

Summary
High dimensional compositional and transcriptional profiling of heterogeneous brain-infiltrating leukocytes can lead to novel biological and therapeutic discoveries. High-quality single-cell leukocyte preparations are a prerequisite for optimal single cell profiling. Here, we describe a protocol for epitope and RNA-preserving dissociation of adult mouse brains and subsequent leukocyte purification and staining, which is adaptable to homeostatic and pathogenic brains. Leukocyte preparation following this protocol permits exquisite single-cell surface protein and RNA profiling in applications including CyTOF and CITE-seq.
For complete details on the use and execution of this protocol, please refer to Guldner et al. (2020) and Golomb et al. (2020).
Subject areas: Cell isolation, Single Cell, Flow Cytometry/Mass Cytometry, Sequencing, RNA-seq, Immunology, Neuroscience
Graphical abstract
Highlights
-
•
Mouse brain dissociation into single-cell suspension for downstream single-cell profiling
-
•
Enrichment for brain-resident and low-abundant brain-infiltrating leukocytes
-
•
Protocol tested to preserve >40 immune cell surface epitopes
-
•
Compatible with scRNA-seq, flow cytometry, and CyTOF
High dimensional compositional and transcriptional profiling of heterogeneous brain-infiltrating leukocytes can lead to novel biological and therapeutic discoveries. High-quality single-cell leukocyte preparations are a prerequisite for optimal single cell profiling. Here, we describe a protocol for epitope and RNA-preserving dissociation of adult mouse brains and subsequent leukocyte purification and staining, which is adaptable to homeostatic and pathogenic brains. Leukocyte preparation following this protocol permits exquisite single-cell surface protein and RNA profiling in applications including CyTOF and CITE-seq.
Before you begin
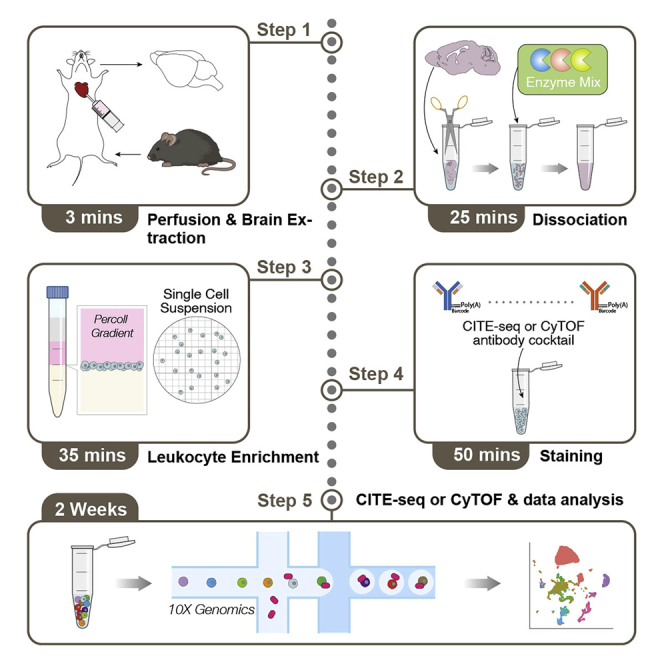
The following protocol (Figure 1A) is for one adult mouse brain (male and female, ages 6 weeks to 70 weeks) with a primary emphasis on preparation for Cellular Indexing of Transcriptome and Epitopes by Sequencing (CITE-seq)/RNA Expression and Protein Sequencing (REAP-seq) and a secondary emphasis on preparation for mass cytometry by time of flight spectrometry (CyTOF) (Bandura et al., 2009; Peterson et al., 2017; Stoeckius et al., 2017). CyTOF and CITE-seq are powerful high dimensional single cell profiling strategies. CyTOF profiles surface epitopes (and in some cases, intracellular epitopes) by detection of metal-conjugated antibodies via time of flight mass spectrometry. CyTOF is predominantly used for immune cell population compositional analyses, and limited functional status may be elucidated by use of antibodies targeting functional markers (for example, PD-L1 or CTLA-4). CITE-seq also profiles surface epitopes, but by means of sequencing oligo-tagged antibodies bound to individual cells, and additionally profiles cellular mRNA by standard 3-prime poly-A capture of mRNA and subsequent sequencing by synthesis. The multi-modal nature of CITE-seq allows both compositional analysis and more detailed functional status determination. Importantly, this protocol can be used for brain dissociation and leukocyte enrichment upstream of several other single cell profiling methods including, but not limited to, standard single cell RNA-sequencing (scRNA-seq) and flow cytometry. Reagents should be scaled to the total number of brain preparations accordingly. Steps listed in ‘Before You Begin’ become increasingly necessary with increasing sample numbers, which is typical for most applications.
Figure 1.

Isolation of brain-infiltrating leukocytes for single cell profiling of epitopes and transcriptomes
(A) Overall protocol schematic.
(B) Images of indicated organs without PBS perfusion (right) and with PBS perfusion (left). Scale bar: 2mm.
(C) Photograph of dissociated brain at the end of enzymatic dissociation (note there are no tissue chunks present).
(D) Photographs of Percoll layering sequences (1-4) and layered Percoll gradient before (lower left) and after (lower middle) centrifugation, with enlarged image pointing out the buffy leukocyte layer post-centrifugation (lower right).
(E) Light microscope image of washed leukocytes post-Percoll gradient centrifugation on a hemocytometer (note the lack of debris and single cell nature of suspension). Scale bar: 100mm.
(F) Bar chart showing the percent of viable cells following brain dissociation, leukocyte enrichment, and cell staining, as determined by the percent of cisplatin negative/low cells of all single cells in CyTOF experiments.
See also Methods video S1.
(G) Bar chart derived from CITE-seq data showing percentage of cells sequenced that were leukocytes (CD45+) from the naive and brain metastasis-burdened brain. Error bars represent SEM.
(H) Stacked bar chart derived from CITE-seq data showing proportions of all identifiable cells sequenced that were leukocytes (CD45+), endothelial cells, pericytes, or metastatic cancer cells from the naive and brain metastasis-burdened brain.
Prepare percoll
Timing: 5 min
-
1.
Prepare ‘working stock’ (WS) Percoll by adding 0.71 mL 10× HBSS (without CaCl2, MgCl2, or CaSO4) to 6.42 mL Percoll. Mix well by inverting or vortexing.
-
2.
Prepare 70% Percoll by adding 1 mL 1× HBSS (without Ca2+, Mg2+, or Phenol Red) to 3.5 mL WS Percoll. Mix well by inverting or vortexing.
-
3.
Prepare ‘37% Red Percoll’ by adding 2.65 mL ‘red 1× HBSS’ (without CaCl2, MgCl2, or CaSO4; with Phenol Red) to 1.85 mL WS Percoll. Mix well by inverting or vortexing.
-
4.
Prepare 30% Percoll by adding 3 mL 1× HBSS (without Ca2+, Mg2+, or Phenol Red) to 1.5 mL WS Percoll. Mix well by inverting or vortexing.
CRITICAL: Percoll solutions should only be prepared the day of use as Percoll solutions can become discolored 24h after preparation, hindering the identification of the buffy leukocyte layer. To increase the speed of preparation on the day of the experiment, we recommended aliquoting pre-measured individual components in separate containers so that they can be quickly mixed when needed on the day of the experiment.
Note: ‘red 1× HBSS’ is used in step 3 to enhance the visibility and thus identification of the buffy leukocyte layer at the interface of the red and clear Percoll layers. 1× HBSS without Phenol Red may be substituted for Phenol Red 1× HBSS, but the visibility of the leukocyte buffy layer will be less obvious.
Note: The Percoll recipes provided here include slightly more volume than necessary for one brain to account for pipetting error.
Prepare blocking buffer, antibody cocktails and staining solutions, and staining/washing buffers
-
5.
Prepare CITE-seq staining and washing buffers according to recipes provided in the Materials and Equipment section. Keep all buffers at 4°C or on ice until use.
-
6.
To prepare blocking buffer, dilute stock mouse FcR Blocking Reagent 1:10 in CITE-seq staining buffer. Keep at 4°C or on ice until use.
-
7.Prepare antibody cocktails. Keep all antibody cocktails at 4°C or on ice until use.
-
a.Prepare CITE-seq antibody cocktail for ∼3–4 × 105 cells by adding 0.25 μL (0.125 μg) of each CITE-seq (oligo-tagged) antibody to 50 μL CITE-seq staining buffer. If multiple samples will be prepared and subsequently pooled and ‘hashed’ to enable multiplexed loading onto the 10× Chromium, first scale up the antibody amounts (minus HTO/“hashing” antibodies) and CITE-seq staining buffer accordingly. Then, aliquot antibody cocktail in 50 μL volumes into individual 1.5 mL tubes. Finally, add 0.3 μl (0.150 μg) unique hashing antibodies, which are oligo-conjugated antibodies that bind ubiquitously expressed proteins MHCI and CD45 and act as sample barcodes, to the appropriate sample tube. For example, add 0.3 μl HTO#1 antibody to sample tube A, 0.3 μl HTO#2 antibody to sample tube B, etcetera (Figure 2A). Antibody pools should be mixed by pipetting up and down.
-
b.Prepare CyTOF antibody cocktail by adding volumes of antibodies indicated in Materials and Equipment section to 50 μL Maxpar Cell Staining Buffer in a 1.5 mL tube (or larger, if needed). Antibody pools should be mixed by pipetting up and down.
-
c.Prepare CyTOF 0.75 μM Cell-ID Cisplatin work solution for dead/live cell identification by diluting 0.5 μL Cell-ID Cisplatin in 2 mL Maxpar PBS.
-
d.Prepare CyTOF iridium intercalator solution for all cell nuclear stain by diluting 0.5 μL Cell-ID Intercalator-Ir in 2 mL Maxpar Fix and Perm Buffer.
-
a.
Note: The recommended antibody amounts in this protocol serve as an excellent starting point that has worked well in our hands, but antibodies should ideally be titrated prior to use. Ideally, titration should be performed with CITE-seq and CyTOF antibodies and optimized by sequencing and CyTOF runs. For CITE-seq, CITE-seq antibody manufacturer BioLegend suggests a less expensive and more convenient option is to perform titration experiments by flow cytometry using fluorescently conjugated antibodies of the same clonal identity, the titration results of which translate well to CITE-seq antibodies.
Figure 2.

CITE-seq quality control
(A) Schematics of antibody staining process.
(B) Representative bioanalyzer electropherograms of CITE-seq antibody (ADT) library showing ADT product peak ~180 bp, which should be the predominant product peak. Smaller peak at ~140 bp is TSO-RT-Oligo product, which could be removed by further SPRI purification.
(C) Representative bioanalyzer electropherograms of RNA library with expected library size of ~300-700 bp.
(D) Representative CellRanger QC summary plot of mean reads per cell versus median genes per cell.
(E) Representative CellRanger QC summary plot of mean reads per cell versus sequencing saturation, with the far right data point indicating full sequencing depth of run.
Prepare items for brain perfusion and extraction
-
8.
Fill a 10 mL syringe with a 25 gauge needle with ice cold 1× PBS (without Ca2+ or Mg2+).
-
9.
Gather and clean bone cutting scissors and regular forceps.
-
10.
Prepare 2 mL collection tube for extracted brain by filling with 1 mL 1× HBSS (without Ca2+, Mg2+, or Phenol Red) and place on ice.
Prepare enzyme dissociation mix
-
11.Prepare stock components of the Multi-Tissue Dissociation Kit I as outlined in manufacturer's datasheet (https://www.miltenyibiotec.com/upload/assets/IM0018263.PDF) and reiterated below.
-
a.Prepare Enzyme D by adding 3 mL serum-free RPMI 1640 to lyophilized Enzyme D vial, inverting continuously until fully dissolved.
-
b.Prepare Enzyme R by adding 2.7 mL serum-free RPMI 1640 to lyophilized Enzyme R vial, inverting continuously until fully dissolved.
-
c.Prepare Enzyme A by adding 1 mL Buffer A to lyophilized Enzyme A vial, inverting continuously until fully dissolved.
-
a.
-
12.
Prepare working enzyme mix by adding 100 μL Enzyme D, 50 μL Enzyme R, 12.5 μL Enzyme A to 2.338 mL 1× D-PBS (with Ca2+ and Mg2+). Prepare immediately prior to beginning brain collection and keep on ice until use.
Note: We have not observed any detrimental effects as a result of not scaling down the components of the working enzyme mix if less than one brain is used. We have found this enzyme mix can be stretched to dissociate as many as 1.5 brains at the expense of slightly longer dissociation time (∼5 min longer).
Note: Unused enzyme aliquots should be immediately stored at −20°C. Enzymes stored at −20°C will remain good for at least 6 months.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CyTOF: anti-CD45-089Y (30-F11) | Fluidigm | Cat#3089005B RRID: N/A |
| CyTOF: rat anti-mouse Ly-6G-141Pr (1A8) | Fluidigm | Cat#3141008B; RRID:AB_2814678 |
| CyTOF: anti-mouse CD11c-142Nd (N418) | Fluidigm | Cat#3142003B; RRID:AB_281473 |
| CyTOF: anti-mouse CD45R-144Nd (RA3-6B2) | Fluidigm | Cat#3144011B; RRID:AB_2811239 |
| CyTOF: anti-mouse CD4-145Nd (RM4-5) | Fluidigm | Cat#3145002B; RRID:AB_2687832 |
| CyTOF: rat anti-mouse CD11b-148Nd (M1/70) | Fluidigm | Cat#3148003B; RRID:AB_2814738 |
| CyTOF: rat anti-mouse CD44-150Nd (IM7) | Fluidigm | Cat#3150018B; RRID:AB_2827882 |
| CyTOF: rat anti-CD25-151Eu (3C7) | Fluidigm | Cat#3151007B; RRID:AB_2827880 |
| CyTOF: anti-mouse CD3e-152Sm (145-2C11) | Fluidigm | Cat#3152004B; RRID:AB_2687836 |
| CyTOF: anti-mouse PD-L1-153Eu (10F.9G2) | Fluidigm | Cat#3153016B; RRID:AB_2687837 |
| CyTOF: anti-CTLA-4-154Sm (UC10-4B9) | Fluidigm | Cat#3154008B RRID: N/A |
| CyTOF: anti-mouse PD-1-159Tb (29F.1A12) | Fluidigm | Cat#3159024B; RRID:AB_2687839 |
| CyTOF: anti-Ly-6C-162Dy (HK1.4) | Fluidigm | Cat#3162014B RRID: N/A |
| CyTOF: anti-mouse CX3CR1-164Dy (SA011F11) | Fluidigm | Cat#3164023B; RRID:AB_2832247 |
| CyTOF: anti-NK1.1-165Ho (PK136) | Fluidigm | Cat#3165018B RRID: N/A |
| CyTOF: rat anti-mouse ckit-166Er (2B8) | Fluidigm | Cat#3166004B; RRID:AB_2801435 |
| CyTOF: rat anti-mouse CD8a-168Er (53-6.7) | Fluidigm | Cat#3168003B; RRID:AB_2811241 |
| CyTOF: anti-CD86-172Yb (GL1) | Fluidigm | Cat#3172016B RRID: N/A |
| CyTOF: anti-I-A/I-E-209Bi (M5/114.15.2) | Fluidigm | Cat#3209006B RRID: N/A |
| CITE-seq: rat anti-CCR2/CD192 (SA203G11) | BioLegend | Cat#150625; RRID:AB_2783122 |
| CITE-seq: rat anti-CD117/c-kit (2B8) | BioLegend | Cat#105843; RRID:AB_2749960 |
| CITE-seq: rat anti-CD11b (M1/70) | BioLegend | Cat#101265; RRID:AB_2734152 |
| CITE-seq: armenian hamster anti-CD11c (N418) | BioLegend | Cat#117355; RRID:AB_2750352 |
| CITE-seq: rat anti-CD172a/SIRPα (P84) | BioLegend | Cat#144033; RRID:AB_2800670 |
| CITE-seq: rat anti-CD25 (PC61) | BioLegend | Cat#102055; RRID:AB_2749982 |
| CITE-seq: rat anti-CD3 (17A2) | BioLegend | Cat#100251; RRID:AB_2750533 |
| CITE-seq: rat anti-CD38 (90) | BioLegend | Cat#102733; RRID:AB_2750556 |
| CITE-seq: rat anti-CD4 (RM4-5) | BioLegend | Cat#100569; RRID:AB_2749956 |
| CITE-seq: rat anti-CD44 (IM7) | BioLegend | Cat#103045; RRID:AB_2734154 |
| CITE-seq: rat anti-CD45 (30-F11) | BioLegend | Cat#103159; RRID:AB_2734156 |
| CITE-seq: rat anti-CD45R/B220 (RA3-6B2) | BioLegend | Cat#103263; RRID:AB_2734158 |
| CITE-seq: rat anti-CD86 (GL-1) | BioLegend | Cat#105047; RRID:AB_2750348 |
| CITE-seq: rat anti-CD8a (53-6.7) | BioLegend | Cat#100773; RRID:AB_2734151 |
| CITE-seq: mouse anti-CD90.1 (OX-7) | BioLegend | Cat#202547; RRID:AB_2783141 |
| CITE-seq: mouse anti-Cx3cr1 (SA011F11) | BioLegend | Cat#149041; RRID:AB_2783121 |
| CITE-seq: rat anti-F4/80 (BM8) | BioLegend | Cat#123153; RRID:AB_2749986 |
| CITE-seq: rat anti-I-A/I-E (M5/114.15.2) | BioLegend | Cat#107653; RRID:AB_2750505 |
| CITE-seq: rat anti-Ly6C (HK1.4) | BioLegend | Cat#128047; RRID:AB_2749961 |
| CITE-seq: rat anti-Ly6G (1A8) | BioLegend | Cat#127655; RRID:AB_2749962 |
| CITE-seq: mouse anti-NK1.1 (PK136) | BioLegend | Cat#108755; RRID:AB_2750536 |
| CITE-seq: rat anti-PD-1 (RMP1-30) | BioLegend | Cat#109123; RRID:AB_2734169 |
| CITE-seq: rat anti-PD-L1 (MIH6) | BioLegend | Cat#153604; RRID:AB_2783125 |
| CITE-seq: rat anti-CD169/Siglec-1 (3D6.112) | BioLegend | Cat#142425; RRID:AB_2783106 |
| CITE-seq: rat anti-Siglec-H (551) | BioLegend | Cat#129615; RRID:AB_275053 |
| CITE-seq: mouse anti-TMEM119 (A16075D) | BioLegend | Cat#853303; RRID:AB_2801201 |
| CITE-seq: mouse anti-XCR1 (Zet) | BioLegend | Cat#148227; RRID:AB_2783120 |
| CITE-seq: rat anti-CD24 (M1/69) | BioLegend | Cat#101841; RRID:AB_2750380 |
| CITE-seq: rat armenian hamster anti-CD103 (2e7) | BioLegend | Cat#121437; RRID:AB_2750349 |
| CITE-seq: rat anti-CD49d (R1-2) | BioLegend | Cat#103623; RRID:AB_2734159 |
| CITE-seq: rat anti-mouse Hashtag 1 antibody (M1/42; 30-F11) | BioLegend | Cat#155801; RRID:AB_2750032 |
| CITE-seq: rat anti-mouse Hashtag 2 antibody (M1/42; 30-F11) | BioLegend | Cat#155803; RRID:AB_2750033 |
| CITE-seq: rat anti-mouse Hashtag 3 antibody (M1/42; 30-F11) | BioLegend | Cat#155805; RRID:AB_2750034 |
| CITE-seq: rat anti-mouse Hashtag 4 antibody (M1/42; 30-F11) | BioLegend | Cat#155807; RRID:AB_2750035 |
| CITE-seq: rat anti-mouse Hashtag 5 antibody (M1/42; 30-F11) | BioLegend | Cat#155809; RRID:AB_2750036 |
| CITE-seq: rat anti-mouse Hashtag 6 antibody (M1/42; 30-F11) | BioLegend | Cat#155811; RRID:AB_2750037 |
| CITE-seq: rat anti-mouse Hashtag 7 antibody (M1/42; 30-F11) | BioLegend | Cat#155813; RRID:AB_2750039 |
| CITE-seq: rat anti-mouse Hashtag 8 antibody (M1/42; 30-F11) | BioLegend | Cat#155815; RRID:AB_2750040 |
| Chemicals, peptides, and recombinant proteins | ||
| Bovine serum albumin | VWR Chemicals | Cat#97061-422 RRID: N/A |
| Cell-ID Cisplatin | Fluidigm | Cat#201064 RRID: N/A |
| Cell-ID Intercalator | Fluidigm | Cat#201192B RRID: N/A |
| Cell Staining Buffer | BioLegend | Cat#20201 RRID: N/A |
| Dulbecco’s PBS (D-PBS), 1×, +Ca, +Mg | Caisson Labs | Cat#PBL02-500M RRID: N/A |
| Ethylenediaminetetra-acetic acid disodium salt dihydrate (EDTA) | VWR Chemicals | Cat#97061-022 RRID: N/A |
| FcR Blocking Reagent, mouse | Miltenyi Biotec | Cat#130-092-575 RRID: N/A |
| Hanks Balanced Salt Solution (HBSS), 1×, -CaCl2, -MgCl2, -MgSO4, +Phenol Red | Thermo Fisher Scientific | Cat#14170161 RRID: N/A |
| Hanks Balanced Salt Solution (HBSS), 1×, -Ca, -Mg, -Phenol Red | Cytiva Life Sciences | Cat#SH30588.02 RRID: N/A |
| Hanks Balanced Salt Solution (HBSS), 10×, -CaCl2, -MgCl2, -MgSO4, -Phenol Red | Thermo Fisher Scientific | Cat#14185052 RRID: N/A |
| Phosphate Buffered Saline (PBS), 1×, -Ca, -Mg, -Phenol Red | Cytiva Life Sciences | Cat#SH3025602 RRID: N/A |
| Maxpar Cell Staining Buffer | Fluidigm | Cat#201068 RRID: N/A |
| MaxPar Fix and Perm Buffer | Fluidigm | Cat#201067 RRID: N/A |
| Maxpar PBS | Fluidigm | Cat#201058 RRID: N/A |
| MaxPar Water | Fluidigm | Cat#201069 RRID: N/A |
| Percoll | GE Healthcare | Cat#17-0891-02 RRID: N/A |
| RPMI 1640 | Thermo Fisher Scientific | Cat#11875093 RRID: N/A |
| Tween 20 | VWR Chemicals | Cat#97062-332 RRID: N/A |
| 1× EQ beads | Fluidigm | Cat#201078 RRID: N/A |
| Critical commercial assays | ||
| Chromium Single Cell 3’ Library and Gel Bead Kit v3 | 10x Genomics | PN-10000092 RRID: N/A |
| Single Cell 3’ GEM Library and Gel Bead Kit v3.1 | 10x Genomics | PN-10000092 RRID: N/A |
| Chromium Chip B Single Cell Kit | 10x Genomics | PN-1000074 RRID: N/A |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 RRID: N/A |
| Multi-tissue Dissociation Kit I | Miltenyi Biotec | 130-110-201 RRID: N/A |
| Deposited data | ||
| Mouse CITE-seq data | (Guldner et al., 2020) | GSE134285 |
| Experimental models: cell lines | ||
| E0771 | CH3 BioSystems | SKU:940001-Vial RRID: N/A |
| Experimental models: organisms/strains | ||
| C57BL/6 | Jackson Laboratories | Stock#:000664 RRID: N/A |
| Oligonucleotides | ||
| Primer: ADT_TruSeq_i7_UDI01 - CAAGCAGAAGACGGCATACG AGAACCGCGGGTGACTGGAGTT CCTTGGCACCCGAGAATTCCA |
This paper | N/A |
| Primer: ADT_TruSeq_i7_UDI04 - CAAGCAGAAGACGGCATACG AGTTGGACTTGTGACTGGAGT TCCTTGGCACCCGAGAATTCCA |
This paper | N/A |
| Primer: ADT-RPI-1 - CAAGCAGAAGA CGGCATACGAGATCGTGATGTGACT GGAGTTCCTTGGCACCCGAGAATTCCA |
This paper | N/A |
| Primer: ADT-RPI-2 - CAAGCAGAAGAC GGCATACGAGATACATCGGTGACTG GAGTTCCTTGGCACCCGAGAATTCCA |
This paper | N/A |
| Primer: HTO_Nextera_i7_UDP01 -CAAG CAGAAGACGGCATACGAGATCGCTCA GTGTGACTGGAGTTCAGACGTGTGC |
This paper | N/A |
| Primer: HTO_Nextera_i7_UDP02 - CAAG CAGAAGACGGCATACGAGATTATCTG ACGTGACTGGAGTTCAGACGTGTGC |
This paper | N/A |
| Primer: HTO_Nextera_i7_UDP03 - CAAG CAGAAGACGGCATACGAGATATATGA GAGTGACTGGAGTTCAGACGTGTGC |
This paper | N/A |
| Primer: HTO_Nextera_i7_UDP04 - CAA GCAGAAGACGGCATACGAGATCTTA TGGAGTGACTGGAGTTCAGACGTGTGC |
This paper | N/A |
| Primer: HTO-N701 - CAAGCAGA AGACGGCATACGAGATTCGCC TTAGTGACTGGAGTTCAGACGT GTGC |
This paper | N/A |
| Primer: HTO-N702 - CAAGCAGAA GACGGCATACGAGATCTAGTACG GTGACTGGAGTTCAGACGTGTGC |
This paper | N/A |
| Software and algorithms | ||
| FlowJo | BD | https://www.flowjo.com |
| R studio | (Ihaka and Gentleman, 1996) | https://rstudio.com |
| CellRanger 3.1.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Cytobank Premium | Beckman Coulter | https://cytobank.org |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com |
| Seurat_3.1.1 | (Satija et al., 2015) | https://satijalab.org/seurat/ |
| Other | ||
| CyTOF2 | Fluidigm | N/A |
| Illumina NovaSeq 6000 | Illumina | N/A |
| 10× Chromium Controller and Accessory Kit | 10x Genomics | PN-120223 |
Materials and equipment
CITE-seq Staining Buffer
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| PBS (1×, without Mg2+ or Ca2+) | 1× | 10 mL |
| BSA | 2.0% | 0.2 g |
| Tween 20 | 0.02% | 2 μL |
| Total | n/a | 10 mL |
Store at 4°C for up to one month.
CITE-seq Wash Buffer A
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| PBS (1×, without Mg2+ or Ca2+) | 1× | 10 mL |
| EDTA (0.5 M) | 2.0mM | 40 μL |
| BSA | 2.0% | 0.2 g |
| Tween 20 | 0.02% | 2 μL |
| Total | n/a | 10 mL |
Store at 4°C for up to one month.
CITE-seq Wash Buffer B
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| PBS (1×, without Mg2+ or Ca2+) | 1× | 10 mL |
| EDTA (0.5 M) | 1.0mM | 20 μL |
| BSA | 2.0% | 0.2 g |
| Tween 20 | 0.02% | 2 μL |
| Total | n/a | 10 mL |
Store at 4°C for up to one month.
CITE-seq Wash Buffer C
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| PBS (1×, without Mg2+ or Ca2+) | 1× | 10 mL |
| EDTA (0.5 M) | 0.1mM | 2.0 μL |
| BSA | 1.0% | 0.1g |
| Tween 20 | 0.02% | 2 μL |
| Total | n/a | 10 mL |
Store at 4°C for up to one month.
Working Stock Percoll
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Percoll (aka original stock) | 90% | 6.42 mL |
| 10× HBSS | 1× | 0.71 mL |
| Total | n/a | 7.13 mL |
Store at room temperature (22°C–25°C) for 3 h or less.
70% Percoll
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Working Stock Percoll | 70% (relative to original stock) | 3.5 mL |
| 1× HBSS | 22% | 1.0 mL |
| Total | n/a | 4.5 mL |
Store at room temperature for 3 h or less.
37% Percoll
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Working Stock Percoll | 37% (relative to original stock) | 1.85 mL |
| 1× HBSS (with Phenol Red) | 58% | 2.65 mL |
| Total | n/a | 4.5 mL |
Store at room temperature for 3 h or less.
30% Percoll
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Working Stock Percoll | 30% (relative to original stock) | 1.5 mL |
| 1× HBSS (with Phenol Red) | 66.66% | 3.0 mL |
| Total | n/a | 4.5 mL |
Store at room temperature for 3 h or less.
Cell-ID Cisplatin Working Stock
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Cell-ID Cisplatin | 0.75 μΜ | 0.5 μL |
| Maxpar PBS | n/a | 2 mL |
| Total | n/a | 2.5 mL |
Make up fresh each use.
Cell-ID Intercalator-IR Working Stock
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Cell-ID Intercalator IR | 0.02% | 0.5 μL |
| Maxpar Fix and Perm Buffer | n/a | 2 mL |
| Total | n/a | 2.5 mL |
Make up fresh each use.
CyTOF Antibody Volumes (per 50 μL Maxpar Cell Staining Buffer/sample)
| Component | Working dilution | Amount |
|---|---|---|
| anti-CD45-089Y (30-F11) | 1:100 | 0.5 μL |
| rat anti-CD25-151Eu (3C7) | 1:100 | 0.5 μL |
| anti-CTLA-4-154Sm (UC10-4B9) | 1:100 | 0.5 μL |
| anti-mouse PD-L1-153Eu (10F.9G2) | 1:100 | 0.5 μL |
| anti-mouse CD11c-142Nd (N418) | 1:100 | 0.5 μL |
| anti-mouse CD4-145Nd (RM4-5) | 1:100 | 0.5 μL |
| anti-mouse CD3e-152Sm (145-2C11) | 1:100 | 0.5 μL |
| anti-mouse PD-1-159Tb (29F.1A12) | 1:100 | 0.5 μL |
| CyTOF: rat anti-mouse ckit-166Er (2B8) | 1:100 | 0.5 μL |
| rat anti-mouse CD11b-148Nd (M1/70) | 1:100 | 0.5 μL |
| rat anti-mouse CD44-150Nd (IM7) | 1:50 | 1.0 μL |
| anti-NK1.1-165Ho (PK136) | 1:50 | 1.0 μL |
| CyTOF: anti-mouse CX3CR1-164Dy (SA011F11) | 1:50 | 1.0 μL |
| anti-CD86-172Yb (GL1) | 1:50 | 1.0 μL |
| anti-Ly-6C-162Dy (HK1.4) | 1:200 | 0.25 μL |
| rat anti-mouse Ly-6G-141Pr (1A8) | 1:200 | 0.25 μL |
| anti-I-A/I-E-209Bi (M5/114.15.2) | 1:200 | 0.25 μL |
| rat anti-mouse CD8a-168Er (53-6.7) | 1:200 | 0.25 μL |
| anti-mouse CD45R-144Nd (RA3-6B2) | 1:200 | 0.25 μL |
Make up fresh each use.
CITE-seq Antibody Volumes (per 50 μL CITE-seq Staining Buffer/sample)
| Component | Working dilution | Amount |
|---|---|---|
| rat anti-CCR2/CD192 (SA203G11) | 1:200 | 0.25 μL |
| rat anti-CD117/c-kit (2B8) | 1:200 | 0.25 μL |
| rat anti-CD11b (M1/70) | 1:200 | 0.25 μL |
| armenian hamster anti-CD11c (N418) | 1:200 | 0.25 μL |
| rat anti-CD172a/SIRPα (P84) | 1:200 | 0.25 μL |
| rat anti-CD25 (PC61) | 1:200 | 0.25 μL |
| rat anti-CD3 (17A2) | 1:200 | 0.25 μL |
| rat anti-CD38 (90) | 1:200 | 0.25 μL |
| rat anti-CD4 (RM4-5) | 1:200 | 0.25 μL |
| rat anti-CD44 (IM7) | 1:200 | 0.25 μL |
| rat anti-CD45 (30-F11) | 1:200 | 0.25 μL |
| rat anti-CD45R/B220 (RA3-6B2) | 1:200 | 0.25 μL |
| rat anti-CD86 (GL-1) | 1:200 | 0.25 μL |
| rat anti-CD8a (53-6.7) | 1:200 | 0.25 μL |
| mouse anti-CD90.1 (OX-7) | 1:200 | 0.25 μL |
| mouse anti-Cx3cr1 (SA011F11) | 1:200 | 0.25 μL |
| rat anti-F4/80 (BM8) | 1:200 | 0.25 μL |
| rat anti-I-A/I-E (M5/114.15.2) | 1:200 | 0.25 μL |
| rat anti-Ly6C (HK1.4) | 1:200 | 0.25 μL |
| rat anti-Ly6G (1A8) | 1:200 | 0.25 μL |
| mouse anti-NK1.1 (PK136) | 1:200 | 0.25 μL |
| rat anti-PD-1 (RMP1-30) | 1:200 | 0.25 μL |
| rat anti-PD-L1 (MIH6) | 1:200 | 0.25 μL |
| rat anti-CD169/Siglec-1 (3D6.112) | 1:200 | 0.25 μL |
| rat anti-Siglec-H (551) | 1:200 | 0.25 μL |
| mouse anti-TMEM119 (A16075D) | 1:200 | 0.25 μL |
| mouse anti-XCR1 (Zet) | 1:200 | 0.25 μL |
| rat anti-CD24 (M1/69) | 1:200 | 0.25 μL |
| rat armenian hamster anti-CD103 (2e7) | 1:200 | 0.25 μL |
| rat anti-CD49d (R1-2) | 1:200 | 0.25 μL |
| rat anti-mouse Hashtag 1 antibody (M1/42; 30-F11) | 1:200 | 0.30 μL |
| rat anti-mouse Hashtag 2 antibody (M1/42; 30-F11) | 1:200 | 0.30 μL |
| rat anti-mouse Hashtag 3 antibody (M1/42; 30-F11) | 1:200 | 0.30 μL |
| rat anti-mouse Hashtag 4 antibody (M1/42; 30-F11) | 1:200 | 0.30 μL |
| rat anti-mouse Hashtag 5 antibody (M1/42; 30-F11) | 1:200 | 0.30 μL |
| rat anti-mouse Hashtag 6 antibody (M1/42; 30-F11) | 1:200 | 0.30 μL |
| rat anti-mouse Hashtag 7 antibody (M1/42; 30-F11) | 1:200 | 0.30 μL |
| rat anti-mouse Hashtag 8 antibody (M1/42; 30-F11) | 1:200 | 0.30 μL |
Make up fresh each use.
Stock Enzyme D
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Lyophilized Enzyme D | n/a | 1 vial |
| serum-free RPMI 1640 | n/a | 3 mL |
| Total | n/a | 3 mL |
Store at −20°C for up to one year.
Stock Enzyme R
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Lyophilized Enzyme R | n/a | 1 vial |
| serum-free RPMI 1640 | n/a | 2.7 mL |
| Total | n/a | 2.7 mL |
Store at −20°C for up to one year.
Stock Enzyme A
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Lyophilized Enzyme A | n/a | 1 vial |
| Buffer A | n/a | 1 mL |
| Total | n/a | 1 mL |
Store at −20°C for up to one year.
Enzyme Mix
| Component | Final Concentration or Percentage | Amount |
|---|---|---|
| Enzyme D | n/a | 100 μL |
| Enzyme R | n/a | 50 μL |
| Enzyme A | n/a | 12.5 μL |
| D-PBS | n/a | 2.338 mL |
| Total | n/a | 2.5 mL |
Make up fresh each use.
Alternative Enzyme Dissociation Kits∗
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Neural Tissue Dissociation Kit (P) | Miltenyi Biotec | 130-092-628 |
| Brain Tumor Dissociation Kit | Miltenyi Biotec | 130-095-942 |
∗Demonstrated to destroy some epitopes
Step-by-step method details
Animal perfusion and brain extraction
This step will perfuse the mouse to eliminate intra-vascular blood cells and results in brain extraction.
-
1.
Anesthetize the mouse using methods approved by the institutional regulatory committee.
-
2.
Spray chest of the mouse with water to matte down fur.
-
3.
Perform a lateral incision down the center of the rib cage, cutting the skin.
-
4.
Perform a lateral incision through the rib cage just to the left or right of the sternum up the entire rib cage to fully expose the chest cavity. Take care to not puncture heart, lungs, or other organs while making this excision. Remove as much of the rib cage as necessary to be able to see and hold the heart with forceps.
-
5.
Holding the heart steady with forceps, make a small incision in the right atrium and immediately begin to perfuse the mouse with 10 mL ice-cold 1× PBS (without Mg2+ or Ca2+) by inserting 25 gauge needle into the apex of the left ventricle, slowly depressing the plunger, perfusing the entire 10 mL volume in no less than 1 min.
-
6.
To extract the brain, first cut the vertebral column/spinal cord at the base of the skull (approximately C1-C3 region) with large bone scissors to detach the skull from the body. This cut should expose the brain stem. Next, insert one blade of fine scissors into the brain stem opening, bracing the sharp part of the scissor blade against the left inner part of the skull. Snip the skull laterally from the opening of the brain stem until you reach the left eye of the mouse, resulting in the left side of the skull as an ‘opening point’ and the right side of the skull as a ‘hinge’. Next, using fine scissors or forceps, push the skull cap upwards, using the previous cut as an insertion/lever point. This should completely remove the skull cap, exposing the top of the brain and entire left side of the brain. Finally, slide forceps between the bottom of the brain and skull to prop out the brain. Keep the brain in a 2 mL tube with cold 1× HBSS on ice until the next step.
Optional: Mouse can be perfused with >10mL of ice-cold PBS in step 5, which is recommended if time allows to further ensure removal of intra-vascular leukocytes.
Brain dissociation
This step will dissociate the mouse brain into a single cell suspension.
-
7.Mechanical Dissociation
-
a.Using sharp scissors (iris scissors or similar), thoroughly mince mouse brain in 2 mL tube filled with 1 mL 1× HBSS (without Ca2+, Mg2+, or Phenol Red) until chunks remain that are generally small enough to be pipetted using a p1000 micropipette.
-
b.Triturate (mechanically dissociate) minced brain using a p1000 micropipette to pipette up and down until there is absolutely no resistance upon pipetting upward. Ideally, this should be done with the sample tube sitting in ice or a pre-chilled tube rack.
-
c.Transfer brain slurry contents to 15 mL conical tube and use fresh 1× HBSS to wash out remaining brain slurry or fine chunks from 2 mL tube into 15 mL tube. As it is important to recover as much of the brain slurry as possible, several washes of 1-2 mL 1× HBSS of the 2 mL tube may be performed. Because this will be centrifuged and pelleted in the next step, there should be no concern about the total volume of 1× HBSS in the 15 mL transfer tube.
-
d.Add at least 2 mL 1× HBSS to 15 mL conical tube containing brain slurry.
-
e.Centrifuge for 3 min at 300RCF and 4°C with acceleration and brake set to low settings.
-
a.
-
8.Enzymatic Dissociation
-
a.Aspirate supernatant from the brain cell pellet.
-
b.Resuspend each brain cell pellet in 2.5 mL pre-prepared Multi-Tissue Dissociation Kit I enzyme mix.
-
c.Rotate or rock sample on a rocker or tube rotator at a medium speed for 10 min at 37°C.
-
d.Triturate sample for 30 seconds to 1 min using a p1000 micropipette, eliminating most chunks of tissue. Any remaining chunks will be further dissociated in subsequent steps.
-
e.Rotate or rock sample at a medium speed for 10 min at 37°C.
-
f.Triturate sample for 30 seconds to 1 min using a p1000 micropipette. No tissue chunks should be present at this point (Figure 1C), but if there are, rotate or rock sample at a medium speed for an additional 2–3 min at 37°C, triturate again, and repeat process until no tissue chunks remain.
-
g.Add at least 8 mL ice cold 1× HBSS (without Ca2+, Mg2+, or Phenol Red) to the sample to help slow the enzymatic reaction. If any tissue chunks remain, remove them by filtering the sample through a 40 μm cell strainer and into a new tube.
-
h.Centrifuge for 10 min at 500RCF at 4°C with acceleration and brake set to low settings.
-
a.
Note: We use a large volume of ice cold 1× HBSS in step 8g to help stop the enzymatic reaction. We theorize this large volume should dilute the reaction and the cold temperature will reduce enzymatic activity. We caution against using serum-containing reagents to help stop the reaction as the serum could artificially stimulate leukocytes in the preparation.
Leukocyte purification by percoll gradient centrifugation
This step will purify brain-infiltrating leukocytes, both enriching these cells in a condensed, extractable layer in the Percoll gradient while removing debris such as myelin (Figure 1D and Methods video S1).
-
9.
Aspirate supernatant from cell pellet.
-
10.
Resuspend cell pellet in 3.5 mL 70% Percoll by pipetting up and down. Bring total volume to 4 mL with 70% Percoll.
-
11.
Carefully layer a total of 4 mL of 37% Red Percoll on top of the 70% layer without allowing intermixing of the two Percoll densities. This can be achieved by using a p1000 micropipette and slowly dripping the 37% Percoll onto the sidewall of the conical tube while the conical tube is held at a slight (∼45 degree) angle (Methods video S1).
-
12.
Carefully layer a total of 4 mL of 30% Percoll on top of the 37% Red Percoll layer without allowing intermixing of the two Percoll densities. This can be achieved as described above. The final layered solution should consist of three distinct layers as shown in Figure 1D.
-
13.
Centrifuge sample in a swinging bucket centrifuge for 25 min at 800 RCF at room temperature (23°C–25°C) with the acceleration at the lowest setting and no break. Following centrifugation, there will be a buffy leukocyte layer at the interface of the 37% Red Percoll and clear 70% Percoll layers and a thick myelin layer at the top of the sample (Figure 1D).
-
14.
Using a vacuum aspirator connected to a p1000 micropipette tip, remove layers above buffy leukocyte layer at the interface of the 37% Red Percoll and 70% clear Percoll layers. Perform this step slowly, keeping the micropipette tip at the surface of the liquid (as opposed to submerged) to ensure the myelin layer is thoroughly removed and minimizing disturbance of the buffy leukocyte layer. Once within 1–2 mL of the buffy leukocyte layer, use a p1000 micropipette with a wide-bore tip to gently remove the remaining Percoll above the buffy leukocyte layer.
-
15.
Using a p1000 micropipette with a fresh tip, extract the buffy leukocyte layer and place it in a new 15 mL conical tube. No more than 2 mL of Percoll on the top or bottom of the buffy leukocyte layer should be extracted.
-
16.
Bring the volume of extracted cells up to 15 mL with cold 1× HBSS (without Ca2+, Mg2+, or Phenol Red) and centrifuge for 7 min at 350–400RCF at 4°C with the acceleration and brake set to medium settings.
-
17.
Aspirate supernatant from cell pellet, leaving as minimal of a volume of residual liquid as possible without disturbing the cell pellet. A small aliquot of cells must be taken out to verify the preparation largely consists of debris-free single cells and to obtain a cell count and examine viability (Figure 1E and 1F). Proceed with the subsequent steps only if the cell preparation is a clean, single cell suspension with >80% viability.
Optional: Centrifugation time in step 15 can be reduced to 20 min with no appreciable effect on cell yield.
Optional: The 30% Percoll layer placed in step 12 can be omitted without compromising cell yield or purity. This layer is mostly used to extend the buffer space between the buffy leukocyte layer and myelin debris, reducing the chance of intermixing during leukocyte extraction.
Note: Related to steps 10 and 11, cells can be resuspended in 37% Percoll instead of 70% Percoll and layered as described above with minimal effect on cell yield. This method has been described and evaluated elsewhere (Lee and Tansey, 2013).
Leukocyte blocking, staining, and washing
This step will block non-specific antibody binding and stain leukocytes with selected antibodies.
-
18.CITE-seq Applications
-
a.Resuspend cell pellet in 50 μL pre-prepared Fc Receptor block. Incubate on ice for 20 min.
-
b.Prepare CITE-seq antibody cocktail as shown in Figure 2A. Add 50 μL antibody cocktail to sample, pipetting up and down to mix with the sample. Incubate on ice for 25 min.
-
c.Add at least 5 mL CITE-seq staining buffer to sample and centrifuge for 5 min at 350–400RCF at 4°C with the acceleration and brake set to medium settings.
-
d.Aspirate supernatant from the cell pellet. Resuspend in at least 5 mL CITE-seq Wash Buffer A and centrifuge for 5 min at 350–400RCF at 4°C with the acceleration and brake set to medium settings.
-
e.Aspirate supernatant from the cell pellet. Resuspend in at least 5 mL CITE-seq Wash Buffer B and centrifuge for 5 min at 350–400RCF at 4°C with the acceleration and brake set to medium settings.
-
f.Aspirate supernatant from the cell pellet. If multiple samples were prepared and hashed, combine individual samples in one tube. Resuspend individual sample or pooled samples in 500 μL CITE-seq Wash Buffer C and aliquot ∼40 μL of the sample to perform a cell count. Add at least 4.5 mL CITE-seq Wash Buffer C and centrifuge sample for 5 min at 350–400RCF at 4C with the acceleration and brake set to medium settings. While cells are pelleting, count the aliquoted cells.
-
g.Resuspend cells in CITE-seq staining buffer at a concentration of 1,600 cells/μL for 10X superloading.
-
a.
-
19.CyTOF applications
-
a.Add at least 5 mL Maxpar PBS to cell pellet and centrifuge for 5 min at 350–400RCF at 4°C with the acceleration and brake set to medium settings.
-
b.Aspirate supernatant from the cell pellet. Resuspend in 1 mL Maxpar PBS. Take a small aliquot of cells to count.
-
c.Transfer an aliquot of 1 million cells or fewer in 100 μL or less Maxpar PBS to a new 15 mL tube. Bring the total volume of cell suspension to 100 μL with Maxpar Cell Staining Buffer.
-
d.Add 100 μL of pre-prepared 0.75 μM Cell-ID cisplatin to the cell suspension and mix by pipetting. Incubate cell suspension in Cell-ID cisplatin for 5 min at room temperature.
-
e.Add 2 mL Maxpar Cell Staining Buffer to cell suspension. Centrifuge for 5 min at 350–400RCF at 4°C with the acceleration and brake set to medium settings.
-
f.Aspirate supernatant from the cell pellet. Resuspend cell pellet in 50 μL pre-prepared Fc Receptor block. Incubate on ice for 20 min.
-
g.Add 50 μL or pre-prepared antibody cocktail to sample, pipetting up and down to mix with the sample. Incubate on ice for 25 min.
-
h.Add at least 1 mL Maxpar Cell Staining Buffer to sample and centrifuge for 5 min at 350–400RCF at 4°C with the acceleration and brake set to medium settings.
-
i.Aspirate supernatant from the cell pellet. Resuspend in 150 μL Maxpar PBS and then add 150 μL 3.2% PFA centrifuge for 5 min at 350–400RCF at 4°C with the acceleration and brake set to medium settings. Incubate at room temperature for 20 min.
-
j.Add at least 1 mL Maxpar Cell Staining Buffer to sample and centrifuge for 5 min at 800RCF at room temperature with the acceleration and brake set to medium settings.
-
k.Aspirate supernatant from the cell pellet. Resuspend in 300 μL pre-prepared Cell-ID Cell-ID Intercalator-Ir to label all nucleated cells. Incubate at room temperature for 1 h or 4°C for 16 h.
-
l.Add 1 mL Maxpar Cell Staining Buffer to sample and centrifuge for 5 min at 800RCF at room temperature.
-
m.Aspirate supernatant from the cell pellet. Resuspend in 1 mL Maxpar Water and centrifuge for 5 min at 800RCF at room temperature.
-
n.Aspirate supernatant from the cell pellet. Resuspend in 200 μL Maxpar Water and aliquot a small volume of cell suspension to count. Bring total cell suspension volume up to 1 mL with Maxpar Water and centrifuge for 5 min at 800RCF at room temperature.
-
o.Aspirate supernatant from the cell pellet. If running sample on a CyTOF instrument immediately, resuspend cells at a concentration of 500 cells/μL in Milli-Q water containing 0.1× EQ Beads.
-
a.
Note: Samples prepared for CyTOF can be stored for days to weeks following the first wash post-Cell-ID Intercalator-Ir staining (step 19l). To store, aspirate supernatant following the first wash post-Cell-ID Intercalator-Ir staining, overlay the cell pellet with ∼50 μL Maxpar Water, seal the tube cap with parafilm, and store at 4°C.
Note: For multiplexed analysis, user should consider to barcode each sample using Cell-ID™ 20-Plex Pd Barcoding Kit available from Fluidigm.
10× genomics-based cell partitioning, library preparation, sequencing, and analysis
This step will partition single cells, create a library for sequencing, and sequence the single cell library.
-
20.
For 10× Genomics-based cell partitioning and library preparation, follow current versions of 10× Genomics protocols and CITE-seq protocols (10× Genomics cell partitioning and library preparation protocol. https://assets.ctfassets.net/an68im79xiti/4tjk4KvXzTWgTs8f3tvUjq/2259891d68c53693e753e1b45e42de2d/CG000183_ChromiumSingleCell3__v3_UG_Rev_C.pdf; CITE-seq without hashing library preparation protocol: https://citeseq.files.wordpress.com/2019/02/cite-seq_190213.pdf; CITE-seq with hashing library preparation protocol: https://citeseq.files.wordpress.com/2019/02/cite-seq_and_hashing_protocol_190213.pdf ). The qualities of cDNA, ADT and HTO libraries should be verified using bioanalyzer before proceeding for sequencing. Examples of high quality cDNA, ADT and HTO libraries are presented in Figures 2B and 2C. Specifically, product peaks should be ∼180 bp for ADT libraries and ∼300–700 bp for mRNA libraries, and these should be the predominant peaks within each library (Figures 2B and 2C). If other product peaks are present, libraries may be re-cleaned with SPRI beads to eliminate unwanted fragments and retain the desired fragments.
-
21.
Sequence the CITE-seq library to the desired sequence depth. In the case of CITE-seq, a critical consideration is the ratio of mRNA to antibody libraries sequenced to obtain optimal sequencing depth for both libraries. Although the amount of each library sequenced will vary based on several factors (for example: the flow cell used and the number of individual libraries if samples were hashed). To this end, we have routinely sequenced hashed samples on a NovaSeq platform. Sequencing a CITE-seq library consisting of 9 HTO-hashed/multiplexed individual samples (68% cDNA library, 25% ADT library, 7%, HTO library) on an NovaSeq S1 flow cell routinely provides the following reads: ∼1000 X 106 reads for cDNA, ∼400 X 106 reads for ADT, and ∼100 X 106 reads for HTO. In our experience, with an expected recovery of ∼16,000 single cells per NovaSeq S1 sequencing lane, such sequencing depth is sufficient to reach >4000 genes/cell and >50% cDNA sequencing saturation of each singlet CD45+ immune cell recovered (Figures 2C and 2D). Multiple CITE-seq libraries can be multiplexed and sequenced on Nova-seq S2 flow cell to further reduce the per cell sequencing cost.
-
22.
To analyze data (analyze differential gene expression, generate dimensional reduction plots, etc.) a number of excellent options are available, including Seurat (Satija et al., 2015) (R coding (Ihaka and Gentleman, 1996)), Scanpy (Wolf et al., 2018) (Python coding), and Cytobank (cloud-based software program). A gating strategy for a brain leukocyte CITE antibody panel is displayed in Figure 3A.
Pause point: Amplified cDNA may be stored at −20°C until ready for final sequencing library preparation. Prepared sequencing library may be stored at −20°C until ready for sequencing.
Note: The upstream work of this protocol should be adaptable to most, if not all, forms of cell partitioning and library preparation.
Note: 10× Genomics regularly updates their reagent kits and protocols related to step 20. Be sure to check 10× Genomics reagent kits that will be used for your experiment and check the associated 10× Genomics protocol before proceeding with droplet generation or library preparation. Likewise, check for updates to the CITE-seq library preparation protocol, which is continuously being adjusted.
Note: When performing cell hashing/sample multiplexing using HTO antibodies to enable sample pooling, we routinely use the HTODemux function in the R package Seurat (Satija et al., 2015) to demultiplex HTO hashed samples as part of the analysis performed in step 22. However, we have observed a low frequency of HTO cross-contamination. Assignment of HTO to respective samples needs to interpretated with caution and should be cross-validated with other computational methods.
Figure 3.

RNA and antibody-based epitope expression in brain infiltrating leukocytes
(A) Example of gating leukocyte populations using CITE antibodies. Briefly, CNS native myeloid cells (microglia and BAM) were gated based on both populations lowly expressing CD45 and then differentiated from each other based on higher I-A-I-E and CD38 expression in BAM. Peripheral leukocytes were gated based on high CD45 expression. Within the peripheral leukocyte population: B cells were defined as having CD19 and B220 expression. T cells were defined based on high CD3 expression and low NK1.1 expression, then subpopulations further distinguished on the basis of CD4 and CD8 expression. Myeloid cells were distinguished from adaptive peripheral immune cells by high CD11b expression, and then further subdivided on the basis of Ly6C expression levels and Ly6G expression.
(B) Example of gating leukocyte populations using CyTOF antibodies.
(C) Biaxial plots of CITE epitope expression on the x axis and its corresponding mRNA expression on the y axis. Discordant expression of mRNA and protein can be observed.
(D) UMAPs derived from CITE-seq data clustered by RNA transcription cluster color coded by transcriptional cluster (left) or canonical cell ID determined by CITE-antibody gating (right). While canonically identified cells generally group within the same cluster, there is appreciable dispersion of canonical cell types among clusters, indicating the utility in using CITE antibody tags rather than mRNA alone to identify canonical cell types.
CyTOF data acquisition and analysis
This step will run cell suspensions on a CyTOF instrument to ultimately obtain a count matrix of the number of antibodies bound to each cell.
-
23.
To run cell suspensions on a CyTOF instrument, follow the most up to date user manual of the specific CyTOF instrument. See https://www.fluidigm.com/binaries/content/documents/fluidigm/consumables/pages/mass-cytometry/mass-cytometry/fluidigm%3Aresources/cytof-2-mass-cytometer/fluidigm%3Afile in the case of operating a CyTOF2.
-
24.
To analyze data (perform gating to analyze population differences and surface marker expression differences, generation dimensional reduction plots, etc.) a number of excellent options are available, including FlowJo and Cytobank. A gating strategy for a brain leukocyte CyTOF panel is displayed in Figure 3B.
Expected outcomes
Single cell profiling requires sample preparations of high quality (a single cell suspension with high cell viability and little debris) that are ideally enriched for the cell population(s) of interest. Adopting this protocol, a debris-free, high viability single cell preparation enriched for leukocytes will be obtained (Figures 1E–1H), meeting the pre-requisite for successful single cell profiling. The number of leukocytes obtained will depend on the pathological state of the mouse, but should yield a minimum of ∼3×105 leukocytes regardless of state, with a viability >90% (Figure 1F). This cell yield and viability meet or exceed the minimum requirements of applications such as CyTOF and CITE-seq. Ultimately, leukocyte preparations derived from following this protocol will lead to exquisite single cell profiling in applications such as CITE-seq/REAP-seq and CyTOF, as demonstrated in our recent publications(Golomb et al., 2020; Guldner et al., 2020). Critically, we observed our brain dissociation protocol preserved many common immune cells identifying or functional surface protein epitopes, as evidenced by positive staining of >40 cell surface epitopes for which we stained in CITE-seq experiments, a subset of which are shown in Figures 3A–3C. When comparing mRNA expression and protein expression of a particular gene, we note a level or discordance between mRNA and protein expression levels (Figure 3C). We also observe that cells identified by use of surface marker expression via CITE antibodies show an appreciable level of dispersion across multiple RNA-based clusters on a UMAP (Figure 3D). Together, the data in Figures 3C and 3D demonstrate the importance of incorporating CITE antibodies into scRNA-seq experiments for proper identification of leukocytes based on the field standard of surface proteins, which may have expression levels discordant with their respective mRNAs.
Quantification and statistical analysis
Pearson correlation between the two features displayed on the biaxial plots in Figure 3C is presented in the top left corner of each plot. This analysis was performed in the R package, Seurat (Satija et al., 2015). This protocol is primarily aimed at describing a strategy for isolating brain infiltrating leukocytes for single cell profiling of transcriptomes and epitopes. For further information on statistical analyses for single cell RNA seq and CyTOF datasets, please refer to application specific guidelines.
Limitations
High-dimensional profiling strategies require cell preparations of high quality (a single cell suspension with high cell viability and little debris) that are ideally enriched for the cell population(s) of interest. Further, the single cell suspensions must be stained with optimized concentrations of antibodies to obtain interpretable results and reduce unnecessary overuse of expensive reagents. The protocol described herein has allowed us to prepare high quality single cell brain leukocyte preparations that have resulted in optimal CyTOF and CITE-seq profiling results. The key broad steps in our protocol include brain dissociation, leukocyte enrichment, and cell staining. Although this protocol has yielded results that meet or exceed current standards, like any methodology, this protocol is not without limitations.
The isolation of mouse brain-infiltrating leukocytes described herein has proven successful in our hands in the contexts of homeostasis, aging, and brain metastasis (Golomb et al., 2020; Guldner et al., 2020). Importantly, these represent conditions in which the brain has an abundance of myelin (aging) or large epithelial lesions (brain metastasis) that can present challenges to brain dissociation and the generation of single cell suspensions. Notably, despite the varying brain conditions we have studied, our protocol was adaptable to each scenario without modification. We have not tested this protocol in the context of protein aggregation pathologies or primary brain tumors, which may present unique challenges to dissociation and subsequent generation of single cell suspensions.
Brain dissociation is a critical step in this protocol that lays the foundation for successful downstream leukocyte enrichment, staining, and profiling. We have tested several mechanical (for example: sonication and Dounce homogenization) and enzymatic (for example: papain-based and collagenase-based) methods for brain dissociation, finding that a combination of mechanical (mincing and triturating) and collagenase-based enzymatic (Multi-Tissue Dissociation Kit I) methods provided optimal results. Importantly, we have optimized this dissociation method to not require any specialized lab equipment. However, this method does have limitations in that (1) it is time consuming and (2) it could destroy some surface protein epitopes. In relation to time, 12 mouse brains can be processed using this protocol with two persons within an appropriate amount of time that does not substantially affect cell yield or viability. Unfortunately, besides the antibodies/surface epitopes we validated here, the resistance of epitopes to enzymatic dissociation must be determined empirically, but can easily be accomplished by less expensive methods such as flow cytometry using identical antibody clones. While manual dissociation alone can be accomplished faster and completely on ice, it could damage and kill cells and may not dissociate tissue thoroughly enough, impacting cell yield and sample cleanliness, both of which are important factors for applications such as CyTOF or scRNA-seq. Lastly, it is worth mentioning that enzyme-based dissociation that occurs at elevated temperatures, such as performed in this protocol at 37°C, could potentially artificially activate/alter the transcriptome of leukocytes (Mattei et al., 2020). If this is observed, we recommend exploring other dissociation options that can be accomplished at lower temperatures.
Even in the inflamed brain, leukocytes represent a small proportion of cells relative to neuronal and glial cells, thus making leukocyte enrichment a critical step. Percoll gradient centrifugation as performed in this protocol can remove myelin debris while confining leukocytes to a small, visible band within the Percoll gradient. This method is a relatively fast and efficient way to achieve a debris-free enriched leukocyte population, but could include some contaminating cells. In fact, we observed that brain metastasis cancer cells can be found within the leukocyte buffy layer and represent approximately 15% of the cells in this layer, and endothelial cells and pericytes can compose approximately 7% and 2% of cells in the buffy layer, respectively (Figures 1G and 1H). Still, neurons, astrocytes, and oligodendrocytes) are largely absent from the leukocyte buffy layer, as evidenced by not being identified in sequencing data. Alternative enrichment methods include fluorescent activated cell sorting (FACS) or magnetic-based sorting strategies which have the potential to be more specific, but are at least as time consuming at Percoll gradient centrifugation, require the use of more specialized equipment, and not as adaptable to performing in a parallelized manner, which is important when several samples are being processed.
Single cell profiling is rapidly and continuously evolving, with existing methods being refined and new techniques being invented. Regardless of the changes that single cell profiling undergo, the requirement for high quality single cell preparation upstream of the profiling will remain constant. This protocol has been demonstrated to reliably dissociate the murine brain, whether homeostatic, aged, or tumor-burdened, into a single cell suspension, and highly enrich brain-infiltrating leukocytes. We have subsequently performed CyTOF and CITE-seq on leukocytes prepared based on the protocol described herein, achieving high quality results, as shown in the figures of this protocol, and our previous publications (Golomb et al., 2020; Guldner et al., 2020).
Troubleshooting
Problem 1
Chunks remain after 20–25 min of enzymatic dissociation (step 8).
Potential solution
Obtaining a tissue suspension free of tissue chunks after enzymatic dissociation requires sufficient upstream preparation, namely, very fine mincing of the brain and thorough trituration after mincing and in the middle of enzymatic dissociation. The brain should initially be minced and triturated upon extraction so that there is no resistance when pipetting the dissociated brain slurry using a P1000 micropipette. Additionally, trituration in the middle of enzymatic dissociation should be performed until nearly all brain chunks are broken up, and titration after enzymatic dissociation should be performed until no more than ∼6 tiny brain chunks remain.
Problem 2
Percoll layers blend together during layering (steps 11 and 12).
Potential solution
Percoll layers tend to blend together due to intermixing during the process of Percoll layering. Slow, gentle placement of less dense Percoll layers on top of the denser Percoll layers is the key to prevent the blending of layers. Additionally, during layering, it can be helpful to (1) tilt tube at a 45 degree angle and (2) drip Percoll dropwise down the sidewall of the tube ∼5 mm away from the surface of the Percoll already in the tube.
Problem 3
Accidental myelin contamination (step 15).
Potential solution
When aspirating myelin, the thick milky-colored layer at the top of all the Percoll layers, and the Percoll above the buffy leukocyte layer, if not all of the myelin was aspirated from the side wall of the conical tube, it may slide down the sidewall of the tube and mix with the buffy leukocyte layer. This issue primarily lies within insufficient removal of the myelin layer. To aid in complete myelin removal, either (1) take extra care to aspirate all of the myelin from the sidewall of the tube and/or (2) add 2 mL 1× HBSS above the 30% Percoll layer before centrifugation. This HBSS layer will remain on top of the myelin layer after centrifugation and aid in cleaner aspiration of myelin.
Problem 4
Cells do not pellet after first percoll wash (step 16).
Potential solution
If too much extra Percoll was extracted with the buffy leukocyte layer (step 14), there may be insufficient dilution of the Percoll in the subsequent washing step, resulting in a cushion of dense Percoll between the bottom of the conical tube and the cells after centrifugation. More specifically, there will be a buffy layer suspended a little above the tip of the conical tube rather than a cell pellet. To avoid this issue, take care not to extract more than ∼3 mL of Percoll total. If this Percoll cushion forms, carefully extract the suspended cells (still look like a buffy layer) in as minimal of a volume as possible and either (1) re-wash or (2) proceed with the next step. As Percoll may interfere with subsequent steps, we advise to re-wash an ensure Percoll is largely removed from the cells.
Problem 5
Unexpected leukocyte activation (step 22).
Potential solution
If leukocytes, especially microglia from a naive brain, display an unexpectedly activated transcriptome, it may be due to any part of the procedure. It is important to work on ice when possible throughout this procedure, as heat could artificially activate leukocytes. If working on ice to the maximum possible does not resolve the artificial activation issue, other dissociation options that avoid incubation at higher temperature should be explored.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Siyuan Zhang (szhang8@nd.edu).
Materials availability
This study did not generate new materials.
Data and code availability
This protocol did not generate new data or employ custom code.
Acknowledgments
This work was funded by NIH R01 CA194697-01 (S.Z.), NIH R01 CA222405 - 01A1 (S.Z.), Notre Dame CRND Catalyst Award (S.Z. and I.H.G.), and NIH CTSI core facility pilot grants (S.Z.). We would like to acknowledge and thank the Dee Family Endowment (S.Z.). We are grateful for the use of the following core facilities and their members’ technical insight: Notre Dame Genomics and Bioinformatics Core Facility, Notre Dame Freimann Life Sciences Center, and Indiana University Simon Cancer Center Core Facility.
Author contributions
I.H.G., Q.W., and S.Z. designed experimental protocols. I.H.G., S.M.G., Q.W., E.W., and S.Z. performed experiments. I.H.G., S.M.G., E.W., and S.Z. analyzed data. I.H.G., S.M.G., E.W., and S.Z. wrote and revised the protocol. S.Z. supervised the study. All authors reviewed the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100537.
Contributor Information
Ian H. Guldner, Email: iguldner@stanford.edu.
Siyuan Zhang, Email: szhang8@nd.edu.
References
- Bandura D.R., Baranov V.I., Ornatsky O.I., Antonov A., Kinach R., Lou X., Pavlov S., Vorobiev S., Dick J.E., Tanner S.D. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009;81:6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- Golomb S.M., Guldner I.H., Zhao A., Wang Q., Palakurthi B., Aleksandrovic E.A., Lopez J.A., Lee S.W., Yang K., Zhang S. Multi-modal single-cell analysis reveals brain immune landscape plasticity during aging and gut microbiota dysbiosis. Cell Rep. 2020;33:108438. doi: 10.1016/j.celrep.2020.108438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldner I.H., Wang Q., Yang L., Golomb S.M., Zhao Z., Lopez J.A., Brunory A., Howe E.N., Zhang Y., Palakurthi B. CNS-native myeloid cells drive immune suppression in the brain metastatic niche through Cxcl10. Cell. 2020;183:1234–1248.e25. doi: 10.1016/j.cell.2020.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R., Gentleman R. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 1996;5:299–314. [Google Scholar]
- Lee J.-K., Tansey M.G. Microglia isolation from adult mouse brain. Methods Mol. Biol. 2013;1041:17–23. doi: 10.1007/978-1-62703-520-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei D., Ivanov A., van Oostrum M., Pantelyushin S., Richetto J., Mueller F., Beffinger M., Schellhammer L., Vom Berg J., Wollscheid B. Enzymatic dissociation induces transcriptional and proteotype bias in brain cell populations. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21217944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson V.M., Zhang K.X., Kumar N., Wong J., Li L., Wilson D.C., Moore R., McClanahan T.K., Sadekova S., Klappenbach J.A. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017;35:936–939. doi: 10.1038/nbt.3973. [DOI] [PubMed] [Google Scholar]
- Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This protocol did not generate new data or employ custom code.