

Abstract
Histones are highly posttranslationally modified proteins that regulate gene expression by modulating chromatin structure and function. Acetylation and methylation are the most abundant histone modifications, with methylation occurring on lysine (mono-, di-, and trimethylation) and arginine (mono- and dimethylation) predominately on histones H3 and H4. In addition, arginine dimethylation can occur either symmetrically (SDMA) or asymmetrically (ADMA) conferring different biological functions. Despite the importance of histone methylation on gene regulation, characterization and quantitation of this modification have proven to be quite challenging. Great advances have been made in the analysis of histone modification using both bottom-up and top-down mass spectrometry (MS). However, MS-based analysis of histone posttranslational modifications (PTMs) is still problematic, due both to the basic nature of the histone N-terminal tails and to the combinatorial complexity of the histone PTMs. In this report, we describe a simplified MS-based platform for histone methylation analysis. The strategy uses chemical acetylation with d0-acetic anhydride to collapse all the differently acetylated histone forms into one form, greatly reducing the complexity of the peptide mixture and improving sensitivity for the detection of methylation via summation of all the differently acetylated forms. We have used this strategy for the robust identification and relative quantitation of H4R3 methylation, for which stoichiometry and symmetry status were determined, providing an antibody-independent evidence that H4R3 is a substrate for both Type I and Type II PRMTs. Additionally, this approach permitted the robust detection of H4K5 monomethylation, a very low stoichiometry methylation event (0.02% methylation). In an independent example, we developed an in vitro assay to profile H3K27 methylation and applied it to an EZH2 mutant xenograft model following small-molecule inhibition of the EZH2 methyltransferase. These specific examples highlight the utility of this simplified MS-based approach to quantify histone methylation profiles.
Keywords: cancer epigenetics, mass spectrometry, histone modification, arginine methylation, lysine methylation, chemical derivatization
Abbreviations: ADMA, asymmetric dimethyl arginine; DMA, dimethyl arginine; DMSO, dimethyl sulfoxide; KMT, lysine methyltransferase; MMA, monomethyl arginine; NCE, normalized collision energy; PRM, parallel reaction monitoring; PRMT, protein arginine methyltransferase; PTM, posttranslational modification; SDMA, symmetric dimethyl arginine; XIC, extracted ion chromatogram
Graphical Abstract
Highlights
-
•
Simplification of histone complexity for analysis of lysine and arginine methylation.
-
•
Improved sensitivity for the analysis of dimethylarginine symmetry.
-
•
Accurate ratio of symmetric and asymmetric H4R3 dimethylarginine in cancer cells.
-
•
Catalog of accessible histone methyl marks to facilitate assay development.
In Brief
A simple derivatization strategy that uses chemical acetylation with d0-acetic anhydride to collapse all the differently acetylated histone forms into one, greatly reduces complexity, and improves sensitivity for the detection of methyl marks. A major advantage of this approach is demonstrated by the ability to reproducibly determine the ratio of symmetric and asymmetric dimethylarginine in cancer cells. This work also demonstrates that the approach can facilitate the site-specific location and relative ratio of closely spaced methyl marks.
In eukaryotic cells, DNA is wound tightly around a core of histone proteins, H2A, H2B, H3, and H4, to form nucleosomes, the basic subunit of a dynamic polymer called chromatin. Chromatin undergoes various structural changes during the life cycle of a cell, and these changes play an important role in transcription, DNA replication, and DNA repair. More than being just spools on which the DNA is wound, the histone proteins themselves are integral to the various structures of the chromatin. The N-termini of the four core histones are highly disordered and projecting out from chromatin-like tails on the nucleosomes. The influence of histones on chromatin structure is mediated largely by the extensive range of posttranslational modifications (PTM) found on these tails (1). Acetylation, methylation, phosphorylation, and ubiquitination, among others, are found in various combinations on multiple residues primarily located along the N-terminal tails regulating interactions with DNA and providing binding sites for transcriptional machinery (2, 3). While it is undeniable that histone modifications contribute to the regulation of gene expression, a complete understanding of how this occurs is still under development (4, 5).
Among the various modifications occurring on histones, acetylation and methylation are the most common (6). Unlike acetylation, which only exists in one form, methylation can take the form of mono-, di-, or trimethylation depending on the residue and specific site. Arginine dimethylation can occur either symmetrically (SDMA) or asymmetrically (ADMA) conferring distinct biological effects (7). In the human genome, there are 24 lysine methyltransferases (KMT) and seven protein arginine methyltransferases (PRMTs) that have been demonstrated to methylate histones on at least 11 lysine and arginine residues, including H3R2, K4, R8, K9, R17, R26, K27, K36 and K79, and H4R3 and K20, as well as others (8, 9, 10). Because lysine residues can be acetylated or methylated, and because they occur quite frequently in the sequences of the histone tails, the combinatorial mixture of these two modifications alone generates enormous complexity in the number of possible isoforms that can exist for any given histone. Complicating this further is that lysine can be mono-, di-, or trimethylated and that the stoichiometry for any of these lysine modifications can vary widely. For example, total lysine acetylation at any given site on histone H4 has been reported to vary from 28 to 64%, while total lysine acetylation on histone H3 ranges from less than 1% to greater than 75% (11). In most biological contexts, total lysine methylation at H3K27 or H4K20 is greater than 90% while total lysine methylation at H3K4 is only approximately 7% (11). Arginine residues in the histone tail occur less frequently and can be both mono- and dimethylated. Methylation stoichiometry on canonical histone arginine is very low. Total methylation on H4R3 has been reported to be less than half a percent (11) and in spite of extensive profiling efforts by several groups (11, 12, 13, 14), identification of arginine methylation at any site on histone H3 or on histone H4 R17and R19 has only been accomplished using site-specific antibodies.
Mass spectrometry (MS) has become an indispensable tool for the study of protein PTMs (15). However, histones by the very nature of their amino acid sequences are problematic for most of the commonly used approaches to study PTMs by mass spectrometry. Trypsin, which enzymatically cleaves proteins after lysine (K) and arginine (R) residues, is most often used in peptide-based bottom-up MS. However, histones are highly enriched in these two basic residues and therefore trypsin digestion generates peptides that might be either too small for detection in the mass spectrometer or too hydrophilic to be recovered from RP HPLC. In addition, because the majority of PTMs are clustered along the histone tails, the complexity of the PTM landscape is concentrated over a limited number of peptides. Chemical modification has been widely used to facilitate bottom-up MS analysis of histones and to overcome some of the difficulties intrinsic to the complexity of histone PTMs. Derivatization of lysine residues restricts trypsin to cleaving only at arginine residues, generating longer peptides compatible with liquid chromatography–mass spectrometry (LC-MS). Most commonly, histones have been derivatized using propionic anhydride (16, 17, 18, 19, 20), which results in propionylation of all unmodified and monomethylated lysine residues as well as the protein N-terminus. Propionylation also increases the hydrophobicity of the peptides from the histone N-terminal tail, improving recovery from RP-HPLC. Acetylation using deuterated acetic anhydride has also been used for the bottom-up MS analysis of histone PTMs (21, 22, 23, 24). Major drawback of these strategies is that the combinatorial complexity of the histone PTM mixture remains unchanged after derivatization. This complexity makes data interpretation extremely challenging and prone to false identification of PTMs. In addition, detection of less abundant modifications, such as arginine methylation, is made more difficult by the distribution of the signal across the complexity of the accompanying lysine methylation and acetylation profile.
Here we present a strategy for the identification and quantitation of histone methylation that utilizes nondeuterated acetic anhydride to fully acetylate all free lysine residues prior to digestion with trypsin and analysis by LC-MS/MS. The result of this chemical derivatization is that in vivo acetylation is indistinguishable from in vitro acetylation, thus reducing a major source of complexity in the histone population while at the same time facilitating the use of trypsin to produce peptides compatible with LC-MS analysis. We demonstrate the utility of this strategy by identifying lysine and arginine methylation in a pool of cancer cell lines and by developing robust, quantitative profiling assays for methylation at H4R3, H4K5, and H3K27.
Experimental procedures
Experimental Design
The overall experimental design comprised a set of method development experiments to evaluate conditions necessary for efficient derivatization of nuclear cell extracts, an initial screen step building a database of accessible histone methyl marks, and finally a screen for specific methyl marks in various cancer cell lines and the development of two Tier 3 assays to evaluate the methyl state of specific histone marks in response to inhibition of protein methyl transferase enzymes. Details on biological and technical replicates for each of the Tier 3 assays are indicated in their respective sections or figures.
Cell Lines and Culture Conditions
The KARPAS-422 cell line was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ; Germany). Pfeiffer, Z-138, MDA-MB-468, and Toledo lines were obtained from the American Type Culture Collection (ATCC). KARPAS-422 and Pfeiffer cell lines were grown in RPMI-1640 medium (Gibco) with HEPES supplemented with 20% Fetal Bovine Serum (FBS; Sigma Aldrich), 1% Glutamax (Life Technologies), and 1% Sodium Pyruvate (Life Technologies) at 37 °C with 5% CO2. Toledo and MDA-MB-468 cells were cultured with RPMI-1640 medium supplemented with 10% FBS. Z-138 cells were cultured with IMDM medium (Gibco) supplemented with 10% horse serum (Gibco). Every 3–4 days, when the cell cultures reached approximately 70–90% confluence, cells were split by dilution with fresh media. Cell culture density was determined using a Vi-Cell Analyzer (Beckman Coulter). For experimental assays, cells were plated to maintain subconfluence for the duration of the assay and maintained at 37 °C with 5% CO2 for a minimum of 16 h prior to compound dosing.
Overexpression of PRMT5 and MEP50 by BacMam Infection in MDA-MB-468 Cells
BacMam viruses were engineered to transiently express either PRMT5 or MEP50 and were stored at 4 °C protected from light. The BacMam constructs expressed untagged, full-length human PRMT5 (NCBI reference NP_006100) or MEP50 (NCBI reference NP_077007) cDNA in mammalian cells using the pHTBV1mcs3 vector. A mixture of 25% by volume PRMT5 BacMam, 25% by volume MEP50 BacMam, and 50% by volume RPMI-1640 plus 10% FBS culture media was added to adherent MDA-MB-468 cells that had achieved approximately 60% confluence (seeded 24–72 h before infection depending on initial seeding density). After 7–24 h of infection, the viral suspension was aspirated, and cells were washed once with DPBS. Fresh culture media containing dimethyl sulfoxide (DMSO), 100 nM GSK3203591 (PRMT5 inhibitor), 2 μM GSK3368712 (type I PRMT inhibitor), or a combination of the inhibitors at these concentrations was added and cells were returned to a 37 °C with 5% CO2 for 2 days. Duplicate pellets for each condition were harvested by scraping followed by centrifugation, and pellets were frozen at –80 °C until processing. One pellet from each pair was used for western blot analysis to confirm expected overexpression of PRMT5 (Santa Cruz, sc-59650) and MEP50 (Cell Signaling Technologies, 2823S) as well as target engagement resulting from compound treatment if applicable (data not shown). Target engagement for PRMT5 inhibition was evaluated by western blot to confirm SDMA reduction in GSK3203591-treated samples compared with DMSO-treated samples (SDMA antibody: Cell Signaling Technologies, 13138BF, clone #D2C3D6). Target engagement for Type I PRMT inhibition was evaluated by western blot to confirm ADMA reduction in GSK3368712-treated samples compared with DMSO-treated samples (ADMA antibody: Cell Signaling Technologies, 13522S). The remaining pellet was used for nuclear extraction and methylation profiling, as described below.
Compound Dosing
EZH2 inhibitor Tazemetostat was synthesized in-house and verified for purity and identity by HPLC, LC-MS, and NMR. Liquid stocks were prepared from powder. Liquid stocks were prepared from powder in carboxymethylcellulose for mouse studies. PRMT5 inhibitor GSK3203591 and Type I PRMT inhibitor GSK3368712 were also synthesized in-house and were prepared from powder at 20 mM or 40 mM in 100% DMSO (Sigma) and were maintained at –20oC without light restriction. Compound dosing was carried out at the specified concentration for 48–72 h, after which cells were harvested by centrifugation, washed with DPBS, and cell pellets were frozen at –80 °C.
In vivo Tumor Inoculation and Treatments
All studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee at GSK (protocol AUP0690). Female beige SCID mice (CB17,B6-Prkdc<scid>Lyst<bg>/Crl) obtained from Charles River Labs (Wilmington, MA) and housed in pathogen-free conditions were injected subcutaneously in the left rear flank with 5.0 × 106 Pfeiffer cells suspended in 100% Matrigel. Compound dosing was administered orally at 10 ml/kg by individual body weight with vehicle or Tazemetostat at 30 and 80 mg/kg as indicated for 11 days. Tumors were excised, snap frozen in liquid nitrogen, and stored at –80 °C until histone extraction.
Nuclear Extraction for H3K27 and H4R3 Methylation Profiling
Frozen pellets were thawed on wet ice, and nuclear enrichment was performed using the CelLytic NuCLEAR Extraction Kit (Sigma, NXTRACT) using the hypotonic lysis buffer with added detergent as per kit instructions. The resulting nuclear pellet was resuspended in 1 ml 0.2 M sulfuric acid (H2SO4) and sonicated two times for 5 s using a Branson Sonifier 150 at 5 W. Histones were extracted overnight at 4 °C under rotation. The extracted proteins were centrifuged at 16,100g at 4 °C for 10 min, and the supernatant precipitated with 10% trichloroacetic acid for 30 min on wet ice and pelleted by centrifugation at 13,200g for 10 min at 4 °C. The pellets were washed twice with ice-cold acetone and allowed to air dry for up to 2 h before resuspending in molecular-grade H2O and freezing overnight at –80 °C. The samples were thawed on wet ice and sonicated two times for 10 s using a Branson Sonifier 150 at 5W to ensure full resuspension. Protein concentrations were determined by BCA protein assay before further analysis.
Sample Preparation for H3K27 Methylation Profiling of In vivo Tumor Samples
Histone proteins were extracted from tumor samples using the Epigentek Total Histone Extraction Kit per the manufacturers instructions. Frozen tumor was homogenized in prelysis buffer containing protease and phosphatase inhibitors, centrifuged, and resuspended in lysis buffer. After incubation, samples were centrifuged, supernatant was transferred to a new tube where balance buffer and DTT were added, and samples were stored at –80 °C. To remove any buffer prior to the acetylation step, samples were thawed on wet ice, precipitated with 10% TCA, and processed as in the above section.
Lysine Acetylation of Nuclear Acid Extracts
Cellular nuclear acid extracts (8 μg) in water (same for propionylation), were diluted to 40 μl with 50 mM NH4HCO3 pH 8.0 and treated with 40 μl of freshly prepared 25% acetic anhydride in acetonitrile at room temperature for 15 min. Samples were flash frozen and dried under reduced pressure (Speedvac). To remove potential serine, threonine, and tyrosine acetylation, dried samples were treated with 50 μl of 50% hydroxylamine and adjusted to pH 12.0 by the addition of 15 μl 30% NH4OH and incubated at room temperature for 20 min (it should be noted that hydroxylamine will remove ADP ribosylation on D/E residues). Samples were then flash frozen and dried.
Lysine Propionylation of Nuclear Acid Extracts
Lysine propionylation was carried out following the protocol described in Sidoli, Bhanu (25). Briefly, 8 μg of cellular nuclear acid extract was diluted to 40 μl with 50 mM NH4HCO3 pH 8.0 and treated with 10 μl of freshly prepared 25% propionic anhydride in acetonitrile. To re-establish pH 8.0, 8 μl of 30% NH4OH was added. Samples were vortexed and incubated at room temperature for 15 min. Following incubation, samples were flash frozen and dried. Dried samples were reconstituted in 40 μl 50 mM NH4HCO3 pH 8.0, and a second propionylation reaction was performed as described above. Following the second propionylation reaction, samples were flash frozen and dried.
Trypsin Digestion of Nuclear Extracts
Dried acetylated or propionylated nuclear extracts were reconstituted in 8 μl in 50 mM NH4HCO3 pH 8.0 (1 μg/μl). Trypsin (Promega) was reconstituted in 50 mM NH4HCO3 pH 8.0 at 0.1 μg/μl and 800 ng added to each sample for a 1:10 E:S ratio (w/w). The digestion was incubated for 3 h at 37 °C and stopped by the addition of 1.6 μl 1% formic acid, 0.5% TFA.
Sep-Pak C18 Desalting
Digested samples were desalted using a Sep-Pak tC18 μElution Plate (Waters) following the manufacturer's instructions. After elution in 60% acetonitrile, 0.1% TFA, samples were dried down and reconstituted in 50 μl 0.1% formic acid, 0.05% TFA. Samples were briefly sonicated, vortexed, centrifuged, and transferred to autosampler vials for LC-MS/MS analysis.
Mass Spectrometry
Tryptic peptides from acetylated or propionylated nuclear preparations (1/20 of each sample corresponding to 0.4 μg) were injected on an Easy-nLC 1000 UHPLC system (Thermo Scientific). The nanoLC was interfaced to a Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific). Tryptic peptides were loaded on a 2 cm × 75 μm Acclaim PepMap 100 C18 trapping column (Thermo Scientific) and separated on a 25 cm × 75 μm, PepMap C18, 2 μm particle column (Thermo Scientific) using a 60 min gradient of 2–30% acetonitrile, 0.2% formic acid and a flow of 300 nl/min. LC-MS/MS-based peptide sequencing was performed by data-dependent analysis (DDA) (Full MS 400–2000 Da at 70,000 resolution, MS AGC target 1e6, MS Maximum IT 200 ms, followed by MS/MS top ten HCD fragmentation, normalized CE 30, Isolation window 1.2 m/z, fixed first mass 145 m/z, 17,500 resolution, MS/MS AGC target 5e4, and MS/MS Maximum IT 200 ms). Alternatively, samples were analyzed using Parallel Reaction Monitoring (PRM) methods targeting potentially methylated histone peptides (MS/MS stepped normalized CE 28, 31, and 34, Isolation window 1.2 m/z, fixed first mass 145 m/z, 17,500 resolution, AGC target 2e5, and Maximum IT 200 ms).
Data Analysis
Peak lists were generated from uninterpreted tandem MS spectra using either Mascot Distiller (2.7.1.0) or Mascot Daemon (2.6.0) and searched for peptide matches using Mascot (2.6.0) (26) against a database of histone sequences extracted from the UniProt_Human_2014_.03 database with a 5 ppm mass tolerance for peptide precursors and 20 mDa mass tolerance for fragment ions. For acetylated samples, the following modifications were selected: acetylation on Lys and on the protein N-terminus as fixed modifications; methylation on Arg, dimethylation on Lys and Arg, and trimethylation on Lys as variable modifications. Methylation on Lys was taken into account by selecting methyl+acetyl=propionylation (indicating acetylation on a monomethylated Lys) as a variable modification. For propionylated samples, the following modifications were selected as variable: acetylation on Lys and on the protein N-termini; propionylation on Lys and on the protein N-termini; methylation on Arg, dimethylation on Lys and Arg, and trimethylation on Lys. Monomethylation on Lys was taken into account by selecting butyrylation (indicating propionylation on a monomethylated Lys) as variable modification.
Histone methylated peptides preliminarily identified by data-dependent LC-MS/MS analysis were targeted by PRM analysis to confirm the site of methylation and to identify all isoforms corresponding to a specific modified peptide. PRM data were searched using the settings described above. To minimize erroneous isoform identifications, Mascot results were validated using the following rules: only identification with Mascot identification score >30 and identification with more than two PSM were considered. In cases where two charge states for the same peptide were targeted, only results relative to the charge state that resulted in the highest Mascot ID score were considered. All possible isobaric modifications or localization isoforms within a Mascot Delta score >8 (27) were manually validated.
Relative quantification of histone modifications was determined, as commonly reported, by measuring the peak area of the precursor corresponding to a specific modified peptide versus the sum of the peak areas corresponding to all other observed modified and unmodified forms of the peptide (25, 28).
Preparation of Histone H4 Synthetic Peptides and Measurement of MS Response Factors
In total, 1.4 nmol each of the synthetic histone H4 peptides (supplemental Fig. S1) (AnaSpec) was diluted to 70 μM in 100 mM NH4HCO3 pH 8.0 and treated with 20 μl of freshly prepared 25% acetic anhydride in acetonitrile. Samples were vortexed and incubated at 37 °C for 15 min, after which they were flash frozen and dried. Acetylated peptides were digested using 20 μl trypsin in 50 mM NH4HCO3 pH 8.0 at 0.1 μg/μl for 3 h at 37 °C. Digested synthetic peptides were desalted by ZipTip C18 and eluted in 15 μl 60% acetonitrile, 0.1% TFA. Samples were flash frozen, dried, reconstituted in 40 μl 0.1% formic acid, 0.05% TFA, and transferred to autosampler vials for LC-MS/MS analysis.
To determine the response factors for the various methylated forms, a 1:1:0.5:0.5 mixture of the synthetic H4 peptides was generated by combining 10 nmol each of the unmethylated and monomethylated with 5 nmol each of both the asymmetrically and symmetrically dimethylated peptide standards. The mixture was acetylated and digested with trypsin as described above. A ten-point fivefold serial dilution was generated from 10 pmol/μl to 5.12 amol/μl in a 50 ng/μl HeLa digest background. Samples were injected on an Easy-nLC 1000 UHPLC system (Thermo Scientific). The nanoLC was interfaced to a Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific). Tryptic peptides were loaded on a 2 cm × 75 μm Acclaim PepMap 100 C18 trapping column (Thermo Scientific) and separated on a 25 cm × 75 μm, PepMap C18, 2 μm particle column (Thermo Scientific) using a 60 min gradient of 2–30% acetonitrile, 0.2% formic acid, and a flow of 300 nl/min. Full-scan MS data 400–1800 Da at 35,000 resolution, MS AGC target 3e6, MS Maximum IT 200 ms were acquired and XICs for peptides H4R3 (4–17) 0 MA (m/z 719.9099), (1–17) MMA (m/z 599.0023 [3+], and 897.9989 [2+]) and (1–17) DMA (m/z 603.6736 [3+] and 905.0070 [2+]) were used to calculated the relative MS response.
Optimization of Diagnostic Fragment Ion Production for Histone H4R3 ADMA and SDMA
Acetylated synthetic histone H4R3ADMA (1–17) and H4R3SDMA (1–17) (10 μM in 0.1% formic acid, 0.05% TFA) were brought to 16% CH3CN and infused at 300 nl/min into a Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific). Ionization was achieved using a Nanospray Flex ion source (Thermo Scientific) fitted with a stainless steel nano-bore emitter (Thermo Scientific) with a positive Spray Voltage of 2400V. MS-based peptide sequencing data were acquired using a PRM method targeting the triply charged H4R3DMA (1–17) precursor ion at 603.6746 and ramping the normalized collision energy (NCE) from 20 to 60 using steps of two every 6 s.
Results and Discussion
Chemical Acetylation as a Derivatization Strategy for the Analysis of Histone Methylation
We explored the use of chemical acetylation with unlabeled acetic anhydride to mitigate the analytical challenges associated with the MS analysis of histone methylation. We reasoned that while conserving all the benefits of the propionyl chemical modifications (e.g., creating more hydrophobic, LC-MS compatible tryptic peptides), chemical acetylation with unlabeled reagent would collapse all the many in vivo acetylated forms of a given histone methyl mark into a single chemical species, simplifying data analysis and increasing the chances of detecting less abundant methyl marks via summation of the intensities of all the in vivo and in vitro fully acetylated forms.
The benefits of this strategy can be illustrated by considering arginine methylation on histone H4. The N-terminal tail of histone H4 (residues 1–20) has been reported to contain a large number of modifications (29), with acetyl and methyl being the most common. Acetylation has been reported at the N-terminus and at K5, K8, K12, K16, and K20. Methylation has been reported on R3, K5, K8, K12, K16, and K20 (6) (Fig. 1A). Data from a published top-down analysis of histone H4 identified 68 isoforms of H4 containing various combinations of these methyl and acetyl marks (14). The N-terminal tail (residues 1–20) was detected with 0–5 acetyl groups and up to four methyl groups distributed across R3 and K20 to yield a total of 54 unique methylated forms (supplemental Table S1). While this study might not be comprehensive for all the possible isoforms of H4 (known methyl marks K5me1, K8me1, K12me1, and K16me1 were not detected), it clearly highlights the level of complexity associated with a single histone protein. Using chemical acetylation, however, where all the unmodified and monomethylated lysine residues and the unmodified protein N-terminus are acetylated, all the differently in vivo acetylated forms of each methyl state would collapse into a single species (supplemental Table S1). For example, in the analysis described by Phanstiel et al. (14), monomethyl R3 (R3me1) is always detected in the presence of dimethyl K20 (K20me2) in 12 different forms that bear 0–5 acetylation groups (Fig. 1B). To measure changes in R3me, we would need to profile 12 differently modified peptides. Chemical acetylation collapses these 12 peptides into one species (Fig. 1B). Likewise, each different K20 methyl mark can be monitored using a single fully acetylated sequence (Fig. 1C). To quantitate any given H4 methyl mark identified by top-down analysis, all of the unmodified and differently modified forms of each methyl state need to be taken in consideration (25, 28). However, by using chemical acetylation, we have reduced the number of modified forms to monitor from 68 to 6 (Fig. 1C).
Fig. 1.

Chemical acetylation facilitates the analysis of histone methylation.A, histone H4 amino acid sequence. Commonly detected modifications are indicated. B, H4 (1–20) peptides containing methylation (me1 and me2) at R3 as reported by top-down MS analysis (14). Chemical acetylation collapses all the differently acetylated form into one. C, chemical acetylation collapses the 68 differently modified forms of H4 (1–20) detected in Phanstiel et al. (14) into six fully acetylated species.
Identification of Histone Methylation in Human Cancer Cell Lines
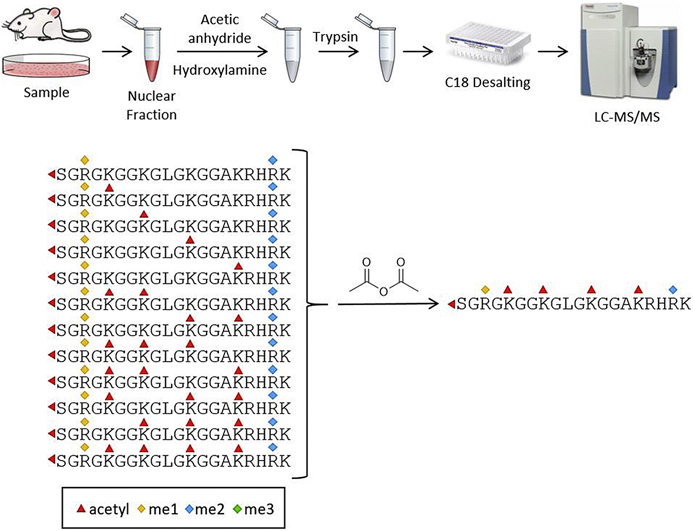
To put our strategy into practice, we developed a simplified protocol for chemical acetylation of cellular nuclear extracts using unlabeled acetic anhydride (Fig. 2). Due to the higher reactivity of acetic anhydride relative to propionic anhydride, our protocol uses a single addition of reagent and does not require pH adjustment following addition of the reagent. Under these conditions, the reaction proceeds to near completion. We monitored the completeness of the reaction by measuring the percent acetylation on four histone peptides from the nuclear extract and found that the conversion was >99.5% (Table 1). We compared the extent of acetylation to the extent of propionylation using a commonly employed propionylation protocol for histones (double reagent addition and incubation after pH adjustment) (25) and found that for these four peptides the extent of the acetylation reaction is equivalent to or better than the propionylation reaction.
Fig. 2.

Schematic workflow for the analysis of histone methylation using chemical acetylation.
Table 1.
Completeness of chemical acetylation and propionylation of histones in nuclear extracts determined by percent derivatization of various histone H3 and H4 tryptic peptides
| Peptide | Sequence | Max number of acetyl Lys | % Acetylation | % Propionylation |
|---|---|---|---|---|
| H4 (25–36) | DNIQGITKPAIR | 1 | 99.8 | 98.4 |
| H4 (80–93) | KTVTAMDVVYALKR | 2 | 99.5 | 99.9 |
| H4 (69–79) | DAVTYTEHAKR | 1 | 99.9 | 98.8 |
| H3 (74–84) | EIAQDFKTDLR | 1 | 99.7 | 96.9 |
After deacetylation of serine, threonine, and tyrosine residues using hydroxylamine, samples are digested with trypsin and analyzed by data-dependent LC-MS/MS analysis. We found that, predictably, the acetylated peptides generally eluted in front of their propionylated counterparts, but that otherwise there was no apparent difference in the chromatography of histone tryptic peptides regardless of whether acetylation or propionylation was chosen (supplemental Fig. S1).
Raw spectra are searched against a Human Histone database using Mascot. Because of the demonstrated completion of the acetylation reaction (Table 1), acetylation on lysine residues can be set as fixed modification. To account for all possible methylation events, we searched for mono-, di-, and trimethyl lysine and mono- and dimethyl arginine. Because monomethylated lysine residues are chemically acetylated, this mark was searched as a [methyl + acetyl] modification. Other than acetylation, we reasoned that any other modifications would be of such low stoichiometry as to make a negligible contribution to the quantitation of the methyl marks and could, therefore, be disregarded.
To validate our proposed strategy, we applied it to a pool of nuclear extracts prepared from equimolar aliquots of four cancer cell lines (Z-138, Toledo, HeLa, and MDA-MB-468). After chemical acetylation, the nuclear extract pool was digested with trypsin and analysed by DDA LC-MS/MS. Mascot searches using the parameters described led to the identification of 34 potentially methylated peptides from either histones H3 or H4. For the sake of simplicity, here we have not considered results from histones H1 and H2. We confirmed the methylation status of these peptides by targeted MS/MS using PRM methods. The PRM data were searched using Mascot with the results being filtered using stringent parameters (see experimental section) to minimize erroneous methyl mark identification or localization. For select peptides described below, spectra were manually validated to confirm specific site(s) of modification. We used the data from the PRM LC-MS/MS analysis of the fully acetylated nuclear extract pool to build the list of confirmed methylated peptides and methyl marks (Table 2). These data provided us with a catalog of methyl marks that were readily detectable using our protocol, the peptide sequence(s) in which they were likely to be found, and other methyl marks that occurred on the same sequence.
Table 2.
Confirmed H3 and H4 methylated peptides and methyl marks detected by bottom-up analysis of a chemically acetylated pool of nuclear extracts from Z-138, Toledo, HeLa, and MDA-MB-468 cancer cell lines
| Histone | Peptide | m/z | Charge | Peptide sequence | Methyl | Methylated residue(s) | Mascot score |
|---|---|---|---|---|---|---|---|
| H3 | 1–17 | 656.7093 | 2+ | ARTKQTARKSTGGKAPR | 2 | K4me2 | 41 |
| 9–17 | 500.2812 | 2+ | KSTGGKAPR | 1 | K9me1 | 52 | |
| 486.2839 | 2+ | KSTGGKAPR | 2 | K9me2 | 40 | ||
| 493.2941 | 2+ | KSTGGKAPR | 3 | K9me3 | 62 | ||
| 500.2812 | 2+ | KSTGGKAPR | 1 | K14me1 | 45 | ||
| 507.2901 | 2+ | KSTGGKAPR | 2 | K9me1K14me1 | 43 | ||
| 18–26 | 542.8272 | 2+ | KQLATKAAR | 1 | K18me1 | 41 | |
| 542.8272 | 2+ | KQLATKAAR | 1 | K23me1 | 48 | ||
| 73–83 | 696.3587 | 2+ | EIAQDFKTDLR | 1 | K79me1 | 88 | |
| 682.3641 | 2+ | EIAQDFKTDLR | 2 | K79me2 | 57 | ||
| H3.1 | 27–40 | 787.4420 | 2+ | KSAPATGGVKKPHR | 1 | K27me1 | 106 |
| 787.4420 | 2+ | KSAPATGGVKKPHR | 1 | K36me1 | 100 | ||
| 773.4452 | 2+ | KSAPATGGVKKPHR | 2 | K27me2 | 104 | ||
| 773.4452 | 2+ | KSAPATGGVKKPHR | 2 | K36me2 | 82 | ||
| 780.4518 | 2+ | KSAPATGGVKKPHR | 3 | K27me3 | 102 | ||
| 794.4489 | 2+ | KSAPATGGVKKPHR | 2 | K27me1K36me1 | 106 | ||
| 780.4518 | 2+ | KSAPATGGVKKPHR | 3 | K27me1K36me2 | 88 | ||
| 780.4518 | 2+ | KSAPATGGVKKPHR | 3 | K27me2K36me1 | 89 | ||
| 766.4553 | 2+ | KSAPATGGVKKPHR | 4 | K27me2K36me2 | 74 | ||
| 787.4616 | 2+ | KSAPATGGVKKPHR | 4 | K27me3K36me1 | 99 | ||
| 773.4645 | 2+ | KSAPATGGVKKPHR | 5 | K27me3K36me2 | 62 | ||
| H 3.3 | 27–40 | 795.4405 | 2+ | KSAPSTGGVKKPHR | 1 | K27me1 | 109 |
| 795.4405 | 2+ | KSAPSTGGVKKPHR | 1 | K36me1 | 109 | ||
| 781.4429 | 2+ | KSAPSTGGVKKPHR | 2 | K27me2 | 44 | ||
| 781.4429 | 2+ | KSAPSTGGVKKPHR | 2 | K36me2 | 76 | ||
| 788.4500 | 2+ | KSAPSTGGVKKPHR | 3 | K27me3 | 102 | ||
| 802.4475 | 2+ | KSAPSTGGVKKPHR | 2 | K27me1K36me1 | 109 | ||
| 788.4500 | 2+ | KSAPSTGGVKKPHR | 3 | K27me2K36me1 | 54 | ||
| 788.4500 | 2+ | KSAPSTGGVKKPHR | 3 | K27me1K36me2 | 77 | ||
| 774.4533 | 2+ | KSAPSTGGVKKPHR | 4 | K27me2K36me2 | 57 | ||
| 795.4579 | 2+ | KSAPSTGGVKKPHR | 4 | K27me3K36me | 89 | ||
| 781.4630 | 2+ | KSAPSTGGVKKPHR | 5 | K27me3K36me2 | 52 | ||
| H4 | 1–17 | 897.9998 | 2+ | SGRGKGGKGLGKGGAKR | 1 | R3me1 | 81 |
| 905.0074 | 2+ | SGRGKGGKGLGKGGAKR | 2 | R3me2 | 41 | ||
| 4–17 | 726.9166 | 2+ | GKGGKGLGKGGAKR | 1 | K5me1 | 67 | |
| 726.9166 | 2+ | GKGGKGLGKGGAKR | 1 | K8me1 | 46 | ||
| 726.9166 | 2+ | GKGGKGLGKGGAKR | 1 | K12me1 | 69 | ||
| 726.9166 | 2+ | GKGGKGLGKGGAKR | 1 | K16me1 | 51 | ||
| 20–35 | 640.7178 | 3+ | KVLRDNIQGITKPAIR | 1 | K20me1 | 51 | |
| 631.3854 | 3+ | KVLRDNIQGITKPAIR | 2 | K20me2 | 73 | ||
| 68–92 | 998.1979 | 3+ | DAVTYTEHAKRKTVTAMDVVYALKR | 3 | K77me3 | 61 |
We used these data as a foundation for the development of assays to profile specific methyl marks as described below. Relative quantitation of specific methyl marks is commonly determined by measuring the intensity of the precursor for a modified peptide relative to the sum of the intensities for all observed modified and unmodified forms of the peptide (25, 28). Quantitation of relevant methyl marks becomes increasingly challenging as more modified forms of the peptide are detected and need to be taken into consideration for quantitation. As shown in the examples below, collapsing all in vivo acetylated forms into one fully acetylated form greatly facilitates the detection and quantitation of histone methylation.
Chemical Acetylation Facilitates Quantitative Profiling of H4R3 Methylation
PRMTs catalyze the addition of methyl groups to the side chain of arginine. Arginine residues can be either mono- (MMA) or dimethylated (DMA). Histone H4R3 has been reported to be a substrate for PRMT-dependent dimethylation and functions in transcriptional regulation. Due to the apparent low stoichiometry of MMA and DMA on H4R3 (<1%) (12, 14) and the overall complexity of PTMs on H4, the detection and quantitation of H4R3 MMA and DMA levels have been extremely challenging. Most of the published data on the state of H4R3 methylation has been generated using site-specific antibodies (30, 31, 32, 33), which can suffer from cross-reactivity and epitope occlusion (34). Our data from the pooled nuclear extract showed that H4R3 could be readily detected as both MMA and DMA. To profile changes in the levels of these marks, we performed bottom-up MS analysis on nuclear extracts from Z-138 cells after chemical acetylation and digestion with trypsin. We identified two fully acetylated H4 peptides containing amino acids 1–17 (SGRGKGGKGLGKGGAKR) with either MMA or DMA on R3 (Table 3). We found no evidence of methylation at any other site on this peptide. When R3 is unmodified, trypsin will cleave at this residue generating the peptide 4-17 (GKGGKGLGKGGAKR). Here we found three 4-17 peptides, which contained 0, 1, and 2 methyl groups (Table 3). When calculating the relative abundance of H4R3 methyl forms, the 4-17 peptides must be accounted for since they represent the zero-methyl state of R3.
Table 3.
N-terminal H4 peptides from Z-138 cell nuclear extract identified by data-dependent LC-MS/MS following derivatization with either acetic or propionic anhydride
| Peptide | Sequence | Acetyl | Methyl |
|---|---|---|---|
| In vitro acetylation | |||
| 1–17 | SGRGKGGKGLGKGGAKR | 5 | 1 |
| 1–17 | SGRGKGGKGLGKGGAKR | 5 | 2 |
| 4–17 | GKGGKGLGKGGAKR | 4 | 0 |
| 4–17 | GKGGKGLGKGGAKR | 4 | 1 |
| In vitro propionylation | |||
| 1–17 | SGRGKGGKGLGKGGAKR | 4 | 0 |
| 1–17 | SGRGKGGKGLGKGGAKR | 3 | 0 |
| 1–17 | SGRGKGGKGLGKGGAKR | 3 | 1 |
| 1–17 | SGRGKGGKGLGKGGAKR | 2 | 0 |
| 1–17 | SGRGKGGKGLGKGGAKR | 2 | 1 |
| 1–17 | SGRGKGGKGLGKGGAKR | 1 | 0 |
| 1–17 | SGRGKGGKGLGKGGAKR | 1 | 1 |
| 1–17 | SGRGKGGKGLGKGGAKR | 1 | 2 |
| 1–17 | SGRGKGGKGLGKGGAKR | 0 | 0 |
| 4–17 | GKGGKGLGKGGAKR | 4 | 0 |
| 4–17 | GKGGKGLGKGGAKR | 3 | 0 |
| 4–17 | GKGGKGLGKGGAKR | 2 | 0 |
| 4–17 | GKGGKGLGKGGAKR | 1 | 0 |
| 4–17 | GKGGKGLGKGGAKR | 0 | 0 |
| 4–17 | GKGGKGLGKGGAKR | 0 | 1 |
Precursors were sequenced by targeted PRM to determine the sites of modification reported in supplemental Table S2.
To further validate our proposed strategy for the analysis of H4R3 methylation, we conducted a parallel experiment using propionylation of the Z-138 nuclear extract and compared the output of the two derivatization experiments. While propionylation successfully increases the hydrophobicity of the histone peptides and limits trypsin activity to cleave only at arginine residues, the extraordinary complexity of the peptide mixture persists. Due to the many in vivo acetylated forms, the propionylated sample yielded a total of 15 unique methyl and acetyl combinations of the 1-17 and 4-17 peptides (Table 3). Using PRM methods, we targeted the 15 different propionylated precursors and found that many of these unique combinations are, in fact, a combination of several isoforms. In total up to 21 isoforms were identified using a predefined set of stringent Mascot parameters (see Experimental section). Three of these contained monomethylation at R3 (supplemental Table S2). H4R3 DMA was not identified in the propionylated sample. To accurately quantify changes at R3, all 21 propionylated isoforms, regardless of the type of modification, would need to be validated, assayed, and their intensities summed.
After chemical acetylation, only two forms each of methylated peptides 1-17 and 4-17 were detected (Table 3). Whereas the PRM data showed that methylation on the 1-17 peptides was confined to R3 as MMA and DMA, the monomethylation on peptide 4-17 was found to be a combination of several isoforms where the single methyl group is potentially distributed over all four different lysine residues. This additional complexity, however, accounts for only ca. 0.1% of the total amount of peptide 4-17, as most of the peptide is in the unmethylated state (further characterization of the 4-17 methyl isoforms as relating to the detection of H4K5 is described below). Because the singly and doubly methylated isoforms account for so little of the total 4-17 sequence, we ignored these when determining the percent of R3 in the unmethylated state. On the contrary, the in vivo acetylated forms of the unmethylated state have been reported with stoichiometries ranging 1–30% (14, 35) and so in the case of a propionylated sample, they could not be disregarded for the purpose of determining an accurate methyl stoichiometry.
After complete chemical acetylation, the percent of MMA and DMA at R3 was calculated for several cancer cell lines using the unmethylated 4-17 peptide to represent total unmethylated R3. Cell extracts from Z-138, MDA-MB-468, and Toledo cells were prepared in triplicate and analyzed in duplicate by MS. Apparent stoichiometries were corrected for ionization efficiency differences derived from fully acetylated synthetic peptides. We determined the stoichiometry in these cells to be between 0.3 and 0.6% for MMA and 0.02 and 0.06% for DMA (Fig. 3). H4R3 DMA has been difficult to quantify in MS-based experiments and often goes undetected due to the low abundance of the mark and the large number of modified forms spanning the H4 1-17 sequence. Using our strategy, we easily identified and quantified H4R3 DMA levels down to 0.02% of the total R3. The facile analysis of the H4R3 methylation profile clearly illustrates the advantages of the chemical acetylation protocol for the identification and quantitation of low abundance methyl marks.
Fig. 3.

Quantitation of H4R3 MMA and DMA in cancer cell lines following chemical acetylation and MS analysis. Two biological replicates were analyzed for Z-138 and MDA-MB-468 cells. Errors bars constitute the standard error of six technical replicates. Note the different y axis scales for the relative stoichiometries H4R3 MMA versus DMA.
Quantitative Profiling of Histone H4R3 DMA Symmetry
Arginine dimethylation can occur either symmetrically (SDMA) where the two methyl groups are on the two different guanidino nitrogen of arginine or asymmetrically (ADMA) where both methyl groups are added to the same guanidino nitrogen (Fig. 4A). Different PRMTs catalyze the formation of the two different symmetries. Type I PRMTs such as PRMT1 catalyze ADMA, whereas type II PRMTs such as PRMT5 catalyze MMA and SDMA. These marks confer different biological effects in cells (7). For example, histone H4R3 ADMA has been described as a transcriptional activator, whereas H4R3 SDMA is classified as a transcriptional repressor (7). PRMT1 catalyzes asymmetric dimethylation of R3, and this has been correlated with a number of diseases, particularly various cancers (36). H4R3 has also been reported to be a substrate for PRMT5, which is overexpressed in a wide variety of cancers where it suppresses the expression of tumor suppressor genes and contributes to increased proliferation (37). Because of the improved sensitivity for the detection of H4R3 DMA gained through full chemical acetylation, we were able to determine the symmetry of DMA at H4R3 in different cell lines and to observe the effect of PRMT inhibitors on the nature of that symmetry.
Fig. 4.

Determination of H4R3 DMA symmetry by MS. A, diagnostic neutral loss fragment ions used to discriminate between ADMA and SDMA. B, equimolar mixture of fully acetylated synthetic H4R3 peptides 1–17 ADMA and SDMA. ADMA and SDMA are discriminated by monitoring the neutral loss fragments on ions b4–b6 (combined).
Under suitable conditions, fragmentation of SDMA and ADMA in the mass spectrometer produces unique neutral loss fragment ions, which can be monitored to distinguish between the two species (38, 39). Using an appropriate collision energy, ADMA fragments to produce ions resulting from the neutral loss of dimethylamine (Δ 45.0578) from either the precursor or a sequence-specific fragment ion. On the other hand, fragmentation through the same bond on SDMA produces ions that result from the neutral loss of methylamine (Δ 31.0422) (Fig. 4A). In order to quantify levels of ADMA and SDMA in cancer cells, we used synthetic peptides to optimize MS conditions for the maximal production and detection of the two types of neutral losses. Commercially available H4 N-terminal peptides unmodified, monomethylated, and dimethylated either symmetrically or asymmetrically on R3 were fully acetylated and digested with trypsin to produce unmodified H4 4-17 peptide and peptide 1–17 containing R3 MMA, ADMA, or SDMA (supplemental Fig. S2). We prepared equimolar mixtures of the fully acetylated unmodified, mono, and both dimethylated H4R3 peptides and analyzed them by LC-MS to determine the relative ionization efficiency of the four components (supplemental Fig. S2). Based upon the integrated peak areas for each peptide, normalization factors were determined relative to the unmodified peptide (3.0 for MMA and 0.9 for DMA) and used to correct the H4R3 data reported throughout. Linearity of the MS response for these peptides was observed over the range of intensities expected for the unmodified, mono, and both dimethylated H4R3 peptides in MDA-MB-468 cells (supplemental Fig. S2). We first attempted to maximize the production of the two neutral loss species from the precursor ion. However, under no conditions tested were we able to efficiently produce these ions. We did observe a weak neutral loss from several ions of the b series. Scanning the NCE on the Orbitrap from 20 to 60 while monitoring the production of neutral losses from the ions b4–b6 of the synthetic peptides, we determined that an NCE of 38 gave the highest yield of the three fragments. It should be noted that this NCE is significantly higher than the value (NCE = 30) used in a typical data-dependent experiment. Using the higher NCE, we were able to increase the signal of the neutral loss fragment ions approximately threefold (supplemental Fig. S3). While the need for a higher collision energy might be specific for this peptide, optimization of the collision energy is necessary for best neutral loss production. The need for higher collision energy for better production for the ADMA/SDMA diagnostic ions has recently been reported as a more general rule (40). Monitoring the neutral loss fragments from ions b4–b6, we could clearly discriminate between H4R3 ADMA and SDMA (supplemental Fig. S4). We analyzed a 1:1 mixture of the acetylated ADMA and SDMA synthetic peptides by PRM using the optimized NCE of 38 and found that the two species produced the neutral loss ions with a comparable efficiency. Interestingly, we also observed a subtle retention time shift between the two peptides (Fig. 4B).
Combining our chemical acetylation strategy with the optimized collision energy parameters, we analyzed nuclear extracts from MDA-MB-468WT cells to determine the baseline methyl state of H4R3. We found that MMA and DMA represented ca. 0.2% and 0.015%, respectively, of the total H4R3 population (Fig. 5A and supplemental Fig. S4). Histone H4R3 has been reported to be a substrate of both Type I and Type II PRMTs (41, 42, 43). However, using PRM data acquisition and monitoring selected neutral loss ions for ADMA and SDMA, we found that H4R3 DMA was only present in these cells in the asymmetric form (Fig. 5B and supplemental Fig. S5). Only after overexpression of exogenous PRMT5 and the cofactor MEP50 in MDA-MB-468 cells (MDA-MB-468PRMT5) was H4R3 SDMA detected (Fig. 5B and supplemental Fig. S5). These cells showed a threefold increase in H4R3 MMA and fourfold increase in total H4R3 DMA (0.7% for MMA and 0.06% for DMA, respectively) (Fig. 5A). These data are in good agreement with reports that PRMT5 catalyzes the addition of both MMA and DMA (44, 45). Finally, neutral loss ions from H4R3 DMA clearly show the presence of both ADMA and SDMA. To our knowledge, this is the first antibody-independent evidence that H4R3 is a substrate for both Type I and Type II PRMTs (Fig. 5B).
Fig. 5.

H4R3 ADMA and SDMA profiling in MDA-MB-468WTand MDA-MB-468PRMT5cells.A, quantitation of relative stoichiometry of H4R3 MMA and DMA. Overexpression of PRMT5 in MDA-MB-468 cells led to a threefold and a fourfold increase in H4R3 MMA and DMA, respectively. Error bars represent the standard error of two biological replicates. Note different scales for H4R3 MMA and DMA relative stoichiometries. B, monitoring of H4R3 ADMA/SDMA diagnostic ions. Only H4R3 ADMA was detected in MDA-MB-468WT. After overexpressing PRMT5 and MEP50, both H4R3 ADMA and SDMA were detected. C, effect of type I PRMT and PRMT5 inhibitors on H4R3 MMA and DMA. Error bars represent the standard error of two biological replicates. Note different scales for the MMA and DMA relative stoichiometries. D, effect of type I PRMT and PRMT5 inhibitors on H4R3 ADMA and SDMA as determined by monitoring of the diagnostic neutral loss on b4–b6 ions.
Selective or combination inhibition of Type I or Type II PRMTs has been linked to anticancer activity and considered as a potential therapeutic for oncology (46, 47, 48). We used the strategy described herein to monitor the effect of PRMT inhibitors on H4R3 in PRMT5-overexpressing MDA-MB-468 cells treated with DMSO, a Type I PRMT inhibitor, or a PRMT5 inhibitor alone or in combination. Analysis of H4R3 MMA and DMA levels showed that while PRMT5i inhibited both MMA and DMA, Type I PRMTi only inhibited H4R3 DMA (Fig. 5C). These findings agree with reports showing that MMA is mainly a product of Type II PRMT activity. As expected, the combination of the inhibitors affected the levels of both MMA and DMA. In addition, we determined the H4R3 ADMA/SDMA ratio before and after compound treatment to confirm inhibitor specificity and prove target engagement. While the DMSO-treated sample contains an equimolar amount of ADMA and SDMA (Fig. 5D), treatment with PRMT5i completely inhibits the formation of H4R3 SDMA, and treatment with Type I PRMTi fully inhibits formation of ADMA. In this experiment, combination treatment decreased DMA to levels that did not allow discrimination between ADMA and SDMA. Due to the very limited stoichiometry of H4R3 DMA (<0.02%), we believe this in-depth analysis would likely not have been possible without the improved sensitivity gained by collapsing the various acetylated forms of the H4 peptide 1-17.
Methylation Profile for Histone H4K5, K12, and K16
Across the same histone H4 sequence (1–17), the four lysines at K5, K8, K12, and K16 are well known to be acetylated (49), and there are reports that these residues are methylated as well (Figure 1A) (6). Histone H4K5 has been reported to be methylated in response to SMYD3 activity (35). H4K12 was shown to be monomethylated in vitro on nucleosomes by KMT9. Depletion of KMT9 in prostate cancer cells results in decreased endogenous H4K12me1 levels (50). In our experiments with Z-138 cells, we found methylation at one of these four sites only when there was no modification on R3, and trypsin produced the peptide 4-17. This shorter peptide was found in only two forms, unmethylated and monomethylated. In our propionylation experiment, analysis by PRM of the 4–17 plus methyl precursor showed this peptide to be a combination of 15 acetyl and methyl isoforms (see supplemental Table S2). The various acetylated forms of this peptide frequently coelute, making quantitation of any one isoform and the less abundant methyl forms difficult (51). Using chemical acetylation, we collapsed the various acetylated forms into one, so that the only variable in the heterogeneity is in the site of lysine methylation. In Z-138 cells, we determined that the monomethylated forms of H4 4–17 account for ca. 0.1% of the total. Using PRM we analyzed the m/z 726.9181 precursor for the monomethylated 4-17 peptide and with manual validation of the MS/MS spectra, identified monomethylation at K5, K12, and K16. Fragment ions unique to K5me1 and K16me1, along with a significant chromatographic separation of the K12me1 species, allowed us to unambiguously characterize these three sites (supplemental Fig. S6). There has been only a single report of H4K5 methylation, (35), but here we were able to clearly confirm K5me1 and differentiate it from the other sites (Fig. 6). Whereas H4K12 accounts for ca. 65% of the total monomethylation, methylation on K5 accounts for only ca. 20% of the total monomethylated forms, corresponding to ca. 0.02% of total peptide 4-17. In these cells, we were unable to unambiguously identify methylation on K8 in the monomethylated 4–17 peptide; however, in a single peptide containing two methyl units, we found evidence for K8me in conjunction with K5me (data not shown). While this analysis is specific to Z-138 cells, it is likely that the composition and stoichiometry of methylation at these sites may differ in other cells.
Fig. 6.

Monomethylation profile for histone H4 K5, K12, and K16 from Z-138 cancer cells. Extracted ion chromatograms from PRM analysis of m/z 726.9178, the [M+2H]2+ precursor ion for the tryptic peptide H4 (4–17) + me1. These ions confirm methylation on different lysine residues within the 4–17 peptide. The stoichiometry for K5me1 was calculated using the intensity of the unmethylated y11 ion (m/z 1154.6698), which is unique for K5me1. The stoichiometry for K16me1 was calculated using the intensity of the methylated y5 ion (m/z 544.3232), which is unique for K16me1. The remaining methylation was assigned to K12me1, which is identified by a combination of the unmethylated y5 ion (m/z 530.3056) and the methylated y6 ion (m/z 714.4268). No unequivocal evidence was found for methylation on K8. Labeled spectra are shown in supplemental Fig. S6.
Quantitation of H3K27me3 as a Biomarker of EZH2 Activity
Trimethylation of histone H3 at K27 (H3K27me3) is strongly associated with transcriptional repression, but is generally considered to be readily reversible (52). As such, it has an important role in the expression of genes involved in lineage commitment and differentiation, where genes expression is dynamically regulated. The role of H3K27me3 in differentiation is also linked to the genesis and maintenance of tumor cells. EZH2, as part of the Polycomb Repressive Complex 2 (PRC2), catalyzes mono-, di-, and trimethylation of H3K27. EZH2 is overexpressed in multiple cancers (53) and gain-of-function mutations in EZH2 lead to an accumulation of H3K27me3 due to the enzyme's increased catalytic efficiency with dimethylated substrates. Because of its central role in modulating H3K27me3 levels, EZH2 is an important target for drug discovery. To efficiently profile the methylation status of H3K27 as part of a campaign to discover selective inhibitors of EZH2, we developed an MS-based assay as an orthogonal approach to existing antibody-based methods. Following chemical derivatization of all the unmodified and monomethylated lysine residues, H3K27 is found in the H3 peptide 27-40 (KSAPATGGVKKPHR). Lysine residues at K27, K36, and K37 have been reported to be acetylated and mono, di, and trimethylated in various combinations (29). Using propionylation, we identified peptide 27-40 in KARPAS-422 cells with 0–2 acetyl groups and up to six methyl groups distributed over K27, K36 and K37 for a total of 44 potentially different acetylated and methylated forms (data not shown). After extensive manual validation, we determined the actual number of modified forms of this peptide in these cells to be 21 (supplemental Table S3).
Collapsing the acetylated forms of H3 27-40 via chemical acetylation yielded a single acetyl form carrying 0–5 methyl units in 14 different isoforms with methylation on K27 and K36 only (Fig. 7 and supplemental Fig. S7). We identified two isoforms K36me3 and K27meK36me3 that were not found in our nuclear catalog (Table 3), likely because the pool of nuclear extracts used to build the catalog did not contain KARPAS-422 cells. H3K27me3 was found in combination with unmodified, mono- and dimethylated K36 but never K36me3. Summing the intensity of all 14 differently methylated forms of the peptide, we obtained an accurate relative quantitation of H3K27me3 levels in response to various inhibitors.
Fig. 7.

H3K27 methylation profile in KARPAS-422 cells and Pfeiffer EZH2 mutant xenografts. Extracted ion chromatogram (A) of 14 different methylated forms (B) of the H3 27-40 peptide from KARPAS-422 cells (2+ and 3+ precursors, supplemental Table S4). Identification of each respective isoform is based on inspection of individual tandem mass spectra (supplemental Fig. S7), allowing us to differentiate the various methylated forms of H3K27 and H3K36. C, monitoring these different extracted ion chromatograms allows accurate relative quantitation of the methyl status on H3K27 from Pfeiffer EZH2 mutant xenografts before and after treatment with the EZH2 inhibitor tazemetostat (57). Error bars represent the standard error of two (vehicle) or three (drug treatment) biological replicates.
EZH2 undergoes recurrent somatic mutation in a subset of diffuse large B-cell lymphomas leading to a gain-of-function phenotype due to a change in the enzyme substrate preference. While wild-type EZH2 is enzymatically most active with unmethylated and monomethylated H3K27-containing peptides, EZH2 Y641 and A677 mutants prefer a dimethylated H3K27 substrate leading to an accumulation of H3K27me3 at the expense of H3K27me2. This finding has been demonstrated biochemically and in cells using both antibody and MS-based methods (54, 55, 56).
Additionally, H3K27 methylation levels can be modulated biochemically, in vitro or in vivo, using small-molecule inhibitors of EZH2 (57, 58). The methodology described herein was utilized to characterize the H3K27 methylation profile in Pfeiffer EZH2 mutant xenografts treated in vivo for 11 days with vehicle or the EZH2 inhibitor, tazemetostat (57) (Fig. 7C). H3K27 was predominantly trimethylated in tumor from vehicle-treated animals consistent with previously published findings using site-specific western blot assays in EZH2 wild-type and mutant cell lines (58) and provides further validation of this MS method. Following EZH2 inhibition, H3K27me3 levels decrease from ca. 46% to 14% at the highest dose tested, with a concomitant increase in H2K27me0/1/2. Overall, these data support the use of this simplified MS strategy to assess changes in histone methylation in a variety of experimental settings.
Conclusions
There is a well-established link between histone lysine methylation and the development and progression of a wide range of cancers (10). More recently it has become clear that histone arginine methylation plays a key role in the pathogenesis of multiple cancers by hijacking processes that facilitate cancer cell survival (9). As such, the enzymes that catalyze the addition and removal of these modifications have become high-priority targets for drug discovery efforts (9, 59). Highly selective and sensitive assays that can measure changes in the site occupancy of these modification are critical enablers of these efforts. Whereas more than a thousand site-specific antibodies are available for many of the most relevant histone modifications (60), it has now become clear that many of these reagents suffer from a lack of selectivity including off-target recognition, neighboring PTM effects, and the inability to distinguish the various methyl forms (60, 61, 62, 63). For this reason, MS has become the gold standard to measure and validate changes in histone marks (11, 64, 65), particularly methylation (66, 67). In this report we described a simple strategy that uses chemical acetylation with d0-acetic anhydride to not only improve the size and chromatographic properties of histone tryptic peptides, but also most importantly reduce the combinatorial complexity of acetylation on a given sequence, thereby facilitating the identification and quantitative analysis of histone methyl marks. This was particularly notable in the case of Histone H4R3 dimethylation, where our strategy provided the selectivity and sensitivity to quantify changes in symmetric and asymmetric dimethylation where the levels of dimethyl arginine were as low as 0.02% of the overall H4R3 occupancy. In the same region of the H4 sequence, we were further able to deconvolute monomethylation on four closely spaced lysine residues, where the overall level of the modification was approximately 0.1%.
A caveat of our proposed strategy, beyond the limitations imposed by any bottom-up strategy, such as the inability to distinguish some sequence variants, is that an understanding of PTM cross talk between acetylation and methylation is lost. For instance, it is reported that whereas H4K5 acetylation promotes ADMA at H4R3, it inhibits SDMA at the same site, and that acetylation at H3K9 and H3K14 inhibits methylation at H3R8 (68). If the goal of the experiment is to understand this cross-coordination, then certainly a different strategy should be employed. On the other hand, our approach does not limit the ability to understand cross talk between different methyl sites or between methylation and any other PTM not obscured by chemical acetylation of lysine residues.
Our goal in this work was to establish a mechanism for rapidly developing highly sensitive assays for selective analysis of specific histone methyl marks. Using a pool of cancer cell extracts, we were able to build a library of identifiable methyl marks and the sequences in which they could be readily identified. There is work yet to be done. Arginine methylation remains particularly difficult to identify. While we were able to efficiently identify all forms of H4R3, other sites on H4 (R17, R19) and H3 (R2, R8, R17, R26) remain refractory. As was the case with H3K36me3, which we only found in Z-138 cells, it's likely that certain arginine marks may be specific to certain cell lines or limited to certain regions of the chromatin. In this case a more focused effort will be necessary to interrogate these marks.
Data Availability
The MS raw data and complete Mascot search results from which Table 1, Table 2, Table 3, supplemental Fig. S3 and Figures 6, 7, and supplemental Fig. S4 are derived have been deposited to the MassIVE data repository under the identifier MSV000086146 (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp). Spectra identifying modified peptides can be viewed using the Mascot browser.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
Author contributions
F. Z., M. T. M., O. B., and R. S. A. designed the experiments. F. Z., C. D. W., A. D. P., C. J. Q., and S. V. G. performed the experiments, analyzed the data, and contributed to Material and Methods. F. Z., C. D. W., M. T. M., and R. S. A. wrote the article. All the authors reviewed and approved the article.
Edited by Alma Burlingame
Supplemental data
Supplemental Figures S1 to S7 and Tables S1 to S3
References
- 1.Strahl B.D., Allis C.D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Henikoff S., Shilatifard A. Histone modification: Cause or cog? Trends Genet. 2011;27:389–396. doi: 10.1016/j.tig.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Castillo J., Lopez-Rodas G., Franco L. Histone post-translational modifications and nucleosome organisation in transcriptional regulation: Some open questions. Adv. Exp. Med. Biol. 2017;966:65–92. doi: 10.1007/5584_2017_58. [DOI] [PubMed] [Google Scholar]
- 5.Lawrence M., Daujat S., Schneider R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016;32:42–56. doi: 10.1016/j.tig.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 6.Zhao Y., Garcia B.A. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb. Perspect. Biol. 2015;7:a025064. doi: 10.1101/cshperspect.a025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun L., Wang M., Lv Z., Yang N., Liu Y., Bao S., Gong W., Xu R.M. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. U. S. A. 2011;108:20538–20543. doi: 10.1073/pnas.1106946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murn J., Shi Y. The winding path of protein methylation research: Milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 2017;18:517–527. doi: 10.1038/nrm.2017.35. [DOI] [PubMed] [Google Scholar]
- 9.Jarrold J., Davies C.C. PRMTs and arginine methylation: Cancer's best-kept secret? Trends Mol. Med. 2019;25:993–1009. doi: 10.1016/j.molmed.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Husmann D., Gozani O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019;26:880–889. doi: 10.1038/s41594-019-0298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leroy G., Dimaggio P.A., Chan E.Y., Zee B.M., Blanco M.A., Bryant B., Flaniken I.Z., Liu S., Kang Y., Trojer P., Garcia B.A. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin. 2013;6:20. doi: 10.1186/1756-8935-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pesavento J.J., Bullock C.R., LeDuc R.D., Mizzen C.A., Kelleher N.L. Combinatorial modification of human histone H4 quantitated by two-dimensional liquid chromatography coupled with top down mass spectrometry. J. Biol. Chem. 2008;283:14927–14937. doi: 10.1074/jbc.M709796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas C.E., Kelleher N.L., Mizzen C.A. Mass spectrometric characterization of human histone H3: A bird's eye view. J. Proteome Res. 2006;5:240–247. doi: 10.1021/pr050266a. [DOI] [PubMed] [Google Scholar]
- 14.Phanstiel D., Brumbaugh J., Berggren W.T., Conard K., Feng X., Levenstein M.E., McAlister G.C., Thomson J.A., Coon J.J. Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4093–4098. doi: 10.1073/pnas.0710515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffiths J.R., Unwin R.D. John Wiley and Sons; Hoboken, NJ: 2017. Analysis of Posttranslational Modifications by Mass Spectrometry. [Google Scholar]
- 16.Garcia B.A., Mollah S., Ueberheide B.M., Busby S.A., Muratore T.L., Shabanowitz J., Hunt D.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007;2:933–938. doi: 10.1038/nprot.2007.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nie L., Shuai L., Zhu M., Liu P., Xie Z.F., Jiang S., Jiang H.W., Li J., Zhao Y., Li J.Y., Tan M. The landscape of histone modifications in a high-fat diet-induced obese (DIO) mouse model. Mol. Cell Proteomics. 2017;16:1324–1334. doi: 10.1074/mcp.M117.067553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilczek C., Chitta R., Woo E., Shabanowitz J., Chait B.T., Hunt D.F., Shechter D. Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J. Biol. Chem. 2011;286:42221–42231. doi: 10.1074/jbc.M111.303677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J., Hu Y., Yu Y., Zhang L., Yang P., Jin H. Quantitative analysis of post-translational modifications of histone H3 variants during the cell cycle. Anal. Chim. Acta. 2019;1080:116–126. doi: 10.1016/j.aca.2019.06.028. [DOI] [PubMed] [Google Scholar]
- 20.Sidoli S., Kori Y., Lopes M., Yuan Z.F., Kim H.J., Kulej K., Janssen K.A., Agosto L.M., Cunha J., Andrews A.J., Garcia B.A. One minute analysis of 200 histone posttranslational modifications by direct injection mass spectrometry. Genome Res. 2019;29:978–987. doi: 10.1101/gr.247353.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feller C., Forne I., Imhof A., Becker P.B. Global and specific responses of the histone acetylome to systematic perturbation. Mol. Cell. 2015;57:559–571. doi: 10.1016/j.molcel.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Smith C.M., Gafken P.R., Zhang Z., Gottschling D.E., Smith J.B., Smith D.L. Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Anal. Biochem. 2003;316:23–33. doi: 10.1016/s0003-2697(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 23.D'Urzo A., Boichenko A.P., van den Bosch T., Hermans J., Dekker F., Andrisano V., Bischoff R. Site-specific quantification of lysine acetylation in the N-terminal tail of histone H4 using a double-labelling, targeted UHPLC MS/MS approach. Anal. Bioanal. Chem. 2016;408:3547–3553. doi: 10.1007/s00216-016-9431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.ElBashir R., Vanselow J.T., Kraus A., Janzen C.J., Siegel T.N., Schlosser A. Fragment ion patchwork quantification for measuring site-specific acetylation degrees. Anal. Chem. 2015;87:9939–9945. doi: 10.1021/acs.analchem.5b02517. [DOI] [PubMed] [Google Scholar]
- 25.Sidoli S., Bhanu N.V., Karch K.R., Wang X., Garcia B.A. Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: From histone extraction to data analysis. J. Vis. Exp. 2016 doi: 10.3791/54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan Z.F., Lin S., Molden R.C., Garcia B.A. Evaluation of proteomic search engines for the analysis of histone modifications. J. Proteome Res. 2014;13:4470–4478. doi: 10.1021/pr5008015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savitski M.M., Lemeer S., Boesche M., Lang M., Mathieson T., Bantscheff M., Kuster B. Confident phosphorylation site localization using the Mascot Delta Score. Mol. Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.003830. M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sidoli S., Garcia B.A. Characterization of individual histone posttranslational modifications and their combinatorial patterns by mass spectrometry-based proteomics strategies. Methods Mol. Biol. 2017;1528:121–148. doi: 10.1007/978-1-4939-6630-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang H., Lin S., Garcia B.A., Zhao Y. Quantitative proteomic analysis of histone modifications. Chem. Rev. 2015;115:2376–2418. doi: 10.1021/cr500491u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Q., Rank G., Tan Y.T., Li H., Moritz R.L., Simpson R.J., Cerruti L., Curtis D.J., Patel D.J., Allis C.D., Cunningham J.M., Jane S.M. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 2009;16:304–311. doi: 10.1038/nsmb.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y., Lu Q., Li C., Wang X., Jiang L., Huang L., Wang C., Chen H. PRMT1 regulates the tumour-initiating properties of esophageal squamous cell carcinoma through histone H4 arginine methylation coupled with transcriptional activation. Cell Death Dis. 2019;10:359. doi: 10.1038/s41419-019-1595-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou X., Wang W., Du C., Yan F., Yang S., He K., Wang H., Zhao A. OGG1 regulates the level of symmetric dimethylation of histone H4 arginine-3 by interacting with PRMT5. Mol. Cell Probes. 2018;38:19–24. doi: 10.1016/j.mcp.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Girardot M., Hirasawa R., Kacem S., Fritsch L., Pontis J., Kota S.K., Filipponi D., Fabbrizio E., Sardet C., Lohmann F., Kadam S., Ait-Si-Ali S., Feil R. PRMT5-mediated histone H4 arginine-3 symmetrical dimethylation marks chromatin at G + C-rich regions of the mouse genome. Nucleic Acids Res. 2014;42:235–248. doi: 10.1093/nar/gkt884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Onder O., Sidoli S., Carroll M., Garcia B.A. Progress in epigenetic histone modification analysis by mass spectrometry for clinical investigations. Expert Rev. Proteomics. 2015;12:499–517. doi: 10.1586/14789450.2015.1084231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Aller G.S., Reynoird N., Barbash O., Huddleston M., Liu S., Zmoos A.F., McDevitt P., Sinnamon R., Le B., Mas G., Annan R., Sage J., Garcia B.A., Tummino P.J., Gozani O. Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics. 2012;7:340–343. doi: 10.4161/epi.19506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCabe M.T., Mohammad H.P., Barbash O., Kruger R.G. Targeting histone methylation in cancer. Cancer J. 2017;23:292–301. doi: 10.1097/PPO.0000000000000283. [DOI] [PubMed] [Google Scholar]
- 37.Xiao W., Chen X., Liu L., Shu Y., Zhang M., Zhong Y. Role of protein arginine methyltransferase 5 in human cancers. Biomed. Pharmacother. 2019;114:108790. doi: 10.1016/j.biopha.2019.108790. [DOI] [PubMed] [Google Scholar]
- 38.Rappsilber J., Friesen W.J., Paushkin S., Dreyfuss G., Mann M. Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal. Chem. 2003;75:3107–3114. doi: 10.1021/ac026283q. [DOI] [PubMed] [Google Scholar]
- 39.Brame C.J., Moran M.F., McBroom-Cerajewski L.D. A mass spectrometry based method for distinguishing between symmetrically and asymmetrically dimethylated arginine residues. Rapid Commun. Mass Spectrom. 2004;18:877–881. doi: 10.1002/rcm.1421. [DOI] [PubMed] [Google Scholar]
- 40.Hartel N.G., Liu C.Z., Graham N.A. Improved discrimination of asymmetric and symmetric arginine dimethylation by optimization of the normalized collision energy in liquid chromatography-mass spectrometry proteomics. J. Proteome Res. 2020;19:3123–3129. doi: 10.1021/acs.jproteome.0c00116. [DOI] [PubMed] [Google Scholar]
- 41.Bedford M.T., Clarke S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pal S., Vishwanath S.N., Erdjument-Bromage H., Tempst P., Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell Biol. 2004;24:9630–9645. doi: 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strahl B.D., Briggs S.D., Brame C.J., Caldwell J.A., Koh S.S., Ma H., Cook R.G., Shabanowitz J., Hunt D.F., Stallcup M.R., Allis C.D. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 44.Branscombe T.L., Frankel A., Lee J.H., Cook J.R., Yang Z., Pestka S., Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 2001;276:32971–32976. doi: 10.1074/jbc.M105412200. [DOI] [PubMed] [Google Scholar]
- 45.Bedford M.T., Richard S. Arginine methylation an emerging regulator of protein function. Mol. Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Gerhart S.V., Kellner W.A., Thompson C., Pappalardi M.B., Zhang X.P., Montes de Oca R., Penebre E., Duncan K., Boriack-Sjodin A., Le B., Majer C., McCabe M.T., Carpenter C., Johnson N., Kruger R.G. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 2018;8:9711. doi: 10.1038/s41598-018-28002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fedoriw A., Rajapurkar S.R., O'Brien S., Gerhart S.V., Mitchell L.H., Adams N.D., Rioux N., Lingaraj T., Ribich S.A., Pappalardi M.B., Shah N., Laraio J., Liu Y., Butticello M., Carpenter C.L. Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell. 2019;36:100–114 e25. doi: 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 48.Mohammad H.P., Barbash O., Creasy C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019;25:403–418. doi: 10.1038/s41591-019-0376-8. [DOI] [PubMed] [Google Scholar]
- 49.Sterner D.E., Berger S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metzger E., Wang S., Urban S., Willmann D., Schmidt A., Offermann A., Allen A., Sum M., Obier N., Cottard F., Ulferts S., Preca B.T., Hermann B., Maurer J., Greschik H. KMT9 monomethylates histone H4 lysine 12 and controls proliferation of prostate cancer cells. Nat. Struct. Mol. Biol. 2019;26:361–371. doi: 10.1038/s41594-019-0219-9. [DOI] [PubMed] [Google Scholar]
- 51.Yuan Z.F., Lin S., Molden R.C., Cao X.J., Bhanu N.V., Wang X., Sidoli S., Liu S., Garcia B.A. EpiProfile quantifies histone peptides with modifications by extracting retention time and intensity in high-resolution mass spectra. Mol. Cell Proteomics. 2015;14:1696–1707. doi: 10.1074/mcp.M114.046011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trojer P., Reinberg D. Facultative heterochromatin: Is there a distinctive molecular signature? Mol. Cell. 2007;28:1–13. doi: 10.1016/j.molcel.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 53.Bracken A.P., Pasini D., Capra M., Prosperini E., Colli E., Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sneeringer C.J., Scott M.P., Kuntz K.W., Knutson S.K., Pollock R.M., Richon V.M., Copeland R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 2010;107:20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCabe M.T., Graves A.P., Ganji G., Diaz E., Halsey W.S., Jiang Y., Smitheman K.N., Ott H.M., Pappalardi M.B., Allen K.E., Chen S.B., Della Pietra A., 3rd, Dul E., Hughes A.M., Gilbert S.A. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27) Proc. Natl. Acad. Sci. U. S. A. 2012;109:2989–2994. doi: 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ott H.M., Graves A.P., Pappalardi M.B., Huddleston M., Halsey W.S., Hughes A.M., Groy A., Dul E., Jiang Y., Bai Y., Annan R., Verma S.K., Knight S.D., Kruger R.G., Dhanak D. A687V EZH2 is a driver of histone H3 lysine 27 (H3K27) hypertrimethylation. Mol. Cancer Ther. 2014;13:3062–3073. doi: 10.1158/1535-7163.MCT-13-0876. [DOI] [PubMed] [Google Scholar]
- 57.Kuntz K.W., Campbell J.E., Keilhack H., Pollock R.M., Knutson S.K., Porter-Scott M., Richon V.M., Sneeringer C.J., Wigle T.J., Allain C.J., Majer C.R., Moyer M.P., Copeland R.A., Chesworth R. The importance of being me: Magic methyls, methyltransferase inhibitors, and the discovery of tazemetostat. J. Med. Chem. 2016;59:1556–1564. doi: 10.1021/acs.jmedchem.5b01501. [DOI] [PubMed] [Google Scholar]
- 58.McCabe M.T., Ott H.M., Ganji G., Korenchuk S., Thompson C., Van Aller G.S., Liu Y., Graves A.P., Della Pietra A., 3rd, Diaz E., LaFrance L.V., Mellinger M., Duquenne C., T ian X., Kruger R.G. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 59.McGrath J., Trojer P. Targeting histone lysine methylation in cancer. Pharmacol. Ther. 2015;150:1–22. doi: 10.1016/j.pharmthera.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Rothbart S.B., Dickson B.M., Raab J.R., Grzybowski A.T., Krajewski K., Guo A.H., Shanle E.K., Josefowicz S.Z., Fuchs S.M., Allis C.D., Magnuson T.R., Ruthenburg A.J., Strahl B.D. An interactive database for the assessment of histone antibody specificity. Mol. Cell. 2015;59:502–511. doi: 10.1016/j.molcel.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rothbart S.B., Lin S., Britton L.M., Krajewski K., Keogh M.C., Garcia B.A., Strahl B.D. Poly-acetylated chromatin signatures are preferred epitopes for site-specific histone H4 acetyl antibodies. Sci. Rep. 2012;2:489. doi: 10.1038/srep00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez-Burgos L., Peters A.H., Opravil S., Kauer M., Mechtler K., Jenuwein T. Generation and characterization of methyl-lysine histone antibodies. Methods Enzymol. 2004;376:234–254. doi: 10.1016/S0076-6879(03)76016-9. [DOI] [PubMed] [Google Scholar]
- 63.Bock I., Dhayalan A., Kudithipudi S., Brandt O., Rathert P., Jeltsch A. Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics. 2011;6:256–263. doi: 10.4161/epi.6.2.13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beck H.C., Nielsen E.C., Matthiesen R., Jensen L.H., Sehested M., Finn P., Grauslund M., Hansen A.M., Jensen O.N. Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell Proteomics. 2006;5:1314–1325. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 65.Schwammle V., Sidoli S., Ruminowicz C., Wu X., Lee C.F., Helin K., Jensen O.N. Systems level analysis of histone H3 post-translational modifications (PTMs) reveals features of PTM crosstalk in chromatin regulation. Mol. Cell Proteomics. 2016;15:2715–2729. doi: 10.1074/mcp.M115.054460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohammad F., Weissmann S., Leblanc B., Pandey D.P., Hojfeldt J.W., Comet I., Zheng C., Johansen J.V., Rapin N., Porse B.T., Tvardovskiy A., Jensen O.N., Olaciregui N.G., Lavarino C., Sunol M. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017;23:483–492. doi: 10.1038/nm.4293. [DOI] [PubMed] [Google Scholar]
- 67.Tvardovskiy A., Schwammle V., Kempf S.J., Rogowska-Wrzesinska A., Jensen O.N. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 2017;45:9272–9289. doi: 10.1093/nar/gkx696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fulton M.D., Brown T., Zheng Y.G. Mechanisms and inhibitors of histone arginine methylation. Chem. Rec. 2018;18:1792–1807. doi: 10.1002/tcr.201800082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures S1 to S7 and Tables S1 to S3
Data Availability Statement
The MS raw data and complete Mascot search results from which Table 1, Table 2, Table 3, supplemental Fig. S3 and Figures 6, 7, and supplemental Fig. S4 are derived have been deposited to the MassIVE data repository under the identifier MSV000086146 (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp). Spectra identifying modified peptides can be viewed using the Mascot browser.