

Abstract
Mason‐Pfizer monkey virus protease (PR) was crystallized in complex with two pepstatin‐based inhibitors in P1 space group. In both crystal structures, the extended flap loops that lock the inhibitor/substrate over the active site, are visible in the electron density either completely or with only small gaps, providing the first observation of the conformation of the flap loops in dimeric complex form of this retropepsin. The H‐bond network in the active site (with D26N mutation) differs from that reported for the P21 crystal structures and is similar to a rarely occurring system in HIV‐1 PR.
Keywords: active site architecture, aspartic protease, dimer, flap structure, inhibitor, Mason‐Pfizer monkey virus, M‐PMV, retropepsin, retrovirus
Short abstract
1. INTRODUCTION
Retroviral proteases (PR) (retropepsins) function as C 2 homodimers 1 with the active site composed of two DTG triads, one from each subunit. The Asp side chains serve the catalytic role, and are positioned in a nearly coplanar orientation. 2 The orientation is preserved in a common D → N mutation, which suppresses the autoproteolytic activity. Mason‐Pfizer monkey virus (M‐PMV) PR is unique among retropepsins with a shift of the dimerization equilibrium toward the monomeric state, 3 as confirmed by a crystallographic study 4 , 5 and different active site conformation. 6 In all described M‐PMV PR structures (PDB IDs 6s1v, 6s1w, 6s1u, 3sqf) the active site is mutated. The side chains of the Asn26 residues are moved apart and twisted, forming additional H‐bonds with water molecules that are further coordinated by main‐chain carbonyls. Such a H‐bond system can only be found in two HIV‐1 PR models (PDB IDs 5kr1 7 and 1n49 * 8 ), albeit in only one subunit per dimer. An additional non‐catalytic water molecule within the M‐PMV PR active site bridges the Asn26 Oδ1 and Gly28 N atoms. Another distinguishing feature is the structure of the flap loops, which in dimeric retropepsin‐inhibitor complexes lock over the active site, but in M‐PMV PR show different conformational behavior. In the previously reported dimeric M‐PMV PR structures 6 the flaps were disordered with fragments not defined by the electron density, and the disorder was coupled with poor definition of the inhibitor molecules. In the apo form one flap is fully defined and uniquely curled into the active site while the other is entirely disordered.
Here, we report the first crystal structures of M‐PMV PR dimer with mostly well‐ordered flaps and clearly defined inhibitors. The enzyme has a new active site conformation, similar to a rarely seen HIV‐1 PR state.
2. RESULTS AND DISCUSSION
2.1. Overall conformation
The M‐PMV PR complexes with inhibitors 1 and 2, hereinafter 3M1 and 3M2, respectively, crystallized isomorphously in P1 space group (Table I). The asymmetric unit contains two homodimeric proteins (A/B and C/D) with inhibitors (chains F and G, respectively) locked in the active sites (Figure 1). The overall protein conformation agrees with the template retropepsin fold 1 and is similar to that of the dimers crystallized in space group P21. 6 An important difference is the stabilization of flap loops of chains B and D, and the concomitant stabilization of the inhibitors. There is also a different active‐site H‐bond network. The differences between subunits are correlated with the intrinsic asymmetry of the inhibitors. In each enzyme, the N‐terminus of the inhibitor interacts with chain B or D (hereinafter N‐subunits, Figure 1), while the C‐terminus of the inhibitor (with higher B‐factors) has only sparse contacts with chain A or C (C‐subunits). This subunit differentiation is supported by rmsd analysis (Table SI).
TABLE I.
Data collection and structure refinement statistics
| Structure | 3M1 | 3M2 |
|---|---|---|
| Data collection | ||
| Space group | P1 | P1 |
| Cell dimensions | ||
| a/b/c (Å) | 29.07/67.89/69.74 | 29.30/67.62/69.72 |
| α/β/γ (°) | 77.1/83.3/83.2 | 76.8/83.9/83.6 |
| Temperature (K) | 100 | 100 |
| X‐ray source | DESY X13 | BESSY BL14.2 |
| Wavelength (Å) | 0.81500 | 0.82657 |
| Resolution (Å) | 42.87–2.43 (2.58–2.43) a | 32.42–1.93 (2.05–1.93) |
| R int b | 0.062 (0.363) | 0.070 (0.637) |
| R meas c | 0.071 (0.421) | 0.095 (0.860) |
| <I/σI> | 15.51 (3.83) | 9.40 (1.49) |
| CC1/2 | 99.9 (93.1) | 99.7 (55.5) |
| Reflections | ||
| Measured | 75,102 (11,724) | 83,558 (13,381) |
| Unique | 19,062 (3,023) | 37,694 (6,043) |
| Multiplicity | 3.94 (3.79) | 2.22 (2.21) |
| Completeness (%) | 98.8 (97.7) | 97.0 (96.3) |
| Wilson B‐factor (Å2) | 39.9 | 27.2 |
| Refinement | ||
| No. reflections, work/test set | 18,089/954 | 36,685/999 |
| R/R free d | 0.1796/0.2362 | 0.1768/0.2110 |
| Protein molecules in ASU | 4 (two dimers) | 4 (two dimers) |
| No. atoms (non‐H) | ||
| Protein | 3,426 | 3,435 |
| Inhibitor | 124 | 128 |
| Water | 170 | 309 |
| PEG/acetate | 32/0 | 32/12 |
| <B > (Å2) | ||
| Protein | 49.0 | 35.9 |
| Inhibitor | 75.5 | 37.1 |
| Water | 47.8 | 43.6 |
| PEG/acetate | 73.4/− | 53.0/67.9 |
| Rmsd from ideal | ||
| Bond lengths (Å) | 0.008 | 0.008 |
| Bond angles (°) | 1.09 | 1.11 |
| Ramachandran statistics (%) | ||
| Favored/allowed/outliers | 96/4/0 | 98/2/0 |
| PDB code | 7bgu | 7bgt |
Values in parentheses are for the highest resolution shell.
R int = ΣhklΣi|I i(hkl) – <I(hkl)>|/ΣhklΣi I i(hkl), where I i(hkl) is the ith measurement of the intensity of reflection hkl and <I(hkl)> is the mean intensity of reflection hkl.
R meas = Σhkl [n/(n‐1)]0.5 Σi|I i(hkl) – <I(hkl)>|/ΣhklΣi I i(hkl), where I i(hkl) is the ith measurement of the intensity of reflection hkl, <I(hkl)> is the mean intensity of reflection hkl, and n is the number of observations of intensity I(hkl) (redundancy).
R = Σhkl║F o│‐│F c║/ Σhkl |F o│, where F o and F c are the observed and calculated structure factors, respectively. R free was calculated analogously for ~1000 randomly selected reflections excluded from refinement.
FIGURE 1.
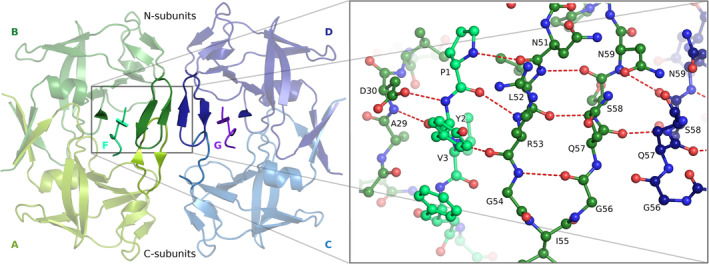
Structural elements forming the intermolecular β‐sheet in 3M2. The A/B dimer is shown in two shades of green and the inhibitor F in turquoise; the C/D dimer is shown in two shades of blue and the inhibitor G in magenta. The zoom‐in view on the right shows the H‐bond pattern
2.2. Different active site conformations
The Asn26 side chain has different conformation in the N‐/C‐subunits (Figure 2(a)). In the N‐subunits it assumes the common position found in other retropepsins, while in the C‐subunits it is shifted and twisted, as seen in all previous M‐PMV PR structures. The water molecule buried deep between the active site loops in the monoclinic structures is now absent. A similar asymmetric active site conformation is only found in two other retropepsins (Figure 2(b)), both of HIV‐1 PR with the same D25N mutation (PDB IDs 5kr1, 1n49).
FIGURE 2.
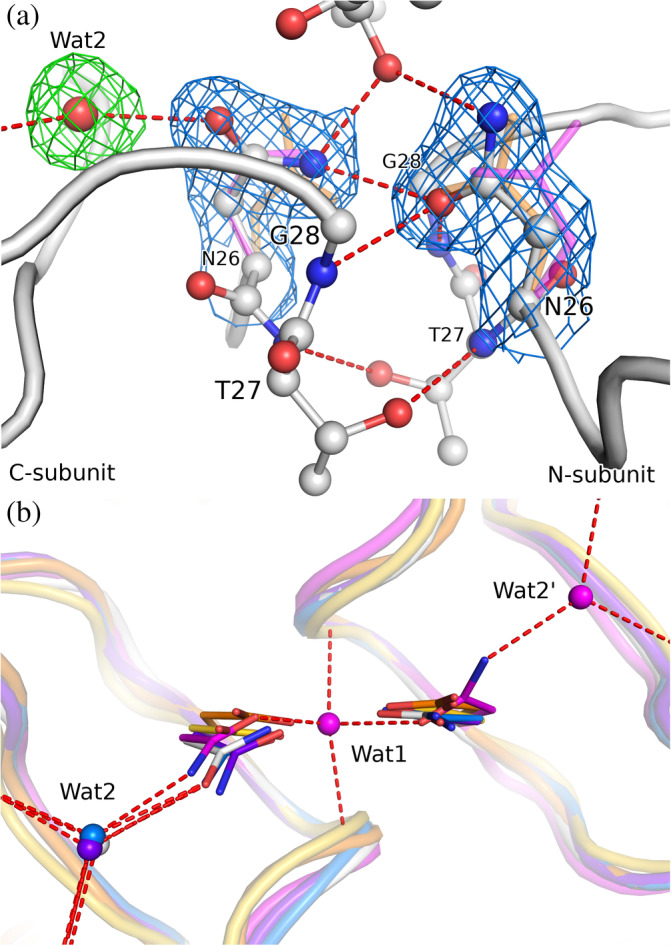
(a) The active site of 3M2 (chains A/B, white) with H‐bond network including a water molecule. The side chains of the Asn26 residues are shown in 2mF o–DF c electron density (blue) contoured at 1.0σ, the water molecule (Wat2) is shown in mF o‐DF c polder electron density (green) contoured at 3.0σ. For comparison, the Asp25 side chain of aligned HIV‐1 PR (PDB ID 4hvp, 19 orange) and Asn26 of a different (P21) crystal form of M‐PMV PR (6s1v, magenta) are shown. (b) A perpendicular view of the active site comparing the conformation of the catalytic Asp (or Asn mutation) residues, together with H‐bonded water sites. Wat1 is a water molecule buried between active site loops in the monoclinic structure 6s1v; Wat2 interacts with the main‐chain carbonyls and the catalytic Asp/Asn side chain. The superposition comprises 3M2 (white), M‐PMV PR crystallized in P21 (6s1v, magenta), HIV‐1 PR: holo (4hvp, orange) and apo form (3hvp, 20 yellow) plus two complexes with water molecules within H‐bond distance of the active site (5kr1, blue; 1n49, purple)
2.3. Well defined inhibitor
There are two dimers in each asymmetric unit; thus a total of four inhibitor molecules were modeled and verified by polder maps 9 (Figure S1, Table SII). The electron density is superior in the case of the 3M2 structure. Except for the C‐terminal residue where 1 and 2 differ in sequence and in conformation, the two inhibitors have largely the same conformation (Figure S2) and protein contacts. As an exception, Ala3 in molecule F of 3M2 is H‐bonded to a water molecule via its C=O group. In retropepsin complexes with peptidic ligands, a tetrahedral water molecule bridges the interaction between two inhibitor/substrate C=O groups and the NH amides at the tips of the flaps. 10 In the present structures, an analogous water molecule interacts with only one C=O group and one flap residue (Ile55 of chain B). The OH group of PSA that mimics the carbonyl group of a substrate is pointed toward the active site with H‐bonds to the side chains of both Asn26 residues.
2.4. Ordered flap loops
The flap loops in the present structures are significantly better defined than in all previously described dimeric complexes of M‐PMV PR. However, an asymmetry in the flap quality is still present: the flaps of the N‐subunits are better defined than in C‐subunits. This effect is enhanced by the additional interaction between the flaps of the two N‐subunits in the asymmetric unit, with the formation of an intermolecular β‐sheet (Figure 1). This secondary structure extends deeper into the protein molecule, incorporating the N‐terminus of the inhibitor and Ala29. The side chain of Asp30 additionally complements this structure. Flaps that are not involved in this interaction (with the exception of chain A of 3M2) are partially not defined in the electron density: Gly56‐Asn59 of chain C in 3M2, and Gly56‐Gln57 of chain A and Ile55‐Ser58 of chain C in 3M1. The hairpin β‐sheet within the flap is partially preserved in the C‐subunits in the case of chains A of both structures.
3. CONCLUSIONS
The 3M2 model shows the structure of M‐PMV PR in the holo form with fully defined flaps. The subunits that interact with the N‐terminus of the peptidomimetic inhibitor have superior quality of the flap region. The active‐site loops of those subunits have conformation that is typical for retropepsins. The subunits that bind the C‐terminus of the inhibitor have less ordered flaps and their active‐site loops are similar to those reported for M‐PMV PR before; however, the water molecule deeply buried between the active‐site loops is absent. The subunit asymmetry is reinforced by intermolecular β‐sheet interaction between the well‐ordered flaps. Flap asymmetry and unique inhibitor polarity were also observed in a different crystal form of M‐PMV PR and are, therefore, not crystal packing artifacts. A variety of flap conformations were also observed for HIV‐1 PR, including two holo conformations described as close and tightly close, 11 which can occur even within one crystal structure. Our results show that also the active site of holo M‐PMV PR can display different conformational states. However, they are found with very similar inhibitors or in free enzyme and may show an inactive form, especially in view of the D26N mutation, which changes the active‐site H‐bond pattern.
4. MATERIALS AND METHODS
4.1. Cloning, expression, and purification
Mutations C7A/D26N/C106A were introduced into the previously described plasmid pBPS13ATG using Quick Change Site Directed Mutagenesis Kit 12 (Agilent Technologies Inc., Santa Clara, California) and verified by DNA sequencing. Expression of C7A/D26N/C106A M‐PMV PR was performed in Escherichia coli BL21 (DE3) as described. 12 The protein was isolated from inclusion bodies by solubilization in 8 M urea and was renatured by stepwise dialysis against 50 mM Tris pH 7.0, 1 mM EDTA and 0.05% mercaptoethanol. Pure M‐PMV PR was obtained by batch ion exchange chromatography (QAE‐Sephadex A 25, Sigma‐Aldrich, St. Louis, Missouri) and was characterized by sedimentation‐equilibrium analytical ultracentrifugation. 12
4.2. Crystallization
Before crystallization, the protein at pH 7.4 (in 10 mM Tris buffer) was incubated with 1.2‐fold excess (relative to PR dimer) of the inhibitor with the sequence of Pro‐(O‐Me)Tyr‐Val‐PSA‐Ala‐Met‐Thr (1) or with 1.4‐fold excess of Pro‐Tyr‐Val‐PSA‐Ala‐Met‐His (2). (O‐Me)Tyr is Oη‐methylated tyrosine and PSA denotes (3S, 4S)‐4‐amino‐3‐hydroxy‐5‐phenylpentanoic acid. Crystallizations were set up using hanging‐drop vapor‐diffusion at 292 K by mixing 1 μl of 8.5 mg ml−1 protein solution with 1 μl reservoir solution (0.1 M sodium citrate pH 5.6, 6% PEG 8000, 5% propan‐2‐ol) for complex 1 (3M1), or by mixing 2 μl of 4.7 mg ml−1 protein solution with 1 μl reservoir solution (0.1 M imidazole pH 7.7, 0.6 M sodium acetate) for complex 2 (3M2). Single crystals appeared within 2 weeks and were harvested after 3 months.
4.3. X‐ray data collection and processing
Both crystals were cryoprotected in mother liquor supplemented with PEG 400. X‐ray diffraction data were collected at 100 K using synchrotron radiation. The diffraction data were processed and scaled using the XDS package. 13 Crystal and data collection parameters are given in Table I. Raw X‐ray diffraction images were deposited in the MX‐RDR Repository at the Interdisciplinary Centre for Mathematical and Computational Modeling of the University of Warsaw, Poland, with the following DOIs: 10.18150/C9DYSH (3M1); 10.18150/1HQGWO (3M2).
4.4. Structure solution and refinement
The crystal structures of 3M1 and 3M2 were determined by molecular replacement in Phaser 14 using the coordinates of one subunit of dimeric M‐PMV PR (PDB ID 6s1v 6 ). Structural refinements were carried out in PHENIX 15 and Coot 16 was used for modeling in electron density maps, as well as to build the inhibitor molecules and to validate the solvent structure. TLS groups (five for 3M1, six for 3M2) were generated with the TLSMD server. 17 Stereochemical restrains for the amide bonds of the PSA residue (Val‐PSA and PSA‐Ala) of the inhibitors were created using JLigand. 18 The refinement statistics are listed in Table I. The coordinate and structure factor files were deposited at the Protein Data Bank with PDB IDs: 7bgu (3M1) and 7bgt (3M2).
AUTHOR CONTRIBUTIONS
Stanislaw Wosicki: Investigation; visualization; writing‐original draft. Maciej Kazmierczyk: Investigation. Miroslaw Gilski: Data curation; investigation; software; supervision; validation; writing‐review & editing. Helena Zabranska: Investigation; writing‐review & editing. Iva Pichova: Conceptualization; funding acquisition; investigation; project administration; resources; supervision; writing‐review & editing. Mariusz Jaskolski: Conceptualization; investigation; project administration; supervision; writing‐review & editing.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Figure S1 Conformation of the inhibitor molecules F (A) and G (B) of 3M1, and F (C) and G (D) of 3M2, shown in 2mF o–DF c electron density (blue) contoured at 1.0σ and mF o–DF c polder electron density (green) contoured at 3.0σ.
Figure S2. Superposition of all inhibitor molecules: from the 3M1 structure together with dimers A/B (red) and C/D (yellow); and from the 3M2 structure together with dimers A/B (green) and C/D (purple). The non‐scissile bond is indicated by arrow.
Table SI. Rmsd values (Å) for Cα alignments of all subunits of 3M1 and 3M2 structures. Numbers of superposed Cα pairs are shown in parentheses.
Table SII. Quality of the inhibitor molecules indicated by the structure acronym and chain code as in the PDB file. The polder correlation coefficients were calculated with PHENIX. Average B‐factors (Å2) are shown for the inhibitor molecules, their N‐ and C‐terminal fragments, and the corresponding dimeric M‐PMV PR molecules (Protein).
ACKNOWLEDGMENT
Diffraction data were collected at EMBL/DESY Hamburg and at BESSY/Helmholtz‐Zentrum Berlin.
Wosicki S, Kazmierczyk M, Gilski M, Zabranska H, Pichova I, Jaskolski M. Crystal structures of inhibitor complexes of M‐PMV protease with visible flap loops. Protein Science. 2021;30:1258–1263. 10.1002/pro.4072
Funding information ASCR, Grant/Award Number: RVO 61388963
Endnote
In the previous paper 6 we incorrectly reported that the active site of 1n49 is not mutated, while in fact it contains the same D25N mutation.
Contributor Information
Miroslaw Gilski, Email: mirek@amu.edu.pl.
Iva Pichova, Email: iva.pichova@uochb.cas.cz.
Mariusz Jaskolski, Email: mariuszj@amu.edu.pl.
REFERENCES
- 1. Miller M, Jaskólski M, Rao JKM, Leis J, Wlodawer A. Crystal structure of a retroviral protease proves relationship to aspartic protease family. Nature. 1989;337:576–579. [DOI] [PubMed] [Google Scholar]
- 2. Dunn BM, Goodenow MM, Gustchina A, Wlodawer A. Retroviral proteases. Genome Biol. 2002;3:REVIEWS3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Veverka V, Bauerova H, Zabransky A, et al. Three‐dimensional structure of a monomeric form of a retroviral protease. J Mol Biol. 2003;333:771–780. [DOI] [PubMed] [Google Scholar]
- 4. Khatib F, DiMaio F, FoldIt Contenders Group , et al. Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat Struct Mol Biol. 2011;18:1175–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gilski M, Kazmierczyk M, Krzywda S, et al. High‐resolution structure of a retroviral protease folded as a monomer. Acta Cryst D. 2011;67:907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wosicki S, Gilski M, Zabranska H, Pichova I, Jaskolski M. Comparison of a retroviral protease in monomeric and dimeric states. Acta Cryst D. 2019;75:904–917. [DOI] [PubMed] [Google Scholar]
- 7. Liu Z, Huang X, Hu L, et al. Effects of hinge‐region natural polymorphisms on human immunodeficiency virus‐type 1 protease structure, dynamics, and drug pressure evolution. J Biol Chem. 2016;291:22741–22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prabu‐Jeyabalan M, Nalivaika EA, King NM, Schiffer CA. Viability of a drug‐resistant human immunodeficiency virus type 1 protease variant: structural insights for better antiviral therapy. J Virol. 2003;77:1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liebschner D, Afonine PV, Moriarty NW, et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Cryst D. 2017;73:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jaskolski M, Tomasselli AG, Sawyer TK, et al. Structure at 2.5‐.ANG. Resolution of chemically synthesized human immunodeficiency virus type 1 protease complexed with a hydroxyethylene‐based inhibitor. Biochemistry. 1991;30:1600–1609. [DOI] [PubMed] [Google Scholar]
- 11. Palese LL. Conformations of the HIV‐1 protease: a crystal structure data set analysis. Biochim Biophys Acta Proteins Proteom. 2017;1865:1416–1422. [DOI] [PubMed] [Google Scholar]
- 12. Zabranska H, Tuma R, Kluh I, et al. The role of the S‐S bridge in retroviral protease function and virion maturation. J Mol Biol. 2007;365:1493–1504. [DOI] [PubMed] [Google Scholar]
- 13. Kabsch W. XDS. XDS Acta Cryst D. 2010;66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liebschner D, Afonine PV, Baker ML, et al. Macromolecular structure determination using X‐rays, neutrons and electrons: recent developments in Phenix. Acta Cryst D. 2019;75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Cryst D. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Painter J, Merritt EA. TLSMD web server for the generation of multi‐group TLS models. J Appl Cryst. 2006;39:109–111. [Google Scholar]
- 18. Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN. JLigand: a graphical tool for the CCP4 template‐restraint library. Acta Cryst D. 2012;68:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller M, Schneider J, Sathyanarayana BK, et al. Structure of complex of synthetic HIV‐1 protease with a substrate‐based inhibitor at 2.3 a resolution. Science. 1989;246:1149–1152. [DOI] [PubMed] [Google Scholar]
- 20. Wlodawer A, Miller M, Jaskolski M, et al. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV‐1 protease. Science. 1989;245:616–621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Conformation of the inhibitor molecules F (A) and G (B) of 3M1, and F (C) and G (D) of 3M2, shown in 2mF o–DF c electron density (blue) contoured at 1.0σ and mF o–DF c polder electron density (green) contoured at 3.0σ.
Figure S2. Superposition of all inhibitor molecules: from the 3M1 structure together with dimers A/B (red) and C/D (yellow); and from the 3M2 structure together with dimers A/B (green) and C/D (purple). The non‐scissile bond is indicated by arrow.
Table SI. Rmsd values (Å) for Cα alignments of all subunits of 3M1 and 3M2 structures. Numbers of superposed Cα pairs are shown in parentheses.
Table SII. Quality of the inhibitor molecules indicated by the structure acronym and chain code as in the PDB file. The polder correlation coefficients were calculated with PHENIX. Average B‐factors (Å2) are shown for the inhibitor molecules, their N‐ and C‐terminal fragments, and the corresponding dimeric M‐PMV PR molecules (Protein).