

Summary
We describe an 18-year-old woman with several month’s history of a 12 x 7 mm palpable mammary nodule, that was hypoechoic, with regular margins and vascularization areas by ultrasound. A fibroadenoma was hypothesized (American College of Radiology BI-RADS 3). A 14 G needle biopsy was performed, showing a LC proliferation suspected for LCH of a lymph node, with florid dermatopathic lymphadenopathy in differential diagnosis. The multidisciplinary team of the breast clinic decided to perform a lumpectomy and a diagnosis of LCH involving an intra-mammary lymph node was made. Langerhans cells (LC) are dendritic cells characterized by grooved nuclei, irregular nuclear contours, and abundant cytoplasm, that normally reside in the skin and mucosal surfaces. They were positive for CD1a, langerin/CD207, and S100 by immunohistochemistry. Langerhans cell histiocytosis (LCH) is a clonal proliferation of histiocytes that is thought to be neoplastic in most cases. Reactive LC can be distinguished from LCH by cyclin D1 immunostaining, which is positive only in LCH. About 50% of cases have BRAF V600E mutations. The revised classification of histiocytes divides LCH in subtypes: LCH SS (single system), LCH lung positive, LCH Multiple System/Risk Organ negative and LCH Multiple System/Risk Organ positive. Localized disease can progress to multisystem involvement. The diagnosis of LCH is based on clinical and radiological findings in combination with histopathological, immunophenotypic or ultrastructural analyses identifying tissue infiltration by LC. It is recommended that biopsy confirmation of suspected LCH be performed in all cases. Lymph nodes may be the only site of disease or a part of multisystem involvement by LCH. The histologic differential diagnosis is discussed.
Key words: breast, lymph node, langerhans cell, histiocytosis, adolescent
Introduction
Histiocytoses are rare disorders characterized by the accumulation of cells thought to be derived from dendritic cells (DCs) or macrophages. Their clinical behavior ranges from mild to disseminated and, sometimes, life-threatening forms.
Langerhans cells (LC) are dendritic cells that normally reside in the skin and mucosal surfaces. After LC encounter antigen, they migrate to lymph nodes and present antigen to T cells 1. LC are round-oval cells with characteristic linear grooves or lobed nuclei, fine chromatin and delicate nuclear membrane, with inconspicuous nucleoli and abundant pale eosinophilic cytoplasm.
Langerhans cell histiocytosis (LCH) is a clonal proliferation of bone-marrow derived dendritic cells that is thought to be neoplastic in most cases. Neoplastic LC are uniformly positive for CD1a, langerin/CD207, and S100 and frequently express CD68. About half of cases express lysozyme and CD45/LCA 2,3. T-cell and B-cell lineage markers, CD30, and follicular dendritic cell markers are negative. However, approximately 30% of cases have detectable clonal IgH, IgK, or TCR rearrangements. Langerin/CD207 is a component of Birbeck granules, which are identified on electron microscopy and are characteristic of LCH 3. Birbeck granules are thought to be components of the endocytic recycling pathway 4-8. Positivity for CD1a, CD207 and S100 protein is mandatory for the diagnosis of LCH 9,10. About 50% of cases have BRAF V600E mutations, identical to those found in hairy cell leukemia 8,11. However, identification of BRAF V600E mutation is not specific for LCH as this mutation has been seen in 54% of Erdheim-Chester disease (ECD) cases. BRAF mutation alone may be necessary but not sufficient for the development of LCH. Additional genetic abnormalities are required for tumorigenesis: a high frequency of MAP2K1 (mitogen-activated protein kinase pathway) mutations (33-50%) with BRAF mutations has also been found 12,13.
The revised classification of histiocytes consist of five groups of diseases: (a) Langerhans-related; (b) cutaneous and mucocutaneous; (c) malignant histiocytoses; (d) Rosai-Dorfman disease (RDD); and (e) haemophagocytic lympho-histiocytosis and macrophages activation syndrome. The Langerhans related group is further divided in to (a) LCH with subtypes as LCH SS (single system), LCH lung positive, LCH Multiple System/Risk Organ negative and LCH Multiple System/Risk Organ positive; (b) indeterminate cell histiocytosis; (c) Erdheim-Chester disease; and (d) mixed ECD and LCH 14. The Langerhans/non-Langerhans dichotomy has become questionable as nearly 20% of patients with Erdheim-Chester disease (ECD) also have LCH lesions 15. Moreover, both diseases have clonal mutations involving genes of the MAPK pathway in > 80% of cases 16,17. In addition, both conditions may be associated with similar clinical complications such as diabetes insipidus and/or neurodegenerative disease.
The annual incidence of LCH in children younger than 15 years of age is around 5 to 9x106 and 1x106 in patients older than 15 years of age 18,19. The disease is more common in childhood, particularly within the first decade of life, with a male predilection (about 3 to 1) 8,10. Rare cases of familial LCH have been reported 20, but no genetic susceptibility has been identified to date. LCH includes a broad spectrum of clinical manifestations in children and adults, ranging from self-healing lesions to life-threatening disseminated disease. The stage of disease at presentation is the most powerful prognostic factor. Patients with unifocal LCH (stage I) has about 99% survival probability. In contrast, high mortality occurs in children with multisystem disease (stage III or IV), which requires chemotherapy 10,12,13. Localized disease can progress to multisystem involvement. Moreover, treatment failure in low-risk patients is associated with an increased risk of late complications 21.
The diagnosis of LCH is based on clinical and radiological findings in combination with histopathological, immunophenotypic or ultrastructural analyses identifying tissue infiltration by LC. It is recommended that biopsy confirmation of suspected LCH be performed in all cases. In LCH, tumor cells are admixed with variable numbers of eosinophils and, histiocytes, both multinucleated and osteoclast-type cells, xanthomatous macrophages, neutrophils, small lymphocytes, and sparse plasma cells. Eosinophils can form micro-abscesses with central necrosis and in some cases presence of Charcot-Leyden crystals. In early lesions, LC predominate, along with eosinophils and neutrophils. In late lesions, the LC are decreased in number, with increased of lipid-laden (foamy) histiocytes and fibrosis 8,10.
LCH may be associated with Hodgkin and non-Hodgkin lymphomas, myeloid leukemia and non-haematological malignant neoplasms. In patients with LCH associated with acute myeloid leukemia or other myeloid neoplasms, the LC show often clonal relationship to the leukemia 8,10,13. In lymph nodes, tumor-associated LCH is most often associated with classic Hodgkin lymphoma, less frequently with diffuse large B-cell lymphoma, mantle cell lymphoma, and peripheral T-cell lymphomas. Tumor associated LCH is composed of cytological and phenotypical LC, present in small foci confined to lymph nodes sinuses. LC are associated with a lower number of histiocytes, eosinophils, and small lymphocytes than in fully developed LCH. Necrosis is rare. Abnormal LC recruitments potentially mimicking LCH occur in the lymph node paracortex in dermatopathic lymphadenitis. These reactive LC can be distinguished from LCH by Cyclin D1 immunostaining, that results positive only in LCH 22.
Materials and methods
Fixation tissue is carried out in 10% neutral buffered formalin for 24 hours. Once tissue is embedded in paraffin, a 3-4 micron tissue section is cut onto charged glass slides.
The detection system for immunostaining is BOND Polymer Refine Detection on staining platform LEICA BOND III for this antibodies: Pan-cytokeratin (AE1/AE3) clone AE1/AE3 (Cell Marque) 1:1000, 10 min at 37°C with Bond Epitope Retrieval Enzyme; S100 protein polyclonal (Leica) 1:1000, CD3 clone LN10 (Leica) 1:100, CD20 clone L26 (Leica) 1:100, CK14 clone LL002 (Leica) 1:50, myeloperoxidase polyclonal (Dako) 1:2500, p63 clone 7Jul (Leica) 1:100, langerin/CD207 antibody clone 12D6 (Leica) 1:50 (test made in Laboratory of ASST Brescia), with 30 min at 100°C in Bond Epitope Retrieval Solution 1 used to antigen retrieval, while 100°C Bond Epitope Retrieval Solution 2 used for antibody CD68R/PG-M1 clone PG-M1 (Dako) 1:500 30 min, CD30 clone Ber-H2 (Menarini) 1:50 40 min and 20 min of antigen retrieval for cyclin D1 clone SP4 (Cell Marques) 1:100. The Roche’s antibodies, ready to use, are detected on platform BenchMark ULTRA using UltraView Universal DAB Detection Kit, 64 min of antigen retrieval with Cell Conditioning 1 and incubation 32 min with this antibodies: CD1a clone 010 and CD14 clone EPR3653, while for antibody BRAF clone VE1 using Optiview DAB IHC Detection Kit, 64 min Cell Conditioning 1, incubation 40 min and amplification signal using Optiview Amplification Kit.
Case report
An 18-year-old woman was referred to the breast clinic of our hospital with a palpable mammary nodule lasting several months. She was submitted to a bilateral breast ultrasound. In the central-external quadrant of the right breast a hypoechoic nodule, with regular margins and vascularization areas, measuring 12 x 7 mm, was found. A fibroadenoma was hypothesized (American College of Radiology BI-RADS 3). The young woman was subjected to needle biopsy (14 G).
The biopsy (Fig. 1) showed small, mature, lymphocytes, a reactive secondary follicle, numerous eosinophilic granulocytes and a large number of medium sized round-oval cells, with grooved, folded, indented, or lobed nuclei, with fine chromatin, small nucleoli, delicate nuclear membrane, and abundant pale eosinophilic cytoplasm, suspicious for Langerhans cells. Immunohistochemical stainings showed phenotype consistent with LC: expression of S100 protein, and CD1a. Immunostainings for CD14, CD68R/PG-M1, CD30, CD3, CD20, cytokeratin (AE1/AE3 and CK14), myeloperoxidase, and p63 were negative. We arrived at a diagnosis of LC proliferation suspected for LCH of a lymph node, with florid dermatopathic lymphadenopathy in differential diagnosis.
Figure 1.

Needle biopsy (14 G). (A) Lymphoid tissue with pale areas (hematoxylin-eosin). (B) Medium sized cells, with folded, indented nuclei, fine chromatin, small nucleoli, delicate, and abundant pale eosinophilic cytoplasm, with a lot of eosinophilic granulocytes (hematoxylin-eosin). (C) CD68R/PG-M1 (immunohistochemistry) positive in occasional macrophages. (D) Myeloperoxidase (immunohistochemistry) positive in granulocytes. (E) S100 protein (immunohistochemistry) positive in Langerhans cells. (F) CD1a (immunohistochemistry) positive in Langerhans cells.
The multidisciplinary team of the breast clinic decided to perform surgical removal of the lesion. Three weeks later the woman performed a lumpectomy. The laboratory received a 2 x 1.8 x 1 cm breast tissue, containing a 1 cm oval nodule, with regular margins. On histology examination the nodule was a lymph node. The lymph node biopsy (Fig. 2) showed preserved architecture, with extensive involvement of the sinuses and paracortical regions by LC, mixed with many eosinophils, focally with micro-abscesses with central necrosis, macrophages, neutrophils, small lymphocytes, and sparse plasma cells. LC showed the same immunophenotype already detected in the biopsy. Moreover, LC expressed langerin/CD207 and Cyclin D1. Immunohistochemical staining for BRAF V600E mutation was negative. No large atypical or “sternbergoid” cells or granulomas were found. A diagnosis of LCH involving an intra-mammary lymph node was made. Clinical and instrumental evaluation of the disease spread is in progress.
Figure 2.
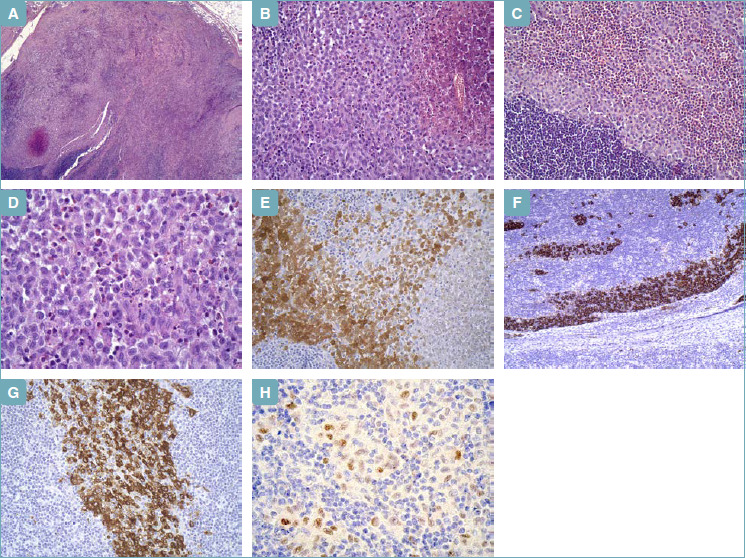
Lumpectomy. (A) Lymph node with pale areas, with central necrosis (hematoxylin-eosin). (B) Medium sized round-oval cells, with grooved, folded, indented nuclei, fine chromatin, small nucleoli, delicate nuclear membrane, and abundant pale eosinophilic cytoplasm, in aggregates with central necrosis (hematoxylin-eosin). (C) Langerhans cells mixed with a large number of eosinophilic granulocytes (hematoxylin-eosin). (D) Langerhans cells at higher magnification (hematoxylin-eosin). (E) S100 protein (immunohistochemistry) positive in Langerhans cells. (F) CD1a (immunohistochemistry) positive in Langerhans cells (intra-sinusoidal distribution). (G) Langerin/CD207 (immunohistochemistry) positive in Langerhans cells. (H) Cyclin D1 (immunohistochemistry) positive in Langerhans cells.
Discussion
Breast masses cause concern for both the patient and their family, although malignancy represents less than 1% of lesions in women ≤ 18 years of age. Fibroadenoma tends to occur at an early age, most commonly in adolescents and less commonly in post-menopausal women. The incidence of fibroadenoma decreases with increasing age and is generally found before 30 years of age. It is estimated that 10% of the world’s female population suffers from fibroadenoma during their lifetime. Sanders et al. describe 1,050 pediatric patients ≤ 18 years who underwent diagnostic breast ultrasound between 2004 and 2016 23. One hundred thirty patients underwent 160 core biopsies of solid lesions: benign lesions were 99%, of which 135 (84.3%) were fibroadenomas. One malignancy was diagnosed, a B cell lymphoma. Two hundred three patients underwent surgical excision for 266 discrete lesions, and 89% were fibroadenomas 23. Langerhans cell histiocytosis can compromise any organ, especially skin, bone and lungs. Breast involvement is very rare, generally within systemic disease, with involvement of multiple systems 24-26. Very rarely has LCH been described in associated with breast carcinoma 27 or can mimic Paget’s disease of the nipple 28.
Lymph nodes may be the only site of disease or as part of multisystem involvement by LCH. Lymphadenopathy refers to lymph nodes greater than 1 cm or with abnormal consistency. In primary care practice, the annual incidence of unexplained lymphadenopathy is 0.6% 29. Only 1.1% of these cases are related to malignancy, but this percentage increases with advancing age: 4% of patients older than 40 years vs 0.4% of those younger than 40 years 30. Moreover, other risk factors for malignancy are duration of lymphadenopathy greater than four to six weeks, generalized lymphadenopathy (two or more regions involved), male sex, node not returned to baseline after eight to 12 weeks, supraclavicular location, systemic signs: fever, night sweats, weight loss, hepatosplenomegaly, white race 29,30. Lymphadenopathy can be caused by various clinical conditions. Etiologies of lymphadenopathy can be remembered with the MIAMI mnemonic: malignancies (e.g. leukemia, lymphoma, metastasis), infections (infection: e.g. Bartonella, tuberculosis, staphylococcus or streptococcus infection, syphilis; viral infection: e.g. cytomegalovirus, Epstein-Barr virus, herpes zoster, human immunodeficiency virus; fungal infection; toxoplasmosis), autoimmune disorders (e.g. systemic lupus erythematosus, rheumatoid arthritis, Still disease, Sjögren syndrome), miscellaneous and unusual conditions (e.g. Castleman disease, Kikuchi lymphadenitis, sarcoidosis), and iatrogenic causes 29,30. Ultrasonography should be used as the initial imaging modality for patients presenting with a lymphadenopathy with or without fever. The American College of Radiology recommends ultrasonography as the initial imaging choice for lymphadenopathy in children up to 14 years of age and computed tomography for those older than 14 years 31. If the diagnosis is still uncertain, biopsy is recommended 31.
The histologic hallmark of LCH is the presence of proliferating LC in the appropriate cellular milieu. The histologic differential diagnosis includes especially granulomatous lymphadenitis, Kikuchi-Fujimoto disease, histiocytic disorders and dermatopathic lymphadenopathy. Unlike LCH, dermatopathic lymphadenopathy is characterized by expansion of the paracortex by interdigitating dendritic cells, small lymphocytes, LC, rare eosinophils, and macrophages, sometimes containing pigment. Patients with LCH of skin may have dermatopathic lymphadenopathy in regional lymph nodes, that should not be over interpreted as LCH involving lymph node. Interdigitating dendritic cells are positive for S100 protein, but negative for CD1a and langerin/CD207, whereas the LC are positive for all three markers. Reactive LC can be distinguished from LCH by Cyclin D1 immunostaining, that results positive only in LCH 22. Shanmugam et al. showed cyclin D1 expression by immunohistochemistry in all LCH cases tested (39/39). Most cases (22/39; 56%) showed strong cyclin D1 expression in the majority (≥ 50%) of LC. Only a few cases (6/39; 15%) showed cyclin D1 expression in a small subset of LC (< 20%). The authors concluded that cyclin D1 is ubiquitously expressed in LCH, while it is not significantly expressed in reactive LC proliferations in lymph node or skin 22. Therefore, cyclin D1 immunohistochemistry may be useful in excluding non-neoplastic mimics of LCH. Interdigitating dendritic cell sarcoma is very rare tumor, cytologically malignant, with a high mitotic rate. The cells of interdigitating dendritic cell sarcoma express S100 protein, but are negative for CD1a and langerin/CD207. Rosai-Dorfman disease expands lymph node sinuses, as when involvement by LCH is focal, but the histiocytes have centrally located round nuclei and small nucleoli, and can show emperipolesis. Rarely, LCH and Rosai-Dorfman disease coexist in the same lymph node. Granulomatous lymphadenopathies, such as cat scratch disease sarcoidosis, and tuberculosis are different from LCH by morphology and immunohistochemistry. Lymph nodes involved by Kikuchi-Fujimoto disease show a paracortical proliferation of histiocytes, with scattered “crescentic histiocytes”, plasmacytoid dendritic cells (CD123 positive), centered by apoptotic cells and necrosis without neutrophils in more advanced cases. The presence of cells with features of LC associated with the expression of selected immunohistochemical markers allow the diagnosis of LCH also on cytological samples. Phulware et al. 32 described 47 cases of LCH diagnosed on cytological material and fine-needle aspiration (FNA). The site of FNA was the lymph node in 29 cases.
In conclusion, we describe an unusual case of suspected fibroadenoma by breast ultrasound, in a 18-year-old woman, which on lumpectomy was a LCH involving an intra-mammary lymph node.
Figures and tables
References
- 1.Barres ML, Merad M, Allen CE. Progress in understanding yhe pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X? Br J Haematol 2015;169:3-13. https://doi.org/10.1111/bjh.13247 10.1111/bjh.13247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, et al. Langerhans’ – cell histiocytosis (histiocytosis X) – a clonal proliferative disease. N Engl J Med 1994;331:154-60. https://doi.org/10.1056/NEJM199407213310303 10.1056/NEJM199407213310303 [DOI] [PubMed] [Google Scholar]
- 3.Pileri SA, Jaffe R, Facchetti F, et al. Swerdlow SH, Campo E, Lee Harris N, et al., eds. Histiocytic and dendritic cell neoplasms. WHO classification of tumours of haematopoietic and lymphoid tissues (revised 4th edition). Lyon: IARC; 2017, pp. 465-482. [Google Scholar]
- 4.Chikwava K, Langerin Jaffe R. (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disorders. Pediatr Dev Pathol 2004;7:607-14 https://doi.org/10.1007/s10024-004-3027-z 10.1007/s10024-004-3027-z [DOI] [PubMed] [Google Scholar]
- 5.Mc DR, Ziylan U, Spehner D, et al. Birbeck granules are subdomains of endosomal recycling compartment in human epidermal Langerhans cells, which form where Langerin accumulates. Mol Biol Cell 2002;13:317-35. https://doi.org/10.1091/mbc.01-06-0300 10.1091/mbc.01-06-0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sholl LM, Hornick JL, Pinkus JL, et al. Immunohistochemical analysis of langerin in langerhans cell histiocytosis and pulmonary inflammatory and infectious diseases. Am J Surg Pathol 2007;31:947-52. https://doi.org/10.1097/01.pas.0000249443.82971.bb 10.1097/01.pas.0000249443.82971.bb [DOI] [PubMed] [Google Scholar]
- 7.Thepaut M, Valladeau J, Nurisso A, et al. Structural studies of langerin and birbeck granule: a macromolecular organization model. Biochemistry 2009;48:2684-98. https://doi.org/10.1021/bi802151w 10.1021/bi802151w [DOI] [PubMed] [Google Scholar]
- 8.Willemze R, Berti E, Facchetti F, et al. Elder DE, Massi D, Scolyer RA, et al., eds. WHO classification of skin tumours. Tumour of haematopoietic and lymphoid origin. Lyon: IARC; 2018, pp. 223-290. [Google Scholar]
- 9.Facchetti F, Lonardi S, Vermi W, et al. Updates in histiocytic and dendritic cell proliferations and neoplasms. Diagn Histopath 2019;25:217-228. https://doi.org/10.1016/j.mpdhp.2019.04.001 10.1016/j.mpdhp.2019.04.001 [DOI] [Google Scholar]
- 10.Medeiros LJ, O’Malley DP, Caraway NP, et al. Tumors of the lymph nodes and spleen. AFIP Atlas of Tumor Pathology 2017; Series 4. [Google Scholar]
- 11.Shanmugam V, Griffin GK, Jacobsen ED, et al. Identification of diverse activating mutations of the RAS-MAPK pathway in histiocytic sarcoma. Mod Pathol 2019;32:830-43. https://doi.org/10.1038/s41379-018-0200-x 10.1038/s41379-018-0200-x [DOI] [PubMed] [Google Scholar]
- 12.Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med 2018;379:856-68. https://doi.org/10.1056/NEJMra1607548 10.1056/NEJMra1607548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Facchetti F, Pileri SA, Lorenzi L, et al. Histiocytic and dendritic cell neoplasms: what have we learnt by studying 67 cases. Virchows Arch 2017;471:467-89. https://doi.org/10.1007/s00428-017-2176-1 10.1007/s00428-017-2176-1 [DOI] [PubMed] [Google Scholar]
- 14.Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127:2672-81. https://doi.org/10.1182/blood-2016-01-690636 10.1182/blood-2016-01-690636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hervier B, Haroche J, Arnaud L, et al. French Histiocytoses Study Group. Association of both Langerhans cell histiocytosis and Erdheim-Chester disease linked to the BRAFV600E mutation. Blood 2014;124:1119-26. https://doi.org/10.1182/blood-2013-12-543793 10.1182/blood-2013-12-543793 [DOI] [PubMed] [Google Scholar]
- 16.Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116:1919-23. https://doi.org/10.1182/blood-2010-04-279083 10.1182/blood-2010-04-279083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov 2016;6:154-65. https://doi.org/10.1158/2159-8290.CD-15-0913 10.1158/2159-8290.CD-15-0913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer 2008;51:71-5. https://doi.org/10.1002/pbc.21498 10.1002/pbc.21498 [DOI] [PubMed] [Google Scholar]
- 19.Stalemark H, Laurencikas E, Karis J, et al. Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr Blood Cancer 2008;51:76-81. https://doi.org/10.1002/pbc.21504 10.1002/pbc.21504 [DOI] [PubMed] [Google Scholar]
- 20.Aricò M, Haupt R, Russotto VS, et al. Langerhans cell histiocytosis in two generations: a new family and review of the literature. Med Pediatr Oncol 2001;36:314-6. https://doi.org/10.1002/1096-911X(20010201)36:2<314::AID-MPO1072>3.0.CO;2-1 [DOI] [PubMed] [Google Scholar]
- 21.Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical workup, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 2013;60:175-84. https://doi.org/10.1002/pbc.24367 10.1002/pbc.24367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shanmugam V, Craig JW, Hornick JL, et al. Cyclin D1 is expressed in neoplastic cells of Langerhans cell histiocytosis but not reactive Langerhans cell proliferations. Am J Surg Pathol 2017;41:1390-6. https://doi.org/10.1097/PAS.0000000000000897 10.1097/PAS.0000000000000897 [DOI] [PubMed] [Google Scholar]
- 23.Sanders LM, Sharma P, El Madany M, et al. Clinical breast concerns in low-risk pediatric patients: practice review with proposed recommendations. Pediatr Radiol 2018;48:186-95. https://doi.org/10.1007/s00247-017-4007-6 10.1007/s00247-017-4007-6 [DOI] [PubMed] [Google Scholar]
- 24.Rodrigues C, Santos P, Simões D, Valério MO, Calhaz-Jorge C. Langerhans cell histiocytosis and breast. Acta Med Port. 2011;24 Suppl 3:713-4. [PubMed] [Google Scholar]
- 25.Castro-González E, Bastida J, Rivero-Vera JC, et al. Cutaneous and breast Langerhans cell sarcoma. J Eur Acad Dermatol Venereol 2016;30:e33-4. https://doi.org/10.1111/jdv.13282 10.1111/jdv.13282 [DOI] [PubMed] [Google Scholar]
- 26.Ahuja A, Uppe A, Nair G. Multisystem involvement of Langerhans cell histiocytosis. J Assoc Physicians India 2018;66:75-6. [PubMed] [Google Scholar]
- 27.O’Kane D, Jenkinson H, Carson J. Langerhans cell histiocytosis associated the breast carcinoma successfully treated with topical imiquimod. Clin Exp Dermatol 2009;34:e829-32. https://doi.org/10.1111/j.1365-2230.2009.03569.x 10.1111/j.1365-2230.2009.03569.x [DOI] [PubMed] [Google Scholar]
- 28.Ansari B, Purdie CA, Brown DC. Adult Langerhans cell histiocytosis mimicking Paget’s disease of the nipple. Breast J. 2005;11:281-2. https://doi.org/10.1111/j.1075-122x.2005.21464.x 10.1111/j.1075-122x.2005.21464.x [DOI] [PubMed] [Google Scholar]
- 29.Fijten GH, Blijham GH. Unexplained lymphadenopathy in family practice. An evaluation of the probability of malignant causes and the effectiveness of physicians’ workup. J Fam Pract 1988;27:373-6. [DOI] [PubMed] [Google Scholar]
- 30.Gaddey HL, Riegel AM. Unexplained lymphadenopathy: evaluation and differential diagnosis. Am Fam Physician 2016;94:896-903. [PubMed] [Google Scholar]
- 31.American College of Radiology. ACR Appropriateness Criteria: neck mass/adenopathy. https://acsearch.acr.org/docs/69504/Narrative/
- 32.Phulware RH, Guleria P, Iyer VK, et al. Cytological diagnosis of Langerhans cell histiocytosis: a series of 47 cases. Cytopathology 2019;30:413-8. https://doi.org/10.1111/cyt.12709 10.1111/cyt.12709 [DOI] [PubMed] [Google Scholar]