

Abstract
Maltose binding protein (MBP) is used in recombinant protein expression as an affinity and solubility tag. The monoclonal antibody B48 binds MBP tightly and has no cross‐reactivity to other proteins in an Escherichia coli lysate. This high level of specificity suggested that MBP contains an epitope that could prove useful as a purification and visualization tag for proteins expressed in E. coli. To discover the MBP epitope, a co‐crystal structure was determined for MBP bound to its antibody and four amino acids of MBP were identified as critical for the binding interaction. Fusions of various fragments of MBP to the glutathione S‐transferase protein were engineered in order to identify the smallest fragment still recognized by the α‐MBP antibody. Stabilization of the epitope via mutational engineering resulted in a minimized 14 amino‐acid tag.
Keywords: affinity purification, epitope, maltose binding protein, MBP
Short abstract
PDB Code(s): 7JTR;
1. INTRODUCTION
Maltose binding protein (MBP), encoded by the malE gene in Escherichia coli, is a 44‐kD monomeric periplasmic protein. Discovered in 1974, 1 MBP is a component of the maltodextrin‐binding ABC transporter. The structure of MBP reveals two globular domains, separated by a groove containing the maltodextrin binding site. 2 When E. coli is grown on maltose as the sole carbon source, MBP is expressed at high levels, 3 as it binds tightly to maltodextrin and facilitates its import via the MalFGK2 transporter. 4 This tight ligand‐binding feature of MBP allowed for its development as a protein purification tag. 5 Amino‐terminal translational fusions of MBP to a target protein have several advantages: (1) increased level of expression, (2) ease of purification, and (3) increased solubility. For these reasons, MBP has been developed as a kit to purify and characterize proteins expressed in periplasmic and cytoplasmic compartments of E. coli. One component of the kit is the antibody B48, a monoclonal IgG directed against MBP that is produced from mouse hybridomas cell lines. 6 Translational fusions of MBP to target proteins can be readily detected in western blots of E. coli lysates using the B48 antibody (here in referred to as α‐MBP‐IgG). Unexpectedly, the α‐MBP‐IgG was found to be highly specific to MBP with little to no cross reactivity to the E. coli proteome. This specificity could therefore potentially be exploited to develop an affinity tag to detect proteins, especially in context of recombinant proteins expressed in E. coli.
In this article, we describe the crystal structure of MBP in complex with an scFv version of α‐MBP‐IgG. This structure has allowed us to identify four amino acids (EKDT) that are primarily responsible for the recognition of MBP by the α‐MBP‐IgG. We have used truncational studies and helix‐stabilizing mutants in order to successfully minimize the binding epitope to a 14 amino‐acid long peptide centered on these four key residues. Herein, we demonstrate the utility of this minimal epitope as a novel protein‐detection and affinity‐purification tag. This novel tag should be of use to the research community by expanding the protein detection and purification tools available.
2. RESULTS
2.1. α‐MBP antibody has low cross‐reactivity to E. coli proteome
Proteins fused to MBP can be detected using the α‐MBP‐IgG, produced from hybridoma cells. This antibody recognizes MBP with high affinity (K D = 10 nM; see Figure S1). In order to compare MBP as a potential epitope tag to other commercially available tags, the model protein glutathione S‐transferase (GST) was tagged with various epitopes (GST‐His, GST‐Myc, GST‐HA, GST‐Flag) and their affinity and specificity of recognition were compared to that of the GST‐MBP fusion. Soluble E. coli lysates expressing GST with various tags were probed in a western blot with antibodies against the tags. Western blot detection of GST using α‐MBP‐IgG compared favorably to the other established tags (Figure 1). This prompted us to search and identify the specific epitope in MBP and investigate its properties as a novel protein affinity tag.
FIGURE 1.

Comparison of MBP to other commercially available affinity tags. GST fusions to MBP, His, Flag, Myc, and HA tag were expressed in E. coli and soluble lysates were serially diluted (lanes 1–10), separated by size in SDS‐PAGE and analyzed by western blots using epitope specific antibodies (a) or by coomassie staining to show the amount of protein loaded (b). Minimum dilution necessary to specifically detect GST fusion in western blots and the corresponding total protein lysate is indicated by boxes. Protein ladder is labeled (M). GST, glutathione S‐transferase; MBP, maltose binding protein
2.2. Truncation and mutation studies to discover the MBP epitope
In order to identify the amino acids that code for the MBP epitope, various deletions of MBP were constructed and the presence of the epitope was evaluated in a western blot using α‐MBP‐IgG antibody. The epitope was not detected in amino‐terminally truncated MBP, strongly indicating that the epitope is present in the protein's first 39 amino acids. Expression of the 1–39 region alone did not result in the detection of a functional epitope, but we reasoned that this might reflect the difficulties associated with expressing and folding small peptides. Therefore, we fused the same fragment to the C‐terminus of GST, which allowed the epitope to be detected with the α‐MBP antibody (Figure S2). Interestingly, when this same fragment was fused N‐terminally to a truncated paramyocin polypeptide from Dirofilaria immitis (aka paramyosin ΔSal), the fusion was not expressed well, suggesting that at least in some cases, the 1–39 fragment can hinder translation when fused at the amino terminus. Further deletions of the 1–39 fragment in GST fusions did not provide additional insights into the location of the epitope (data not shown).
Careful inspection of the 1–39 region in the MBP crystal structure 7 revealed a short loop (26–39) protruding into the solution, which might harbor a surface‐exposed epitope. To further probe this region and identify the exact location of the epitope, alanine scanning of the 26–39 region was conducted. Amino acids in the 26–39 region were mutated to alanine, with the hope that mutations within the epitope would result in decreased signals in a western blot. The signal was markedly reduced by the F27A mutation, but because the F27 side chain is nestled in a hydrophobic depression on the protein surface, this effect likely represents perturbation of the protein structure. None of the remaining mutations, which all involve more surface‐exposed residues, had a strong impact upon binding (data not shown).
2.3. Identification of the MBP epitope using the crystal structure of the MBP:scFv complex
Attempts were made to use phage display to identify a peptide that would bind the monoclonal α‐MBP‐IgG, but these yielded no success (data not shown). Since efforts to discover the epitope using truncation studies stalled after identifying the 1–39 region of MBP, we decided to co‐crystallize the α‐MBP‐IgG with MBP to discover the exact amino acids involved in the MBP::IgG interaction. Initial attempts to prepare co‐crystals using commercially available IgG (NEB cat# E8032) failed, due most likely to the flexibility of the IgG hinge region. Surprisingly, the antibody proved to be extremely sensitive to reducing agents and attempts at making Fab fragments using papain resulted in the complete reduction of the α‐MBP‐IgG antibody. F(ab')2 fragments were then prepared using pepsin digestion, but no crystals were obtained from trials using MBP in complex with these fragments. Therefore, in order to circumvent the technical issues in generating Fab fragments from full length IgG, we decided to convert the full‐length IgG to a single‐chain scFv, expressing the light chain (LC) and the VH and CH1 domains of the heavy chain (HC) as a single polypeptide fused by a flexible linker.
A plasmid expressing the α‐MBP‐scFv was transformed into SHuffle cells, a genetically engineered E. coli strain capable of catalyzing disulfide bond formation in its cytoplasm. 8 The scFv was expressed and purified as described in the Methods section, resulting in yields of 1 mg/L. Intriguingly, although the α‐MBP scFv was expressed in the cytoplasm of SHuffle cells—a compartment that lacks MBP—the final purified scFv eluted bound to MBP. Presumably, during protein purification the cytoplasmic α‐MBP‐scFv bound to periplasmic MBP released during cell lysis. The final purified α‐MBP‐scFv::MBP complex was co‐crystallized and its structure determined at 2.5 Å resolution (Figure 2).
FIGURE 2.
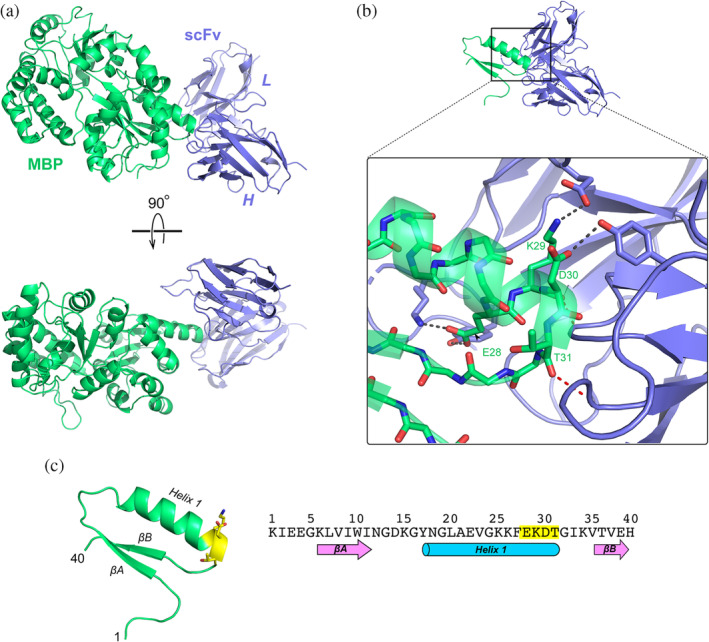
The co‐crystal structure of MBP::αMBP‐scFv reveals the principle binding epitope. (a) Crystal structure of MBP (green) in complex with αMBP‐scFv (blue), shown in two orthogonal views. (b) Close‐up view of the interface between MBP and αMBP‐scFv (this panel is shown in the same orientation as the upper view in panel (a)). The principle residues of MBP that interact with the antibody are E28, K29, D30, and T31. Key hydrogen bonds and salt bridges are shown as dashed lines. (c) Amino‐acid sequence of the first 40 residues of MBP and the structure of the corresponding region. This region includes the first two beta strands (βA and βB) and Helix 1. The epitope EKDT is highlighted in yellow. MBP, maltose binding protein
The crystal asymmetric unit contains four independent copies of the α‐MBP‐scFv::MBP complex; all four copies of the complex adopt essentially the same structure, in which the C‐terminal end of MBP's helix 1 is inserted into the cleft between the heavy‐ and light‐chain lobes of the scFv, where it packs against CDR 3 of the heavy‐chain lobe (Figure 2). Four amino acids on the MBP molecule make the majority of the interactions with the scFv, and thus comprise the main epitope for recognition: E28, K29, D30, and T31. These residues form a network of hydrogen bonds and salt bridges with the scFv. The side chain of MBP E28 forms a salt bridge with the scFv K165 sidechain, and also hydrogen bonds with the side‐chain hydroxyl of scFv Y167; both of the residues lie within CDR 1 of the LC. MBP K29 forms backbone–backbone and sidechain–sidechain hydrogen bonds with L100 and Y103, respectively, on CDR3 of the scFv, as well participating in an ionic interaction with E190 on the light‐chain CDR2. The side chain of MBP D30 is within hydrogen‐bonding distance of the side‐chain hydroxyl of scFv Y103; however, this interaction is only seen in one of the four complexes within the asymmetric unit. The backbone carbonyl of MBP T31 forms a hydrogen bond with the backbone amide of scFv Y33, on CDR 1 of the HC.
2.4. Amino terminal minimization of the MBP epitope
Previous attempts at discovering the MBP epitope by expressing short fragments of MBP alone reached a minimum size limit with the 1–39 fragment. To further minimize the 39 amino‐acid fragment, truncation fusions of 1–39 peptide to the C‐terminus of glutathione S‐transferase (GST) protein were constructed and the functional presence of the epitope was interrogated with the α‐MBP‐IgG in a western blot analysis.
Truncations of the N‐terminal 1–39 fragment to 12–39, 15–39, 16–39, and 17–39 fragments revealed a functional MBP epitope tag minimized to 23 amino acids (YNGLAEVGKKFEKDTGIKVTVEH) where the four underlined residues in the sequence correspond to the four key residues identified from the crystal structure (Figure 3(a)). However, with decreasing lengths of the peptide, the apparent binding of α‐MBP‐IgG to the N‐terminal fragments of MBP became weaker (Figure 3(c)), even though the expression levels of the peptide fusions to GST remained relatively constant (Figure 3(b)).
FIGURE 3.

N‐terminal minimization of the MBP epitope. (a) Sequence and structure of the first 40 amino acids of the MBP protein (green) fused C‐terminally to the GST (blue). Dotted arrow indicates the truncated region (Δ), where the numbered residues of truncation are underlined and indicated with dotted lines. The EKDT epitope is highlighted in yellow. Soluble extracts of E. coli expressing GST‐MBP fusions were analyzed in an SDS‐PAGE and visualized by coomassie staining (b) or in a western blot of the same gel probed with α‐MBP IgG antibody (c). M, molecular‐weight marker; MBP, full‐length MBP; and dash (−), empty vector negative control. GST, glutathione S‐transferase; MBP, maltose binding protein
2.5. Helix‐promoting mutations stabilize the MBP epitope
We reasoned that the decreased epitope recognition associated with smaller fragments resulted from a failure to fold, resulting in the conformational loss of the epitope. To compensate for loss of stabilizing interactions, several mutations were engineered into the minimal 18–39 fragment to promote solubility and helix formation (Figure 4(a)). These mutations were chosen by inspection of the MBP‐scFv crystal structure and consisted of a leucine‐to‐arginine substitution at residue 20, as this leucine is buried in the full MBP structure but exposed in the 1–39 peptide, and three helix‐promoting mutations, G19A, V23L, and G24A.
FIGURE 4.

Helix‐promoting mutations help stabilize the epitope. (a) Helix‐promoting mutations (G19A, V23L, G24A, underlined) and a hydrophilicity‐promoting mutation (L20R) were introduced (red side chains) to various peptides containing the EKDT epitope (yellow), fused to GST. Structural models of the mutant constructs are shown. Soluble cell lysates of E. coli expressing wild type (wt) or mutant (*) constructs were separated by SDS‐PAGE and visualized with Coomassie blue (b) or with α‐MBP antibody in a western blot (c), revealing the ~29 kDa fusion constructs (arrow). M, molecular‐weight marker; MBP, full‐length MBP; and dash (−), empty vector negative control. GST, glutathione S‐transferase; MBP, maltose binding protein
When additional amino‐terminal truncations were made in the 18–39 fragment with and without helix‐promoting mutations, we observed that the mutations proved critical for formation of the binding epitope. While the 18–39 fragment could form the epitope and the mutations increased its stability, the much shorter 22–39 fragment was completely dependent on the presence of the mutations for the formation of the epitope (Figure 4(b)).
2.6. Carboxy terminal minimization of the MBP epitope
To further shorten the epitope‐presenting fragment, carboxyl‐terminal truncation fusions of the 22–39* peptide (containing the helix‐promoting mutations) were constructed and fused to the C‐terminus of GST. The functional presence of the EKDT epitope was then interrogated with α‐MBP‐IgG in a western blot. The fragments constructed corresponded to the 22–37*, 22–36*, 22–35*, and 22–34* peptides. The results defined a functional MBP epitope tag minimized to a 14‐amino‐acid 22–35* fragment (ELAKKFEKDTGIKV) (Figure 5). In order to quantify the intensity of the observed protein bands, digital analysis was conducted using ImageJ and the results reflect the observed intensities (Supplementary Table S4). This minimized fragment that still displays the functional epitope was named the mm epitope, for minimal MBP epitope.
FIGURE 5.

C‐terminal minimization of the MBP epitope. (a) Sequence and structure of the 22–39 MBP peptide containing the helix‐promoting mutations V23L and G24A (red side chain and underlined) fused to GST (blue); and the EKDT epitope is highlighted in yellow. Dotted arrow indicates the residues truncated (Δ). Soluble lysates expressing various GST‐MBP fragment fusions (lanes 3–7) were separated by SDS‐PAGE and visualized either by coomassie staining (b) or by α‐MBP IgG in a western blot (c). M, molecular‐weight marker; MBP, full‐length MBP; and dash (−), empty vector negative control. The minimal MBP epitope (mm) 22–35 is indicated (lane 4). GST, glutathione S‐transferase; MBP, maltose binding protein
2.7. Use of the mm epitope as a general affinity tag
In order to demonstrate the functionality of the mm epitope as a generally useful tag, the 15‐amino acid mm tag was inserted both at the N‐ and C‐termini of GST and GFP and its functionality was interrogated with α‐MBP‐IgG in a western blot. Fusions to full‐length MBP were constructed as well to provide a comparison (Figure 6). Western‐blot analysis of soluble lysates of E. coli expressing various fusions indicate that mm tag is detected at varying levels, depending on the protein to which it is fused and on the terminus to which it is attached. The mm tag is detected at a higher level when fused at the GST N‐terminus, as opposed to the C‐terminus (Figure 6(b), lanes 5 and 6), even though the fusions are expressed at similar levels (Figure 6(a), lanes 5 and 6). In contrast, the mm tag is detected at a higher level when fused C‐terminally to GFP (Figure 6(b), lanes 7 and 8), even though the N‐terminal fusion is expressed at much higher levels (Figure 6(a), lanes 7 and 8). To confirm that this result accurately reflects the recognition of the N‐terminally‐tagged GFP by the antibody, we used isothermal titration calorimetry to measure the affinity of this interaction (Figure S4). The K D was found to be quite modest (~40 μM), which is consistent with the inefficient recognition suggested by the western blotting. Because other N‐terminally‐tagged proteins can be efficiently recognized by the antibody, these results indicate some variation in the ability of the mm tag to fold and be detected, depending both on its position and on the cargo protein to which it is fused. Since the C‐terminal fusion of full‐length MBP to GFP also results in significant decrease in expression levels (Figure 6(a), lanes 3 and 4), the reduced levels of expression are probably not due to mm, but rather reflect the properties of GFP.
FIGURE 6.

Characterization of the minimal 22–35* MBP “mm” epitope. Soluble extracts were prepared from E. coli expressing GST and GFP fusions with either the minimal 22–35* MBP peptide (mm) or full‐length MBP (the mm peptide contains the helix‐promoting mutations V23L and G24A). mm and MBP fusions were constructed at both the amino and carboxy termini of the two proteins. Proteins were separated by size using SDS‐PAGE and visualized by Coomassie staining (a) or by α‐MBP IgG in a western blot (b). Expected molecular weights: GST‐MBP (69.4 kD); GST‐mm (27.1 kD); GFP‐MBP (66.8 kD); and GFP‐mm (28.4 kD). Expected sizes of the fusions are indicated by arrows and the protein ladder is indicated by M
2.8. Affinity purification of mm‐tagged GST and GFP
To further characterize and expand the use of the mm epitope as an affinity tag for protein purification, GST tagged at its C‐terminus with mm or MBP was expressed and purified via antibody affinity. E. coli cells expressing GST‐MBP or GST‐mm fusion constructs were harvested and the fusion proteins were purified using magnetic beads conjugated to α‐MBP‐IgG (Figure 7(a)). Both fusions expressed well and at comparable levels. Similar levels of purified GST were obtained for both the MBP and mm fusions, while mm‐tagged GST was purified with fewer impurities (Figure 7(c)).
FIGURE 7.

Affinity purification of GST using MBP or mm. (a) Schematic representation of the purification of GST‐MBP and GST‐mm fusions, using αMBP‐IgG conjugated magnetic beads. (b) Predicted final structure of the mm tag (ELAKKFEKDTGIKV). To create a model, helix‐promoting mutations V23L and G24A were modeled using Coot, and images of the minimal epitope (amino acids 22–35 from the crystal structure 7JTR) were rendered using PyMol. The EKDT epitope is highlighted in yellow and the sidechains of the helix promoting mutations V23L and G24A are shown. (c) SDS‐PAGE analysis of soluble lysates expressing GST‐MBP or GST‐mm (lanes 1–2) and elution of the same lysates using αMBP‐IgG magnetic beads (lanes 3–4) were visualized by coomasssie staining. Expected molecular weights of the fusions for GST‐MBP (69.4 kD) and GST‐mm (27.1 kD) are shown. GST, glutathione S‐transferase; MBP, maltose binding protein
3. DISCUSSION
MBP has been used as an affinity protein fusion tag and can significantly improve the solubility of its fused cargo. Since the inception of the MBP protein‐purification kit in 1991, a key component has been αMBP‐IgG, used to identify and purify the MBP fusion of interest. Through empirical observations, it has been noted that the recognition of αMBP‐IgG has been very specific to MBP with little cross‐reactivity to E. coli proteome. This premise led us to discover the epitope of αMBP‐IgG and characterize its use as (1) an affinity detection tag, and (2) a purification tag. The final epitope was minimized by N‐ and C‐terminal truncations, coupled with amino‐acid substitutions that promoted helix formation, resulting in a 14‐amino acid peptide (ELAKKFEKDTGIKV) with EKDT comprising the core epitope. This final affinity tag was named mm for minimal MBP epitope.
Although the αMBP‐IgG is able to recognize both the mm tag and full‐length MBP with high efficiency, it is unclear whether the mm tag can promote the folding of its fused cargo, as MBP does. However, due to its small size (1.6 kDa vs. 40.7 kDa for mature, full‐length MBP) the mm tag should be less intrusive in structural and biochemical studies and be less likely to inhibit the enzymatic activity of its fused cargo, as has been observed on some occasions. 9
Common epitope tags such as His, Myc, HA, and Flag have been reviewed extensively. 10 , 11 The first use of epitope tagging was described by Munro and Pelham in 1984, 12 followed shortly thereafter by the introduction of the Flag tag, 13 HA tag, 14 His tag, 15 and Myc tag. 16 Since then, many additional affinity tags have been designed and developed. However, due to the complexity of modern molecular biology and the increasing demand for multiplex studies in synthetic biology, the demand for new and improved affinity tags has not diminished. The mm epitope described in this study is the latest tool to be added to the researcher's toolbox.
The mm tag is larger (1.6 kDa) and more basic (isoelectric point [pI] = 5.22) than many commonly used tags (His6: 0.84 kDa, pI = 7.21; Myc: 1.2 kDa, pI = 4.00; HA: 1.1 kDa, pI = 3.56; and Flag: 1.0 kDa, pI = 3.97). Similar to the HA and Myc tags, the mm tag was also identified from a protein epitope recognized by a monoclonal antibody, while the Flag tag contains a designed sequence, against which monoclonal antibodies were raised. Importantly, to our knowledge, none of these affinity tags were developed with their specificity against the E. coli proteome in mind. For this reason, the mm tag could be very useful for studies requiring high levels of specificity combined with low levels of non‐specific cross‐reactivity, especially in the context of recombinant protein expression in E. coli.
This study describes the initial identification of the αMBP‐IgG mm epitope and its demonstration as a protein detection and purification tag. Further engineering and characterization can be accomplished. For example, duplication of the Flag tag sequence has been demonstrated to improve affinity, and similar repetition of the mm tag may improve affinity. Also, the Flag tag was engineered to be more hydrophilic than other common epitope tags and therefore less likely to denature or inactivate proteins to which it is fused, and additional substitutions may increase the hydrophilicity of mm tag. Finally, the 22–34 fragment, which apparently does not fold properly (Figure 5(c), lane 3), can potentially be used to capture helix‐promoting events. For example, by engineering cysteines into a non‐folding mm tag, the tag would become sensitive to oxidation events that promote disulfide bonds that stabilize the correct fold. When coupled with the αMBP‐IgG, this would then become a redox‐sensitive tag.
4. MATERIALS AND METHODS
4.1. E. coli strains and plasmids
Bacterial strains and plasmids used in this work are described in Table S1 and were constructed using standard molecular and genetic techniques. 17 The primer sequences are listed in Table S2. All PCR reactions were carried out using Q5 DNA polymerase (NEB, cat#M0492), at the annealing temperatures recommended by the manufacturer's web site (http://tmcalculator.neb.com/#!/main) and extension times of 30s per kb. The results of plasmid constructions were confirmed by DNA sequencing.
4.2. Construction of α‐MBP scFv plasmid
A duplex oligonucleotide encoding the α‐MBP‐scFv was designed by linking the VH and VL regions of the α‐MBP genes in pET21b‐cyclonal‐MBP 6 with a sequence encoding a (GGGGS)x3 linker between the two. An expression plasmid for the scFv was creating by assembling the duplex oligonucleotide with pET28a cut with NcoI and NdeI using the NEBuilder HiFi Assembly kit, creating pET28a‐αMBP scFv. A second expression plasmid was constructed by assembling a PCR fragment made using primers 1 and 2 with pET28a cut with NcoI and XhoI using the NEBuilder HiFi Assembly kit, creating pET28a‐αMBP‐scFv‐His. The plasmids were used to transform SHuffle T7 Express (NEB, Cat#C3029), creating MB4852 and MB4905.
4.3. Construction of Gst‐MBP plasmids
The plasmid pGEX‐5X1‐C‐MBP 1–39 was constructed by combining a PCR fragment encoding amino acids 1–19 of MBP produced using primers 3 and 4 and pMAL‐vc5X as a template with a PCR of pGEX‐5X1 (GE Life Sciences) using primers 5 and 6 using NEBuilder HiFi DNA assembly (NEB Cat#E5520). Truncations of the resulting plasmid were made using the Q5 Site‐directed mutagenesis kit and primer 6 plus primer (7–17). Helix‐promoting and hydrophilicity mutations G19A, L20R, V23L, and G24A were added to pGEX‐5X1‐MBP 1–39 by assembling a pGEX‐5X1 PCR fragment using primers 6 and 18 with oligo 2, a duplex g‐block (IDT) using NEBuilder HiFi DNA assembly. Truncations to make pGEX‐5X1‐MBP 18–39* to pGEX‐5X1‐MBP 22–39* were made by Q5 site‐directed mutagenesis (NEB, Cat#E0554) using primers 19 plus (20–24). Truncations from the C‐terminus of pGEX‐5X1‐MBP 22–39* were made by Q5 site‐directed mutagenesis using primers 30 plus primers (25–29).
The plasmid pGEX‐5X1 N‐mm was constructed by assembling an MBP 22–35* PCR fragment made with primers 31 and 32 using pGEX‐5X1‐MBP 22–35* as a template, with a pGEX‐5X1 PCR fragment made with primers 33 and 34 by NEBuilder HiFi assembly. The plasmid pGEX‐5X1‐N‐MBP was constructed by assembling a malE PCR fragment made with primers 36 and 37 using MG1655 genomic DNA as a template, with a pGEX‐5X1 PCR fragment made with primers 33 and 35, by NEBuilder HiFi assembly. The plasmid pGEX‐5X1‐C‐MBP was constructed by assembling pGEX‐5X1 cut with BamHI and EagI with a malE PCR fragment made with primers 38 and 39, by NEBuilder HiFi assembly. All primers used in constructing plasmids are listed in the Table S2.
4.4. Construction of sfGFP‐MBP plasmids
All the pDSW204‐sfGFP plasmids were constructed by assembling DNA fragments using NEBuilder HiFi assembly. The plasmid pDSW204‐mm‐sfGFP was assembled from three fragments: a pDSW204 PCR fragment made with primers 40 and 41, oligonucleotide 2, a duplex oligo g‐block of malE 22–35*, and a sfGFP PCR fragment made with primers 42 and 43 using pDHL584 as a template. The plasmid pDSW204‐sfGFP‐mm was assembled from three fragments: pDSW204 cut with NcoI and XbaI, a sfGFP PCR fragment made with primers 44 and 45 using pDHL584 as a template, and a malE 22–35* PCR fragment made with primers 46 and 47 using pGEX‐5X1‐MBP 22–35* as a template. The pDSW204‐MBP‐sfGFP plasmid was assembled from three fragments: pDSW204 cut with NcoI and XbaI, a full‐length malE PCR fragment made using primers 48 and 49 using MG1655 genomic DNA as a template, and a sfGFP fragment made using primers 50 and 51 and pDHL584 as a template. The pDSW204‐sfGFP‐MBP plasmid was assembled from three fragments: pDSW204 cut with NcoI and XbaI, a sfGFP PCR fragment made with primers 52 and 53 using pDHL584 as a template, and a full‐length malE PCR fragment made using primers 54 and 55 using MG1655 genomic DNA as a template.
4.5. Construction of an alternate mmGFP expression plasmid
Because the sfGFP protein with an N‐terminal mm tag expressed poorly, an alternate plasmid pETHSUL‐mm‐sfGFP was constructed by inserting the tagged sequence into the pETHSUL vector. 18 The insert was amplified from plasmid pDSW204‐mm‐sfGFP using primers 64 and 65 and inserted into the BseRI‐cleaved pETHSUL vector by NEBuilder HiFi assembly.
4.6. Construction of pGEX plasmids with other tags
The plasmids for producing Gst fused to the His, Myc, hemagglutinin (HA) and Flag tags were constructed as follows. pGEX‐5X1 was cut with BamHI and EagI. Duplex oligos of each tag were created by annealing oligos 56 and 57 (His), 58 and 59 (Myc), 60 and 61 (HA) and 62 and 63 (Flag) at 10 mM each by boiling for 5 m in 10 mM Tris, 150 mM NaCl, then letting the oligos cool slowly to room temperature in a 200 ml water bath. The duplex oligos made in this way contained 4 base 5′ overhangs complementary to the cut ends of the pGEX‐5x1. One ul of each annealed oligo was ligated to 1 ul of the cut pGEX‐5X1 (approx. 1 ng) using T4 DNA ligase (NEB, Cat#M0202), and the mixture used to transform NEB 10‐beta cells (NEB, Cat#C3019). Plasmids were prepared from the transformants and used to transform PR700.
4.7. Culture growth conditions
Cells were grown in LB (10 g/L soy peptone, 5 g/L yeast extract, 5 g/L NaCl, NaOH to pH 7.2) at 30°C in the presence of appropriate antibiotics and were induced with 1 mM isopropyl‐β‐D‐thiogalactopyranoside (IPTG).
4.8. Production and purification of α‐MBP‐scFv
Six 2 L flasks containing 1 L of LB medium +200 mg/L ampicillin were inoculated with 5 ml of an overnight culture of MB4905 grown in the same medium. The cultures were grown at 30°C shaking at 250 rpm for about 8 h. At this point, IPTG was added, the temperature was lowered to 22°C, and incubation was continued about 16 h. Cells were harvested by centrifugation at 4000 x g for 30 m, and the pellets were resuspended in 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA. The resuspended cells were frozen at −20°C, then thawed in a cold‐water bath and the six samples combined. The sample was placed in a metal beaker in an ice/water bath and sonicated 2 x for 5 min, 30 s on and 10 s off. The crude extract was clarified by centrifugation at 27,000 x g for 30 m and the supernatant decanted. The supernatant was applied to a plastic column (Biorad Econo‐Pac) packed with 2 ml Ni‐NTA resin (Qiagen Superflow #30410). The column was washed with 50 ml of 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 20 mM imidazole and eluted with 10 ml of 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 250 mM imidazole.
A 2 ml amylose column was prepared in a plastic column (Bio‐Rad Poly‐Prep) and equilibrated with 40 ml of 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA. The eluate from the Ni‐NTA column was applied, washed with 100 ml of 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA and eluted with 10 ml of 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% maltose.
4.9. Protein purification
GST‐MBP fusion constructs were purified using glutathione magnetic agarose beads following the reagents protocol (Cat#. Pierce 78,601). The mm‐sfGFP protein was purified by subtractive immobilized‐metal chromatography as described. 18
4.10. Crystallography
When purified, the scFv was found to be tightly bound to MBP, which presumably was derived from the periplasm and encountered the scFv after cell lysis. This MBP‐scFv complex (in 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, & 0.5% maltose) was concentrated to 10.9 mg/ml. Microbatch drops were set up under Al's Oil 19 using 0.5 ul of protein +0.5 ul of precipitant solution; the precipitant solution used was 0.2 M ammonium sulfate (unbuffered) in 20% (wt/vol) PEG‐4000. Trays were incubated at 4°, and crystalline rods with maximum dimensions 10 x 10 x 50 microns were observed within 4–5 days. Crystals were harvested using polyimide loops (Mitegen), dipped briefly into a cryoprotectant, and flash‐cooled in liquid nitrogen. The cryoprotectant solution was prepared by mixing three volumes of glycerol with seven volumes of precipitant solution.
Diffraction data were measured at beamline 24‐ID‐E of the Advanced Photon Source. The crystal was maintained at ca. 100 K during data collection. Data were processed using XDS, 20 and the structure was determined using Phaser. 21 Molecular replacement probes for MBP and the antibody fragment were derived from PDB entries 1ANF and 2GKI, respectively. The asymmetric unit contains two independent copies of the MBP‐scFv complex. Maltose was omitted from the probe coordinate set, but strong density was nevertheless seen for the ligand in the binding sites of both MBP molecules, providing confidence in the quality of the initial phases. The structure was refined using the PHENIX suite, 22 using torsion‐angle NCS restraints; refinement cycles were alternated with manual rebuilding in Coot. 23 Data collection and refinement statistics are given in Table S3. The final structure has been deposited in the Protein Data Bank (accession code 7JTR).
4.11. SDS‐PAGE and Western Blot
Protein samples were electrophoresed on Novex 10%–20% precast gradient gels (ThermoFisher #XP10202BOX). For western blotting, proteins were transferred on PVDF membrane using a Bio‐Rad Trans‐Blot Turbo (#1704150) and Transfer Packs (#1704157) according to the manufacturer's directions. Primary antibodies used were, from NEB, α‐MBP monoclonal antibody (#E8032, 1:10,000 dilution); from Cell Signaling Technology, HA‐Tag (6E2) mouse mAb (#2367S), Flag Tag rabbit Ab (#2368S), His‐Tag (27E8) mouse mAb (#2366S), GST (26H1) mouse mAb (#2624S), myc‐Tag (9B11) mouse mAb (#2276S). Secondary anitibodies were from Cell Signaling Technology, anti‐rabbit IgG DyLight 680 Conjugate (#5366P) and anti‐mouse IgG DyLight 680 Conjugate (#5470P). Antibodies were used at 1:1000 dilution suggested by the manufacturer.
4.12. Isothermal titration calorimetry
Purified mm‐sfGFP and α‐MBP‐IgG were dialyzed exhaustively against 25 mM sodium phosphate, pH 7.4 + 100 mM NaCl. The antibody was placed in the cell of a MicroCal ITC200 calorimeter at a concentration of 10 uM and titrated with a syringe solution of 325 uM mm‐sfGFP, at a constant temperature of 25°C.
4.13. Surface plasmon resonance interaction analysis
All binding assays were performed on a ProteOn XPR36 SPR Protein Interaction Array System (Bio‐Rad Laboratories, Hercules, CA). The instrument temperature was set at 25°C for all kinetic analyses. ProteOn GLC sensor chips were preconditioned with two short pulses (10 s) each of 50 mM NaOH, 100 mM HCl, and 0.5% sodium dodecyl sulfide. Then the system was equilibrated with PBS‐T buffer (20 mM sodium phosphate, 150 mM NaCl, and 0.005% polysorbate 20, pH 7.4). The surface of a GLC sensor chip was activated with a 1:10 dilution of a 1:1 mixture of 1‐ethyl‐3‐(3‐ dimethylaminopropyl) carbodiimide hydrochloride (0.2 M) and sulfo‐N‐hydroxysuccinimide (0.05 M). Immediately after chip activation, the anti‐MBP antibody was prepared at a concentration of 0.01 mg/L in 10 mM sodium acetate, pH 5.0 and injected across ligand flow channels for 17 s at a flow rate of 30 μl/min. Then, after unreacted protein had been washed out, excess active ester groups on the sensor surface were capped by a 5 min injection of 1 M ethanolamine HCl (pH 8.0) at a flow rate of 30 μl/min so that 150 response units (RU) of ligand was immobilized on the sensor chip surface. A reference surface was similarly created by immobilizing a non‐specific protein (IgG b12 anti HIV‐1 gp120; obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti‐HIV‐1 gp120 Monoclonal [IgG1 b12] from Dr. Dennis Burton and Carlos Barbas) and was used as a background to correct non‐specific binding. For direct binding analysis, serial dilutions of the proteins were prepared in the running buffer (PBS, 0.005% polysorbate 20, pH 7.4) and injected at a flow rate of 100 μl/min, for a 1‐min association phase, followed by up to a 10‐min dissociation phase using the “one shot kinetics” capability of the ProteOn instrument. Data were analyzed using the ProteOn Manager Software version 3.0 (BioRad). The responses from the reference flow cell were subtracted to account for the nonspecific binding and injection artifacts. The equilibrium dissociation constant (K D) for the interaction, derived from a minimum of three experiments, was calculated in ProteOn Manager Version 3.1.0.6 (Bio‐Rad, Hercules, CA), using the equilibrium analysis function.
CONFLICT OF INTEREST
Na Ke, Guoping Ren, Mehmet Berkmen, and Paul Riggs are owners of New England Biolabs Stock.
AUTHOR CONTRIBUTIONS
Marine Lénon: Data curation; writing‐review & editing. Na Ke: Data curation; writing‐review & editing. Guoping Ren: Data curation; writing‐review & editing. Megan E. Meuser: Data curation. Patrick J. Loll: Data curation; supervision; writing‐original draft; writing‐review & editing. Paul Riggs: Conceptualization; data curation; writing‐original draft; writing‐review & editing. Mehmet Berkmen: Conceptualization; formal analysis; writing‐original draft; writing‐review & editing.
Supporting information
Figure S1 SPR analysis of the interaction between MBP and the α‐MBP antibody. (a) Representative sensorgrams for the interaction between the analyte (MBP) and immobilized α‐MBP antibody. Colored lines show experimental response data for an MBP concentration series (1:5 serial dilutions, starting at 1 μM). Black lines show a global fit to the concentration series. (b) Plot of equilibrium response versus analyte concentration. (c) Table showing kinetic parameters and dissociation constant KD derived from the global ‐t shown in panel A
Figure S2 Detection of the epitope in the N‐terminal 1–39 fragment. (a) Soluble lysates of E. coli expressing the N‐terminal fragment of MBP (1–39, 28 kD). The MBP fragment was expressed as a C‐terminal fusion to GST (Lane 1, 28 kD); as an N‐terminal fusion to delta‐Sal (Lane 2, 36,457 kD); or unfused (Lane 3, 4.5 kD). Proteins were separated by SDS‐PAGE and visualized by Coomassie staining or (b) by α‐MBP IgG in a western blot. The expected GST::1–39 fusion protein at 28 kDa is indicated by an arrow
Figure S3 Alanine scanning of amino acids in region 26–39 of MBP. (a) Soluble lysates of E. coli expressing alanine substitutions mutants of MBP were separated by size in SDS‐PAGE and visualized in a western blot using MBP‐IgG and compared to wild type MBP (wt) or stained with Coomassie blue. The EKDT epitope is indicated.
Figure S4 ITC analysis of the interaction between α‐MBP‐IgG and mm‐sfGFP. The estimated KD value is 37 uM.
Table S1 Bacterial strains and plasmids utilized in this study
Table S2 Primers and oligonucleotides used in this study
Table S3 Data collection and refinement statistics
Table S4 Protein band intensity
ACKNOWLEDGEMENTS
The authors would like to thank the creative, collaborative scientific environment that the late Don Comb has built and maintained by NEB management. The authors are also grateful to Kushol Gupta for assistance with the calorimetry experiments.
Lénon M, Ke N, Ren G, et al. A useful epitope tag derived from maltose binding protein. Protein Science. 2021;30:1235–1246. 10.1002/pro.4088
REFERENCES
- 1. Kellermann O, Szmelcman S. Active transport of maltose in Escherichia coli K12. Involvement of a "periplasmic" maltose binding protein. Eur J Biochem. 1974;47:139–149. [DOI] [PubMed] [Google Scholar]
- 2. Spurlino JC, Lu GY, Quiocho FA. The 2.3‐a resolution structure of the maltose‐ or maltodextrin‐binding protein, a primary receptor of bacterial active transport and chemotaxis. J Biol Chem. 1991;266:5202–5219. [DOI] [PubMed] [Google Scholar]
- 3. Dietzel I, Kolb V, Boos W. Pole cap formation in Escherichia coli following induction of the maltose‐binding protein. Arch Microbiol. 1978;118:207–218. [DOI] [PubMed] [Google Scholar]
- 4. Davidson AL, Shuman HA, Nikaido H. Mechanism of maltose transport in Escherichia coli: transmembrane signaling by periplasmic binding proteins. Proc Natl Acad Sci U S A. 1992;89:2360–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Riggs P. Expression and purification of maltose‐binding protein fusions. Curr Protoc Mol Biol. 2001; Chapter 16:Unit16.6:1–14. [DOI] [PubMed] [Google Scholar]
- 6. Robinson MP, Ke N, Lobstein J, et al. Efficient expression of full‐length antibodies in the cytoplasm of engineered bacteria. Nat Commun. 2015;6:8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quiocho FA, Spurlino JC, Rodseth LE. Extensive features of tight oligosaccharide binding revealed in high‐resolution structures of the maltodextrin transport/chemosensory receptor. Structure. 1997;5:997–1015. [DOI] [PubMed] [Google Scholar]
- 8. Lobstein J, Emrich CA, Jeans C, Faulkner M, Riggs P, Berkmen M. Shuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microbial Cell Fact. 2012;11:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Z, Li Y, Sun X, Yuan Q. Improvement of expression level of polysaccharide lyases with new tag GAPDH in E. coli . J Biotechnol. 2016;236:159–165. [DOI] [PubMed] [Google Scholar]
- 10. Kimple ME, Brill AL, Pasker RL. Overview of affinity tags for protein purification. Curr Protoc Protein Sci. 2013;73(Unit–9.9.). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4527311/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pina AS, Batalha IL, Roque AC. Affinity tags in protein purification and peptide enrichment: an overview. Methods Mol Biol. 2014;1129:147–168. [DOI] [PubMed] [Google Scholar]
- 12. Munro S, Pelham HR. Use of peptide tagging to detect proteins expressed from cloned genes: deletion mapping functional domains of Drosophila hsp 70. EMBO J. 1984;3:3087–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hopp TP, Prickett KS, Price VL, et al. A short polypeptide marker sequence useful for recombinant protein identification and purification. Nat Biotechnol. 1988;6:1204–1210. [Google Scholar]
- 14. Field J, Nikawa J, Broek D, et al. Purification of a RAS‐responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol Cell Biol. 1988;8:2159–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hochuli E, Bannwarth W, Döbeli H, Gentz R, Stüber D. Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent. Nat Biotechnol. 1988;6:1321–1325. [Google Scholar]
- 16. Kolodziej PA, Young RA. Epitope tagging and protein surveillance. Methods Enzymol. 1991;194:508–519. [DOI] [PubMed] [Google Scholar]
- 17. Sambrook J, Fritsch EF, Maniatis . Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory, 1989. [Google Scholar]
- 18. Weeks SD, Drinker M, Loll PJ. Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr Purif. 2007;53:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D'Arcy A, Elmore C, Stihle M, Johnston JE. A novel approach to crystallising proteins under oil. J Crystal Growth. 1996;168:175–180. [Google Scholar]
- 20. Kabsch W. XDS. Xds Acta Cryst D. 2010;66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: A comprehensive python‐based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Cryst D. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 SPR analysis of the interaction between MBP and the α‐MBP antibody. (a) Representative sensorgrams for the interaction between the analyte (MBP) and immobilized α‐MBP antibody. Colored lines show experimental response data for an MBP concentration series (1:5 serial dilutions, starting at 1 μM). Black lines show a global fit to the concentration series. (b) Plot of equilibrium response versus analyte concentration. (c) Table showing kinetic parameters and dissociation constant KD derived from the global ‐t shown in panel A
Figure S2 Detection of the epitope in the N‐terminal 1–39 fragment. (a) Soluble lysates of E. coli expressing the N‐terminal fragment of MBP (1–39, 28 kD). The MBP fragment was expressed as a C‐terminal fusion to GST (Lane 1, 28 kD); as an N‐terminal fusion to delta‐Sal (Lane 2, 36,457 kD); or unfused (Lane 3, 4.5 kD). Proteins were separated by SDS‐PAGE and visualized by Coomassie staining or (b) by α‐MBP IgG in a western blot. The expected GST::1–39 fusion protein at 28 kDa is indicated by an arrow
Figure S3 Alanine scanning of amino acids in region 26–39 of MBP. (a) Soluble lysates of E. coli expressing alanine substitutions mutants of MBP were separated by size in SDS‐PAGE and visualized in a western blot using MBP‐IgG and compared to wild type MBP (wt) or stained with Coomassie blue. The EKDT epitope is indicated.
Figure S4 ITC analysis of the interaction between α‐MBP‐IgG and mm‐sfGFP. The estimated KD value is 37 uM.
Table S1 Bacterial strains and plasmids utilized in this study
Table S2 Primers and oligonucleotides used in this study
Table S3 Data collection and refinement statistics
Table S4 Protein band intensity