

Abstract
It has been increasingly accepted that microRNA (miRNA) can both activate and suppress gene expression, directly or indirectly, under particular circumstances. Yet, a systematic study on the switch in their interaction pattern between activation and suppression and between normal and cancer conditions based on multi-omics evidences is not available. We built miRactDB, a database for miRNA–gene interaction, at https://ccsm.uth.edu/miRactDB, to provide a versatile resource and platform for annotation and interpretation of miRNA–gene relations. We conducted a comprehensive investigation on miRNA–gene interactions and their biological implications across tissue types in both tumour and normal conditions, based on TCGA, CCLE and GTEx databases. We particularly explored the genetic and epigenetic mechanisms potentially contributing to the positive correlation, including identification of miRNA binding sites in the gene coding sequence (CDS) and promoter regions of partner genes. Integrative analysis based on this resource revealed that top-ranked genes derived from TCGA tumour and adjacent normal samples share an overwhelming part of biological processes, which are quite different than those from CCLE and GTEx. The most active miRNAs predicted to target CDS and promoter regions are largely overlapped. These findings corroborate that adjacent normal tissues might have undergone significant molecular transformations towards oncogenesis before phenotypic and histological change; and there probably exists a small yet critical set of miRNAs that profoundly influence various cancer hallmark processes. miRactDB provides a unique resource for the cancer and genomics communities to screen, prioritize and rationalize their candidates of miRNA–gene interactions, in both normal and cancer scenarios.
Keywords: microRNA activation, pan-cancer, correlation, genetic mechanism, epigenetic regulation
Significance miRactDB, the first resource for comprehensive annotation of miRNA–gene relation, provides expression profiles, prognostic landscapes and potential mechanisms of action across multiple cancer and healthy tissue types.
Introduction
Cancer is a disease initiated and driven by a series of genetic and epigenetic aberrations in the genome [1, 2]. Among them, microRNAs (miRNAs) are small noncoding RNAs recognized as an epigenetic modulator that plays a critical role in post-transcriptional regulation of many protein-coding and noncoding genes [3]. The discovery of hundreds of distinct miRNAs has profoundly changed our understanding of the epigenetic control of gene expression in both health and disease conditions, especially cancer [4, 5]. Public databases and resources provided useful platforms for retrieving miRNA-related information on a particular aspect. TargetScan [6], miRBase [7] and miRDB [8] focus on prediction of miRNA targets or annotation of the miRNAs and targets separately, but not emphasizing their real interactions. Other databases, such as miRTarBase [9], StarBase [10] and miRwalk [11], work on the miRNA–gene interactions, but they are presented in a general sense, not in cancer context. The oncomiR [12] and miRCancerdb [13] databases calculate correlations based on cancer genomics data, but limited to the correlation, not involving their biological meaning or molecular mechanism, and for individual cancers rather than pan-cancer. Therefore, an integrative resource for miRNA–gene interactions with associated biological significance and potential mechanisms in a pan-cancer and tumour-normal comparative way is still in pressing need, as it’s critical for prioritizing and rationalizing candidates in cancer research.
We recently performed a comprehensive study on the miRNA–gene relations across multiple cancers based on The Cancer Genome Atlas (TCGA) [14], in which we paid greater attention to the miRNA–gene positive correlation due to increasing evidence from in vitro experiments [15–17]. However, we presented findings in a general manner and in cancer conditions alone, but did not go into the depth of the pattern switch between positive and negative, between normal and tumour and between tissue and cell line conditions. In this study, we furthered our investigation on TCGA tumour sample (TCGA_t) data by integrating three additional datasets: the TCGA normal sample (TCGA_n), the Cancer Cell Line Encyclopedia (CCLE) and the Genotype-Tissue Expression (GTEx, disease-free tissue) databases. We inspected the expression correlations of all possible miRNA–gene pairs in each of the four datasets, compared the correlation pattern and prevalence across tissues/cell lines between these four datasets and explored their biological significance in respective scenarios. Taking all of our previous and current analysis results together, we developed miRactDB (miRNA–gene interaction database), a versatile online platform that profiles any queried miRNA–gene pair in multiple facets: the basic biographic information for the miRNA and gene, their detailed correlation pattern in all four datasets and evidences for five hypothesized molecular mechanisms.
The current miRactDB includes 21 475 426 miRNA–gene pairs across 31 cancer types and 20 normal tissue types involving 8375 patients from TCGA; 10 979 878 pairs across 25 tissue origins involving 941 cell lines from CCLE; and 13 829 868 pairs across 30 non-disease healthy tissues involving 11 688 samples from GTEx. To support the hypotheses for positive correlation, we curated, processed and analysed additional information from public resources into the database. Specifically, we incorporated 591 host genes and their intragenic miRNAs based on GENCODE [18]. We obtained 198 312 records of conserved targeting of 321 miRNAs on 13 035 genes from TargetScan. We also curated 2705 transcription factors (TF) with their potential target genes from 6 previous studies to account for the co-transcription hypothesis. In addition, we downloaded H3K27ac ChIP-seq data from ENCODE [19] for 64 human samples spanning 15 tissues and calculated the signal intensity surrounding all miRNAs and genes. Finally, we for the first time scanned the CDS and promoter region [transcription start site (TSS) ± 2 kb] of 16 748 genes for binding sites for 1694 different miRNAs and identified about 10 million confident binding sites for each scenario.
With miRactDB, the users can access a variety of annotations and analysis results, including miRNA and gene summary, expression level over tumour progression stage, correlation switch between conditions (tumour and adjacent normal, cancer cell line, non-disease healthy), intragenic miRNA–host gene profiles, evidence of indirect activation and co-transcription, local super-enhancer enrichment, miRNA binding sites along whole gene structure (especially CDS and promoter) and clinical relevance in terms of survival rate and hazard ratio (HR). Based on this plentiful resource, we performed an example integrative analysis and obtained multiple new insights about miRNA regulation, including its pattern switch between conditions, and novel functioning mechanisms through binding to gene CDS and promoter regions. In summary, miRactDB represents a unique resource and platform for the community to efficiently screen, prioritize, rationalize and visualize their candidates of miRNA–gene interactions, especially in the sense of miRNA-associated gene upregulation and in the pan-cancer manner.
Materials and methods
Data acquisition and miRNA–gene correlation profiling for TCGA, CCLE and GTEx
We downloaded the miRNA (IlluminaHiSeq miRNASeq, Level 3) and gene (IlluminaHiSeq RNASeqV2, Level 3) expression data, as well as survival information of each cancer from TCGA data portal (https://portal.gdc.cancer.gov/). We excluded three cancers, including FPPP (FFPE Pilot Phase II), GBM (glioblastoma multiforme) and LAML (acute myeloid leukaemia), due to small sample size or platform inconsistency. The pan-cancer analyses eventually consisted of 1046 miRNAs and 20 531 genes resulting in 21 475 426 miRNA–gene pairs based on 8375 patient samples across 31 TCGA cancer types. We dealt with the TCGA tumour (TCGA_t) and normal (TCGA_n) samples separately.
CCLE expression data were downloaded from the Broad Institute data portal [20]. For miRNAs with both 3′- and 5′-strand, we only kept the one with higher average expression across all samples. After that, we overlapped them with TCGA data and obtained 598 miRNAs and 18 361 genes across 941 cell lines derived from 25 types of tissues. GTEx (v7) expression data were downloaded from portal https://gtexportal.org/. Taking overlap with TCGA data, we eventually obtained expression data for 754 miRNAs and 18 342 genes across 11 688 samples spanning 30 different tissue types. A summary of sample size for each individual tissue type in respective datasets is provided in Table S1A–C.
We calculated the Spearman correlation of each miRNA–gene pair (log2 transformed) and set the significance criterion as |R| > 0.1 and Hochberg adjusted P-value <0.05. Due to the large number of miRNA–gene pairs tested in each dataset, a cutoff of 0.05 with Hochberg adjustment corresponds to a much smaller cutoff of original P-value (typically <10−8) in the Spearman correlation analysis. We set a higher P-value threshold for TCGA_n and CCLE to compensate their small sample size (Table S1D). This does not affect the downstream analysis since we ranked the pairs by their cancer coverage and only picked the top pairs for further study. We also extracted and parsed miRNA summary information from miRBase [21] and TargetScan [4] and gene information from GenBank [22].
Functional annotation of miRNAs and genes in top correlations
We conducted functional annotation of the top miRNAs and genes on two aspects: biological and clinical significance. For the biological meaning, we performed KEGG signalling pathway [23] and gene ontology (GO) term enrichment analysis on the top-ranked pairs in the correlation analysis. Briefly, we first extracted the top 200 unique genes from the ranked correlation table for each dataset (Table S2), followed by enrichment analysis with the extracted genes for each dataset, respectively, using the R package clusterProfiler [24]. Note that these top 200 genes of respective datasets (TCGA_t, TCGA_n, CCLE and GTEx) were extracted from the same background genes (n = 18 342) overlapped across the 4 datasets (with genes not in this overlapped gene set skipped). The KEGG and GO databases adopted in this study include a total of 7925 and 18 678 genes, respectively. Then, we compared the top enriched pathways (Fisher exact test P < 0.05) of genes from different datasets. To investigate the clinical relevance of each miRNA and gene, we conducted survival analysis on patients of different groups characterized by their miRNA and gene expression. Briefly, patients were approximately equally divided into three nonoverlapped groups based on their miRNA or gene expression, high-middle-low (Hi-Mi-Lo), followed by a log-rank test to compare the overall survival probability of different groups. Results were visualized by Kaplan–Meier plot for each cancer type. To further explore the clinical relevance of the paired miRNA and gene as a cofactor, we performed both univariate and multivariate Cox regression analysis to obtain the HR profile of the queried pair using the R package survivalAnalysis.
Identification of miRNAs with host genes
We downloaded the gene annotations of 27 423 genes, including 19 902 protein-coding genes and 7521 long noncoding RNAs (lncRNAs), from GENCODE (GTF v25, hg38) [18]. We obtained annotations of 1881 human miRNAs from miRBase (hsa.gff3) [25]. We curated intragenic miRNAs which are embedded in another protein-coding or noncoding gene according to their genomic coordinates from the annotation file. In total, 591 miRNAs were found to be located inside a specific gene (‘host’ gene), of which 451 pairs were covered in the TCGA data. These 591 host genes contain 474 protein-coding genes and 117 lncRNAs. We also checked the conservation information of the partner miRNA from TargetScanHuman (release 7.2).
Generation of indirect miRNA–gene interactions
We checked two types of indirect miRNA–gene interactions for those significant positive pairs covering at least 10 cancer types (PanCan10), involving 17 753 pairs consisted of 347 miRNAs and 3017 genes. First, for each pair, since the miRNA might upregulate the partner gene by inhibiting its upstream suppressor, we explored the double-negative patterns by screening all genes that satisfy two criteria: (i) the candidate gene is negatively correlated with both the constituent miRNA and gene of the pair, and (ii) the candidate gene is a conserved target of the constituent miRNA predicted by TargetScanHuman [6]. Second, since a positively correlated miRNA–gene pair might also be regulated by shared transcription activators, we detected the double-positive patterns by screening genes satisfying the following two criteria: (i) the candidate gene is positively correlated with both the constituent miRNA and gene, and (ii) the candidate gene is a transcription factor (TF) of the constituent gene predicted by at least one of the six published databases [14].
H3K27ac enrichment detection in miRNA and gene locus
We investigated the super-enhancer formation profile surrounding each miRNA and gene TSS with a modified ROSE pipeline [26]. We achieved this by quantifying the ChIP-seq signal intensity of H3K27ac in each miRNA and gene’s region based on 64 samples across 15 human tissues from ENCODE [19]. In the present work, we calculated the ROSE score for all miRNAs and genes in all ENCODE samples and drew each miRNA/gene (across samples) with a bar plot and built into the dataset for query and download.
Identification of miRNA binding sites in CDS and promoter regions
We downloaded the CDS sequence of all genes from GENCODE (release 30). We extracted the sequence of the TSS ± 2 kb region of each gene with the R package BSgenome.Hsapiens.UCSC.hg38. We downloaded the miRNA family information from TargetScanHuman v7.2 (miR_Family_Infor.txt) and extracted the records for human (Homo sapiens) (n = 2064). Then, we scanned potential binding sites of each miRNA on the CDS and promoter regions of all genes using the open script targetscan.pl provided by TargetScan. We scanned 8715 and 8596 gene promoters of positive and negative strand separately. After scanning, we parsed the miRNA family information and mapped the families back to associated individual miRNAs.
Drug and disease information
The drug–gene interactions (DTIs) were extracted from the DrugBank (April 2018, version 5.1.0) [27]. The duplicated DTI pairs were excluded. All drugs were grouped using the Anatomical Therapeutic Chemical (ATC) classification system codes. Disease gene information was extracted from DisGeNET (June 2018, version 4.0) [28]. We also checked published work regarding drug–miRNA relation including SM2MiR [29] and disease–miRNA association including HMDD (v3.2) [30] and miR2Disease [31] and integrated relevant information into miRactDB.
Database architecture
The miRactDB system is based on a three-tier architecture: client, server and database. It includes a user-friendly web interface, Perl’s DBI module and MySQL database. This database was developed on the MySQL 3.23 with the MyISAM storage engine.
Results
Overview of miRactDB
The database consists of five major components (Figure 1). For a queried miRNA–gene pair, miRactDB first generates a page with four lookup tables summarizing the correlation profile across tissue/cell types in TCGA_t, TCGA_n, CCLE and GTEx datasets, respectively. The grids of significant positive and negative correlations are marked red and green, and nonsignificant ones are coloured in grey. On this page, clicking the hyperlink on the miRNA–gene pair name in the lookup table will open a new page with various annotations we curated and analysed for this pair in pan-cancer/tissue. Figure 1A shows three representative pairs with consistent positive, consistent negative and dual direction correlations, respectively.
Figure 1.
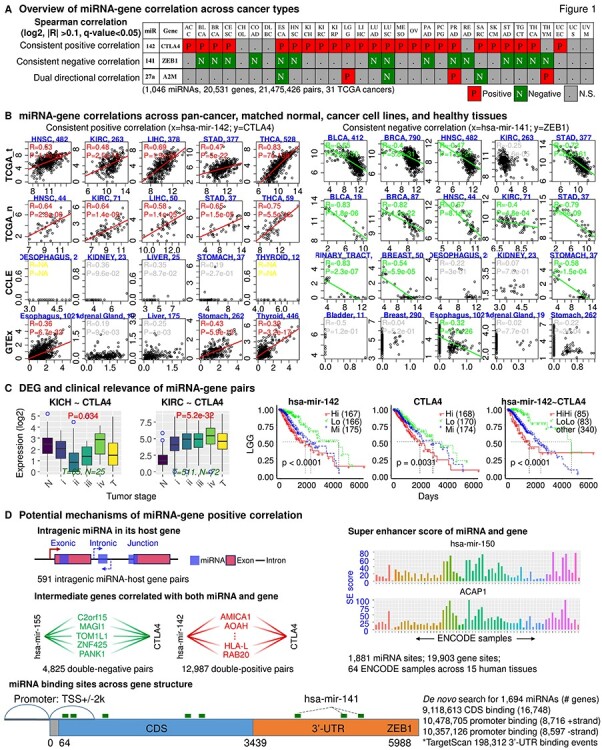
Overview of miRactDB. (A) General output upon miRNA–gene query: correlation profile across TCGA cancers. Representative results are shown for consistent positive and negative correlations, as well as correlation with dual direction in different cancers. N.S., not significant. (B) Example correlation patterns for miR-142~CTLA4 (left) and miR-141~ZEB1 (right) across representative cancer/tissue types in TCGA (both tumour and normal), CCLE (cancer cell line) and GTEx (non-disease tissue). (C) Left, gene expression along normal and different tumour stages; right, clinical relevance by survival analysis based on individual or combined miRNA–gene expression. (D) Evidence for five hypotheses regarding miRNA-associated/miRNA-directed gene activation as indicated in each panel: intragenic miRNA–host gene co-expression; double-negative and double-positive indirect regulation; co-regulation by same epigenetic factor based on ENCODE ChIP-seq for H3K27ac; and direct binding and upregulation by scanning potential miRNA binding sites on the coding and promoter regions of queried gene with TargetScan script.
On the new page as abovementioned, the correlation chart visualizes the detailed correlation profile of the pair across cancer/tissue types in TCGA_t, TCGA_n, CCLE and GTEx separately with related statistic information such as coefficient, P-value and number of samples for each cancer/tissue type. Figure 1B shows the correlation of two example pairs, miR-142~CTLA4 and miR-141~ZEB1, in representative tissue/cell types in the four datasets. Negligible expression level of CTLA4 is observed in cancer cell lines, which is not surprising since CLTA4 functions as an immune checkpoint and is supposed to express only in regulatory T cells [32]. Although the miR-142~CTLA4 pair shows significantly positive correlation in most GTEx tissues, the majority of samples in each tissue type expresses considerably low CTLA4 level. This indicates that regulatory T cells are not intensively present in the healthy tissues compared to tumour samples. Tissue/cell type, number of samples and statistics of Spearman correlation (R and P-value) are provided in each panel. Note that in this illustrative figure, one dot represents one tumour sample instead of one patient for the TCGA data. Although it is generally infeasible to evaluate all significant pairs with existing evidences, some of the detected pairs have been verified by previous in vitro/in vivo studies. For instance, the top positive pair miR-196b~HOXA10 (Table S2) has been reported to co-express, and their overexpression results in poor prognosis in patients with gastric cancer [33]. In another two positive pairs miR-483~IGF2 and miR-675~H19, miR-483 was shown to upregulate its host gene IGF2 [34, 35], and miR-675 was identified as the functional regulatory unit of its host lncRNA gene H19 [36]. The negative correlation of miR-141~ZEB1 and its contribution to EMT (epithelial to mesenchymal transition) has consistently been confirmed by previous research [37, 38]. Taken together, these previous studies concurred the biological and clinical significance of the detected miRNA–gene correlations that are conserved across multiple cancer types.
miRactDB illustrates miRNA and gene expression across patient groups of progressive tumour stage for each cancer type, with boxplot (Figure 1C, left). It also profiles the expression difference between tumour and matched normal samples by student’s t-test. The clinical relevance of the constituent miRNA and gene is illustrated by Kaplan–Meier survival curves (Figure 1C, right). Univariate and multivariate Cox regression results of the pair are shown with bar plot (see web page).
miRactDB provides evidence for five potential mechanisms underlying the positive correlation of the queried pair (Figure 1D): (i) the miRNA is intragenic and imbedded in its host gene; (ii) the miRNA activates the gene by inhibiting its upstream suppressor (double-negative); (iii) the miRNA and its partner gene are co-regulated by same transcription factors (double-positive); (iv) the miRNA and its partner gene bear similar H3K27ac enrichment, i.e. co-regulated by the same histone modifications (super-enhancer); and (v) the miRNA upregulates the gene by directing binding to its gene structure (especially promoter or enhancer region). In total, we curated 591 intragenic miRNA–host gene pairs, with their genomic coordinates and species conservation. For the double-negative and double-positive mechanisms, the current version of miRactDB collected analysis results for the 3017 PanCan10 genes. For the direct binding hypothesis, we scanned the potential binding sites on the CDS and promoter regions, not covering the enhancer region at this stage, and removed the 6mer site type, retaining three binding types: 7mer-m8, 7mer-1a and 8mer-1a. The eventual number of binding sites in CDS, promoter and 3′-untranslated region (3′-UTR) are summarized in Figure 1D. These five evidences will be provided in separate sections with table and/or figure on the page.
Significant miRNA–gene interactions in miRactDB
We investigated the correlation patterns of miRNA–gene pairs across four big databases: TCGA tumour (TCGA_t), TCGA normal (TCGA_n), cancer cell line and non-disease tissue (GTEx). Figure 2A summarizes the numbers of significant pairs and their tissue coverage in each dataset. It should be noted that no pairs passed the test in CCLE due to insufficient samples, including SALIVARY_GLAND (n = 2) and SMALL_INTESTINE (n = 1), and we kept blank columns in Table S2. The correlation between miRNAs and genes exemplifies both common and cancer type-specific patterns in all datasets, consistent with previous studies on other cancer genomics topics [39, 40]. As shown in Figure 2A, miRNA–gene pairs exemplify varied cancer coverage in both positive and negative correlations in all datasets. While the majority of miRNA–gene pairs are either only positively or only negatively correlated in all cancers, a considerable part of pairs shows positive correlation in some cancers and negative correlation in others. It is notable that in comparison with negative correlation, the positive correlations are much more prevalent and consistent across cancer types in TCGA_t, CCLE and GTEx. However, this tendency is not significant in TCGA_n. For instance, while the most conservative negative correlation pairs are present in only 16 TCGA cancer types, a large number of pairs display positive correlation in more than 20 cancer types. We also noticed that miRNA–gene pairs showed polarized distribution along the correlation direction in the cancer conditions (TCGA_t and CCLE) compared to normal counterparts (TCGA_n and GTEx). Specifically, whereas the pairs in TCGA_t and CCLE tend to keep their correlation in one direction (either positive or negative) across cancer types, those in GTEx and TCGA_n present more uniform distribution in both correlation directions.
Figure 2.
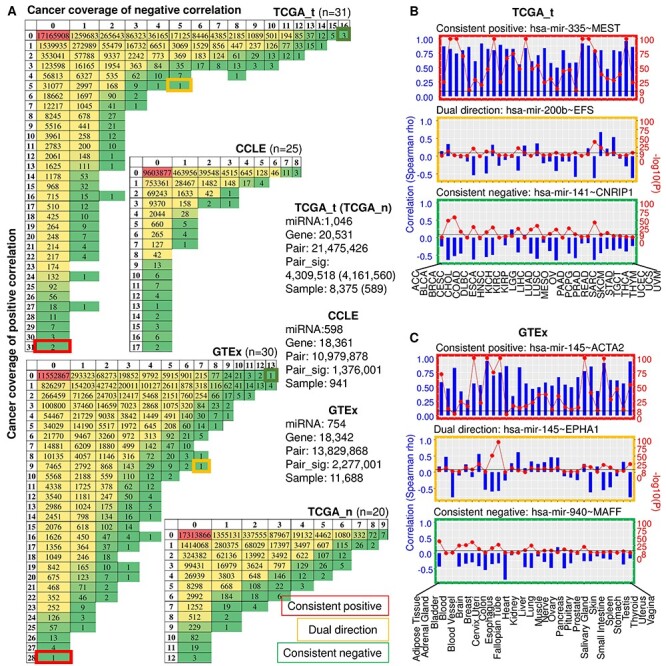
A summary of significant miRNA–gene correlations in TCGA, CCLE and GTEx. (A) Number of significantly positive and negative miRNA–gene correlations grouped by cancer coverage. In CCLE and GTEx, only miRNAs and genes overlapped with those of TCGA were included. Sample numbers refer to the overlap of samples with miRNA and gene expression data. (B and C) Representative top-ranked positive, negative and dual correlations for TCGA (B) and GTEx (C) are shown across cancer/tissue types. Spearman correlation coefficient is depicted by blue bar and P-value by red solid circle.
Figure 2B and C shows representative cases of miRNA–gene correlation with distinct correlation patterns across cancer types. While miR-335~MEST is positively correlated in all 31 TCGA cancer types, miR-141~CNRIP1 shows consistent negative correlation in 16 cancers, and miR-200b~EFS switches correlation direction in different cancer types (Figure 2B). Similar pattern variation in miRNA–gene correlation can be detected in GTEx, although in different pairs. Specifically, miR-145~ACTA2 is significantly positively correlated in 28 out of 30 tissue types, and miR-940~MAFF shows negative correlation in 13 tissues, while miR-145~EPHA1 switches correlation direction across tissue types (Figure 2C). Taken together, these results revealed previously unrecognized heterogeneity in miRNA–gene correlation pattern in all studied scenarios.
Functional annotation of miRNAs and genes from significant correlations
To check both the biological and clinical significance of the top miRNA–gene correlations (Table S2), we first compared the constituent miRNAs and genes from different datasets. While the top miRNA–gene pairs calculated from different datasets have negligible overlap, the constituent miRNAs and genes are largely overlapped, especially between TCGA tumour and normal samples (Figure 3A). Most of the overlapped miRNAs have been previously reported as cancer-associated miRNAs, including the oncogenic miR-17-92 family (miR-17, miR-18a, miR-19a/b, miR-92a) [41] and miR-30 family (miR-30b/c-2/d) [42] and the tumour-suppressor miR-141/200 family [37, 38].
Figure 3.
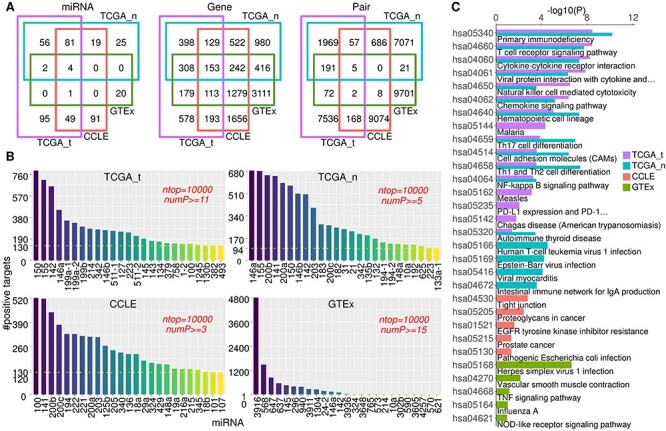
Comparison of miRNAs and genes from top-ranked correlations and their biological significance in TCGA, CCLE and GTEx. (A) Venn diagram showing the overlap between different datasets for miRNAs, genes and pairs as indicated for the top 10 000 positive pairs from four datasets. (B) Top miRNAs ranked by the number of its positively correlated genes counted from the top 10 000 positive pairs in different datasets. numP refers to cancer coverage of the positive pair. (C) KEGG signalling pathway enrichment profiles for the top 200 genes in different datasets. With cutoff P-value <0.05, top 16 pathways for TCGA_t and TCGA_n and top 5 pathways for CCLE and GTEx are shown. See also Figure S2 and Table S3.
We ranked the miRNAs by the number of their positive targets based on the top 10 000 positive pairs in each dataset (Table S2). The TCGA_t, TCGA_n and CCLE datasets displayed similar distribution pattern, which is distinct from the healthy tissue GTEx (Figure 3B). Specifically, the number of positive targets for the top 25 miRNAs gradually diminishes in TCGA_t, TCGA_n and CCLE, while it drops sharply in GTEx. Actually, miR-3916 alone dominates the top 10 000 positive correlations in GTEx. It is also notable that a bunch of miRNAs ranked top 25 in both TCGA_t and TCGA_n sets, including miR-150, miR-155, miR-142 and miR-146a, which were absent from CCLE and GTEx.
We examined the biological meaning of the genes derived from top positive correlations with KEGG pathway enrichment analysis. To do this, we focused on the top 200 genes obtained from each dataset. While genes from CCLE and GTEx are enriched in different KEGG pathways that are both independent of those for TCGA, the genes from TCGA_t and TCGA_n datasets share a surprisingly high proportion of signalling pathways. Specifically, the top genes are most enriched in tight junction pathway in CCLE and herpes simplex virus 1 infection pathway in GTEx. The top pathways shared by TCGA_t and TCGA_n mainly cover the immune cell differentiation and response and cytokine–receptor interactions, including primary immunodeficiency, T-cell receptor signalling and Th1/2/17 cell differentiation (Figure 3C, see Table S3 for complete enrichment profiles). Interestingly, TCGA_t covers the ‘PD-L1 expression and PD-1 checkpoint pathway in cancer’ pathway, implying that genes critical for immunotherapy response are also present in the top positive miRNA–gene correlations. These results indicate that the immune-related signalling is significantly activated in both tumour and adjacent normal tissues, which is distinct from the scenario in cancer cell lines (CCLE) and non-disease healthy tissues (GTEx).
We inspected the clinical relevance of the top miRNAs and genes by survival and Cox regression analysis. As mentioned above, survival analysis were performed by log-rank test and visualized by Kaplan–Meier curves. To further explore how the miRNA–gene pair as a co-effector changes the clinical relevance of each individual factor, we conducted both univariate and multivariate Cox regression analysis with regard to patient survival time on all miRNA–gene pairs. We found that the HR of many miRNAs and genes was changed between univariate and multivariate analysis, indicating that the miRNA–gene interaction altered their role as a single prognostic factor. Table S4 lists the Cox regression results for the three representative miRNA–gene pairs: miR-196b~HOXA10, miR-150~ITK and miR-142~CTLA4. miR-196b and HOXA10 switched from a tumour-suppressor role (HR < 1) in univariate analysis to an oncogene role (HR > 1) in multivariate analysis in pheochromocytoma and paraganglioma (PCPG). And this switch was also present in the miR-150~ITK pair. In contrast, the HR of miR-142 and CTLA4 was dramatically reduced in the multivariate analysis in thymoma (THYM). These results verified that the interactions of a miRNA–gene pair might alter the role of each component in patient prognosis.
Interpreting the binding sites on gene CDS and promoter regions
miRactDB provides the miRNA binding sites across whole gene body structure, including promoter, CDS and 3′-UTR. Since binding sites on 3′-UTR are adopted from TargetScan database, here we only summarize and interpret the scanning results for CDS and promoter regions. For each pair, we counted the number of binding sites of three different types (7mer-m8, 7mer-1a and 8mer) separately and then multiplied the three numbers by different scalars (1, 1.25 and 1.5, respectively) to account for their relative binding affinity. These scalars only reflect the relative potency of binding. After that, we added the three weighted numbers together as the overall binding affinity score for this queried pair. We then constructed a table based on these binding scores, with rows and columns corresponding to the miRNAs and genes, respectively, and each grid representing their binding affinity score. To obtain the most active miRNAs, we first sorted the rows based on the sum score of each row and then sorted the columns based on the sum score of each column. Figure 4 shows the top 10 miRNAs and top 20 genes in the affinity score table for CDS binding (Figure 4A), promoter of plus-strand gene (Figure 4B) and promoter of minus-strand gene (Figure 4C). While the top genes for the 3 scenarios are very different, the top 10 miRNAs are the same and ranked in the same order, indicating that the small set of top miRNAs can actively bind to both the CDS and promoter regions of (typically different) genes. We noticed three members of the mucin gene family ranked top 5 in the CDS binding table, including MUC5B, MUC12 and MUC4. This might be partially attributable to their superlong gene sequence (with 39K, 49K and 65K bases, respectively). Here, we did not normalize the binding score to gene length, since we focused on comparing the activity of the miRNAs rather than genes and normalization won’t change the rank of the miRNAs.
Figure 4.
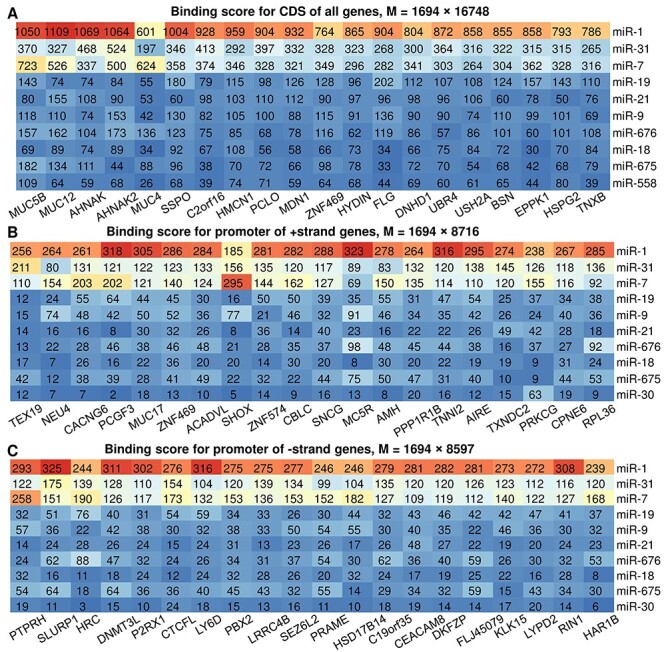
A summary of miRNA binding sites on gene CDS and promoter regions. (A) Top miRNAs (n = 10) and genes (n = 20) ranked by binding score calculated on the coding region for all genes. M refers to the total number of miRNAs and genes in the whole table. (B and C) Top miRNAs (n = 10) and genes (n = 20) ranked by binding score calculated on the promoter region for genes of positive (B) and negative (C) strand.
Discussion
miRactDB addressed the long-term scarcity of an integrative resource facilitating comprehensive characterization of any designated miRNA–gene pair, including its correlation, biological and clinical significance and potential molecular mechanisms, in a pan-cancer and normal-cancer comparative manner. Although miRactDB was originally designed to focus on the special yet surprisingly prevalent case—the positive miRNA–gene correlation across cancer types—we calculated correlations of all possible pairs and collected both positive and negative correlations with statistical significance in TCGA, CCLE and GTEx databases. In addition, we for the first time illustrated the expression level of each miRNA and gene in patients grouped by tumour stage (including adjacent normal sample), enabling the biologists to clearly see the dynamic change of expression over tumour progression for any cancer type.
Since miRNAs have been widely reported to activate gene expression by directly binding to the promoter regions of the target gene, we conducted a complete de novo scanning on the TSS ± 2 kb region of each gene for complementarity with the well-established TargetScan tool. We also scanned the coding sequence (CDS) region of each gene. By removing the less effective 6mer binding type, the retained scanning results are expected to provide promising candidates for further experimental investigation. From the scanning results on promoter and CDS region (Figure 4), we frequently observed that one miRNA can target on multiple genes and one gene bears binding sites for multiple miRNAs, which is well consistent with the 3′-UTR binding scenario. Future work might consider two aspects: first, the species conservation can be taken into account as did by the original TargetScan working on 3′-UTR; second, other regions such as the enhancer and 5′-untranslated region (5′-UTR) can be also scanned to identify conservative binding sites for corresponding miRNAs.
It should be noted that in the present study, the specific cutoffs for Spearman correlation coefficient |R| and P-value are not essential, as we performed function/mechanism analysis only on the very top pairs (top 200 genes) ranked by significance level, which turned out to be stringent enough in both |R| and P-value (Table S2). A major purpose of this study is to provide the community with the miRNA–gene correlation profiles across cancer/cell line/tissue types in different scenarios (TCGA, CCLE and GTEx) for inquiry. We intended to include as many nontrivial pairs as possible in the database. For this purpose, we provided the actual |R| and P-values on the output figures for the users to decide for significance. Despite that, we further explored how the cutoff of |R| affects the number of miRNA–gene pairs detected as significant by checking on a range of thresholds for |R|, from 0 to 1, with an increment of 0.1 (Figure S1). While the number of significant pairs generally decreases with the increase of cutoff of |R| as expected, the numbers kept very well until |R| = 0.3. We even observed a considerable number of pairs remained until |R| = 0.6 (Figure S1, left), and the median |R| of the top 10 000 pairs stayed at the level of 0.4–0.6 for all scenarios (Figure S1, right). This is not surprising since, although we set a relatively loose criterion on the coefficient |R|, we imposed a very strict restriction on the significance level (Methods).
The current miRactDB can be improved on two aspects. First, the current version only focuses on the PanCan10-positive correlations for double-negative and double-positive mechanisms. This includes 16 554 pairs involving 329 miRNAs and 2786 genes (Spearman correlation), which are sufficient to cover most important miRNAs and genes participating in the core biological processes of cancer hallmarks. The present work could be extended by searching the intermediate genes for the remaining pairs based on our proposed pipeline. Second, the present work only explores miRNA–gene interactions and their pattern shift between tumour and normal samples; future work might integrate other genomic alternations such as somatic mutations [2, 39] to better understand the role of positive correlations in human tumorigenesis. This is reasoned by the fact that genetic mutations in the regulatory regions such as 3′-UTR, promoter, enhancer or even CDS regions might influence the miRNA binding and subsequent gene regulation [43, 44]. We will continue to extend and improve it based on further analysis of the miRNA–gene interactions and prospective users’ feedbacks.
Our integrative analysis represents an insightful example of leveraging this comprehensive resource to test interesting hypotheses. Particularly, we for the first time corroborated the hypothesis regarding the gradual oncogenic transformation of histologically normal tissues [45] from the angle of miRNA–gene interactions (Figure 3). Furthermore, with de novo scanning on the CDS and promoter regions of partner genes for miRNA binding sites, we for the first time revealed that the top active miRNAs for CDS and promoter binding are largely overlapped, indicating that a small group of miRNAs dominates the regulatory networks across various biological processes (Figure 4). Cox regression analysis results showed that miRNA–gene interactions substantially altered the role of each component of the pair in patient prognosis in particular cancer types. Although targeted experiments are warranted to confirm these findings obtained from high-throughput data, the highly consistent patterns detected in large-scale, cross-platform datasets pose high priority for further investigation. We believe that the unique features of miRactDB as elucidated above make it much useful and critical for the community of both cancer biology and genomics.
Key Points
miRactDB represents an unprecedentedly comprehensive study on the pattern switch in miRNA–gene interaction between activation and suppression and between normal and cancer conditions based on multi-omics data. The current version of miRactDB includes 21 475 426 miRNA–gene pairs across 31 cancer types and 20 normal tissue types involving 8375 patients from TCGA; 10 979 878 pairs across 25 tissue origins involving 941 cell lines from CCLE; and 13 829 868 pairs across 30 non-disease healthy tissues involving 11 688 samples from GTEx.
miRactDB provides the first resource of evidences for potential mechanisms underlying the conserved positive miRNA–gene correlation: (i) the miRNA is intragenic and imbedded in its host gene; (ii) the miRNA activates the gene by inhibiting its upstream suppressor (double-negative); (iii) the miRNA and its partner gene are co-regulated by the same transcription factors (double-positive); and (iv) the miRNA and its partner gene bear similar H3K27ac enrichment, i.e. co-regulated by the same histone modifications (super-enhancer).
miRactDB for the first time identifies miRNA binding sites along the whole gene structure besides 3′-UTR, especially in the coding sequence (CDS) and promoter regions of partner genes. We further revealed that top active miRNAs tend to bind both CDS and promoters of different genes.
Supplementary Material
Hua Tan received his PhD degree in Mathematics from Beijing Normal University, China, in 2011. His research interests include complex system modelling, pan-cancer genomics and machine learning in biomedicine.
Pora Kim received her PhD degree in Bioinformatics from Ewha Womans University, Korea, in 2013. Her research interests are bioinformatics, computational biology and computational cancer genomics.
Peiqing Sun received his PhD degree in Cancer Biology from University of Iowa, United States, in 1995. His research interests include cellular defence and metabolism, aberrant signalling pathways in tumour cells, cell growth and survival, molecular cancer epidemiology, gene–environment interactions and cancer control.
Xiaobo Zhou received his PhD degree in Applied Mathematics from Beijing University, China, in 1998. His research interests are bioinformatics, systems biology, imaging informatics and clinical informatics.
Funding
National Institute of Health (grants GM123037 and U01AR069395); NIH (grants R01CA172115 and R01CA131231 to P.S.).
Conflict of Interest statement
The authors declare no potential conflicts of interest.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 2. Tan H, Bao J, Zhou X. A novel missense-mutation-related feature extraction scheme for 'driver' mutation identification. Bioinformatics 2012;28:2948–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–97. [DOI] [PubMed] [Google Scholar]
- 4. Friedman RC, Farh KK, Burge CB, et al. . Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009;19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dhawan A, Scott JG, Harris AL, et al. . Pan-cancer characterisation of microRNA across cancer hallmarks reveals microRNA-mediated downregulation of tumour suppressors. Nat Commun 2018;9:5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Agarwal V, Bell GW, Nam JW, et al. . Predicting effective microRNA target sites in mammalian mRNAs. elife 2015;4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res 2019;47:D155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 2015;43:D146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chou CH, Shrestha S, Yang CD, et al. . miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 2018;46:D296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang JH, Li JH, Shao P, et al. . starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res 2011;39:D202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods 2015;12:697. [DOI] [PubMed] [Google Scholar]
- 12. Wong NW, Chen Y, Chen S, et al. . OncomiR: an online resource for exploring pan-cancer microRNA dysregulation. Bioinformatics 2018;34:713–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahmed M, Nguyen H, Lai T, et al. . miRCancerdb: a database for correlation analysis between microRNA and gene expression in cancer. BMC Res Notes 2018;11:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan H, Huang S, Zhang Z, et al. . Pan-cancer analysis on microRNA-associated gene activation. EBioMedicine 2019;43:82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007;318:1931–4. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y, Fan M, Zhang X, et al. . Cellular microRNAs up-regulate transcription via interaction with promoter TATA-box motifs. RNA 2014;20:1878–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang V, Place RF, Portnoy V, et al. . Upregulation of cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res 2012;40:1695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harrow J, Frankish A, Gonzalez JM, et al. . GENCODE: the reference human genome annotation for the ENCODE project. Genome Res 2012;22:1760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barretina J, Caponigro G, Stransky N, et al. . The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Griffiths-Jones S, Grocock RJ, van Dongen S, et al. . miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006;34:D140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benson DA, Karsch-Mizrachi I, Lipman DJ, et al. . GenBank. Nucleic Acids Res 2005;33:D34–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kanehisa M, Furumichi M, Tanabe M, et al. . KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017;45:D353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu G, Wang LG, Han Y, et al. . clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014;42:D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loven J, Hoke HA, Lin CY, et al. . Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wishart DS, Feunang YD, Guo AC, et al. . DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pinero J, Bravo A, Queralt-Rosinach N, et al. . DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res 2017;45:D833–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu X, Wang S, Meng F, et al. . SM2miR: a database of the experimentally validated small molecules' effects on microRNA expression. Bioinformatics 2013;29:409–11. [DOI] [PubMed] [Google Scholar]
- 30. Huang Z, Shi J, Gao Y, et al. . HMDD v3.0: a database for experimentally supported human microRNA-disease associations. Nucleic Acids Res 2019;47:D1013–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jiang Q, Wang Y, Hao Y, et al. . miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 2009;37:D98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Syn NL, Teng MWL, Mok TSK, et al. . De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol 2017;18:e731–41. [DOI] [PubMed] [Google Scholar]
- 33. Lim JY, Yoon SO, Seol SY, et al. . Overexpression of miR-196b and HOXA10 characterize a poor-prognosis gastric cancer subtype. World J Gastroenterol 2013;19:7078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu M, Roth A, Yu M, et al. . The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev 2013;27:2543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Veronese A, Lupini L, Consiglio J, et al. . Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res 2010;70:3140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Keniry A, Oxley D, Monnier P, et al. . The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol 2012;14:659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cursons J, Pillman KA, Scheer KG, et al. . Combinatorial targeting by microRNAs co-ordinates post-transcriptional control of EMT. Cell Syst 2018;7:77–91 e7. [DOI] [PubMed] [Google Scholar]
- 38. Gregory PA, Bert AG, Paterson EL, et al. . The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008;10:593–601. [DOI] [PubMed] [Google Scholar]
- 39. Tan H, Bao J, Zhou X. Genome-wide mutational spectra analysis reveals significant cancer-specific heterogeneity. Sci Rep 2015;5:12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vogelstein B, Papadopoulos N, Velculescu VE, et al. . Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ 2013;20:1603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Su W, Hong L, Xu X, et al. . miR-30 disrupts senescence and promotes cancer by targeting both p16(INK4A) and DNA damage pathways. Oncogene 2018;37:5618–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tan H. The association between gene SNPs and cancer predisposition: correlation or causality? EBioMedicine 2017;16:8–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tan H. On the protective effects of gene SNPs against human cancer. EBioMedicine 2018;33:4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aran D, Camarda R, Odegaard J, et al. . Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat Commun 2017;8:1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.