

Abstract
Novel polymyxin derivatives are often classified as having either direct activity against Gram negative pathogens, or as compounds inactive in their own right, which through permeabilization of the outer membrane act as potentiators of other antibiotics. Here we report the systematic investigation of the influence of lipophilicity on microbiological activity (including against strains with reduced susceptibility to polymyxins), potentiation of rifampicin, and in vitro toxicity within a series of next-generation polymyxin nonapeptides. We demonstrate that the lipophilicity at the N-terminus and amino acids 6 and 7 in the cyclic peptide core is interchangeable, and that activity, ability to potentiate, and cytotoxicity all appear to be primarily driven by overall lipophilicity. Our work also suggests that the characterization of a polymyxin molecule as either a direct acting compound or a potentiator is more of a continuum, that is strongly influenced by lipophilicity rather than as a result of fundamentally different modes-of-action.
Keywords: polymyxin, antimicrobial resistance, multidrug-resistant bacteria, lipophilicity, potentiator, Gram-negative
For Table of Contents only:
In recent years polymyxins have been re-established as an important antibiotic class for therapy of infections caused by Gram-negative bacteria resistant to other antibiotic agents1,2. Although some alternative treatments have recently been introduced3, especially for those bacteria where resistance is primarily due to expression of serine β-lactamases4, there remain very few treatment options for organisms producing metallo β-lactamases, and for many isolates of Pseudomonas aeruginosa and especially Acinetobacter baumannii. Polymyxins are thus likely to remain an important class of antibiotics for some years to come.
A series of research and development programmes has been carried out aiming to improve on the currently marketed polymyxins, polymyxin B (PMB, 1) and polymyxin E (PME, colistin, 2)5–8 (Figure 1). Much attention has been paid to reduction of toxicity, especially nephrotoxicity, but there has also been interest in potentiator compounds which lack direct antibacterial activity whilst retaining the ability to potentiate the activity of other antibiotics, likely by enhancing their uptake across the outer membrane9. The restoration of activity against polymyxin-resistant bacteria has also been an area of some interest10.
Figure 1. Structures of Polymyxin B (1), Colistin (2), and aminoacyl nonapeptides 3-47 of general structure A, B and C. Compounds 3-47 are shown in descending order of lipophilicity (ALogP).
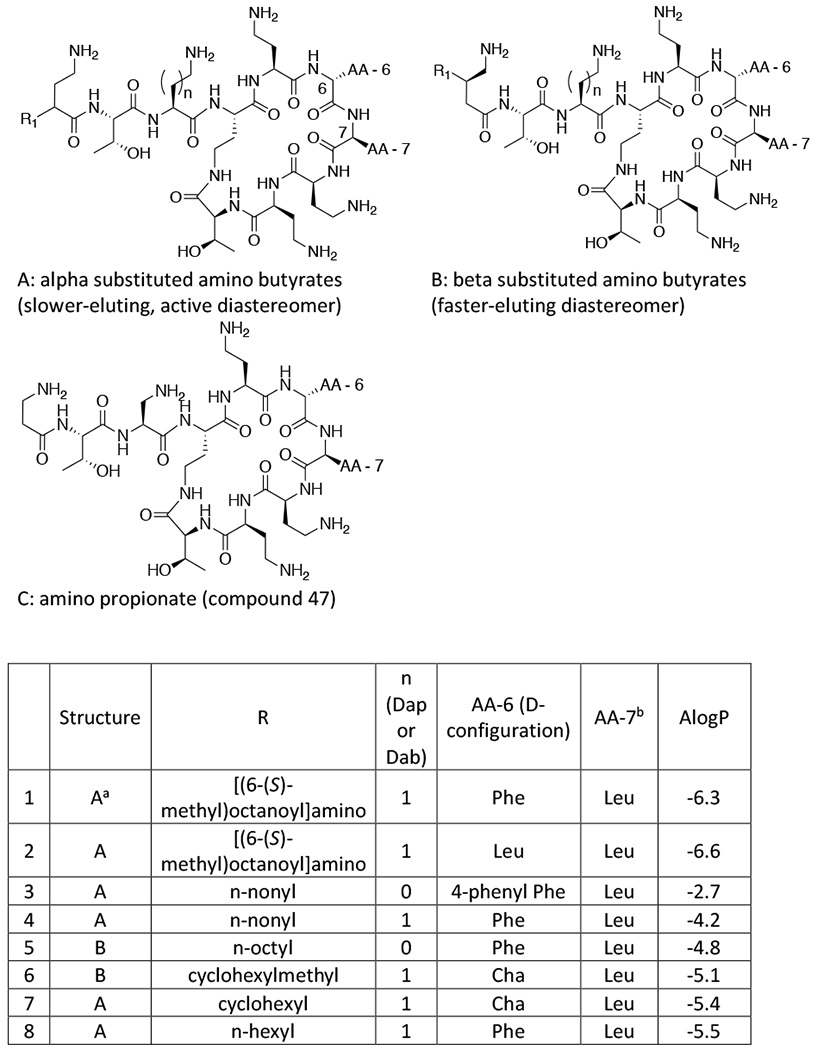
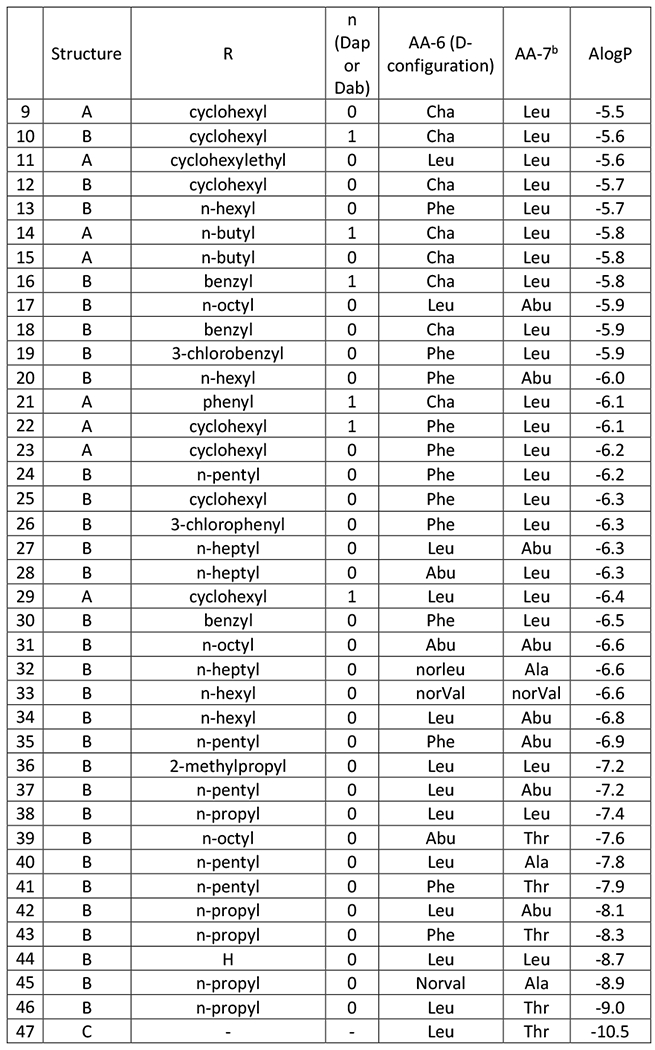
aFor compounds of structure A, the slower eluting diastereomer from reverse phase HPLC (for details see Supplementary Information) was consistently more active than the faster eluting diastereomer and all data is for the slower eluting isomer8.
bFor compounds of structure B the faster-eluting diastereomer was evaluated, which is believed to have the stereochemistry as shown by correlation with SPR206 (26)8. For compounds 34 and 36 the N-terminal acid stereochemistry was confirmed by independent synthesis (see Supplementary Information).
The first step in the antimicrobial action of polymyxins is an interaction with the lipid A component of lipopolysaccharide (LPS) in the Gram-negative outer membrane. Nmr studies11 indicate charge interactions between the primary amine moieties of polymyxin B and the phosphate groups of the LPS , while the N-terminal aliphatic chain and lipophilic residues D-phenylalanine and leucine form hydrophobic interactions with the lipid regions. Thus, polymyxins behave as amphiphilic molecules, comprising a charged face of the diaminobutyric acid (DAB) residues and lipophilic face consisting of the N-terminal group, together with positions 6 and 7. Interaction with LPS appears to disrupt the outer membrane integrity so as to potentiate the activity of hydrophobic antibiotics, which would not normally enter Gram-negative bacteria. However, the subsequent steps in the action of polymyxins leading to cell death are poorly understood12–13.
It is clear that both charge and lipophilicity are important for the antibacterial activity of polymyxins. A survey of attempts to reduce the toxicity of the polymyxin molecule unfortunately suggests that these same features are also the drivers for toxicity. Nevertheless, the molecular mechanisms of activity and toxicity are undoubtedly divergent and this offers some scope for careful manipulation of the therapeutic index, leading to some success in developing active, but less toxic polymyxin derivatives5–8.
The Vaara group for example took the approach of reducing the number of positive charges in the molecule, resulting in compounds which retained antimicrobial activity with reduced cytotoxicity both in vitro and in vivo 6. Earlier it is known from the same group that reducing lipophilicity with shortened or absent N-terminal fatty acyl moieties results in molecules which are less toxic than polymyxin B or colistin (eg PMBN14). Whilst lacking direct antibacterial activity, these molecules retain the ability to potentiate the activity of antibiotics which would not readily cross the Gram-negative outer membrane such as rifampicin, novobiocin or even vancomycin9,14. The mechanistic underpinnings of the ability of certain polymyxin molecules to exhibit potentiation activity without direct antibacterial activity are not well understood. However, it may be that such molecules have the ability to make the initial interaction with LPS in the outer membrane, but do not go on to disrupt the inner membrane15. In an alternative approach we have recently identified a series of polymyxin analogues which retain five positive charges, one of which forms part of a key aminobutyrate N-terminus on a polymyxin nonapeptide (for general structures, see Figure 1). These compounds demonstrate a promising ratio of activity to toxicity in vitro and in vivo; one such analogue, SPR206 (26, Figure 1), is currently undergoing clinical evaluation8. Reducing both lipophilicity and charge may be expected to reduce toxicity still further and indeed SPR741 with only three positive charges, and lacking a lipophilic tail, has recently been demonstrated to be well-tolerated in clinical Phase-1 studies though like PMBN lacks direct antibacterial activity16,17.
Resistance to polymyxins primarily involves the modification of the LPS structure so as to either reduce polymyxin binding or reduce the impact of polymyxin binding. The most common modifications involve reduction of the negative charge on the LPS molecule by conjugation with 4’-amino-arabinose or phosphoethanolamine18. It is reasonable to speculate that the loss of the charge interaction between polymyxin and LPS might be complemented by an increase in lipophilic interactions and certain more lipophilic polymyxin analogues do have improved activity against polymyxin-resistant strains10.
Indeed, certain other cationic-lipophilic natural products which are more lipophilic than the polymyxins (eg octapeptins) also demonstrate activity against polymyxin-resistant strains19. When considering structure-activity studies on the polymyxins it is often asserted that a particular moiety on the polymyxin molecule is responsible for a particular biological property eg the N-terminal fatty acyl chain is important for activity and in its absence, molecules lack direct activity (though retaining the ability to potentiate the activity of other antibiotics). However, our own studies in the area have led us to conjecture that, as long as the key interactions are fulfilled, the in vitro properties of polymyxins are primarily driven by overall physicochemical properties and changes in one moiety can often be compensated for by changes elsewhere in the molecule.
In order to further our hypothesis and provide fundamental information to support the development of improved polymyxin analogues, we have carried out a systematic survey, within our series of amino butyrate-containing nonapeptides with five positive charges, of the influence of lipophilicity (of the lipophilic ‘face’ of the molecule) on microbiological activity (including against strains with reduced susceptibility to polymyxins), potentiation of other antibiotics, and in vitro toxicity. We expect the learning of the underlying trends to be applicable to other polymyxin derivatives.
Results and Discussion:
In order to investigate systematically the influence of lipophilicity on the biological properties of a range of polymyxin derivatives all possessing five positive charges, we synthesised a series of polymyxin nonapeptides acylated at the N-terminus by 4-aminobutyric acid, which was substituted at either the 2- or 3-position by a variety of alkyl, cycloalkyl or aryl moieties (Fig. 1). In order to achieve a broad range of lipophilicities, the side chains of amino acids 6 and 7 in the heptapeptide ring were also varied.
Two main routes were utilised to prepare this series of compounds; semi-synthesis from the corresponding polymyxin nonapeptide or solid-phase peptide synthesis. Scheme 1 shows the general semi-synthetic route from polymyxin B (1) or colistin (2) via the corresponding heptapeptide core (I)8. Where the amino acid at position 6 (polymyxin numbering, Figure 1) corresponded to cyclohexylalanine the nonapeptide (IIc) was prepared by reduction of the phenylalanine residue, while biphenylalanine-containing nonapeptide (IId) was prepared from Polymyxin B via bromination and subsequent Suzuki coupling using our recently-published procedures20.
Scheme 1.
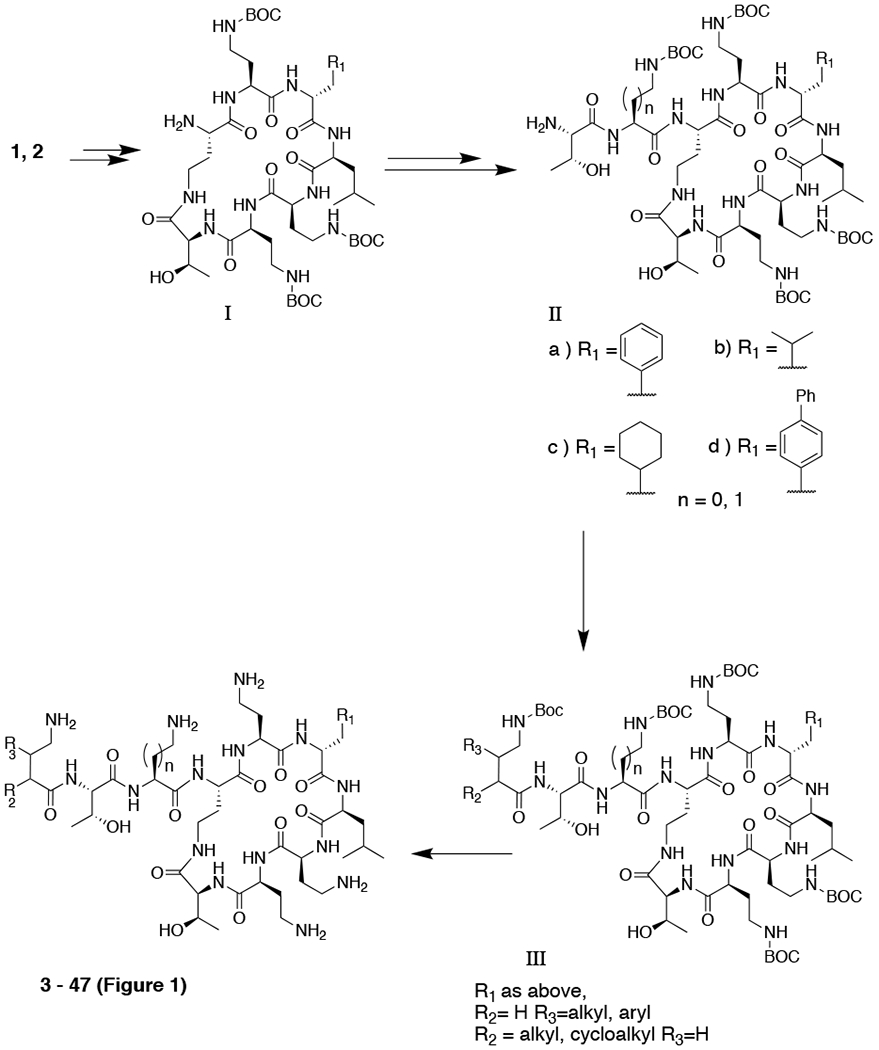
Semi-synthesis of Polymyxin Derivatives
Solid-phase peptide synthesis, utilised for compounds in which non-PMB or PME amino acids were present at positions 6 or 7 (Figure 1), used FMOC chemistry with CBZ protection of the side-chain amines, as described by Vaara et al21. Compounds 8, 16, 23-26 and 30 have already been reported by Brown et al8, 20.
In considering how best to compare the relative lipophilicity of the novel compounds, conventional measures of lipophilicity estimation, for example reverse-phase high-performance liquid-chromatography (RP-HPLC) at physiological pH of 7.4, were not appropriate due to the highly basic nature of the compounds for which highly acidic RP-HPLC conditions are required (see Methods). Likewise, to avoid any issues with calculation of the distribution coefficient, LogD7.4 due to the presence of five amine groups, and since all derivatives had the same number of charges, we chose to calculate logP prior to synthesis as an estimate of the relative lipophilicity of the derivatives. The calculated octanol-water partition coefficient AlogP22, was utilized (accessed through Biovia Insight for Excel 201723) and, for the compounds described, gave a good correlation with RP-HPLC retention time as an indicator of relative lipophilicity (details tabulated in Supplementary data).
Compounds were tested as single diastereomers as depicted in Figure 1 which corresponds to the diastereomer previously shown to be associated with improved activity and/or lower cytotoxicity compared with the other isomer8. All compounds were at least 90% pure by 1HNMR and HPLC, except for the following: 5 (84%), 29 (86%), 38 (85%), 44 (85%), 45 (84%), 46 (84%), 47 (85%).
Dependence of activity against polymyxin-susceptible strains on overall lipophilicity
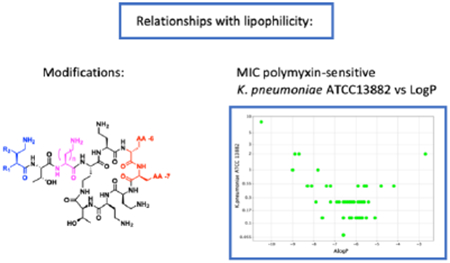
The left-hand columns of Table 1 show the activity of compounds 3-47, PMB and colistin, against polymyxin-susceptible strains of Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii. The analogues 3 – 47 are ordered according to their lipophilicity. Close inspection shows a V-shaped relationship between the MIC values and ALogP around an optimum region which is depicted for selected bacterial strains in Figure 2. It is possible that the increase in MIC values at high lipophilicity is at least partially due to binding of these compounds to plastic surfaces (and therefore less free compound available to exert microbiological activity) as has been previously described for polymyxins24, 25 and would expect to be exacerbated with more lipophilic derivatives. Indeed, a comparison between MIC determinations carried out in different microtiter plate types supports this suggestion (supplementary data, Table S1).
Table 1.
Antibacterial activity (MIC, μg/ml) of Polymyxin B (1), Colistin (2) and aminoacyl nonapeptides 3-47 versus polymyxin-susceptible and polymyxin-resistant strains; cytotoxicity against the HK-2 cell line (IC50 expressed relative to polymyxin B). Compounds 3-47 are shown in descending order of lipophilicity (ALogP)
| compound | AlogP | E.coli ATCC 25922 | K.pneumonia ATCC 13882 | P.aeruginosa ATCC 27853 | A.baumannii NCTC 13301 | A.baumannii BAA-747 | E.coli IHMA940398 | E. coli IHMA558090 | K. pneumoniae IHMA580884 | K.pneumoniae IHMA520329 | K.pneumoniae IHMA652780 | P.aeruginosa IHMA644636 | P.aeruginosa IHMA517175 | A.baumannii IHMA 517303 | A.baumannii IHMA 851735 | IC50 rel to PMB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (PMB) | −6.3 | 0.25 | 0.25 | 0.5 | 0.25 | 0.25 | 16 | 4 | 8 | 64 | 4 | 32 | 8 | >64 | 4 | 1 |
| 2 (Colistin) | −6.6 | 0.25 | 0.25 | 1 | 0.5 | 0.25 | 16 | 4 | 16 | >64 | ND | 32 | 64 | >64 | ND | 2.6 |
| 3 | −2.7 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 1 | 2 | 2 | 2 | ND | 0.2 |
| 4 | −4.2 | 0.5 | 0.5 | 1 | 0.25 | 0.5 | 4 | 1 | 0.5 | 2 | 0.5 | 2 | 2 | 8 | ND | ND |
| 5 | −4.8 | ND | 0.5 | 0.5 | 0.25 | 0.5 | 4 | 1 | 4 | 8 | ND | 1 | 1 | 8 | ND | ND |
| 6 | −5.1 | 0.125 | 0.125 | 0.25 | 0.25 | 0.125 | 8 | 2 | 2 | 8 | 0.5 | 8 | 4 | 64 | ND | 1.3 |
| 7 | −5.4 | 0.06 | 0.25 | 0.25 | 0.03 | 0.25 | 16 | 4 | 2 | 16 | 1 | 8 | 4 | >64 | ND | 1.1 |
| 8 | −5.5 | 0.06 | 0.125 | 0.125 | 0.06 | 0.125 | 16 | 2 | 1 | 4 | 1 | 32 | 8 | >64 | ND | 1.1 |
| 9 | −5.5 | 0.125 | 0.5 | 0.5 | 0.125 | 0.125 | 16 | 4 | 2 | 8 | 0.5 | 4 | 2 | 64 | ND | 4.1 |
| 10 | −5.6 | 0.125 | 0.5 | 0.25 | 0.25 | 0.125 | 32 | 8 | 8 | >64 | 8 | 8 | 2 | >64 | ND | 2.1 |
| 11 | −5.6 | 0.125 | 0.25 | 0.125 | ND | 0.06 | 8 | 1 | 2 | 32 | 1 | 8 | 4 | 64 | ND | 4.8 |
| 12 | −5.7 | 0.25 | 0.25 | 0.25 | ND | 0.125 | 8 | 1 | 2 | 16 | 0.5 | 2 | 2 | 64 | ND | ND |
| 13 | −5.7 | 0.125 | 0.125 | 0.06 | 0.06 | 0.06 | 8 | 2 | 8 | >64 | 8 | 8 | 4 | ND | ND | 6.0 |
| 14 | −5.8 | 0.125 | 0.25 | 0.25 | 0.06 | 0.06 | 16 | 8 | 8 | 32 | 2 | 8 | 4 | >64 | ND | 2.0 |
| 15 | −5.8 | 0.125 | 0.125 | 0.5 | 0.125 | 0.06 | 8 | 4 | 4 | 16 | 1.5 | 4 | 4 | >32 | ND | ND |
| 16 | −5.8 | 0.125 | 0.125 | 0.25 | ND | 0.125 | 32 | 8 | 16 | >64 | 4 | 32 | 8 | >64 | ND | 2.4 |
| 17 | −5.9 | 0.125 | 0.125 | 0.06 | ND | 0.03 | ND | 2 | 4 | ND | ND | ND | ND | ND | ND | 11.3 |
| 18 | −5.9 | 0.125 | 0.25 | 0.25 | 0.25 | 0.03 | ND | 2 | ND | ND | 2 | 8 | 4 | 32 | ND | 3.2 |
| 19 | −5.9 | 0.5 | 0.5 | 0.25 | ND | 0.5 | 8 | 4 | 16 | >64 | ND | 32 | 8 | >64 | ND | 5.2 |
| 20 | −6.0 | 0.125 | 0.125 | 0.06 | ND | 0.03 | ND | 1 | 4 | ND | ND | ND | ND | ND | ND | 6.8 |
| 21 | −6.1 | 0.125 | 0.25 | 0.25 | ND | 0.125 | 32 | 8 | 8 | >64 | 4 | 16 | 8 | 32 | ND | 3.2 |
| 22 | −6.1 | 0.25 | 0.25 | ND | ND | 0.25 | ND | 16 | ND | ND | ND | ND | ND | ND | ND | ND |
| 23 | −6.2 | 0.06 | 0.125 | 0.125 | 0.125 | 0.06 | 16 | 4 | 4 | 32 | 1 | 16 | 8 | >64 | ND | 3.8 |
| 24 | −6.2 | 0.125 | 0.25 | 0.125 | 0.06 | 0.125 | 16 | 4 | 16 | 32 | 4 | 16 | 8 | 64 | 0.5 | 5.9 |
| 25 | −6.3 | 0.06 | 0.125 | 0.06 | 0.06 | 0.125 | 16 | 4 | 16 | >64 | 4 | 16 | 8 | >64 | ND | 6.9 |
| 26 | −6.3 | 0.125 | 0.125 | 0.25 | 0.06 | 0.125 | 32 | 8 | 16 | 64 | ND | 32 | 8 | >64 | ND | 11.6 |
| 27 | −6.3 | 0.06 | 0.125 | 0.125 | ND | 0.03 | 8 | 4 | 8 | >64 | ND | 64 | 16 | >64 | ND | 18.0 |
| 28 | −6.3 | 0.25 | 0.25 | 0.06 | ND | 0.06 | ND | 16 | 32 | ND | ND | ND | ND | ND | ND | ND |
| 29 | −6.4 | 0.25 | 0.25 | 0.125 | ND | 0.25 | ND | 16 | ND | ND | 16 | ND | ND | ND | ND | ND |
| 30 | −6.5 | 0.25 | 0.25 | 0.125 | 0.125 | 0.25 | 32 | 8 | 64 | >64 | ND | 64 | 8 | >64 | ND | 12.0 |
| 31 | −6.6 | 0.25 | 0.125 | 0.125 | ND | 0.06 | ND | 16 | >64 | ND | ND | ND | ND | ND | ND | ND |
| 32 | −6.6 | 0.125 | 0.06 | 0.06 | 0.25 | 0.03 | 32 | 8 | 32 | >64 | ND | >64 | 64 | >64 | ND | 65.3 |
| 33 | −6.6 | 0.125 | 0.125 | 0.125 | ND | 0.03 | ND | 8 | 16 | ND | ND | ND | ND | ND | ND | ND |
| 34 | −6.8 | 0.125 | 0.25 | 0.06 | 0.06 | 0.06 | 32 | 8 | 16 | >64 | ND | >64 | 64 | >64 | ND | >68 |
| 35 | −6.9 | 0.125 | 0.25 | 0.25 | 0.06 | 0.06 | 32 | 16 | 32 | >64 | ND | >64 | ND | >64 | 0.5 | 12.5 |
| 36 | −7.2 | 1 | 0.5 | 0.5 | 0.5 | 0.5 | 64 | 64 | >64 | 64 | ND | >64 | ND | >64 | >8 | >65.3 |
| 37 | −7.2 | 0.125 | 0.25 | 0.25 | 0.125 | 0.125 | 64 | 32 | 32 | >64 | ND | >64 | ND | >64 | 1 | 20 |
| 38 | −7.4 | 1 | 0.5 | 1 | 1 | ND | >64 | ND | >64 | >64 | ND | >64 | 64 | >64 | ND | ND |
| 39 | −7.6 | 0.25 | 0.125 | 0.06 | ND | 0.06 | ND | 32 | ND | >64 | ND | ND | ND | ND | ND | ND |
| 40 | −7.8 | 0.25 | 1 | 0.25 | 0.25 | ND | >64 | ND | ND | >64 | ND | >64 | >64 | >64 | 2 | >68.4 |
| 41 | −7.9 | ≤0.125 | 0.25 | ≤0.125 | ≤0.125 | 0.125 | 64 | 16 | 32 | >64 | ND | >64 | ND | >64 | 2 | ND |
| 42 | −8.1 | 0.5 | 0.5 | 0.5 | 1 | ND | >64 | ND | ND | 16 | ND | >64 | 2 | >64 | >8 | ND |
| 43 | −8.3 | 0.25 | 0.5 | 0.25 | 2 | ND | >64 | ND | ND | >64 | ND | >64 | >64 | >64 | >8 | ND |
| 44 | −8.7 | 0.25 | 2 | 1 | 4 | ND | >64 | ND | ND | >64 | ND | >64 | >64 | >64 | >64 | ND |
| 45 | −8.9 | 1 | 2 | 0.5 | 2 | ND | >64 | ND | ND | >64 | ND | >64 | >64 | >64 | 8 | >68.4 |
| 46 | −9.0 | 1 | 1 | 0.5 | 4 | ND | >64 | ND | ND | >64 | ND | >64 | >64 | >64 | >8 | ND |
| 47 | −10.5 | 4 | >8 | 1 | >8 | ND | >64 | ND | ND | >64 | ND | >64 | >64 | >64 | >8 | ND |
Figure 2.
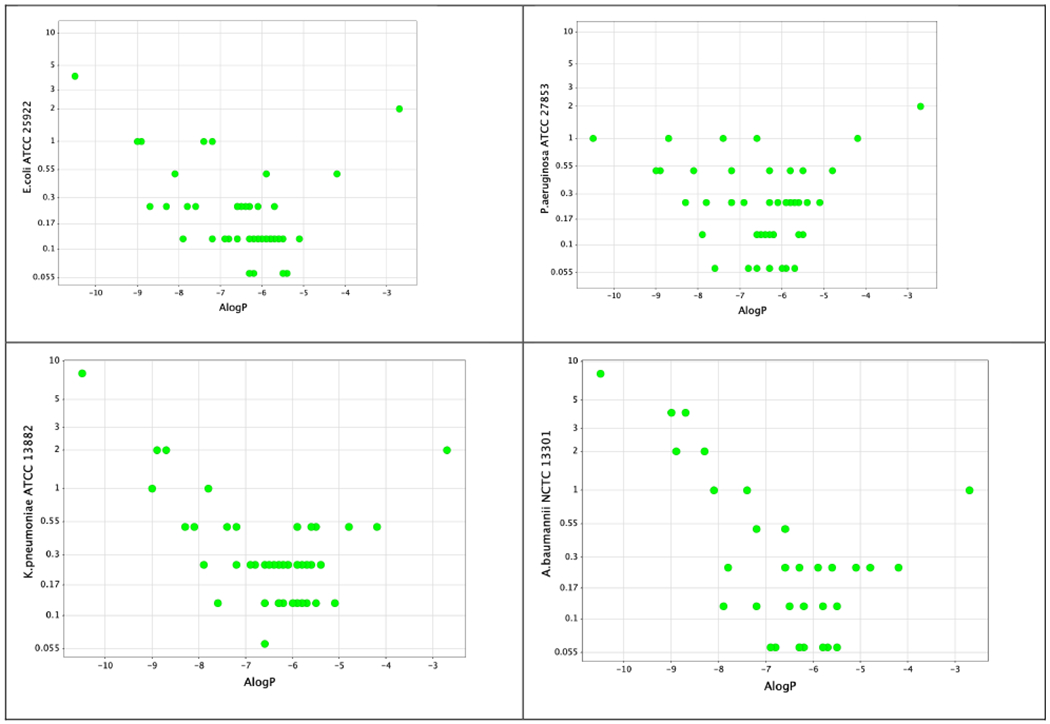
MIC versus ALogP for compounds 1-47 against polymyxin-susceptible strains of E. coli, K. pneumoniae, P. aeruginosa and A. baumannii
Alpha and beta branched N-terminal groups on the same nonapeptide scaffold and having the same N-terminal alkyl group showed equivalent activity, for example compounds 23 (cyclohexyl alpha branched) and 25 (cyclohexyl beta branched). Further, the exact structure of the N-terminal alkyl group did not seem important for activity as different moieties of the same lipophilicity on the same nonapeptide scaffold showed equal potency, for example compounds 24 (n-pentyl), 25 (cyclohexyl) and 26 (3-chlorophenyl).
The overall dominant effect of lipophilicity on microbiological activity was further demonstrated in compounds with the same lipophilicity showing equivalent potency regardless of whether the lipophilicity resided primarily at the N-terminus or in the core cyclic peptide. For example, compound 15 with a short α-n-butylaminobutyrate at the N-terminus, cyclohexylalanine at AA-6 and leucine at AA-7, had very similar activity to compound 17 with the more lipophilic β-n-octylaminobutyrate at the N-terminus but a less lipophilic cyclic core comprising leucine at AA-6 and aminobutyrate at AA-7.
There was, however, a drop-off in microbiological activity at the lower end of the lipophilicity scale irrespective of whether the reduction in lipophilicity had occurred predominantly at the N-terminus or at amino acids 6 or 7. Interestingly, the decline in activity with decreasing lipophilicity occurred earlier with A. baumannii (ALogP around −8) than with the other strains (ALogP around −8.5). Notably, even compounds without a lipophilic chain at the N-terminus (eg. 44, N-terminal aminobutyryl colistin nonapeptide) retained some antimicrobial activity.
Dependence of activity against strains with reduced susceptibility to polymyxins on overall lipophilicity
Table 1 also shows the antimicrobial activity of the same series of compounds against strains that exhibit reduced susceptibility to polymyxins. As expected, analogues with higher degrees of lipophilicity had superior activity against these strains, as also illustrated for selected strains in Figure 3.
Figure 3.
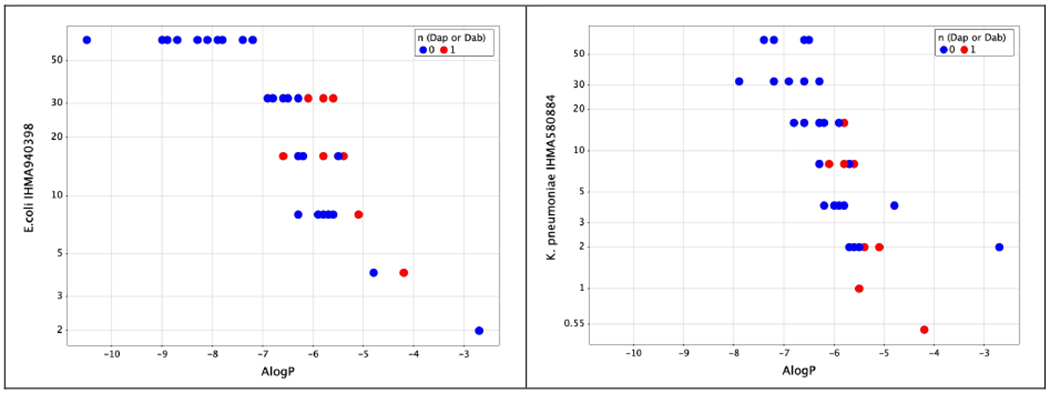
MIC versus ALogP for compounds 1-47 against selected strains of E.coli and K.pneumoniae with reduced susceptibility to polymyxins, coloured on Dap or Dab residue adjacent to the heptapeptide core.
Compounds such as 3 had essentially equivalent activity against polymyxin-susceptible strains and strains with reduced susceptibility. This profile has also been observed with a range of lipophilic lipopeptides reported by Velkov et al.10 and with natural lipophilic lipopeptides such as the octapeptins19.
Compound 3 was examined for efficacy against A. baumannii NCTC13301 in a neutropenic mouse thigh efficacy model but exhibited no significant effect at a dose of 20mg/kg, while PMB produced a greater than 2 log10 drop in colony forming units (CFU) from the pre-treatment level under the same conditions (supplementary data; Figure S1). This disparity in in vivo effect is not reflected in in vitro activity (MIC 1 μg/mL for compound 3 vs 0.25 μg/mL for PMB) and is instead very likely due to the high protein binding of compound 3 (>99% by equilibrium dialysis).
By analogy with the compounds reported by Velkov et al10, compound 3 has a highly lipophilic moiety at the 6-position (biphenylglycine). However, this does not appear to be an essential feature for microbiological activity as derivatives with high lipophilicity positioned instead at the N-terminal alkyl chain (for example compounds 4 and 5) also exhibit potent activity against strains with reduced susceptibility to polymyxins.
As well as lipophilicity, a diaminopropionate group (Dap, Figure 1 , n=0) as opposed to diaminobutyrate group (Dab, Figure 1, n=1) at the position adjacent to the cyclic core, was a determinant of microbiological activity against less susceptible strains, as previously noted by Magee et al.26 and in our own work8. Figure 3 shows that, generally Dap-3 containing compounds exhibit better activity than Dab-3 variants for a given AlogP value. Nevertheless, activity against resistant strains fell away markedly at lower ALogP and for compounds with an ALogP value below −7 the majority of less susceptible strains were profoundly resistant (MIC > 64μg/ml).
The earlier observation that the lipophilicity of the N-terminus and of the core of the molecule can be “balanced” against each other also appeared to hold for activity against polymyxin resistant strains. For example, in the alpha substituted amino butyrates (Figure 1) compound 9 with the cyclohexyl N-terminus and cyclohexylalanine at position 6 exhibited the same microbiological activity as compound 11 with a more lipophilic cyclohexylethyl N-terminus combined with a less lipophilic leucine at position 6. Similarly, in the beta substituted amino butyrate series, the cyclohexyl compound 25 on the Phe-6, Leu-7 scaffold had similar activity to the n-heptyl 27 on the Leu-6 Abu-7 scaffold.
Effect of lipophilicity on the ability of polymyxin analogues to potentiate rifampicin
Rifampicin is a highly lipophilic macrocyclic antibiotic which acts as an inhibitor of bacterial DNA-directed RNA polymerase (RNAP), located in the cytoplasm of the bacterial cell. Rifampicin shows potent enzyme inhibition of RNAP from both Gram positive and Gram negative pathogens27 and demonstrates excellent whole-cell activity against Gram positive strains. Activity is weaker against Gram negative strains however, which is thought to be as a result of poor penetration into the bacterial cell. While rifampicin alone exhibits generally poor activity against Gram negative strains, in the presence of PMB (or the inactive PMBN, data not shown) excellent activity is obtained, even against strains with reduced susceptibility to the polymyxin alone. (Table 2A).
Table 2A.
Antibacterial activity of rifampicin, PMB alone and PMB in the presence of 1μg/ml Rifampicin versus polymyxin-susceptible and polymyxin-resistant strains
| compound | AlogP | E.coli ATTC 25922 | K.pneumoniae ATCC 13882 | P.aeruginosa ATCC 27853 | A.baumannii NCTC 13301 | E.coli IHMA940398 | E.coli CDF1 | K.pneumoniae IHMA580884 | K.pneumoniae IHMA520329 | P.aeruginosa IHMA644636 | P.aeruginosa IHMA517175 | A.baumannii IHMA517303 | A.baumanni IHMA 851735 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rifampicin | N/A | 4 | 16 | 16 | 4 | 8 | 8 | 16 | 32 | 16 | 16 | 2 | 4 |
| PMB (1) | −6.3 | 0.25 | 0.125 | 0.25 | 0.125 | 16 | 4 | 8 | 64 | 64 | 8 | >64 | 4 |
| PMB (1) plus 1μg/ml Rifampicin | −6.3 | ≤0.015 | 0.06 | 0.125 | ≤0.015 | 0.06 | 0.06 | 0.06 | 0.125 | 0.5 | 0.25 | 0.125 | ≤0.015 |
Table 2B shows the antimicrobial activity profile of selected derivatives (compounds 24, 26, 35-38 and 40-47) against both polymyxin-susceptible and polymyxin-resistant strains in the presence of a fixed sub-MIC concentration of rifampicin of 1 μg/ml. Remarkably, the ability to potentiate the activity of rifampicin against polymyxin-susceptible strains was maintained even by compound 47, the least lipophilic derivative made for this study.
Table 2B.
Antimicrobial activity of selected derivatives in the presence of rifampicin (1μg/ml) versus polymyxin-susceptible and polymyxin-resistant strains
| compound | AlogP | E.coli ATCC 25922 | K.pneumonia ATCC 13882 | P.aeruginosa ATCC 27853 | A.baumannii NCTC 13301 | E.coli IHMA940398 | E.coli CDF1 | K.pneumoniae IHMA580884 | K.pneumoniae IHMA520329 | P.aeruginosa IHMA644636 | P.aeruginosa IHMA517175 | A.baumannii IHMA517303 | A.baumannii IHMA 851735 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 | −6.2 | ≤0.015 | 0.03 | 0.06 | ≤0.015 | 0.03 | 0.06 | 0.06 | 0.06 | 0.5 | 0.25 | ≤0.015 | ≤0.015 |
| 26 | −6.3 | ≤0.015 | 0.06 | 0.125 | ≤0.015 | 0.125 | ND | ND | 0.06 | 1 | ND | 0.03 | ND |
| 35 | −6.9 | ≤0.015 | 0.06 | 0.125 | ≤0.015 | 0.125 | 0.125 | 0.06 | 0.06 | 2 | 0.5 | 0.06 | 0.03 |
| 36 | −7.2 | ≤0.015 | 0.06 | 0.25 | ≤0.015 | 0.25 | 0.25 | 0.25 | 0.125 | 2 | 1 | 0.25 | 0.06 |
| 37 | −7.2 | ≤0.015 | 0.06 | 0.25 | ≤0.015 | 0.125 | 0.125 | 0.125 | 0.03 | 4 | 1 | 0.125 | 0.03 |
| 38 | −7.4 | 0.03 | 0.125 | 0.5 | ≤0.015 | 1 | 1 | 0.5 | 0.125 | 4 | 2 | 0.5 | 0.125 |
| 40 | −7.8 | ≤0.015 | 0.125 | 0.25 | ≤0.015 | 1 | 1 | 0.5 | 0.06 | >8 | 8 | 1 | 0.125 |
| 41 | −7.9 | ≤0.015 | 0.25 | 0.125 | ≤0.015 | 0.25 | 0.25 | 0.25 | 0.125 | 4 | 1 | 0.25 | 0.03 |
| 42 | −8.1 | ≤0.015 | 0.06 | 0.5 | 0.03 | 4 | 2 | ND | 0.125 | >8 | 8 | 2 | 0.5 |
| 43 | −8.3 | ≤0.015 | 0.06 | 0.25 | ≤0.015 | 4 | 2 | ND | 0.06 | >8 | 8 | 2 | 0.5 |
| 44 | −8.7 | 0.03 | 0.125 | 1 | 0.03 | 8 | 4 | 4 | 0.25 | >8 | 8 | 2 | 0.5 |
| 45 | −8.9 | 0.06 | 0.125 | 0.5 | 0.06 | 1 | 2 | 1 | 0.5 | 8 | 8 | 2 | 0.5 |
| 46 | −9.0 | ≤0.015 | 0.06 | 0.5 | 0.03 | 8 | 4 | 4 | 0.125 | >8 | >8 | 4 | 2 |
| 47 | −10.5 | 0.06 | 0.25 | 1 | 0.125 | >8 | >8 | >8 | 0.5 | >8 | >8 | >8 | 8 |
Generally, the ability of these molecules to potentiate the activity of rifampicin decreased with decreasing ALogP, but with different ‘cut-offs’ for different strains. Surprisingly, this was not primarily driven by the degree of resistance to polymyxins as reflected by the MIC for PMB, but was strain dependent. For instance, the ability of our novel polymyxin analogues in combination with rifampicin to achieve an MIC ≦ 2μg/ml against both polymyxin-resistant E. coli strains assessed, was maintained until an ALogP value of between −8 and −9. Surprisingly, potentiation was maintained for molecules of all ALogP against the profoundly resistant K. pneumoniae IHMA520329 (PMB MIC = 64μg/ml), but only down to around ALogP −8 for the moderately resistant K.pneumoniae IHMA580884 (PMB MIC = 8μg/ml). Potentiation of rifampicin activity against polymyxin-resistant P. aeruginosa was least tolerant of reduced lipophilicity, ‘failing’ at an ALogP value of around −7.2 to −7.4. As with E. coli, the polymyxin-resistant A. baumannii strains assessed were susceptible to potentiation of rifampicin by molecules with an ALogP value down to around −9. It should be noted, however, that these strains were relatively susceptible to rifampicin alone.
Effect of lipophilicity on in vitro cytotoxicity of polymyxin analogues
Table 1 also shows the cytotoxicity of these novel compounds against a human renal proximal tubule epithelial cell line (HK-2) expressed relative to the IC50 value for polymyxin B. In general, there was a clear trend towards reduced cytotoxicity with less lipophilic compounds, which is illustrated in Figure 4.
Figure 4.
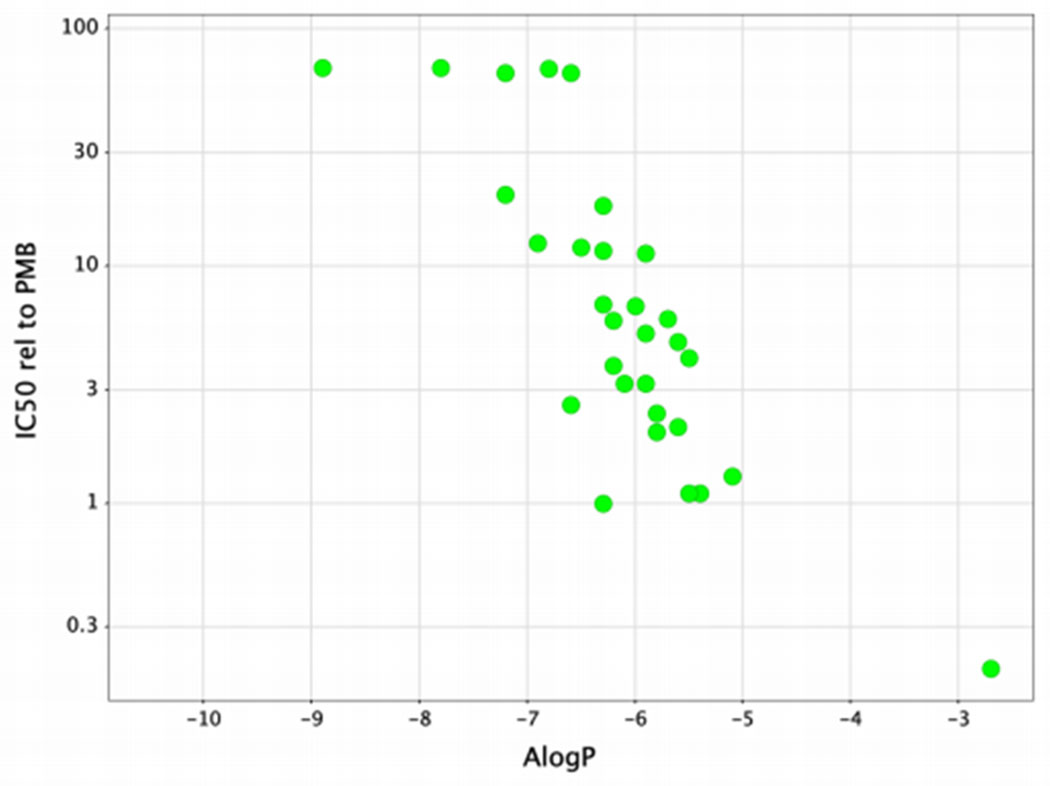
Cytotoxicity versus ALogP for compounds 1-47
Effect of a less lipophilic scaffold on in vivo toxicity.
As we have reported previously8, the in vivo toxicity of polymyxins is not solely related to the measured cytotoxicity, but also to the propensity of the compound to accumulate in kidney tissue. Early members of our nonapeptide series, with an N-terminal heterocycle showed high kidney accumulation, which led to high in vivo nephrotoxicity despite lower in vitro cytotoxicity than PMB (data not shown). Compounds with a branched amino butyrate, in particular, the beta branched compounds of general structure B, (Figure 1) showed more moderate kidney exposure. Of these, compound 26 (SPR206), which has an 11.6-fold improvement in cytotoxicity and an AUC of 850 μg.h/g in renal tissue during the period 4 -16 hr after a 17.2 mg/kg subcutaneous dose in mouse, similar to that of 688 μg.h/g for PMB, has already been shown to have significantly reduced renal toxicity compared to PMB in both mouse and Cynomolgus monkey and is currently undergoing clinical evaluation8.
We decided to evaluate the renal toxicity of compound 34 as a representative of molecules of this structural type but with less lipophilicity in the cyclic peptide core. Compound 34 has a >68-fold improvement in cytotoxicity relative to PMB. However, after a single 17.2mg/kg subcutaneous dose in mouse, compound 34 has an AUC from 4 – 16 hr of 1,545 μg.h/g ie more than twice that of PMB under similar conditions. In repeat dose evaluations, compound 34 was administered to mice in 4 doses over 24 hr or 12 doses over 4 days (three times per day) after which urine was collected for 24 hr for biomarker analysis and mice were euthanised for investigation of kidney histopathology. Compound 34 was administered at doses of 25, 50 and 75 mg/kg, whereas PMB as a comparator was administered at doses of 12.5 and 25 mg/kg (25 mg/kg is the maximum PMB dose tolerated in this regimen). Polymyxin B showed significant histopathology changes in the kidneys at the 25 mg/kg dose level (plasma AUCinf 57 μg.h/ml), after both the 24 hr and 4 day studies. In contrast, compound 34 showed no histopathological lesions at the same dose (25 mg/kg) and only minimal lesions at 50 mg/kg (plasma AUCinf 113 μg.h/ml). Histopathology at 75 mg/kg (plasma AUCinf 215 μg.h/ml) was similar to PMB at 25 mg/kg (Table 3). Urinary biomarker levels aligned well with histopathology and were suggestive of similar renal toxicity for compound 34 to PMB at >4-fold plasma exposure (Table S2). The marked reduction in intrinsic cytotoxicity of Compound 34 relative to PMB (>68-fold lower IC50 against HK-2) appears therefore to compensate for the higher drug levels of compound 34 in the kidney achieved at equivalent doses to PMB.
Table 3.
Renal toxicity of Compound 34 relative to PMB in a mouse model
| Parameter | Dose# (mg/kg) | Duration | Histopathology score – number of animals | |||
|---|---|---|---|---|---|---|
| Normal | Minimal | Mild | Moderate | |||
| Vehicle | N/A | 24hr | 5 | 0 | 0 | 0 |
| 4 days | 5 | 0 | 0 | 0 | ||
| PMB | 12.5 | 24hr | 5 | 0 | 0 | 0 |
| 4 days | 4 | 1 | 0 | 0 | ||
| PMB | 25 | 24hr | 0 | 2 | 2 | 1* |
| 4 days | 0 | 2 | 3 | 0 | ||
| Compound 34 | 25 | 24hr | 5 | 0 | 0 | 0 |
| 4 days | 5 | 0 | 0 | 0 | ||
| Compound 34 | 50 | 24hr | 2 | 3 | 0 | 0 |
| 4 days | 1 | 4 | 0 | 0 | ||
| Compound 34 | 75 | 24hr | 0 | 3 | 2 | 0 |
| 4 days | 0 | 3 | 1 | 1* | ||
dose expressed as mg free base/kg
deceased
Conclusions
Whilst there have been many investigations into the SAR of polymyxin analogues and the influence of lipophilicity on the biological properties of such molecules, there has not been a systematic investigation within a consistent series of analogues. Further, interpretation has often been made in the context of changes to individual moieties in the absence of a consideration of the overall physicochemical properties of the molecule.
For instance, it is reported that the shortening of the N-terminal moiety below a certain length leads to molecules with much reduced activity but which retain the ability to potentiate other antibiotics9,26. However, in our series of aminoacyl nonapeptides we have shown that short N-terminal moieties can support potent activity providing the overall lipophilicity of the molecule is within an optimal range.
The interchangeability of lipophilicity at the N-terminus and amino acids 6 and 7 in the cyclic peptide core is an important medicinal chemistry principle that should enable the design and development of improved polymyxins. We consider that these three moieties act in concert to interact with the lipophilic region of the outer membrane and hence the lipophilicity may be distributed between these three moieties in different ways. Likewise, the cytotoxicity of the polymyxins appears to be primarily driven by overall lipophilicity rather than by the contribution of any individual moiety.
Our work also suggests that the characterization of a polymyxin molecule as either a direct acting compound or a potentiator is more of a continuum, that is strongly influenced by the LogP, rather than as a result of fundamentally different modes-of-action. It appears that potentiation may require less penetration into the lipophilic region of the outer membrane than is required for direct activity and that again this is reflected in the lipophilicity of amino acids 6,7 and the N-terminal chain in total rather than any individual moiety.
As regards the results with less-susceptible strains, the derivatives studied here all contain five positive charges, and are thus able to disrupt the charge-interactions around the phosphate groups of LPS. We suggest that, due to modification of the phosphate groups in the LPS of less susceptible strains18, hydrophobic interactions are a more important component of binding to these strains. Thus, compounds with the greatest lipophilicity in this region (amino acid 6, 7 and N-terminus) show the greatest activity against strains less susceptible to polymyxins. Again less lipophilicity appears to be needed for potentiation as described above.
We show therefore that as lipophilicity decreases, there is a gradual impact on antimicrobial properties affecting first the direct activity against less susceptible strains, followed by the ability to potentiate activity against these same strains, and eventually direct activity against susceptible strains. Even the least lipophilic derivatives we have made in this series (completely lacking a lipophilic N-terminus) retain some direct activity against susceptible strains and are highly potent potentiators of rifampicin.
Methods
Antimicrobial susceptibility testing
The in vitro antimicrobial activity (MIC) of compounds 1 – 47 was determined against Escherichia coli ATCC25922, CDF1 (mcr-1)29, IHMA558090 and IHMA940398 (last three have reduced susceptibility to polymyxins); Klebsiella pneumoniae ATCC13882, IHMA580884, IHMA520329 and IHMA652780 (last three have reduced susceptibility to polymyxins); Pseudomonas aeruginosa ATCC27853, IHMA517175 and IHMA644636 (last two have reduced susceptibility to polymyxins); and Acinetobacter baumanii ATCC BAA-747, IHMA517303 and IHMA851735 (last two have reduced susceptibility to polymyxins) by broth microdilution using cation-adjusted Mueller-Hinton broth (Oxoid, CM0405) according to CLSI guidelines30. Polypropylene microtiter plates were used as they have been shown to bind polymyxins less strongly than polystyrene plates24,25.
In vitro cytotoxicity
Mammalian cell toxicity was measured using confluent monolayers of the human HK-2 proximal tubule epithelial cell line. Compounds were incubated with cells for 24h at 37ºC in 5% CO2 using a top concentration of 1,000 or 3,000 μg/mL with semi-log dilutions to give a 9-point concentration range. Cell viability was measured using resazurin blue. Compound concentration values were plotted as log values to enable a dose-response curve to be fitted. The bottom of the curve was constrained to zero and IC50 values were determined using GraphPad Prism. The relative cytotoxicity is reported as the ratio of the IC50 of test compound to that of PMB in the same experiment (Horizon Discovery Ltd.).
Determination of kidney drug levels
Kidney drug levels were determined after dosing subcutaneously to mouse at 17.2 mg/kg as described in Brown et al.8
In vivo nephrotoxicity
Renal toxicity in male CD-1 mice (n = 5) was determined for Compound 34 (25, 50, 75 mg/kg/dose) in comparison with PMB (12.5, 25 mg/kg/dose) (Charles River Laboratories Inc.). Compounds were dosed subcutaneously three times per day (8 hr apart) for either 24 hr (4 doses) or 4 days (12 doses). Immediately after the last dose, animals were transferred to metabolic cages for collection of urine for 24hr after which animals were sacrificed for histopathology. Levels of urinary biomarkers, KIM-1, cystatin C, albumin, β2 microglobulin and NGAL, as well as creatinine, in the urine were measured by Charles River standard analytical methods. Biomarker levels were expressed relative to the urinary creatinine level (supplementary data). Experiments were conducted in accordance with The Guide for the Care and Use of Laboratory Animals and The Current International Council on Harmonisation (ICH) Harmonised Tripartite Guidelines and generally accepted procedures for the testing of pharmaceutical compounds.
Synthesis
The synthesis and characterisation of all non-commercially available carboxylic acids used in the preparation of the final compounds are given in the Supporting Information. Compounds 3-16, 18-19, 21-26, 29-10, 36, 38 and 44 were prepared according to the procedure described by Brown et al8. Compounds 17, 20, 27-28, 31-35, 37, 39-43, and 45-47 were prepared by solid phase peptide synthesis, exemplified below by the synthesis of compound 34. Characterisation of all other final compounds is given in the Supplementary Information.
Analytical HPLC was performed on all final compounds on an Agilent 1100 System with a Phenomenex Hyperclone C18 BDS 5 μm (4.6 mm x 150 mm) column, eluted with appropriate water/acetonitrile gradients containing 0.15% TFA, with detection at 210 and 254 nm. All reagents used for chemical synthesis were purchased from commercially available sources and used without further purification. Preparative HPLC was performed on a Gilson preparative HPLC system using a Waters Sunfire C18 OBD 5 μm (19 mm x 150 mm) column eluted with appropriate water/acetonitrile gradients containing 0.15% TFA, with detection at 210 nm. 1H NMR spectra were recorded at 400 MHz on a Mercury 400 NMR spectrometer (Agilent Technologies). 13C nmr were recorded at 100 MHz on a Varian INOVA NMR spectrometer . Chemicals shifts (δ) are reported in ppm downfield from TMS. Coupling constants J are recorded in Hertz (Hz). Mass spectra were recorded on an LCQ DecaXP mass spectrometer with +ve ion electrospray ionisation, and with Waters Xevo G2-S Tof.
Analytical HPLC conditions for Compound 34:
| Column: | Phenomenex Hyperclone C18 BDS 5 μm × 4.6 mm × 150 mm |
| Mobile phase: | A: water/acetonitrile 90/10, v/v, 0.15% TFA. |
| B: acetonitrile/water 90/10, v/v, 0.15% TFA | |
| Flow rate: | 1 mL/min |
| Gradient: | |
| Time (mins) | % mobile phase A |
| 0 | 100% |
| 20 | 40% |
| 21 | 0% |
| 23 | 0% |
| 23.5 | 100 |
| 25 | 100 |
| Detection: | 210, 254 nm |
| Injection volume: 20 μL | |
General method of Solid Phase peptide synthesis:
Synthesis of the protected linear peptide (residues 1-9 and N-terminal group) was carried out on an automated peptide synthesizer using standard Fmoc solid phase peptide chemistry. Specifically, synthesis was undertaken using Fmoc-Thr(tBu)-PEG-PS resin as starting material. Coupling of the Fmoc-amino acids with CBZ protection on the terminal amino groups was performed using 5 molar equivalents (relative to resin loading) of Fmoc amino acid and HATU in DMF with activation in situ, using 10 molar equivalents of DIPEA. Fmoc deprotection was performed using 20% piperidine in dimethylformamide. BOC was used as the orthogonal protecting group on the Dab involved in cyclisation.
The resin-bound linear peptide was treated with TFA/TIS/H2O (96/2/2v/v) for 2hrs to reveal the Dab residue involved in cyclisation, and to cleave the peptide from the resin. This material was cyclised using PyBop/HOBt/NMM (4/4/8 molar equivalents relative to the initial loading) in DMF for 3h. The crude material was partially evaporated, taken up acetonitrile/water and lyophilised overnight. The CBZ groups were then removed using 10% Pd/C in Acetic acid/MeOH/water (5/4/1 v/v).
General method of preparation of acetate salts:
AG1-X2 resin (Bio-Rad Laboratories Ltd) acetate form 200-400—mesh, was regenerated by washing with 10% aqueous acetic followed by 1% aqueous acetic acid, and placed in a fritted cartridge. A solution of the compound as a TFA salt in water was applied to the column, using a loading of 30 g resin to 1g TFA salt, and the column allowed to drip under gravity, eluting with water. Product-containing fractions were combined and lyophilised to a white solid.
[(3R)-3-(aminomethyl)nonanoyl]-Thr-Dap-Cyclo[Dab-Dab-DLeu-Abu-Dab-Dab-Thr] (34)
Solid phase peptide synthesis was carried out as described in the general method using 3-({[(benzyloxy)carbonyl]amino}methyl)nonanoic acid at the N-terminal. The crude deprotected product was then purified by preparative HPLC. Fractions containing the faster-eluting diastereomer were collected and lyophilised to afford the title compound as the TFA salt. The material was converted to the acetate salt as described in the general method, followed by lyophilisation to afford the title compound as the acetate salt as a white solid. 1H NMR (400 MHz, D2O): δ (ppm) 0.77 – 0.88 (12H, m), 1.11 – 1.38 (16H, m), 1.50 – 1.68 (4H, m), 1.78 – 2.28 (25H, m, includes 1.85, s, OAc), 2.44 (2H, d, J 6.6 Hz), 2.90-3.13 (9H, m), 3.21-3.33 (2H, m), 3.42 (1 H, dd, J 4.6, 13.4 Hz), 4.14 (1H, d, J 4.6 Hz), 4.16-4.27(7H, m), 4.35 (1H, d, J 4.1 Hz), 4.47 (1H, dd, J 5.0, 9.3 Hz). 13C NMR (101 MHz, D2O): δ(ppm) 181.20, 175.23, 174.89, 174.38, 173.27, 172.90, 172.53, 172.09, 171.40, 170.15, 66.94, 66.33, 59.47, 59.15, 55.01, 52.86, 52.69, 51.88, 51.73, 50.97, 50.77, 42.77, 39.84, 39.62, 37.53, 36.54, 36.38, 36.17, 36.04, 33.50, 30.94, 30.64, 30.45, 29.64, 28.42, 28.26, 27.88, 25.40, 24.37, 23.92, 23.18, 21.93, 20.69, 19.12, 18.75, 13.38, 9.76. m/z (+ve ESI) 529 [M+2H]2+, 100%. HRMS (Tof) 1056.7014 C47H89N15O12 require 1056.6887. HPLC retention time: (using analytical HPLC conditions shown above) 7.8 min.
34. Alternative synthesis:
The synthetic method was carried out as described above, using the optically pure (3R)-3-({[(benzyloxy)carbonyl]amino}methyl)nonanoic acid. m/z (+ve ESI) 529 [M+2H]2+ . NMR and HPLC retention time identical to an authentic sample prepared above.
Supplementary Material
Acknowledgments
We thank Mr S. Sandhu, Alta Bioscience, Redditch, UK for solid-phase peptide synthesis. Chiral separations were carried out at Reach Separations Ltd, Nottingham, UK, and at WuXi Apptec (Shanghai) Ltd, P.R.China. Chiral synthesis and scale-up chemistry was carried out at Avista, Durham, USA, and Asymchem, P.R. China. In-life pharmacokinetics were carried out at Pharmidex Pharmaceutical Services, Ltd, UK. In vivo efficacy experiments were performed at Evotec (UK) Ltd, Alderley Park, Macclesfield, Cheshire, UK. Cytotoxicity assays were performed at Horizon Discovery Ltd, Cambridge, UK. Renal toxicity studies were carried out at MPI Research, Mattawan, USA. Bacterial strains with reduced susceptibility to Polymyxin B were obtained from IHMA Europe Sàrl, Monthey, Switzerland.
Funding
This work was partially supported by grants awarded to Cantab Anti-infectives Ltd by Innovate UK under the Biomedical Catalyst scheme, award references 101357 and 102866. This work has been funded in part with US Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract HHSN272201500014C.
Footnotes
Conflict of Interest Statement:
The authors declare the following competing financial interest(s): P.B. and M.J.D. are consultants to Spero Therapeutics.
Syntheses of carboxylic acids, assignment of stereochemistry of beta branched series, effect of microtiter plate on MIC values, efficacy of compound 34 in neutropenic mouse thigh model, biomarker data for mouse renal toxicity study, and RP-HPLC retention times and characterisation of all tested compounds.
References
- (1).Michalopoulos AS, Tsiodras S, Rellos K, Mentzelopoulos S, Falagas ME (2005) Colistin treatment in patients with ICU-acquired infections caused by multiresistant Gram-negative bacteria: the renaissance of an old antibiotic. Clin. Microbiol. Infect 11 (2), 115–121. [DOI] [PubMed] [Google Scholar]
- (2).Nation RL, Li J, Cars O, Couet W, Dudley MN, Kaye KS, Mouton JW, Paterson DL, Tam VH, Theuretzbacher U, Tsuji BT, Turnidge JD (2015) Framework for optimisation of the clinical use of colistin and polymyxin B: the Prato polymyxin consensus. Lancet Infect. Dis 15 (2), 225–234. [DOI] [PubMed] [Google Scholar]
- (3).Karaiskos I, Lagou S, Konstantinos P, Rapti X, Poulakou G (2019) The “Old” and the “New” Antibiotics for MDR Gram-Negative Pathogens: For Whom, When, and How. Front. Public Health doi. 10.3389/fpubh.2019.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tehrani KHME, Martin NI (2018) β-lactam/β-lactamase inhibitor combinations: an update. Medchemcomm. 9 (9), 1439–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Brown P, Dawson MJ (2017) Development of new polymyxin derivatives for multi-drug resistant Gram-negative infections. J. Antibiotics (Tokyo) 70 (4), 386–394. [DOI] [PubMed] [Google Scholar]
- (6).Vaara M (2018) New polymyxin derivatives that display improved efficacy in animal infection models as compared to polymyxin B and colistin. Med. Res. Rev 38 (5), 1661–1673. [DOI] [PubMed] [Google Scholar]
- (7).Velkov T, Roberts KD (2019) Discovery of polymyxin-like antibiotics. In Li J et al. (eds) Polymyxin antibiotics: from laboratory bench to bedside. Advances in Experimental Medicine and Biology 1145, doi. 10.1007/978-3-030-16373-0_20. [DOI] [PubMed] [Google Scholar]
- (8).Brown P, Abbott E, Abdulle O, Boakes S, Coleman S, Divall N, Duperchy E, Moss S, Rivers D, Simonovic M, Singh J, Stanway S, Wilson A, Dawson MJ (2019) Design of next generation polymyxins with lower toxicity: the discovery of SPR206. ACS Infect. Dis 5 (10), 1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vaara M (2019) Polymyxin Derivatives that Sensitize Gram-Negative Bacteria to Other Antibiotics. Molecules 24, 249; doi: 10.3390/molecules24020249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Velkov T, Roberts KD, Nation RL, Wang J, Thompson PE, Li J (2014) Teaching ‘Old’ Polymyxins New Tricks: New-Generation Lipopeptides Targeting Gram-Negative ‘Superbugs’. ACS Chem. Biol 9 (5), 1172–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mares J, Kumaran S, Gobbo M, Zerbe O (2009) Interactions of lipopolysaccharide and polymyxin studies by NMR spectroscopy. J. Biol. Chem 284, 17, 11498–11506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Velkov T, Roberts KD, Nation RL, Thompson PE, Li J (2013) Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol. 8, 6, 711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Trimble MJ, Mlynarcik P, Kolar M, Hancock REW (2016) Polymyxin: alternative mechanisms of action and resistance. Cold Spring Harb. Perspect. Med 6, 10, doi: 10.1101/cshperspect.a025288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vaara M (1992) Agents that increase the permeability of the outer membrane. Microbiol. Rev 56 (3), 395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).French S, Farha M, Ellis MJ, Sameer Z, Cote J-P, Cotroneo N, Lister T, Rubio A, Brown ED (2019) Potentiation of antibiotics against Gram-negative bacteria by polymyxin B analogue SPR741 from unique perturbation of the outer membrane. ACS Infect. Dis 10.1021/acsinfecdis.9b00159 [DOI] [PubMed] [Google Scholar]
- (16).Corbett D, Wise A, Langley T, Skinner K, Trimby E, Birchall S, Dorali A, Sandiford S, Williams J, Warn P, Vaara M, Lister T (2017) Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741 Antimicrob Agents Chemother. 2017. July 25;61(8):e00200–17. doi: 10.1128/AAC.00200-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Eckburg PB, Lister T, Walpole S, Keutzer T, Utley L, Tomayko J, Kopp E, Farinola N, Coleman S (2019). Safety, tolerability, pharmacokinetics, and drug interaction potential of SPR741, an intravenous potentiator, after single and multiple ascending doses and when combined with β-lactam antibiotics in healthy subjects. Antimicrob Agents Chemother 63:e00892–19. 10.1128/AAC.00892-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Moffatt JH, Harper M, Boyce JD (2019) Mechanisms of polymyxin resistance. Adv. Exp. Med. Biol 1145, 55–71. [DOI] [PubMed] [Google Scholar]
- (19).Velkov T, Gallardo-Godoy A, Swarbrick JD, Blaskovich MAT, Elliott AG, Ham M, Thompson PE, Roberts KD, Huang JX, Becker B, Butler MS, Lash LH, Henriques ST, Nation RL, Sivanesan S, Sani M-A, Separovic F, Mertens H, Bulach D, Seemann T, Owen J,. Li J, Cooper MA (2018) Structure, function, and biosynthetic origin of octapeptin antibiotics active against extensively drug resistant Gram-negative bacteria. Cell Chem. Biol 25 (4), 380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brown P, Abdulle O, Boakes S, Moss S, Simonovic M, Stanway S, Wilson A, Dawson M (2020). Direct modifications of the cyclic peptide Polymyxin B leading to analogues with enhanced in vitro antibacterial activity. Bioorg. Med Chem Lett, 30, 127163. 10.1016/j.bmcl.2020.127163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Vaara M, Vaara T and Tyrrell JM (2017) Structure–activity studies on polymyxin derivatives carrying three positive charges only reveal a new class of compounds with strong antibacterial activity. Peptides 91, 8–12. DOI: 10.1016/j.peptides.2017.03.002 [DOI] [PubMed] [Google Scholar]
- (22).Ghose AK, Viswanadhan VN, and Wendoloski JJ,(1998) Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragment Methods: An Analysis of AlogP and CLogP Methods.” J. Phys. Chem. A, 1998, 102, 3762–3772). [Google Scholar]
- (23).Biovia Insight for Excel 2017. (Dassault Systemes). [Google Scholar]
- (24).Sharafi T, Ardebili A (2019) Plastic binding feature of polymyxins: the effect on MIC susceptibility measurements. Infect. Drug Resist 12, 2649–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Karvanen M, Malmberg C, Lagerbäck P, Friberg LE, Otto Cars O (2017) Colistin Is Extensively Lost during Standard In Vitro Experimental Conditions. Antimicrob. Agents Chemother 61, (11) e00857–17; DOI: 10.1128/AAC.00857-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Magee TV, Brown MF, Starr JT, Ackley DC, Abramite JA, Aubrecht J, Butler A, Crandon JL, Dib-Hajj F, Flanagan ME, Granskog K, Hardink JR, Huband MD, Irvine R, Kuhn M, Leach KL, Li B, Lin J, Luke DR, MacVane SH, Miller AA, McCurdy S, McKim JM Jr., Nicolau DP, Nguyen T-T, Noe MC, O’Donnell JP, Seibel SB, Shen Y, Stepan AF, Tomaras AP, Wilga PC, Zhang L, Xu J, Chen JM (2013) Discovery of Dap-3 polymyxin analogues for the treatment of multidrug-resistant Gram-negative nosocomial infections. J. Med. Chem 56, 5079–5093. [DOI] [PubMed] [Google Scholar]
- (27).Sarubbi E, Monti F, Corti E, Miele A and Selva E (2004). Mode of action of the microbial metabolite GE23077, a novel potent and selective inhibitor of bacterial RNA polymerase. European J. Biochemistry, 271: 3146–3154. 10.1111/j.1432-1033.2004.04244.x [DOI] [PubMed] [Google Scholar]
- (28).Vaara M, Siikanen O, Apajalahti J, Fox J, Frimodt-Moller N, He H, Poudyal A, Li J, Nation RL, Vaara T (2010) A novel polymyxin derivative that lacks the fatty acyl tail and carries only three positive charges has strong synergism with agents excluded by the intact outer membrane. Antimicrob. Agents Chemother 54, 3341–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jayol A, Kieffer N, Poirel L, Guerin F, Guneser D, Cattoir D, Nordmann P (2018) Evaluation of the rapid polymyxin NP test and its industrial version for the detection of polymyxin-resistant Enterobacteriaceae. Diagnost. Microbiol. Infect. Dis 92, 90–94. [DOI] [PubMed] [Google Scholar]
- (30).Clinical and Laboratory Standards Institute (2012). Methods for dilution antimicrobial susceptibility test for bacteria that grow aerobically: approved standard, 9th ed. CLSI document M07-A9. CLSI, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.