilizing a 3 μg/kg/day (5 days on 2 days off) sargramostim regimen. In total, 6 PD subjects were enrolled, and of those, 5 met study entry criteria. All were recruited from the Omaha metropolitan area, assessed for three months for clinical status and baseline immune, hematological, and metabolic profiling, and treated for 12 months between January 2019 to July 2020. Eligibility criteria were 35 to 85 years of age with PD signs and symptoms that included asymmetric bradykinesia, resting tremor, and/or muscle rigidity persisting for longer than three years with less than stage four on the Hoehn and Yahr disease scale. Exclusion criteria included poor venous access, inability to undergo leukapheresis, use of a wheelchair, walker, and/or cane, multiple system atrophy, corticobasal degeneration, unilateral Parkinsonism of >3 years, prior head injury, stroke, brain surgery including deep brain stimulation, a family history of >1 blood relative with PD, mental illness, cognitive impairment, autoimmune, systemic inflammatory or hematologic diseases, current treatment with neuroleptics or lithium, past treatment with sargramostim, past immunosuppressive treatments, and known allergies to colony-stimulating factors or yeast-derived products.
2.2. Ethics
The research study protocol (IRB Protocol 839-18) was approved by the UNMC Institutional Review Board. Subjects were identified and referred to the Clinical Research Center (CRC) by their primary care physician. Subjects were enrolled after informed consent was obtained by the study physician following Good Clinical Practice guidelines. No randomization or blinding was performed, as all study subjects were on treatment. The trial is registered at ClinicalTrials.gov, identifier: NCT03790670.
2.3. Procedures
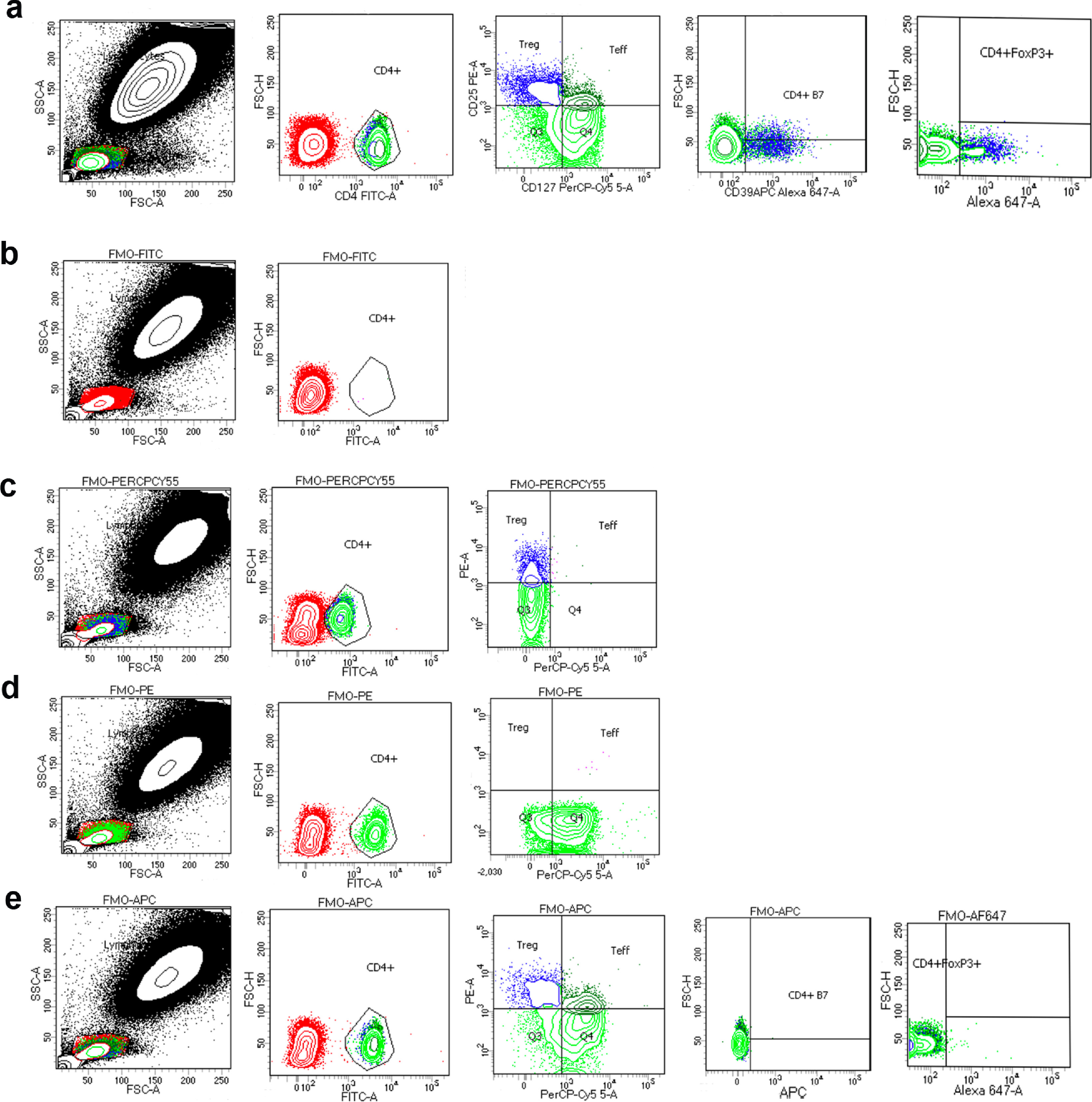
The current study (Phase 1b) was designed for safety and tolerability assessment for direct comparison with a previously published (Phase 1a) study in which PD subjects self-administered sargramostim at 6 μg/kg/day for two months (NCT01882010). For the current study, PD subjects underwent three pre-treatment monthly interval appointments to determine baseline immune, hematologic, and metabolic profiles (Supplemental Tables 2–4, baseline column). On visit three (month 0), subjects initiated self-administered sargramostim at 3 ug/kg/day (five days on, two days off) subcutaneously for 12 months, returning for clinical assessments every four weeks. Prior to treatment initiation and at two and six months after initiation, subjects underwent leukapheresis to allow for peripheral blood lymphocyte enrichment to obtain sufficient numbers of CD4+ Tregs to complete analyses for DNA methylation assessment, flow cytometric analysis, and Treg functional assessments. Peripheral blood samples, physical examinations, and MDS-UPDRS Parts I-IV assessments were completed during each appointment. Anti-parkinsonian therapies (carbidopa-levodopa) were continued during the course of study without modification for 4/5 subjects (Table 1). The primary neurologist (PS) performed MDS-UPDRS III assessments in the “ON” state at the same time of day for each study visit. Observable and/or clinical adverse events discovered during physical examination were recorded directly by the study neurologist such as elevated white blood cell counts or site injection reactions. Subjects were also provided with an “adverse event log” for events occurring between visits. The severity of the adverse event and likelihood of relationship to treatment were determined by the study neurologist. Study drug was withheld during the drug holiday for two days prior to each clinical visit, except for during leukapheresis visits (months 2–6). On these visits, subjects did not undergo the two-day drug holiday, and blood was harvested on day five of treatment. WBC counts with differentials, immunocyte numbers, CD4 and CD8 T cell percentages and ratios, and comprehensive blood chemistry profiles were monitored for safety. Lastly, peripheral blood cells were stained with fluorescently-conjugated monoclonal antibodies against CD4 (FITC or AF700; RRID: AB_395751 and AB_396943), CD127 (PerCP-Cy5.5; RRID: AB_1645548), CD25 (PE; RRID: AB_400203), FOXP3 (AF647; RRID: AB_1645411), Helios (AF-488; RRID: AB_10661895), CD152/CTLA-4 and/or iCTLA-4 (APC; RRID: AB_398615), CD95/FAS/Apo1 (APC; RRID: AB_398659), CD39 (APC; RRID: AB_1645459), CD31 (AF647; RRID: AB_397020), CD27 (APC; RRID: AB_1645457), CD45RA (AF700; RRID: AB_1727496), CD45RO (APC; RRID: AB_398673), CCR7 (PE-Cy7; AB_396765), Integrin β7 (APC; RRID: AB_398490) (all from BD Biosciences, San Jose, CA), and CD49d (PE-Cy7; RRID: AB_10643278) (BioLegend Inc., San Diego, CA), with isotype-matched antibodies and fluorescence minus one (FMOs) serving as negative controls. For intracellular markers, cells were permeabilized using the Intracellular Fixation & Permeabilization Buffer Set (eBioscience; catalog # 00-5523-00). Extracellular and intracellular labels were examined with an LSR II flow cytometer (BD Biosciences) and analyzed using BD FACSDiva software (RRID: SCR_001456). A representative gating strategy for T cell subset determination is depicted in Supplemental Fig. 1a and Supplemental Fig. 2. CD4+ T cell phenotypes and lymphocyte profiles were compared with those of the previous Phase 1a study [13].
Table 1.
Demographics.
| PD Subjectsa |
||
|---|---|---|
| N | Mean (SD) | |
| Age (years) | 5 | 64 (5) |
| Time Since Diagnosis (years) | 5 | 8 (5) |
| UPDRS Part III Score | 5 | 20 (5) |
| N | Percentage | |
| Male Sex | 5 | 100 |
| Caucasian Race | 5 | 100 |
| Anti-Parkinsonian Therapy: | ||
| Carbidopa-Levodopa 25-100 mgb | 3 | 60 |
| Carbidopa-Levodopa 50-200 mg | 1 | 20 |
| Carbidopa-Levodopa 23-95 mg | 1 | 20 |
Demographic data taken from subjects at the time of enrollment
Subject 2001 began anti-parkinsonian therapy on month 8
2.4. Outcomes
The primary study endpoint was drug safety and tolerability assessed by clinical signs and symptoms, complete blood counts with differential, comprehensive blood chemistry profiles, physical examination, and MDS-UPDRS Part III scores. Hematological profiles were performed by the hospital's clinical diagnostics laboratory, and one neurologist performed all clinical examinations including blood pressure, pulse, temperature, skin, lung, heart, and abdomen evaluations as well as MDS-UPDRS assessments in the “ON” state. Adverse events were recorded and scored based on severity of event as mild (1), moderate (2), or severe (3). Events were also scored in relation to drug treatment as unrelated (1), unlikely (2), possible (3), probable (4), or definitely related (5) [13]. Mild events caused minimal discomfort or concern and did not interfere with daily activities. Moderate events were defined as discomfort, inconvenience, or concerns ameliorated with simple therapeutic measures. Severe adverse events were defined as discomfort or incapacitation that may require prescription drug therapy, other treatments, or interventions. No events required interruption of treatment. A data and safety monitoring board of UNMC physicians and faculty monitored safety outcomes and advised study investigators during the course of study. Secondary outcomes were immune phenotype and function, DNA methylation status, and gene and proteome analyses. Methodology for secondary outcomes can be found in supplemental methods.
2.5. Statistical analysis
Sample size estimates of five PD subjects were determined to provide 80% power and to afford an increased score of 1.63 (32%) in baseline immune response using a two-sided Wilcoxon test assuming normal distribution. The immune response profile score, a measure of immune deficit compared to baseline control, was calculated from a group of 20 PD patients from a previous clinical study wherein results of CBC/diff, FACS, and functional assays of peripheral blood lymphocytes and T cells were utilized to provide an immunological score profile that comprised an overall adaptive immune response profile [8]. The mean (± SD) immune response score was 5.4 ± 0.88, thus an n of 5 is sufficient to detect a 1.63 difference for α=0.05 and power of 0.80 (Shapiro-Wilk W = 0.9326, p= 0.1723). Parametric ANOVA analyses were used as the data set proved to be normally distributed and homoscedasticity was determined by Levene tests. Therefore, to assess the effect of sargramostim over time and before and after treatment, all five subjects were utilized. Statistical analysis was performed using GraphPad Prism 8.0 software (La Jolla, CA; RRID: SCR_002798) and Statistica v13.3 (Tibco Software, Palo Alto, CA; RRID: SCR_014213). All values are expressed as mean ± SD. When applicable, differences in between-group means were analyzed using one-way ANOVA followed by Dunnett's post hoc test. The dependent variable for ANOVA and Mann-Whitney U analyses are shown on the Y-axis for each experimental parameter and the grouping variable is by subject or by month on treatment; both variables are provided in the figure legends. T-tests were performed for each experimental parameter to compare the means of the baseline with the means for the cumulative time on sargramostim and multiple p values were adjusted for false discovery rate (FDR) at 5% using the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli [14]. Significant differences for these studies, including peripheral blood profiles, MDS-UPDRS Part III motor assessments, flow cytometric analysis, methylation status of the forkhead box P3 Treg-specific demethylated region (FOXP3 TSDR), and gene and protein expression analyses were selected at p < 0.05. For adverse event profiles ANOVA, Fisher's Exact, and/or Mann-Whitney U analyses were performed. All other correlation analyses were determined using Pearson product-moment correlation coefficients, best-fit lines were determined using linear regression, p values were determined for r values greater than 0.25, and multiple p values adjusted for FDR at 5% by the procedure of Benjamini et al. [14].
2.6. Role of funding source
The funding sources had no role in the study design or data collection, analysis, interpretation, or writing of the manuscript. The corresponding author had access to all data sets and takes complete responsibility for the published manuscript.
3. Results
3.1. Demographics and baseline immune profiles
Collectively, six PD subjects were enrolled and assessed for eligibility. One subject was excluded due to poor venous access, and the remaining five subjects continued on study for baseline evaluations and treatment (Table 1). All remaining subjects (n = 5) were Caucasian males, 57–69 years of age with a mean of 64 years and have been diagnosed with PD for 3–15 years with a mean of 8 years. All subjects displayed complete blood counts, blood chemistry profiles, and immune cell ratios within normal reference values as a requirement for study participation (Supplemental Tables 2–4, baseline column). Due to the wide range of disease duration and potential impact of disease severity on baseline immune profile and treatment-related responses, subjects with abnormal baseline values were excluded. All but one subject began sargramostim therapy while on their anti-Parkinson's medications, as indicated in Table 1. These medications were maintained and continued during the course of study. The remaining subject began anti-Parkinsonian treatment at month 8 post-sargramostim initiation.
3.2. Safety, tolerability, and adverse event profiles
Lowering drug dose and extending treatment to 12 months was safe and generally well-tolerated. However, all subjects reported at least one adverse event over the course of the study, with the majority reporting elevated WBC counts (5/5, 100%), injection site reactions (4/5, 80%), fall with injury (3/5, 60%), and GI tract problems/nausea (3/5, 60%) (Table 2). Less frequently reported adverse events included pain in the upper torso and extremities, chest pain/discomfort, muscle weakness, headache, infection, dyskinesia, and skin and eye problems (1/5, 20% and/or 2/5, 40%). Compared to reported adverse events associated with our Phase 1a study using 6 μg/kg/day for 56 days of treatment [13], subjects administered 3 μg/kg/day (5 days on, 2 days off) for 56 days experienced fewer injection site reactions and rashes, less pain in the chest, upper torso, lower torso, and extremities, and less itching, muscle soreness, and weakness. Subjects also displayed significantly less adverse events per subject per month as well as lower severity of adverse events (Table 2 and Fig. 1). Similarly, continued treatment for 12 months, resulted in diminished adverse event profiles. All adverse events in this study were considered mild/moderate with no severe or serious adverse events reported to be associated with treatment and no withdrawals. Two severe adverse events, a viral infection and leg cramping, were reported over the course of 12 months but were deemed to be unrelated to treatment. This is in contrast to the prior study wherein three severe adverse events were reported, and one serious adverse event that led to study withdrawal of one subject [13]. As expected, sargramostim increased levels of WBC, lymphocytes, monocytes, eosinophils, and neutrophils (Supplemental Table 2) and slight increases in CD3+ and CD4+ T cells (Supplemental Table 3). In addition to lower adverse event profiles, comprehensive metabolic panels for subjects showed no significant increases compared to baseline (Supplemental Table 4) which validated the safety and tolerability of this low dosage regimen of sargramostim.
Table 2.
Incdence and severity of adverse events.
| Sargramostim Phase 1a 6 μg/kg, qd, 2 mos (n = 10) | Sargramostim Phase 1b 3 μg/kg, 5d/wk, 2 mos (n = 5) | Sargramostim Phase 1b 3 μg/kg, 5d/wk, 12 mos (n = 5) | ||||
|---|---|---|---|---|---|---|
| Adverse Eventa,bfor each subject | Number | Percentage | Number | Percentage | Number | Percentage |
| Any adverse event | 10 | 100 | 5 | 100 | 5 | 100 |
| Any severe adverse events | 3 | 30 | 0 | 0 | 2 | 40 |
| Any serious adverse events | 1 | 10 | 0 | 0 | 0 | 0 |
| Adverse event leading to withdrawal | 4 | 40 | 0 | 0 | 0 | 0 |
| Possible relationship to drug/placebo | 10 | 100 | 5 | 100 | 5 | 100 |
| Definitive relationship to drug/placebo | 7 | 70 | 3 | 60 | 3 | 60 |
| Category, Subjects reporting | ||||||
| 1 Abnormal Laboratory | 10 | 100 | 1 | 20 | 5 | 100 |
| 2 Injection site reaction | 10 | 100 | 4 | 80 | 4 | 80 |
| 3 Chest pain or discomfort | 4 | 40 | 1 | 20 | 1 | 20 |
| 4 Pain, upper torso & extremities | 7 | 70 | 0 | 0 | 1 | 20 |
| 5 Pain, lower torso & extremities | 3 | 30 | 0 | 0 | 0 | 0 |
| 6 Pain, other than exctremities | 0 | 0 | 0 | 0 | 0 | 0 |
| 7 Rash, other than injection site | 4 | 40 | 0 | 0 | 0 | 0 |
| 8 Itching, other than injection site | 2 | 20 | 0 | 0 | 0 | 0 |
| 9 Edema, other than injection site | 1 | 10 | 0 | 0 | 0 | 0 |
| 10 Shortness of breath, wheezing | 3 | 30 | 0 | 0 | 0 | 0 |
| 11 Headache | 2 | 20 | 1 | 20 | 1 | 20 |
| 12 Fatigue | 2 | 20 | 0 | 0 | 0 | 0 |
| 13 Chills, fever | 2 | 20 | 0 | 0 | 0 | 0 |
| 14 Infection, any | 2 | 20 | 0 | 0 | 2 | 40 |
| 15 GI tract, nausea, vomiting | 3 | 30 | 0 | 0 | 3 | 60 |
| 16 Muscle, soreness, weakness | 3 | 30 | 1 | 20 | 2 | 40 |
| 17 Equilibrium | 1 | 10 | 0 | 0 | 0 | 0 |
| 18 Inury, fall | 2 | 20 | 1 | 20 | 3 | 60 |
| 19 Skin, not infection | 1 | 10 | 0 | 0 | 1 | 20 |
| 20 Cardiovascular, hematological | 2 | 20 | 0 | 0 | 0 | 0 |
| 21 Neurological, psychological, dyskinesia | 2 | 20 | 1 | 20 | 2 | 40 |
| 22 Ophthalmological | 1 | 10 | 0 | 0 | 2 | 40 |
| 23 Sleep anomalies | 1 | 10 | 0 | 0 | 0 | 0 |
| Median | Mean ± SD | Median | Mean ± SD | Median | Mean ± SD | |
| Total adverse events/subject | 13.5 | 15.1 ± 8.5 | 3.0 | 3.2 ± 1.3 | 17.0 | 16.2 ± 5.5 |
| Total adverse events/subject/mo | 6.8 | 7.6 ± 1.2 | 0.25 | 0.27 ±0.11 | 1.4 | 1.4 ± 0.5 |
| Severity of adverse eventsc | 1.7 | 1.6 ± 0.3 | 1.0 | 1.2 ± 0.3 | 1.2 | 1.3 ± 0.3 |
| Likelihood of drug-relatedc | 3.75 | 3.5 ± 0.7 | 4.2 | 3.6 ± 1.0 | 3.0 | 2.9 ± 1.0 |
p < 0.05 vs Phase 1a, Mann-Whitney U test.
Reported adverse events since the initiation of drug.
More than 2 adverse advents per patient may have been reported.
Detemined by attending physician.
Fig. 1.

Adverse events (AE) comparing two clinical trials of sargramostim. PD subjects were administered sargramostim (Leukine®, human recombinant GM-CSF) in a previous Phase 1a (Ph 1a) (n = 10) and current Phase 1b (Ph 1b) clinical trial (n = 5). Subjects in the Ph 1a trial received 6 μg/kg of sargramostim every day for 2 months. In a proof-of-concept study to attenuate AE frequency and severity and extend administration, subjects in the Ph 1b trial received 3 μg/kg sargramostim on a 5 day on/2 day off regimen for 12 months. (a) Total number of adverse events (AEs) per subject recorded during the 2- and 12- month interventional period (Total) normalized on a monthly basis (Total/Mo). (b) AE severity scored on a scale of 1–3 severity as (1) mild, (2) moderate, or (3) severe. Mild events cause minimal discomfort or concern, may require minimal or no treatment, and do not interfere with daily activities. Moderate events were defined as causing discomfort, inconvenience, or concerns which were ameliorated with simple therapeutic measures. Severe adverse events were defined as causing discomfort or incapacitation that require prescription drug therapy or other treatments or interventions by medical personnel. Differences in means (± SD) for Total AE/Subject, Total AE/Subject/Mo, and Severity of AEs between Ph Ia vs Ph 1b trials were determined by Student's t-test and p values annotated above the pair-wise comparisons. (c) Graphical representation displaying reported AE based on AE severity (y axis), AE category (z axis), and day of treatment (x axis) for the Ph 1a trial. (d) Graphical representation displaying reported AE based on AE severity (y axis), AE category (z axis), and day of treatment (x axis) for the Ph 1b study. (c and d) AE categories are defined in Table 2.
3.3. MDS-UPDRS Part III motor assessments
MDS-UPDRS Part III scores were monitored over three months prior to initiating treatment to establish baseline motor function for disease progression monitoring. No worsening of motor function scores was observed for any subject during the course of treatment (Fig. 2). Compared to baseline, sargramostim treatment resulted in an overall decrease in MDS-UPDRS Part III scores for all subjects over time (Fig. 2a). Large variation in raw scores led to non-significant findings (Fig. 2b), however, significance was achieved comparing baseline to cumulative scores (Fig. 2c). Comparison of individual subject baseline and treatment scores demonstrated that 60% (3/5) of subjects displayed decreased MDS-UPDRS Part III scores following sargramostim initiation (Fig. 2c). Normalization of MDS-UPDRS Part III score as a change from baseline showed a profound decrease by three months and was significantly enhanced by eight months (Fig. 2d and 2e). Similar to total UPDRS readings, 60% (3/5) of subjects experienced significant diminution from baseline at all times during sargramostim treatment (Fig. 2f).
Fig. 2.

MDS-Unified Parkinson's Disease Rating Scale (UPDRS) Part III motor assessment before and during sargramostim treatment. Prior to treatment, subjects (n = 5) underwent at least three separate baseline evaluations (at month -4, -3, -2, and/or -1 before initiation) and then began drug administration (3 ug/kg per day, 5 days on/2 days off). After treatment initiation, subjects were evaluated by the study neurologist once/month for 6 months and once/ every 2 months thereafter for 12 months. (a) Raw UPDRS Part III scores over time grouped for individual subjects (2001, 2003, 2004, 2005, 2006). (b) Total mean UPDRS Part III scores grouped by time of treatment. Blue dashed lines indicate mean baseline measurement. (c) Mean UPDRS Part III scores ± SD grouped by combined (All) and individual subjects. Specific p values are indicated above each subject. Baseline values are represented as blue circles and sargramostim treatment is represented as red squares. (d) Change from baseline in UPDRS Part III scores over time grouped for individual subjects. (e) Mean change from baseline ± SD in UPDRS Part III scores grouped by time of treatment. (f) Mean change from baseline in UPDRS Part III scores ± SD grouped by combined (All) and individual subjects. Specific p values are indicated above each subject. Baseline values are represented as blue circles and sargramostim treatment is represented as red squares. Significant differences (± SD) in baseline and treated means were determined by Student's t-test with p values denoted above comparisons. Differences in means ± SD over time were also determined by one-way ANOVA where p ≤ 0.05 compared to baseline (b).
3.4. Immune modulation and peripheral biomarker evaluations
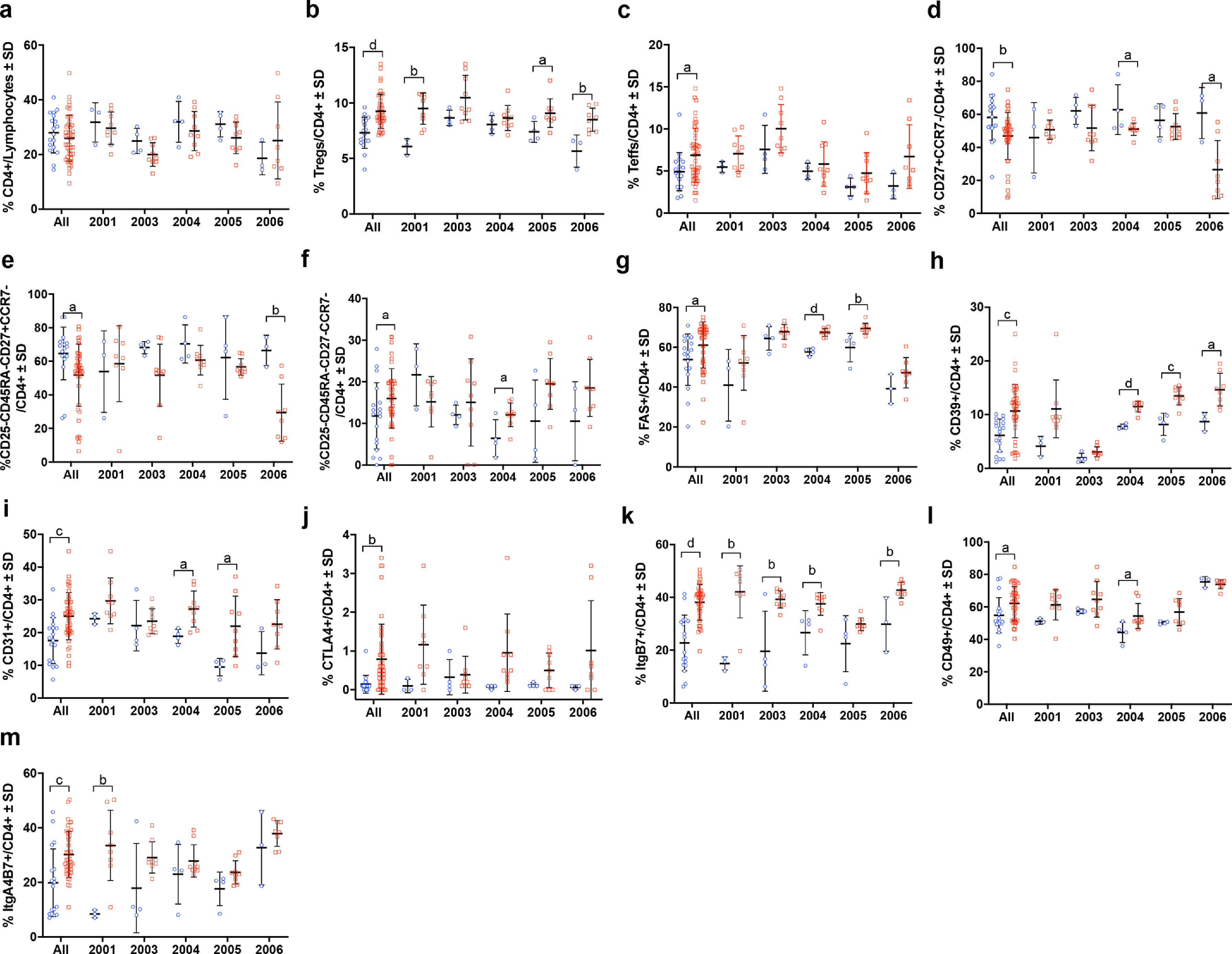
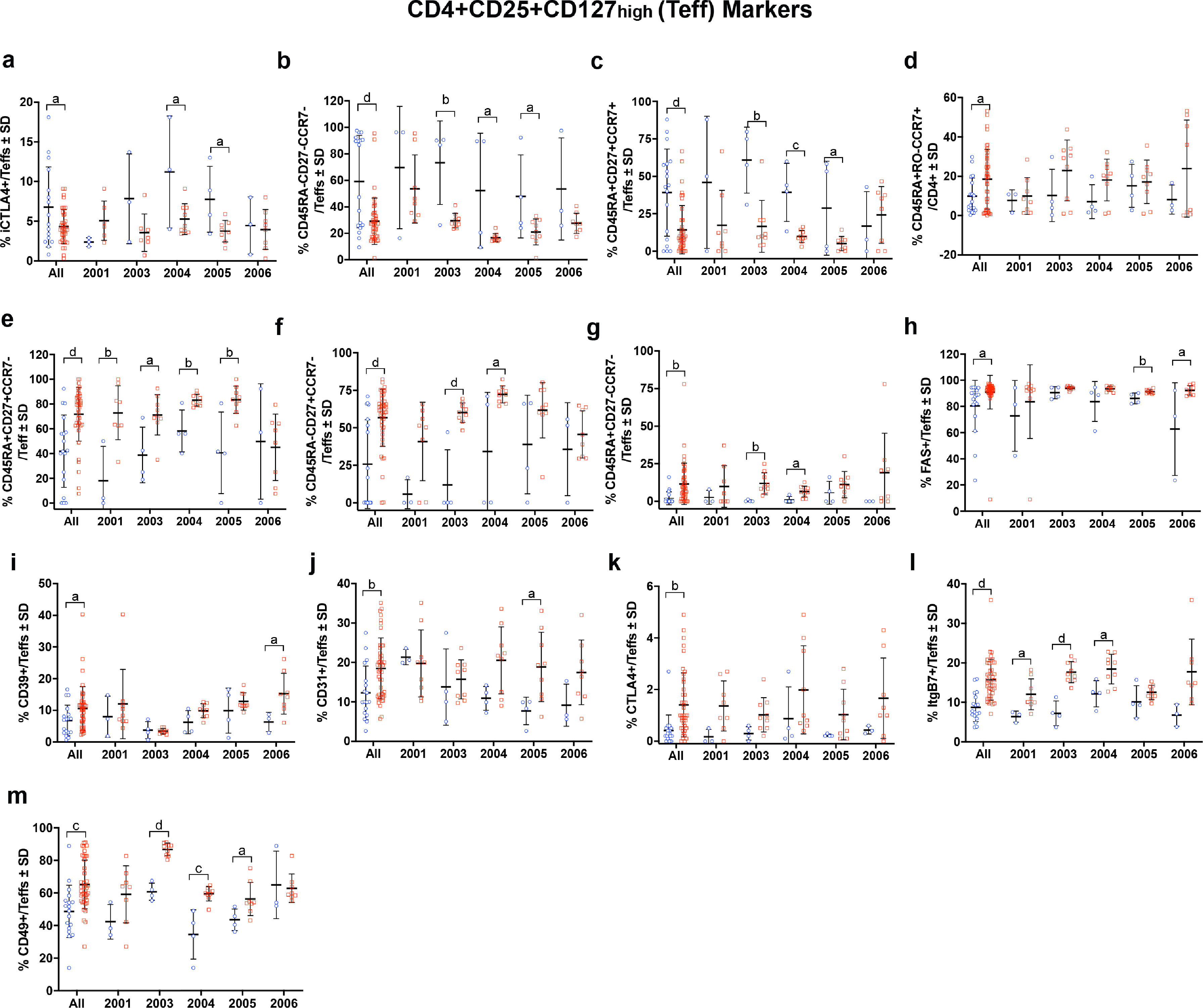
As T cell subsets have been shown to correlate with disease severity, we assessed the immunomodulatory effects of sargramostim on phenotypic and functional CD4+ T cell biomarkers [10,13,15]. Flow cytometric analysis revealed no change in total CD4+ lymphocytes and a significant sustained increase in CD4+CD127lowCD25+ Tregs and transient increase in CD4+CD127highCD25+ Teffs (Fig. 3a–c). Among CD4+ lymphocytes, frequencies of cells expressing FOXP3, Helios, CD31, CTLA, ItgB7, and ItgA4B7 were also increased significantly during treatment (Fig. 3d–i). Subsequent evaluation of Treg subsets also indicated elevated levels of CD45RO, CD31, CD49, CTLA4, and ItgB7 (Fig. 4a–e). Evaluation of peripheral biomarkers before and after treatment on an individual basis and within the Teff population showed similar patterns (Supplemental Figs. 3–6).
Fig. 3.

Flow cytometric analysis of CD4+ peripheral blood populations over time. Quantification of frequencies for the following dependent variables: (a) CD4+ lymphocytes, (b) CD4+CD127lowCD25+ Tregs, (c) CD4+CD127highCD25+ Teffs, (d) CD4+FOXP3+, (e) CD4+FOXP3+HELIOS+, (f) CD4+CD31+, (g) CD4+CTLA+, (h) CD4+ItgB7+, (i) CD4+ItgA4B7+ over the course of treatment. Differences in means (± SD) for each dependent variable grouped by time on treatment were determined by one-way ANOVA and Dunnett's post hoc test where p ≤ 0.05 compared to baseline (*).
Fig. 4.

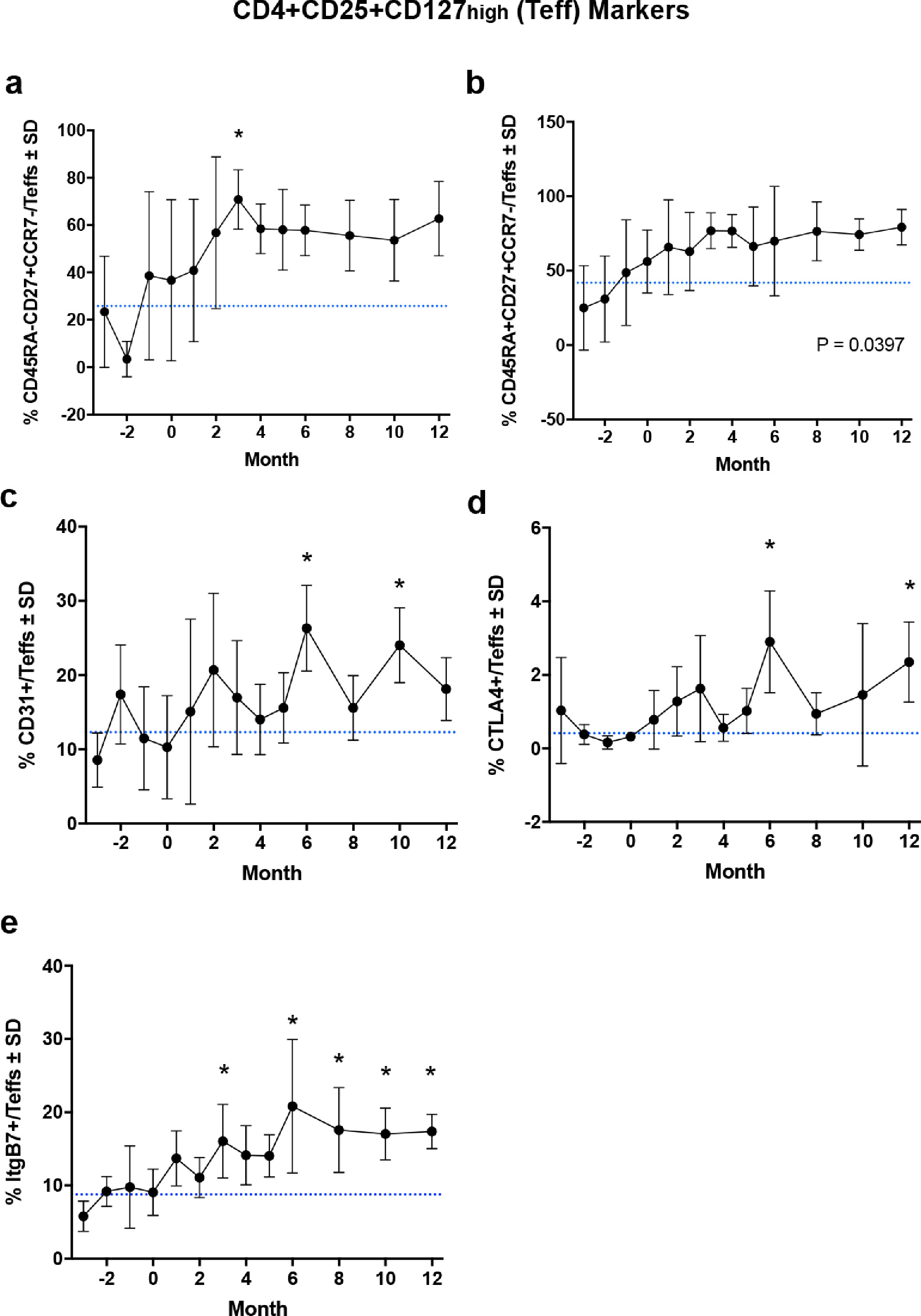
Flow cytometric analysis, immunosuppressive function, and FOXP3 Treg-Specific Demethylated Region (TSDR) methylation status of CD4+CD127lowCD25+ Treg subsets within CD4+ peripheral blood lymphocytes. Quantification for the dependent variables included (a) CD45RA-RO+ Treg, (b) CD31+ Treg, (c) CTLA+ Treg, (d) CD49+ Treg, and (e) ItgB7+ Treg subset frequencies within peripheral blood over time. Difference in means (± SD) for each dependent variable grouped by time of treatment were determined by one-way ANOVA and Dunnett's post hoc test where p ≤ 0.05 compared to baseline (*). (f) Percent demethylation (± SD) within the TSDR of the FOXP3 intron from isolated Tregs before and at 2 and 6 months after initiation of sargramostim treatment. Differences in means (± SD) were determined by one-way ANOVA where p ≤ 0.05 compared to baseline (*). (g) Quantification of Treg-mediated suppression (± SD) of Tresp (CD4+CD25-) proliferation at various Tresp:Treg ratios. Treg-mediated suppression is calculated as % Inhibition = 1 – (% Proliferating Tresp:Treg ÷ % Proliferating Stimulated Tresp Alone) and is reported as percent inhibition. Linear regression analysis indicates r2 ≥ 0.81, p < 0.0001 for all lines and significant elevation (p < 0.0001) from baseline (blue) compared to each month of treatment. (h) Correlation analysis for percent TSDR demethylation and corresponding Treg-mediated inhibition (Treg activity, AUC) at baseline (blue), 2-month (red), and 6-month (green) during sargramostim treatment, indicating a direct correlation of TSDR demethylation and Treg activity with r = 0.3212, p = 0.0004.
From the same Treg isolates, we evaluated the methylation status of the FOXP3 TSDR at baseline following sargramostim treatment. Results indicated that 65% of the TSDR was demethylated at baseline compared to highly methylated DNA controls (Fig. 4f). Sargramostim treatment significantly increased the level of demethylation by 20% at 2 months and maintained demethylation levels at six months which is indicative of stable FOXP3 expression and suppressive Treg phenotype during treatment. Therefore, we next assessed the effect of sargramostim treatment on Treg suppressive activities. By 2 months, Treg function was significantly enhanced compared to baseline, and potentiation of Treg function was maintained over the 12-month study (Fig. 4g). Moreover, we confirmed that levels of Treg-mediated activity and FOXP3 TSDR demethylation were positively correlated (r = 0.3212, p = 0.0004), thus verifying the effect of sargramostim on FOXP3 TSDR-directed Treg immunosuppressive capacity (Fig. 4h).
3.4.1. Association of peripheral T cell biomarkers with Treg cell function and MDS-UPDRS Part III scores
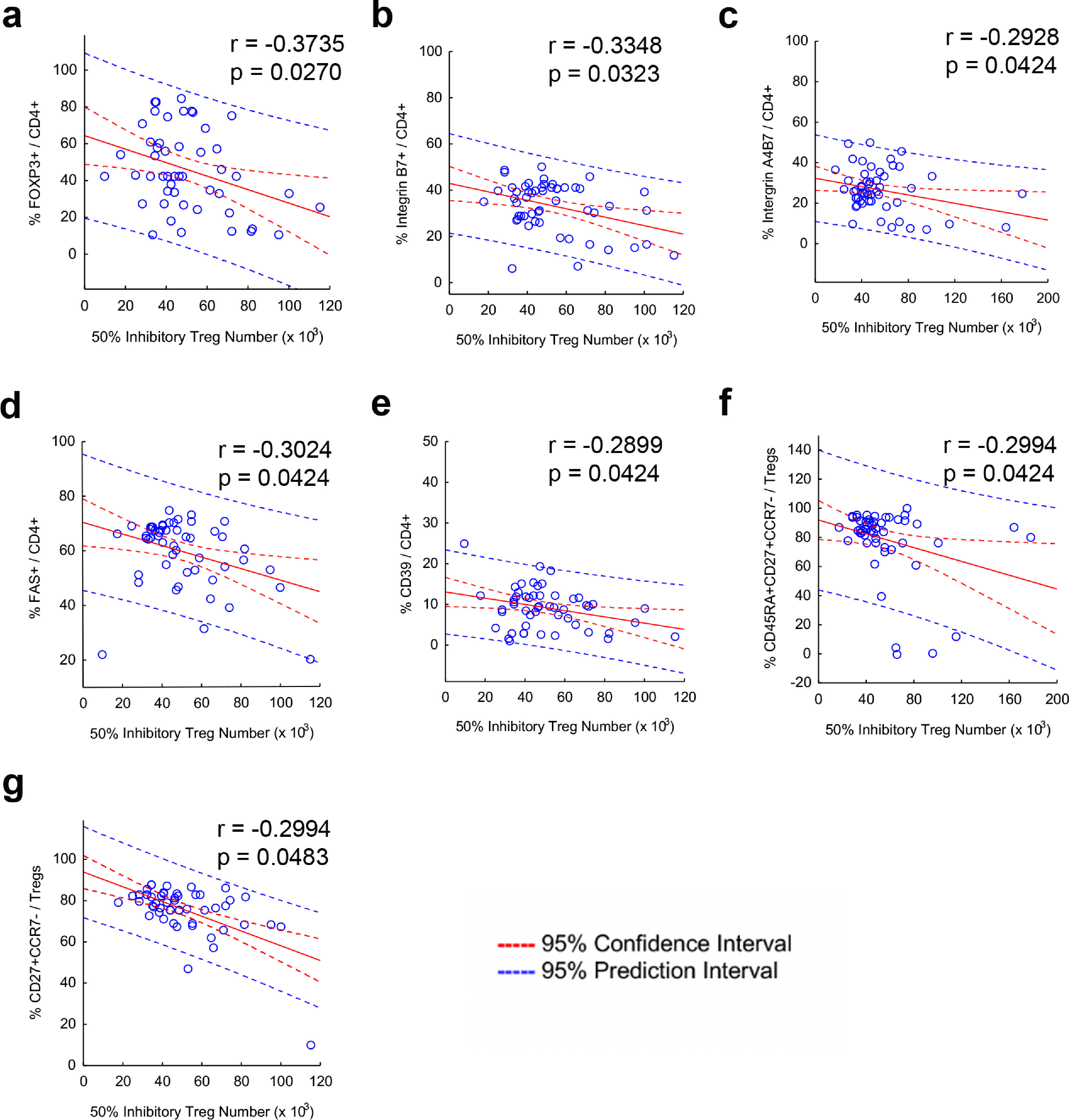
Next, we assessed the effect of sargramostim on treatment biomarker expression with Treg-mediated immunosuppressive function (Fig. 5). First, Treg activity was positively associated with increased co-expression of ItgB7, FOXP3, FAS, CD27, and CD45RA (Fig. 5a–h). Second, negative correlations were shown between increasing MDS-UPDRS Part III scores and diminished frequencies of FAS+ CD4+ T cells and Treg subsets that co-express ItgB7, CD45RA, and CD27 with the lack of CCR7 (Fig. 6a–d), suggesting that motor function is improved with increased Treg subset levels and greater suppressive activity. Indeed, this was confirmed by positive correlation of increased Treg function as measured by lower number of Tregs required to yield 50% suppression and diminished MDS-UPDRS Part III scores (Fig. 6e, Supplemental Fig. 7).
Fig. 5.

Elevated peripheral blood markers are associated with enhanced Treg function. Correlation analyses are depicted for the dependent variables including AUC for Treg activity and (a) %Integrin B7+CD4+ T cells, (b) %FOXP3+CD4+ T cells, (c) %FAS+CD4+ T cells, (d) %CD45RA-RO+CD4+ T cells, (e) %CD27+CCR7- Treg, (f) %Integrin B7+ Treg, (g) %Integrin A4B7+ Treg, and (h) %CD45RA-CD27+CCR7- Treg. For all correlation analyses, regression bands are indicated by dashed lines that encompass the 95% confidence intervals (red) and 95% prediction values (blue). Pearson r and p values are depicted on individual graphs. Data are displayed as scatter plots using the percentage of peripheral blood marker in either the total CD4 population or the Treg population against Treg activity, AUC. Correlations were determined using Pearson product–moment correlation coefficients, p values determined for correlation coefficients greater than 0.25, and the resulting 16 determinations adjusted for FDR. Best-fit lines were determined by linear regression.
Fig. 6.

Elevated peripheral blood markers and enhanced Treg function are associated with decreased UPDRS Part III scores. Correlation analyses for the dependent variables that included UPRS Part III scores and (a) %FAS+ CD4+ T cells, (b) %Integrin B7+ Tregs, (c) %CD45RA+CD27+CCR7- Tregs, (d) %CD27+CCR7- Tregs, and (e) Treg activity as determined by 50% Inhibitory Treg number. For all correlation analyses, regression bands are indicated by dashed lines that encompass the 95% confidence intervals (red) and 95% prediction values (blue). The Pearson r and p values are depicted on individual graphs. Data are displayed as scatter plots using the percentage of peripheral blood marker in total CD4 population, Treg population, or Treg function as determined about 50% inhibitory Treg number against UPDRS Part III scores. Correlations were determined using Pearson product-moment correlation coefficients, p values determined for correlation coefficients greater than 0.25, and the resulting 39 determinations adjusted for FDR. Best-fit lines were determined using linear regression.
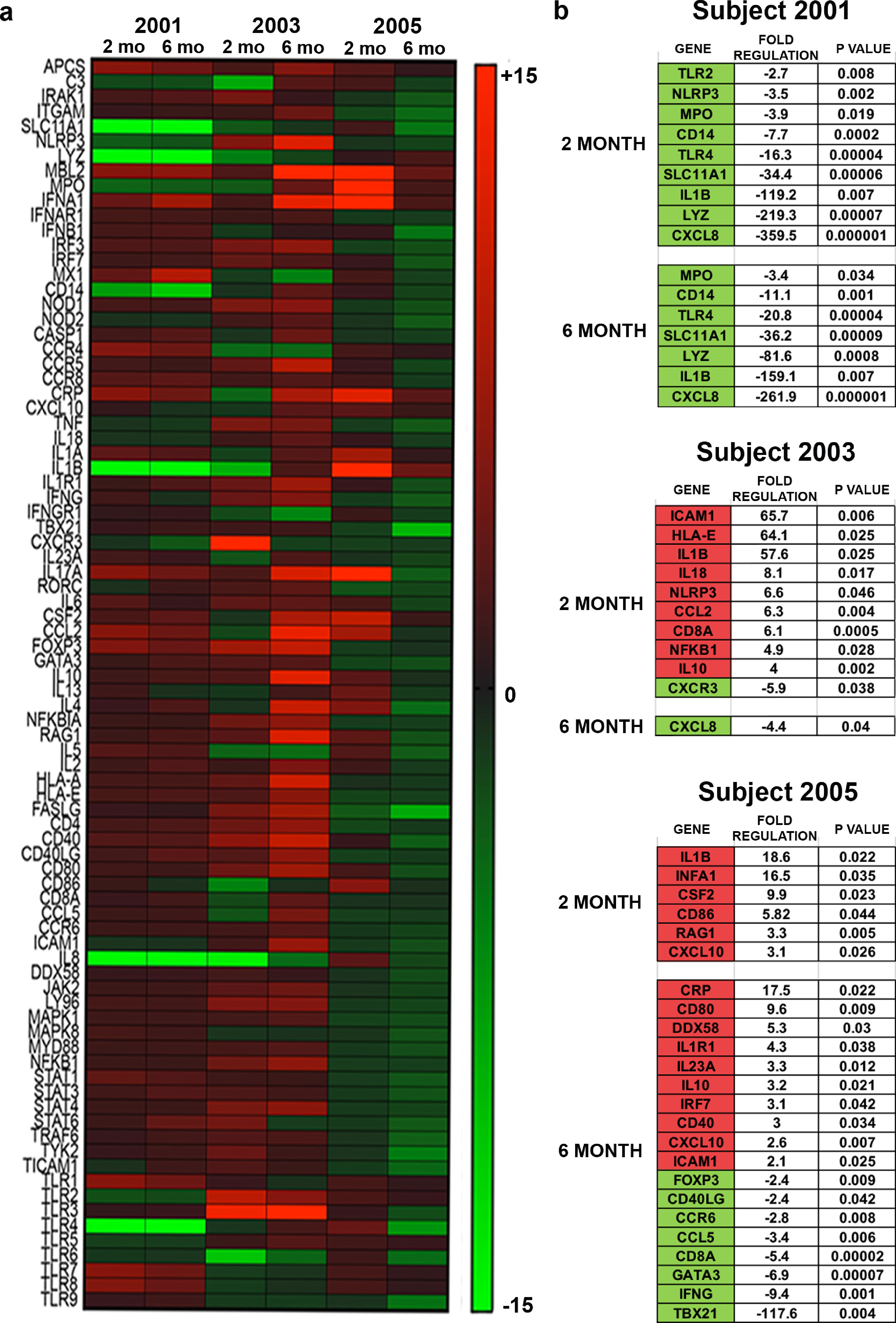
3.5. Proteomic profile of peripheral blood lymphocytes (PBLs)
Before sargramostim initiation and at 2 and 6 months of sargramostim treatment, PBLs isolated by leukapheresis were subjected to transcriptomic (Supplemental Fig. 8) and proteomic analyses. Expression of over 2500 proteins were identified and quantified for analysis. Among these proteins, 785 and 152 proteins were significantly differentially expressed at 2 and 6 months of sargramostim treatment, respectively. Volcano plots highlighting the proteins whose expression was significantly down-regulated (green) or upregulated (red) post-treatment as compared with pre-treatment are shown in Fig. 7. IPA comparison analysis of canonical pathways altered at both 2 and 6 months of treatment showed activation of 25 pathways (Supplemental Table 5) identified by selected association with cellular immune response signaling, neuroinflammation signaling, and PD signaling pathways. Interestingly, 15 out of 25 pathways showed a downregulation of calcineurin and/or nuclear factor kappa B (NF-κB) expression at 2 months, but not 6 months after treatment (Supplemental Table 5).
Fig. 7.

Differential proteomic analysis of peripheral blood lymphocytes at 2 and 6 months after treatment. Volcano plots showing the fold change (treatment versus baseline) plotted against the p value highlighting significantly changed proteins (red – upregulation and green – downregulation; p ≤ 0.05 and an absolute fold change of 2). The vertical lines correspond to the absolute fold change of 2, and the horizontal line represents a p value of 0.05.
4. Discussion
Treatment with sargramostim at 3 μg/kg/day (5 days on, 2 days off) for 12 months was generally well-tolerated. Commonly reported adverse events were associated with known side effects including increased injection site reactions, elevated WBC counts, and bone pain [16]. No serious or severe adverse events were reported as being linked to drug administration and no subjects were withdrawn from treatment, further supporting the drug's safety and tolerability for this indication. Chronic treatment also did not result in disease worsening in any subject as determined by MDS-UPDRS Part III scores before and during treatment. With dopamine replacement therapies, UPDRS Part III scores increase in a linear fashion by an average of 2.4 points per year over the course of five years [17]. In the current study, which includes adjunctive sargramostim, four subjects collectively decreased MDS-UPDRS part III scores, and one subject maintained baseline scores. While UPDRS part III motor assessment to evaluate clinical efficacy can be subjective and has inherent limitations due to variation, it remains the gold standard in PD for evaluation of motor dysfunction during clinical assessments [18]. However, clinical efficacy utilizing motor improvement evaluations in PD are difficult to assess due to the known placebo effect in this disease population [19]. We previously observed this effect in placebo-treated subjects for six weeks post-treatment initiation in our initial Phase 1 clinical trial [13]. Although, by seven weeks, placebo controls returned to baseline UPDRS Part III scores, while scores of sargramostim-treated subjects continued to decline and remained below baseline values and those of placebo-treated subjects until discontinuation. Therefore, we believe the effect observed over the course of a year is noteworthy, but cannot be verified until a larger-scale, placebo-controlled study powered for clinical efficacy is performed.
Low-dose sargramostim positively altered immune function, shifted T cell phenotypes, and enhanced treatment-induced biomarker levels that were associated with lowered MDS-UPDRS Part III scores. Treg and Teff frequencies were increased within one month after sargramostim initiation with parallel increases in CD4+ T cell biomarkers. These biomarkers were associated with cell homing, such as Itgβ7 and Itgα4β7, as well as signaling and anti-inflammatory mechanisms for Treg-mediated immunosuppression that include FAS, CD49, CD31, FOXP3, CTLA4, and CD39. Increased levels of Itgβ7 and Itgα4β7 are likely associated with increased migratory capacity of T cells homing to sites of inflammation and MAdCAM-1 within the gut [20,21], but can also bind to vascular CAM-1 (VCAM-1) under inflammatory conditions [22], playing a role in progression of chronic forms of neurodegenerative disease [23]. Moreover, Tregs expressing Itgα4β7 display higher suppressive function than those lacking expression by enhancing IL-10 secretion and inducing other regulatory-like T cell phenotypes [21]. We also observed increases in IL-10 gene expression by PBLs from sargramostim-treated subjects suggesting an anti-inflammatory role for sargramostim. Blockade of Itgβ7 is also linked to increased inflammation due to impaired homing and migration of Tregs, further strengthening the notion that upregulation of integrin biomarkers is beneficial for repair or replenishment of dysfunctional Treg populations as found in PD [24], [25], [26].
Expression of CD49 may reflect a maturation biomarker for T cells as development of functionally mature Tregs have been linked to expression of CD49 [27]. Absence of CD31 or platelet endothelial cell adhesion molecule (PECAM-1) expression is associated with Treg dysfunction [28]. Low frequency of CD31+ Tregs has been connected to decreased FOXP3 expression in coronary heart disease and Treg dysfunction in multiple sclerosis [28,29]. Similarly, increased FOXP3 expression is indicative of enhanced immunosuppressive function, as its presence is required to maintain a stable suppressive Treg phenotype [30]. Along with FOXP3 expression, Tregs utilize CTLA4 and CD39 to maintain suppressive capability [31]. Both are considered to be aligned with potent mechanisms of immunosuppression. CTLA4 expression controls antigen presentation by inhibiting co-stimulation via CD80/CD86 blockade [32,33]. CD39 is an ectonucleotidase that converts ATP into AMP that can metabolically starve surrounding cells, thus stunting cellular division [34]. CD73, another Treg-associated ectonucleotidase, converts AMP to adenosine that can interact with purinergic receptors on Teff to elevate intracellular cAMP and suppress proliferation [34,35]. CD39+ Tregs also maintains strong suppression and functional stability in the presence of inflammatory stimuli such as IL-1β and IL-6, which are both upregulated in PD [9,36,37]. Specifically, within Treg and Teff subsets, we also observed increased CCR7, CD27, and CD45RO expression following sargramostim treatment. Increased CCR7 and CD27 expression is associated with migration and enhanced function of effector memory-like Tregs and with maintenance of Treg circulation within the periphery [38], [39], [40]. Lastly, we observed a significant elevation in CD45RA-CD45RO+ Tregs which is indicative of memory and past activation, as well as enhanced suppressive function, suggesting that sargramostim is inducing Tregs with high proliferative capacity, potentially leading to the observed increased cell frequency [41]. With increased Treg subsets, CD4+CD127high CD25+ Teffs were also increased in the first months post-treatment; however, increased Teff prevalence was not observed after four months. Although, Teffs expressing CD27, CTLA4, ItgB7, and CD31 were significantly elevated with treatment. CD27 promotes cell survival, and increased expression of CTLA4 and ItgB7 is likely associated with cell activation. Also, the presence of CD31+ Teffs has been positively associated with lower UPDRS Part III scores [8]. Therefore, the potential effect of concomitantly inducing these effector populations alongside Tregs does not appear to be deleterious nor have a negative impact on Treg function and the overall immunosuppressive phenotype afforded by sargramostim treatment. Additionally, GM-CSF is known to expand other immunosuppressive cell populations such as myeloid-derived suppressor cells (MDSCs), regulatory B cells, and/or tolerogenic dendritic cells [42], [43], [44], [45], [46], [47]. These populations were not evaluated in the current study, but it should be noted that any clinical effects observed may also be mediated, in part, by the presence and induction of this immunoregulatory population as well. Pre-clinical evaluations utilizing the MPTP mouse model of PD indicate the ability of GM-CSF to induce tolerogenic bone marrow-derived dendritic cells that likely contribute to the increased presence of Tregs following treatment [47]. Furthermore, a clinical study investigating immunoregulatory cell populations within 32 PD subjects indicated decreased levels of suppressor and activated Tregs, IL-10 producing CD8+ Tregs, and tolerogenic dendritic cells, further supporting the notion that PD subjects have an impaired ability to suppress proinflammatory responses [48]. Therefore, the potential effect of GM-CSF on other regulatory phenotypes would open an exciting avenue to explore for future investigation.
Concordant with the observed Treg and Teff biomarker increases, sargramostim treatment enhanced Treg-mediated immunosuppressive function that was maintained over the course of the study. Previously, Tregs isolated from PD subjects showed impaired ability to suppress Teff proliferation that correlated with increased disease severity [8]. Treg deficiency has been associated with increased disease progression in Alzheimer's disease, ALS, stroke, traumatic brain injury, and multiple sclerosis [4]. These reports and our works suggest that controlling neuroinflammation via Treg induction or enhancement may be a promising therapeutic avenue for the clinic [13,49,50]. Here, we show that sargramostim treatment induces Tregs and restores Treg function via increased demethylation of FOXP3 TSDR and enhanced expression of biomarkers necessary to maintain a suppressive phenotype. This was indicated by the correlation between Treg activity, methylation status, and levels of peripheral T cells expressing Treg biomarkers. Demethylation of the TSDR is responsible for maintaining stable FOXP3 expression and Treg function, while hypermethylation of the TSDR is associated with Treg dysfunction in other diseases [51], [52], [53]. This study shows that sargramostim treatment leads to hypomethylation and increased FOXP3 levels, positively impacting and restoring Treg function. However, although significantly elevated throughout the course of treatment, Treg function peaked 2 months post-drug initiation, slowly decreasing in effectiveness over time. This decrease may be due to decreased capacity for induction of Tregs over the extended treatment, exhaustion of bone-marrow derived cell production, and/or the presence of neutralizing anti-drug antibodies [13]. Previously, low levels of anti-sargramostim antibodies within the serum were detected one month after treatment.
Overall, transcriptomic and proteomic analyses of PBLs during sargramostim treatment revealed an activated phenotype (Fig. 7, Supplemental Fig. 7, and Supplemental Table 7). Gene dysregulations involved both pro- and anti-inflammatory mediators, suggesting that low-dose sargramostim treatment results in immune activation within the PBL population similar to observations within the CD4+CD25- Teff and Treg subsets isolated in our previous high-dose treatment [13]. However, the lower dosage sargramostim regimen reported here also resulted in increased IL-10 gene expression at both 2 and 6 months after treatment initiation, supporting the immunosuppressive biomarker expression observed in flow cytometric analysis and Treg function. Previous findings also support the notion that Tregs require immune activation and presence of an inflammatory response to function properly and maintain a highly suppressive phenotype [7,54]. Therefore, the immune activation observed here may be responsible for the induced Treg function and phenotype following sargramostim treatment. Similarly, proteomic analysis also indicated that sargramostim treatment downregulates calcineurin expression in nine pathways within 2 months of treatment. High calcineurin activity is found to drive a toxic response in the presence of high α-synuclein levels in PD [55]. Additionally, activation of the nuclear factor of activated T cells (NFAT) pathway which plays important roles in T cell activation and modulation of immune responses is altered [56]. Most NFAT proteins are known to be regulated by calcineurin, and altered calcineurin/NFAT activation has been linked to the pathology of several neurodegenerative diseases including PD [57,58]. Thus, reducing calcineurin activity during sargramostim treatment suggests a mechanism by which sargramostim can provide a protective outcome in PD subjects. Secondly, the NF-κB pathway, which serves as a central mediator of inflammation, was significantly downregulated in 11 pathways. In both microglia and astroglia, activation of NF-κB, along with other proinflammatory transcription factors, leads to the transcription of several proinflammatory molecules [59,60] that contribute or are causal to the loss of dopaminergic neurons in MPTP-intoxicated mice and PD patients [61,62]. Additional studies show that inhibition of NF-κB activation reduces the induction of proinflammatory molecules and significantly protects nigrostriatal neurons against MPTP-induced neurodegeneration [63]. Therefore, reduction of NF-κB activity by sargramostim treatment may reduce the inflammation-mediated neurodegeneration and provide a consequent protective effect in PD patients.
4.1. Limitations of the study
As stated, this study was designed as an open-label, unblinded pilot investigation seeking to evaluate the safety and tolerability of a reduced dosing regimen for an extended time in PD. Therefore, an inherent limitation is the lack of a placebo control arm with limited subject entry. However, utilization of the subject baseline evaluations allowed for before and after treatment comparisons and timed evaluations. Secondly, the study contains a broad variability in baseline UPDRS scores, times since diagnosis, and variable immune profiles. This includes Treg and Teff numbers, lymphocyte ratios, and T cell functional assessments. To account for this variability, we utilized each subject as their own control to assess treatment-induced alterations. However, it is possible that evaluation in a more homogeneous population, such as early verse late disease, would yield variable outcomes. Lastly, the lack of female participants and start dates for anti-Parkinsonian therapies during study are further limitations. All limit stratification of treatment effects, interpretation of sex differences, evaluation of potential drug-drug effects or assessment of its therapeutic use in drug naïve subjects. Therefore, although statistically significant data is offered and these data sets are potentially meaningful, any confirmation of neurological improvements observed with treatment require careful verification in a larger, Phase II placebo-controlled study designed for drug efficacy.
5. Conclusions
Taken together, and even with inherent limitations, our findings support the safety and tolerability of extended treatment with sargramostim using a low-dose and discontinuous regimen. Treatment was well-tolerated and resulted in decreased frequency and severity of adverse events compared to a higher dose and continuous regimen. The lower dose regimen also resulted in stable MDS-UPDRS Part III scores indicating no worsening of disease, and observed alterations in MDS-UPDRS Part III scores were associated with increased expression of Treg phenotypes and immunosuppressive function, thus suggesting a potential role of Treg function in diminution of disease progression. These results are intriguing and provide the basis for larger scale assessments to determine clinical efficacy of a reduced sargramostim regimen within the PD population. The study supports the notion that use of immunomodulators to induce and/or expand Tregs, shift Teff phenotype, and enhance immunosuppression in neurodegenerative disease affects neuroimmune interactions and has the potential to slow disease outcome. The study also helps to support the idea of utilizing Tregs as a therapeutic target, which forms the basis for future clinical assessment.
Data sharing statement
The datasets generated for this study and full study protocol are available upon request from the corresponding author.
Declaration of Competing Interest
The authors declare no conflicts of interest.
Acknowledgments
The authors would like to thank the participants and family members for their willingness to participate and dedication to the study as well as those who supported our research efforts. We greatly appreciate the time and effort put in by staff within our investigational pharmacy services, the Great Plains Center for Clinical and Translational Research, and Red Cross Apheresis Center for assistance with drug dispensation, subject scheduling, and sample collection. We also thank the members of our data safety monitoring board including R. Gregory Bociek, MD, Kenneth Follett, MD, Daniel Murman, MD, Gleb Haynatzki, PhD, and Christopher Kratochvil, MD for review and discussion of the work, as well as Farah Shahjin and Jatin Machhi for assistance with proteomic sample preparation. The authors would also like to thank the UNMC Flow Cytometry Research Facility for exceptional flow cytometric analysis and support, the INBRE grant from NIH (2P20GM103427) for supporting a site license to EndNote software, the UNMC elutriation core for isolation of subject monocyte and lymphocyte populations, and the UNMC Epigenomics Core Facility for assessing methylation status of isolated DNA samples. The authors would also like to acknowledge the professional services of UNMC Mass Spectrometry & Proteomics Core and Bioinformatics and Systems Biology Core for assistance with proteomic analysis. Lastly, we express our gratitude to the funding sources that made this work possible, Partner Therapeutics and the University of Nebraska Foundation, which includes community donations from the Carol Swarts, M.D. Emerging Neuroscience Research Laboratory, the Margaret R. Larson Professorship, the Eisenberg Parkinson's Research Fund, and the Frances and Louie Blumkin and Harriet Singer Research Foundations. We also thank the Vice Chancellor's Office of the University of Nebraska Medical Center for Core Facility support.
Footnotes
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.ebiom.2021.103380.
Appendix. Supplementary materials
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Schwab A.D., Thurston M.J., Machhi J. Immunotherapy for Parkinson's disease. Neurobiol Dis. 2020;137 doi: 10.1016/j.nbd.2020.104760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olanow C.W., Kieburtz K., Schapira A.H. Why have we failed to achieve neuroprotection in Parkinson's disease? Ann Neurol. 2008;64(Suppl 2):S101–S110. doi: 10.1002/ana.21461. [DOI] [PubMed] [Google Scholar]
- 3.Guzman-Martinez L., Maccioni R.B., Andrade V., Navarrete L.P., Pastor M.G., Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol. 2019;10:1008. doi: 10.3389/fphar.2019.01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Machhi J., Kevadiya B.D., Muhammad I.K. Harnessing regulatory T cell neuroprotective activities for treatment of neurodegenerative disorders. Mol Neurodegener. 2020;15(1):32. doi: 10.1186/s13024-020-00375-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosloski L.M., Kosmacek E.A., Olson K.E., Mosley R.L., Gendelman H.E. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J Neuroimmunol. 2013;265(1-2):1–10. doi: 10.1016/j.jneuroim.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson K.E., Kosloski-Bilek L.M., Anderson K.M. Selective VIP receptor agonists facilitate immune transformation for dopaminergic neuroprotection in MPTP-intoxicated mice. J Neurosci. 2015;35(50):16463–16478. doi: 10.1523/JNEUROSCI.2131-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds A.D., Banerjee R., Liu J., Gendelman H.E., Mosley R.L. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J Leukoc Biol. 2007;82(5):1083–1094. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- 8.Saunders J.A., Estes K.A., Kosloski L.M. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson's disease. J Neuroimmune Pharmacol. 2012;7(4):927–938. doi: 10.1007/s11481-012-9402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chao Y., Wong S.C., Tan E.K. Evidence of inflammatory system involvement in Parkinson's disease. Biomed Res Int. 2014;2014 doi: 10.1155/2014/308654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindestam Arlehamn C.S., Dhanwani R., Pham J. Alpha-synuclein-specific T cell reactivity is associated with preclinical and early Parkinson's disease. Nat Commun. 2020;11(1):1875. doi: 10.1038/s41467-020-15626-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kustrimovic N., Comi C., Magistrelli L. Parkinson's disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J Neuroinflammation. 2018;15(1):205. doi: 10.1186/s12974-018-1248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olson K.E., Namminga K.L., Schwab A.D. Neuroprotective activities of long-acting granulocyte-macrophage colony-stimulating factor (mPDM608) in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-intoxicated mice. Neurotherapeutics. 2020;17(4):1861–1877. doi: 10.1007/s13311-020-00877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gendelman H.E., Zhang Y., Santamaria P. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson's disease trial. NPJ Parkinsons Dis. 2017;3:10. doi: 10.1038/s41531-017-0013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benjamini Y., Krieger A.M., Yekutieli D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika. 2006;93:491–507. [Google Scholar]
- 15.Sulzer D., Alcalay R.N., Garretti F. T cells from patients with Parkinson's disease recognize alpha-synuclein peptides. Nature. 2017;546(7660):656–661. doi: 10.1038/nature22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baldo B.A. Side effects of cytokines approved for therapy. Drug Saf. 2014;37(11):921–943. doi: 10.1007/s40264-014-0226-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holden S.K., Finseth T., Sillau S.H., Berman B.D. Progression of MDS-UPDRS scores over five years in de novo Parkinson disease from the Parkinson's progression markers initiative cohort. Mov Disord Clin Pract. 2018;5(1):47–53. doi: 10.1002/mdc3.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evers L.J.W., Krijthe J.H., Meinders M.J., Bloem B.R., Heskes T.M. Measuring Parkinson's disease over time: The real-world within-subject reliability of the MDS-UPDRS. Mov Disord. 2019;34(10):1480–1487. doi: 10.1002/mds.27790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quattrone A., Barbagallo G., Cerasa A., Stoessl A.J. Neurobiology of placebo effect in Parkinson's disease: what we have learned and where we are going. Mov Disord. 2018;33(8):1213–1227. doi: 10.1002/mds.27438. [DOI] [PubMed] [Google Scholar]
- 20.DeNucci C.C., Pagan A.J., Mitchell J.S., Shimizu Y. Control of alpha4beta7 integrin expression and CD4 T cell homing by the beta1 integrin subunit. J Immunol. 2010;184(5):2458–2467. doi: 10.4049/jimmunol.0902407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stassen M., Fondel S., Bopp T. Human CD25+ regulatory T cells: two subsets defined by the integrins alpha 4 beta 7 or alpha 4 beta 1 confer distinct suppressive properties upon CD4+ T helper cells. Eur J Immunol. 2004;34(5):1303–1311. doi: 10.1002/eji.200324656. [DOI] [PubMed] [Google Scholar]
- 22.Swerlick R.A., Lee K.H., Li L.J., Sepp N.T., Caughman S.W., Lawley T.J. Regulation of vascular cell adhesion molecule 1 on human dermal microvascular endothelial cells. J Immunol. 1992;149(2):698–705. [PubMed] [Google Scholar]
- 23.Kanwar J.R., Harrison J.E., Wang D. Beta7 integrins contribute to demyelinating disease of the central nervous system. J Neuroimmunol. 2000;103(2):146–152. doi: 10.1016/s0165-5728(99)00245-3. [DOI] [PubMed] [Google Scholar]
- 24.Sun H., Kuk W., Rivera-Nieves J., Lopez-Ramirez M.A., Eckmann L., Ginsberg M.H. Beta7 integrin inhibition can increase intestinal inflammation by impairing homing of CD25(hi)FoxP3(+) regulatory T cells. Cell Mol Gastroenterol Hepatol. 2020;9(3):369–385. doi: 10.1016/j.jcmgh.2019.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klann J.E., Kim S.H., Remedios K.A. Integrin activation controls regulatory T cell-mediated peripheral tolerance. J Immunol. 2018;200(12):4012–4023. doi: 10.4049/jimmunol.1800112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann J., Huehn J., de la Rosa M. Expression of the integrin alpha Ebeta 7 identifies unique subsets of CD25+ as well as CD25- regulatory T cells. Proc Natl Acad Sci U S A. 2002;99(20):13031–13036. doi: 10.1073/pnas.192162899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan X., Moltedo B., Mendoza A. CD49b defines functionally mature Treg cells that survey skin and vascular tissues. J Exp Med. 2018;215(11):2796–2814. doi: 10.1084/jem.20181442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang L., Zheng Y., Yuan X. Decreased frequencies and impaired functions of the CD31(+) subpopulation in Treg cells associated with decreased FoxP3 expression and enhanced Treg cell defects in patients with coronary heart disease. Clin Exp Immunol. 2017;187(3):441–454. doi: 10.1111/cei.12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haas J., Korporal M., Balint B., Fritzsching B., Schwarz A., Wildemann B. Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4(+)CD25(+)FOXP3(+)CD31(+) T-cells in patients with multiple sclerosis. J Neuroimmunol. 2009;216(1-2):113–117. doi: 10.1016/j.jneuroim.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Lu L., Barbi J., Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol. 2017;17(11):703–717. doi: 10.1038/nri.2017.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shevyrev D., Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol. 2019;10:3100. doi: 10.3389/fimmu.2019.03100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker L.S. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun. 2013;45:49–57. doi: 10.1016/j.jaut.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wing J.B., Ise W., Kurosaki T., Sakaguchi S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity. 2014;41(6):1013–1025. doi: 10.1016/j.immuni.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Borsellino G., Kleinewietfeld M., Di Mitri D. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110(4):1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 35.Deaglio S., Dwyer K.M., Gao W. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6):1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu J., Ni X., Pan X. Human CD39(hi) regulatory T cells present stronger stability and function under inflammatory conditions. Cell Mol Immunol. 2017;14(6):521–528. doi: 10.1038/cmi.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stojakovic A., Paz-Filho G., Arcos-Burgos M., Licinio J., Wong M.L., Mastronardi C.A. Role of the IL-1 pathway in dopaminergic neurodegeneration and decreased voluntary movement. Mol Neurobiol. 2017;54(6):4486–4495. doi: 10.1007/s12035-016-9988-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borst J., Hendriks J., Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol. 2005;17(3):275–281. doi: 10.1016/j.coi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Menning A., Hopken U.E., Siegmund K., Lipp M., Hamann A., Huehn J. Distinctive role of CCR7 in migration and functional activity of naive- and effector/memory-like Treg subsets. Eur J Immunol. 2007;37(6):1575–1583. doi: 10.1002/eji.200737201. [DOI] [PubMed] [Google Scholar]
- 40.Cowan J.E., McCarthy N.I., Anderson G. CCR7 controls thymus recirculation, but not production and emigration, of Foxp3(+) T cells. Cell Rep. 2016;14(5):1041–1048. doi: 10.1016/j.celrep.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booth N.J., McQuaid A.J., Sobande T. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J Immunol. 2010;184(8):4317–4326. doi: 10.4049/jimmunol.0903781. [DOI] [PubMed] [Google Scholar]
- 42.Park M.Y., Lim B.G., Kim S.Y., Sohn H.J., Kim S., Kim T.G. GM-CSF promotes the expansion and differentiation of cord blood myeloid-derived suppressor cells, which attenuate xenogeneic graft-vs.-host disease. Front Immunol. 2019;10:183. doi: 10.3389/fimmu.2019.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamilton J.A., Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34(2):81–89. doi: 10.1016/j.it.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Pulendran B., Banchereau J., Burkeholder S. Flt3-ligand and granulocyte colony-stimulating factor mobilize distinct human dendritic cell subsets in vivo. J Immunol. 2000;165(1):566–572. doi: 10.4049/jimmunol.165.1.566. [DOI] [PubMed] [Google Scholar]
- 45.Bhattacharya P., Thiruppathi M., Elshabrawy H.A., Alharshawi K., Kumar P., Prabhakar B.S. GM-CSF: an immune modulatory cytokine that can suppress autoimmunity. Cytokine. 2015;75(2):261–271. doi: 10.1016/j.cyto.2015.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheng J.R., Quan S., Soliven B. CD1d(hi)CD5+ B cells expanded by GM-CSF in vivo suppress experimental autoimmune myasthenia gravis. J Immunol. 2014;193(6):2669–2677. doi: 10.4049/jimmunol.1303397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schutt C.R., Gendelman H.E., Mosley R.L. Tolerogenic bone marrow-derived dendritic cells induce neuroprotective regulatory T cells in a model of Parkinson's disease. Mol Neurodegener. 2018;13(1):26. doi: 10.1186/s13024-018-0255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alvarez-Luquin D.D., Arce-Sillas A., Leyva-Hernandez J. Regulatory impairment in untreated Parkinson's disease is not restricted to Tregs: other regulatory populations are also involved. J Neuroinflammation. 2019;16(1):212. doi: 10.1186/s12974-019-1606-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beers D.R., Zhao W., Wang J. ALS patients' regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight. 2017;2(5):e89530. doi: 10.1172/jci.insight.89530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faridar A., Thome A.D., Zhao W. Restoring regulatory T-cell dysfunction in Alzheimer's disease through ex vivo expansion. Brain Commun. 2020;2(2):fcaa112. doi: 10.1093/braincomms/fcaa112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schreiber L., Pietzsch B., Floess S. The Treg-specific demethylated region stabilizes Foxp3 expression independently of NF-kappaB signaling. PLoS One. 2014;9(2):e88318. doi: 10.1371/journal.pone.0088318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson M.R., Enose-Akahata Y., Massoud R. Epigenetic modification of the FoxP3 TSDR in HAM/TSP decreases the functional suppression of Tregs. J Neuroimmune Pharmacol. 2014;9(4):522–532. doi: 10.1007/s11481-014-9547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimazu Y., Shimazu Y., Hishizawa M. Hypomethylation of the Treg-specific demethylated region in FOXP3 Is a hallmark of the regulatory T-cell subtype in adult T-cell Leukemia. Cancer Immunol Res. 2016;4(2):136–145. doi: 10.1158/2326-6066.CIR-15-0148. [DOI] [PubMed] [Google Scholar]
- 54.Reynolds A.D., Stone D.K., Hutter J.A., Benner E.J., Mosley R.L., Gendelman H.E. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol. 2010;184(5):2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caraveo G., Auluck P.K., Whitesell L. Calcineurin determines toxic versus beneficial responses to alpha-synuclein. Proc Natl Acad Sci U S A. 2014;111(34):E3544–E3552. doi: 10.1073/pnas.1413201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hermann-Kleiter N., Baier G. NFAT pulls the strings during CD4+ T helper cell effector functions. Blood. 2010;115(15):2989–2997. doi: 10.1182/blood-2009-10-233585. [DOI] [PubMed] [Google Scholar]
- 57.Hogan P.G., Chen L., Nardone J., Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17(18):2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 58.Kipanyula M.J., Kimaro W.H., Seke Etet P.F. The emerging roles of the calcineurin-nuclear factor of activated T-lymphocytes pathway in nervous system functions and diseases. J Aging Res. 2016;2016 doi: 10.1155/2016/5081021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X., Jana M., Dasgupta S. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277(42):39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dasgupta S., Jana M., Liu X., Pahan K. Role of very-late antigen-4 (VLA-4) in myelin basic protein-primed T cell contact-induced expression of proinflammatory cytokines in microglial cells. J Biol Chem. 2003;278(25):22424–22431. doi: 10.1074/jbc.M301789200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagatsu T., Mogi M., Ichinose H., Togari A. Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Transm Suppl. 2000;(60):277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 62.Mogi M., Harada M., Kondo T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180(2):147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- 63.Ghosh A., Roy A., Liu X. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2007;104(47):18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.