

Abstract
Gene regulatory network is a complicated set of interactions between genetic materials, which dictates how cells develop in living organisms and react to their surrounding environment. Robust comprehension of these interactions would help explain how cells function as well as predict their reactions to external factors. This knowledge can benefit both developmental biology and clinical research such as drug development or epidemiology research. Recently, the rapid advance of single-cell sequencing technologies, which pushed the limit of transcriptomic profiling to the individual cell level, opens up an entirely new area for regulatory network research. To exploit this new abundant source of data and take advantage of data in single-cell resolution, a number of computational methods have been proposed to uncover the interactions hidden by the averaging process in standard bulk sequencing. In this article, we review 15 such network inference methods developed for single-cell data. We discuss their underlying assumptions, inference techniques, usability, and pros and cons. In an extensive analysis using simulation, we also assess the methods’ performance, sensitivity to dropout and time complexity. The main objective of this survey is to assist not only life scientists in selecting suitable methods for their data and analysis purposes but also computational scientists in developing new methods by highlighting outstanding challenges in the field that remain to be addressed in the future development.
Keywords: gene regulatory network, single-cell data, RNA sequencing, scRNA-seq, simulation studies
1 Introduction
Gene regulatory networks (GRNs) consist of interactions between regulators that influence the biological processes of living organisms. These networks with dynamic interactions regulate gene expression of different cell types in different developmental stages in a spatio-temporal manner. Understanding the role of each gene and their relationships with others in those GRNs is the key to interpret biological processes at molecular levels. GRNs are expected to benefit many fields from basic biological research to practical applications, such as medicine and drug design [1]. Therefore, reconstruction and analysis of GRNs have become effective methods for studying underlying mechanisms that drive genotypes to phenotypes. A large number of computational tools [2–13] have been developed to predict such GRNs using gene expression data from bulk population sequencing technologies, which accumulate expression profile from all cells in a tissue. These methods have proved to be useful tools for studying transcriptional interactions [14–19]. However, as bulk sequencing technologies mainly measure the averaged gene expression across cell populations, they are insufficient for studying heterogeneous systems such as early developmental stages and complex tissues. Networks constructed from bulk data are unable to describe the gene relationships in specific cell types in different developmental stages. The critical question now is whether we can discover GRNs that determine cell fate, control cell differentiation and drive transitions of cell types.
Recent advances in biochemistry and sequencing technologies have enabled the monitoring of biological systems and complex tissues at the resolution of individual cells [20–22]. This allows us to deeper analyze the GRN that controls cell differentiation and specification by dissecting complex tissues into distinct components (cell types) and inspecting their biological and molecular characteristic using their spatial and temporal position in the cell population. However, one cannot simply apply GRN methods developed for bulk sequencing to the analysis of single-cell data. This is due to several reasons. First, bulk analysis methods are typically designed to assess changes in bulk samples across conditions and thus cannot comprehensively assess differences between cell types in a spatial and temporal manner. Second, these methods are not efficient in coping with high levels of sparsity (dropouts) and the large number of cells in single-cell data. Therefore, a fast-growing number of GRN inference methods have been developed specifically for the analysis of scRNA-seq data.
Thus far, there have been numerous studies that utilize GRNs constructed from scRNA-seq data to determine the role of transcriptional regulators in cell fate decisions [23–30] that are important in understanding and explaining the cellular heterogeneity in both normal and dysfunctional tissues [31]. This comprehensive decomposition and monitoring of complex tissues hold enormous potential in both developmental biology and clinical research. However, at the current stage, the construction of GRNs mostly contributes to identifying interactions that happen in specific cell types. The applications of these methods have not yet presented breakthroughs in real-world setting studies. This comes from the difficulty in dealing with technical limitations in the scRNA-seq platforms and the heterogeneity of single-cell data. The validation of the output networks and computational challenges also raises questions about their reliability. Nevertheless, it is expected that GRN inference methods can be applied to a wide range of real-world applications from identifying disease biomarkers and pathways to network medicine and drug design [1]. Despite the importance of GNR inference techniques in single-cell data analysis, there is no survey that comprehensively reviews existing methods and discusses their applicability, current challenges and opportunities and future perspectives.
Here, we review 15 computational methods that were specifically developed for GRN inference using single-cell data. We recapitulate and discuss each method from the following perspectives: availability, user-friendliness, underlying assumption and network inference techniques. We classify these methods into four categories according to the way the network is constructed: (i) boolean model, (ii) differential equation, (iii) gene correlation and (iv) correlation ensemble over pseudo-time. In Section 2, we describe in detail the availability, implementation, documentation and user-friendliness of each method. In Section 3, we categorize and discuss the methods according to the underlying assumption, data transformation and inference techniques being used. In Section 4, we systematically compare the methods using simulation. We assess each method based on three essential metrics: (i) accuracy in reconstructing reference networks using scRNA-seq data, (ii) sensitivity to different levels of dropout/sparsity and (iii) time complexity. In Section 5, we discuss the limitations and outstanding challenges that the current GRN inference methods are facing. We also provide detailed descriptions and analysis pipelines of individual methods in Supplementary Materials (Supplementary Note and Tables S1 and S2).
This is the first article that provides such an in-depth review and discussion about GRN inference using scRNA-seq data. Chen et al. [32] provide a simulation study to assess the performance of some GRN methods. However, they include only three methods that are single-cell specific and do not provide an in-depth discussion of the underlying techniques. Fiers et al. [33] recapitulate the GRN inference methods from the application perspectives without going into details of the analysis techniques. In addition, many of the methods mentioned are not available for download. In contrast, here we provided a comprehensive review of the most prominent single-cell specific GRN methods that are available. This survey is expected to benefit life scientists who wish to choose a method that is most suitable for their data. At the same time, this survey also benefits computational scientists who wish to know the limitations of existing methods in order to develop new methods and infrastructure addressing the current shortcomings.
2 Implementation and usability
Table 1 shows the 15 methods that we review in this article. Although more GRN methods for single-cell analysis have been published, we select only those that have available and executable software. Tools that are not accessible by the research communities are excluded from this survey, including ACTION [49], WASABI [50] and some others [51, 52]. In Table 1, we show the tools’ available hyperlink, programming language, software interface (graphic user interface, command line or executable scripts), references to their original articles, number of citations, license and rating for overall usability. The number of citations (Google Scholar) partially reflects how each tool is received or known among the research community. We believe that these meta-data are essential for users to be aware of before investing time to understand the method and to conduct any analysis.
Table 1.
Computational tools for GRN inference using single-cell data. Each method is categorized according to the core model they use to construct the GRN.  Tools with a web-based interface.
Tools with a web-based interface.  Tools provided as scripts that are executable in the command line.
Tools provided as scripts that are executable in the command line.  Tools distributed as stand-alone packages. (*) Inference Snapshot provides a mixture of scripts written in C++ and Matlab. This requires users to switch between the two environments to finish any single analysis. Tools with an undeclared license (None) are free for noncommercial use without redistribution. (**) SCENIC is free for noncommercial use and has its own license agreement. The usability of each software ranges from 1 (
Tools distributed as stand-alone packages. (*) Inference Snapshot provides a mixture of scripts written in C++ and Matlab. This requires users to switch between the two environments to finish any single analysis. Tools with an undeclared license (None) are free for noncommercial use without redistribution. (**) SCENIC is free for noncommercial use and has its own license agreement. The usability of each software ranges from 1 ( ) to 5 (
) to 5 ( ). This score indicates how easy it is for users to analyze an scRNA-seq dataset using a tool. The higher the score (and the darker the color), the easier it is to use. A method with a score of 5 means that it is very easy to start an analysis with the method
). This score indicates how easy it is for users to analyze an scRNA-seq dataset using a tool. The higher the score (and the darker the color), the easier it is to use. A method with a score of 5 means that it is very easy to start an analysis with the method
| Method | Availability | Code | Pkg. | Ref. | Year | Cit. | License | Usability |
|---|---|---|---|---|---|---|---|---|
| Boolean model | ||||||||
| Boolean Pseudotime | https://github.com/fionahamey/Pseudotime-network-inference | Python |

|
[34] | 2017 | 23 | Apache 2.0 |

|
| BTR | https://cran.r-project.org/package=BTR | R |

|
[35] | 2016 | 23 | GPL-3 |

|
| SCNS | https://github.com/swoodhouse/SCNS-GUI | F# |

|
[36] | 2018 | 5 | MIT |

|
| Differential equation | ||||||||
| Inference Snapshot | https://www.helmholtz-muenchen.de/fileadmin/ICB/software /inferenceSnapshot.zip | C++/Matlab | * | [37] | 2015 | 80 | None |

|
| SCODE | https://github.com/hmatsu1226/SCODE | R/Julia/Ruby |

|
[38] | 2017 | 34 | MIT |

|
| SCOUP | https://github.com/hmatsu1226/SCOUP | C++ |

|
[39] | 2016 | 24 | MIT |

|
| Gene correlation | ||||||||
| Empirical Bayes | https://github.com/ananth-pallaseni/EmpiricalBayes.jl | Julia |

|
[40] | 2018 | 2 | MIT |

|
| Information Measures | https://github.com/Tchanders/InformationMeasures.jl | Julia |

|
[41] | 2017 | 61 | MIT |

|
| NLNET | https://cran.r-project.org/package=nlnet | R |

|
[42] | 2016 | 1 | GPL 2 2 |

|
| SINCERA | https://research.cchmc.org/pbge/sincera.html | R |

|
[43] | 2015 | 115 | GPL-3 |

|
| SCENIC | https://github.com/aertslab/SCENIC | R/Python |

|
[44] | 2017 | 149 | Free
|

|
| Correlation ensemble over pseudotime | ||||||||
| LEAP | https://cran.r-project.org/package=LEAP | R |

|
[45] | 2016 | 18 | GPL-2 |

|
| SINCERITIES | http://www.cabsel.ethz.ch/tools/sincerities.html | R/Matlab |

|
[46] | 2017 | 20 | BSD |

|
| SCIMITAR | https://github.com/dimenwarper/scimitar | Python |

|
[47] | 2017 | 8 | None |

|
| SCINGE | https://github.com/gitter-lab/SCINGE | Matlab |

|
[48] | 2019 | 1 | MIT |

|
Most of these methods are free for noncommercial use and can be modified and redistributed. These methods can be included in other analysis pipelines or can be modified to serve the users’ purpose, given that the users follow the terms given in the license. Among the 15 methods, Inference Snapshot and SCIMITAR are two license-free methods that are free for any analysis. However, users who want to redistribute the software or to embed these methods in their workflow need permission from the authors of the tools.
Regarding programming language, R and Python are still the favorite languages used to implement GRN inference methods (11 out of 15). Julia is a relatively new programming language that is promising for scientific and numerical computing with high performance. Tools that are provided as an R package or Julia package can be easily installed through their package manager. Tools that come with C++ components need to be compiled. Among the 15 methods, three are provided as Matlab scripts. Note that while R, Python, Julia and C++ are open-source and can be freely downloaded for most operation systems, Matlab usually requires an expensive license.
Among the 15 methods, only SCNS provides a web-based interface. The interface is simple and only provides handy tools for selecting the input, viewing and visualizing the output. It uses Adobe Flash to visualize the network. Since Adobe Flash is deprecated, the software may not be able to function in the near future. The remaining 14 tools are provided in the form of stand-alone package or executable scripts that can be run on the command line. The three methods, Boolean Pseudotime, SCODE and SCOUP, provide a command-line interface for users to perform analysis. Although it is convenient to perform an analysis using these tools, combining these with other packages might require additional implementations to make it work smoothly between different environments. Inference Snapshot is the only method that performs its analysis using a mixture of scripts using multiple programming languages. In order to complete an analysis, users have to switch from Matlab to C++ to complete their analysis. In addition, this method hardcodes the input and parameters, i.e. users have to modify the code to change the name of the input file or to change any parameters.
We also provide ratings for the usability of the surveyed tools. For each method, we provide a score for each of the following categories: (i) tutorial, (ii) documentation, (iii) code quality, (iv) user-friendliness and (v) completion rate. First, we check if the authors provide a detailed and easy-to-follow tutorial for their tool. Boolean Pseudotime, BTR, SCNS, SCODE, SINCERA, SCENIC and SCIMITAR are the best in providing high-quality tutorials, whereas Inference Snapshot, Empirical Bayes, Information Measures, SINCERITIES and SCINGE provide only short scripts as examples of how to perform the analysis. Second, we check if each of the functions and parameters is correctly documented with details. Methods distributed as R packages (BTR, NLNET, SINCERA, SCENIC and LEAP) provide the most detailed documentation. In contrast, Inference Snapshot and SCIMITAR provide only minimum comments on function parameters. Third, we assess the quality of the code regarding its structure, testability (e.g. unit test), compatibility (with different operation systems, dependencies, compilers) and reliability (how frequent the software crashes). Methods that are bundled as packages can be installed easily (Boolean Pseudotime, Empirical Bayes, Information Measures, NLNET, SINCERA and SCENIC). In contrast, SCNS, SCIMITAR and SCINGE provide executable files and scripts that require users to manually resolve conflicted and missing dependencies. Inference Snapshot even requires users to compile C++ and to switch between command-line and Matlab environment to finish an analysis. This method is not included in our performance assessment because we were not able to execute any analysis.
Fourth, we assess how easy it is for the users to perform an analysis using their own data, e.g. preparing the input expression and creating additional required inputs. For Boolean Pseudotime, BTR, SCODE, Information Measures, NLNET, SINCERA and LEAP, users can easily provide the expression matrix as input. In contrast, SCINGE requires users to prepare data in specific formats while SCOUP requires users to compute the summary statistics of the distribution of each gene (mean and variance). Finally, we assess the methods based on completion rate using 139 datasets from our simulation studies with varying numbers of genes (20 to 3000), samples (200 to 1000) and sparsity levels (30–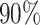 ). SCODE, SCENIC and LEAP are the only methods that have
). SCODE, SCENIC and LEAP are the only methods that have 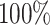 completion rate. We were not able to finish any analysis using Inference Snapshot. Among the 14 methods tested, BTR has the lowest completion rate (
completion rate. We were not able to finish any analysis using Inference Snapshot. Among the 14 methods tested, BTR has the lowest completion rate (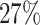 ) since it cannot finish analyses with more than 30 genes. The overall usability score of each method is shown in the last column of Table 1. SCODE, Information Measures, NLNET, SCENIC and LEAP have the highest overall usability rating (5/5), whereas Inference Snapshot (1/5), SCIMITAR (2/5) and SCINGE (2/5) have the lowest rating. Supplementary Table S1 provides more details about the usability of each method while Supplementary Table S2 provides the input and workflow.
) since it cannot finish analyses with more than 30 genes. The overall usability score of each method is shown in the last column of Table 1. SCODE, Information Measures, NLNET, SCENIC and LEAP have the highest overall usability rating (5/5), whereas Inference Snapshot (1/5), SCIMITAR (2/5) and SCINGE (2/5) have the lowest rating. Supplementary Table S1 provides more details about the usability of each method while Supplementary Table S2 provides the input and workflow.
3 Methods
In general, GRN inference methods aim at capturing the network dynamics that explain the underlying regulatory states in different cellular compartments and conditions. Each GRN inference model follows an explicit assumption about the regulatory dynamics that can be observed through the changes in expression data. The overall workflow of GRN inference methods is presented in Figure 1. The input includes a scRNA-seq expression matrix in which rows represent genes and columns represent cells (or vice versa). Due to computational limitation, all GRN methods start with a gene filtering step, which narrows the analysis to genes with high variability or genes that are of users’ interest (pre-defined genes). Depending on the assumption of the regulatory dynamics and the inference technique used, the filtered data are then transformed into necessary structure/format, such as binary values (boolean model), pseudo-time data or gene correlation. These methods then utilize different techniques to infer the ultimate network. The output graph of these methods can be directed or undirected and be represented in different forms such as continuous adjacency matrix, binary adjacency matrix, list of ranked edges or boolean functions.
Figure 1 .
The overall workflow of GRN inference methods. The methods start with filtering genes based on their variability or a priori knowledge. They next construct intermediate data depending on the modeling and data assumption and then infer the network. The output of these methods can be either co-expression networks which are undirected from top selected connections or directed networks with regulatory relationships between genes. To evaluate the constructed networks, each method adopts different validation techniques, including using simulation, enrichment analysis, literature support, and expert interpretation and conducting additional laboratory experiments.
Unlike bulk sequencing, single-cell data provides the expression of individual cells, many of which are from different developmental stages. This makes it possible to arrange cells in a temporal manner so that one can model the expression changes of individual genes over time [53]. Among the 15 methods surveyed, 8 methods use time-ordering information. These include all methods in the differential equation and correlation ensemble over pseudo-time categories, as well as the Boolean Pseudotime method. In order to infer the GRN, these methods require the pseudo-time data in addition to the gene expression.
It is worth noting that the data processing and gene filtering steps are not included in most of the provided source code/software. All tools except SCENIC and SINCERA do not provide code or instructions for the gene filtering step while the analysis results reported in the reference articles involve this step. Regarding time-trajectory, only Inference Snapshot and SCIMITAR provide the code for producing the time-ordering data from the gene expression. The other six methods require users to provide the time-series data as part of the input without any detailed instruction. In order to perform analysis using these six methods (Boolean Pseudotime, SCODE, SCOUP, LEAP, SINCERITIES, SCINGE), users have to use third-party software to infer the time trajectory.
Regarding validation, there are numerous techniques that have been utilized to assess the performance of network inference methods: simulation, enrichment analysis, literature support, expert interpretation and laboratory validation. In simulated studies, the ground truth (reference networks) is known and is used to assess the quality of the constructed networks. In enrichment analysis, the constructed networks are enriched against existing knowledge bases (KEGG, STRING, GO terms, etc.) in order to find biological processes and pathways that are relevant to the underlying condition. In literature support, authors often search for evidence in the literature that supports their findings. In expert interpretation, experts in the field (which are often co-authors) interpret the results and provide insights based on their knowledge and expertise. In laboratory experiments, authors usually perform Chip-seq sequencing to detect the binding sites and confirm the interactions between genes.
In the next parts of this section, we describe the methods in each category, together with their strengths and weaknesses. We also provide a detailed description and analysis procedure of each method in Supplemental Table S2 and Supplementary Note.
3.1 Boolean model
Methods in this category utilize boolean network and functions to model the gene regulations. In the boolean network, the relations between genes are expressed as logical operations. Nodes and edges represent the genes and their topology while the boolean functions represent gene expression depending on the expression of other genes. In the boolean model, the expression of a gene is either 0 (unexpressed) or 1 (expressed).
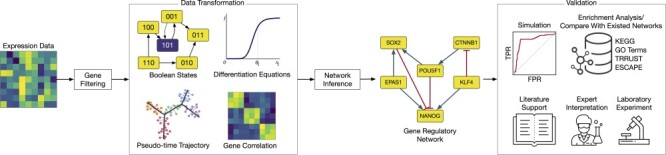
Here, we review three methods—BTR, SCNS and Boolean Pseudotime Inference—that utilize the boolean networks and boolean update functions to construct the GRN. The general workflow of these methods is shown in Figure 2. The methods start by binarizing the expression data to construct the state space for the model, in which each cell represents a state (a boolean vector of gene expression). The initial state of the model can be either randomly generated (BTR), provided by users (SCNS) or from the temporal ordering of the data (Boolean Pseudotime). Each method applies a different approach to iteratively optimize the boolean states from the initial states. BTR implements a state-space scoring function to calculate the distance between the state space and its expression value. A hill-climbing strategy is used to minimize this distance. BTR outputs a network with activation and repression edges.
Figure 2 .
The overall workflow of methods using the boolean model. (i) These methods first binarize the gene expression data and then generate the initial boolean states. (ii) The methods optimize the states of the model with respect to the binary values. (iii) The methods output the GRN with activation and repression edges or a set of boolean functions.
Boolean Pseudotime and SCNS find the shortest path from the initial cell to the end cell and then use this path to restrict the search of the boolean optimizer model in order to reduce the computational complexity. Both Boolean Pseudotime and SCNS use diffusion maps to represent the expression data in a lower dimension and then use the breath-first search (SCNS) or Dijkstra’s algorithm (Boolean Pseudotime) to find the shortest path from the initial cell to the target cell. While SCNS limits the update function to one activator and one inhibitor, Boolean Pseudotime allows users to customize these parameters. An increase in the number of activators and inhibitors may result in more complex boolean rules and computational complexity. Both methods use Z3 solver [54] to check the satisfiability of the provided logical formulas and to obtain a list of optimal logical functions (output).
While BTR outputs the network as a graph object, Boolean Pseudotime and SCNS only output the optimized boolean rules. BTR can also be used to optimize an existing putative regulatory network. Although Boolean Pseudotime uses diffusion maps in their analysis to infer pseudotime ordering, the software package requires users to input external time ordering data which can be inferred from any method of their preference. It also performs correlation analysis (described in their manuscript but not implemented in the package) to filter potential gene relationships before feeding to the network inference model.
By binarizing the gene expression values to show relations between genes, the model requires fewer parameters and thus avoid overfitting. As boolean models usually define a threshold to binarize the gene expression and dropouts are likely to happen to low or moderate expression values [55], binarization makes boolean models less sensitive to dropouts. It also gives the advantage that closely related cell states can be detected easily by finding small bit changes in the boolean representation of gene expression. Boolean models can also easily represent self-activation/inhibition in a GRN which might be a problem for some models such as correlation-based models. However, the binarization may lead to loss of information which might be useful during method execution. In addition, since the boolean methods need to optimize the boolean rules through the state transition, the more genes are included in the analysis, the more cells are required to build a connected state transition graph.
3.2 Differential equation
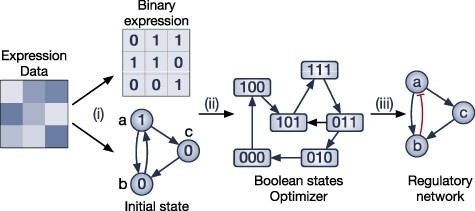
Methods using differential equations describe gene expression dynamics as a function of the expression of other genes and environmental factors. Differential equations are used to model the changing rate of expression data through time given a set of parameters. In other words, the time ordering of cells is required to complete the analysis using differential equations. Here, we discuss three methods that applied either ordinary differential equations (ODEs) or stochastic differential equations (SDEs) to infer the connections between genes. These methods are Inference Snapshot, SCODE and SCOUP. The overall workflow of the methods in this category is described in Figure 3. The pseudo-time trajectory data can be generated using embedded functions in each method (Inference Snapshot and SCOUP) or by using an external package (SCODE uses monocole). SCODE and SCOUP are the two methods that construct the GRN directly from gene expression data with timestamps. Inference Snapshot, on the other hand, uses an external package, GENIE3 [4], to infer the initial network and then uses the inferred timestamp data to further refine the relationships from the GENIE3 network (see Section 3.3 for the description of GENIE3).
Figure 3 .
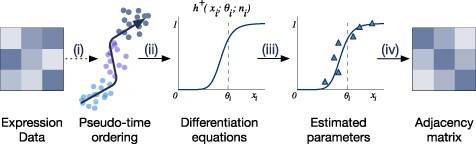
The overall workflow of methods using differential equations. (i) Pseudo-temporal ordering of cells is inferred using external software or embedded functions. (ii) The methods use differential equations to describe the relationship between genes with respect to the inferred time. (iii) Parameters used in the model are estimated using different optimization techniques. (iv) Using the optimized parameters, the relationship between genes are inferred from the declared differential equations to output an affinity matrix of the GRN.
To construct time-ordering data, Inference Snapshot first uses a nonlinear dimension reduction approach, diffusion maps, to reduce the dimension of the input data. It then clusters the obtained data by manually selecting a starting cell and a final cell and then uses the Dijkstra algorithm to find the shortest path between them. For each branch constructed from the previous ad hoc clustering, the method uses Wanderlust algorithm [56] to construct time-series data using the original high-dimension space of cells belonging to this branch. On the other hand, SCODE and SCOUP find the minimal spanning tree to assign a pseudo-time to each cell. While SCODE uses Monocle which uses independent components analysis to reduce data dimension and finds the minimal spanning tree on it, SCOUP uses principal components analysis to reduce the dimension of the data. SCOUP constructs minimum spanning trees from an initial cell to all other cells to build the initial ordering of the cells. The initial cell is a dummy point with the mean of the normal distributions for each gene. Users are required to input an estimation of these distributions.
To construct the network from time-ordering data, SCODE and SCOUP respectively introduce ODEs and SDEs to calculate the correlation between genes. SCODE represents the correlation between genes as a variable that controls the differentiation of gene expression levels over time. By modeling the gene expression level of one gene at a certain time point as a linear dependency on other gene expression levels, SCODE uses linear regression to estimate the correlation matrix. SCOUP, on the other hand, models the differentiation of gene expression levels of each gene over time as a continuous and stochastic diffusion process, i.e. Ornstein–Uhlenbeck (OU) process. In this model, the expression of one gene at a certain time point can be estimated by the normal distribution of the current OU process. The correlations between genes are obtained using 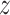 value calculated for all cells.
value calculated for all cells.
Inference Snapshot only uses differential equations to determine the uncertain regulatory edge signs. It uses GENIE3 to construct the network and then applies the ODE to describe the temporary evolution of a target gene depended on an input gene. The input dynamics are determined by Hill-type functions, which are nonlinear functions that describe the transcription phase. The values of the Hill-type functions increase when the input is an activator and decrease when the input is an inhibitor. The parameters of the model are estimated using Markov chain Monte Carlo. At the end of the analysis, while SCODE and SCOUP output a correlation matrix representing genes relationships that can be easily visualized using third-party network visualization application, Inference Snapshot only outputs estimated parameters for the model.
Among the three methods, only SCODE assumes that dependencies between genes are linear. This may oversimplify the realistic relationship between genes, especially in large GRNs. The method also does not account for the stochastic nature of gene expression data, which may reduce the quality of the inferred GRN, especially when the number of samples is low. The quality of the network can be improved by increasing the number of cells. However, an increase in the number of cells can exponentially increase the computational complexity of the method. This is due to the large number of parameters used in the model. This applies to the other two methods as well. Therefore, methods in this category are most suitable for the inference of small-size networks.
3.3 Gene correlation
The overall workflow of these methods is illustrated as in Figure 4. Here, we review five methods—Empirical Bayes, Information Measures, NLNET, SINCERA and SCENIC—that focus on modeling the pair-wise relationship between genes using different correlation metrics: Empirical Bayes and Information Measures use techniques from information theory; NLNET defines the correlation as distance based on conditional ordered list; SINCERA uses low-order partial correlation; and SCENIC utilizes a tree-based algorithm to estimate the connectivity between genes. These metrics are designed to capture not only the information shared between two genes but also the effects of other genes. While the other four methods are designed to infer only the GRN, SINCERA provides a comprehensive pipeline for multiple purposes, including data processing, clustering, GRN inference and network enrichment.
Figure 4 .
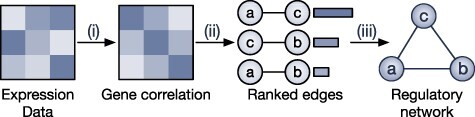
The overall workflow of methods using gene expression correlation. (i) The methods first initialize the weights of the edges by calculating the expression correlation for each gene pair. (ii) The methods perform a hypothesis testing to estimate the significance of each edge and then remove edges that are considered insignificant using a predefined significance threshold. (iii) The methods output the largest connected component.
Empirical Bayes and Information Measures were developed by the same research laboratory, in which Empirical Bayes is claimed to be an improved version of Information Measures. The two methods employ information theory to infer the relationship between genes. In particular, the methods use partial information decomposition (PID) [57] to model the information provided by a set of source variables (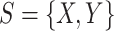 ) about another target variable (
) about another target variable (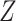 ), partitioned into redundant, synergistic and unique information. Among these, the unique information of
), partitioned into redundant, synergistic and unique information. Among these, the unique information of 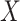 (or
(or 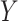 ) is the unique information provided by only
) is the unique information provided by only 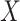 (or
(or 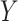 ). The methods then aggregate the unique information provided by
). The methods then aggregate the unique information provided by 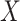 and
and 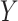 for each of the gene
for each of the gene 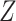 , normalized by the mutual information. This is termed as the proportional unique contribution (PUC).
, normalized by the mutual information. This is termed as the proportional unique contribution (PUC).
To quantify the confidence of the calculated PUC values, the two methods estimate the distribution of the PUC values under the null and then compute the confidence scores. For Information Measures, the confidence score is calculated as 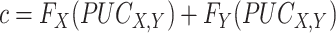 where
where 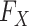 is the cumulative distribution of all the PUC scores involving the gene
is the cumulative distribution of all the PUC scores involving the gene 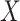 . Empirical Bayes provides an additional step to smooth the empirical distributions using a regression-based mode-matching method [58]. The methods output a ranked list of edges using the obtained confidence scores.
. Empirical Bayes provides an additional step to smooth the empirical distributions using a regression-based mode-matching method [58]. The methods output a ranked list of edges using the obtained confidence scores.
In the NLNET method, the correlation between two genes is defined as the distance based on conditional ordered list (DCOL) [59], in which the distance from gene 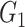 to gene
to gene 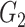 depends on the order of expressions through all samples in
depends on the order of expressions through all samples in 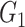 . This distance metric, therefore, is not symmetrical and is claimed to capture both linear and nonlinear relationships. Unlike the PUC used in Empirical Bayes and Information Measures, this distance metric used by NLNET does not account for the fact that a gene in a real biological network may interact with more than one gene. In this perspective, SINCERA, which makes use of the low-order partial correlation that can involve more than two genes in the measurement, is a more realistic way to present interactions of a complex network. In the SINCERA method, the correlation between gene
. This distance metric, therefore, is not symmetrical and is claimed to capture both linear and nonlinear relationships. Unlike the PUC used in Empirical Bayes and Information Measures, this distance metric used by NLNET does not account for the fact that a gene in a real biological network may interact with more than one gene. In this perspective, SINCERA, which makes use of the low-order partial correlation that can involve more than two genes in the measurement, is a more realistic way to present interactions of a complex network. In the SINCERA method, the correlation between gene 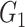 and gene
and gene 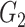 given a third gene
given a third gene 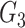 is the correlation between the residuals resulting from the linear regression of
is the correlation between the residuals resulting from the linear regression of 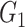 with
with 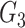 and that of
and that of 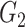 with
with 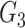 . SINCERA calculates the coefficients of the target gene and the conditional gene of the regression using the least square estimator. By using this correlation, SINCERA assumes the linear dependency between genes.
. SINCERA calculates the coefficients of the target gene and the conditional gene of the regression using the least square estimator. By using this correlation, SINCERA assumes the linear dependency between genes.
The fifth method, SCENIC, adopts a regression-tree-based method (GENIE3) to construct the network. Although GENIE3 was developed for bulk RNA-seq data, it has been applied to single-cell analysis. Instead of inferring the relationship of genes in gene pairs (NLNET) or gene triplets (Empirical Bayes, Information Measures and SINCERA), GENIE3 takes into account the interaction of an arbitrary number of genes in one calculation and can capture the nonlinear dependencies between genes. For each gene 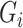 from the expression matrix, GENIE3 generates a random forest where the output is the expression in all samples of
from the expression matrix, GENIE3 generates a random forest where the output is the expression in all samples of 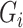 , and the input includes the expression of other genes
, and the input includes the expression of other genes 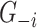 . The trees of each gene are then ensembled to obtain the ‘importance measure’ (IM) of each gene in
. The trees of each gene are then ensembled to obtain the ‘importance measure’ (IM) of each gene in 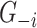 to gene
to gene 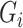 . This indicates how likely a connection exists between two genes.
. This indicates how likely a connection exists between two genes.
The computed correlation matrices from the three methods (NLNET, SINCERA, and SCENIC) are often too complex to provide a meaningful GRN network. Therefore, these methods perform a hypothesis testing or enrichment to filter out edges that are not significant. NLNET computes the null distributions of DCOL using permutation. The distribution of DCOL from gene 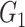 to gene
to gene 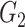 is obtained by randomly ordering the expression values in
is obtained by randomly ordering the expression values in 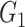 500 times. The method then corrects the
500 times. The method then corrects the  -values using false discovery rate (FDR) and removes edges using a predefined threshold. SINCERA computes the null distribution for each by setting the coefficient of the target gene to zero. SINCERA calculates the
-values using false discovery rate (FDR) and removes edges using a predefined threshold. SINCERA computes the null distribution for each by setting the coefficient of the target gene to zero. SINCERA calculates the  -values for each edge using t-test and then filters out connections with
-values for each edge using t-test and then filters out connections with  -value larger than a predefined threshold. Instead of comparing with the null distribution of the scoring function, SCENIC performs an enrichment on the output gene list to identify candidate transcription factors and DNA motifs that are overrepresented. Only enriched transcript factors and their predicted targets are retained in the final network.
-value larger than a predefined threshold. Instead of comparing with the null distribution of the scoring function, SCENIC performs an enrichment on the output gene list to identify candidate transcription factors and DNA motifs that are overrepresented. Only enriched transcript factors and their predicted targets are retained in the final network.
The output networks from NLNET, Empirical Bayes and Information Measures are undirected. With SINCERA, users can follow their pipeline to perform an enrichment to determine transcript factors and target genes. SCENIC also turns the undirected network obtained from GENIE3 to a directed network using the motif enrichment. SCENIC’s output contains different connected modules called regulon consisting of a transcript factor and its potential direct targets.
In general, methods in this category measure the global similarity of the transcriptomic profile between genes. This is especially helpful when the data are homogeneous or just have a few cell types. However, when it comes to data that are heterogeneous with multiple different cell types in different developmental stages, these methods may ignore connections that present strong correlations in one stage but not in other stages.
The performance and time complexity of these methods vary depending on the metric used in calculating the correlation. Mutual information, DCOL and regression tree are expected to capture the nonlinear relationship between genes and to account for the stochastic nature of gene expression data. In principle, the computational complexity of Empirical Bayes, Information Measures and SCENIC greatly depends on the number of genes since they need to calculate the PID for every gene triplets or build a random forest for each gene. For NLNET, since it sorts gene expression in each calculation, its computational complexity mostly depends on the sample size. On the other hand, the conditional dependence metric used by SINCERA is unsophisticated and cannot capture nonlinear relationships between genes.
Methods in this category give a good estimation of how likely a connection can be established between genes. However, one common drawback is that they are unable to infer the regulations between genes (e.g. activation, repression). SCENIC and SINCERA work around this bottleneck by performing an enrichment with known regulatory networks in existing databases.
3.4 Correlation ensemble over pseudo-time
Methods in this category also use gene correlation to represent the pair-wise relationship between genes. However, these methods take into account the fact that the relationship between genes may change depending on the developmental stages. These methods calculate the gene correlation in smaller time windows and then combine the correlation matrices using an ensemble strategy. Here, we review four methods—LEAP, SCIMITAR, SINCERETIES and SCINGE—that exploit the time trajectory information to construct the GRNs.
The overall workflow of these methods is illustrated as in Figure 5. All four methods take the single-cell data as input. LEAP, SCINGE and SINCERETIES also require users to provide the time ordering of the cells. All four methods output a correlation matrix indicating weights and signs of the regulation, which can be easily imported to any third-party software for network visualization.
Figure 5 .
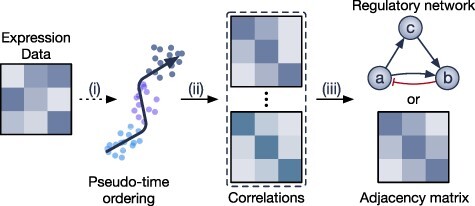
The overall workflow of methods that calculate genes correlation based on pseudo-time ordering. (i) The methods either infer the pseudo-temporal ordering of the cells or require users to provide the time ordering. (ii) The methods divide the data into smaller time windows and then calculate the gene correlation for each time window. (iii) The methods then aggregate (ensemble strategy) multiple correlation matrices into one single adjacency matrix that represents the GRN.
Among the four methods, only SCIMITAR implements a pseudo-time inference method in the pipeline to obtain the time ordering directly from the expression data. SCIMITAR models the data using a continuous multivariate Gaussian mixture model and then estimates the parameters using the expectation-maximization (EM) algorithm. The EM algorithm estimates the parameters of each distribution and how likely a cell belongs to each distribution. SCIMITAR computes the correlation matrices for each pseudo-time from the covariance matrices of the mixture model. The method then computes a similarity matrix by calculating the distance between the covariance matrices. Spectral clustering is then used to determine the developmental stages across the time trajectory using the similarity matrix. For each developmental stage, the method outputs the consensus network by averaging the correlation matrices in this stage.
The other three methods require users to provide the time ordering of the cells as part of the input. They all first generate multiple overlapping windows along the time axis, calculate the gene correlation in each time window and then merge the correlation matrices using different strategies. LEAP simply uses the Pearson correlation to calculate the correlation between genes for each time window. It then combines all correlation matrices by gathering the maximum correlation for all gene pairs over all time windows. SCINGE uses a regression model to determine the correlation between two genes in a time window. For each target gene, the method utilizes the kernel-based Granger causality regression to calculate the correlation (i.e. edge or connection) of the gene to all other genes. For ensemble, the method uses the Borda count aggregation method [60], which favors connections that are consistently ranked high by multiple Granger tests, to rank the connection between two genes over time. In contrast to the other three methods, SINCERETIES does not calculate the correlation between genes in each time window. For each gene, it constructs a differentiation vector that represents the expression change over time. It then uses the Granger causality regression to infer the interaction between genes using the differentiation vectors.
By using timestamp data to construct the relationship between genes, methods in this category aim at finding prominent groups of genes that have similar expression patterns over time. These methods infer the temporary correlation between each gene pair in each time window rather than calculating one single correlation for the whole time trajectory. This is especially helpful when the data are noisy or when some unexpected events affect the expression of the genes in a certain time window.
The performance (accuracy) of each method in this category heavily depends on the accuracy of the time ordering, which is not always available. This information is usually inferred by pseudo-time inference methods. Most of these time trajectory inference methods, including Monocle and Wanderlust, require other prior information such as start cells, end cells and the number of branches. However, this information may not be available for users, making it difficult to construct a reliable time trajectory. In addition, the GRN methods in this category are not able to handle multiple-branching cellular trajectories that correspond to different cell fate decisions. These methods forcefully merge all branches into one linear developmental path, which may compromise the accuracy of the network. Finally, these methods use Pearson correlation and Granger causality to infer the relationship between genes. However, biological systems are complex and might have nonlinear gene interactions. Regarding computational complexity, LEAP has a clear advantage due to its simplicity in calculating the correlation.
4 Simulation studies
In this section, we systematically compare the methods based on three essential metrics: (i) accuracy in reconstructing reference networks using scRNA-seq data, (ii) sensitivity to dropout rate and sparsity and (iii) time complexity.
We generated 139 simulated datasets using known reference networks and then assess the performance of the network inference methods based on the three metrics listed above. We used the curated human network in the TRRUST database [61, 62] as the reference network and the GeneNetWeaver [63] as the software to generate scRNA-seq datasets. GeneNetWeaver has become a widely used tool to synthesize data from a given network for the purpose of evaluation [35, 41, 46]. This software was also used to generate gold-standard data for DREAM4 [64] and DREAM5 [65] international competitions. Briefly, GeneNetWeaver uses ODEs and SDEs to synthesize data measured at different time points. After generating the datasets, we used the surveyed methods to construct the networks and then compared the constructed networks against the reference networks. For each constructed network, the receiver operating characteristic (ROC) curve is generated by plotting the true positive rate against the false positive rate at different thresholds when comparing the edges of the constructed network with the reference network. The quality of each constructed network is assessed using the area under the ROC curve (AUROC). A constructed network has an AUROC of 1 if it is identical to the reference network.
We assessed the performance of each method in three different scenarios. In the first scenarios, we generated datasets with 200 samples and varying numbers of genes (20, 500, 1000, 2000 and 3000). Each setting has 20 replicates. We also simulated dropouts (at 30% sparsity) using weighted random sampling, in which low-expression values will have a higher chance to be dropout. We used the surveyed methods to analyze the datasets and assess their performance by comparing the constructed networks against the ground truth (reference networks). In the second scenarios, we generated 25 datasets with different levels of dropout/sparsity—30%, 50%, 70%, 80% and 90%—5 datasets per different gene number and dropout level. We then used the surveyed methods to analyze the datasets and then assess their accuracy. In the third scenario, we increased the number of genes and samples and measured the running time of each method. First, we set the number of samples to 200 and increase the number of genes from 20 to 30, 50, 100, 200, 500, 1000, 2000 and 3000. Second, we set the number of genes to 20 and increase the number of samples from 200 to 400, 600, 800 and 1000.
The analysis was performed on a desktop with 128GB of RAM and AMD Ryzen Threadripper 2950X 16-Core Processor. We experienced difficulties in running Inference Snapshot and BTR. Inference Snapshot has poor documentation and requires users to switch back and forth among multiple environments (MATLAB, C++, Linux shell). In addition, the parameters and input are hardcoded in the source code that needs to be compiled. Therefore, we excluded Inference Snapshot from our analysis. Regarding BTR, the method iteratively constructs the network but the algorithm does not always converge. When analyzing networks with 20 genes, the method sometimes does not converge (11 out of 20 times) and runs for two weeks without any sign of stopping. This method does not converge at all for datasets with 30 genes or more. Therefore, we report NA for BTR in datasets with 30 genes or more. Among the 14 methods tested, only four methods (BTR, SCNS, SCENIC and SCIMITAR) are able to use multiple cores. For these methods, we used eight cores to analyze the simulated datasets.
Figure 6 shows the distribution of the AUROC values in the first scenario (100 simulated datasets with 20 to 3000 genes, 200 samples and 30% dropout rate). For datasets with 20 genes, most methods produce networks that have AUROC values greater than 0.5 and with the peak performance at 0.62 (Information Measures). These results are also consistent with those reported by Chen and Mar [32]. When the number of genes in the network increases from 20 to 3000, eight methods are not able to finish the analysis: BTR does not converge for datasets with 30 genes or more, SCNS cannot analyze more than 64 genes; Boolean Pseudotime and SINCERA do not converge when analyzing datasets with 500 genes; SCOUP, SCINGE, SINCERITIES and Empirical Bayes crash when analyzing datasets with 500, 1000, 2000 and 3000 genes, respectively. Only six methods are able to analyze all datasets: SCODE, Information Measures, NLNET, SCENIC, LEAP and SCIMITAR. However, the AUROC values of these methods drop drastically when the number of genes increases. Especially, when the number of genes reaches 3000, all six methods have the AUROC value close to 0.5, which is the expected AUROC value of randomly generated networks. Overall, SCENIC has the best median AUROC values in all cases (20, 500, 1000, 2000 and 3000 genes).
Figure 6 .
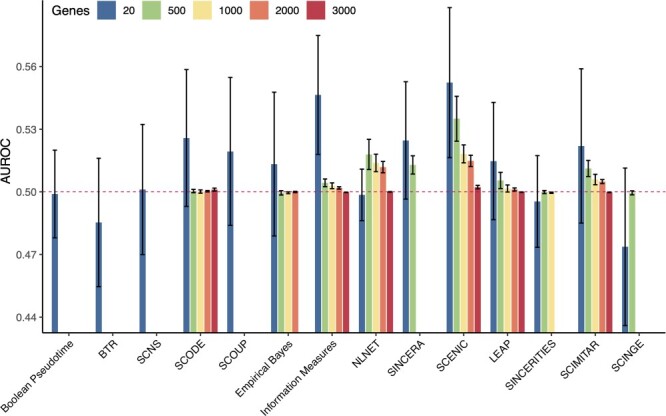
Performance of 14 GRN inference methods using 100 simulated datasets with 200 samples and varying number of genes (20, 500, 1000, 2000 and 3000). For each specific number of genes, we generated 20 datasets, reconstructed the networks using the GRN inference methods and compared the constructed networks against the ground truth (reference networks). The horizontal axis shows the methods while the vertical axis shows the AUROC values that represent the accuracy of the methods. Only six methods are able to analyze the datasets with 3000 genes: SCODE, Information Measures, NLNET, SCENIC, LEAP and SCIMITAR. In all scenarios, SCENIC has the highest median AUROC values.
Figure 7 shows the AUROC values obtained in the second scenario (20 simulated datasets with 20 genes, 200 samples and varying sparsity levels). Most methods were able to analyze all datasets except SINCERA and SINCERITIES, which crash when the sparsity level goes beyond 50% and 80%, respectively. Among the remaining methods, nine methods (Boolean Pseudotime, BTR, SCNS, Empirical Bayes, Information Measures, NLNET, LEAP, SCIMITAR and SCINGE) have the AUROC values that fluctuate at 0.5 or below when the sparsity levels increase from 30% to 90%. Only three methods that have AUROC values consistently above 0.5: SCODE, SCOUP and SCENIC. Among these three methods, SCOUP is the most stable with very low variability (standard deviation of 0.005 compared with 0.013 and 0.026 of SCODE and SCENIC, respectively).
Figure 7 .
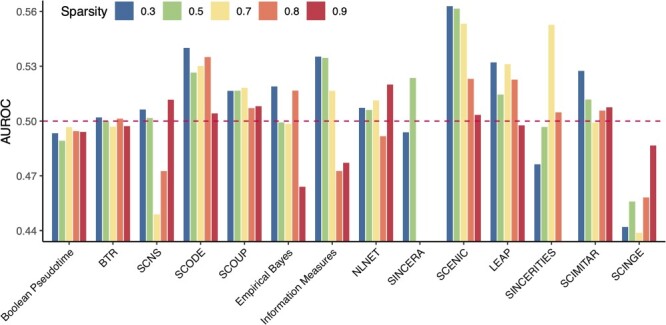
Performance of network inference methods with different levels of sparsity using 25 simulated datasets (5 datasets per sparsity level). The horizontal axis shows the methods while the vertical axis shows the AUROC values that represent the quality of the constructed networks. At each level of sparsity, we show the mean AUROC of five datasets for each method. SCOUP is the most stable method. It produces AUROC values that are consistently above 0.5 with very low variability across five sparsity levels.
The running time of each method is shown in Figure 8 (14 datasets with varying numbers of genes and samples) and Supplementary Tables S3 and S4 (Supplementary Note). In Figure 8, the vertical axes represent the running time in 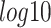 scale of minutes. As we explained above, BTR does not converge for networks with 30 genes or more. The method SCNS does not support the analysis of any network with 64 genes or more. Even for datasets with 50 genes, it takes the method more than two hours to finish the analysis. SINCERA and Boolean Pseudotime can be very slow when analyzing large networks. It takes SINCERA 15 hours and Boolean Pseudotime a week to analyze one dataset with 200 genes. The other four methods, SCOUP, SCINGE, SINCERITIES and Empirical Bayes produce errors when the number of genes reaches 500, 1000, 2000 and 3000, respectively. When the number of samples increases, only the running time of SCIMITAR seems to be affected drastically. The running time of SCIMITAR increases from ten minutes to seven hours when the number of samples increases from 200 to 800. This method crashes for the dataset with 1000 samples. Overall, LEAP and NLNET are the fastest and can finish every single analysis in minutes.
scale of minutes. As we explained above, BTR does not converge for networks with 30 genes or more. The method SCNS does not support the analysis of any network with 64 genes or more. Even for datasets with 50 genes, it takes the method more than two hours to finish the analysis. SINCERA and Boolean Pseudotime can be very slow when analyzing large networks. It takes SINCERA 15 hours and Boolean Pseudotime a week to analyze one dataset with 200 genes. The other four methods, SCOUP, SCINGE, SINCERITIES and Empirical Bayes produce errors when the number of genes reaches 500, 1000, 2000 and 3000, respectively. When the number of samples increases, only the running time of SCIMITAR seems to be affected drastically. The running time of SCIMITAR increases from ten minutes to seven hours when the number of samples increases from 200 to 800. This method crashes for the dataset with 1000 samples. Overall, LEAP and NLNET are the fastest and can finish every single analysis in minutes.
Figure 8 .
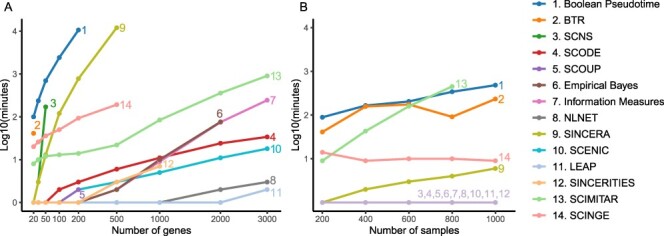
Running time of network inference methods with varying numbers of genes (panel A) and samples (panel B) in the 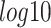 scale of minutes. Overall, LEAP and NLNET are the fastest methods that can finish every single analysis in minutes.
scale of minutes. Overall, LEAP and NLNET are the fastest methods that can finish every single analysis in minutes.
In conclusion, regarding the accuracy, SCENIC is the best method with the highest AUROC values in most simulation studies. Regarding time complexity, LEAP and NLNET are the best methods that can finish every single analysis in minutes (even for datasets with 3000 genes or 1000 samples). Regarding sensitivity to sparsity, SCOUP is the most stable method. It produces AUROC values that are consistently above 0.5 with very low variability across different sparsity levels.
5 Challenges and future development
Although the field of GRN inference for single-cell is rapidly growing, it is still relatively new. The application of these methods is not yet presented in many real-world-setting studies. This may come from the difficulty in dealing with technical limitations in the scRNA-seq platforms or the heterogeneity of single-cell data. The validation of the output networks and computational challenges also raise questions about their applicability. Here, we discuss such problems that need to be addressed for new GRN inference methods in the future.
Dropout and batch effects Single-cell RNA sequencing technologies allow us to decompose complex tissues at the single-cell resolution. However, dropout events and batch effects present significant challenges for single-cell analysis, including GRN inference. Compared with RNA-seq data from a bulk cell population, the scRNA-seq data is usually sparser with a higher level of noise. It is common that more than 50% entries of the gene expression matrix obtained from scRNA-seq are equal or close to zero. This rate can be as high as 70% in many datasets [66, 67]. The high dropout rate means that the true expressions of many genes are not captured. GRN methods usually exclude genes with a high dropout rate, leading to the potential of removing important genes from the network. In addition, dropout events alter the distribution of the gene expression, making many mathematical models inaccurate.
One solution to this challenge is to utilize imputation methods. Many methods have been developed recently to impute dropout values in single-cell data [66, 68–71]. These methods may help improve the data quality before conducting GRN inference analysis. However, these methods also have limitations and may introduce false signals. First, distinguishing the truly unexpressed genes from those caused by sequencing error is not trivial. Second, the accuracy of the imputed data is difficult to validate.
At the same time, batch effects may have negative impacts on the performance and accuracy of the inferred GRNs [72]. Even though many studies have reported the severity of batch effects in both bulk RNA-seq [73] and scRNA-seq [67] analyses, most GRN methods completely ignore the potential impacts of batch effects. Among the 15 methods, only SCENIC takes into account this problem by performing cross-species comparisons. However, this requires users to prepare sequencing data of similar tissues from different species which is not always available. One possible solution is to use statistical tools to correct for batch effects before performing analysis, which is thoroughly discussed in other review articles [67, 73].
Heterogeneity and stochastic factors Besides the limitations of sequencing platforms, the heterogeneous and stochastic nature of gene expression in individual cells also present additional challenges to any single-cell analysis. The biological heterogeneity of single-cell data may come from multiple sources, including genotypic factors caused by random mutations and phenotypic factors caused by internal cellular interactions (gene networks and products) and environmental change [74]. Additional sources of variation can come from technical heterogeneity, including study design, sample collection, storage, sample preparation, assays and sequencing technologies. These sources of variation result in batch effects and different signal detection rates among cells and genes [75, 76]. This leads to variation and heterogeneity among both biological and technical replicates.
Although several GRN inference methods claim to deal with the stochastic aspects of gene expression using modeling and simulation, characterizing the role of stochasticity in GRNs remains a significant challenge. The effects of the stochastic information in gene expression data can be reduced when the number of cells increases. However, most current GRN methods are designed to analyze datasets with a small or moderate number of cells, making this challenge remained to be addressed. In addition, to reduce their computational complexity, GRN methods often focus the analysis on genes with high variability. However, genes with high variability, which may due to the heterogeneity of the cells and stochastic nature of the gene expression, may not be the genes that drive cell transition progress [77, 78].
Temporal ordering of cells Eight out of 15 methods in this review heavily depend on the temporal ordering of cells. Since it is currently impossible to generate real time-ordering single-cell data, these methods rely on statistical inference to construct a pseudo-time trajectory from the input expression data. Time-trajectory inference methods usually order the cells along a trajectory topology, which can be cycled, linear, multifurcating or complex graph structure [53]. Time trajectories also may not exist in cell types that cells are equally matured. However, all of the eight GRN methods reviewed here are designed to handle only the linear time-trajectory. For methods that require users to input time-ordering data, users need to manually remove unwanted branches. Other methods that embed pseudo-time inference in their pipeline assume that the developmental stages follow a linear and continuous trajectory. The inability to handle nonlinear time trajectory may heavily affect the accuracy of these methods. Foremost, most single-cell studies try to identify the mechanisms that drive the cells in the state transition progress and especially when the cell fate decisions are made [28, 79, 80]. Unable to deal with branching time trajectories makes it challenging to infer the regulatory network that decides the cell fates using these methods.
Network complexity The purpose of constructing GRNs is to gain insights into the regulation of genes and/or transcription factors during cell state transition. Therefore, the networks are usually cell-type specific. However, most GRN inference methods (except SCIMITAR) produce one single network containing all possible interactions. This might be useful in small-scale experiments, in which only one or very few cell types and genes are involved. For complex tissues, however, one large network is difficult to interpret and is insufficient to reveal the underlying mechanisms of cell transitions. It fails to explain the dynamics happen in different cell types or different developmental stages. In this case, reporting different networks for different stages of cell transitions will help us to better understand how gene regulations drive cell state transitions. Future methods will need to accurately separate cell stages from single-cell expression data to generate a comprehensive network.
Computational complexity With the rapid advancement of sequencing technologies, the number of cells available in each dataset increases exponentially. For example, the number of cells has increased from hundreds to millions or tens of millions in recent years [81]. With this pace, current GRN methods will not be able to analyze new datasets in a reasonable time. Scalability will be a critical challenge in the development of future methods. This problem presents a bottleneck for not only GRN inference methods but also for any analysis methods developed for single-cell data. Unlike other analyses that can utilize dimension reduction techniques to reduce the complexity, GRNs methods need to analyze all features/genes to provide a comprehensive gene network. To reduce the complexity, current analysis pipelines either limit the analysis to a predefined gene list or to the top differentially expressed (DE) genes. However, by using a predefined gene list, we assume that the underlying mechanisms are limited to prior knowledge about the genes. This often leads to just reconfirming the already known information. On the other hand, by using only DE genes, we may eliminate genes that act like connecting nodes between DE genes or components. Future methods need to address the scalability bottleneck in order to generate more comprehensive networks.
Validation Validation and performance assessment of the GRN inference methods is the most challenging problem. Most of the surveyed methods use simulation to assess their performance. These methods usually start with one known or simulated network and then generate the gene expression data that follows the regulation defined by the network. For example, BTR, Information Measures and SINCERITIES used GeneNetWeaver [63] to synthesize transcriptome data from a given GRN and then added artificial dropouts to the output to simulate the sparsity of single-cell data. Other methods (Inference Snapshot, NLNET, SCODE, SCOUP) developed a dedicated model to generate simulated data. The advantage of simulation is that the underlying mechanisms are known and thus can be used to assess the accuracy of the constructed networks. However, simulations are generally unsophisticated and often fail to account for the real and complex biology. In addition, the simulated data may follow certain distributions and patterns that favor the authors’ hypothesis and assumption, leading to biased analyses and conclusions.
Another way for performance assessment is to use real single-cell data and then compare the constructed networks with prior knowledge. In these analyses, the authors usually seek support from experts in the field, or find supporting evidence from the literature. Some authors compare the GRNs with networks from databases that were curated from small-scale experiments (e.g. TRRUST [61, 62], RegulonDB [82], ESCAPE [83] and CODEX [84]). However, for real biological data, the actual mechanisms involved at the cell level are never fully known. When seeking supporting evidence from experts or literature, researchers might be influenced by the observer-expectancy effect [85]. This might lead to biased conclusions. When seeking evidence from curated networks, researchers often overlook the limitations of the existing databases. First, the above-mentioned databases were constructed using bulk RNA-seq data. These databases do not provide cell-type-specific GRNs. Second, the curated networks only cover a moderate or small number of transcription factors, genes or known interactions. For example, the number of human genes in the TRRUST database is approximately 3000 while the number of human protein-encoding genes is estimated to be around 20 000 [86]. Although a number of single-cell atlases have been built [87, 88], there is no network database constructed to reflect gene regulations in different developmental stages and cell types. The lack of experimental evidence supporting the resulted GRNs may limit the GRN methods from discovering novel regulatory modules.
As the field matures, the existence of such network databases will provide us with a more reliable knowledge for validating GRN inference methods. At the same time, generating gold standard datasets that can be used to benchmark the accuracy of the resulted GRNs will also greatly boost the reliability of GRN inference methods and their usefulness. Given these conditions, GRN methods will be more reliable and will significantly contribute to the causal map of molecular interactions. The resulted networks then can be applied to a wide range of real-world applications from disease biomarkers and pathways to network medicine and drug design [1].
Conclusion
In this survey, we discuss 15 GRN inference methods developed dedicatedly for scRNA-seq data analysis. At the time of this survey, all of these tools are publicly available as a stand-alone package or script. Our main objective is to help potential users, especially life scientists, to choose a method that is most suitable for their available data and analysis purpose. At the same time, this review will also help computational scientists in identifying the shortcomings of existing approaches in order to develop new methods that address current limitations. To accomplish these objectives, we first rank the methods based on their usability and friendliness so that users can easily get started with the tools. We then discuss in-depth the hypothesis and the techniques each method uses to model the GRN. We divide the methods into four different categories according to the underlying inference techniques: (i) boolean model, (ii) differential equation, (iii) gene correlation and (iv) correlation ensemble over pseudotime. We finally discuss the challenges that future methods need to address to generate more accurate and comprehensive GRN from single-cell data.
In conclusion, GRN inference methods are useful tools for mapping molecular interactions to form meaningful biological networks that reflect the internal mechanisms of living organisms. Networks generated from single-cell data have the potential of unraveling gene regulatory interactions in the level of individual cells at specific developmental stages. However, the field of GRN inference using single-cell data is relatively new and there are technical, biological, and computational challenges that remain to be addressed. Current methods are sensitive to technical noise and are not sophisticated enough to cope with the complex nature of the regulatory network from single-cell data. These methods are also designed to work with a limited number of genes and are not ready for the rapidly increasing of number of cells generated in single-cell data. Most importantly, the lack of standard benchmarks in assessing the performance of these mathematical models raises questions about the reliability of the constructed GRN. As the field develops and more information becomes available to validate the networks, GRN inference methods using single-cell data will be promising tools not only in discovering missing components of GRNs in the cell level but also in a wide range of real-world applications such as identifying disease biomarker and pathways, designing drugs and precision medicine.
Key Points
Single-cell data allow us to construct and analyze gene regulatory networks (GRNs) of heterogeneous cell types in different developmental stages.
This article reviews and discusses in-depth 15 GRN inference methods using scRNA-seq from the following perspectives: their availability, usability, hypothesis and inference techniques.
This article points out outstanding challenges of GRN inference methods and discusses future directions regarding data quality, benchmarking and computational complexity.
This article will assist life scientists in selecting suitable methods for their analysis purposes, as well as computational scientists in identifying shortcomings of current methods.
This article provides simulation studies to assess the performance of GRN inference methods based on three different metrics: accuracy, robustness against dropout and time complexity.
Supplementary Material
Hung Nguyen is a PhD student in the Department of Computer Science and Engineering at the University of Nevada, Reno, Nevada, USA. His research interests include cancer subtyping, network inference and single-cell data analysis.
Duc Tran is a PhD student in the Department of Computer Science and Engineering at the University of Nevada, Reno, Nevada, USA. His research interests include machine learning, and single-cell data analysis.
Bang Tran is a PhD student in the Department of Computer Science and Engineering at the University of Nevada, Reno, Nevada, USA. His research interests include single-cell clustering and imputation.
Bahadir Pehlivan is an MS student in the Department of Computer Science and Engineering at the University of Nevada, Reno, Nevada, USA.
Tin Nguyen is an assistant professor in the Department of Computer Science and Engineering at the University of Nevada, Reno, Nevada, USA. He is also the head of the Bioinformatics Laboratory at UNR. His research focuses on developing machine learning and statistical methods that can be applied to bioinformatics and computational biology.
Funding
This work was partially supported by NASA under grant number 80NSSC19M0170, by NIH NIGMS under grant number GM103440, and by NSF under grant number 2001385. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of any of the funding agencies.
References
- 1.Emmert-Streib F, Dehmer M, Haibe-Kains B. Gene regulatory networks and their applications: understanding biological and medical problems in terms of networks. Front Cell Dev Biol 2014; 2:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nat Rev Microbiol 2010; 8(10): 717–29. [DOI] [PubMed] [Google Scholar]
- 3.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9(1): 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huynh-Thu VA, Irrthum A, Wehenkel L, et al. Inferring regulatory networks from expression data using tree-based methods. PLoS One 2010; 5(9): 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faith JJ, Hayete B, Thaden JT, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol 2007; 5(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mordelet F, Vert J-P. SIRENE: supervised inference of regulatory networks. Bioinformatics 2008; 24(16): i76–82. [DOI] [PubMed] [Google Scholar]
- 7.Haury A-C, Mordelet F, Vera-Licona P, et al. TIGRESS: trustful inference of gene regulation using stability selection. BMC Syst Biol 2012; 6(1): 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Margolin AA, Nemenman I, Basso K, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 2006; 7(Suppl 1): S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shafi A, Nguyen T, Peyvandipour A, et al. GSMA: an approach to identify robust global and test gene signatures using meta-analysis. Bioinformatics 2019; 36(2): 487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shafi A, Nguyen T, Peyvandipour A, et al. A multi-cohort and multi-omics meta-analysis framework to identify network-based gene signatures. Front Genet 2019; 10:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen T, Shafi A, Nguyen T-M, et al. NBIA: a network-based integrative analysis framework–applied to pathway analysis. Nat Sci Rep 2020; 10:4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen T, Diaz D, Tagett R, et al. Overcoming the matched-sample bottleneck: an orthogonal approach to integrate omic data. Nat Sci Rep 2016; 6:29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz D, Donato M, Nguyen T, et al. MicroRNA-augmented pathways (mirAP) and their applications to pathway analysis and disease subtyping. Pac Symp Biocomput 2016; 22:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acquaah-Mensah GK, Agu N, Khan T, et al. A regulatory role for the insulin- and BDNF-linked RORA in the hippocampus: implications for Alzheimer’s disease. J Alzheimers Dis 2015; 44(3): 827–38. [DOI] [PubMed] [Google Scholar]
- 15.Jin M, Aibar S, Ge Z, et al. Identification of novel direct targets of Drosophila Sine oculis and eyes absent by integration of genome-wide data sets. Dev Biol 2016; 415(1): 157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X, Zheng G, Shan L, et al. Reconstruction of gene regulatory network related to photosynthesis in Arabidopsis thaliana. Front Plant Sci 2014; 5:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen H, Shrestha S, Tran D, et al. A comprehensive survey of tools and software for active subnetwork identification. Front Genet 2019; 10:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen T, Mitrea C, Draghici S. Network-based approaches for pathway level analysis. Curr Protoc Bioinformatics 2018; 61(1): 8–25. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen T-M, Shafi A, Nguyen T, et al. Identifying significantly impacted pathways: a comprehensive review and assessment. Genome Biol 2019; 20(1): 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saliba A-E, Westermann AJ, Gorski SA, et al. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res 2014; 42(14): 8845–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shields CW, IV, Reyes CD, López GP. Microfluidic cell sorting: a review of the advances in the separation of cells from debulking to rare cell isolation. Lab Chip 2015; 15(5): 1230–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanevski J, Nguyen T, Truong B, et al. Predicting cellular position in the Drosophila embryo from single-cell transcriptomics data. bioRxiv 2019;796029. [Google Scholar]
- 23.Sun N, Yu X, Li F, et al. Inference of differentiation time for single cell transcriptomes using cell population reference data. Nat Commun 2017; 8(1): 1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Churko JM, Garg P, Treutlein B, et al. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat Commun 2018; 9(1): 4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Jenjaroenpun P, Bhinge A, et al. Single-cell gene expression analysis reveals regulators of distinct cell subpopulations among developing human neurons. Genome Res 2017; 27(11): 1783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stumpf PS, MacArthur BD. Machine learning of stem cell identities from single-cell expression data via regulatory network archetypes. Front Genet 2019; 10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buganim Y, Faddah DA, Cheng AW, et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012; 150(6): 1209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moignard V, Woodhouse S, Haghverdi L, et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat Biotechnol 2015; 33(3): 269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo G, Luc S, Marco E, et al. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell Stem Cell 2013; 13(4): 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moignard V, Macaulay IC, Swiers G, et al. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat Cell Biol 2013; 15(4): 363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalerba P, Kalisky T, Sahoo D, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol 2011; 29(12): 1120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S, Mar JC. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinformatics 2018; 19(1): 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fiers MWEJ, Minnoye L, Aibar S, et al. Mapping gene regulatory networks from single-cell omics data. Brief Funct Genomics 2018; 17(4): 246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamey FK, Nestorowa S, Kinston SJ, et al. Reconstructing blood stem cell regulatory network models from single-cell molecular profiles. Proc Natl Acad Sci U S A 2017; 114(23): 5822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lim CY, Wang H, Woodhouse S, et al. BTR: training asynchronous Boolean models using single-cell expression data. BMC Bioinformatics 2016; 17(1): 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woodhouse S, Piterman N, Wintersteiger CM, et al. SCNS: a graphical tool for reconstructing executable regulatory networks from single-cell genomic data. BMC Syst Biol 2018; 12(1): 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ocone A, Haghverdi L, Mueller NS, et al. Reconstructing gene regulatory dynamics from high-dimensional single-cell snapshot data. Bioinformatics 2015; 31(12): i89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsumoto H, Kiryu H, Furusawa C, et al. SCODE: an efficient regulatory network inference algorithm from single-cell RNA-seq during differentiation. Bioinformatics 2017; 33(15): 2314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumoto H, Kiryu H. SCOUP: probabilistic model based on the Ornstein–Uhlenbeck process to analyze single-cell expression data during differentiation. BMC Bioinformatics 2016; 17(1): 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan TE, Pallaseni A, Babtie AC, et al. Empirical Bayes meets information theoretical network reconstruction from single cell data. bioRxiv 2018;264853. [Google Scholar]
- 41.Chan TE, Stumpf MP, Babtie AC. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst 2017; 5(3): 251–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu H, Li P, Zhu M, et al. Nonlinear network reconstruction from gene expression data using marginal dependencies measured by DCOL. PLoS One 2016; 11(7): e0158247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo M, Wang H, Potter SS, et al. SINCERA: a pipeline for single-cell RNA-seq profiling analysis. PLoS Comput Biol 2015; 11(11): e1004575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aibar S, González-Blas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017; 14(11): 1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Specht AT, Li J. LEAP: constructing gene co-expression networks for single-cell RNA-sequencing data using pseudotime ordering. Bioinformatics 2016; 33(5): 764–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papili Gao N, Ud-Dean SM, Gandrillon O, et al. SINCERITIES: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics 2017; 34(2): 258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cordero P, Stuart JM. Tracing co-regulatory network dynamics in noisy, single-cell transcriptome trajectories. Pac Symp Biocomput 2017;576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deshpande A, Chu L-F, Stewart R, et al. Network inference with granger causality ensembles on single-cell transcriptomic data. bioRxiv 2019;534834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohammadi S, Ravindra V, Gleich DF, et al. A geometric approach to characterize the functional identity of single cells. Nat Commun 2018; 9(1): 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonnaffoux A, Herbach U, Richard A, et al. WASABI: a dynamic iterative framework for gene regulatory network inference. BMC Bioinformatics 2019; 20(1): 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herbach U, Bonnaffoux A, Espinasse T, et al. Inferring gene regulatory networks from single-cell data: a mechanistic approach. BMC Syst Biol 2017; 11(1): 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei J, Hu X, Zou X, et al. Reverse-engineering of gene networks for regulating early blood development from single-cell measurements. BMC Med Genomics 2017; 10(5):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saelens W, Cannoodt R, Todorov H, et al. A comparison of single-cell trajectory inference methods. Nat Biotechnol 2019; 37(5): 547–54. [DOI] [PubMed] [Google Scholar]
- 54.De Moura L, Bjørner N. Z3: an efficient SMT solver. In: International Conference on Tools and Algorithms for the Construction and Analysis of Systems. Springer, Berlin-Heidelberg, Germany, 2008, 337–40. [Google Scholar]
- 55.Kharchenko PV, Silberstein L, Scadden DT. Bayesian approach to single-cell differential expression analysis. Nat Methods 2014; 11(7): 740–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bendall SC, Davis KL, Amir E, et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 2014; 157(3): 714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams PL, Beer RD. Nonnegative decomposition of multivariate information. 2010; arXiv.
- 58.Efron B. Size, power and false discovery rates. Ann Statist 2007; 35(4): 1351–77. [Google Scholar]
- 59.Yu T, Peng H. Hierarchical clustering of high-throughput expression data based on general dependences. IEEE ACM T Comput Biol Bioinform 2013; 10(4): 1080–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Erp M, Schomaker L. Variants of the borda count method for combining ranked classifier hypotheses. In: The Seventh International Workshop on Frontiers in Handwriting Recognition. Nijmegen Institute for Cognition and Information, Amsterdam, 2000, 443–52. [Google Scholar]
- 61.Han H, Shim H, Shin D, et al. TRRUST: a reference database of human transcriptional regulatory interactions. Sci Rep 2015; 5:11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han H, Cho J-W, Lee S, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res 2017; 46(D1): D380–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schaffter T, Marbach D, Floreano D. GeneNetWeaver: in silico benchmark generation and performance profiling of network inference methods. Bioinformatics 2011; 27(16): 2263–70. [DOI] [PubMed] [Google Scholar]
- 64.Greenfield A, Madar A, Ostrer H, et al. DREAM4: combining genetic and dynamic information to identify biological networks and dynamical models. PLoS One 2010; 5(10): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marbach D, Costello JC, Küffner R, et al. Wisdom of crowds for robust gene network inference. Nat Methods 2012; 9(8): 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li WV, Li JJ. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat Commun 2018; 9:997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hicks SC, Townes FW, Teng M, et al. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 2017; 19(4): 562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Dijk D, Sharma R, Nainys J, et al. Recovering gene interactions from single-cell data using data diffusion. Cell 2018; 174(3): 716–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gong W, Kwak I-Y, Pota P, et al. DrImpute: imputing dropout events in single cell RNA sequencing data. BMC Bioinformatics 2018; 19:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang M, Wang J, Torre E, et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods 2018; 15(7): 539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tran B, Tran D, Nguyen H, et al. RIA: a novel regression-based imputation approach for single-cell RNA sequencing. In: 2019 11th International Conference on Knowledge and Systems Engineering (KSE). IEEE, Da Nang, Vietnam, 2019, 1–9. [Google Scholar]
- 72.Goh WWB, Wang W, Wong L. Why batch effects matter in omics data, and how to avoid them. Trends Biotechnol 2017; 35(6): 498–507. [DOI] [PubMed] [Google Scholar]
- 73.Luo J, Schumacher M, Scherer A, et al. A comparison of batch effect removal methods for enhancement of prediction performance using MAQC-II microarray gene expression data. Pharmacogenomics J 2010; 10(4): 278–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goldman SL, MacKay M, Afshinnekoo E, et al. The impact of heterogeneity on single-cell sequencing. Front Genet 2019; 10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hicks SC, Townes FW, Teng M, et al. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 2017; 19(4): 562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tung P-Y, Blischak JD, Hsiao CJ, et al. Batch effects and the effective design of single-cell gene expression studies. Sci Rep 2017; 7(1): 39921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bar-Even A, Paulsson J, Maheshri N, et al. Noise in protein expression scales with natural protein abundance. Nat Genet 2006; 38(6): 636–43. [DOI] [PubMed] [Google Scholar]
- 78.Maamar H, Raj A, Dubnau D. Noise in gene expression determines cell fate in Bacillus subtilis. Science 2007; 317(5837): 526–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo G, Huss M, Tong GQ, et al. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell 2010; 18(4): 675–85. [DOI] [PubMed] [Google Scholar]
- 80.Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014; 32(4): 381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017; 8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Santos-Zavaleta A, Salgado H, Gama-Castro S, et al. RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res 2018; 47(D1): D212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu H, Baroukh C, Dannenfelser R, et al. ESCAPE: database for integrating high-content published data collected from human and mouse embryonic stem cells. Database 2013; 2013: 1–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sánchez-Castillo M, Ruau D, Wilkinson AC, et al. CODEX: a next-generation sequencing experiment database for the haematopoietic and embryonic stem cell communities. Nucleic Acids Res 2014; 43(D1): D1117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sackett DL. Bias in analytic research. J Chronic Dis 1979; 32(1–2): 51–63. [DOI] [PubMed] [Google Scholar]
- 86.Ezkurdia I, Juan D, Rodriguez JM, et al. Multiple evidence strands suggest that there may be as few as 19 000 human protein-coding genes. Hum Mol Genet 2014; 23(22): 5866–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davie K, Janssens J, Koldere D, et al. A single-cell transcriptome atlas of the aging Drosophila brain. Cell 2018; 174(4): 982–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rozenblatt-Rosen O, Stubbington MJ, Regev A, et al. The human cell atlas: from vision to reality. Nature 2017; 550(7677): 451–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.