

Abstract
The NanoString RNA counting assay for formalin-fixed paraffin embedded samples is unique in its sensitivity, technical reproducibility and robustness for analysis of clinical and archival samples. While commercial normalization methods are provided by NanoString, they are not optimal for all settings, particularly when samples exhibit strong technical or biological variation or where housekeeping genes have variable performance across the cohort. Here, we develop and evaluate a more comprehensive normalization procedure for NanoString data with steps for quality control, selection of housekeeping targets, normalization and iterative data visualization and biological validation. The approach was evaluated using a large cohort (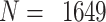 ) from the Carolina Breast Cancer Study, two cohorts of moderate sample size (
) from the Carolina Breast Cancer Study, two cohorts of moderate sample size (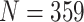 and
and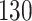 ) and a small published dataset (
) and a small published dataset (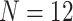 ). The iterative process developed here eliminates technical variation (e.g. from different study phases or sites) more reliably than the three other methods, including NanoString’s commercial package, without diminishing biological variation, especially in long-term longitudinal multiphase or multisite cohorts. We also find that probe sets validated for nCounter, such as the PAM50 gene signature, are impervious to batch issues. This work emphasizes that systematic quality control, normalization and visualization of NanoString nCounter data are an imperative component of study design that influences results in downstream analyses.
). The iterative process developed here eliminates technical variation (e.g. from different study phases or sites) more reliably than the three other methods, including NanoString’s commercial package, without diminishing biological variation, especially in long-term longitudinal multiphase or multisite cohorts. We also find that probe sets validated for nCounter, such as the PAM50 gene signature, are impervious to batch issues. This work emphasizes that systematic quality control, normalization and visualization of NanoString nCounter data are an imperative component of study design that influences results in downstream analyses.
Keywords: NanoString nCounter expression, gene expression normalization, quality control, data visualization
Introduction
The NanoString nCounter platform offers a targeted strategy for gene expression quantification using a panel of up to 800 genes without requiring cDNA synthesis or amplification steps [1]. The technology offers advantages in sensitivity, technical reproducibility and strong robustness for profiling formalin-fixed, paraffin-embedded (FFPE) samples [2]. Given these advantages, nCounter is increasingly used for longitudinal studies involving FFPE samples carried out over several years [3] and diagnostic assays in clinical settings [4, 5].
Proper normalization and quality control (QC) of gene expression is necessary prior to statistical analysis to reduce unwanted variation that may be associated with technical batches or RNA degradation from sample fixation [6, 7]. While some sources of variation can be enumerated a priori (e.g. different research centers, batches over time or RNA preservation methods), not all can be captured. In all cases, it is advisable to define a QC and normalization pipeline to detect and account for technical variation in downstream statistical modeling. All normalization methods deal with a trade-off between bias that needs correction and bias or variance that may be introduced in normalization [8].
Many approaches have been developed to normalize nCounter data. NanoString provides two forms of normalization in its commonly used nSolver Analysis Software [9]: (i) a graphical user interface with optional background correction and positive-control and housekeeping gene normalization and (ii) the Advanced Analysis tool, which draws on the NormqPCR R package [10, 11] to select co-expressed housekeeping genes prior to normalization. The NanoStringNorm package implements the nSolver algorithms in R [12]. The R packages NanoStringDiff and RCRnorm use hierarchical modeling methods that incorporate information from the positive, negative and housekeeping controls for normalization [13, 14]. The NACHO R package proposes a simple QC and visualization pipeline that precedes normalization using either NanoStringNorm or NanostringDiff [15], though, without postnormalization visualization to assess normalization quality. When technical replicates are available, a method from Molania et al., Remove Unwanted Variation-III (RUV-III), can be used along with an iterative normalization process where several parameters (i.e. number of housekeeping genes, number of detected outliers and number of dimensions of technical noise) are tuned with relevant visual and biological checks [7]. RUV-III normalization frequently outperformed nSolver normalization by more efficiently removing technical sources of variation while preserving biological variation [7]. Since many cohorts do not have technical replicates, we extend Molania et al.’s iterative framework using RUVSeq [6–8], a precursor of RUV-III.
Here, we provide a framework for the QC and normalization of mRNA expression count data from the NanoString nCounter platform, using a large dataset (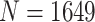 ) of breast tumor expression from the Carolina Breast Cancer Study (CBCS) and three other cohorts of differing sample size (N
) of breast tumor expression from the Carolina Breast Cancer Study (CBCS) and three other cohorts of differing sample size (N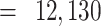 and
and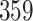 ). We illustrate some of the pitfalls in the nSolver method of background correction and positive control normalization, provide an alternative approach that uses RUVSeq [6, 8] and benchmark our framework against other normalization methods [9, 13, 14]. We find that, especially in longitudinal, multiphase or multisite cohorts, RUVSeq outperforms nSolver in removing differences across technical sources of variation. Lastly, we provide quality checks for normalization and outline the impact of proper normalization on inference for biological associations and expression-based disease subtyping.
). We illustrate some of the pitfalls in the nSolver method of background correction and positive control normalization, provide an alternative approach that uses RUVSeq [6, 8] and benchmark our framework against other normalization methods [9, 13, 14]. We find that, especially in longitudinal, multiphase or multisite cohorts, RUVSeq outperforms nSolver in removing differences across technical sources of variation. Lastly, we provide quality checks for normalization and outline the impact of proper normalization on inference for biological associations and expression-based disease subtyping.
Materials and methods
Data collection
We used four cohorts with nCounter gene expression data to evaluate differences between normalization procedures. Cohort details and the normalization parameters for each cohort are given below and summarized in Supplementary Table S1 available online at https://academic.oup.com/bib.
CBCS gene expression data
The CBCS is a multiphase cohort of women with breast cancer in North Carolina. Samples were collected during three study phases: Phase 1 (1993–1996), Phase 2 (1996–2001) and Phase 3 (2008–2013). Paraffin-embedded tumor blocks were reviewed and assayed for gene expression using the NanoString nCounter system as discussed previously [3, 16, 17]. Study phase gives the relative age of the tumor block. In total, 1649 samples from patients with invasive breast cancer from CBCS, across all three study phases, were analyzed on a custom panel of 417 genes. All assays were performed in the Translational Genomics Laboratory (TGL) at the University of North Carolina at Chapel Hill (UNC). After QC and normalization, 1264 samples remained in the nSolver-normalized data, and 1219 samples remained in the RUVSeq-normalized data. This dataset was used to benchmark against NanoStringDiff [13] and RCRnorm [14], using the same 1264 samples in the nSolver-normalized set.
Bladder tumor gene expression data
FFPE Biospecimens from 42 samples of nonmuscle invasive bladder cancer from UNC (Chapel Hill, NC) and 88 samples from a study conducted by the Memorial Sloan Kettering Cancer Center (New York, NY) with nonmuscle invasive bladder cancer were analyzed. RNA was isolated using the RNeasy FFPE Kit (Qiagen) at UNC, and NanoString assays were performed at the TGL at UNC using a custom codeset consisting of 440 endogenous and 6 housekeeping genes. After QC and normalization, 86 samples remained in both the nSolver-normalized and RUVSeq-normalized datasets.
Kidney tumor gene expression data
This study includes 359 samples from patients with clear cell renal cell carcinoma with fresh-frozen tissue collected as part of a large case-control study of kidney cancer conducted in central and eastern Europe [18]. Slides for each case were reviewed by a pathologist to assess tumor stage and grade [19]. Manual microdissection was performed to remove nontumor tissue. Frozen sections were placed directly in Trizol reagent (Invitrogen, Carlsbad, CA) and homogenized for 2 minutes on ice, and RNA was isolated using the manufacturer’s protocol. NanoString assays were performed at UNC TGL using a custom codeset consisting of 62 endogenous and 6 housekeeping genes commonly studied in kidney cancer. After QC and normalization, 331 samples remained in both the nSolver- and RUVSeq-normalized data.
Sabry et al. gene expression data
We downloaded raw RCC files from Sabry et al. [20] from the NCBI Gene Expression Omnibus (GEO) with accession number GSE130286 and imported them using functions in NanoStringQCPro [21]. This dataset comprised 12 samples, all of which remained after normalization with both procedures. The dataset measured 706 endogenous genes with 40 housekeeping genes from the NanoString nCounter Human Myeloid Innate Immunity Panel [20].
QC and normalization
The full QC and normalization process using nSolver and RUVSeq is summarized in Figure 1, starting with familiarization of the raw data (Figure 1(1)), technical QC (Figure 1(2)), prenormalization assessment of housekeeping genes (Figure 1(3)) and data visualization to detect problematic samples and assess whether flagged samples should be removed (Figure 1(4)). Normalization is performed with either nSolver or RUVSeq (Figure 1(5)), and the processed expression data are assessed for validity through relevant visualization and biological checks (Figure 1(6)). If validation is unsatisfactory and technical variation is still present, this process is iterated.
Figure 1.
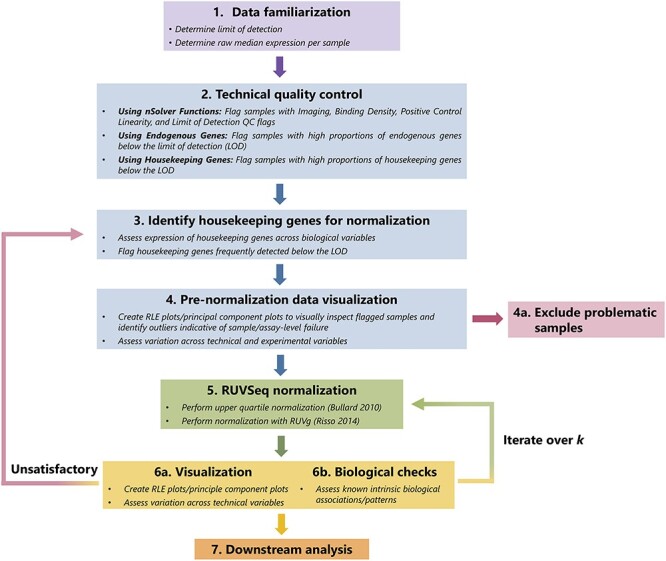
Graphical summary of RUVSeq normalization pipeline. The QC and normalization process starts with familiarization with the data (Step 1) and technical QC to flag samples with potentially poor quality (Step 2). After a set of housekeeping genes are selected (Step 3), important unwanted technical variables are also investigated through visualization techniques (Step 4). Problematic samples (e.g. those that are flagged multiple times in technical QC checks) are excluded. Next, the data are normalized using upper quartile normalization and RUVSeq (Step 5), and the normalized data are visualized to assess the removal of unwanted technical variation and retention of important biological variation (Step 6). Steps 3—6 are iterated until technical variation is satisfactorily removed, changing the set of housekeeping genes or the number of dimensions of unwanted technical variation (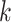 ) estimated using RUVSeq. These data can then be used for downstream analysis (Step 7).
) estimated using RUVSeq. These data can then be used for downstream analysis (Step 7).
Technical QC flags
nSolver provides QC flags to assess the quality of the data for imaging, binding density, linearity of the positive controls and limit of detection (LOD). The definition and implementation of this QC are summarized in nSolver [9] and NanoStringNorm [12] documentation. We mark any sample that is flagged in at least one of these four QC assessments as technical QC. We use these QC flags in both nSolver normalization and RUVSeq normalization.
Below LOD QC
We use high proportions of both endogenous and housekeeping genes below the LOD as a QC flag to assess reduced assay or sample quality. The per-sample LOD is defined as the mean of the counts of negative control probes for a given sample. We assessed the percent of counts below the LOD in the housekeeping genes per sample to flag both poor quality samples and housekeeping genes with problems in their measurement. We used samples with all housekeeping genes above the LOD as a reference group to determine the regular distribution of genes below the LOD. Samples were flagged if (i) they had more than one housekeeping gene below the LOD and (ii) the percent of endogenous genes below the LOD was greater than the top quartile of the distribution of percent below LOD in the reference group.
Housekeeping gene assessment
Housekeeping genes serve two purposes: (i) for QC purposes to remove samples with overall poor quality and (ii) for assessing the amount of technical variation present in the normalization procedure. NanoString documentation suggests that ideal housekeeping genes are highly expressed, have similar coefficients of variation and have expression values that correlate well with other housekeeping genes across all samples [9, 12]. Because of these definitions, these targets will ideally vary only due to the level of technical variation present. RUVSeq relies on housekeeping genes, i.e. genes not influenced by the condition of interest (e.g. cancer subtype), with no assumptions on co-expression of all housekeeping genes. To assess the potential for housekeeping correction to introduce bias, housekeeping genes were assessed for differential expression across a primary biological covariate of interest (estrogen receptor status in CBCS, tumor stage in the kidney and bladder cancer data and treatment groups in Sabry et al. [20]) using negative binomial regression on the raw counts from the MASS package [22].
nSolver normalization
Background correction
NanoString guidelines suggest background correction [9, 12] by either subtraction or thresholding for an estimated background noise level for experiments in which low expressing targets are common, or when the presence of a transcript has an important research implication [7, 12]. Data from all four cohorts considered do not necessarily fall under this criterion, and accordingly, we did not background correct by either method. To demonstrate the effect of background correction, we tested nSolver-normalized gene expression with and without background thresholding in CBCS using relative log expression (RLE) plots.
Positive control and housekeeping gene-based normalization
The arithmetic mean of the geometric means of the positive controls for each lane was computed and then divided by the geometric mean of each lane to generate a lane-specific positive control normalization factor [9, 12]. The counts for every gene were multiplied by their lane-specific normalization factor. To account for any noise introduced into the nCounter assay by positive normalization, the housekeeping genes were used similarly as the positive control genes to compute housekeeping normalization factors to scale the expression values [9, 12]. NanoString flagged samples with large housekeeping gene scaling factors (we call this a housekeeping QC flag) and large positive control scaling factors (positive QC flag), but note that samples with these flags simply indicate that a sample is divergent from other samples in the dataset and do not necessarily require removal. Prenormalization visualization (Figure 1(4)) is important for confirming the inclusion or removal of these samples.
RUVSeq normalization pipeline
Normalization
The RUVSeq-based normalization process (Figure 1(5)), an alternative approach to nSolver normalization, proceeds following QC and housekeeping assessment. Distributional differences were scaled between lanes using upper-quartile normalization [23]. Unwanted technical factors were estimated in the resulting gene expression data with the RUVg function from RUVSeq [8]. Unwanted variation was estimated using the final set of endogenous housekeeping genes on the NanoString gene expression panel [24, 25]. In general, the number of dimensions of unwanted variation to remove was chosen by iteratively normalizing the data for a given number of dimensions and checking for the removal of known technical factors already identified in the raw expression data (e.g. study phase) and the presence of key biological variation (e.g. bimodality of ESR1 expression in the CBCS breast cancer data where estrogen receptor (ER) status is a known predominant feature). Further details about choosing this dimension are given by Gagnon-Bartsch et al. and Risso et al. [6, 8]. DESeq2 was used to compute a variance stabilizing transformation of the original count data [25], and estimated unwanted variation was removed using the removeBatchEffects function from limma [26]. Ultimately, we removed 1, 1, 3 and 1 dimensions of unwanted variation from CBCS, kidney cancer, bladder cancer and the Sabry et al. datasets, respectively. RLE plots, principal component analysis and heatmaps were used to detect any potential outliers before and after normalization.
Alternative normalization methods for benchmarking
Using CBCS data, we compared the normalized datasets from nSolver, RUVSeq, NanoStringDiff [13] and RCRnorm [14] with the raw data through visualization methods outlined above (Figure 1(1)–(4), RLE plots and scatter plots of principal components over important technical and biological sources of variation). Details about these methods are provided in Supplementary Table S2 available online at https://academic.oup.com/bib.
Downstream analyses
We used several data visualization or benchmarking methods for each cohort.
Silhouette width analysis in CBCS
Silhouette width, a measure used to assess how similar a sample is to its own group (i.e. study phase) as compared to other groups, was used to determine the impact of the two normalization procedures on technical and biological variation [27]. Many samples with large silhouettes can be interpreted as indicating that the different study phases are distinct and that a batch effect is still present in the data.
eQTL analysis in CBCS
We assessed the additive relationship between the gene expression values and germline genotypes with linear regression analysis using MatrixEQTL [28], applying the same linear model as detailed in previous work [29]. Briefly, for each gene and SNP in our data, we constructed a simple linear regression, where the dependent variable is the scaled expression of the gene with zero mean and unit variance, the predictor of interest is the dosage of the alternative allele of the SNP and the adjusting covariates are the top five principal components of the genotype matrix. We considered both cis- (SNP is less than 0.5 Mb from the gene) and trans-eQTLs in our analysis. We adjusted for multiple testing via the Benjamini-Hochberg procedure [30].
PAM50 subtyping in CBCS
We classified each subject into PAM50 subtypes using the procedure summarized by Parker et al. [31, 32]. Briefly, for each sample, we computed the Euclidean distance of the log-scale expression values for the 50 PAM50 genes to the PAM50 centroids for each of the molecular subtypes. Each sample was classified to the subtype with the minimal distance [31]. The PAM50 genes were clustered hierarchically for both samples and genes and visualized in heatmaps. Subtype concordance was assessed between normalization methods excluding normal-like cases.
RNA-seq normalization and distance correlation analysis in CBCS
We obtained a separate set of samples (not included in the analysis described above) from CBCS with both RNA-seq and nCounter expression (on a different codeset of 166 genes). We followed a standard RNA-seq normalization process with DESeq2 [25], using the median of ratios method to estimate scaling factors [24]. We calculated the distance correlation and conducted a multivariate permutation test of independence between the RNA-seq data set (subset to the overlapping genes on the NanoString codeset) with each of the nSolver-normalized and RUVSeq-normalized nCounter data using the energy package [33]. The distance correlation and associated permutation test allow for detection of nonindependence across multivariate datasets of different distribution.
Differential expression analysis with Sabry et al. dataset [20]
We conducted differential expression analysis to compare both normalization methods in the Sabry et al. dataset [20] using DESeq2 [25] and adjusting for multiple testing with the Benjamini-Hochberg [30] procedure. We compared differential expression across IL-2–primed NK cells versus NK cells alone and CTV-1-primed NK cells for 6 hours versus NK cells alone.
Results
We evaluated the ability of normalization methods to remove technical variation while retaining biologically meaningful variation across four cohorts of differing sample size and varying sources of technical bias (see Supplementary Table S1 available online at https://academic.oup.com/bib). Known sources of technical variation included age of sample (study phase) and different study sites. The cohorts varied in preservation methods; two cohorts used fresh-frozen specimens, while two used archival FFPE specimens. The number of genes measured for both endogenous genes and housekeeping genes also varied by study. In addition, some studies used validated and optimized code sets for specific gene signatures versus a more general code set.
In cohorts with large technical biases, RUVSeq provided superior normalization with more robust removal of technical variation and provided stronger biological associations compared to other normalization methods. In two of the datasets, we found that downstream analyses performed on data normalized with nSolver and RUVSeq detected substantially different biological associations. However, when few strong technical biases were present or if a validated and optimized code set (e.g. PAM50 genes) was used, nSolver and RUVSeq performed comparably.
Case study: CBCS
Evaluation of background correction
Background thresholding led to increased per-sample variance, while per-sample medians remained relatively similar (see Supplementary Figure S1A available online at https://academic.oup.com/bib). The distributions of per-sample median expression values were more right-skewed (greater mean than median) when using background thresholding prior to normalization compared to not using background thresholding (see Supplementary Figure S1B available online at https://academic.oup.com/bib). Based on this analysis, we did not perform background correction prior to normalization for all cohorts analyzed.
Quality assessment of expression levels using LOD of housekeeping genes
We used the housekeeping genes to assess if the lack of expression of endogenous genes was due to biology or due to technical failures. We compared the level of missing endogenous genes in samples with all housekeeping genes present to those with increasing number of housekeeping genes below LOD. There was a strong positive correlation for increasing proportions of genes below the LOD in both the endogenous and housekeeping genes (Figure 2A, see Supplementary Figure S2 available online at https://academic.oup.com/bib). Samples with higher numbers of genes below the LOD were from earlier phases of CBCS (i.e. Phase 1 from 1993 to 1996 and Phase 2 from 1996 to 2001) and thus associated with sample age (see Figure 2A, Supplementary Figure S3 available online at https://academic.oup.com/bib). Samples with a higher proportion of endogenous genes below the LOD had increased numbers of QC flags as well (see Supplementary Figure S2 available online at https://academic.oup.com/bib).
Figure 2.
QC and normalization validation in CBCS. (A) Boxplot of percent of endogenous genes below the LOD ( -axis) over varying numbers of the 11 housekeeping genes below LOD (
-axis) over varying numbers of the 11 housekeeping genes below LOD ( -axis), colored by CBCS study phase. Note that the
-axis), colored by CBCS study phase. Note that the 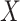 -axis scale is decreasing. (B) Kernel density plots of deviations from median per-sample log2-expression from the raw, nSolver-, RUVSeq-, NanoStringDiff- and RCRnorm-normalized expression matrices, colored by CBCS study phase. (C) Plots of the first principal component (
-axis scale is decreasing. (B) Kernel density plots of deviations from median per-sample log2-expression from the raw, nSolver-, RUVSeq-, NanoStringDiff- and RCRnorm-normalized expression matrices, colored by CBCS study phase. (C) Plots of the first principal component (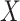 -axis) versus second principal component (
-axis) versus second principal component (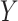 -axis) colored by ER subtype of the raw, nSolver-, RUVSeq-, NanoStringDiff- and RCRnorm-normalized expression data. (D) Violin plots of the distribution of per-sample silhouette values, as calculated to study phase, using raw, nSolver-, RUVSeq-, NanoStringDiff- and RCRnorm-normalized expression. The boxplot shows the 25% quartile, median and 75% quartile of the distribution, and the plotted triangle shows the mean of the distribution.
-axis) colored by ER subtype of the raw, nSolver-, RUVSeq-, NanoStringDiff- and RCRnorm-normalized expression data. (D) Violin plots of the distribution of per-sample silhouette values, as calculated to study phase, using raw, nSolver-, RUVSeq-, NanoStringDiff- and RCRnorm-normalized expression. The boxplot shows the 25% quartile, median and 75% quartile of the distribution, and the plotted triangle shows the mean of the distribution.
Evaluation of normalization methods
We benchmarked RUVSeq and nSolver with two other normalization methods, NanoStringDiff [13] and RCRnorm [14]. We observed differences across the four normalization strategies (described in Supplemental Table S2 available online at https://academic.oup.com/bib), namely greater remaining technical variation using nSolver and NanoStringDiff than RCRnorm and RUVSeq (Figure 2B–D). A large portion of the variation in the raw expression could be attributed to study phase (see Supplementary Figure S4A available online at https://academic.oup.com/bib). While all methods reduced study phase associated variation compared to the raw data, there were considerable differences in the deviations from the median log-expressions in the nSolver- and NanoStringDiff-normalized expression that are not present in the RUVSeq- and RCRnorm-normalized data (Figure 2B). The nSolver and NanoStringDiff methods retained technical variation, either not fully corrected or re-introduced during the nSolver normalization process.
We examined the ability of each normalization method to retain biological variation. ER status is one of the most important clinical and biological features in breast cancer and is used for determining course of treatment [34, 35]. ER status drives many of the molecular classifications [36–38] and even drives separate classification of breast tumors in TCGA’s pan-cancer analysis of 10 000 tumors [39]. In the raw expression, variation due to ER status was captured in PC2 rather than PC1 (study age); however, after RUVSeq-normalization, ER status was reflected predominantly in PC1 (Figure 2C). In the nSolver-, NanoStringDiff- and RCRnorm-normalized data, ER status was shared between PC1 and PC2, suggesting that unresolved technical variation was still present. RUVSeq demonstrated effective removal of technical variation and boosting of the true biological signal. The PAM50 molecular subtypes [31], which are also linked with ER status, were also clearly separated by PC1 for RUVSeq-normalized data, but this was not thess case for nSolver-, NanoStringDiff-, or RCRnorm-normalization (see Supplementary Figure S4B available online at https://academic.oup.com/bib). These results suggest that RUVSeq-normalization best balances the removal of technical variation with the retention of important axes of biological variation, with RCRnorm showing better performance than nSolver and NanoStringDiff, but not superior to RUVSeq. A significant disadvantage of RCRnorm is its computational cost: RCRnorm was unable to run on the CBCS dataset (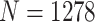 after QC) on a 64-bit operating system with 8 GB of installed RAM, requiring RCRnorm-normalization to be performed on a high-performance cluster. We summarize the maximum memory used by the method in CBCS in Supplemental Table S2 available online at https://academic.oup.com/bib.
after QC) on a 64-bit operating system with 8 GB of installed RAM, requiring RCRnorm-normalization to be performed on a high-performance cluster. We summarize the maximum memory used by the method in CBCS in Supplemental Table S2 available online at https://academic.oup.com/bib.
We used silhouette width to assess extent of unwanted technical variation from study phase remaining by the normalization methods. Larger positive silhouette values indicate within-group similarity (i.e. samples clustering by study phase). Per-sample silhouettes across the alternatively normalized datasets showed that RUVSeq best addressed the largest source of technical variation identified in the raw data (Figure 2D, see Supplementary Figure S5A available online at https://academic.oup.com/bib) while also not removing a significant portion of biological variation (see Supplementary Figure S5B available online at https://academic.oup.com/bib). NanoStringDiff also demonstrated less similarity of samples across study phase similar to RUVSeq but removed biologically relevant similarity of samples grouped by ER status. Due to the performance of NanoStringDiff and computational limitations of RCRnorm, for subsequent analyses and datasets, we only illustrate differences between nSolver- and RUVSeq-normalized data.
Genomic analyses and expression profiles across normalization methods
We evaluated the impact of normalization choice on downstream analyses including eQTLs, PAM50 molecular subtyping, known expression patterns and similarity to RNA-seq data. In a full cis-trans eQTL analysis accounting for race and genetic-based ancestry, we found considerably more eQTLs using nSolver as opposed to RUVSeq, thresholding at nominal 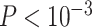 (2050 versus 1143). We identified strong cis-eQTL signals in both normalized datasets; however, stronger FDR values were identified with RUVSeq (Figure 3A, densely populated around the 45° line). We observed considerably more trans-eQTLs using nSolver, including a higher proportion of trans-eQTLs across various FDR-adjusted significance levels (Figure 3B, see Supplementary Figures S6 and S7 available online at https://academic.oup.com/bib). We suspected that spurious trans-eQTLs may have resulted from residual technical variation in expression data, which was confounded with study phase, subsequently being identified as a QTL due to ancestry differences across study phase. In cross-chromosomal trans-eQTL analysis, distributions of absolute differences in minor allele frequency (MAF) for trans-eSNPs across women of African and European ancestry were wide for both methods (see Supplementary Figure S7 available online at https://academic.oup.com/bib). However, we observed substantially more trans-eSNPs with moderate absolute MAF differences across study phase with nSolver, compared to RUVSeq. This provides some evidence for the presence of residual confounding technical variation in the nSolver-normalized expression data leading to spurious trans-eQTL results (with a directed acyclic graph for this hypothesis in Supplementary Figure S8 available online at https://academic.oup.com/bib), though we cannot confirm this with eQTL analysis alone.
(2050 versus 1143). We identified strong cis-eQTL signals in both normalized datasets; however, stronger FDR values were identified with RUVSeq (Figure 3A, densely populated around the 45° line). We observed considerably more trans-eQTLs using nSolver, including a higher proportion of trans-eQTLs across various FDR-adjusted significance levels (Figure 3B, see Supplementary Figures S6 and S7 available online at https://academic.oup.com/bib). We suspected that spurious trans-eQTLs may have resulted from residual technical variation in expression data, which was confounded with study phase, subsequently being identified as a QTL due to ancestry differences across study phase. In cross-chromosomal trans-eQTL analysis, distributions of absolute differences in minor allele frequency (MAF) for trans-eSNPs across women of African and European ancestry were wide for both methods (see Supplementary Figure S7 available online at https://academic.oup.com/bib). However, we observed substantially more trans-eSNPs with moderate absolute MAF differences across study phase with nSolver, compared to RUVSeq. This provides some evidence for the presence of residual confounding technical variation in the nSolver-normalized expression data leading to spurious trans-eQTL results (with a directed acyclic graph for this hypothesis in Supplementary Figure S8 available online at https://academic.oup.com/bib), though we cannot confirm this with eQTL analysis alone.
Figure 3.
eQTL analysis in CBCS. (A) Cis-trans plots of eQTL results from nSolver-normalized (left) and RUVSeq-normalized data with chromosomal position of eSNP on the  -axis and the transcription start site of associated gene in the eQTL (eGene) on the
-axis and the transcription start site of associated gene in the eQTL (eGene) on the 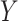 -axis. Points for eQTLs are colored by FDR-adjusted
-axis. Points for eQTLs are colored by FDR-adjusted 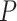 -value of the association. The dotted line provides a 45° reference line for cis-eQTLs. (B) Number of cis- (left) and trans-eQTLs (right) across various FDR-adjusted significance levels. The number of eQTLs identified in nSolver-normalized data is shown in red and the number of eQTLs identified in RUVSeq-normalized data is shown in blue.
-value of the association. The dotted line provides a 45° reference line for cis-eQTLs. (B) Number of cis- (left) and trans-eQTLs (right) across various FDR-adjusted significance levels. The number of eQTLs identified in nSolver-normalized data is shown in red and the number of eQTLs identified in RUVSeq-normalized data is shown in blue.
We compared each normalization method for the ability to classify breast cancer samples into PAM50 intrinsic molecular subtype using the classification scheme outlined by Parker et al. [31]. Our PAM50 subtyping calls were robust across normalization methods with 91% agreement and a Kappa of 0.87 [95% CI (0.85, 0.90)] (see Supplemental Table S3 available online at https://academic.oup.com/bib). Among discordant calls, approximately half had low confidence values from the subtyping algorithm, and half had differences in correlations to centroids less than 0.1 between the discordant calls (data not shown). Most of these discordant calls were among HER2-enriched, luminal B and luminal A subtypes, which are molecularly similar [40].
We observed noticeable differences between the RUVSeq- and nSolver-normalized gene expression when visualized after hierarchical clustering via heatmaps, similar to the principal component analysis. Using this method, we identified 14 additional samples with strong technical errors in the nSolver-normalized data not previously marked by QC flags (see Supplementary Figure S9 available online at https://academic.oup.com/bib), emphasizing the need for postnormalization data visualization. In early breast cancer clustering papers, the first major division was by ER status separating basal-like and HER2-enriched molecular subtypes (predominantly ER-negative) from luminal A and B molecular subtypes (predominantly ER-positive) [31]. This pattern was observed in RUVSeq-data but only partially preserved with nSolver normalization (see Supplementary Figure S9 available online at https://academic.oup.com/bib). Rather, nSolver data clustering was driven by a combination of ER status and study phase. Study phase dominated two of the groups and were formed by Phase 1 and Phase 3 samples, respectively—samples with a 10+ year difference in age.
Lastly, we compared normalization choices for NanoString data to RNA-seq data performed on the same samples. CBCS collected RNA-seq measurements for 70 samples that have data on a different nCounter codeset (162 genes instead of 417) and RNA-seq normalized using standard procedures. A permutation-based test of independence using the distance correlation [33, 41] revealed that the distance correlation between the RNA-seq and nSolver data was small and near 0 (distance correlation = 0.051, 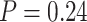 ) while the distance correlation between the RNA-seq and RUVSeq- data was larger (distance correlation = 0.36,
) while the distance correlation between the RNA-seq and RUVSeq- data was larger (distance correlation = 0.36, 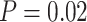 ). The permutation-based test rejected the null hypothesis of independence (distance correlation of zero for unrelated datasets) between RUVSeq-normalized nCounter data and RNA-seq data but fails to reject the null hypothesis for nSolver-normalization nCounter and RNA-seq data. We conclude that RUVSeq produced normalized data with closer relation to the RNA-seq, in terms of distance correlation and test of independence, compared to nSolver.
). The permutation-based test rejected the null hypothesis of independence (distance correlation of zero for unrelated datasets) between RUVSeq-normalized nCounter data and RNA-seq data but fails to reject the null hypothesis for nSolver-normalization nCounter and RNA-seq data. We conclude that RUVSeq produced normalized data with closer relation to the RNA-seq, in terms of distance correlation and test of independence, compared to nSolver.
Case study: differential expression analysis in natural killer cells
We looked at the impact of the two normalization methods in a small cohort (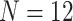 ) on DE analysis across natural killer (NK) cells primed for tumor-specific cells and cytokines from Sabry et al. [20]. RLE plots before and after normalization showed minor differences between the two normalization methods (see Supplementary Figure S10 available online at https://academic.oup.com/bib).
) on DE analysis across natural killer (NK) cells primed for tumor-specific cells and cytokines from Sabry et al. [20]. RLE plots before and after normalization showed minor differences between the two normalization methods (see Supplementary Figure S10 available online at https://academic.oup.com/bib).
Using DESeq2 [25], we identified genes differentially expressed in NK cells primed by CTV-1 or IL-2 cytokines compared to unprimed NK cells at FDR-adjusted 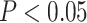 . The two normalization methods led to a different number of differentially expressed genes with a limited overlap of significant genes by both methods (Figure 4A). The raw
. The two normalization methods led to a different number of differentially expressed genes with a limited overlap of significant genes by both methods (Figure 4A). The raw 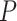 -value histograms from differential expression analysis using nSolver-normalized expression exhibited a slope toward 0 for
-value histograms from differential expression analysis using nSolver-normalized expression exhibited a slope toward 0 for 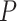 -values under 0.3, which can indicate issues with unaccounted-for correlations among samples [42], such as residual technical variation. The distributions of
-values under 0.3, which can indicate issues with unaccounted-for correlations among samples [42], such as residual technical variation. The distributions of 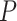 -values using the RUVSeq-normalized data were closer to uniform throughout the range [0,1] for most genes (Figure 4B). While the log2-fold changes were correlated between the two normalization procedures, the genes found to be differentially expressed only with nSolver-normalized data tended to have large standard errors with RUVSeq-normalized data and therefore not statistically significant using RUVSeq (Figure 4C). These differences in DE results emphasize the importance of properly validating normalization prior to downstream genomic analyses.
-values using the RUVSeq-normalized data were closer to uniform throughout the range [0,1] for most genes (Figure 4B). While the log2-fold changes were correlated between the two normalization procedures, the genes found to be differentially expressed only with nSolver-normalized data tended to have large standard errors with RUVSeq-normalized data and therefore not statistically significant using RUVSeq (Figure 4C). These differences in DE results emphasize the importance of properly validating normalization prior to downstream genomic analyses.
Figure 4.
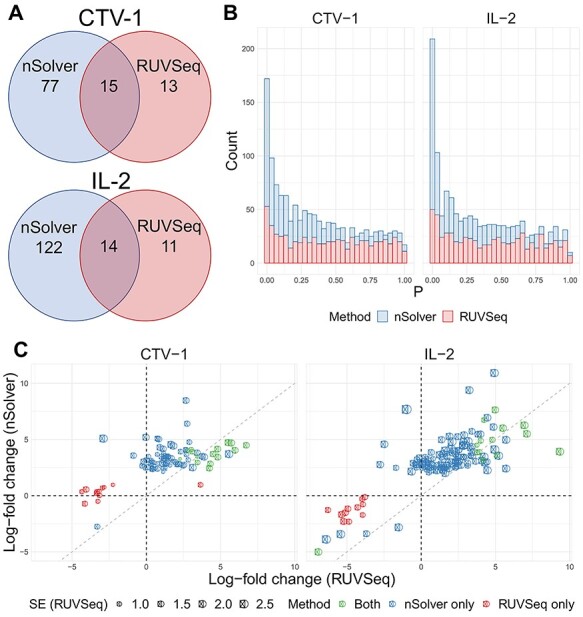
Differential expression analysis from Sabry et al. [20]. (A) Venn diagram of the number of differentially expressed genes using nSolver-normalized (blue) and RUVSeq-normalized data (red) across comparisons for IL-2-primed (top) and CTV-1-primed NK cells (bottom). (B) Raw 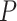 -value histograms for differential expression analysis using nSolver-normalized (blue) and RUVSeq-normalized (red) data across the two comparisons. (C) Scatterplots of log2-fold changes from differential expression analysis using RUVSeq-normalized data (
-value histograms for differential expression analysis using nSolver-normalized (blue) and RUVSeq-normalized (red) data across the two comparisons. (C) Scatterplots of log2-fold changes from differential expression analysis using RUVSeq-normalized data (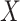 -axis) and nSolver-normalized data (
-axis) and nSolver-normalized data (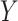 -axis) for any gene identified as differentially expressed in either one of the two datasets. Points are colored by the datasets in which that given gene was classified as differentially expressed. The size of point reflects the standard error of the effect size as estimated in the RUVSeq-normalized data.
-axis) for any gene identified as differentially expressed in either one of the two datasets. Points are colored by the datasets in which that given gene was classified as differentially expressed. The size of point reflects the standard error of the effect size as estimated in the RUVSeq-normalized data.  and the 45° lines are provided for reference.
and the 45° lines are provided for reference.
Case study: bladder cancer gene expression
RUVSeq reduced technical variation (study site) while maintaining the biological variation (tumor grade). RUVSeq data showed the most homogeneity in per-sample median deviation of log-expressions compared to raw and nSolver data (Figure 5A). The first principal component of nSolver data had significant differences by study sites, which was not present in RUVSeq data (Figure 5B). In addition, there was a stronger biological association with tumor grade in the first principal component of expression using RUVSeq data (Figure 5C).
Figure 5.
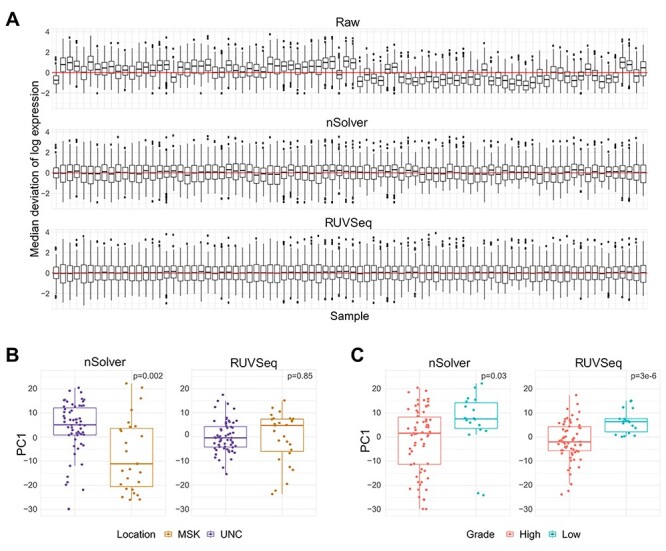
Normalization differences in bladder cancer dataset. (A) RLE plot from bladder cancer dataset, ordered temporally from oldest to newest sample. (B) Boxplot of first principal component of expression by tumor collection site (location) across nSolver- (left) and RUVSeq-normalized (right) data. (C) Boxplot of first principal component of expression by tumor grade across nSolver- (left) and RUVSeq-normalized (right) data.
Case study: kidney cancer gene expression
We only found subtle differences in the deviations from the median expression between the normalization procedures for the kidney cancer dataset (Figure 6A). This cohort did not have the same known technical variables observed in the other cohorts such as study site or sample age, and the RNA came from fresh-frozen material (see Supplementary Table S1 available online at https://academic.oup.com/bib). We evaluated normalization methods on a source of technical variation, DV300, the proportion of RNA fragments detected at greater than 300 base pairs as a source of technical variation and tumor stage as a biological variable of interest. The first two principal components colored by level of DV300 (Figure 6B) and tumor stage (Figure 6C) showed little difference across the two normalization methods. When there were limited sources of technical variation and a robust, high quality dataset, we found both normalization methods performed equally well.
Figure 6.
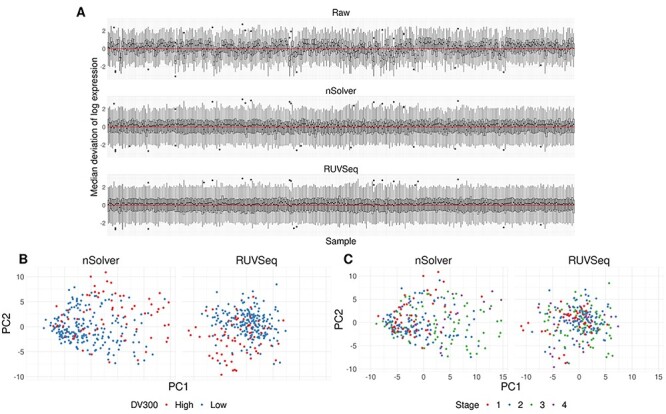
Equal performance of normalization procedures in kidney cancer dataset. (A) RLE plot of per-sample deviations from the median for raw, nSolver- and RUVSeq-normalized data. (B) Scatter plot of the first and second principal component of nSolver- (left) and RUVSeq-normalized (right) expression, colored by high and low DV300. (C) Scatter plot of the first and second principal component of nSolver- (left) and RUVSeq-normalized (right) expression, colored by tumor stage.
Discussion
Proper normalization is imperative in performing correct statistical inference from complex gene expression data. Here, we outline a sequential framework for NanoString nCounter RNA expression data, which provides both QC checks, considerations for choosing housekeeping genes and iterative normalization with biological validation using both NanoString’s nSolver software [9, 12] and RUVSeq [6, 8]. We show that RUVSeq provided a superior normalization to nSolver on three out of four datasets by more efficiently removing sources of technical variation, while retaining robust biological associations. We also benchmark RUVSeq-normalization with two other normalization methods implemented in R and show that RUVSeq outperformed all methods in reducing technical variation.
We observed that normalization methods were sensitive to the quality and the set of housekeeping genes. Several genes thought to behave exclusively in a ‘housekeeping’ fashion in fact associate with biological variables under certain conditions [43] or across different tissue types [44]. A careful validation of housekeeping gene stability on a case-by-case basis and separately for new studies, considering both technical and biological sources of variation in each dataset, is therefore imperative for an optimized normalization procedure.
We developed a quality metric to assess sample quality: samples with high proportions of genes detected below the LOD in both endogenous genes and housekeepers were indicative of either low-quality samples or reduced assay efficiency. Sample age was correlated with higher proportions of genes below the LOD in both endogenous and housekeeping genes, which was likely due to RNA degradation over time. We stress that missing counts in endogenous genes alone does not suggest poor sample quality in the absence of additional QC flags but could represent genes not expressed and therefore not detected under certain biological conditions or cell types. An example includes using an immuno-oncology gene panel in a tumor sample with little to no immune cell infiltration. Conversely, many samples with counts below the LOD in both endogenous genes and housekeepers had additional QC flags including those derived from nSolver’s assessment of data quality. We excluded these samples for analysis in both the nSolver- and RUVSeq-based procedures.
nSolver-normalized data was prone to residual unwanted technical variation when there were known technical biases, such as in CBCS and the bladder example. We checked for known biological associations that are intrinsic to the sample, as in eQTL analysis, to judge the performance of the normalization process [45, 46]. A full cis-trans eQTL analysis using nSolver- and RUVSeq-normalized data showed a strong cis-eQTL signal in data from both normalization methods. We found significantly more trans-eQTLs with the nSolver-normalized data (Figure 3). However, many of the trans-eSNPs for the loci found with nSolver-normalized data tended to have moderate MAF differences across phase, leading us to suspect they were spurious associations driven by residual technical variation in gene expression (see Supplementary Figure S8 available online at https://academic.oup.com/bib). Such spurious associations from population stratification have been described in many previous studies of eQTL analysis [47–50].
The choice of normalization procedure is less of a concern in cohorts with minimal sources of technical variation or in nCounter targeted gene panels that have been optimized for robust measurement across preservation methods. In the CBCS breast cancer cohort, we identified significant differences in gene expression between normalization methods across the entire gene set (417 total genes). However, PAM50 subtyping was robust across the two normalization procedures. The genes in the PAM50 classifier were selected due to their consistent measurement in both FFPE and fresh frozen breast tissues [31], suggesting that robustly measured genes may be less affected by different normalization procedures. Furthermore, we see minimal differences in residual technical variation in the kidney cancer dataset and the Sabry et al. dataset, both of which were measured on either robustly validated genes or nCounter panels. The kidney cancer example had newer, fresh-frozen specimens that were profiled using a small and well-validated set of genes important in that cancer type. This dataset gives an opportunity to stress the importance of the general principles of normalization: as Gagnon-Bartsch et al. and Molania et al. recommend [6, 7], normalization should be a part of scientific process and should be approached iteratively with visual inspection and biological validation to tune the process. One normalization procedure is not necessarily applicable to all datasets and must be re-evaluated on each dataset.
In conclusion, we outline a systematic and iterative framework for the normalization of NanoString nCounter expression data. Even without background correction, a technique which has been shown to impair normalization of microarray expression data [51, 52], we believe that relying solely on positive control and housekeeping gene-based normalization may result in residual technical variation after normalization. Here, we show the merits of a comprehensive procedure that includes sample QC checks including the addition of new checks, assessments of housekeeping genes, normalization with RUVSeq [6, 8] and data analysis with popular count-based R/Bioconductor packages, as well as iterative data visualization and biological validation to assess normalization. Researchers must pay close attention to the normalization process and systematically assess pipelines that best suit each dataset.
Availability
Relevant R code for these analyses is freely bundled into an R package on Github: https://github.com/bhattacharya-a-bt/NanoNormIter. R code to recreate the Sabry et al. analysis and a tutorial for the iterative framework is also provided: https://github.com/bhattacharya-a-bt/CBCS_normalization/ [53]. Summary statistics for eQTL analysis are available at https://github.com/bhattacharya-a-bt/CBCS_TWAS_Paper [54], as a part of Bhattacharya et al. [29].
CBCS genotype datasets analyzed in this study are not publicly available as many CBCS patients are still being followed and accordingly CBCS data are considered sensitive; the data are available from M.A.T upon reasonable request. Raw and normalized expression data from CBCS Raw and normalized expression data from CBCS is available from the NCBI Gene Expression Omnibus with accession number GSE148426. Data from the bladder and kidney cancer datasets may be provided by the authors upon reasonable request.
Accession Numbers
Raw RCC files for nCounter expression from Sabry et al. [20] are available on NCBI GEO with the accession numbers GSE130286. Raw and normalized expression data from CBCS will be available on GEO upon publication. For replication prior to publication, these data can be requested from the authors.
Key Points
The NanoString nCounter RNA counting assay, an attractive option in archived samples, has suboptimal QC and normalization pipelines.
We provide an iterative framework for nCounter data with steps for QC, normalization and visualization/validation using RUVSeq.
Using four real datasets, we show that our framework eliminates technical variation more reliably than other methods, including NanoString’s provided software nSolver, without diminishing biological variation.
We stress that QC and normalization must be emphasized in study design and evaluated using proper visualization and other checks, or else results in downstream analyses may be biased.
Supplementary Material
Acknowledgement
We thank the CBCS participants and volunteers. We thank Halei Benefield, Xiaohua Gao, Erin Kirk, Linnea Olsson and Jessica Tse for their invaluable support during the research process.
Arjun Bhattacharya is a doctoral candidate in Biostatistics at the University of North Carolina at Chapel Hill.
Alina M. Hamilton is a doctoral candidate in Pathology and Laboratory Medicine at the University of North Carolina at Chapel Hill.
Helena Furberg is an associate attending epidemiologist at Memorial Sloan Kettering Cancer Center.
Eugene Pietzak is a urological surgeon at Memorial Sloan Kettering Cancer Center.
Mark P. Purdue is a senior investigator in the Division of Cancer Epidemiology and Genetics, National Cancer Institute.
Melissa A. Troester is a professor of Epidemiology and Pathology and Laboratory Medicine at the University of North Carolina at Chapel Hill.
Katherine A. Hoadley is an assistant professor of Genetics at the University of North Carolina at Chapel Hill.
Michael I. Love is an assistant professor of Biostatistics and Genetics at the University of North Carolina at Chapel Hill.
Funding
Susan G. Komen® provided financial support for CBCS study infrastructure. Funding was provided by the National Institutes of Health, National Cancer Institute (P01-CA151135, P50-CA05822, U01-CA179715 to M.A.T.). M.I.L. is supported by P01-CA142538 and P30-ES010126. K.A.H. is supported by a Komen Career Catalyst (CCR16376756). A.M.H. is supported by 1T32GM12274 (National Institute of General Medical Sciences). The TGL is supported in part by grants from the National Cancer Institute (3P30CA016086) and the University of North Carolina at Chapel Hill University Cancer Research Fund. The kidney cancer study and gene expression analysis were supported by the Intramural Research Program of the National Institutes of Health and the National Cancer Institute.
The funders had no role in the design of the study, collection, analysis or interpretation of the data, the writing of the manuscript or the decision to submit the manuscript for publication.
References
- 1. Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008;26:317–25. [DOI] [PubMed] [Google Scholar]
- 2. Veldman-Jones MH, Brant R, Rooney C, et al. Evaluating robustness and sensitivity of the NanoString technologies nCounter platform to enable multiplexed gene expression analysis of clinical samples. Cancer Res 2015;75:2587–93. [DOI] [PubMed] [Google Scholar]
- 3. Troester MA, Sun X, Allott EH, et al. Racial differences in PAM50 subtypes in the Carolina Breast Cancer Study. J Natl Cancer Inst 2018;110:176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wallden B, Storhoff J, Nielsen T, et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics 2015;8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vieira AF, Schmitt F. An update on breast cancer multigene prognostic tests-emergent clinical biomarkers. Front Med 2018;5:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gagnon-Bartsch JA, Speed TP. Using control genes to correct for unwanted variation in microarray data. Biostatistics 2012;13:539–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Molania R, Gagnon-Bartsch JA, Dobrovic A, et al. A new normalization for Nanostring nCounter gene expression data. Nucleic Acids Res 2019;47:6073–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Risso D, Ngai J, Speed TP, et al. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 2014;32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. NanoString Technologies. nSolverTM 4.0 Analysis Software. 2018;5–98.
- 10. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:1–12; research0034.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perkins JR, Dawes JM, McMahon SB, et al. ReadqPCR and NormqPCR: R packages for the reading, quality checking and normalisation of RT-qPCR quantification cycle (Cq) data. BMC Genomics 2012;13:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Waggott D, Chu K, Yin S, et al. Gene expression NanoStringNorm: an extensible R package for the pre-processing of NanoString mRNA and miRNA data. Bioinforma Appl Note 2012;28:1546–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang H, Horbinski C, Wu H, et al. NanoStringDiff: a novel statistical method for differential expression analysis based on NanoString nCounter data. Nucleic Acids Res 2016;44:gkw677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jia G, Wang X, Li Q, et al. Rcrnorm: an integrated system of random-coefficient hierarchical regression models for normalizing nanostring ncounter data. Ann Appl Stat 2019;13:1617–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Canouil ML, Bouland GA, Lie Bonnefond A, et al. NACHO: an R package for quality control of NanoString nCounter data. Bioinformatics 2020;36:970–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. D’Arcy M, Fleming J, Robinson WR, et al. Race-associated biological differences among luminal A breast tumors. Breast Cancer Res Treat 2015;152:437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hall IJ, Moorman PG, Millikan RC, et al. Comparative analysis of breast cancer risk factors among African-American women and white women. Am J Epidemiol 2005;161:40–51. [DOI] [PubMed] [Google Scholar]
- 18. Brennan P, Van Der Hel O, Moore LE, et al. Tobacco smoking, body mass index, hypertension, and kidney cancer risk in central and eastern Europe. Br J Cancer 2008;99:1912–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moore LE, Nickerson ML, Brennan P, et al. Von Hippel-Lindau (VHL) inactivation in sporadic clear cell renal cancer: associations with germline VHL polymorphisms and etiologic risk factors. PLoS Genet 2011;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sabry M, Zubiak A, Hood SP, et al. Tumor- and cytokine-primed human natural killer cells exhibit distinct phenotypic and transcriptional signatures. PLoS One 2019;14:e0218674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nickles D, Sandmann T, Ziman R, et al. (2015) NanoStringQCPro: Quality metrics and data processing methods for NanoString mRNA gene expression data. R package version 1.20.0. [Google Scholar]
- 22. Venables WN, Ripley BD. Modern Applied Statistics with S Springer, 2002. [Google Scholar]
- 23. Bullard JH, Purdom E, Hansen KD, et al. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 2010;11:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rousseeuw PJ. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math 1987;20:53–65. [Google Scholar]
- 28. Shabalin AA. Gene expression matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012;28:1353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhattacharya A, García-Closas M, Olshan AF, et al. A framework for transcriptome-wide association studies in breast cancer in diverse study populations. Genome Biol 2020;57:21(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple. Source J R Stat Soc Ser B 1995;57:289–300. [Google Scholar]
- 31. Parker JS, Mullins M, Cheang MCU, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gendoo DM, Ratanasirigulchai N, Schröder MS, et al. (2020) genefu: Computation of Gene Expression-Based Signatures in Breast Cancer. R package version 2.20.0, http://www.pmgenomics.ca/bhklab/software/genefu. [DOI] [PMC free article] [PubMed]
- 33. Székely GJ, Rizzo ML. The energy of data. Annu Rev Stat Its Appl 2017;4:447–79. [Google Scholar]
- 34. Dai X, Xiang L, Li T, et al. Cancer hallmarks, biomarkers and breast cancer molecular subtypes. J Cancer 2016;7:1281–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elizabeth M, Hammond H, Hayes DF, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol 2010;28:2784–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012;486:346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perou CM, Sørile T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52. [DOI] [PubMed] [Google Scholar]
- 38. Sørlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 2003;100:8418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018;173:291–304e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Picornell AC, Echavarria I, Alvarez E, et al. Breast cancer PAM50 signature: correlation and concordance between RNA-Seq and digital multiplexed gene expression technologies in a triple negative breast cancer series. BMC Genomics 2019;20:452:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res 1967;27:209–20. [PubMed] [Google Scholar]
- 42. Breheny P, Stromberg A, Lambert J. P-value histograms: inference and diagnostics. High-Throughput 2018;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sikand K, Singh J, Ebron JS, et al. Housekeeping gene selection advisory: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin are targets of miR-644a. PLoS One 2012;7:e47510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barber RD, Harmer DW, Coleman RA, et al. GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol Genomics 2005;21:389–95. [DOI] [PubMed] [Google Scholar]
- 45. Raulerson CK, Ko A, Kidd JC, et al. Adipose Tissue Gene Expression Associations Reveal Hundreds of Candidate Genes for Cardiometabolic Traits Am J Hum Genet 2019;7(4):773–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aguet F, Brown AA, Castel SE, et al. Genetic effects on gene expression across human tissues. Nature 2017;550:204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee C. Genome-wide expression quantitative trait loci analysis using mixed models. Front Genet 2018;9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang N, Wang M, Jia T, et al. A robust statistical method for association-based eQTL analysis. PLoS One 2011;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hyun MK, Ye C, Eskin E. Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics 2008;180:1909–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mao W, Hausler R, Chikina M.. DataRemix: A Universal Data Transformation for Optimal Inference from Gene Expression Datasets. bioRxiv 2019;1–8. [DOI] [PMC free article] [PubMed]
- 51. Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003;4:249–64. [DOI] [PubMed] [Google Scholar]
- 52. Freytag S, Gagnon-Bartsch J, Speed TP, et al. Systematic noise degrades gene co-expression signals but can be corrected. BMC Bioinformatics 2015;16:309: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bhattacharya A (2020, April 9). bhattacharya-a-bt/CBCS_normalization: Code and summary results for “An approach for normalization and quality control for NanoString RNA expression data” (Version v1.0). Zenodo. 10.5281/zenodo.3746885 [DOI] [PMC free article] [PubMed]
- 54. Bhattacharya A (2019) bhattacharya-a-bt/CBCS_TWAS_Paper: Code, models, and results for CBCS TWAS Paper (Version v1.0). Zenodo. 10.5281/zenodo.3407384. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.