

Abstract
Virulence factors (VFs) enable pathogens to infect their hosts. A wealth of individual, disease-focused studies has identified a wide variety of VFs, and the growing mass of bacterial genome sequence data provides an opportunity for computational methods aimed at predicting VFs. Despite their attractive advantages and performance improvements, the existing methods have some limitations and drawbacks. Firstly, as the characteristics and mechanisms of VFs are continually evolving with the emergence of antibiotic resistance, it is more and more difficult to identify novel VFs using existing tools that were previously developed based on the outdated data sets; secondly, few systematic feature engineering efforts have been made to examine the utility of different types of features for model performances, as the majority of tools only focused on extracting very few types of features. By addressing the aforementioned issues, the accuracy of VF predictors can likely be significantly improved. This, in turn, would be particularly useful in the context of genome wide predictions of VFs. In this work, we present a deep learning (DL)-based hybrid framework (termed DeepVF) that is utilizing the stacking strategy to achieve more accurate identification of VFs. Using an enlarged, up-to-date dataset, DeepVF comprehensively explores a wide range of heterogeneous features with popular machine learning algorithms. Specifically, four classical algorithms, including random forest, support vector machines, extreme gradient boosting and multilayer perceptron, and three DL algorithms, including convolutional neural networks, long short-term memory networks and deep neural networks are employed to train 62 baseline models using these features. In order to integrate their individual strengths, DeepVF effectively combines these baseline models to construct the final meta model using the stacking strategy. Extensive benchmarking experiments demonstrate the effectiveness of DeepVF: it achieves a more accurate and stable performance compared with baseline models on the benchmark dataset and clearly outperforms state-of-the-art VF predictors on the independent test. Using the proposed hybrid ensemble model, a user-friendly online predictor of DeepVF (http://deepvf.erc.monash.edu/) is implemented. Furthermore, its utility, from the user’s viewpoint, is compared with that of existing toolkits. We believe that DeepVF will be exploited as a useful tool for screening and identifying potential VFs from protein-coding gene sequences in bacterial genomes.
Keywords: virulence factor prediction, recognition, sequence analysis, machine learning, deep learning, ensemble learning
Introduction
The spread of infectious diseases caused by pathogenic bacteria is a major cause of human and animal mortality [1–3], and predictions for how dire this problem will grow due to increases in drug-resistant bacteria are being revised upwards [4]. The pathogenesis associated with a bacterial infection can be complex but, in many cases, is primarily driven by virulence factors (VFs): proteins produced by the bacterium that enable it to persist, grow and do damage to the tissues of its human or animal host [3, 5, 6]. Indeed, the presence or absence of a VF can be the difference between two closely related species: Bacillus anthracis (which causes anthrax) and Bacillus cereus (a non-harmful soil bacterium) differ chiefly in a set of VFs encoded by plasmids found in B. anthracis [7]. Escherichia coli and Vibrio cholerae are not usually dangerous to humans, but acquisition of one or a few genes makes them deadly pathogens [8–10]: the primary VF in enterohemorrhagic E. coli O157:H7 is a shiga-like toxin that directly causes the kidney failure that can lead to death [9] and, in V. cholerae, acquisition of the gene encoding cholera toxin transduces the otherwise harmless species to be a highly virulent pathogen causing cholera [10].
In addition to toxins, other VFs mediate a vast array of functions, including mediating bacterial adhesion to host tissues, modulation of the immune system by various means, lysis of red blood cells, etc. Effectively identifying VFs has been suggested as a means to devise novel drug/vaccine targets for preventing and treating infectious diseases [11–13]. However, the great diversity in the structure and function of VFs should preclude the use of computational methods as means to finding VFs from large-scale protein sequences. It was therefore initially surprising to see the successful publication of several key studies in this area [11, 14–21] (summarized in Table S1).
One strategy was to greatly narrow the focus of the VF types being addressed. The software program SPAAN was developed to predict a single class of VF, the adhesins, using a neural network based on five different features (amino acid frequencies, multiplet frequencies, dipeptide frequencies, charge composition and hydrophobic composition) pertinent to the frequencies or compositions of amino acids, and achieved a considerable accuracy [16]. In the second study, a broader range of VFs were used to construct an inspiring bi-layer cascade support vector machine (SVM) using the stacking strategy for predicting VFs together with sequence-based and position-specific scoring matrix (PSSM)-based features, and a user-friendly web server was developed and called VirulentPred [11]. Further three algorithms were proposed to predict virulent proteins, and while their performance was only marginally better than VirulentPred, this independently supported the prospect that the VFs of diverse structure, function and origins could be detected with a single tool [14, 15, 17]. Later, Zheng et al. [18] proposed a novel network-based method based on the protein–protein interaction networks to predict VFs in the proteomes of six bacteria species. This method achieved a high-prediction performance on their benchmark tests; however, it was not expanded and implemented as a universal VF prediction predictor and, therefore, could not be further validated and applied in general and practical scenarios. Another VF prediction method took into account the protein–protein interactions based on the STRING database and KEGG pathways [21]. This method was validated through analysis of three specific species recorded in the VFDB [22]. In 2014, the MP3 method [19], implemented as both standalone tool and web server, combined an SVM classifier trained with sequence-based features and the hidden Markov model to predict virulent proteins. MP3 showed a better performance over VirulentPred in three different datasets. More recently, Rentzsch et al. [20] proposed (termed as PBVF) effective negative data selection strategies to construct novel and more diverse data set. Based on this, they explored various SVM-based and random forest (RF)-based classifiers, a direct sequence similarity (seqsim)-based method and their combinations for VF prediction. They showed that seqsim plays an unparalleled important role in VF identification. Its combination with other features could lead to performance improvement when being incorporated to machine learning (ML) models. In addition, the performance of the seqsim-based method turned out to be much better than that of MP3 based on the same training data set.
With the recent, rapid advances of artificial intelligence, an increasingly diverse range of deep learning (DL) and classical ML algorithms have been developed. This provides opportunities to develop and apply new methods to address the VF prediction problem. In addition, the new VF database (Victors) [3] and another two databases (VDFB [22] and Pathosystems Resource Integration Center (PATRIC) [23, 24]) provide comprehensive data sources for constructing ML models to enable better VF prediction from sequence data.
In this study, we present DeepVF, a DL-based hybrid framework for accurately identifying VFs from genome sequence data. We assembled an up-to-date benchmark dataset by collecting positive samples from multiple recently published VF databases [3, 22, 25] and negative samples from PBVF [20]. DeepVF employs four classical ML methods and three DL algorithms (Figure 1): to this end, we constructed a total of 62 baseline models and effectively integrated them using the stacking strategy inspired by previous work [11]. For classical ML, a comprehensive set of traditional ML features (including sequence-based features, physicochemical property-based features and evolutionary information-based features) was extracted to represent the characteristic features of VFs. Accordingly, four commonly used classical ML algorithms [26–29], including RF, SVM, extreme gradient boosting (XGBoost) and multilayer perceptron (MLP), were utilized to construct such ML baseline models. For DL, three different DL techniques were employed, namely convolutional neural networks (CNNs), long short-term memory networks (LSTMs) and deep neural networks (DNNs). Sequence-to-vector encoding was used to train these individual DL baseline models. Next, the prediction labels of the baseline models (including ML baseline models and DL baseline models) were used as inputs to train the final meta model using the XGBoost algorithm. We show that the final meta model that is based on the stacking strategy achieves a better overall performance compared with the baseline models on cross-validation and independent tests. Moreover, DeepVF achieves significantly a better performance than several existing state-of-the-art methods for VF prediction in terms of ACC (0.812), F-value (0.807), Matthew’s correlation coefficient (MCC) (0.624) and AUC (0.896) on the independent test. The results highlight the effectiveness of the proposed hybrid framework. As an implementation of this method, we also established a user-friendly online web server of DeepVF, which is publicly accessible at http://deepvf.erc.monash.edu/. The utility of DeepVF is further compared with that of other existing toolkits, focusing on user guidance, data requirements, computational costs and output interpretation. We anticipate that DeepVF will greatly facilitate users’ efforts for screening and mining more novel putative VFs from sequence information in bacterial pathogens.
Figure 1.
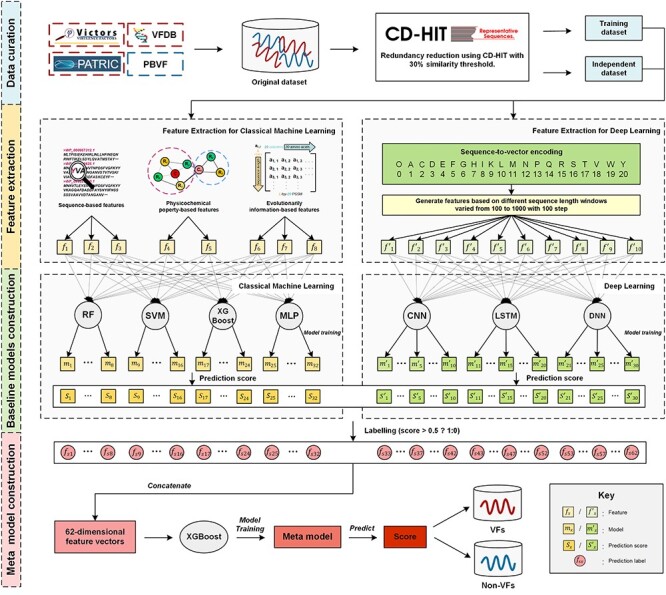
An overview of the ML- and DL-based hybrid framework of DeepVF for predicting VFs in bacterial pathogens. Schematic displaying the four stages in construction of DeepVF.
Methods
The overall workflow of the development of DeepVF (Figure 1) can be summarized by the following steps, which include: (i) data collection and curation; (ii) feature extraction; (iii) model training and optimization and (iv) integrative model construction.
Data collection and curation
To construct an up-to-date dataset, we collected 9749 VFs from the three public databases (Victors [3], VDFB [22] and PATRIC [25]) associated with several bacterial pathogens. For the negative samples, we extracted 66 982 non-VFs from PBVF [20], which was constructed based on the proposed effective negative data selection strategy. Next, the CD-HIT [30] program was applied to cluster the sequences and remove the sequence redundancy in the positive and negative datasets, respectively. The sequence identity threshold was set as 0.3 to remove the sequence redundancy (Table S2). As a result, the final non-redundant dataset contained 3576 VFs and 4910 non-VFs. Among these, 576 VFs and 576 non-VFs were randomly selected as the independent test dataset, while the remaining were used as the training dataset (3000 VFs and 4334 non-VFs). To solve the data imbalance problem, we constructed five balanced training datasets (each of them contained 3000 VFs and 3000 non-VFs) by randomly selecting the same number of non-VFs as that of VFs.
Feature extraction
Comprehensive and effective feature extraction is a key to construct stable and reliable models with a competitive performance. In view of the differences between classical ML and DL algorithms, two different types of feature extraction methods were used in this study. A detailed description of these methods is provided in the following subsections.
Feature extraction for classical ML
We extracted three major groups of features, including sequence-based features, physicochemical property-based features and evolutionary information-based features [26, 28, 29, 31, 32] to systematically explore the representative and specific patterns of VF proteins.
Group 1: Sequence-based feature group. Three features primarily describe the frequencies or compositions of sequence elements, namely amino acid composition (AAC), dipeptide composition (DPC) and dipeptide deviation from expected mean (DDE) [33].
Amino Acid composition. AAC defines the frequency of an amino acid type present in a protein sequence [34], which can be calculated as follows:
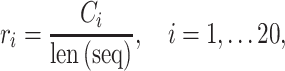 |
(1) |
where 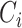 denotes the number of occurrences of the
denotes the number of occurrences of the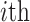 amino acid,
amino acid, 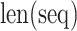 is the length of the query sequence and
is the length of the query sequence and 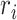 is the ratio of amino acid
is the ratio of amino acid 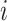 in the whole sequence. As a result, a 20-dimensional feature vector is generated.
in the whole sequence. As a result, a 20-dimensional feature vector is generated.
Dipeptide composition. DPC measures the relevant frequencies of all possible dipeptides in a given protein sequence [29]. Using DPC, any protein sequence can be encoded as a 400-dimensional feature vector, in which each element 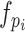 can be calculated as follows:
can be calculated as follows:
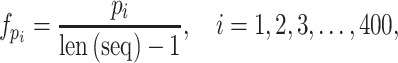 |
(2) |
where 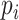 and
and 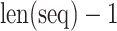 denote the number of occurrences of dipeptide
denote the number of occurrences of dipeptide 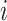 in the sequence and the total number of dipeptides in a sequence of length
in the sequence and the total number of dipeptides in a sequence of length 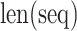 , respectively.
, respectively.
Dipeptide deviation from expected mean. DDE [33] generates a 400-dimensional feature vector from a protein sequence in three steps. The first step is to calculate the DPC 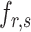 as follows:
as follows:
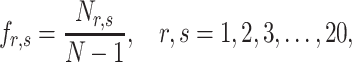 |
(3) |
where 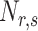 is the number of the dipeptide
is the number of the dipeptide 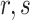 in the sequence.
in the sequence.  represents the total number of dipeptides in the sequence of length
represents the total number of dipeptides in the sequence of length 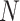 . Second, the theoretical mean
. Second, the theoretical mean 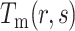 and theoretical variance
and theoretical variance 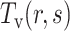 for the dipeptide
for the dipeptide 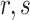 can be calculated by
can be calculated by
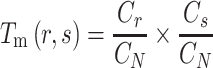 |
(4) |
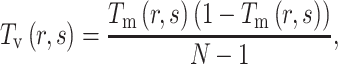 |
(5) |
where 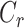 and
and 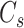 are the numbers of codons coded for the corresponding amino acids, respectively.
are the numbers of codons coded for the corresponding amino acids, respectively. 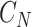 is the total number of possible codons that are not any of the three stop codons (64–3 = 61). Finally, the DDE of the dipeptide
is the total number of possible codons that are not any of the three stop codons (64–3 = 61). Finally, the DDE of the dipeptide 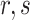 can be calculated as
can be calculated as
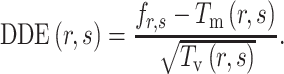 |
(6) |
Group 2: Physicochemical property-based features. Physicochemical property-based features describe the statistical information pertinent to the physicochemical properties of amino acids in a protein sequence.
Pseudo-AAC. Pseudo-ACC (PAAC) [35] has been successfully applied to solve various problems in bioinformatics tasks that are related to protein sequence analysis [36]. In this algorithm, let 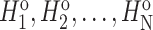 denote the numbers of N original property values of 20 natural amino acids. We obtain
denote the numbers of N original property values of 20 natural amino acids. We obtain 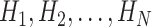 using the following formula:
using the following formula:
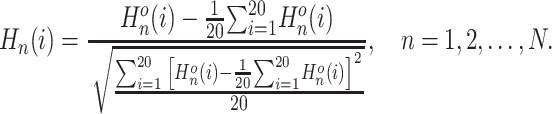 |
(7) |
For amino acids  and
and 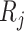 , their correlation can be defined as
, their correlation can be defined as
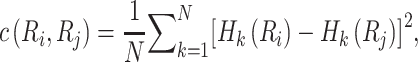 |
(8) |
where 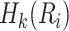 is the kth property of
is the kth property of 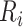 . For a sequence of length L, the sequence order-correlated factors
. For a sequence of length L, the sequence order-correlated factors 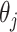 can be defined as
can be defined as
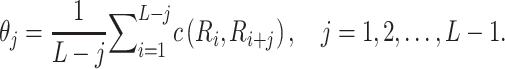 |
(9) |
Accordingly, a (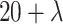 (
(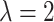 ))-dimensional vector can be obtained by
))-dimensional vector can be obtained by
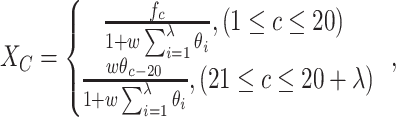 |
(10) |
where 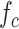 denotes the frequency of amino acid
denotes the frequency of amino acid 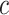 in the sequence, while
in the sequence, while 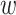 is the weighting factor for the sequence-order with the default value of 0.1.
is the weighting factor for the sequence-order with the default value of 0.1.
Quasi-sequence order. Quasi-sequence order (QSO) describes the probability of occurrence of amino acids in the protein sequence based on two distance matrices, namely the Schneider–Wrede and the Grantham distance matrices [37]. For any given distance matrix (either Schneider–Wrede or Grantham), we calculate
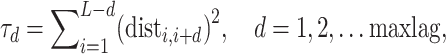 |
(11) |
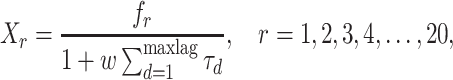 |
(12) |
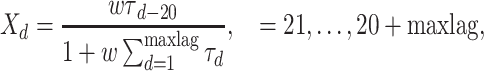 |
(13) |
where L is the length of the sequence, 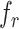 denotes the normalized occurrence for amino acid
denotes the normalized occurrence for amino acid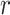 ,
, 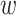 is the weighting factor with the default value of 0.1,
is the weighting factor with the default value of 0.1, 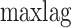 is set as 10 and
is set as 10 and 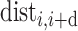 denotes the distance between amino acids at the
denotes the distance between amino acids at the 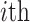 position and
position and 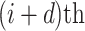 position according to the distance matrix. As a result, a (
position according to the distance matrix. As a result, a (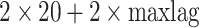 )-dimensional vector will be obtained for the sequence.
)-dimensional vector will be obtained for the sequence.
Group 3: Evolutionary information-based features. Evolutionary information in the form of PSSM can add potentially useful value to the analysis of protein sequences [38–40]. The PSSM describes the characteristics of each amino acid at different positions in the protein sequence. It was shown that the use of PSSM can make a difference in many biological classification problems. In this work, we performed the PSI-BLAST search against the uniref50 database with the parameters 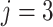 and
and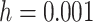 to get the PSSM profiles. In this way, a sequence with the length of N will generate an N
to get the PSSM profiles. In this way, a sequence with the length of N will generate an N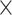 20 matrix as there are 20 native amino acid types. Accordingly, three types of evolutionary information-based features are represented through different forms of PSSM transformation [39], which include PSSM-composition [40], S-FPSSM [41] and Residue Probing Method (RPM)-PSSM [42].
20 matrix as there are 20 native amino acid types. Accordingly, three types of evolutionary information-based features are represented through different forms of PSSM transformation [39], which include PSSM-composition [40], S-FPSSM [41] and Residue Probing Method (RPM)-PSSM [42].
PSSM-composition. PSSM-composition [40] represents a row transformation of the original PSSM, by summing and averaging the row vectors that belong to the same type of amino acid. Thus, a 20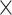 20 matrix can be obtained using the following formula:
20 matrix can be obtained using the following formula:
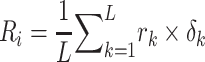 |
(14) |
subject to
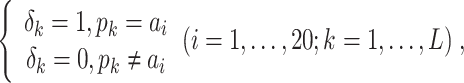 |
(15) |
where 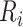 denotes the
denotes the 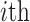 row of the transformed matrix,
row of the transformed matrix, 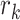 describes the
describes the 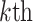 row of the original PSSM,
row of the original PSSM, 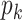 is the
is the 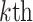 amino acid in the original sequence,
amino acid in the original sequence, 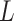 is the length of the sequence and
is the length of the sequence and 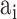 denotes the
denotes the 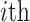 amino acid of the 20 natural amino acids.
amino acid of the 20 natural amino acids.
S-FPSSM. S-FPSSM [41] extracts evolutionary information from a formulated matrix FPSSM obtained from the original PSSM profile, by setting all scores less than 0–0 and all positive scores greater than n to n (n = 7). Based on the FPSSM, the S-FPSSM can be calculated as follows:
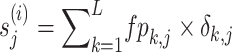 |
(16) |
subject to
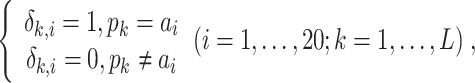 |
(17) |
where 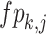 denotes the element of the
denotes the element of the 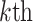 row and the
row and the 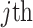 column of the FPSSM,
column of the FPSSM, 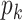 denotes the
denotes the 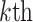 residue in the original protein sequence, while
residue in the original protein sequence, while 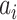 denotes the
denotes the 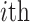 natural amino acid. As a result, the sequence is encoded as a 400-dimensional vector.
natural amino acid. As a result, the sequence is encoded as a 400-dimensional vector.
RPM-PSSM. RPM-PSSM transforms the original PSSM by borrowing the probe concept employed in microarray technologies [42]. RPM-PSSM first transforms the original PSSM profile into a new ‘filtered’ matrix (named as PPSSM) by setting all negative elements to zero. Then, the RPM-PSSM features can be calculated based on the PPSSM using the similar formula as the PSSM-composition
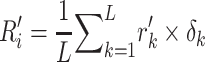 |
(18) |
subject to
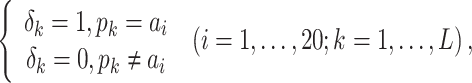 |
(19) |
where 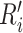 denotes the
denotes the 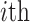 row of the resultant matrix,
row of the resultant matrix, 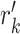 denotes the
denotes the 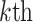 row of the PPSSM,
row of the PPSSM, 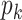 denotes the
denotes the 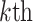 residue in the original protein sequence and
residue in the original protein sequence and 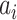 denotes the
denotes the 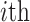 native type of amino acid. Similarly, a 400-dimensional feature vector is obtained.
native type of amino acid. Similarly, a 400-dimensional feature vector is obtained.
Feature extraction for DL
Compared with classical ML algorithms, it is often not straightforward for DL to identify useful and relevant information from complex features for making prediction, especially considering the limited volume of data. Alternatively, the sequence-to-vector encoding has been successfully applied to DL modelling in order to address a wide range of biological sequence analysis tasks [43–45]. It conveniently converts protein sequences into numerical vectors, which can subsequently serve as the input to train DL models. Here, the 20 native types of amino acids in protein sequences are mapped into 1, 2, 3, …, 20 according to the alphabetical order. As can be seen from Figure S1, the lengths of the protein sequences in the dataset were mostly distributed within the 1000 limit. In view of this, we extracted different dimensional features based on different sequence length windows, ranging from 100 to 1000, with an interval of 100. Specifically, when the length of a protein sequence was less than the specified window size, the corresponding numeric vectors were padded with 0 at the tail. As a result, 10 feature vectors in different dimensions (i.e., L100, L200, …, and L1000) were generated for constructing the DL models, based on different window sizes.
Model training and optimization
In this work, four classical ML methods (RF, SVM, XGBoost and MLP) and three popular DL algorithms (CNN, LSTM and DNN) were employed. Their detailed implementations and parameter optimizations are introduced below.
Classical ML
RF [46] integrates multiple decision trees based on the bagging strategy. It has been successfully applied to address a variety of research questions in bioinformatics and computational biology with excellent effectiveness and stability [26, 29, 47, 48]. In this study, the RF algorithm was implemented using the randomForest package [49] in R. The number of randomly selected features (mtry) was optimized, and the number of decision trees was set to 1000. The parameter tuning was based on the training dataset, and the tuned parameter was then applied to conduct all experiments in this study. These apply to the following ML parameter tuning (if necessary).
SVM has been shown to achieve reasonably good performance on small-scale datasets with many successful applications to biological classification problems [11, 28, 29]. Training of SVM models involves the optimization of two essential parameters, i.e. the Cost C and Gamma γ. In this study, we trained the SVM models with the Gaussian radial basis kernel using the e1071 package (https://cran.r-project.org/web/packages/e1071/) in R. The parameters C ∈ {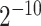 ,
, 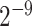 , …, 1,
, …, 1, 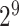 ,
, 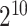 } and Gamma γ ∈ {
} and Gamma γ ∈ {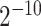 ,
, 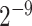 , …, 1,
, …, 1, 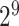 ,
,  } were optimized by grid search.
} were optimized by grid search.
As an effective implementation and extension of gradient boosting decision tree, XGBoost is specifically designed to handle industrial-scale, ‘big’ data and parallel computing [50]. XGBoost can generate a strong learning model by linearly integrating weak learning models composed of decision trees. As such, it has been widely applied to solve a variety of bioinformatics tasks [27, 51, 52]. In this study, XGBoost was implemented using the xgboost package (https://cran.r-project.org/web/packages/xgboost) in R language. We performed a random search [53] to tune the multiple parameters (Table S3) by maximizing the AUC value based on 5-fold cross validation test.
MLP is a feed forward artificial neural network that consists of multiple layers, i.e. the input layer, output layer and one or more non-linear layers (hidden layers). MLP has demonstrated its usefulness by achieving superior performance in a number of classification problems, including secreted protein classification [29], molecular classification [54] and automated cancer diagnosis [55]. Using the R implementation of Keras (https://keras.rstudio.com/), we trained the MLP model with three hidden layers, with the number of nodes for each hidden layer set to 256 and a dropout rate of 0.05. To prevent overfitting, the value of epochs was set to 30. In addition, we defined the callbacks function, which allows the early termination of the iteration while preserving the optimal model.
Deep learning
With the explosive growth of biological data in recent years, the advantages and utility of DL techniques have been demonstrated by numerous applications in biological research [56–61].
CNN is a class of feed forward neural networks with convolutional computation and deep structure. In CNNs, each neuron connects only to a small number of neurons in the upper layer, instead of all neurons. This architecture is able to achieve a better learning effect by retaining, as much as possible, the important parameters (often a much smaller number) while removing unimportant parameters [62]. In recent years, many biomedical classification problems have been addressed using CNNs, such as DNA–protein binding prediction [63], protein solubility prediction [44], somatic mutation detection [64] and cancer imaging analysis [65, 66]. In this work, the embedding layer was added to the first layer (output_dim: 256) with a dropout rate of 0.01. Next, the obtained symbols were fed into a one-dimensional convolutional layer (filters: 64, kernel_size: 16, activation: relu and strides: 1). This layer creates a convolution kernel that utilizes 64 filters to intensify the representation of valid features. The default global max pooling layer employs an undersampling strategy to reduce the dimensionality of these features, compress the number of data points and parameters and improve the fault tolerance of the model to avoid potential overfitting. Finally, the hidden layer was added with a total of 256 nodes and a dropout rate of 0.01.
A recurrent neural network (RNN) is a type of directed (one-way) recursive neural network in which all nodes are connected by chains. The RNN has been demonstrated to be superior on dealing with different bioinformatics tasks [43, 47, 67, 68]. As a common type of RNN, LSTM has been successfully applied to protein subcellular localization prediction [69] and antimicrobial peptide recognition [43] with an outstanding ability to ‘recognize’ and ‘forget’ gap-separated patterns [70]. Here, we substituted the hidden layer in the abovementioned CNN with the LSTM layer (units: 128, return_sequences: FALSE, dropout: 0.01) to construct a LSTM model, which can identify effective gap-separated patterns from sequences.
Characterized by multiple hidden layers, DNN has been successfully applied in bioinformatics and computational biology in recent years [71–73]. In this study, the first layer in the DNN was an embedding layer with the output_dim of 256 and the dropout rate of 0.01. Following the embedding layer was a default global average pooling layer, which was used to replace traditionally fully connected layers to avoid potential overfitting [74]. Finally, three hidden layers, each of which contained 64 units, were added to construct the final architecture of DNN.
All the three aforementioned neural networks were implemented by the Keras package in R using a sequential model. The models were compiled with the ‘adam’ optimizer using 30 epochs (loss: binary_crossentropy, metrics: accuracy). The callbacks function was defined similarly to the one used by the MLP.
Integrative model construction
Numerous previous studies have indicated that ensemble models are able to achieve significantly improved performance over their baseline models [26, 28, 29, 31, 32, 75–78]. To this end, we constructed a number of baseline models and integrated them into a single stable hybrid model using the stacking strategy. A detailed description is provided in the following subsections.
Construction of baseline models
As described in Sections 2.2 and 2.3, eight different types of features (AAC, DPC, DDE, PAAC, QSO, PSSM-composition, S-FPSSM and RPM-PSSM) were used to construct models based on four classical ML algorithms (RF, SVM, XGBoost and MLP), while 10 features (L100, L200, …, L1000) were used to construct models based on three DL algorithms (CNN, LSTM and DNN). In total, 62 baseline models were constructed, which included 32 (4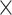 8) ML baseline models plus 30 (3
8) ML baseline models plus 30 (3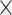 10) DL baseline models. To construct each baseline model (Figure S2), the training dataset was randomly partitioned into 10 equally sized subsets. One subset was retained as the validation set, whereas the remaining subsets were used to construct a subset-based model. This procedure was repeated 10 times to generate 10 subset-based models and to ensure that each sequence in the training set would receive one prediction score. Finally, the prediction scores of all the 62 baseline models were obtained for the training dataset. Specifically, the prediction score of the baseline model on the independent dataset was calculated by averaging the prediction scores of 10 subset-based models (Figure S2).
10) DL baseline models. To construct each baseline model (Figure S2), the training dataset was randomly partitioned into 10 equally sized subsets. One subset was retained as the validation set, whereas the remaining subsets were used to construct a subset-based model. This procedure was repeated 10 times to generate 10 subset-based models and to ensure that each sequence in the training set would receive one prediction score. Finally, the prediction scores of all the 62 baseline models were obtained for the training dataset. Specifically, the prediction score of the baseline model on the independent dataset was calculated by averaging the prediction scores of 10 subset-based models (Figure S2).
Generation of a new and informative feature vector
A new and informative feature vector was generated based on 62 prediction scores as follows. For a given protein sequence P, its 62 prediction scores generated by 62 baseline models were converted to 62 labels, which can be represented by
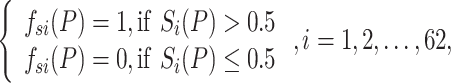 |
(20) |
where 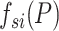 and
and 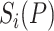 denote the prediction label and prediction score of the ith baseline model, respectively. Then, a new feature vector
denote the prediction label and prediction score of the ith baseline model, respectively. Then, a new feature vector 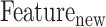 can be generated by concatenating the 62 prediction labels
can be generated by concatenating the 62 prediction labels
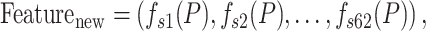 |
(21) |
Finally, the protein sequence P was converted to a 62-dimensional feature vector.
Establishment of a meta model using XGBoost
A meta model was constructed using XGBoost based on the new 62-dimensional feature vector. Unlike those models constructed with straightforward ensemble strategies (such as average scoring and majority voting), this stacking strategy enables an automatic exploration of different baseline models. Through intelligent integration of their respective strength without human intervention, the final metal model can potentially provide a better and more stable performance [11, 79–81]. Moreover, XGBoost also allows the feature importance analysis in a statistically sound manner, to quantify the impact of each baseline model on the performance of the final meta model (data shown in the Section Effect of baseline models on the performance of the final meta model). This will allow us to better characterize and understand the contribution of each of the features and individual baseline models to the performance improvement of the final meta model.
Performance evaluation
In this study, five performance measures, including sensitivity (SN), specificity (SP), accuracy (ACC), F-value and MCC, are employed to quantify the performance of the constructed models according to the confusion matrix [82]. These measures are defined as follows:
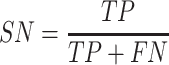 |
(22) |
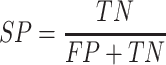 |
(23) |
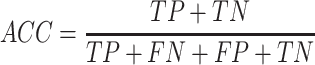 |
(24) |
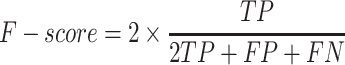 |
(25) |
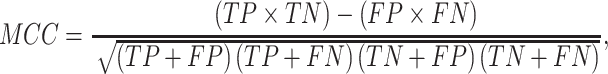 |
(26) |
where TP, TN, FP and FN represent the numbers of true positives, true negatives, false positives and false negatives, respectively. In addition, we also plotted the receiver operating characteristic (ROC) curves to visualize the comprehensive performance of the models and calculated the area under ROC curve (AUC). The higher the AUC value, the better the model performance.
Experimental results
Performance evaluation based on 10-fold cross-validation test
In this section, we conducted, analyzed and compared the performance results using the training dataset on the 10-fold cross-validation test. In this work, all 10-fold cross-validation tests were conducted based on five balanced training datasets, and the results were averaged as the final performance. The results are provided in Tables 1, 2 and S4–S9 and also shown in Figures 2, 3 and S3.
Table 1.
Performance comparison of baseline models with combined features (seqsim incorporated) for predicting VFs on 10-fold cross-validation test
| Model | Encoding | SN | SP | ACC | F-value | MCC |
|---|---|---|---|---|---|---|
| RF | AAC | 0.839 ± 0.017 | 0.812 ± 0.023 | 0.825 ± 0.014 | 0.827 ± 0.014 | 0.651 ± 0.027 |
| DPC | 0.799 ± 0.033 | 0.809 ± 0.030 | 0.804 ± 0.018 | 0.802 ± 0.020 | 0.608 ± 0.036 | |
| DDE | 0.820 ± 0.028 | 0.780 ± 0.022 | 0.800 ± 0.019 | 0.803 ± 0.020 | 0.600 ± 0.037 | |
| PAAC | 0.838 ± 0.017 | 0.812 ± 0.023 | 0.825 ± 0.015 | 0.827 ± 0.015 | 0.651 ± 0.029 | |
| QSO | 0.829 ± 0.017 | 0.817 ± 0.022 | 0.823 ± 0.013 | 0.824 ± 0.014 | 0.646 ± 0.026 | |
| PSSM-composition | 0.763 ± 0.025 | 0.907 ± 0.015 | 0.835 ± 0.015 | 0.822 ± 0.018 | 0.677 ± 0.029 | |
| S-FPSSM | 0.796 ± 0.022 | 0.863 ± 0.018 | 0.830 ± 0.015 | 0.823 ± 0.017 | 0.661 ± 0.030 | |
| RPM-PSSM | 0.765 ± 0.029 | 0.890 ± 0.018 | 0.828 ± 0.017 | 0.816 ± 0.020 | 0.661 ± 0.033 | |
| SVM | AAC | 0.821 ± 0.018 | 0.764 ± 0.025 | 0.792 ± 0.017 | 0.798 ± 0.018 | 0.586 ± 0.033 |
| DPC | 0.738 ± 0.023 | 0.738 ± 0.025 | 0.738 ± 0.016 | 0.738 ± 0.015 | 0.476 ± 0.032 | |
| DDE | 0.742 ± 0.021 | 0.725 ± 0.028 | 0.734 ± 0.016 | 0.736 ± 0.015 | 0.468 ± 0.032 | |
| PAAC | 0.823 ± 0.019 | 0.761 ± 0.025 | 0.792 ± 0.015 | 0.798 ± 0.016 | 0.585 ± 0.029 | |
| QSO | 0.808 ± 0.021 | 0.754 ± 0.024 | 0.781 ± 0.014 | 0.787 ± 0.014 | 0.563 ± 0.029 | |
| PSSM-composition | 0.792 ± 0.029 | 0.836 ± 0.025 | 0.814 ± 0.019 | 0.810 ± 0.020 | 0.629 ± 0.038 | |
| S-FPSSM | 0.797 ± 0.026 | 0.776 ± 0.028 | 0.786 ± 0.021 | 0.788 ± 0.021 | 0.573 ± 0.042 | |
| RPM-PSSM | 0.760 ± 0.034 | 0.830 ± 0.024 | 0.795 ± 0.018 | 0.787 ± 0.021 | 0.591 ± 0.035 | |
| XGBoost | AAC | 0.836 ± 0.017 | 0.813 ± 0.024 | 0.825 ± 0.015 | 0.827 ± 0.015 | 0.650 ± 0.029 |
| DPC | 0.845 ± 0.016 | 0.802 ± 0.024 | 0.823 ± 0.015 | 0.827 ± 0.016 | 0.648 ± 0.030 | |
| DDE | 0.843 ± 0.016 | 0.814 ± 0.020 | 0.829 ± 0.013 | 0.831 ± 0.015 | 0.658 ± 0.026 | |
| PAAC | 0.832 ± 0.015 | 0.816 ± 0.021 | 0.824 ± 0.014 | 0.825 ± 0.015 | 0.649 ± 0.028 | |
| QSO | 0.838 ± 0.017 | 0.820 ± 0.019 | 0.829 ± 0.013 | 0.831 ± 0.014 | 0.659 ± 0.025 | |
| PSSM-composition | 0.833 ± 0.017 | 0.885 ± 0.017 | 0.859 ± 0.012 | 0.855 ± 0.013 | 0.719 ± 0.024 | |
| S-FPSSM | 0.841 ± 0.019 | 0.871 ± 0.019 | 0.856 ± 0.015 | 0.854 ± 0.016 | 0.713 ± 0.029 | |
| RPM-PSSM | 0.835 ± 0.018 | 0.874 ± 0.019 | 0.854 ± 0.014 | 0.852 ± 0.015 | 0.710 ± 0.028 | |
| MLP | AAC | 0.858 ± 0.027 | 0.741 ± 0.032 | 0.800 ± 0.012 | 0.811 ± 0.014 | 0.604 ± 0.024 |
| DPC | 0.855 ± 0.032 | 0.754 ± 0.035 | 0.804 ± 0.013 | 0.813 ± 0.015 | 0.613 ± 0.025 | |
| DDE | 0.804 ± 0.038 | 0.804 ± 0.046 | 0.804 ± 0.012 | 0.804 ± 0.013 | 0.610 ± 0.023 | |
| PAAC | 0.856 ± 0.030 | 0.743 ± 0.028 | 0.800 ± 0.013 | 0.810 ± 0.016 | 0.604 ± 0.026 | |
| QSO | 0.849 ± 0.036 | 0.764 ± 0.036 | 0.806 ± 0.013 | 0.814 ± 0.016 | 0.616 ± 0.025 | |
| PSSM-composition | 0.819 ± 0.026 | 0.886 ± 0.025 | 0.852 ± 0.009 | 0.847 ± 0.011 | 0.707 ± 0.019 | |
| S-FPSSM | 0.801 ± 0.039 | 0.876 ± 0.034 | 0.839 ± 0.014 | 0.832 ± 0.018 | 0.680 ± 0.028 | |
| RPM-PSSM | 0.826 ± 0.031 | 0.863 ± 0.034 | 0.844 ± 0.008 | 0.841 ± 0.010 | 0.691 ± 0.017 | |
| CNN | L100 | 0.565 ± 0.094 | 0.680 ± 0.092 | 0.623 ± 0.015 | 0.596 ± 0.047 | 0.251 ± 0.029 |
| L200 | 0.606 ± 0.093 | 0.683 ± 0.084 | 0.645 ± 0.018 | 0.627 ± 0.047 | 0.295 ± 0.033 | |
| L300 | 0.608 ± 0.097 | 0.689 ± 0.093 | 0.649 ± 0.014 | 0.630 ± 0.047 | 0.303 ± 0.029 | |
| L400 | 0.601 ± 0.085 | 0.711 ± 0.078 | 0.657 ± 0.016 | 0.633 ± 0.043 | 0.318 ± 0.030 | |
| L500 | 0.610 ± 0.092 | 0.704 ± 0.085 | 0.657 ± 0.018 | 0.637 ± 0.044 | 0.321 ± 0.035 | |
| L600 | 0.610 ± 0.081 | 0.711 ± 0.078 | 0.660 ± 0.019 | 0.639 ± 0.038 | 0.326 ± 0.037 | |
| L700 | 0.619 ± 0.067 | 0.700 ± 0.078 | 0.660 ± 0.018 | 0.644 ± 0.027 | 0.324 ± 0.036 | |
| L800 | 0.608 ± 0.069 | 0.711 ± 0.071 | 0.659 ± 0.016 | 0.639 ± 0.030 | 0.324 ± 0.033 | |
| L900 | 0.598 ± 0.087 | 0.724 ± 0.084 | 0.661 ± 0.017 | 0.635 ± 0.042 | 0.329 ± 0.032 | |
| L1000 | 0.615 ± 0.085 | 0.705 ± 0.085 | 0.660 ± 0.015 | 0.641 ± 0.036 | 0.326 ± 0.030 | |
| LSTM | L100 | 0.566 ± 0.107 | 0.674 ± 0.112 | 0.621 ± 0.018 | 0.594 ± 0.052 | 0.249 ± 0.038 |
| L200 | 0.565 ± 0.084 | 0.727 ± 0.077 | 0.647 ± 0.019 | 0.612 ± 0.045 | 0.300 ± 0.036 | |
| L300 | 0.567 ± 0.117 | 0.723 ± 0.114 | 0.645 ± 0.018 | 0.609 ± 0.055 | 0.302 ± 0.035 | |
| L400 | 0.589 ± 0.061 | 0.707 ± 0.071 | 0.648 ± 0.023 | 0.625 ± 0.031 | 0.301 ± 0.046 | |
| L500 | 0.557 ± 0.092 | 0.737 ± 0.086 | 0.647 ± 0.020 | 0.609 ± 0.046 | 0.304 ± 0.036 | |
| L600 | 0.544 ± 0.082 | 0.754 ± 0.070 | 0.649 ± 0.024 | 0.605 ± 0.052 | 0.308 ± 0.046 | |
| L700 | 0.522 ± 0.114 | 0.758 ± 0.098 | 0.640 ± 0.034 | 0.584 ± 0.077 | 0.294 ± 0.066 | |
| L800 | 0.552 ± 0.102 | 0.745 ± 0.092 | 0.648 ± 0.026 | 0.606 ± 0.058 | 0.307 ± 0.050 | |
| L900 | 0.535 ± 0.091 | 0.763 ± 0.080 | 0.649 ± 0.028 | 0.600 ± 0.058 | 0.311 ± 0.054 | |
| L1000 | 0.560 ± 0.102 | 0.748 ± 0.093 | 0.654 ± 0.028 | 0.613 ± 0.062 | 0.318 ± 0.051 | |
| DNN | L100 | 0.580 ± 0.057 | 0.694 ± 0.054 | 0.637 ± 0.017 | 0.614 ± 0.028 | 0.277 ± 0.035 |
| L200 | 0.598 ± 0.063 | 0.719 ± 0.050 | 0.659 ± 0.017 | 0.635 ± 0.034 | 0.321 ± 0.031 | |
| L300 | 0.594 ± 0.065 | 0.729 ± 0.057 | 0.661 ± 0.017 | 0.635 ± 0.033 | 0.328 ± 0.032 | |
| L400 | 0.632 ± 0.060 | 0.706 ± 0.060 | 0.669 ± 0.016 | 0.655 ± 0.027 | 0.341 ± 0.031 | |
| L500 | 0.620 ± 0.054 | 0.730 ± 0.050 | 0.675 ± 0.019 | 0.655 ± 0.027 | 0.353 ± 0.036 | |
| L600 | 0.613 ± 0.058 | 0.734 ± 0.062 | 0.674 ± 0.019 | 0.651 ± 0.027 | 0.352 ± 0.036 | |
| L700 | 0.599 ± 0.063 | 0.744 ± 0.058 | 0.672 ± 0.019 | 0.645 ± 0.031 | 0.349 ± 0.035 | |
| L800 | 0.606 ± 0.060 | 0.736 ± 0.063 | 0.671 ± 0.019 | 0.647 ± 0.029 | 0.348 ± 0.036 | |
| L900 | 0.604 ± 0.061 | 0.740 ± 0.059 | 0.672 ± 0.018 | 0.647 ± 0.030 | 0.349 ± 0.034 | |
| L1000 | 0.601 ± 0.070 | 0.741 ± 0.070 | 0.671 ± 0.018 | 0.644 ± 0.033 | 0.349 ± 0.034 |
Note: Values are expressed as mean ± standard deviation. The best performance value for each metric across different encoding methods in each model is highlighted in bold.
Table 2.
Performance comparison of different models trained based on different ensemble strategies on the 10-fold cross-validation test
| Ensemble strategy | Model | SN | SP | ACC | F-value | MCC |
|---|---|---|---|---|---|---|
| Stacking | ML model | 0.836 ± 0.021 | 0.916 ± 0.017 | 0.876 ± 0.013 | 0.871 ± 0.015 | 0.755 ± 0.027 |
| DL model | 0.623 ± 0.036 | 0.739 ± 0.030 | 0.681 ± 0.020 | 0.661 ± 0.024 | 0.365 ± 0.038 | |
| Hybrid model | 0.837 ± 0.023 | 0.918 ± 0.015 | 0.878 ± 0.014 | 0.872 ± 0.016 | 0.758 ± 0.027 | |
| Average scoring | ML model | 0.847 ± 0.017 | 0.854 ± 0.020 | 0.851 ± 0.013 | 0.850 ± 0.014 | 0.701 ± 0.026 |
| DL model | 0.616 ± 0.033 | 0.739 ± 0.031 | 0.678 ± 0.019 | 0.656 ± 0.022 | 0.358 ± 0.037 | |
| Hybrid model | 0.789 ± 0.023 | 0.865 ± 0.019 | 0.827 ± 0.015 | 0.820 ± 0.016 | 0.657 ± 0.030 | |
| Majority voting | ML model | 0.845 ± 0.016 | 0.846 ± 0.019 | 0.845 ± 0.012 | 0.845 ± 0.013 | 0.691 ± 0.025 |
| DL model | 0.595 ± 0.035 | 0.765 ± 0.028 | 0.681 ± 0.018 | 0.650 ± 0.023 | 0.367 ± 0.034 | |
| Hybrid model | 0.743 ± 0.022 | 0.874 ± 0.020 | 0.809 ± 0.016 | 0.795 ± 0.017 | 0.622 ± 0.032 |
Note: Values are expressed as mean ± standard deviation. The best performance value is highlighted in bold.
Figure 2.
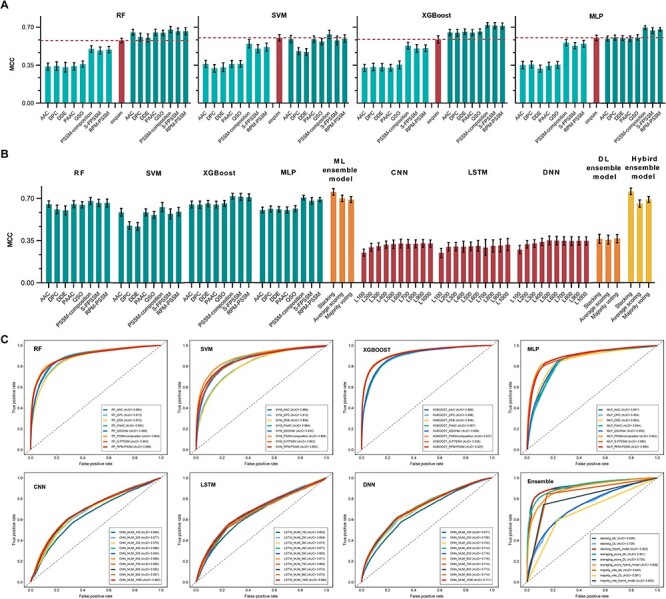
Performance comparison of different models. (A) Performance comparison between the models trained using the original features (appearing at the left, light green bars within each panel) and those trained using the combined features (i.e. the seqsim feature incorporated; appearing at the right, dark green bars within each panel). The red bar in the middle of each panel represents the model trained using the seqsim feature; (B) Performance comparison between baseline models (trained using the combined features) and their ensemble models based on 10-fold cross-validation test; (C) ROC curves of 62 baseline models (trained using the combined features) and their ensemble models using different ensemble strategies based on 10-fold cross-validation tests.
Figure 3.
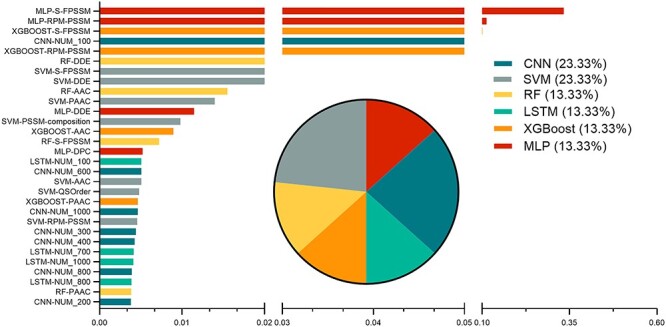
The effect of the top 30 selected features on the predictive performance of the meta XGBoost model. Bar chart lists the Gain values of these features, while the pie chart shows the proportions of the top 30 features according to the six different ML and DL algorithms.
Performance evaluation of baseline models
We assessed the performance of baseline models with a default threshold of 0.5 by performing the 10-fold cross-validation test on the training dataset.
The results show that models trained with the seqsim feature clearly outperformed those trained with traditional ML features across different ML algorithms (Table S4). This observation highlighted the importance of the seqsim feature, which is consistent with previous work [20] where the combination of seqsim and DPC was demonstrated to achieve an improved performance.
Here, we further explored whether combining the seqsim feature could boost the performance of VF prediction in general. To generate the seqsim feature, we used the BLAST+ software [83] to search a query protein against the positive and negative training sets with an E-value of 10, respectively. Finally, we generated a two-dimensional feature vector (seqsim) with the highest bitscores against the positive and negative training sets. The seqsim feature was incorporated into each of traditional ML features to retrain the models. Specially, the combined features were normalized to a range of 0 and 1, when training SVM models, as their performance are easily influenced by features with large variations [26, 29]. As shown in Figure 2A, the performance of most ML models trained with combined features (detailed in Table 1) generally improved compared with those trained with traditional ML features or only the seqsim feature (detailed in Table S4). In particular, the XGBoost model trained with the combined PSSM-composition and seqsim feature achieved the best performance with an AAC of 0.859, an F-value of 0.855, an MCC of 0.719 and an AUC value of 0.927, respectively (Table 1 and Figure 2B and C). These observations again confirmed the generalized capability of the seqsim feature for enhancing the VF prediction performance of ML models.
Among these models trained with the combined features, in most cases, the performances of the models trained using evolutionary information-based features (e.g., PSSM-composition, S-FPSSM and RPM-PSSM) were found to be superior to those of the models trained using the other two groups of features. The results are consistent with a number of previous studies which show that the evolutionary information-based features can be used as primary information for predicting different protein attributes [14, 15, 28, 29, 31, 32, 38, 40]. From the algorithmic point of view, MLP and XGBoost achieved an overall better performance than RF and SVM (Table 1 and Figure 2B and C).
Hereafter, ML baseline models adopted in this work refer to the model trained with the combined features instead of the traditional ML features if not explicitly specified.
In the case of DL algorithms, DNN outperformed CNN and LSTM trained with different features in most cases. This is expected as the DNN models with three hidden layers were able to mine deeper patterns and characteristics from sequences. In contrast, the CNN models only recognized local sequence motif, while LSTM models captured relatively remote interactions. Specifically, the DNN model trained using 500-dimensional features achieved the overall best performance with an AAC of 0.675, an F-value of 0.655, an MCC of 0.353 and an AUC value of 0.716. Those results indicate that DNN has certain advantages in analyzing biological sequence data and training more accurate models [73, 84]. Generally, as the feature dimension increased, the performance of a model gradually increased and then reached a stable plateau (Figure S3). The overall best performance for CNN, LSTM and DNN was achieved in feature dimensions of 900, 1000 and 500, respectively (Table 1). We also re-trained the three DL models using their optimal features but with different dropout rates ranging from 0.01 to 0.3 and compared their performance based on 10-fold cross-validation test. The results indicate that, with the increase of dropout rate, the performance of DL models did not have any obvious change (Table S5). Among them, the dropout rate of 0.01 showed more stable performance compared with the others and exhibited general fitness among various DL models.
In addition to the threshold of 0.5, we also investigated the effect of different thresholds (ranging from 0.3 to 0.7) on the performance of models. The experimental results showed that models with the threshold of 0.5 (Table 1) achieved more stable performance, with a better balance of SN and SP, than those with other thresholds (Table S6). Therefore, if not explicitly specified, the default threshold was set to 0.5 in this work. This is also the default setting on the DeepVF server, but with an option for users to customize their own thresholds for specific demands.
We found that the effectiveness of DL algorithms was not as good as that of classical ML algorithms, which is reflected by the comparatively lower performance measures (Table 1 and Figure 2). The possible reason is that DL can better leverage very large datasets (typically millions of samples) to achieve high performance [56]; however, it is difficult for DL algorithms to extract highly useful information just from a limited number of sequences. Moreover, the complex features (e.g. sequence-based features, physicochemical property-based features and evolutionary information-based features), which can be effectively used to construct ML models, lead to underfitting in DL models due to the constraints of limited data. Having that said, it might be possible for the DL models to identify specific patterns from sequences via self-learning without human intervention. Without manually assuming how a model could contribute to the final ensemble predictor, we integrated both ML and DL models to construct a hybrid ensemble model. The hybrid ensemble model will determine on its own how to utilize its baseline models based on their contributions.
The performance of ensemble models
To examine the performance of the ensemble models trained based on the stacking strategy, we designed and performed three sets of experiments: 32 ML, 30 DL and all 62 baseline models were integrated to establish three different ensemble models, which were termed as ML model, DL model and hybrid model, for brevity, respectively. As shown in Figure 2 and Tables 1 and 2, it is apparent that ML and DL stacking strategy-based ensemble models consistently achieved an overall better performance in terms of all metrics (i.e., SN, SP, ACC, F-value and MCC) compared with their respective baseline models. These results demonstrate that the prediction performance of individual models can indeed be improved by using ensemble models with stacking strategy, as has been shown in a number of previous studies [11, 79, 81]. Moreover, the hybrid model appeared to be the most powerful classifier, which achieved the highest values for SN (0.837), SP (0.918), ACC (0.878), F-value (0.872) and MCC (0.758). This again confirmed that the stacking strategy-based ensemble model can take advantages of individual ML and DL models to make more stable and accurate VF prediction.
We also compared our stacking strategy-based ensemble model with another two commonly used ensemble strategies, namely the average scoring [26, 28, 31, 32] and majority voting [29, 75]. These two strategies either average or vote the prediction scores of ML, DL and all baseline models to construct simplified ensemble models. As shown in Figure 2 and Tables 1 and 2, most of these ensemble models did not obviously improve the prediction performance compared with their corresponding baseline models. In contrast, the stacking strategy-based hybrid model achieved the overall best performance, thereby validating its strengths to utilize individual baseline models based on XGBoost. We also constructed the ensemble meta models using MLP (which achieved the second-best performance amongst ML models) and DNN (which achieved the best performance amongst DL models). The results confirmed XGBoost as the optimal choice for constructing the meta model of VF prediction (Table S7). Using XGBoost, we further constructed the meta model based on the prediction scores instead of the prediction labels of the baseline models. The results (Table S8) showed that the meta model using the prediction label of baseline models outperformed the counterpart model using the raw prediction scores. A possible reason might be that prediction scores are highly variable and too sensitive for the meta-model to learn.
Effect of baseline models on the performance of the final meta model
To better understand the contributions of individual baseline models to the performance of the final meta model, we performed a feature importance analysis of the meta model using the built-in function of XGBoost. The importance of each feature was generated and ranked using the xgb.importance function of the xgboost package (https://cran.r-project.org/web/packages/xgboost) in R language. As each feature used by the meta model represents the output of a baseline model, the importance of a feature directly shows how many contributions the associated baseline model can make to the meta model. Specifically, three measures, namely Gain, Cover and Frequency, were calculated for each feature (Table S9). As a result, we identified the top 30 features and listed their Gain values and the proportions with respect to each of the ML and DL algorithms (Figure 3).
Generally, the better a baseline model performs, the more it contributes to the final meta model (Table 1 and Figure 3). The prediction labels of the MLP baseline models trained using the evolutionary information-based features (S-FPSSM and RPM-PSSM) were the most important amongst all features, suggesting their crucial contribution to VF prediction. Specifically, the prediction label of the MLP baseline model trained using S-FPSSM was ranked with the highest importance with a Gain of 0.585, a Cover of 0.148 and a Frequency of 0.016, respectively. The prediction labels of the CNN and SVM baseline models appeared to contribute more, both achieving the highest percentage (23.33%) amongst all the top 30 features. This observation was not intuitive as they achieved the worst performance among the DL and ML baseline models, respectively (Table 1). In contrast, the prediction labels of DNN models, which achieved the best performance among DL baseline models (Table 1), were not ranked in the top 30 feature list. Similar situations also occurred to the prediction labels of the MLP baseline models, which achieved the best performance among ML baseline models (Table 1) but only accounted for 13.33% of the top 30 features. Altogether, these results illustrate that although the baseline models with a better performance could generally contribute more, those with lower performance may also make an indispensable contribution to the performance of the final meta model. In this way, the stacking strategy-based meta model could make a good use of the seemingly uninformative features to achieve its superior performance, as opposed to other straightforward ensemble models.
Performance validation on the independent test
We first benchmarked and compared the performance of the baseline models with the final hybrid meta model (termed DeepVF) on the independent test dataset. From Tables 3 and S10, several observations can be made: (i) baseline models trained with evolutionary information-based features achieved a better performance compared with other baseline models in most cases; (ii) the performance of DL baseline models was worse than that of ML baseline models, possibly due to limited dataset, and (iii) the final meta model achieved a top-level performance but performed slightly worse than a few baseline models on the independent test. Taking into consideration the performance on both training and independent tests, the hybrid meta model showed a stable performance, highlighting its robustness and excellent generalization ability.
Table 3.
Performance comparison of DeepVF and existing toolkits for predicting VFs on the independent test
| Methods | SN | SP | ACC | F-value | MCC |
|---|---|---|---|---|---|
| VirulentPred | 0.641 | 0.573 | 0.607 | 0.620 | 0.214 |
| MP3 | 0.536 | 0.783 | 0.66 | 0.612 | 0.33 |
| PBVF | 0.774 | 0.814 | 0.794 | 0.790 | 0.589 |
| DeepVF | 0.790 | 0.833 | 0.812 | 0.807 | 0.624 |
| BLAST | 0.682 | 0.818 | 0.750 | 0.732 | 0.505 |
Note: The best performance value for each metric across different toolkits is highlighted in bold.
Moreover, we compared the performance of DeepVF with the BLAST-based baseline predictor and three state-of-the-art methods (i.e. VirulentPred [11], MP3 [19] and PBVF [20]). For VirulentPred and MP3, we tested their webservers using their own default prediction cutoff thresholds. As PBVF [20] did not provide an online webserver for VF prediction, we implemented an RF-based hybrid classifier (which achieved the top performance reported in the PBVF work) by strictly following the original method based on our training datasets. The BLAST-based baseline predictor was constructed based on the seqsim vector [20], which would predict a query protein as positive if the bitscore against the positive training set was higher, and otherwise negative.
As can be seen from Table 3 and Figure S4, DeepVF clearly outperformed the BLAST-based baseline predictor and the three state-of-the-art methods in terms of all performance metrics. In particular, DeepVF achieved an ACC of 0.812, an F-value of 0.807, an MCC of 0.624 and an AUC of 0.896, respectively. Benefitting from the combination of the DPC and seqsim features, PBVF achieved the second-best performance. In contrast, DeepVF stepped further in performance improvement, presumably because it integrated more informative features and models within its hybrid framework. The BLAST-based baseline predictor ranked the third, highlighting the effectiveness of the seqsim feature. MP3 followed the BLAST-based baseline predictor in the performance ranking. This is in accordance with the previous work [20], which showed that the performance of MP3 was far worse than that of PBVF on the same dataset.
As the earliest webserver of its type, VirulentPred performed the worst on the independent test. To make a fair and objective comparison, we retrained the model of DeepVF based on VirulentPred’s training dataset. In this way, we aimed to avoid the potential influence of different training datasets, without re-implementing the algorithm of VirulentPred. We compared the performance of DeepVF and VirulentPred based on the independent test datasets of both VirulentPred and DeepVF. As shown in Table S11, DeepVF achieved a higher (or equal) performance based on the two different independent datasets of VirulentPred and the independent dataset of DeepVF. In the latter case, DeepVF failed to perform the prediction as accurately as it achieved when trained on its own dataset. The decrease in the prediction performance might be attributed to the outdated training dataset of VirulentPred.
Application of DeepVF to genome-scale VF prediction
We further applied our DeepVF approach to perform a genome-wide VF prediction across three bacterial pathogens (including E. coli O157:H7, Streptococcus pneumoniae (strain ATCC BAA-255/R6) and Mycobacterium tuberculosis (strain ATCC 25618/H37Rv)). The BLAST-based baseline predictor was also used for comparison. As the original BLAST-based baseline predictor only provided the prediction label, we upgraded it to a classifier that was able to list the detailed prediction score by following the PBVF method [20]. The overlapping sequences between our training dataset and bacterial genomes were removed, while those sequences shared by our independent dataset and the genomes were retained to measure the predictors’ capabilities.
Upon looking at the top 100 sequences with the highest prediction scores, we found that DeepVF identified a larger number of positive samples (5/1/3 samples) in the independent dataset than the BLAST-based baseline predictor (0/1/0 samples) among the three genomes (Table S12). This suggests that DeepVF achieved a better recall of VFs than the BLAST-based baseline predictor in genome-wide prediction. There were also a few sequences that were predicted as VFs by both DeepVF and the BLAST-based baseline predictor in the top 100 ranks among three genomes (highlighted in Supplementary File S2 and summarized in Table S12). Those sequences would be of particular interest for further experimental validation. For the same purpose, the gene ontology terms of the top 100 proteins predicted by DeepVF in the three genomes are provided in the Supplementary File S2.
For genome-scale prediction, application of a default cutoff threshold of 0.5 often led to a large number of false-positives. To address this, we conducted a series of experiments to determine a proper threshold that could keep the false-positive rate under a low level of 5%. We added the known positives and negatives samples (those included in the training and independent datasets) into the three genomes and then ranked the entire genomes according to the DeepVF prediction scores. Accordingly, there were no false-positive samples across all three genomes when the threshold was set as higher than 0.85 (Supplementary File S2). We further applied this threshold to the three genomes and finally predicted 974, 473 and 508 putative VFs and recalled 11, 1 and 12 experimentally validated VFs (i.e., positive samples appearing in the independent test) among E. coli O157:H7, S. pneumoniae (strain ATCC BAA-255/R6) and M. tuberculosis (strain ATCC 25618/H37Rv), respectively (Supplementary File S2).
Discussion of DeepVF and existing toolkits from the user’s viewpoint
The value of a toolkit lies in its ability to provide users with convenient, fast and high-quality services in practical scenarios. In this section, we discuss DeepVF and two existing toolkits (i.e. VirulentPred and MP3) for predicting VFs from the user’s viewpoint. We particularly focus on four aspects including user guidance, data requirements, computational costs and output interpretation (summarized in Table S13).
Generally, VirulentPred and DeepVF provide sustained service through web servers, while MP3 provides both web server and standalone toolkit. The standalone toolkit could act as a substitution for the MP3 server that often provides intermittent access. Another advantage of such standalone toolkit is that it allows local execution of VF prediction in batches and integration into downstream pipelines.
To improve user experience, MP3 and DeepVF offer a well-documented guidance to facilitate users’ interactions with the servers and interpretation of their prediction results. To use these toolkits, users are required to submit protein sequences in FASTA format, while DeepVF also accepts raw sequences that will be automatically formatted into FASTA format upon submission. Unlike MP3 that has no limitation on the number of submitted sequences, VirulentPred and DeepVF specify the max numbers of 500 and 5000 sequences per submission, respectively. These considerations are based on the fact that their computational costs are relatively high (Table S13). Both of them engage more features (e.g. PSSM-based features) or ML models to achieve better predictive performance (shown in the Section Performance validation on the independent test) with high computational costs incurred. After accomplishing the prediction tasks, VirulentPred and DeepVF present the detailed results at the webpage, while MP3 directly provides weblinks for result download without the online presentation. DeepVF further provides auxiliary functions including output rank, search and filter to enable a quick and preliminary analysis of the prediction results. These functionalities cater for users’ common needs, which often require ranking of the top candidate VFs or nomination of certain proteins of interest that are worthy of further experimental validation.
Conclusion
In this work, we have developed DeepVF, which is a DL-based hybrid framework for accurate identification of VFs in bacterial pathogens. To establish an effective and accurate prediction model, we first collected an up-to-date and high-quality dataset from recently published literature and public databases. Based on this dataset, we explored a variety of features for both classical ML and DL algorithms: four classical ML and three DL algorithms were employed to construct 62 baseline models using proposed effective features. To maximize the utility of each baseline model, we effectively combined them to obtain the final hybrid meta model based on the stacking strategy. Extensive benchmarking experiments show that the final hybrid meta model achieved a higher performance compared with baseline models, thereby demonstrating the effectiveness of this strategy. When compared with three existing state-of-the-art methods (i.e. VirulentPred, MP3 and PBVF) on the independent test, DeepVF was more effective and outperformed the other methods for VF prediction, with an ACC of 0.812, an F-value of 0.807, an MCC of 0.624 and an AUC of 0.896, respectively. The application of DeepVF to three bacterial genomes led to the in silico identification of 24 experimentally validated VFs and 1955 putative VFs, the latter of which could be worthy of further experimental investigations. Using the proposed hybrid ensemble model, we implemented DeepVF as a publicly available web server. We further discussed and compared its practical utility with the existing toolkits from the user’s viewpoint. We anticipate that DeepVF will be used as a powerful tool to expedite the discovery of novel putative VFs for various bacterial species and therefore facilitate the community-wide effort for in-depth interrogation and characterization of mechanisms of VFs. Moreover, the proposed DL-based hybrid framework of DeepVF can serve as a useful template to guide future developments of prediction models for other classification problems in bioinformatics and computational biology.
Key Points
We propose a hybrid framework termed DeepVF and implement it as a user-friendly web server for accurate identification of virulence factors from sequence information.
DeepVF trains a number of baseline models with heterogeneous features based on four classical machine learning algorithms and three deep learning algorithms and integrates them within the hybrid framework using the stacking strategy.
Extensive benchmarking experiments demonstrate that DeepVF achieves a superior performance of VF prediction compared with its constituent baseline models and the existing state-of-the-art toolkits.
Supplementary Material
Acknowledgements
We would like to express our sincere gratitude to the four anonymous reviewers for their constructive comments and suggestions, which have considerably improved the presentation of our work. This work was financially supported by grants from the National Natural Science Foundation of China (61862017), the Natural Science Foundation of Guangxi (2018GXNSFAA138117, 2016GXNSFCA380005), the National Health and Medical Research Council of Australia (NHMRC) (1092262, 1144652 and 1127948), the Australian Research Council (ARC) (LP110200333 and DP120104460), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965), the Collaborative Research Program of Institute for Chemical Research and Kyoto University (2019-32 and 2018-28). TML and AL’s work was supported in part by the Informatics Institute of the School of Medicine at UAB.
Ruopeng Xie is currently a Research Assistant in the Bioinformatics Lab at Guilin University of Electronic Technology. He received his master degree in the School of Computer Science and Information Security, Guilin University of Electronic Technology, China, and his bachelor degree in computer science and technology from Shanghai Business School, China. His research interests are bioinformatics, machine learning and data mining.
Jiahui Li is currently a Research Assistant in the Bioinformatics Lab at the Guilin University of Electronic Technology. She received her master degree from The First Affiliated Hospital of Wenzhou Medical University, China, and was a visiting master student at the Biomedicine Discovery Institute and the Department of Microbiology, Monash University. Her interests cover bacterial secretion mechanism, bacterial antimicrobial resistance, bacteria-phage interaction and bioinformatics.
Jiawei Wang is currently a postdoctoral research fellow in the Biomedicine Discovery Institute and the Department of Microbiology at Monash University, Australia. He received his bachelor degree in software engineering from Tongji University, master degree in computer science from Peking University, China and PhD degree from Monash University. His research interests are computational biology, bioinformatics, machine learning and data mining.
Wei Dai is currently a master student in the School of Computer Science and Information Security, Guilin University of Electronic Technology, China. His research interests are bioinformatics, computational biology, machine learning and ensemble learning.
André Leier is currently an Assistant Professor in the Department of Genetics and the Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham (UAB) School of Medicine, USA. He is also an Associate Scientist in the UAB Comprehensive Cancer Center. He received his Ph.D. in computer science (Dr. rer. nat.) from the University of Dortmund, Germany. His research interests are in biomedical informatics and computational and systems biomedicine.
Tatiana T. Marquez-Lago is an Associate Professor in the Department of Genetics and the Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham (UAB) School of Medicine, USA. Her research interests include multiscale modelling and simulations, artificial intelligence, bioengineering and systems biomedicine. Her interdisciplinary lab studies stochastic gene expression, chromatin organization, antibiotic resistance in bacteria and host–microbiota interactions in complex diseases.
Tatsuya Akutsu received his DEng degree in information engineering in 1989 from the University of Tokyo, Japan. Since 2001, he has been a Professor in the Bioinformatics Center, Institute for Chemical Research, Kyoto University, Japan. His research interests include bioinformatics and discrete algorithms.
Trevor Lithgow is a Professor in the Biomedicine Discovery Institute and the Director of the Centre to Impact AMR at Monash University, Australia. He received his PhD degree in 1992 from La Trobe University. His research interests particularly focus on bacterial cell biology. His lab develops and deploys multidisciplinary approaches, ranging from bioinformatics to biochemical assays to nanoscale imaging to characterize bacterial cell surfaces and to discover and characterize the bacteriophages that kill antibiotic resistant ‘superbugs’.
Jiangning Song is an Associate Professor and the Group Leader in the Biomedicine Discovery Institute and the Department of Biochemistry and Molecular Biology, Monash University, Melbourne, Australia. He is a member of the Monash Centre for Data Science, Monash University. He is also an Associate Investigator at the ARC Centre of Excellence in Advanced Molecular Imaging, Monash University. His research interests include bioinformatics, computational biomedicine, machine learning, functional genomics and enzyme engineering.
Yanju Zhang received her PhD degree at the Leiden Institute of Advanced Computer Science, Leiden University, the Netherlands, in 2011. She is currently a Professor and the Head of the Bioinformatics Group, School of Computer Science and Information Security, Guilin University of Electronic Technology, China. Her research interests are bioinformatics, algorithms and modeling, machine learning, data mining and precision medicine.
References
- 1. Becker K, Hu Y, Biller-Andorno N. Infectious diseases - a global challenge. Int J Med Microbiol 2006;296:179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller RS, Farnsworth ML, Malmberg JL. Diseases at the livestock-wildlife interface: status, challenges, and opportunities in the United States. Prev Vet Med 2013;110:119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sayers S, Li L, Ong E, et al. Victors: a web-based knowledge base of virulence factors in human and animal pathogens. Nucleic Acids Res 2019;47:D693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burnham JP, Olsen MA, Kollef MH. Re-estimating annual deaths due to multidrug-resistant organism infections. Infect Control Hosp Epidemiol 2019;40:112–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Casadevall A, Pirofski L. Host-pathogen interactions: the attributes of virulence. J Infect Dis 2001;184:337–44. [DOI] [PubMed] [Google Scholar]
- 6. Cross AS. What is a virulence factor? Crit Care 2008;12:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Helgason E, Okstad OA, Caugant DA, et al. Bacillus anthracis, Bacillus cereus, and bacillus thuringiensis--one species on the basis of genetic evidence. Appl Environ Microbiol 2000;66:2627–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 2004;68:560–602table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eppinger M, Mammel MK, Leclerc JE, et al. Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc Natl Acad Sci U S A 2011;108:20142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pant A, Das B, Bhadra RK. CTX phage of Vibrio cholerae: genomics and applications. Vaccine 2019,doi: 10.1016/j.vaccine.2019.06.034 [DOI] [PubMed] [Google Scholar]
- 11. Garg A, Gupta D. VirulentPred: a SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinformat 2008;9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weiss RA. Virulence and pathogenesis. Trends Microbiol 2002;10:314–7. [DOI] [PubMed] [Google Scholar]
- 13. Keen EC. Paradigms of pathogenesis: targeting the mobile genetic elements of disease. Front Cell Infect Microbiol 2012;2:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nanni L, Lumini A. An ensemble of support vector machines for predicting virulent proteins. Expert Syst Appl 2009;36:7458–62. [Google Scholar]
- 15. Nanni L, Lumini A, Gupta D, et al. Identifying bacterial virulent proteins by fusing a set of classifiers based on variants of Chou's pseudo amino acid composition and on evolutionary information. IEEE/ACM Trans Comput Biol Bioinform 2012;9:467–75. [DOI] [PubMed] [Google Scholar]
- 16. Sachdeva G, Kumar K, Jain P, et al. SPAAN: a software program for prediction of adhesins and adhesin-like proteins using neural networks. Bioinformatics 2005;21:483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsai C-T, Huang W-L, Ho S-J, et al. Virulent-GO: prediction of virulent proteins in bacterial pathogens utilizing gene ontology terms. Development 2009;1:3. [Google Scholar]
- 18. Zheng LL, Li YX, Ding J, et al. A comparison of computational methods for identifying virulence factors. PLoS One 2012;7:e42517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gupta A, Kapil R, Dhakan DB, et al. MP3: a software tool for the prediction of pathogenic proteins in genomic and metagenomic data. PLoS One 2014;9:e93907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rentzsch R, Deneke C, Nitsche A, et al. Predicting bacterial virulence factors – evaluation of machine learning and negative data strategies. Brief Bioinform 2019, doi: 10.1093/bib/bbz076. [DOI] [PubMed] [Google Scholar]
- 21. Cui W, Chen L, Huang T, et al. Computationally identifying virulence factors based on KEGG pathways. Mol Biosyst 2013;9:1447–52. [DOI] [PubMed] [Google Scholar]
- 22. Liu B, Zheng D, Jin Q, et al. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res 2019;47:D687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mao C, Abraham D, Wattam AR, et al. Curation, integration and visualization of bacterial virulence factors in PATRIC. Bioinformatics 2015;31:252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wattam AR, Davis JJ, Assaf R, et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res 2017;45:D535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. PATRIC v2 FTP Download Site. ftp://ftp.patricbrc.org/patric2/specialty_genes/referenceDBs.
- 26. Zhang Y, Xie R, Wang J, et al. Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework. Brief Bioinform 2019;20:2185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu J, Shi S, Zhang F, et al. PredGly: predicting lysine glycation sites for Homo sapiens based on XGboost feature optimization. Bioinformatics 2019;35:2749–56. [DOI] [PubMed] [Google Scholar]
- 28. Wang J, Yang B, Leier A, et al. Bastion6: a bioinformatics approach for accurate prediction of type VI secreted effectors. Bioinformatics 2018;34:2546–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J, Yang B, An Y, et al. Systematic analysis and prediction of type IV secreted effector proteins by machine learning approaches. Brief Bioinform 2019;20:931–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang Y, Niu B, Gao Y, et al. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26:680–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang J, Li J, Yang B, et al. Bastion3: a two-layer ensemble predictor of type III secreted effectors. Bioinformatics 2019;35:2017–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y, Yu S, Xie R, et al. PeNGaRoo, a combined gradient boosting and ensemble learning framework for predicting non-classical secreted proteins. Bioinformatics 2020;36:704–12. [DOI] [PubMed] [Google Scholar]
- 33. Saravanan V, Gautham N. Harnessing computational biology for exact linear B-cell epitope prediction: a novel amino acid composition-based feature descriptor. OMICS 2015;19:648–58. [DOI] [PubMed] [Google Scholar]
- 34. Li K, Xu C, Huang J, et al. Prediction and identification of the effectors of heterotrimeric G proteins in rice (Oryza sativa L.). Brief Bioinform 2017;18:270–8. [DOI] [PubMed] [Google Scholar]
- 35. Shen HB, Chou KC. PseAAC: a flexible web server for generating various kinds of protein pseudo amino acid composition. Anal Biochem 2008;373:386–8. [DOI] [PubMed] [Google Scholar]
- 36. Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 2001;43:246–55. [DOI] [PubMed] [Google Scholar]
- 37. Chou KC. Prediction of protein subcellular locations by incorporating quasi-sequence-order effect. Biochem Biophys Res Commun 2000;278:477–83. [DOI] [PubMed] [Google Scholar]
- 38. An Y, Wang J, Li C, et al. Comprehensive assessment and performance improvement of effector protein predictors for bacterial secretion systems III, IV and VI. Brief Bioinform 2018;19:148–61. [DOI] [PubMed] [Google Scholar]
- 39. Wang J, Yang B, Revote J, et al. POSSUM: a bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics 2017;33:2756–8. [DOI] [PubMed] [Google Scholar]
- 40. Zou L, Nan C, Hu F. Accurate prediction of bacterial type IV secreted effectors using amino acid composition and PSSM profiles. Bioinformatics 2013;29:3135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zahiri J, Yaghoubi O, Mohammad-Noori M, et al. PPIevo: protein-protein interaction prediction from PSSM based evolutionary information. Genomics 2013;102:237–42. [DOI] [PubMed] [Google Scholar]
- 42. Jeong JC, Lin X, Chen XW. On position-specific scoring matrix for protein function prediction. IEEE/ACM Trans Comput Biol Bioinform 2011;8:308–15. [DOI] [PubMed] [Google Scholar]
- 43. Veltri D, Kamath U, Shehu A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018;34:2740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Khurana S, Rawi R, Kunji K, et al. DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics 2018;34:2605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jurtz VI, Johansen AR, Nielsen M, et al. An introduction to deep learning on biological sequence data: examples and solutions. Bioinformatics 2017;33:3685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Breiman L. Random forests. Mach Learn 2001;45:5–32. [Google Scholar]
- 47. Chen Z, Liu X, Li F, et al. Large-scale comparative assessment of computational predictors for lysine post-translational modification sites. Brief Bioinform 2019;20:2267–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pouyan MB, Kostka D. Random forest based similarity learning for single cell RNA sequencing data. Bioinformatics 2018;34:i79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liaw A, Wiener M. Classification and regression by RandomForest. R News 2002;2:18–22. [Google Scholar]
- 50. Chen T, Guestrin C. Xgboost: A scalable tree boosting system. In: Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining. ACM, San Francisco, 2016, 785–94. [Google Scholar]
- 51. Zhang L, Ai H, Chen W, et al. CarcinoPred-EL: novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci Rep 2017;7:2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Babajide Mustapha I, Saeed F. Bioactive molecule prediction using extreme gradient boosting. Molecules 2016;21:983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bergstra J, Bengio Y. Random search for hyper-parameter optimization. J Mach Learn Res 2012;13:281–305. [Google Scholar]
- 54. Wang Z, Wang Y, Xuan J, et al. Optimized multilayer perceptrons for molecular classification and diagnosis using genomic data. Bioinformatics 2006;22:755–61. [DOI] [PubMed] [Google Scholar]
- 55. Demir C, Gultekin SH, Yener B. Augmented cell-graphs for automated cancer diagnosis. Bioinformatics 2005;21(Suppl 2):ii7–12. [DOI] [PubMed] [Google Scholar]
- 56. Angermueller C, Parnamaa T, Parts L, et al. Deep learning for computational biology. Mol Syst Biol 2016;12:878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Busia A, Jaitly N. Next-Step Conditioned Deep Convolutional Neural Networks Improve Protein Secondary Structure PredictionarXiv preprint arXiv:1702.03865. 2017.
- 58. Di Lena P, Nagata K, Baldi P. Deep architectures for protein contact map prediction. Bioinformatics 2012;28:2449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Singh R, Lanchantin J, Robins G, et al. DeepChrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics 2016;32:i639–48. [DOI] [PubMed] [Google Scholar]
- 60. Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 2015;12:931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuksa PP, Min MR, Dugar R, et al. High-order neural networks and kernel methods for peptide-MHC binding prediction. Bioinformatics 2015;31:3600–7. [DOI] [PubMed] [Google Scholar]
- 62. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 2017;18:851–69. [DOI] [PubMed] [Google Scholar]
- 63. Zeng H, Edwards MD, Liu G, et al. Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics 2016;32:i121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sahraeian SME, Liu R, Lau B, et al. Deep convolutional neural networks for accurate somatic mutation detection. Nat Commun 2019;10:1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Coudray N, Ocampo PS, Sakellaropoulos T, et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat Med 2018;24:1559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kather JN, Pearson AT, Halama N, et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat Med 2019;25:1054–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hamid MN, Friedberg I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics 2019;35:2009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hochreiter S, Heusel M, Obermayer K. Fast model-based protein homology detection without alignment. Bioinformatics 2007;23:1728–36. [DOI] [PubMed] [Google Scholar]
- 69. Sønderby SK, Sønderby CK, Nielsen H, et al. Convolutional LSTM networks for subcellular localization of proteins. In: International Conference on Algorithms for Computational Biology. Springer, Cham, 2015, 68–80. [Google Scholar]
- 70. Schmidhuber J. Deep learning in neural networks: an overview. Neural Netw 2015;61:85–117. [DOI] [PubMed] [Google Scholar]
- 71. Leung MK, Xiong HY, Lee LJ, et al. Deep learning of the tissue-regulated splicing code. Bioinformatics 2014;30:i121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Asgari E, Mofrad MR. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One 2015;10:e0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shi Q, Chen W, Huang S, et al. DNN-Dom: predicting protein domain boundary from sequence alone by deep neural network. Bioinformatics 2019;35:5128–36. [DOI] [PubMed] [Google Scholar]
- 74. Lin M, Chen Q, Yan S. Network in networkarXiv preprint arXiv:1312.4400. 2013.
- 75. Chen XW, Jeong JC. Sequence-based prediction of protein interaction sites with an integrative method. Bioinformatics 2009;25:585–91. [DOI] [PubMed] [Google Scholar]
- 76. Chen W, Xing P, Zou Q. Detecting N(6)-methyladenosine sites from RNA transcriptomes using ensemble support vector machines. Sci Rep 2017;7:40242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wan S, Duan Y, Zou Q. HPSLPred: An ensemble multi-label classifier for human protein subcellular location prediction with imbalanced source. Proteomics 2017;17:1700262. [DOI] [PubMed] [Google Scholar]
- 78. Zou Q, Guo J, Ju Y, et al. Improving tRNAscan-SE annotation results via ensemble classifiers. Mol Inform 2015;34:761–70. [DOI] [PubMed] [Google Scholar]
- 79. Wei L, Zhou C, Chen H, et al. ACPred-FL: a sequence-based predictor based on effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018;34:4007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xiong Y, Wang Q, Yang J, et al. PredT4SE-stack: prediction of bacterial type IV secreted effectors from protein sequences using a stacked ensemble method. Front Microbiol 2018;9:2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang L, Zhang C, Gao R, et al. An ensemble method to distinguish bacteriophage Virion from non-Virion proteins based on protein sequence characteristics. Int J Mol Sci 2015;16:21734–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Azadpour M, McKay CM, Smith RL. Estimating confidence intervals for information transfer analysis of confusion matrices. J Acoust Soc Am 2014;135:EL140–6. [DOI] [PubMed] [Google Scholar]
- 83. Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformat 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lochel HF, Eger D, Sperlea T, et al. Deep learning on chaos game representation for proteins. Bioinformatics 2020;36:272–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.