

CONSPECTUS
Biocatalytic transformations that leverage the selectivity and efficiency of enzymes represent powerful tools for the construction of complex natural products. Enabled by innovations in genome mining, bioinformatics, and enzyme engineering, synthetic chemists are now more than ever able to develop and employ enzymes to solve outstanding chemical problems – one of which is the reliable and facile generation of stereochemistry within natural product scaffolds. In recognition of this unmet need, our group has sought to advance novel chemoenzymatic strategies to both expand and reinvigorate the chiral pool. Broadly defined, the chiral pool comprises cheap, enantiopure feedstock chemicals that serve as popular foundations for asymmetric total synthesis. Among these building blocks, amino acids and enantiopure terpenes, whose core structures can be mapped onto several classes of structurally and pharmaceutically intriguing natural products, are of particular interest to the synthetic community.
In this Account, we summarize recent efforts from our group in leveraging biocatalytic transformations to expand the chiral pool, as well as efforts toward the efficient application of these transformations in natural products total synthesis – the ultimate testing ground for any novel methodology. First, we describe several examples of enzymatic generation of noncanonical amino acids as means to simplify the synthesis of peptide natural products. By extracting amino acid hydroxylases from native biosynthetic pathways, we obtain efficient access to hydroxylated variants of proline, lysine, arginine, and their derivatives. The newly-installed hydroxyl moiety then becomes a chemical handle that can facilitate additional complexity generation, thereby expanding the pool of amino acid-derived building blocks available for peptide synthesis. Next, we present our efforts in enzymatic C–H oxidations of diverse terpene scaffolds, in which traditional chemistry can be combined with strategic applications of biocatalysis to selectively and efficiently derivatize several commercial terpenoid skeletons. The synergistic logic of this approach enables a small handful of synthetic intermediates to provide access to a plethora of terpenoid natural product families. Taken together, these findings demonstrate the advantages of applying enzymes in total synthesis in conjunction with established methodologies, as well as toward the expansion of the chiral pool to enable facile incorporation of stereochemistry during synthetic campaigns.
Graphical Abstract
1. INTRODUCTION
The regio-, chemo-, and stereoselective functionalization of C–H bonds represents a powerful paradigm for the construction and derivatization of complex natural products.5 Given the ubiquity of C–H bonds in organic molecules, the ability to chemically differentiate them to facilitate oxidations, fragment couplings, and other transformations stands as a holy grail in synthetic chemistry. Despite the ongoing expansion of the synthetic repertoire, selective C–H functionalization remains a significant challenge, especially in complex settings.
Recent advances in genome mining and microbial genetics have granted unprecedented access to biosynthetic enzymes, and the ability to heterologously express and isolate them with relative ease has rendered biocatalysis more feasible than ever before.6 Recognizing the inherent potential therein, synthetic chemists have begun to apply enzymes in methodology and synthesis.7 However, the field is still very much in its infancy, and the biocatalytic utility of many enzyme superfamilies, such as Fe- and α-ketoglutarate-dependent dioxygenases (Fe/αKGs), remains largely untapped, especially for natural product synthesis.8 Responding to the demand for widely applicable C–H functionalization strategies, our group has sought to leverage biocatalytic methods for selective C–H oxidation en route to a variety of natural products in the past few years. During this time, contemporaneous efforts from other research groups, such as Narayan,7a Stoltz,7c Sherman,7d Fasan7e and Moore,8a have also provided significant contributions to address various unmet knowledge gaps in the field.
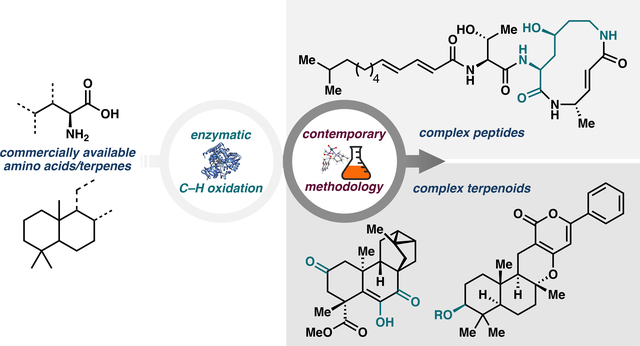
To date, we have focused on the biocatalytic oxidation of readily available compounds as a means to prepare building blocks and synthetic intermediates that are otherwise challenging to construct; this can be viewed as a reinvigoration of the chiral pool approach to natural product synthesis (Figure 1). Drawn upon for decades as a source of inexpensive stereochemical information, the chiral pool comprises a diverse array of enantiopure feedstock chemicals that can be easily converted into useful starting materials or chiral catalysts/reagents.9 Many terpenes and amino acids are commercially available in various stereoforms and thus represent important members of the chiral pool. Much effort has been devoted to selectively derivatizing these compounds, but overriding the innate reactivity of their scaffolds remains a difficult undertaking in organic synthesis.9,10 To bolster the traditional synthetic toolbox, we have advanced a number of biocatalytic methodologies to functionalize amino acid and terpene building blocks, ultimately with an eye toward natural products synthesis.
Figure 1.
Comparison between prior applications of amino acid and terpenes in chiral pool synthesis and the approaches advanced by our group.
2. AMINO ACID FUNCTIONALIZATION
Amino acids represent a highly useful class of organic small molecules, finding practical application as pharmaceuticals and small molecule probes, among many others.10 The twenty canonical amino acids, so called due to their incorporation into proteins during translation, are commercially available and provide traditional methodology and synthesis with valuable sources of stereochemical information. Conversely, noncanonical amino acids (ncAAs) trace their biological origins to post-translational modifications in protein biosynthesis or to secondary metabolic pathways.11 Given that the presence of ncAAs in small molecules can influence biological activity, such compounds are often attractive drug or probe candidates.4,12 The synthetic community has devoted significant effort to establishing concise routes to ncAAs, ultimately in pursuit of rapid access to ncAA-containing natural products and drug scaffolds. Due to the innate chemical challenges posed by amino acids, including the presence of free amino and carboxylate moieties and potentially reactive side chains, as well as the need to set one or more stereogenic centers, construction of ncAAs from existing amino acids or by de novo synthesis remains difficult.11
Therefore, our group has turned to Nature for inspiration, noting that nonribosomal peptide biosynthesis often utilizes hydroxylation as a gateway transformation to synthesize ncAAs.12 We looked to reproduce this process in vitro by (1) directly access hydroxylated amino acid building blocks and (2) preparing additional ncAAs by utilizing the newly-introduced alcohol as a chemical handle for further complexity generation. In the following section, we describe our exploration of biocatalytic hydroxylation as an effective means to derivatize amino acids, as well as our applications of these methods toward syntheses of ncAA-containing natural products.
a. HYDROXYLATION AS A GATEWAY
At the outset of our group, we were aware of several proposed biogeneses of 4-methylproline in nonribosomal peptides that invoked iterative oxidation of leucine, intramolecular amine condensation, and subsequent reduction of the cyclic species.13 Inspired by Nature’s strategy, we sought to replicate and subsequently improve upon this sequence in the flask, first using Fe/αKGs to hydroxylate amino acid scaffolds and then converting the resulting alcohol into other useful functional groups – either via biocatalytic or chemical means – to facilitate more diverse transformations.
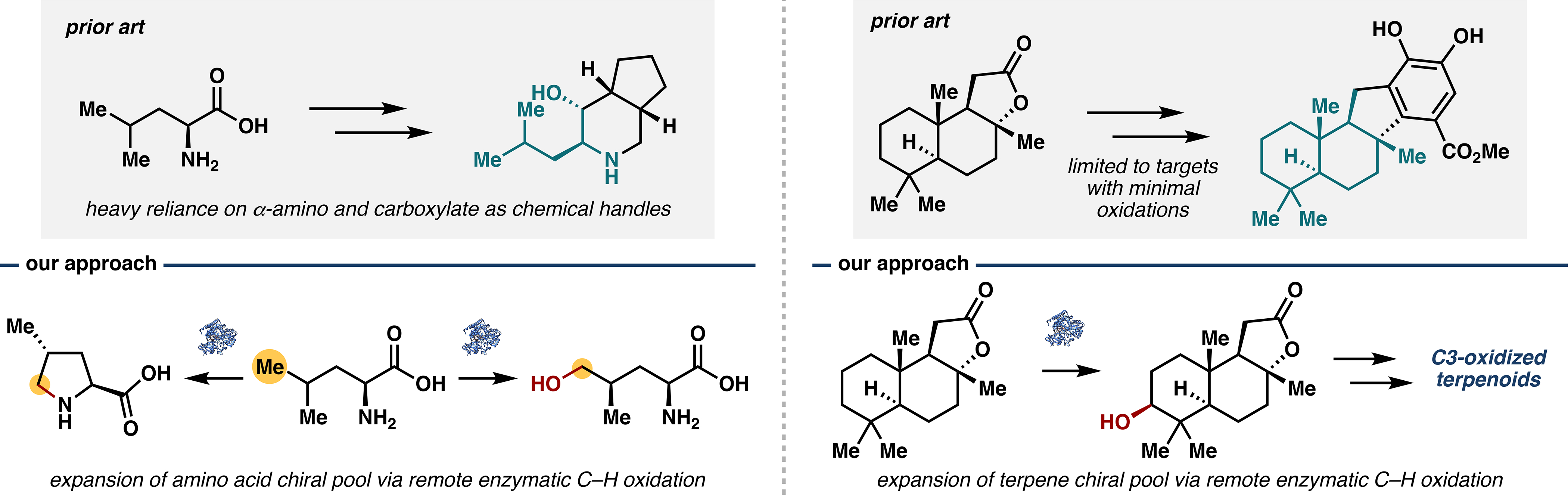
In 2015, the leucine δ-hydroxylase GriE was implicated in the biosynthesis of griselimycin14 – a peptide natural product containing two 4-methylproline motifs – and was later found to work in tandem with zinc-dependent dehydrogenase GriF to effect iterative leucine oxidation and imine formation.15 Looking to obtain a robust biocatalyst for preparative-scale leucine hydroxylation, we acquired pure GriE from heterologous expression in high yield (ca. 100 mg from 1 L of culture) and subjected a large panel of amino acids to hydroxylation in the presence of αKG, ascorbate, FeSO4, and O2.1 GriE readily converted leucine to the corresponding δ-hydroxylated product with complete regio- and diastereoselectivity and high total turnover number (TTN). Several other amino acids were also accepted as substrates, exclusively yielding δ-hydroxylation with complete diastereoselectivity in nearly all cases (Figure 2A). The impressive promiscuity of GriE is complemented by excellent scalability, as reactions in GriE-containing lysate could be run at high substrate concentrations (up to 50 mM in leucine) on gram scale with no decrease in conversion. Other amino acid substrates proceeded in high conversion on 100–200 mg scale, further validating the utility of GriE. During this study, reactions conducted in lysate were found to be more scalable and convenient than with purified enzyme, simply requiring sonication of resuspended cells followed by addition of the appropriate substrates and cofactors (αKG, Fe2+ and ascorbic acid for Fe/αKGs, NAD(P)H for P450s). Subsequent work from our lab has predominantly employed lysate for scaled-up reactions.
Figure 2.
(A) Native function and substrate scope of GriE. (B) Use of GriE in the chemoenzymatic synthesis of manzacidin C (11). (C) Synthesis of various proline analogues via GriE-catalyzed iterative C5 oxidation.
We then sought to implement GriE toward the synthesis of manzacidin C (11), a densely functionalized alkaloid natural product from Hymeniacidon sp.16 A two-step process has been reported to convert lactone 10 to manzacidin C, but efficient, step-economic access to 10 has yet to be achieved.17 We proposed a formal synthesis of 10, wherein biocatalytic hydroxylation would introduce a primary alcohol at C5 and facilitate lactone formation via routine intramolecular cyclization. In light of the substrate-activity relationship of GriE, we envisioned that a masked amine derivative of leucine could be submitted to hydroxylation and later revealed as the amine.
Thus, treatment of leucine with tetrabutylammonium decatungstate (TBADT) and azide 6 under photocatalytic conditions gave azidoleucine 7,18 which was subjected to reaction with GriE to deliver the desired hydroxylated product 8 with >95% conversion. A telescoped hydrogenation/dual Boc protection/selective lactonization procedure then afforded lactone 10 in 41% yield over two steps (Figure 2B). Given the aforementioned two-step elaboration of 10 to the natural product, our route represents a five-step formal synthesis of manzacidin C and a drastic improvement in step economy over prior approaches.19 This improvement, coupled with absolute regio- and stereocontrol, underscores the capability of enzymatic C–H functionalization to streamline synthetic efforts. At the time of publication, this work also comprised the first use of an Fe/αKG-dependent enzyme in natural product synthesis.
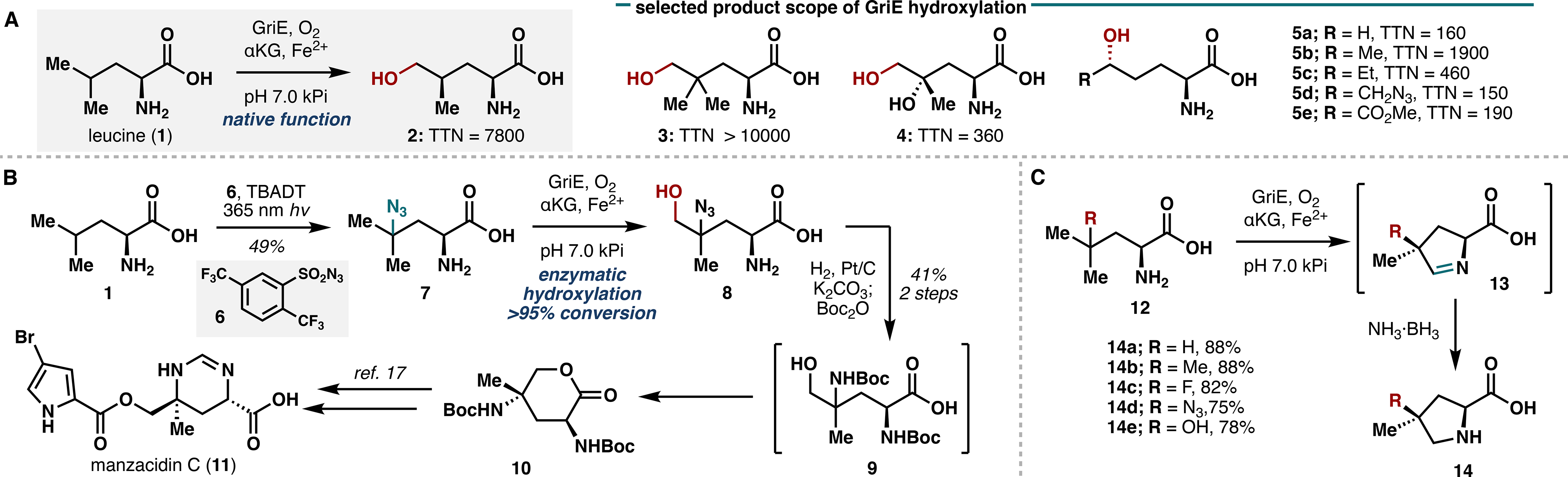
During the characterization of GriE, we discovered that GriE also performs iterative δ-oxidation on γ-methylleucine, which led us to investigate the use of GriE to construct various proline derivatives. Leucine and several related analogues were submitted to a two-step, one-pot sequence of GriE-catalyzed oxidation followed by in situ imine reduction with NH3·BH3, which provided proline analogues 14a–e in high yields and with complete stereocontrol (Figure 2C). This highly efficient protocol stands in contrast to existing chemical methods, which often lack stereocontrol at C4 and require several functional group interconversions. A similar method was devised to access 3-hydroxy-3-methylproline (18) from isoleucine using the Fe/αKGs UcsF and GetF,20 thereby demonstrating the broad applicability of this strategy and laying the groundwork for access to 3-hydroxy-3-methylproline-containing natural products (Figure 3A).
Figure 3.
(A) Chemoenzymatic synthesis of 3-hydroxy-3-methylproline (18) utilizing UcsF and GetF. (B) GriE-enabled synthesis of Fmoc-protected 4-methylproline monomer 21. (C) One-pot solid-phase peptide synthesis of cavinafungin B (22).
To highlight the synthetic utility of our approach, we devised a total synthesis of cavinafungin B (22), an antiviral lipopeptide natural product containing 4-methylproline.21 Having already obtained access to 4-methylproline via action of GriE and subsequent imine reduction (Figure 2B), we performed Fmoc protection of the free amine and executed a solid-phase peptide synthesis (SPPS) campaign featuring an AT(Boc)G-Rink resin linkage. After four rounds of successive PyAOP/NMM-mediated peptide coupling and piperidine-mediated Fmoc deprotection, a sequence of oleic acid coupling, global deprotection, and resin cleavage ultimately gave cavinafungin B in 37% yield over 10 steps (Figure 3C).
Beyond facilitating lactone formation or iterative oxidation, hydroxylation can also serve as a gateway to other functional groups, as illustrated by our synthesis of tambromycin (37),22 a cytotoxic peptide produced by several Streptomyces strains (Scheme 1).23 Comprising four modified amino acid monomers, tambromycin derives its name from the presence of tambroline, a rare pyrrolidine-containing ncAA originating biogenically from lysine. This biosynthetic proposal and previous synthetic efforts toward similar compounds led us to attempt hypoiodite-based C–H amination at the β-position of lysine to deliver the pyrrolidine ring. However, this strategy proved unsuccessful.
Scheme 1.
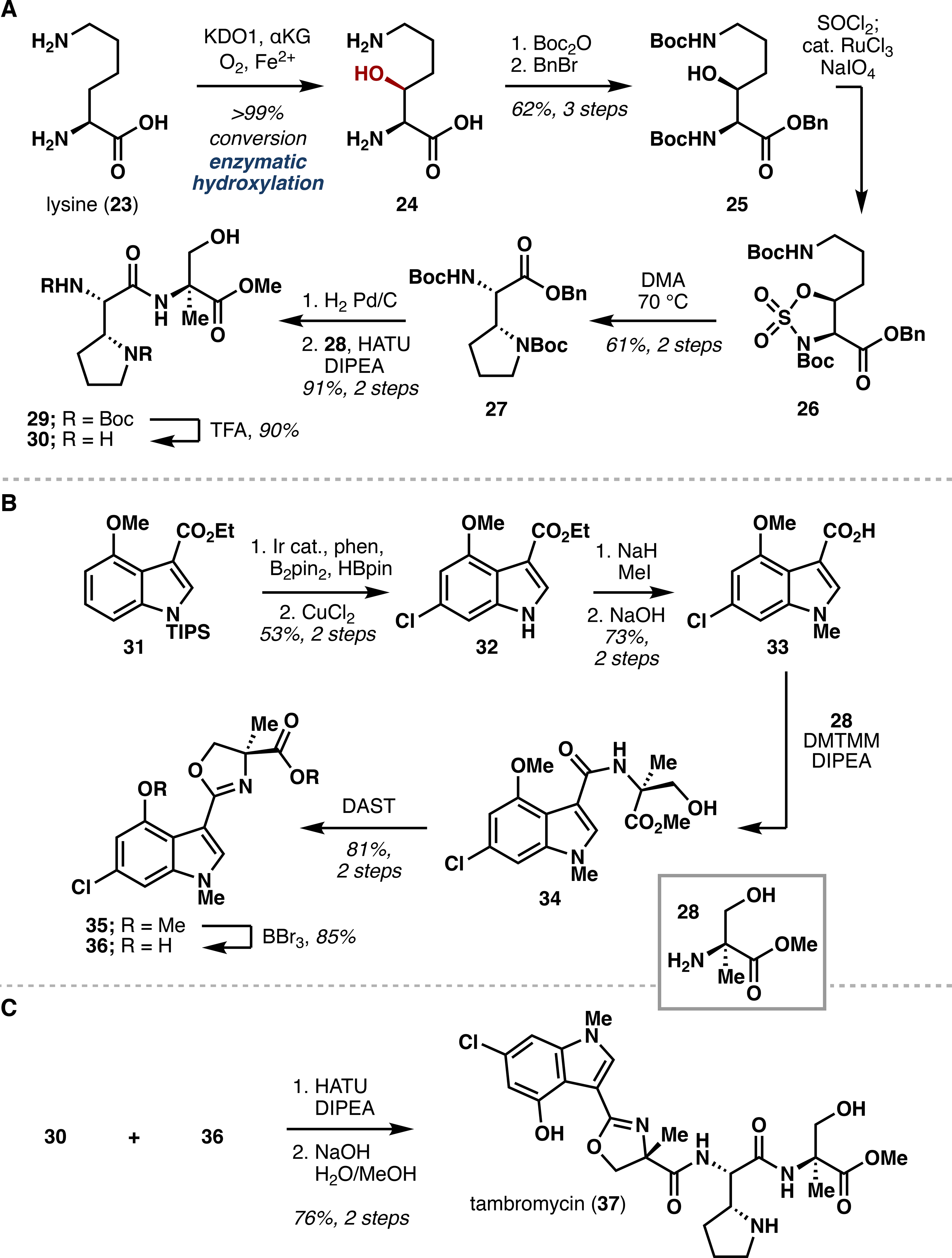
Total Chemoenzymatic Synthesis of Tambromycin (37)
We turned instead to KDO1, an Fe/αKG reported to catalyze β-hydroxylation of lysine,24 envisioning that the pyrrolidine motif could be constructed through a stereocontrolled displacement of the hydroxylysine β-OH by its ε-amine. Initial heterologous expression of KDO1 provided insufficient yield of soluble enzyme, but following co-expression of the molecular chaperones GroES/GroEL,8c,25 reaction with KDO1 allowed for hydroxylation of 4.1 g of lysine from 1 L of expression culture, giving >99% conversion to 24 at high (35 mM) substrate concentration. Following routine protecting group introductions, 25 was converted to sulfamidate 26, which was heated in DMA to cleanly give protected tambroline 27.
Next, a C6-selective C–H borylation/halogenation sequence was chosen to construct the 3,4,6-trisubstituted indole motif of tambromycin.26 Thus, treatment of 31 with B2Pin2 and catalytic [Ir(cod)OMe]2 followed by chlorination with CuCl2 gave indole 32, which was quickly converted to acid 33 after N1 methylation and ester hydrolysis. The remainder of the synthesis followed summarily from elaboration of 33 and tambroline 27, wherein a series of peptide couplings and functional group interconversions gave tambromycin after the liberation of the terminal carboxylic acid from the methyl ester. Out of this work emerged the first total synthesis of tambromycin, empowered by harnessing two C–H functionalization methods in tandem: namely, a gram scale procedure for biocatalytic implementation of KDO1 to hydroxylate the β position of lysine and a modular chemocatalytic approach to synthesize 3,4,6-trisubstituted indoles.
b. HYDROXYLATION AS A DESTINATION
We have also sought to leverage biocatalysis to access natural products that themselves contain hydroxylated ncAAs. In particular, 4-hydroxylysine, 4-hydroxycitrulline, and 4-hydroxyarginine are found in several nonribosomal peptides, though the current chemical state-of-the-art is unable to selectively and efficiently access such functionalities. Thus, we have developed biocatalytic procedures for hydroxylation of the parent amino acids and applied them to the total syntheses of several natural products and derivatives thereof, namely cepafungin I and GE81112 B1.
The syrbactins comprise a family of peptide-polyketide natural products that share a common 12-membered unsaturated macrolactam and display potent inhibitory activity against the eukaryotic 20S proteasome core particle.27 Given the prior success of proteasome inhibitors in the treatment of multiple myeloma,28 the syrbactins are attractive candidates for anticancer applications. Among the family members, cepafungin I (38) was reported to display the highest potency with an IC50 of 4 nM against purified yeast 20S proteasome.29 We noted that the glidobactin/cepafungin biosynthetic gene cluster features the enzyme GlbB,30 which belongs to the PF10014 Fe/αKG family and is likely responsible for formation of the 4-hydroxylysine motif. Motivated to fill a gap in the lysine derivatization literature,31 we sought to characterize GlbB and evaluate its utility as a hydroxylation biocatalyst.
In vitro assay with purified GlbB revealed that the enzyme could hydroxylate the γ-position of lysine with high turnover number (5900 TTN) and excellent diastereoselectivity (>99:1).32 Leucine (γ-hydroxylation) and methionine (S-oxidation) were also accepted as substrates, giving 310 and 330 TTN, respectively (Figure 4A). We next investigated the large-scale preparation of 4-hydroxylysine to ensure adequate material supply for the synthesis of cepafungin I. Co-expression of GlbB with GroES/GroEL25 increased yield of soluble enzyme such that 6–7 g of lysine could be fully converted to 40 with 1 L of lysate. To access a suitable cyclization precursor, Weinreb amide 44 was used in an AlMe3-mediated coupling with lactone 43, providing dipeptide 45 in 90% yield. Upon reduction of the amide and olefination of the resulting aldehyde, enoate 46 was subjected to global deprotection and treated with DMTMMT to effect macrocyclization.33 The macrolactam core (47) was coupled with acid 48 to give cepafungin I in 9 linear steps and 7.9% overall yield (Figure 2B).4
Figure 4.
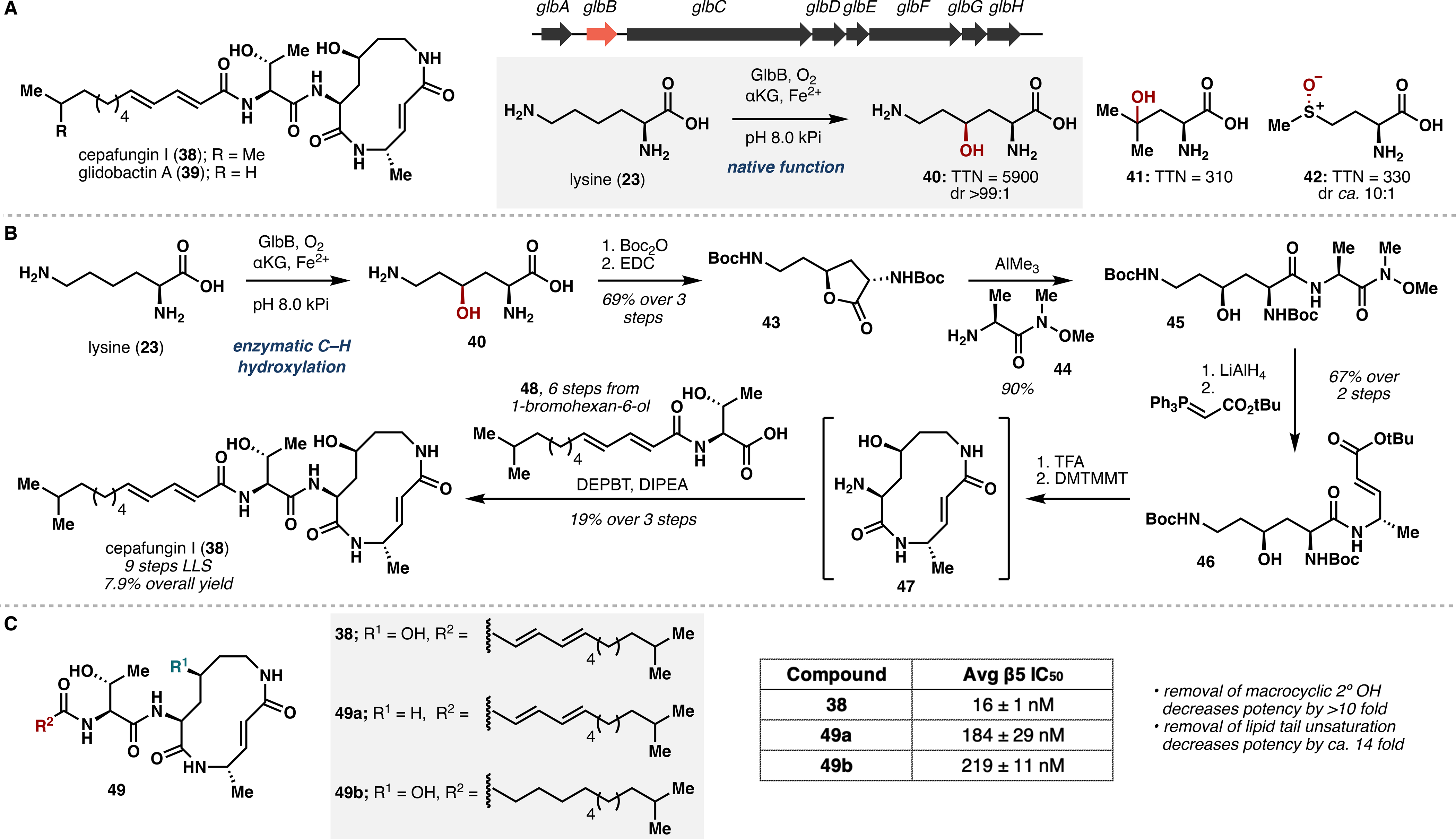
(A) Characterization of lysine hydroxylase GlbB and selected substrate scope. (B) Nine-step total synthesis of cepafungin I empowered by lysine hydroxylation with GlbB. (C) PSMB5 inhibitory assay results with synthetic analogues of 38.
By biocatalytically expanding the amino acid chiral pool, our route allows for concise introduction of the secondary alcohol to the cepafungin macrolactam, thereby addressing the main shortcoming of prior approaches to the natural product. This strategy also enabled biological investigations into cepafungin I through the synthesis of a chemoproteomic probe and several analogues (Figure 4C).4 Competitive proteomic profiling experiments with cepafungin I showed remarkable selectivity for 20S proteasome subunits PSMB2 and PSMB5 with minimal off-target activity. Further PSMB5 inhibitory assays with synthetic analogues suggest the importance of the macrocyclic secondary alcohol and the degree of chain unsaturation of the lipid tail, as analogues varying in these key motifs showed drastically attenuated potency relative to the parent natural product.
In addition to the syrbactins, we also set our sights on the GE81112s, an intriguing family of hydroxylated tetrapeptide natural products (Figure 5A).34 Notable for their dense functionalities and unique antibacterial activity, the GE81112s consist largely of noncanonical amino acids and function as inhibitors of prokaryotic translation. With the exception of the 3-hydroxypipecolic acid unit, which is formed via hydroxylation by the Fe/αKG GetF,35 little is known about the biogenesis of the GE81112 ncAA fragments. Previous studies into the GE81112 biosynthetic gene cluster uncovered a dedicated nonribosomal peptide synthase (NRPS) and several tailoring enzymes, including the aforementioned GetF and a second Fe/αKG GetI.36 Initially proposed to catalyze β-hydroxylation of 2-chlorohistidine,35 GetI represented a potentially useful biocatalyst to generate ncAAs and aid in the synthesis of GE81112 B1.
Figure 5.
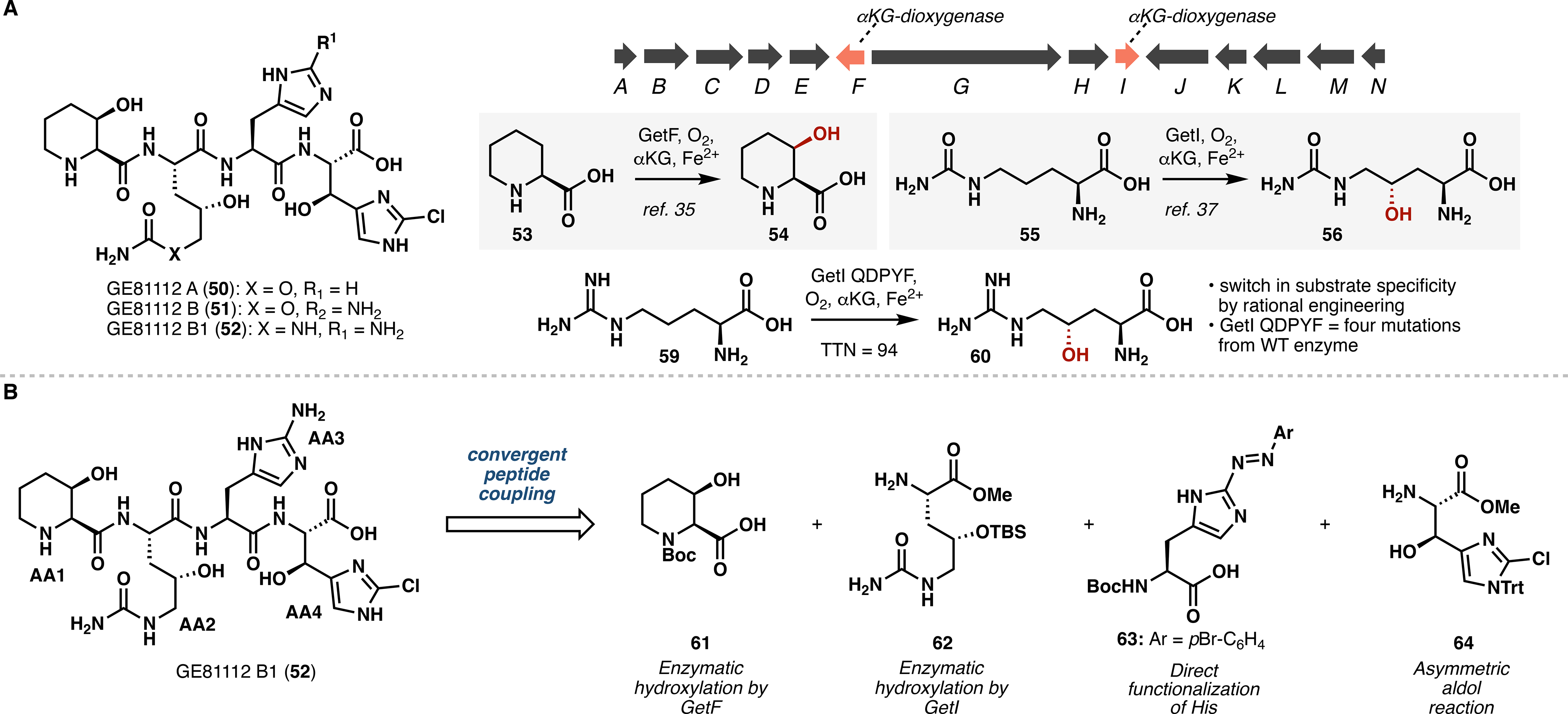
(A) Characterization and rational engineering of GetI. (B) Retrosynthetic analysis of GE81112 B1.
Surprisingly, neither 2-chlorohistidine nor histidine was converted to its corresponding hydroxylation product in reaction with GetI.37 However, the high sequence homology shared by GetI with OrfP38 and VioC,39 two previously characterized Fe/αKG arginine hydroxylases, led us instead to examine citrulline (55) as a substrate. To our delight, LCMS analysis revealed a monohydroxylated product, and subsequent NMR analysis confirmed its identity as 4-hydroxycitrulline (56). At this stage, GetI was noted to exhibit comparable activity on α-amino-δ-carbamoylhydroxyvaleric acid and low levels of hydroxylation activity on arginine. Given that no dedicated arginine γ-hydroxylase was known at the time of this work, this discovery inspired an enzyme engineering campaign to develop such enzyme. Using homology models of OrfP and VioC, we performed sequential site-directed mutagenesis on GetI and obtained variant QDPYF, which was capable of converting arginine to 4-hydroxyarginine with 94 TTN. Preparative scale reaction with E. coli lysates expressing GetI QDPYF afforded nearly complete conversion, and we successfully implemented this process in a brief synthesis of a dipeptide fragment of enduracidin.37
We next targeted the chemoenzymatic synthesis of GE81112 B1 (52),40 which comprises 3-hydroxypipecolic acid (AA1), 4-hydroxycitrulline (AA2), 2-aminohistidine (AA3), and β-hydroxy-2-chlorohistidine (AA4) (Figure 5B). Chemical construction of these monomers is nontrivial, as a previous synthesis of GE81112 A required 7–8 steps to make each fragment and suffered from poor stereocontrol.41 A strategy was thus devised that would showcase the strengths of both biocatalysis and contemporary chemical methodology: Specifically, enzymatic hydroxylations with GetF and GetI were proposed to enable construction of AA1 and AA2, whereas traditional chemistry could grant access to AA3 and AA4.
For the preparation of AA1, co-expression of GetF with GroES/GroEL8c,25 delivered complete conversion of pipecolic acid (53) to 3-hydroxypipecolic acid on 500 mg scale. Upon treatment with Boc2O, protected monomer 65 was obtained in 74% yield over two steps. Toward AA2, GetI facilitated gram-scale conversion of citrulline to 4-hydroxycitrulline and provided, after subsequent protecting group adjustments, fragment 66 in four steps and 41% overall yield. Unmasking of the primary amine 67 from 66 was followed by routine coupling with 65 to give dipeptide 68 after saponification.
Preparation of AA3 proceeded via two-step functionalization of Boc-His-OMe (69), wherein C2 azotisation of its imidazole ring was followed by saponification of the methyl ester to reveal acid 70. Construction of AA4 proved more challenging, as several attempts at an asymmetric aldol reaction were met with failure. Finally, upon optimization of the steric environment, reaction of titanium enolate 71 and aldehyde 72 gave the desired adduct as a single diastereomer in 59% yield after methanolysis. Treatment with aqueous ammonium sulfide cleanly provided the desired amine 73, which was coupled with acid 70 and deprotected under buffered conditions to deliver dipeptide 74.
With 68 and 74 in hand, the corresponding peptide coupling reaction proceeded uneventfully to afford the desired protected tetrapeptide in 66% yield as a single diastereomer. The use of H2 and PtO2 cleanly reduced the azo moiety of AA3 to the corresponding amine, and after ester hydrolysis and global deprotection we successfully isolated the pure TFA salt of GE81112 B1 in 11 steps (longest linear sequence) (Scheme 2). Importantly, our synthesis features an ideal combination of biocatalytic and chemical approaches, wherein the strengths of each paradigm are leveraged to streamline construction of the complex GE81112 scaffold. This route has also enabled synthesis of several analogues, facilitating structure-activity relationship studies and identification of the key pharmacophores of GE81112 B1.40
Scheme 2.
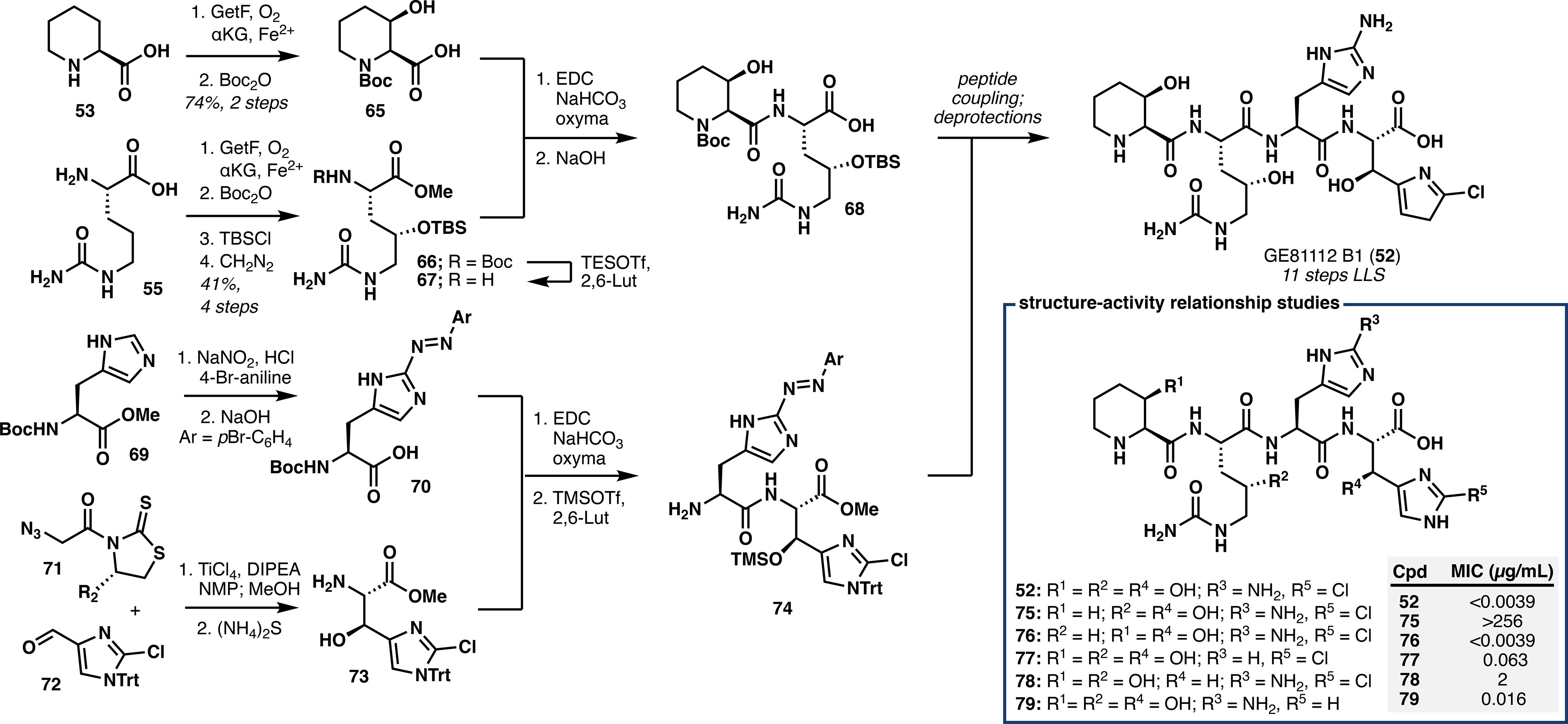
Total Chemoenzymatic Synthesis of GE81112 B1 and Structure-Activity Relationship of Synthetic Analogues.
3. TERPENE FUNCTIONALIZATION
In stark contrast to amino acids, terpenes consist primarily of hydrocarbons in varying degrees of saturation and possess few other functional groups. Despite this, the terpene chiral pool is exceptionally diverse and has provided materials for many total syntheses of terpene-derived natural products.9b Indeed, as sources of carbon framework and stereochemical information, enantiopure terpenes are broadly employed as cheap synthetic building blocks and are generally available in both stereoisomers. Our lab has been increasingly interested in a number of these inexpensive terpene building blocks; namely, sclareolide (89), sclareol (90), and stevioside (121). Available in kilogram quantities for ca. $1/g, these compounds have begun to see application in chiral pool terpene synthesis,42 though their broad use remains limited due to a general inability to regio- and stereoselectively derivatize different positions on their skeletons. Eager to demonstrate the potential of these compounds, our group has leveraged biocatalysis to functionalize positions previously inaccessible to traditional chemistry, thereby reinvigorating these molecules for use in chiral pool synthesis.
a. SCLAREOL AND SCLAREOLIDE OXIDATION: MEROTERPENOIDS
Meroterpenoids constitute a large family of natural products that arise from mixed terpenoid-polyketide biosynthetic pathways (Figure 6A).43 Two subclasses are the α-pyrone meroterpenoids,44 which contain a C3-oxidized drimane unit coupled to a polyketide pyrone, and the meroditerpenoids,45 which bear a C3-oxidized ent-isocopolane motif appended to various aromatic moieties. Traditional synthetic approaches to the C3-oxidized terpene core rely on polyene cyclization from polyprenyl-derived epoxides46 or elaboration of the Wieland-Miescher ketone46 but often suffer from poor stereoselectivity and synthetic efficiency. Moreover, as meroterpenoid natural products possess diverse biological activities, there is considerable demand for concise and modular routes to these compounds and analogues thereof.
Figure 6.
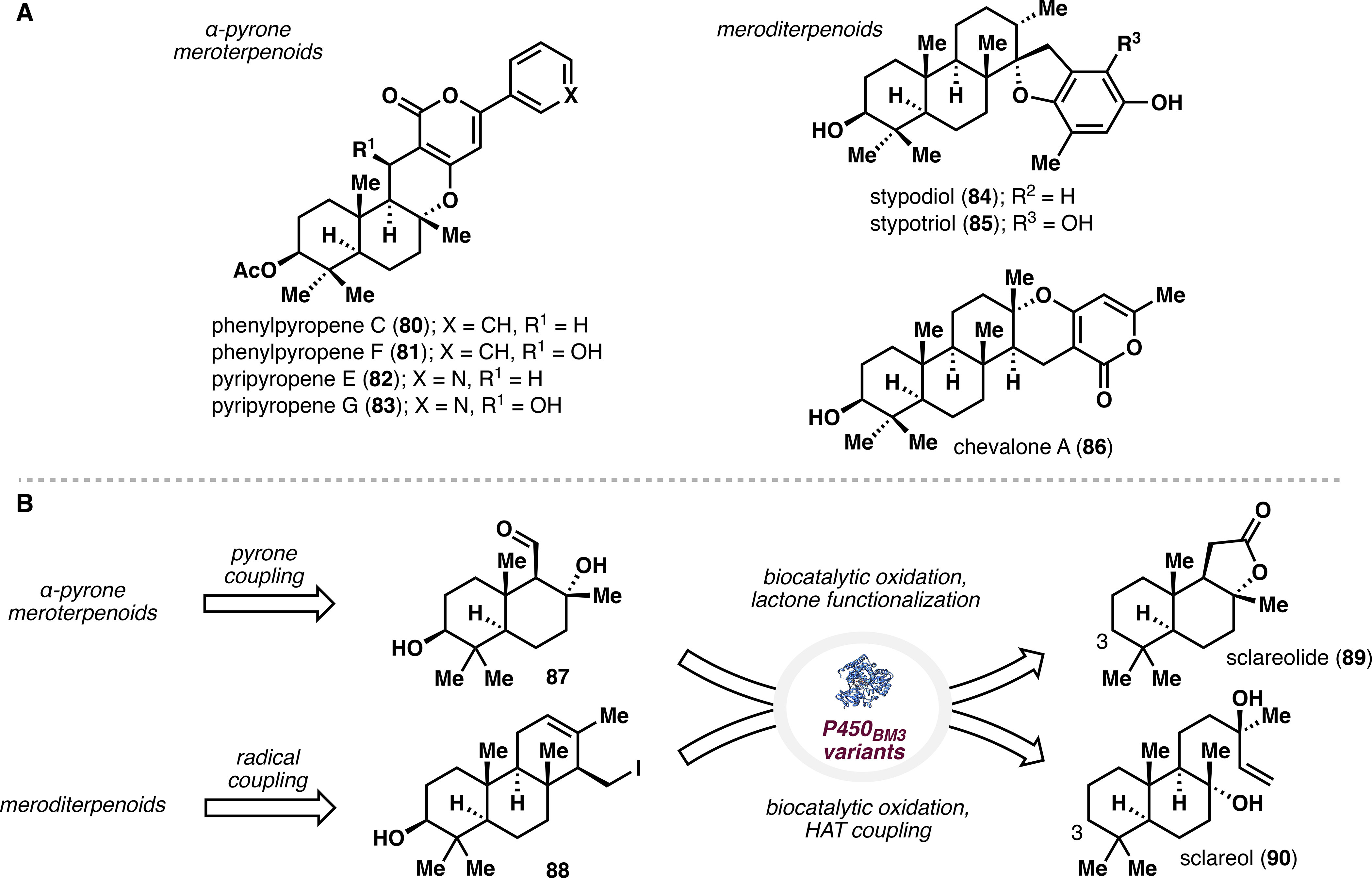
(A) Representative α-pyrone meroterpenoids and meroditerpenoids. (B) Retrosynthetic analysis of the two meroterpenoid families.
We thus sought to implement enzymatic C–H oxidation to access the core terpenoid skeleton and to leverage recent advances in radical chemistry to introduce the polyketide unit. Retrosynthetically, the α-pyrone meroterpenoids could be traced to sclareolide, and the meroditerpenoids could arise in a similar fashion from sclareol (Figure 6B).2 Radical transformations would then effect conjugation of the pendant aromatic functionalities or late-stage modification of the skeleton. We hoped to employ two common intermediates to construct eight oxidized meroterpenoids, thereby highlighting the potential of an expanded chiral pool, wherein increased utility of key starting materials empowers modular synthesis of a number of complex scaffolds.
At this stage, we were aware of a number of P450 monooxygenases reported to catalyze C3 hydroxylation of sclareolide, 48 though their use was limited to 50 mg scale reactions performed under high substrate dilution. A brief enzyme engineering campaign was conducted, wherein several variants of P450BM3 were tested for activity on sclareol and sclareolide. Out of this screen emerged BM3 MERO1, which selectively hydroxylated C3 of sclareolide to >95% conversion and exhibited low levels of analogous activity on sclareol. Importantly, hydroxylation of sclareolide was amenable to large scale, consistently giving 80–90% conversion and 60–70% isolated yield of 91. Moreover, the use of phosphite dehydrogenase Opt13 alongside BM3 MERO1 allowed for substoichiometric use of NADPH, greatly decreasing cost of material preparation.49
Having established the utility of BM3 MERO1, we next sought to access aldehyde 92 as the branching point toward the α-pyrone meroterpenoids. Thus, a four-step tailoring sequence was executed to fragment the lactone moiety of 91 and give 92 in 54% overall yield. A screen of Lewis and Brønsted acids identified phosphoric acid 95 as a suitable catalyst to append the aromatic pyrone moieties (96–98) in a formal [3 + 3] transformation. This strategy enabled access to 99 and 101, whereas 104 followed from initial dehydration of the tertiary alcohol to enal 93. En route to meroterpenoids 102, 103, and 105, chemoselective hydrogen atom transfer (HAT) reduction proved the only viable method to regio- and stereoselectively convert the C9–C11 alkene to the desired hydrogenated products. Lastly, the unique oxidation pattern of 106 required elaboration of 93 to acid chloride 94, followed by Friedel–Crafts acylation and cyclization/reduction for the completion of its synthesis.
In tandem with the above efforts, we also pursued syntheses of several C3-oxidized meroditerpenoids. Due to the poor activity of BM3 MERO1 on sclareol, the skeleton was elaborated in 5 steps to acid 107, and each intermediate was subjected to enzymatic hydroxylation with BM3 MERO1 and other P450BM3 mutants. Acid 107 showed the highest activity, and after further alanine screening we identified variant MERO1 L75A, which provided 62% isolated yield of the C3-hydroxylated species 108 on gram scale. Hydroxylated acid 108 was then transformed to iodide 109, which served as the divergency point in the series. Treatment of 109 with catalytic Ni(II) and partners 111 and 112 successfully afforded the desired coupling products, which each required two further steps to complete the synthesis of taondiol (113) and chevalone A (115), respectively.
Alternatively, access to decaturin E (118) and stypodiol (120) proceeded from diene 110 – obtained via tBuOK-mediated elimination of iodide 109 – through a single-electron-transfer-based (SET) [3 + 2] transformation. Whereas ceric ammonium nitrate (CAN) effected coupling of diene 110 and pyrone 116 to deliver decaturin E, phenol 117 required deployment of electrochemical methodology.50 With coupled product 119 in hand, we turned again to selective HAT reduction, followed by BBr3-mediated demethylation to give stypodiol in 58% yield over two steps (Scheme 3B). Overall, this work demonstrates the efficient merger of enzymatic oxidations and radical-based methodology toward several meroterpenoid natural products and provides the foundation for the synthesis of other drimane-containing structures.
Scheme 3.
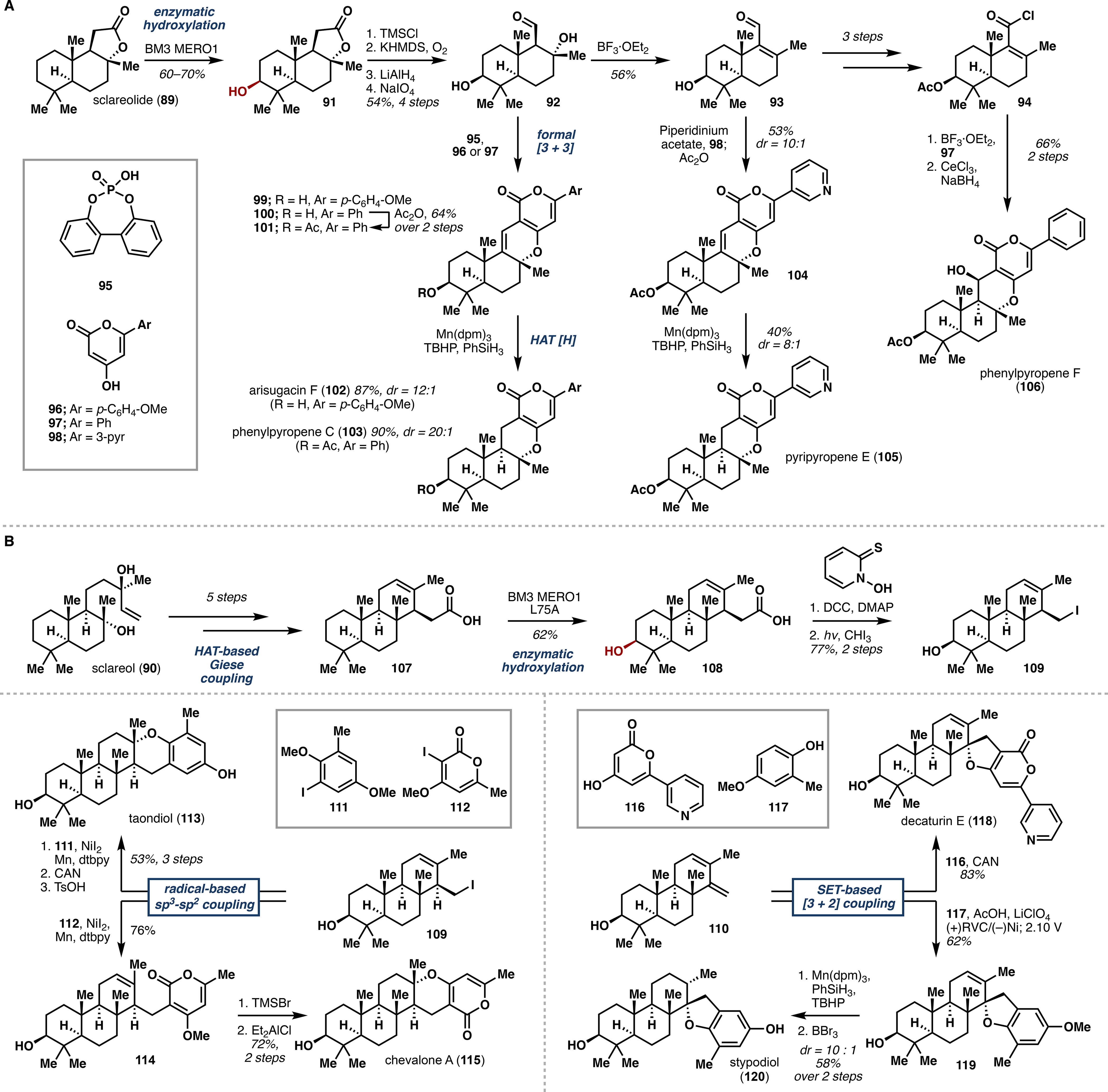
Enzymatic Hydroxylation in (A) Total Synthesis of α-Pyrone Meroterpenoids from Sclareolide and (B) Total Synthesis of Meroditerpenoids from Sclareol.
b. STEVIOSIDE DERIVATIZATION: ENT-KAURANE, ENT-ATISANE, ENT-TRACHYLOBANE DITERPENOIDS
Given the success of our meroterpenoid campaign, we looked to extend this paradigm of chiral pool synthesis to the ent-kaurane, ent-atisane, and ent-trachylobane diterpenoids.51 Arising from unique carbocationic rearrangements of ent-copalyl pyrophosphate,52 these terpenoid families share many structural features but differ in the architecture of their C and D rings. Furthermore, family members display a wide range of biological activities,52 making them appealing targets for medicinal chemistry evaluation and chemical probe development. Previous semisynthetic studies53 inspired us to examine the ent-kaurane diterpene stevioside (121) as a potential starting point for our endeavor. At $0.65/g, stevioside is available in bulk quantities and can be readily converted to the aglycones steviol (122) and isosteviol (137), though synthetic methodologies to selectively functionalize its ent-kaurane skeleton are exceedingly limited. Thus, we sought to develop a biocatalytic C–H oxidation program that would afford rapid access to not only the ent-kauranes, but also the ent-atisanes and ent-trachylobanes through manipulation of the C and D rings.
At the outset, it was crucial to identify enzymes that could selectively oxidize the A, B, and C rings of steviol and related structures. Previous collaboration with the Shen lab in the characterization of the platensimycin biosynthetic pathway (Figure 7), as well as our aforementioned work with P450BM3 variants, revealed several potential enzymes for this purpose.54 After a comprehensive screening campaign, three of these enzymes – P450BM3 variant BM3 MERO1 M177A, the Fe/αKG PtmO6, and the chimeric P450 PtmO5-RhFRed – emerged as promising biocatalysts to effect selective hydroxylation of the A, B, and C rings, respectively, of steviol and ent-kaurenoic acid (Figure 7).3 Critically, each enzyme was amenable to preparative scale and accepted a range of substrates en route to ent-kaurane, ent-atisane, and ent-trachylobane natural products.
Figure 7.
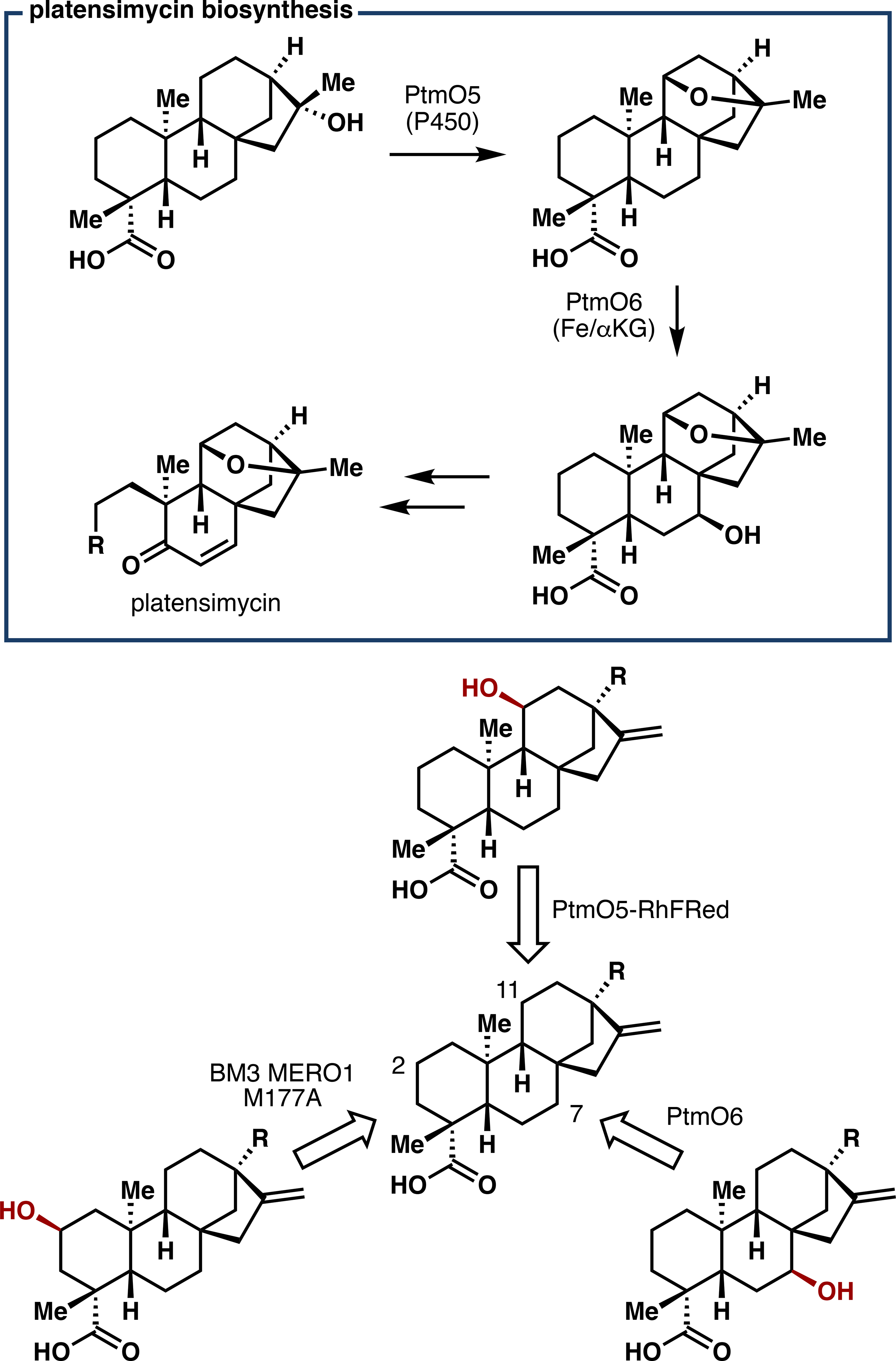
Snapshot of platensimycin biosynthesis involving hydroxylases PtmO5 and PtmO6; proposed application of biocatalytic oxidation to the steviol skeleton.
With three efficient terpene hydroxylases in hand, we first pursued divergent syntheses of mitrekaurenone (126), fujenoic acid (128), and pharboside aglycone (129), each of which would require only B ring oxidation (Scheme 4A). Starting from ent-kaurenoic acid (123), we performed C7 hydroxylation with PtmO6 to obtain secondary alcohol 124 in high yield as a single diastereomer. Toward mitrekaurenone, 124 was oxidized to ketone 125 and submitted to α-oxidation to effect intramolecular lactonization, giving 126 in five steps and 36% overall yield. Alternatively, it was found that ketone 125 could be oxidized by PtmO6 to C6-alcohol 127, which was then treated successively with NaIO4 and DMP to afford fujenoic acid in seven steps and 26% overall yield. Finally, access to pharboside aglycone came in three steps from secondary alcohol 124, featuring methyl esterification, dehydration, and dual dihydroxylation.
Scheme 4.
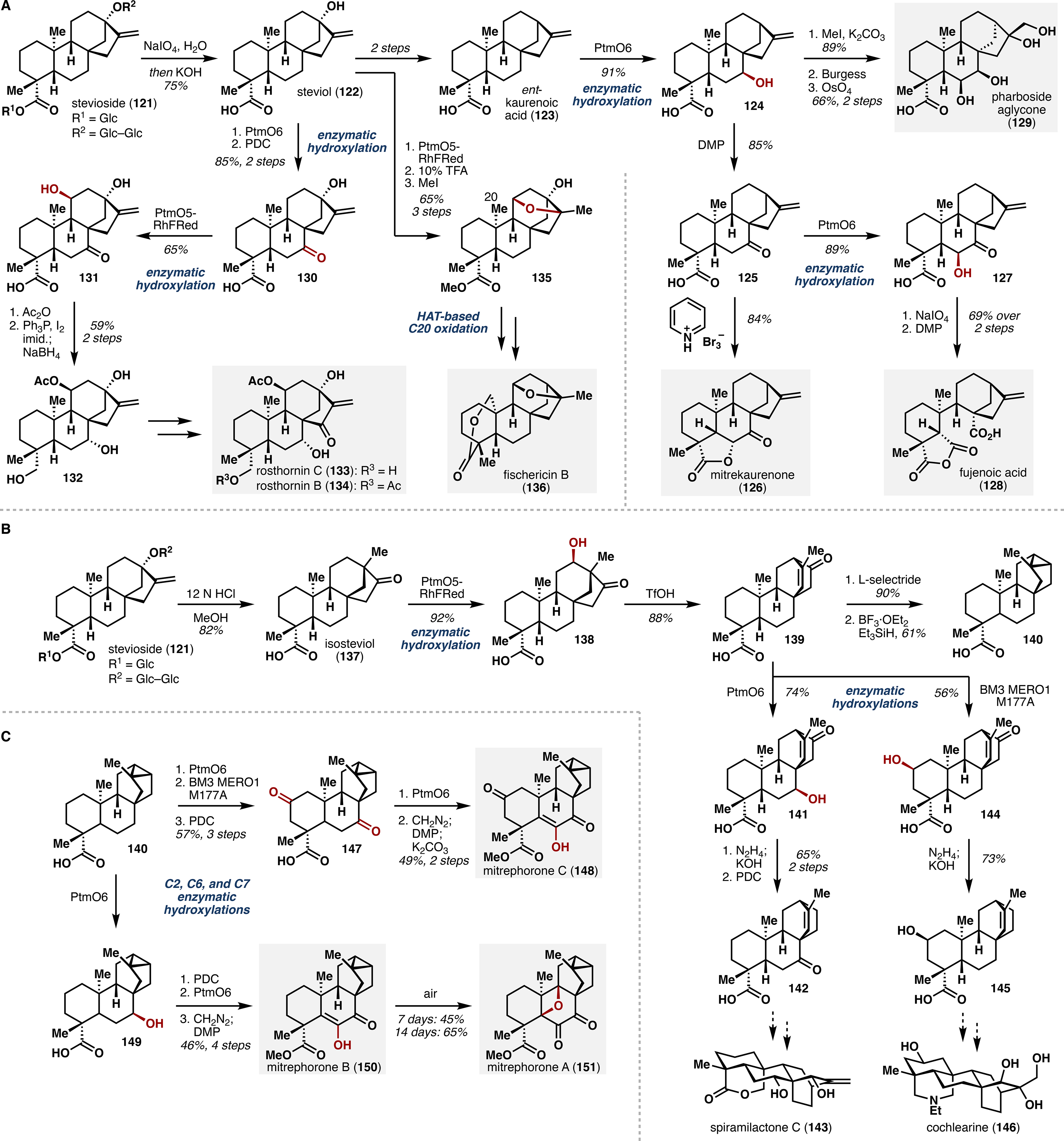
Applications of Enzymatic C–H Oxidation in (A) Total Synthesis of ent-Kaurane Diterpenes. (B) Carbocationic Rearrangement of the ent-Kaurane Skeleton and Synthetic Studies Toward ent-Atisane Natural Products. (C) Total Synthesis of ent-Trachylobane Natural Products.
To further demonstrate the utility of our approach, we targeted rosthornin B (134) and C (133) and fischericin B (136), ent-kauranes containing oxidations on multiple rings (Scheme 4A). Stevioside was first deglycosylated to steviol, which was then subjected to a two-step oxidation sequence involving enzymatic installation of the C7 alcohol with PtmO6 and subsequent PDC oxidation to ketone 130. Gratifyingly, PtmO5-RhFRed acted on 130, selectively introducing the C11 alcohol to give 131, which was elaborated in two further steps to intermediate 132. Following installation of the D-ring enone, synthesis of rosthornin C was completed, and rosthornin B could be accessed via acetylation of the C19 alcohol. Fischericin B, on the other hand, lacks any oxidation on its B ring. Starting again from steviol, a three-step sequence was employed to (1) hydroxylate at C11 with PtmO5-RhFRed, (2) form the corresponding ether, and (3) esterify the C19 carboxylate to give 135 in 65% overall yield. Completion of the synthesis proceeded via selective tailoring steps and HAT-based functionalization at C20, furnishing fischericin B in nine total steps and 25% overall yield.
We next sought to employ a carbocation rearrangement strategy to access the alternative C and D ring architectures of ent-atisane and ent-trachylobane natural products (Scheme 4B).52 In contrast to the ent-kaurane series, entry to these scaffolds began with isosteviol (137), obtained in a single step from stevioside. Stereo- and regioselective hydroxylation at C12 was accomplished in excellent yield with PtmO5-RhFRed to give secondary alcohol 138, which was then treated with trifluoromethanesulfonic acid to effect rearrangement to ent-atisane tetracycle 139. Further validating this approach, 139 could be converted to ent-trachylobane 140 in two additional steps. Thus, the ent-atisane and ent-trachylobane architectures could be accessed in two and four steps, respectively, from isosteviol, ultimately enabled by biocatalytic C–H hydroxylation.
Using 139 as a springboard to ent-atisane natural products, we targeted spiramilactone C (143), which contains further B ring oxidation, and cochleareine (146), which contains further A ring oxidation (Scheme 4B). Our strategy delivered late-stage synthetic intermediates 142 and 145, each of which contains the requisite functionalities to be elaborated to their corresponding target natural products. Analogously, intermediate 140 served as a gateway to the mitrephorone family of ent-trachylobane natural products (Scheme 4C). First, construction of mitrephorone C (148) was initiated by a series of enzymatic and chemical oxidations of 140, wherein BM3 MERO1 M177A and PtmO6 oxidized at C2 and C7, respectively, providing diketone 147 after reaction with PDC. At this juncture, we leveraged the promiscuous C6-hydroxylation activity of PtmO6 to install an α-hydroxyketone that was treated sequentially with diazomethane and DMP to effect methyl esterification and enone formation, yielding mitrephorone C in nine steps and 12% overall yield. Analogously, mitrephorones A and B were accessed by C7 hydroxylation of 140 with PtmO6 to give alcohol 149. PDC oxidation and a second PtmO6-catalyzed hydroxylation delivered an α-hydroxyketone that was elaborated in similar fashion to mitrephorone B (150). Serendipitously, it was discovered that 150 could undergo autooxidation to mitrephorone A (151) over several days, thus completing the synthesis of the mitrephorone family of natural products. Overall, our strategy toward these natural products not only represents a highly efficient, modular synthesis of various terpene scaffolds, but also lays the groundwork for combining biocatalytic and chemical oxidation methodologies in total synthesis.
4. CONCLUSIONS
Inspired by cutting-edge chemical methodology and by Nature’s enduring biosynthetic logic, the above work represents a synthetic paradigm that is distinct from but also complementary to other approaches in chemical synthesis. In particular, the use of enzymatic C–H oxidation to uniquely modify commercial starting materials and provide access to a larger variety of enantiopure building blocks reinvigorates the chiral pool approach to synthesis and, in turn, streamlines synthetic access to a wide variety of natural products. Excitingly, the number of characterized and uncharacterized biosynthetic enzymes is ever growing, providing ample opportunity for biocatalyst discovery and development to further expand this strategy. Though the synthetic repertoire is more complete today than ever before, we hope others are encouraged to view biocatalysis-enabled expansion of the chiral pool as an increasingly viable solution to current and future problems in total synthesis.
Acknowledgments
We are grateful to all past and present members of the Renata laboratory for their contributions to the work outlined above. We acknowledge funding from the National Institutes of Health (grant GM128895) and from The Scripps Research Institute. CNS acknowledges support from the Reba and Nat Newman Endowed Fellowship in the Skaggs Graduate School of Chemical and Biological Sciences.
Biographies
Carter Stout received his B.S. and M.S. in Chemistry from the University of Chicago in 2019, conducting research with Professor Scott Snyder. He is currently a graduate student at Scripps Research under the supervision of Professor Hans Renata, where his research interests include natural products total synthesis, biocatalysis, enzyme engineering, and reaction development.
Hans Renata received his B.A. degree from Columbia University in 2008, conducting research under the tutelage of Professor Tristan H. Lambert. He earned his Ph.D. from The Scripps Research Institute in 2013 under the guidance of Professor Phil S. Baran. After postdoctoral studies with Professor Frances H. Arnold at the California Institute of Technology, he started his independent career at The Scripps Research Institute in 2016. His research focuses on synthetic and biosynthetic studies of natural products and biocatalytic reaction developments.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Zwick CR III; Renata H Remote C–H Hydroxylation by an α-Ketoglutarate-Dependent Dioxygenase Enables Efficient Chemoenzymatic Synthesis of Manzacidin C and Proline Analogs. J. Am. Chem. Soc. 2018, 140, 1165–1169.This study resulted in the functional characterization of leucine 5-hydroxylase GriE and showcased its synthetic utility in the synthesis of rare alkaloid manzacidin C and in the construction of proline derivatives.
- (2).Li J; Li F; King-Smith E; Renata H Merging Chemoenzymatic and Radical-Based Retrosynthetic Logic for Rapid and Modular Synthesis of Oxidized Meroterpenoids. Nat. Chem. 2020, 12, 173–179.This work demonstrated the potential of biocatalytic C–H oxidation as an effective means to functionalize sclareol and sclareolide toward the synthesis of eight oxidized meroterpenoids from a handful of late-stage synthetic intermediates.
- (3).Zhang X; King-Smith E; Dong L-B; Yang L-C; Rudolf JD; Shen B; Renata H Divergent Synthesis of Complex Diterpenes Through a Hybrid Oxidative Approach. Science 2020, 369, 799–806.This effort utilized three terpene hydroxylases to selective oxidize various polycyclic diterpenes and enable the synthesis of nine complex natural products in 10 steps or less from steviol.
- (4).Amatuni A; Shuster A; Adibekian A; Renata H Concise Chemoenzymatic Total Synthesis and Identification of Cellular Targets of Cepafungin I. Cell Chem. Biol. 2020, 27, 1318–1326.This work employed biocatalytic lysine hydroxylation to enable concise synthesis of the potent proteasome inhibitor cepafungin I and analogues, which were then subjected to proteomic analysis to probe structure-activity relationships and confirm biological activity.
- (5).Yamaguchi J; Yamaguchi AD; Itami K C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]
- (6).(a) Walsh CT; Fischbach MA Natural Products Version 2.0: Connecting Genes to Molecules. J. Am. Chem. Soc. 2010, 132, 2469–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Harvey AL; Edrada-Ebel F; Quinn RJ The Re-Emergence of Natural Products for Drug Discovery in the Genomics Era. Nat. Rev. Drug Discovery 2015, 14, 111–129. [DOI] [PubMed] [Google Scholar]; (c) Zhang MM; Qiao Y; Ang EL; Zhao H Using Natural Products for Drug Discovery: The Impact of the Genomics Era. Expert Opin. Drug Discovery 2017, 12, 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Pyser JB; Baker Dockrey SA; Benitez Rodriguez A; Joyce LA; Wiscons RA; Smith JL; Narayan ARH; Stereodivergent, Chemoenzymatic Synthesis of Azaphilone Natural Products. J. Am. Chem. Soc. 2019, 141 (46), 18551–18559. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) King-Smith E; Zwick CR III; Renata H Applications of Oxygenases in the Chemoenzymatic Total Synthesis of Complex Natural Products. Biochemistry 2018, 57, 403–412. [DOI] [PubMed] [Google Scholar]; (c) Loskot SA; Romney DK; Arnold FH; Stoltz BM Enantioselective Total Synthesis of Nigelladine A via Late-Stage C–H Oxidation Enabled by an Engineered P450 Enzyme. J. Am. Chem. Soc. 2017, 139 (30), 10196–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lowell AN; DeMars MD II; Slocum ST; Yu F; Anand K; Chemler JA; Korakavi N; Priessnitz JK; Park SR; Koch AA; Schultz PJ; Sherman DH Chemoenzymatic Total Synthesis and Structural Diversification of Tylactone-Based Macrolide Antibiotics Through Late-Stage Polyketide Assembly, Tailoring, and C–H Functionalization. J. Am. Chem. Soc. 2017, 139 (23), 7913–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kolev JN; O’Dwyer KM; Jordan CT; Fasan R Discovery of Potent Parthenolide-Based Antileukemic Agents Enabled by Late-Stage P450-Mediated C–H Functionalization. ACS Chem. Biol. 2014, 9 (1), 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Chekan JR; McKinnie SMK; Moore ML; Poplawski SG; Michael TP; Moore BS Scalable Biosynthesis of the Seaweed Neurochemical, Kainic Acid. Angew. Chem. Int. Ed. 2019, 58 (25), 8454–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hamed RB; Gomez-Castellanos JR; Henry L; Ducho C; McDonough MA; Schofield CJ The Enzymes of β-Lactam Biosynthesis. Nat. Prod. Rep. 2013, 30, 21–107. [DOI] [PubMed] [Google Scholar]; (c) Klein C; Hüttel W A Simple Procedure for Selective Hydroxylation of L-Proline and L-Pipecolic Acid with Recombinantly Expressed Proline Hydroxylases. Adv. Synth. Catal. 2011, 353 (8), 1375–1383. [Google Scholar]; (d) Zwick CR III; Renata H Harnessing the Biocatalytic Potential of Iron- and α-Ketoglutarate-Dependent Dioxygenases in Natural Product Total Synthesis. Nat. Prod. Rep. 2020, 37, 1065–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Blaser H-U The Chiral Pool as a Source of Enantioselective Catalysts and Auxiliaries. Chem. Rev. 1992, 92, 935–952. [Google Scholar]; (b) Brill ZG; Condakes ML; Ting CP; Maimone TJ Navigating the Chiral Pool in the Total Synthesis of Complex Terpene Natural Products. Chem. Rev. 2017, 117 (18), 11753–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Noisier AFM; Brimble MA C–H Functionalization in the Synthesis of Amino Acids and Peptides. Chem. Rev. 2014, 114 (18), 8775–8806. [DOI] [PubMed] [Google Scholar]
- (11).(a) Gao S-S; Naowarojna N; Cheng R; Liu X; Liu P Recent Examples of α-Ketoglutarate-Dependent Mononuclear Non-Haem Iron Enzymes in Natural Products Biosyntheses. Nat. Prod. Rep. 2018, 35, 792–837. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu L,-F; Meng S; Tang G-L Ferrous Iron and α-Ketoglutarate-Dependent Dioxygenases in the Biosynthesis of Microbial Natural Products. Biochim. Biophys. Acta. 2016, 1864 (5), 453–470. [DOI] [PubMed] [Google Scholar]
- (12).Hedges JB; Ryan KS Biosynthetic Pathways to Nonproteinogenic α-Amino Acids. Chem. Rev. 2020, 120, 3161–3209. [DOI] [PubMed] [Google Scholar]
- (13).(a) Hibi M; Kawashima T; Sokolov PM; Smirnov SV; Kodera T; Sugiyama M; Shimizu S; Yokozeki K; Ogawa J L-Leucine 5-Hydroxylase of Nostoc Punctiforme is a Novel Type of Fe(II)/α-Ketoglutarate-Dependent Dioxygenase That is Useful as a Biocatalyst. Appl. Microbiol. Biotechnol. 2013, 97 (6), 2467–2472. [DOI] [PubMed] [Google Scholar]; (b) Jiang W; Cacho RA; Chiou G; Garg NK; Tang Y; Walsh CT EcdGHK Are Three Tailoring Iron Oxygenases for Amino Acid Building Blocks of the Echinocandin Scaffold. J. Am. Chem. Soc. 2013, 135 (11), 4457–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kling A; Lukat P; Almeida DV; Bauer A; Fontaine E; Sordello S; Zaburannyi N; Herrmann J; Wenzel S,C; König C; Ammerman NC; Barrio MB; Borchers K; Bordon-Pallier F; Brönstrup M; Courtemanche G; Gerlitz M; Geslin M; Hammann P; Heinz DW; Hoffmann H; Klieber S; Kohlmann M; Kurz M; Lair C; Matter H; Nuermberger E; Tyagi S; Fraisse L; Grosset JH; Lagrange S; Müller R Targeting DnaN for Tuberculosis Therapy Using Novel Griselimycins. Science 2015, 348 (6239), 1106–1112. [DOI] [PubMed] [Google Scholar]
- (15).Lukat P; Katsuyama Y; Wenzel S; Binz T; König C; Blankenfeldt W; Brönstrup M; Müller R Biosynthesis of Methyl-Proline Containing Griselimycins, Natural Products with Anti-Tuberculosis Activity. Chem. Sci. 2017, 8, 7521–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kobayashi J; Kanda F; Ishibashi M; Shigemori H Manzacidins A-C, Novel Tetrahydropyrimidine Alkaloids from the Okinawan Marine Sponge Hymeniacidon sp. J. Org. Chem. 1991, 56 (14), 4574–4576. [Google Scholar]
- (17).Namba K; Shinada T; Teramoto T; Ohfune Y Total Synthesis and Absolute Structure of Manzacidin A and C. J. Am. Chem. Soc. 2000, 122 (43), 10708–10709. [Google Scholar]
- (18).Huang X; Groves JT Taming Azide Radicals for Catalytic C–H Azidation. ACS Catal. 2016, 6 (2), 751–759. [Google Scholar]
- (19).(a) Hashimoto T; Maruoka K Synthesis of Manzacidins: A Stage for the Demonstration of Synthetic Methodologies. Org. Biomol. Chem. 2008, 6, 829–835. [DOI] [PubMed] [Google Scholar]; (b) Tran K; Lombardi PJ; Leighton JL An Efficient Asymmetric Synthesis of Manzacidin C. Org. Lett. 2008, 10, 3165–3167. [DOI] [PubMed] [Google Scholar]; (c) Yoshimura T; Kinoshita T; Yoshioka H; Kawabata T Asymmetric Intermolecular Conjugate Addition of Amino Acid Derivatives via Memory of Chirality: Total Synthesis of Manzacidin A. Org. Lett. 2013, 15, 864–867. [DOI] [PubMed] [Google Scholar]; (d) Ichikawa Y; Okumura K; Matsuda T; Nakano K; Kotsuki H Synthesis of Manzacidin A and C: Efficient Construction of Quaternary Carbon Stereocenters Bearing Nitrogen Substituents. Org. Biomol. Chem. 2012, 10, 614–622. [DOI] [PubMed] [Google Scholar]; (e) Tong TMT; Soeta T; Suga T; Kawamoto K; Hayashi Y; Ukaji Y Formal Total Synthesis of Manzacidin C Based on Asymmetric 1,3-Dipolar Cycloaddition of Azomethine Imines. J. Org. Chem. 2017, 82, 1969–1976. [DOI] [PubMed] [Google Scholar]
- (20).Zhang X; Renata H Efficient Chemoenzymatic Synthesis of (2S,3R)-3-Hydroxy-3-Methylproline, A Key Fragment in Polyoxypeptin A and FR225659. Tetrahedron 2019, 75, 3253–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zwick CR III; Renata H A One-Pot Chemoenzymatic Synthesis of (2S,4R)-4-Methylproline Enables the First Total Synthesis of Antiviral Lipopeptide Cavinafungin B. Tetrahedron, 2018, 74, 6469–6473. [Google Scholar]
- (22).Zhang X; King-Smith E; Renata H Total Synthesis of Tambromycin by Combining Chemocatalytic and Biocatalytic C–H Functionalization. Angew. Chem. Int. Ed. 2018, 57, 5037–5041. [DOI] [PubMed] [Google Scholar]
- (23).(a) Goering AW; McClure RA; Doroghazi JR; Albright JC; Haverland NA; Zhang Y; Ju K-S; Thomson RJ; Metcalf WW; Kelleher NL Metabologenomics: Correlation of Microbial Gene Clusters with Metabolites Drives Discovery of a Nonribosomal Peptide with an Unusual Amino Acid Monomer. ACS Cent. Sci. 2016, 2, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Izumikawa M; Kawahara T; Kagaya N; Yamamura H; Hayakawa M; Takagi M; Yoshida M; Doi T; Shin-ya K Pyrrolidine-Containing Peptides, JBIR-126, −148, and −149, from Streptomyces sp. NBRC 111228. Tetrahedron Lett. 2015, 56, 5333–5336. [Google Scholar]
- (24).Baud D; Saaidi P-L; Monfleur A; Harari M; Cuccaro J; Fossey A; Besnard M; Debard A; Mariage A; Pellouin V; Petit J-L; Salanoubat M; Weissenbach J; de Berardinis V; Zaparucha A Synthesis of Mono- and Dihydroxylated Amino Acids with New α-Ketoglutarate-Dependent Dioxygenases: Biocatalytic Oxidation of C–H Bonds. ChemCatChem 2014, 6 (10), 3012–3017. [Google Scholar]
- (25).(a) Betancor L; Fernández M-J; Weissman KJ Improved Catalytic Activity of a Purified Multienzyme from a Modular Polyketide Synthase After Coexpression with Streptomyces Chaperones in Escherichia coli. ChemBioChem 2008, 9 (18), 2962–2966. [DOI] [PubMed] [Google Scholar]; (b) Li J; Zhang X; Renata H Asymmetric Chemoenzymatic Synthesis of Podophyllotoxin and Related Aryltetralin Lignans. Angew. Chem. Int. Ed. 2019, 58, 11657–11660. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Thomas JG; Ayling A; Baneyx F Molecular Chaperones, Folding Catalysts, and the Recovery of Active Recombinant Proteins from E. coli. To Fold or to Refold. Appl. Biochem. Biotechnol. 1997, 66 (3), 197–238. [DOI] [PubMed] [Google Scholar]
- (26).Feng Y; Holte D; Zoller J; Umemiya S; Simke LR; Baran PS Total Synthesis of Verruculogen and Fumitremorgin A Enabled by Ligand-Controlled C–H Borylation. J. Am. Chem. Soc. 2015, 137 (32), 10160–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).(a) Oka M; Nishiyama Y; Ohta S; Kamei H; Konishi M; Miyaki T; Oki T; Kawaguchi H Glidobactins A, B and C, New Antitumor Antibiotics. I. Production, Isolation, Chemical Properties and Biological Activity. J. Antibiot. 1988, 41, 1331–1337. [DOI] [PubMed] [Google Scholar]; (b) Oka M; Yaginuma K; Numata K; Konishi M; Oki T; Kawaguchi H Glidobactins A, B and C, New Antitumor Antibiotics. II. Structure Elucidation. J. Antibiot. 1988, 41, 1338–1350. [DOI] [PubMed] [Google Scholar]; (c) Oka M; Iwata K-I; Nishiyama Y; Kamei H; Konishi M; Oki T; Kawaguchi H Chemical Modification of the Antitumor Antibiotic Glidobactin. J. Antibiot. 1988, 41, 1812–1822. [DOI] [PubMed] [Google Scholar]
- (28).Guerrero-Garcia TA; Gandolfi S; Laubach JP; Hideshima T; Chauhan D; Mitsiades C; Anderson KC; Richardson PG The Power of Proteasome Inhibition in Multiple Myeloma. Expert Rev. Proteomics 2018, 15, 1033–1052. [DOI] [PubMed] [Google Scholar]
- (29).Stein ML; Beck P; Kaiser M; Dudler R; Becker CFW; Groll M One-Shot NMR Analysis of Microbial Secretions Identifies Highly Potent Proteasome Inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 18367–18371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Schellenberg B; Bigler L, Dudler R Identification of Genes Involved in the Biosynthesis of the Cytotoxic Compound Glidobactin from a Soil Bacterium. Environ. Microbiol. 2007, 9, 1640–1650. [DOI] [PubMed] [Google Scholar]
- (31).(a) Izumiya N, Fujita Y; Irreverre F; Witkop B The Synthesis of Erythro-G-Hydroxy-L-Lysine and Its Nonoccurence in Collagen. Biochemistry 1965, 4, 2501–2507. [Google Scholar]; (b) Marin J; Didierjean C; Aubry A; Casimir J-R; Briand J-P; Guichard G Synthesis of Enantiopure 4-Hydroxypipecolate and 4-Hydroxylysine Derivatives from a Common 4,6-Dioxopiperidinecarboxylate Precursor. J. Org. Chem. 2004, 69, 130–141. [DOI] [PubMed] [Google Scholar]; (c) Schmidt U; Kleefeldt A; Mangold R The Synthesis of Glidobactin A. J. Chem. Soc. Chem. Commun. 1992, 22, 1687–1689. [Google Scholar]
- (32).Amatuni A; Renata H Identification of a Lysine 4-Hydroxylase from the Glidobactin Biosynthesis and Evaluation of Its Biocatalytic Potential. Org. Biomol. Chem. 2019, 17, 1736–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kamiński ZJ; Kolesińska B; Sabatino G; Chelli M; Rovero P; Blaszczyk M; Główka ML; Papini AM N-Triazinylammonium Tetrafluoroborates. A New Generation of Efficient Coupling Reagents Useful for Peptide Synthesis. J. Am. Chem. Soc. 2005, 127 (48), 16912–16920. [DOI] [PubMed] [Google Scholar]
- (34).(a) Brandi L; Fabbretti A; La Teana A; Abbondi M; Losi D; Donadio S; Gualerzi CO Specific, Efficient, and Selective Inhibition of Prokaryotic Translation Initiation by a Novel Peptide Antibiotic. Proc. Natl. Acad. Sci. USA 2006, 103 (1), 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brandi L; Lazzarini A; Cavaletti L; Abbondi E; Corti E; Ciciliato L; Gastaldo S; Marazzi A; Feroggio M; Fabbretti A; Maio A; Colombo L; Donadio S; Marinelli F; Losi D; Gualerzi CO; Selva E Novel Tetrapeptide Inhibitors of Bacterial Protein Synthesis Produced by a Streptomyces Sp. Biochemistry 2006, 45, 3692–3702. [DOI] [PubMed] [Google Scholar]
- (35).Mattay J; Hüttel W Pipecolic Acid Hydroxylases: A Monophyletic Clade Among cis-Selective Bacterial Proline Hydroxylases that Discriminates L-Proline. ChemBioChem 2017, 18, 1523–1528. [DOI] [PubMed] [Google Scholar]
- (36).Binz TM; Maffioli SI; Sosio M; Donadio S; Müller R Insights into an Unusual Nonribosomal Peptide Synthetase Biosynthesis: Identification and Characterization of the GE81112 Biosynthetic Gene Cluster. J. Biol. Chem. 2010, 285 (43), 32710–32719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zwick CR III; Sosa MB; Renata H Characterization of a Citrulline 4-Hydroxylase from Nonribosomal Peptide GE81112 Biosynthesis and Engineering of Its Substrate Specificity for the Chemoenzymatic Synthesis of Enduracididine. Angew. Chem. Int. Ed. 2019, 58, 18854–18858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chang C-Y; Lyu S-Y; Liu Y-C; Hsu N-S; Wu C-C; Tang C-F; Lin K-H; Ho J-Y; Wu C-J; Tsai M-D; Li T-L Biosynthesis of Streptolidine Involved Two Unexpected Intermediates Produced by a Dihydroxylase and a Cyclase through Unusual Mechanisms. Angew. Chem. Int. Ed. 2014, 53, 1943–1948; Angew. Chem. 2014, 126, 1974–1979. [DOI] [PubMed] [Google Scholar]
- (39).(a) Ju J; Ozanick SG; Shen B; Thomas MG Conversion of (2S)-Arginine to (2S,3R)-Capreomycidine by VioC and VioD from the Viomycin Biosynthetic Pathway of Streptomyces sp. Strain ATCC11861. ChemBioChem 2004, 5, 1281–1285. [DOI] [PubMed] [Google Scholar]; (b) Yin X; Zabriskie TM VioC is a Non-Heme Iron, α-Ketoglutarate-Dependent Oxygenase that Catalyzes the Formation of 3S-Hydroxy-L-Arginine during Viomycin Biosynthesis. ChemBioChem 2004, 5, 1274–1277. [DOI] [PubMed] [Google Scholar]; (c) Dunham NP; Chang W-C; Mitchell AJ; Martinie RJ; Zhang B; Bergman JA; Rajakovich LJ; Wang B; Silakov A; Krebs C; Boal AK; Bollinger JM Jr. Two Distinct Mechanisms for C–C Desaturation by Iron(II)- and 2-(Oxo)glutarate-Dependent Oxygenases: Importance of α-Heteroatom Assistance. J. Am. Chem. Soc. 2018, 140, 7116–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zwick CR III; Sosa MB; Renata H Modular Chemoenzymatic Synthesis of GE81112 B1 and Related Analogues Enables Elucidation of Its Key Pharmacophores. J. Am. Chem. Soc. 2021, 143 (3), 1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jürjens G; Schuler SMM; Kurz M; Petit S; Couturier C; Jeannot F; Nguyen F; Wende RC; Hammann PE; Wilson DN; Bacqué E; Pöverlein C; Bauer A Total Synthesis and Structural Revision of the Antibiotic Tetrapeptide GE81112A. Angew. Chem. Int. Ed. 2018, 57, 12157–12161. [DOI] [PubMed] [Google Scholar]
- (42).Dixon DD; Lockner JW; Zhou Q; Baran PS Scalable, Divergent Synthesis of Meroterpenoids via “Borono-sclareolide.” J. Am. Chem. Soc. 2012, 134 (20), 8432–8435. [DOI] [PubMed] [Google Scholar]
- (43).Matsuda Y; Abe I Biosynthesis of Fungal Meroterpenoids. Nat. Prod. Rep. 2016, 33, 26–53. [DOI] [PubMed] [Google Scholar]
- (44).Sunazuka T; Omura S Total Synthesis of α-Pyrone Meroterpenoids, Novel Bioactive Microbial Metabolites. Chem. Rev. 2005, 105, 4559–4580. [DOI] [PubMed] [Google Scholar]
- (45).Macías FA; Carrera C; Galindo JCG Brevianes Revisited. Chem. Rev. 2014, 114, 2717–2732. [DOI] [PubMed] [Google Scholar]
- (46).Corey EJ; Noe MC; Lin S A Mechanistically Designed bis-Cinchona Alkaloid Ligand Allows Position- and Enantioselective Dihydroxylation of Farnesol and Other Oligoprenyl Derivatives at the Terminal Isopropylidine Unit. Tetrahedron Lett. 1995, 60, 8741–8744. [Google Scholar]
- (47).(a) Nagamitsu T; Sunazuka T; Obata R; Tomoda H; Tanaka H; Harigaya Y; Omura S; Smith AB III. Total Synthesis of (+)-Pyripyropene A, a Potent, Orally Bioavailable Inhibitor of Acyl-CoA:Cholesterol Acytransferase. J. Org. Chem. 1995, 60, 8126–8127. [Google Scholar]; (b) Abad A; Agulló C; Arnó M; Cuñat AC; Meseguer B; Zaragozá RJ An Efficient Stereoselective Synthesis of Stypodiol and Epistypodiol. J. Org. Chem. 1998, 63, 5100–5106. [Google Scholar]; (c) Takikawa H; Imamura Y; Sasaki M Synthesis and Absolute Configuration of Brevione B, an Allelochemical Isolated from Penicillium sp. Tetrahedron 2006, 62, 39–48. [Google Scholar]
- (48).(a) Zhang K; El Damaty S; Fasan R P450 Fingerprinting Method for Rapid Discovery of Terpene Hydroxylating P450 Catalysts with Diversified Regioselectivity. J. Am. Chem. Soc. 2011, 133, 3242–3245. [DOI] [PubMed] [Google Scholar]; (b) Hall EA; Sarkar MR; Lee JHZ; Munday SD; Bell SG Improving the Monooxygenase Activity and the Regio- and Stereoselectivity of Terpenoid Hydroxylation Using Ester Directing Groups. ACS Catal. 2016, 6, 6306–6317. [Google Scholar]
- (49).McLachlan MJ; Johannes TW; Zhao H Further Improvement of Phosphite Dehydrogenase Thermostability by Saturation Mutagenesis. Biotechnol. Bioeng. 2008, 99, 268–274. [DOI] [PubMed] [Google Scholar]
- (50).Chiba K; Fukuda M; Kim S; Kitano Y; Tada M Dihydrobenzofuran Synthesis by an Anodic [3 + 2] Cycloaddition of Phenols and Unactivated Alkenes. J. Org. Chem. 1999, 64, 7654–7656. [Google Scholar]
- (51).Liu M; Wang W-G; Sun H-D; Pu J-X Diterpenoids From Isodon Species: An Update. Nat. Prod. Rep. 2017, 34, 1090–1140. [DOI] [PubMed] [Google Scholar]
- (52).Hong HJ; Tantillo DJ Formation of Beyerene, Kaurene, Trachylobane, and Atiserene Diterpenes by Rearrangements That Avoid Secondary Carbocations. J. Am. Chem. Soc. 2010, 132, 5375–5386. [DOI] [PubMed] [Google Scholar]
- (53).(a) Cherney EC; Lopchuk JM; Green JC; Baran PS A Unified Approach to ent-Atisane Diterpenes and Related Alkaloids: Synthesis of (–)-Methyl Atisenoate, (–)-Isoatisine, and the Hetidine Skeleton. J. Am. Chem. Soc. 2014, 136, 12592–12595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kobayashi S; Shibukawa K; Hamada Y; Kuruma T; Kawabata A; Masuyama A Synthesis of (–)-Tripterifordin and (–)-Neotripterifordin from Stevioside. J. Org. Chem. 2018, 83, 1606–1613. [DOI] [PubMed] [Google Scholar]
- (54).(a) Rudolf JD; Dong L-B; Zhang X; Renata H; Shen B Cytochrome P450-Catalyzed Hydroxylation Initiating Ether Formation in Platensimycin Biosynthesis. J. Am. Chem. Soc. 2018, 140, 12349–12353. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dong L-B; Zhang X; Rudolf JD; Deng MR; Kalkreuter E; Cepeda AJ; Renata H; Shen B Cryptic and Stereospecific Hydroxylation, Oxidation, and Reduction in Platensimycin and Platencin Biosynthesis. J. Am. Chem. Soc. 2019, 141, 4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]