

Bone marrow serves as an endocrine organ that regulates the pathophysiological function of arteries via MYDGF.
Abstract
Whether bone marrow modulates systemic metabolism remains unknown. Here, we found that (i) myeloid cell–specific myeloid-derived growth factor (MYDGF) deficiency exacerbated vascular inflammation, adhesion responses, endothelial injury, and atherosclerosis in vivo. (ii) Myeloid cell–specific MYDGF restoration attenuated vascular inflammation, adhesion responses and leukocyte homing and alleviated endothelial injury and atherosclerosis in vivo. (iii) MYDGF attenuated endothelial inflammation, apoptosis, permeability, and adhesion responses induced by palmitic acid in vitro. (iv) MYDGF alleviated endothelial injury and atherosclerosis through mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4)/nuclear factor κB (NF-κB) signaling. Therefore, we concluded that MYDGF inhibits endothelial inflammation and adhesion responses, blunts leukocyte homing, protects against endothelial injury and atherosclerosis in a manner involving MAP4K4/NF-κB signaling, and serves as a cross-talk factor between bone marrow and arteries to regulate the pathophysiology of arteries. Bone marrow functions as an endocrine organ and serves as a potential therapeutic target for metabolic disorders.
INTRODUCTION
Atherosclerosis is highly prevalent in older persons, has catastrophic consequences in their quality of life, and increases disability and mortality in this population (1, 2). It is well known that inflammation plays a key role in the development of atherosclerosis (3, 4). This inflammatory process leads to the development of complex lesions or plaques (1, 5). Chronic vascular inflammation increases cell surface adhesion molecule up-regulation in the vessel wall, which is required to mediate monocyte extravasation from blood vessels into the subendothelial space in response to vascular injury (4). On the other hand, anti-inflammatory therapy decreases vascular adhesion molecule expression, delays plaque development, and stabilizes late-stage lesions (4, 6). Of note, endothelial dysfunction is an early pathophysiological change in the development of atherosclerosis that represents a key step in the maintenance of atherosclerosis, and inflammation can also damage the endothelium (7, 8). For these reasons, anti-inflammatory approaches for the treatment of atherosclerosis are actively pursued.
It is well known that bone marrow serves as a hematopoietic organ and is necessary for human survival and health. However, little is known about the other potential benefits of bone marrow on human diseases, such as its effects on metabolic disorders. Our recent one study showed that myeloid-derived growth factor (MYDGF), a secreted protein from bone marrow–derived monocytes and macrophages that is encoded by an open reading frame on chromosome 19 (C19orf10) (9), improves insulin resistance (IR) and glucose/lipid metabolism in diabetic mice (10). Metabolic disorders are closely associated with inflammation, endothelial dysfunction, and atherosclerosis (11, 12). In addition, some other growth factors such as fibroblast growth factor 21 and growth differentiation factor 11 display anti-inflammation effects in atherosclerosis (7, 11). Therefore, we hypothesized that myeloid cell–specific MYDGF may be involved in the regulation of atherosclerosis. Thus, in this study, we first aimed to test whether myeloid cell–specific MYDGF alleviates vascular inflammation and adhesion responses and protects against endothelial injury and atherosclerosis as well as the possible mechanisms involved. Second, we also explored whether MYDGF serves as a cross-talk factor between bone marrow and arteries to regulate the pathophysiology of arteries.
RESULTS
Decreased MYDGF levels and increased inflammation in atherosclerotic patients and mice
Our previous study found that plasma MYDGF declined in diabetic mice (10). Here, circulating MYDGF in carotid atherosclerosis (CAS) subjects was lower than that in controls (table S1). Accordingly, plasma MYDGF, bone marrow MYDGF mRNA and protein, as well as immunofluorescent expression in Western diet (WD)–fed apolipoprotein E knockout mice (AKO) mice (WD for 12 weeks) also decreased compared with those of normal chow diet (NCD)–fed wild-type (WT) mice (table S2 and fig. S1, A to C). Moreover, plasma MYDGF was positively associated with vascular endothelium–dependent dilation in patients and mice with atherosclerosis (fig. S1, D and E). These data indicated that MYDGF may be associated with endothelial dysfunction and atherosclerosis.
Inflammation is a critical factor in triggering or exacerbating atherosclerosis (4, 11). Likewise, our data showed increased inflammation including tumor necrosis factor–α (TNF-α), interleukin-1β (IL-1β) and IL-6, and adhesion molecules including vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-selectin expression in atherosclerotic patients and mice (fig. S1, F to G, and tables S1 and S2), indicating that MYDGF may be related to inflammation. In addition, in accordance with our studies (12, 13), the results also showed increased body weight and worsened lipid metabolism in patients and mice with atherosclerosis (tables S1 and S2).
Myeloid cell–specific MYDGF deficiency is associated with endothelial injury and inflammation in mice
First, we sought to explore the bone marrow integrity in peripheral blood or at the bone marrow in myeloid cell–specific MYDGF knockout (KO) mice. Compared to WT mice, the analysis of peripheral blood cells and distributions of nucleus in both bone marrow and cortical bone from toluidine blue staining of femur sections did not alter in KO mice (table S3 and fig. S2A), indicating that the bone marrow is integrity after myeloid cell–specific MYDGF KO in mice. Second, we found that the expression of MYDGF in the bone marrow of KO mice was completely knocked out, and low abundance expression of MYDGF was found in liver and white blood cells in KO mice (fig. S2B).
Next, we needed to explore the effects of myeloid cell–specific MYDGF deficiency on endothelial injury and inflammation in KO mice after 12 weeks of a WD or NCD, as shown in fig. S3A. The results showed that MYDGF deficiency reduced endothelium-dependent relaxation (by 38.9% in WD-KO mice and 25.1% in NCD-KO mice), increased endothelial apoptosis, and decreased the intact endothelium compared with those of both WD- and NCD-fed WT mice, and these effects were more severe in WD mice than NCD mice (Fig. 1, A to E). It is well known that inflammation accelerates endothelial injury (7, 14). The levels of inflammation (TNF-α, IL-1β, and IL-6) and adhesion molecules (VCAM-1, ICAM-1, and E-selectin) in both plasma and mouse aorta endothelial cells (MAECs) significantly increased in KO mice compared to those of both WD- and NCD-fed WT mice, and the effects were more severe in WD mice than NCD mice (Fig. 1F, fig. S3H, and table S4). In addition, consistent with previous results (10), worse lipid metabolism and increased body weight gain were observed in KO mice than in both WD- and NCD-fed WT mice, and the effects were more severe in WD mice than NCD mice (fig. S3, B to F, and table S4). In addition, larger epididymal white adipose tissue mass in KO mice was found than WT mice (fig. S 3G), and this may contribute to the increased body weight gain in KO mice. However, the fasting blood glucose (FBG), glycosylated hemoglobin (HbA1c), systolic blood pressure, diastolic blood pressure, food intake, total feces mass, or lipid content in the feces among different groups did not differ (table S4). These data indicate that myeloid cell–specific MYDGF deletion is related to endothelial injury and inflammation.
Fig. 1. Myeloid cell–specific MYDGF deficiency is associated with endothelial injury and inflammation in mice.
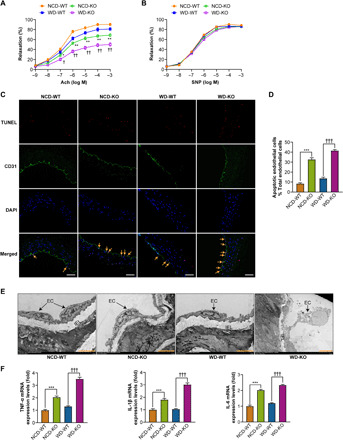
KO and WT mice aged 4 to 6 weeks were divided into four groups (NCD-WT, NCD-KO, WD-WT, and WD-KO mice) and were fed their respective diets for 12 weeks (10 mice in each group). (A and B) The vasodilation responses to (A) acetylcholine (Ach) and (B) sodium nitroprusside (SNP) (n = 6). (C) Representative images of terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining in sections of thoracic aortas. TUNEL (apoptotic cells, red), anti-CD31 (endothelial cells, green), and 4′,6-diamidino-2-phenylindole (DAPI) (nuclei, blue). Arrows indicate CD31/TUNEL colocalization. Scale bars, 200 μm. (D) The percentage of apoptotic endothelial cells (n = 6). (E) Representative electron microscopy images of endothelium. The arrows show endothelial cell (EC); IEL, internal elastic lamina. Scale bars, 50 μm. (F) The mRNA levels of inflammation (TNF-α, IL-1β, and IL-6) in MAECs of mice (n = 8). The data are presented as the means ± SEM. *P < 0.05 versus NCD-WT, **P < 0.01 versus NCD-WT, ***P < 0.001 versus NCD-WT; †P < 0.05 versus WD-WT, †† P < 0.01 versus WD-WT, †††P < 0.001 versus WD-WT
Myeloid cell–specific MYDGF deficiency is associated with atherosclerosis in AKO mice
We rationally questioned whether myeloid cell–specific MYDGF deficiency worsens the late stage of atherosclerosis. Thus, AKO and MYDGF and apolipoprotein E double gene knockout (DKO) mice were fed a WD for 12 weeks. As expected, MYDGF deficiency was related to endothelial dysfunction and increased the en face (3.1-fold) and cross-sectional atherosclerotic lesion area (2.9-fold) (Fig. 2, A to F) in DKO mice. As shown in Fig. 2 (G and H), the relative levels of vascular smooth muscle cells (VSMCs) and collagen were reduced in MYDGF-deficient mice, possibly contributing to the instability of atherosclerotic plaques. Notably, MYDGF deficiency increased the area of macrophage and T lymphocyte infiltration in plaques compared with those of AKO mice. In addition, increased inflammation (TNF-α, IL-1β, and IL-6) and adhesion molecule (VCAM-1, ICAM-1, and E-selectin) expressions were observed in MAECs of MYDGF-deficient mice (Fig. 2, I and J). On the basis of these results, myeloid cell–specific MYDGF deficiency rendered AKO mice more susceptible to atherosclerosis and instability of atherosclerotic plaques.
Fig. 2. Myeloid cell–specific MYDGF deficiency is associated with atherosclerotic plaque formation in AKO mice.
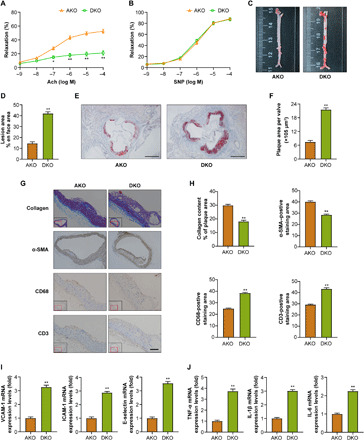
AKO and DKO mice aged 4 to 6 weeks were fed a WD for 12 weeks (10 mice in each group). (A and B) The vasodilatation reaction induced by Ach (A) and SNP (B) (n = 10). (C) Representative images of en face atherosclerotic lesions. (D) Quantitative analysis of (C) (n = 5). (E) Representative images of the cross-sectional area of the aortic root (n = 8). Scale bars, 500 μm. (F) Quantitative analysis of (E). (G) Representative immunohistochemical staining images of VSMCs [α–smooth muscle actin (α-SMA)], collagen (Masson), macrophages (anti-CD68), and T lymphocytes (anti-CD3) in aortic plaques. Scale bar, 100 μm. (H) Quantitative analysis of (G) (n = 8). (I and J) The mRNA levels of adhesion molecules (VCAM-1, ICAM-1, and E-selectin) (I) and inflammation (TNF-α, IL-1β, and IL-6) (J) in MAECs of mice (n = 5). The data are presented as the means ± SEM. *P < 0.05 and **P < 0.001.
Bone marrow transplantation alleviated endothelial injury and inflammation in KO mice
We were interested in endothelial injury and inflammation responses after MYDGF restoration from myeloid cell in KO mice. Initially, we needed to determine whether or not the receptor of MYDGF exists on endothelial cells. Thus, we performed a MYDGF label and tracing experiment in WT mice. The results showed that IRB-NHS-MYDGF binds to endothelium after 24 hours via tail vein injection, indicating the presence of MYDGF receptor on endothelium (fig. S4D). Next, we generated bone marrow chimeric mice to specifically address the protection of MYDGF restoration from myeloid cells on the early stage of atherosclerosis. After KO recipients were transplanted with bone marrow cells (BMCs) from WT mice for 4 weeks, MYDGF began to be expressed in the circulation, and expression was sustained at a high level to week 12 (fig. S4, A and B). Then, the formal bone marrow transplantation (BMT) experiment was performed as shown in fig. S4C. The results showed that transplantation of WT BMCs into WT and KO recipients improved endothelial function, decreased endothelial cell apoptosis, improved endothelial intact (Fig. 3, A to D), reduced inflammation (TNF-α, IL-1β, and IL-6) and adhesion molecule (VCAM-1, ICAM-1, and E-selectin) expression of MAECs, improved IR, and decreased body weight gain (fig. S5, A to H) compared with those of WT and KO recipients that were transplanted with KO BMCs, especially in KO recipients. These data indicated that the restoration of BMC-derived MYDGF inhibits inflammation and adhesion responses and protects against endothelial injury.
Fig. 3. BMT alleviated endothelial injury and atherosclerosis in mice.
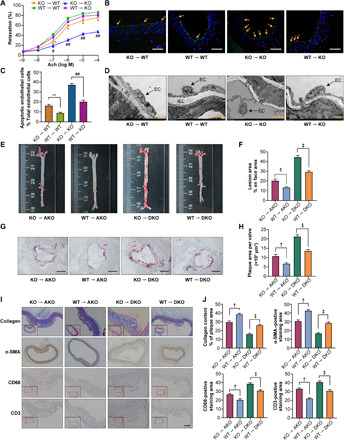
As shown in fig. S4C, BMT was performed, and atherosclerosis was assessed after WD feeding for 12 weeks (10 mice in each group). (A) The aortic vasodilatation induced by Ach in KO mice (n = 10). (B) Representative images of TUNEL staining in sections of thoracic aortas. Scale bars, 200 μm. (C) The percentage of apoptotic endothelial cells (n = 5). (D) Representative electron microscopy images of endothelium in KO mice (n = 5). Scale bars, 50 μm. (E) Representative images of en face atherosclerotic lesion areas in AKO and DKO mice. (F) Quantitative analysis of (E) (n = 5). (G) Representative images of the cross-sectional area of the aortic root in AKO and DKO mice. Scale bars, 500 μm. (H) Quantitative analysis of (G) (n = 8). (I) Representative immunohistochemical staining images of VSMCs, collagen, macrophages, and T lymphocytes in aortic plaques. Scale bar, 100 μm. (J) Quantitative analysis of (I) (n = 5). The data are presented as the means ± SEM. *P < 0.05 versus WT → WT and **P < 0.01 versus WT → WT; #P < 0.05 versus WT → KO and ##P < 0.001 versus WT → KO; †P < 0.01 versus WT → AKO; ‡P < 0.001 versus WT → DKO.
BMT attenuated atherosclerosis in AKO and DKO mice
Then, we hypothesized that MYDGF restoration from myeloid cells displays an anti–late-stage atherosclerotic effect. Therefore, we performed BMT in AKO or DKO recipients as shown in fig. S4C. The WT BMCs that were transplanted into AKO or DKO recipients attenuated atherosclerosis including the en face and cross-sectional atherosclerotic lesion area and improved cellular components in atherosclerotic plaques (Fig. 3, E to J) compared with those in AKO or DKO recipients that were transplanted with KO BMCs. These results revealed that BMC-derived MYDGF is sufficient and required to alleviate atherosclerosis.
Bone marrow–specific overexpression of MYDGF alleviated endothelial injury, atherosclerosis, and inflammation in mice
To further verify that bone marrow–derived MYDGF restoration inhibits inflammation and adhesion responses and attenuates endothelial injury and atherosclerosis in KO, AKO, and DKO mice, we injected adeno-associated viral (AAV)–MYDGF into the bone marrow cavity of mice to establish an animal model of MYDGF expression in bone marrow in situ. MYDGF expression could be detected in circulation 7 days after AAV-MYDGF transfer and lasted for up to 3 weeks in KO mice following a single injection (fig. S6A). Thus, KO mice received intramarrow injection of AAV-MYDGF every 3 weeks for 12 weeks, and the results showed that plasma MYDGF was maintained at a sustained high level (fig. S6B). In parallel, bone marrow MYDGF mRNA and protein levels, as well as the fluorescence expression, in AAV-MYDGF mice were higher than those in AAV–green fluorescent protein (GFP) mice at 12 weeks (fig. S6, C to E). Then, formal experiments including WT, KO + AAV-GFP (KO-GFP), and KO + AAV-MYDGF (KO-MYDGF) groups, as shown in fig. S6F, were performed. The results showed that AAV-MYDGF improved endothelial function, decreased endothelial cell apoptosis (Fig. 4, A to D), reduced inflammation and adhesion molecule expression of MAECs, improved IR, and decreased body weight gain (fig. S7, A to H), compared with those of KO-GFP mice. These data suggested that bone marrow–derived MYDGF alleviates inflammation and endothelial injury.
Fig. 4. The MYDGF overexpression of bone marrow in situ alleviated atherosclerosis.
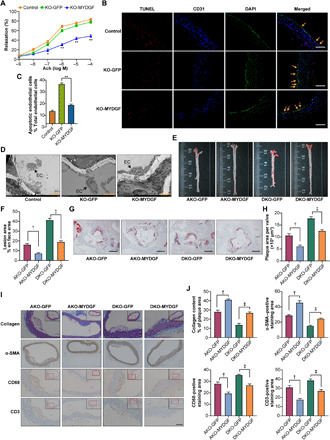
In situ MYDGF overexpression in bone marrow was performed in KO, AKO, and DKO mice aged 4 to 6 weeks. Then, the mice were fed a WD for 12 weeks, and atherosclerosis was assessed at the end of the experiment (10 mice in each group). (A) The aortic vasodilatation induced by Ach in KO mice (n = 10). (B) Representative images of TUNEL staining in sections of thoracic aortas. Scale bars, 200 μm. (C) The percentage of apoptotic endothelial cells (n = 9). (D) Representative electron microscopy images of endothelium. Scale bars, 50 μm. (E) Representative images of en face atherosclerotic lesions. (F) Quantitative analysis of (E) (n = 5). (G) Representative images of the cross-sectional area of the aortic root. Scale bars, 500 μm. (H) Quantitative analysis of (G) (n = 9). (I) Representative immunohistochemical staining images of VSMCs, collagen, macrophages, and T lymphocytes in aortic plaques. Scale bar, 100 μm. (J) Quantitative analysis of (I) (n = 5). The data are shown as the means ± SEM. *P < 0.05 versus KO-GFP and **P < 0.001 versus KO-GFP; †P < 0.001 versus AKO-GFP; ‡P < 0.001 versus DKO-GFP.
Next, to further test whether bone marrow–derived MYDGF blunted atherosclerosis in mice, mice were randomized to four groups [AKO + AAV-GFP (AKO-GFP), AKO + AAV-MYDGF (AKO-MYDGF), DKO + AAV-GFP (DKO-GFP), and DKO + AAV-MYDGF (AKO-MYDGF)], as shown in fig. S6F. As expected, AAV-MYDGF treatment reduced the atherosclerotic lesion area and improved cellular components within atherosclerotic plaques (Fig. 4, E to J) compared with AAV-GFP treatment. These results verified that bone marrow–derived MYDGF attenuated atherosclerosis.
MYDGF overexpression of bone marrow in situ attenuated leukocyte homing in the aortas of DKO mice
Inflammation induces leukocyte homing and macrophage accumulation within aortic plaques (3, 4). Thus, we investigated leukocyte recruitment after MYDGF restoration by MYDGF overexpression of bone marrow in situ in DKO mice that were fed a WD for 12 weeks. First, decreased mRNA expression of macrophage marker genes (F4/80 and CD68) and endothelial-derived chemokines, which contribute to leukocyte homing, was observed in the aortas of DKO + AAV-MYDGF (DKO-MYDGF) mice compared with that of DKO + AAV-GFP (DKO-GFP) mice (Fig. 5, A and B). Second, thioglycolate-stimulated peritoneal exudate cells were extracted from GFP-expressing mice and injected intravenously into DKO-MYDGF and DKO-GFP mice. The GFP-positive cell level was quantified within the aortic roots to assess leukocyte homing (Fig. 5C). A 60% reduction in GFP-positive cells within plaques in DKO-MYDGF mice was found compared with that of DKO-GFP mice (Fig. 5D). Third, leukocyte adhesion molecules ICAM-1 and VCAM-1 are required to mediate leukocyte homing in response to endothelial injury (4). Immunofluorescence (IF) of the aortic arches in DKO mice revealed significantly lower levels of both ICAM-1 and VCAM-1 protein expression after MYDGF restoration (fig. S8, A and B). In addition, the mRNA expression of VCAM-1, ICAM-1, and E-selectin in MAECs of the aorta showed similar changes after MYDGF restoration (fig. S8, C to E). Thus, bone marrow–derived MYDGF inhibits endothelial adhesion responses and alleviates leukocyte homing to and macrophage accumulation within atherosclerotic plaques.
Fig. 5. The MYDGF overexpression of bone marrow in situ decreased the leukocytes homing within aortic plaques from DKO mice.
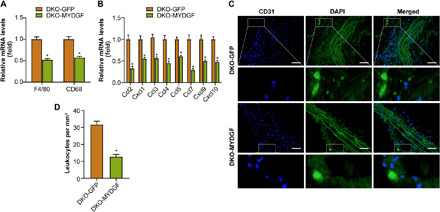
MYDGF overexpression of bone marrow in situ was performed in DKO mice aged 4 weeks, and leukocyte homing was analyzed in DKO-GFP or DKO-MYDGF mice that had been fed a WD for 12 weeks. (A) The mRNA expression of the macrophage markers F4/80 and CD68 in aortas. (B) The mRNA expression of the chemokines in aortas. (C and D) The homing of GFP leukocytes to atherosclerotic plaques 48 hours after intravenous injection into DKO-GFP and DKO-MYDGF mice that were fed a WD for 12 weeks. (C) Fluorescence micrograph of aortic root plaques. The dashed line indicates the plaque border. Inset, magnification of GFP leukocytes. Left, DAPI; middle, GFP; right, merge. Scale bars, 150 μm. (D) Quantification of GFP leukocytes per square millimeter of plaque (n = 5). The data represent the means ± SEM; *P < 0.001.
MYDGF reduced apoptosis, permeability, and inflammation of MAECs induced by palmitic acid
To test the direct effect of MYDGF on the endothelium, we treated MAECs with recombinant MYDGF (rMYDGF; 25-166, Cloud-Clone Corp., Wuhan) in vitro. Because palmitic acid (PA) is an atherosclerosis-relevant stimulus, we used PA as a stimulus for the in vitro experiments (11, 15). First, we determined that rMYDGF (50 ng/ml) for 48 hours are the optimum conditions for the proliferation of MAECs (fig. S9A). Second, the formal experiments showed that a 48-hour treatment with rMYDGF increased the proliferation and migration of MAECs compared with those of the vehicle treatment (fig. S9, B to E). Third, we chose PA (0.4 mM) and 24 hours as the optimum conditions in the following experiments (11). Compared with the vehicle, rMYDGF treatment attenuated endothelial apoptosis, decreased the apoptotic proteins (cleaved caspase-3 and bax) and increased antiapoptotic protein (bcl-2) expression, and decreased endothelial permeability, inflammation (TNF-α, IL-1β, and IL-6), and adhesion molecule (VCAM-1, ICAM-1, and E-selectin) expression as well as nuclear translocation of P (phosphorylated)-p65 in PA-induced MAECs (fig. S10, A to H). Collectively, we concluded that MYDGF induced proliferation and reduced apoptosis, permeability, and inflammation in PA-induced endothelial cells.
MYDGF reduced PA-induced inflammation in RAW264.7 macrophages
Macrophages represent a key cellular component of plaques (16). Our present data showed that MYDGF inhibited leukocyte homing and macrophage accumulation within aortic plaques. Thus, we explored whether MYDGF has a direct effect on macrophages. The results showed that MYDGF reduced the inflammation (TNF-α, IL-1β, and IL-6) induced by PA and reduced migration of macrophages (fig. S11, A to D). Collectively, the benefits of MYDGF on aortic plaques are related to reducing macrophage migration and inflammation.
BMCs from WT mice attenuated endothelial apoptosis and inflammation induced by PA in coculture experiments
To further verify myeloid cell–derived MYDGF as a factor involved in the cross-talk between bone marrow and the artery in vitro, we performed coculture experiments with BMCs and MAECs from WT mice under PA (0.4 mM) stimulation. The results showed that BMCs from WT mice blunted MAEC injury, as evidenced by decreased apoptosis, the Bax/Bcl-2 ratio and expression of cleaved caspase-3, decreased inflammation (TNF-α, IL-1β, and IL-6), and nuclear translocation of P-p65 as well as adhesion molecule (VCAM-1, ICAM-1, and E-selectin) expression in PA-induced MAECs compared to those in MAECs that were cocultured in the absence of BMCs or with the BMCs from KO mice (fig. S12, A to G). These results further supported directly that myeloid cell–derived MYDGF protects against vascular endothelial injury.
MAP4K4/NF-κB signaling is essential for the effects of MYDGF on atherosclerosis
We are still interested in the possible mechanisms for the protective effects of MYDGF on atherosclerosis. Atherosclerosis is a chronic inflammatory disease, and activation of NF-κB contributes to inflammatory reactions (4). Our results showed that MYDGF inhibits endothelial inflammation and adhesion response and blunts leukocyte homing and macrophage accumulation in aortic plaques. Thus, we first measured the NF-κB signal. The results showed that phosphorylated I-κ-B-α (P-IκBα) and nuclear P-p65 increased in MAECs of KO mice compared with WT mice (fig. S13A), while their expressions reduced in MYDGF-replenished mice (fig. S13B). In addition, pretreatment with rMYDGF inhibited PA-induced P-IκBα and nuclear P-p65 in MAECs (fig. S13C). These results showed that NF-κB signaling is involved in the beneficial effects of MYDGF on endothelial inflammation.
NF-κB signaling is regulated by c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinases (p38MAPK), extracellular signal–regulated kinase (ERK), inhibitor of nuclear factor kappa kinase β (IKKβ), and mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) (4, 17, 18). Our results suggested that P (phosphorylated)-MAP4K4 increased in MAECs of KO mice compared with those of WT mice (fig. S13D), while its expression decreased in MYDGF-replenished mice, but the other phosphorylated proteins (JNK, p38MAPK, ERK, and IKKβ) were not affected by MYDGF (fig. S13E). Consistently, pretreatment with rMYDGF not only inhibited PA-induced P-MAP4K4 expression in MAECs but also did not alter the expression of other phosphorylated proteins (JNK, p38MAPK, ERK, and IKKβ) (fig. S13F). These data suggest that MAP4K4 signaling may be involved in the modulation of NF-κB signaling.
Next, to confirm the effect of MAP4K4 signaling in vivo, an endothelial MAP4K4 knockdown mouse (MAP4K4 shRNA-VE-cadherin Cre-expressing mouse, MAP4K4 KD) was generated (Fig. 6A) (4). MAECs from the MAP4K4 KD showed a significant reduction in MAP4K4 mRNA and protein expression compared with the MAP4K4 short hairpin RNA (shRNA) mice that did not express Cre (control) (Fig. 6, B and C), confirming endothelial-specific MAP4K4 KD. MAP4K4 KD mice and control were crossed onto KO mice, and then male MAP4K4 KD/KO and control/KO mice were fed a WD for 12 weeks. Initially, the MAP4K4 KD/KO mice were further confirmed (fig. S14, A and B). Then, the formal experiment results showed that, in MAP4K4 KD/KO mice, less P-IκBα and nuclear P-p65 proteins in MAECs were observed (Fig. 6D); consequently, the endothelial injury and endothelial inflammation were improved in MAP4K4 KD/ KO mice than in control/KO mice (Fig. 6, E to J). These data confirmed that MAP4K4/NF-κB signal is essential for the benefit effects of MYDGF on endothelium.
Fig. 6. Endothelial-specific MAP4K4 KD vitiates MYDGF-mediated protection of endothelial injury in vivo.
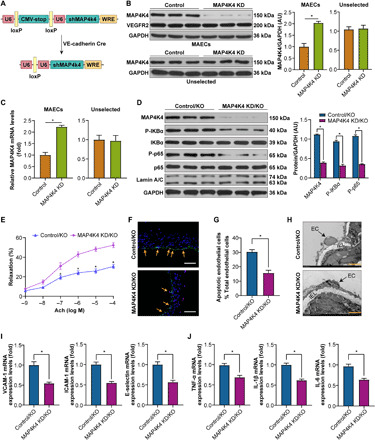
MAP4K4 KD/KO and control/KO mice were fed a WD for 12 weeks, and endothelial injury and NF-κB signaling were investigated. (A) Schematic of the transgenic construct used to generate MAP4K4 KD animals. (B and C) MAECs were derived from MAP4K4 KD and control mice. (B) MAP4K4 and VEGFR2 (vascular endothelial growth factor receptor 2) protein expression in MAECs and unselected cell lysates (n = 3). (C) mRNA levels of MAP4K4 in immune-selected or unselected cells (n = 3). (U6, promoter; WRE, woodchuck hepatitis virus posttranscriptional regulatory element). (D) The expression levels of NF-κB signaling in MAECs of MAP4K4 KD/KO and control/KO mice (n = 6). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (E) The aortic vasodilatation induced by Ach in MAP4K4 KD/KO and control/KO mice (n = 10). (F) Representative images of TUNEL staining in sections of thoracic aortas. (G) The percentage of apoptotic endothelial cells (n = 6). (H) Representative electron microscopy images of endothelium. Scale bars, 50 μm. (I) The mRNA levels of adhesion molecules and (J) inflammation in MAECs of aortas (n = 10). The data are shown as the means ± SEM. *P < 0.001.
In addition, MAP4K4 silencing was further performed in MAECs in vitro (fig. S14, C and D). First, the results showed increased inflammation (TNF-α, IL-1β, and IL-6) and adhesion molecule (VCAM-1, ICAM-1, and E-selectin) expression, increased fluorescein isothiocyanate (FITC)–labeled dextran movement across a monolayer, and monocyte adhesion to an activated endothelial monolayer in PA-induced MAECs, which were blunted when MAP4K4 was silenced. Furthermore, rMYDGF mimicked the roles of siMAP4K4 (fig. S14, E to H), indicating that MYDGF inhibits inflammation and monocyte adhesion to endothelium through MAP4K4 in vitro. Second, the increased NF-κB signal in PA-induced MAECs were blunted when MAP4K4 was silenced or in response to rMYDGF (fig. S15A). Consequently, the increased PA-induced apoptosis was attenuated by siMAP4K4 or rMYDGF intervention in MAECs (fig. S15, B and C), indicating that MYDGF inhibits NF-κB through MAP4K4. Third, an assay using a luciferase reporter containing NF-κB–binding elements in MAP4K4-silenced MAECs was performed as reported previously (19). The results showed that PA-induced NF-κB transcriptional activity was reduced when MAP4K4 was silenced, and rMYDGF mimicked the effects of siMAP4K4 on NF-κB transcriptional activity (fig. S15D). Furthermore, by using p65 chromatin immunoprecipitation (ChIP), the increased p65 binding to the VCAM-1, E-selectin, and IκBα promoters induced by PA was decreased when MAP4K4 was silenced in MAECs, and rMYDGF mimicked the roles of siMAP4K4 on p65 binding, indicating that MYDGF inhibits NF-κB transcriptional and binding activity through MAP4K4 (fig. S15, E to G). Last, the NF-κB inhibitor SC75741 (5 μM for 24 hours) (fig. S14I) mimicked the effects of siMAP4K4 intervention in PA-induced MAECs (fig. S15, A to C), indicating that NF-κB is involved in downstream of MAP4K4. Together, these data suggest that endothelial MAP4K4/NF-κB signaling is essential for the beneficial effects of MYDGF in atherosclerosis.
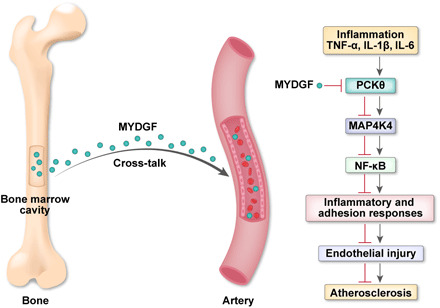
In the last experiments, we sought to explore how MYDGF regulates the phosphorylation of MAP4K4 in MAECs. It is established that MAP4K4 is usually regulated by protein kinase C (PKC) (20, 21). Thus, we determined the expression of PKC isoforms including α, β, θ, and λ. Results showed P (phosphorylated)-PKCθ in the MAECs was increased in KO mice compared with WT mice, while the expression of P-PKCθ in the MAECs was significantly decreased in MYDGF-replenished mice compared with AAV-GFP mice (fig. S16, A and B). However, the expression of P-PKCα, P-PKCβ, or P-PKCλ was not affected by MYDGF (fig. S16, A and B). Besides, rMYDGF treatment in MAECs decreased the expression of P-MAP4K4 and P-IκBα (fig. S16C). In addition, to further verify whether PKCθ is involved in the upstream events of MAP4K4 signaling, we treated MAECs with the PKCθ inhibitor; the results showed that the effects of treatment with 2 μM PKCθ inhibitor for 24 hours strongly mimicked those of rMYDGF intervention, as evidenced by the significantly decreased expression of P-PKCθ, P-MAP4K4, and P-IκBα (fig. S16C). These data suggested that PKCθ is involved in the regulation effects of MYDGF on the phosphorylation of MAP4K4 in MAECs (Fig. 7).
Fig. 7. Schematic showing that MYDGF plays a protective role in atherosclerosis via MAP4K4/NF-κB signaling pathway.
DISCUSSION
The main findings were as follows: (i) Myeloid cell–derived MYDGF inhibited endothelial inflammation and adhesion responses, blunted leukocyte homing and macrophage accumulation in plaques, and alleviated endothelial injury and atherosclerosis in vivo; (ii) myeloid cell–derived MYDGF is a cross-talk factor between bone marrow and arteries that regulates the pathophysiology of arteries; (iii) rMYDGF attenuated endothelial inflammation, apoptosis, permeability, and adhesion responses induced by PA in vitro; and (iv) MAP4K4/NF-κB signaling is essential for the beneficial effect of MYDGF on endothelial injury and atherosclerosis. This study finds that myeloid cell–derived MYDGF inhibited endothelial inflammation and adhesion responses and alleviated endothelial injury and atherosclerosis, and we provided direct evidence for bone marrow as an endocrine organ to regulate the pathophysiological function of arteries via MYDGF.
Endothelial dysfunction is an early pathophysiological change in the development of atherosclerosis (11). Here, our data showed that myeloid cell–derived MYDGF protected endothelial function and decreased endothelial apoptosis in mice. Of note, our results also revealed that bone marrow–specific MYDGF deletion itself is sufficient to induce endothelial injury and inflammation under NCD conditions; the underlying mechanisms remain unknown. The possible explanations are as follows: (i) The bone marrow–specific MYDGF is critical in maintaining the integrity of endothelium under normal conditions; (ii) this inflammation may be secondary to the adiposity under NCD in KO mice. In addition, rMYDGF inhibited endothelial inflammation and adhesion responses and reduced endothelial permeability and apoptosis induced by PA in vitro. Thus, we suggest that myeloid cell–derived MYDGF protects against endothelial injury.
Next, we questioned whether myeloid cell–derived MYDGF alleviates late-stage atherosclerotic lesions. Our data showed that MYDGF reduced the atherosclerotic plaque areas in AKO and DKO mice, indicating that MYDGF ameliorates late-stage lesions in atherosclerosis. Aortic plaques are characterized by increased levels of macrophages and T lymphocytes and reduced levels of collagen and VSMCs (11). Our results revealed that MYDGF improves the cellular components of plaques and decreases leukocyte homing and macrophage accumulation within atherosclerotic plaques. The data indicated that myeloid cell–derived MYDGF attenuates atherosclerosis and improves plaque components to support a stable plaque phenotype.
Atherosclerosis is an inflammatory disease that promotes continual monocyte recruitment in a leukocyte adhesion molecule–dependent manner (4, 22). Here, inflammation and adhesion responses increased in patients and mice with atherosclerosis. Myeloid cell–derived MYDGF reduced endothelial inflammation and adhesion responses and consequently decreased leukocyte homing and macrophage accumulation in plaque. In addition, rMYDGF treatment attenuated inflammation, monocyte adhesion, permeability, and p65 nuclear translocation induced by PA in MAECs. These data indicate that the decreased endothelial inflammation and adhesion responses contributed to the protection of myeloid cell–derived MYDGF to endothelial injury and atherosclerosis.
In accordance with our previous study (10), we also found that MYDGF improved IR and lipid profiles and decreased body weight gain. Thus, improved metabolic profiles also contribute to the antiatherosclerotic effects of MYDGF.
It is important to address the possible pathways by which myeloid cell–derived MYDGF protects against atherosclerosis. Endothelial NF-kB is essential for the expression of leukocyte adhesion molecules, atherosclerosis, and macrophage homing to aortic plaques (4, 18, 23). We confirmed that MYDGF inhibits endothelial NF-kB signaling, as evidenced by decreased endothelial inflammation and adhesion responses, decreased leukocyte homing and macrophage accumulation in plaques, and decreased endothelial expression of P-IκBα and nuclear P-p65. Furthermore, MAP4K4, p38MAPK, ERK, JNK, and IKKβ are upstream molecules of NF-κB signaling (4). Our animal experiments showed that endothelial MAP4K4 is involved in the action of MYDGF on NF-κB signaling, and our in vitro experiments further confirmed these results. However, MYDGF did not affect the other signal protein expression including p38MAPK, ERK, JNK, and IKKβ. Of importance, when MAP4K4 was specifically knocked down in endothelial cells, the activation of NF-κB signaling disappeared, and the downstream events improved. Furthermore, MYDGF restoration or rMYDGF reversed these effects. Notably, when MAP4K4 was silenced in vitro, the increased activity of NF-κB transcription and p65 binding induced by PA were blunted, and rMYDGF reversed these effects. Last, we also found that PKCθ is involved in the beneficial effects of MYDGF that regulates the phosphorylation of MAP4K4 in MAECs. These pieces of evidence confirmed that endothelial MAP4K4/NF-κB signaling is essential for the beneficial effects of myeloid cell–derived MYDGF on atherosclerosis.
In addition, we should comment on the cellular origin of bone marrow–derived MYDGF. It is reported that MYDGF is mainly produced by bone marrow–derived monocytes and macrophages (9), but other BMCs such as hematopoietic stem cells (HSCs), endothelial progenitor cells (EPCs), neutrophils, T cells, and B cells may also partially contribute to the cellular origin of those MYDGF. In addition, many studies showed that HSCs and EPCs from the bone marrow are being recruited to the lesions of artery (24, 25). Thus, both bone marrow and the cells from bone marrow may be involved in the contribution to those MYDGF.
Some limitations should be mentioned here. First, the receptor mediating effects of MYDGF and the biological functions are not identified in our study. Second, we cannot exclude a direct effect of MYDGF in other tissues, such as pancreatic β cell and bone. Last, body weight was reduced upon restoring MYDGF levels; however, the underlying mechanism remained unclear and needed to be further investigated in multiple aspects, especially in adipose metabolism.
In conclusion, our data identified that myeloid cell–derived MYDGF alleviated endothelial inflammation and adhesion response, blunted leukocyte homing and macrophage accumulation in plaques, and protected against endothelial injury and atherosclerosis in a manner involving the MAP4K4/NF-κB pathway (Fig. 7). These results imply the possible translational value of this research in clinical in the future. Considering the result that controlled elevation of MYDGF in the circulation has therapeutic benefits in treating atherosclerosis, endothelial injury, and metabolic disorders, long-acting formulation of MYDGF (e.g., microspheres, nanoparticles, and AAV-mediated stable gene expression) may be developed in the future for the sake of better medical compliance. However, there is still a long way to go to get this treatment to market because there are a lot of clinical trials and regulatory affairs to be finished and because the cost effectiveness analysis of this treatment needs to be further clarified.
MATERIALS AND METHODS
Animal experiments
The study was conducted in accordance with the National Institutes of Health Guidelines for the use of experimental animals and approved by the Animal Ethics Committee of the General Hospital of Central Theater Command (Wuhan, China). WT C57BL/6J, MYDGF-floxed (MYDGFF/F, exons 1-3), and AKO mice were obtained from Shanghai Model Organisms Centre Inc. (Shanghai, China). VE-cadherin Cre transgenic mice [B6.Cg-Tg(Cdh5-cre)7Mlia/J] and LysMCre+ mice, in which the expression of Cre recombinase is under the control of lysosome M promoter, were obtained from the Jackson laboratory (Bar Harbor, ME, USA). MYDGF-floxed mice were bred with LysMCre+ mice to generate myeloid cell–specific KO mice and littermate (MYDGF+/+) control. DKO mice were obtained by mating KO mice with AKO mice. MAP4K4-pSico mice were generated by a lentiviral vector as previously described (4, 26) and then crossed with VE-cadherin Cre transgenic mice to generate endothelial-specific MAP4K4 KD mice. All mice were housed in a pathogen-free environment with a 12-hour light-dark cycle at a controlled temperature (23° ± 1°C) with free access to food and water. From 4 to 6 weeks of age, the mice were fed NCD or WD (40% kcal fat, 43% kcal carbohydrates, and 17% kcal protein; Beijing Hfk Bioscience Co. Ltd., Beijing, China) until the end of the experiment. Body weight, food intake, blood pressure, fecal output, and lipid content in fecal were measured weekly as our previous study (27). At the terminal of the study, mice were fasted overnight and then anesthetized by intraperitoneal injection of pentobarbital sodium (60 mg/kg) and euthanized for blood and tissue samples.
Generation of mice
The original pSico-MAP4K4 lentiviral vector was constructed as described previously (4). A conditional U6 promoter was created by inserting a cytomegalovirus (CMV)–enhanced stop cassette between two loxP sites. Thus, a functional U6 promoter is obtained after Cre excision, which drives the expression of a hairpin targeting MAP4K4 (5′-GCTGTCTGGTGAAGAATTA-3′). Because the polyA tail that is required for CMV–enhanced GFP (EGFP) expression locates in the 3′ SIN-LTR (self-inactivating long terminal repeat), any possibility of EGFP expression in primary tissues and thus any side effects of EGFP expression are excluded. MAP4K4 small interfering RNA (siRNA) transcripts are still produced by U6 promoter, because the 6-nucleotide polyT sequence of the end of the MAP4K4 shRNA antisense sequence is recognized as a termination signal by RNA pol III promoters including U6. Then, the construct was injected into eggs at the one-cell stage, and the two-cell stage eggs were implanted into female pseudo-pregnant C57Bl/6J mice. Next, the mice were bred with C57Bl6/J mice for seven generations. Genomic DNA was extracted from the obtained mice and subjected to polymerase chain reaction (PCR) for genotyping (shRNA primer 5′-CCCGTATGGCTTTCATT–TTCTCC-3′, 5′-AAGGAAGGTCCGCTGGATTGAG-3′).
Histological assays and analysis of peripheral blood cells
IF staining was performed following standard procedures. Bone marrow sections were incubated with MYDGF polyclonal antibody (1:200; Proteintech, #11353-1-AP). After incubation with primary antibodies, the sections were washed with phosphate-buffered saline (PBS) and incubated with the appropriate fluorescent secondary antibodies. Sections were mounted using 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes) and imaged by fluorescent microscopy. IF images were obtained with FluoView FV1000 confocal microscopy (Olympus, Shinjuku, Japan). Regarding the toluidine blue staining, femora were harvested from mice after euthanasia, and 4-μm-thick longitudinally oriented bone sections were stained with toluidine blue for histological analysis. For analysis of peripheral blood cells, the WT and KO mice aged 10 weeks were weighed and anesthetized by intraperitoneal injection of pentobarbital sodium. Blood was collected from the inner canthus vein, EDTA anticoagulation, and the blood count was used to determine the peripheral blood count and classification. We incubated immune cells from peripheral blood with labeled antibodies. Fluorescent-labeled antibody NK1.1-FITC was purchased from BioLegend; CD11b-APC was purchased from Invitrogen; B220-percp, F4/80-PE, CD4-PE-cy7, and CD8a-APC-cy7 were purchased from BD; B220-percp-cy5.5 and CD3-APC were purchased from BioLegend Division; EDTA was purchased from Sigma-Aldrich; and erythrocyte lysate was purchased from Invitrogen. We then added the cells to TruCOUNT tubes (BD Biosciences) and counted them on an LSR II flow cytometer (Becton Dickinson). We analyzed the data with FlowJo software (version 10).
Glucose and insulin tolerance tests and biochemical assays
The glucose tolerance test and insulin tolerance test were performed as described in our studies (10, 11). Blood samples were obtained from mice after a 12-hour fast and stored at −80°C until further analysis. Blood glucose was measured using a blood glucose meter (Bayer Company, Germany) by tail bleeding. The plasma MYDGF level was detected by targeted liquid chromatography–mass spectrometry (LC-MS) assay. Serum levels of insulin, HbA1c, IL-6, IL-1β, TNF-α, ICAM-1, and VCAM-1, as well as E-selectin, were measured using enzyme-linked immunosorbent assay (ELISA) kits (Systems Inc., Minneapolis, USA). Plasma high-density lipoprotein (HDL-C), low-density lipoprotein (LDL-C), triglyceride (TG), total cholesterol (TC), and free fatty acids (FFAs) were determined by colorimetric assays using a commercially available kit (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions.
MYDGF label and tracing
For the synthesis of labeled MYDGF proteins, IRB-NHS-MYDGF was conducted according to the manufacturers’ instructions of the commercial IRB-NHS fluorescence probing (Sciencelight, China) as described in previous reports (28, 29). Briefly, IRB-NHS (10 mg/ml) in 20 ml of dimethyl sulfoxide was added into 4 ml of MYDGF suspension (5 mg/ml) in PBS [0.01 M (pH 7.4)] followed by sonication (50 W). The product was subjected to HiTrap G25 desalting column to remove free IRB-NHS after a 2-hour reaction at 25°C. The amount of immobilized IRB-NHS on MYDGF was determined by measuring unbound IRB-NHS concentrations in the washing solution by a visible spectrophotometry method at 783 nm.
Mice (n = 3) aged 8 weeks were administrated with IRB-NHS-MYDGF [(10 mg/kg, per body weight (b.w.)] via tail vein injection; Sham group (n = 3) aged 8 weeks received IRB-NHS-saline as control. After 24 hours of intervention, the sections of thoracic aortas were stained with monoclonal anti-CD31 (1:100; ABclonal, ab24590) for observing the fluorescence of IRB-NHS-MYDGF and endothelium.
Endothelial function assessment in mice
The endothelial-dependent vasodilation and endothelial cell apoptosis were measured as described in our studies (11, 13). Briefly, the thoracic aortas were cut into 4-mm rings immediately after euthanasia. Aortic rings were precontracted with norepinephrine (10−6 mM), and vasodilation responses were evaluated by cumulative concentration response curves to acetylcholine (Ach; 10−9 to 10−4 mM) and sodium nitroprusside (SNP; 10−9 to 10−4 mM). The endothelium-dependent and endothelium-independent vasodilation were measured.
Analysis of endothelial apoptosis in vivo
According to our previous reports (11, 13), endothelial cell apoptosis in thoracic aortas was detected by double stain with terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (Alexa Fluor 640, 40308ES20, Yeasen Biotech Co. Ltd.) and monoclonal anti-CD31 (1:100; ABclonal, ab24590). Electron microscopy was performed on thoracic segments using ultrathin sections and examined with a Hitachi HT7700 light microscope (Hitachi, Japan).
Aortic staining, blood pressure, and other parameters
The plaque en face area of the whole aortas and cross-sectional area of atherosclerotic plaque from aortic root were stained with Oil Red O (4, 11, 13). To detect target protein expressions, the immunohistochemical analysis was used in serial plaque sections from the aortic arch. Immunohistochemical analysis of CD68 polyclonal antibody (1:200; Boster Biological Technology, BA3638), CD3 monoclonal antibody (1:200; Servicebio, GB13014), and α–smooth muscle actin monoclonal antibody (1:2000; Servicebio, GB13044) were performed. The sections from the aortic arch were additionally stained with monoclonal anti–ICAM-1 BBIG-I1 (1:100) or monoclonal anti–VCAM-1 BBIG-V1 (1:200) (R&D Systems) and rat monoclonal anti-CD31 (1:100; ABclonal, ab24590) and mounted in Prolong Gold with DAPI (Life Technologies). Images were quantified using Image Pro Plus Analysis Software (Media Cybernetics).
Blood pressure was noninvasively measured in animals by the tail-cuff method (Softron BP-98A, Tokyo, Japan). Blood pressure values were averaged from three consecutive measurements under steady-state conditions. Food intake, fecal output, and lipid content in feces were measured as previously described (27).
Bone marrow transplantation
According to our previous report (10), we transplanted BMCs via the tail vein into lethally irradiated (7.5 gray) recipient mice (1 × 106 cells per mouse).
Injection of AAV into the bone marrow cavity
Bone marrow aspirates and biopsies were performed following general anesthesia with pentobarbital sodium (60 mg/kg) in mice. Using sterile technique, marrow was aspirated into 100-μl syringes containing 100 U of sodium heparin. Viral vectors (AAV-MYDGF or AAV-GFP), used for intramarrow delivery, were prepared at a dose of 1 × 1012 viral genomes in a total volume of 5 μl and pushed through a bone marrow aspirate needle once placement was confirmed to be within the marrow cavity. The efficiency of injection of AAV-MYDGF was measured by plasma LC-MS assay and Western blot. The mice received 5 μl of either AAV-MYDGF or AAV-GFP every 3 weeks for 12 weeks (30).
Construction of AAV vectors for mice
The MYDGF (GenBank accession number NM_080837.2) gene sequence was directly synthesized in the pHBAAV-CMV-MCS-3flag-T2A-ZsGreen vector and then cotransfected into AAV-293 cells with pAAV-RC and pHelper plasmids. Large-scale recombinant AAV production, purification, and preparation were described previously (10, 31).
Leukocyte homing
The leukocyte homing assay was performed as described previously (4). Peritoneal exudate cells were stimulated by an intraperitoneal injection of 4% thioglycolate into male C57BL/6-Tg (CAG-EGFP)1Osb/J mice (the Jackson laboratory) aged 8 weeks. After 2 days, three million cells were injected intravenously into DKO-GFP or DKO-MYDGF mice that had been fed a WD for 12 weeks. After 2 days, aortic roots were embedded in OCT (optimal cutting temperature). Total fluorescent cells were counted in 10 × 8-mm sections over a 0.5-mm area, and the cell number was normalized to plaque area.
Cell experiments
Primary MAEC isolation
The primary MAECs were isolated as described by us (11). When needed, the cells were magnetically selected with anti-rat dynal beads (Life Technologies) that had been conjugated to anti-CD31 monoclonal antibodies (ab119339) and anti-CD102 monoclonal antibodies (ab34333). Selected cells were cultured in M199 medium with 20% fetal bovine serum, heparin (10 mg/ml), and endothelial cell growth factor (50 μg/ml) until confluent. Unselected cells were kept as the nonendothelial fraction and cultured until confluent.
Cell culture and apoptosis
MAECs and RAW264.7 macrophages were cultured according to our report (11). All cells were maintained at 37°C under humidified conditions and 5% CO2. Mouse MAECs were cultured in M199 medium (Gibco Laboratories, USA) with 20% fetal bovine serum (Gibco Laboratories, USA), heparin (10 mg/ml), and endothelial cell growth factor (50 μg/ml) (Sigma-Aldrich, USA). MAECs can be determined by fluorescent staining of antibodies against von Willebrand factor. RAW264.7 cells were cultured routinely in Dulbecco’s modified Eagle’s medium (Gibco Laboratories, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 mg/ml).
Analysis of endothelial apoptosis and permeability in vitro
MAEC were pretreated with or without rMYDGF (50 mg/ml) for 48 hours, and an additional treatment with or without PA (0.4 mM for 16 hours; Sigma-Aldrich, USA) was done when needed. Cell apoptosis was determined by flow cytometry with an annexin V–FITC/PI assay (BD, CA, USA) according to the manufacturer’s instructions (11). The endothelial permeability was measured by the In Vitro Vascular Permeability Assay Kit (Millipore) according to the manufacturer’s instructions.
The migration assay for MAECs and RAW264.7 cells
MAECs and RAW264.7 migration were analyzed with Transwell using a modified Boyden’s chamber (pore size, 8 μm) (BD Biosciences, USA) assay. MAECs or RAW264.7 cells were added to the upper compartment of the Transwell chambers (105 cells per chamber). MYDGF for 48 hours or PA for 16 hours was added in the lower chambers. Cells migration was assessed in five random high-power fields (×400) with a light microscope after staining with crystal violet dye.
siRNA-mediated silencing assay
According to our previous reports (10, 31), the scramble (siCon) or siMAP4K4 was used for MAP4K4 silencing assay in MAECs. The siRNA was transfected to silence MAP4K4 in MAECs using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) in Opti-MEM medium (Invitrogen), according to the manufacturer’s instructions. The siRNA sequences are listed in table S5.
Measurements of MAEC p65 nuclear translocation
The practice of IF staining was to observe the standard procedures. Using p65 monoclonal antibody [1:400; Cell Signaling Technology (CST), #8242] incubated sections. After incubated with primary antibodies, the sections using PBS washed sections and using the proper fluorescent secondary antibodies incubated. IF images were obtained with FluoView FV1000 confocal microscopy (Olympus, Shinjuku, Japan).
Coculture experiments
BMCs and MAECs were noncontact cocultured through a Transwell 24-well plate. BMCs were cultured on the floor of the culture plate (lower well), and MAECs were seeded on the Transwell insert (upper well) of a 24-well plate and cultured in a 37°C incubator for 48 hours.
Luciferase assay
The NF-kB luciferase reporter plasmid was constructed by Hanbio (Shanghai, China). Luciferase reporter plasmids or control plasmids were transfected into MAECs with the JET-PEI transfection system (Polyplus). After 24 hours, cells were treated with MYDGF (50 ng/ml) for 48 hours, and PA (0.4 mM) was added for an additional 16 hours before the end. Luciferase and Renilla were determined by the dual-luciferase reporter assay system (Promega) according to the manufacturer’s instructions.
Chromatin immunoprecipitation
MAECs were transfected with siMAP4K4 or siCon. After 12 hours, cells were treated with MYDGF (50 ng/ml) for 48 hours, and PA (0.4 mM) was added for an additional 16 hours before the end. ChIP was performed using the SimpleChIP enzymatic chromatin IP kit (CST) according to the manufacturer’s instructions. The p65 antibody (ab19870) or immunoglobulin G (ab2410) was used for p65 immunoprecipitations. The primers for IκBα, VCAM-1, and E-selectin are listed in table S5. Real-time PCR (RT-PCR) was used to amplify the target genes.
THP-1 monocyte adhesion
MAECs were transfected with siCon or siMAP4K4 for 12 hours before the adhesion experiments. THP-1 monocytes were incubated in M199 medium and labeled fluorescently with calcein-AM (BD) according to the kit insert 30 min before the adhesion experiments. MAECs were treated with MYDGF (50 ng/ml) for 48 hours before the end of adhesion assay and added PA (0.4 mM) for another 16 hours of treatment before the end of the experiment. In total, 1 × 106 THP-1 cells were incubated with the treated MAECs for 30 min at 37°C before the end. Nonadherent THP-1 cells were removed by washing, and the cells were fixed, and the remaining cells were observed with fluorescence microscopy at 488 nm. At least seven random fields were quantified in each condition as the average number of adherent cells in each field.
Real-time PCR
RT-PCR was performed as previously described (12, 31) and conducted on the Applied Biosystems Prism 7000 sequence detection system, with specific primers described in table S5. Relative changes in mRNA levels among groups were determined with 2-ΔΔCt method. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the housekeeping gene to normalize the expressions of the target genes. The thermal cycling conditions for RT-PCR were 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, 60°C for 30 s, and 70°C for 30 s.
Western blotting
Antibodies against the following proteins were used: MYDGF (1:1000; Proteintech, #11353-1-AP), phosphorylated JNK (Thr183/Tyr185) (1:2000; CST, #9255), JNK (1:1000; CST, #9252), phosphorylated MAP4K4 (Ser631) (1:1000; Abcam, #yb-5492R), MAP4K4 (1:1000; CST, #9948), phosphorylated IKKβ (Ser176/180) (1:1000; CST, #2697), IKKβ (1:1000; CST, #2684), phosphorylated p65 (Ser468) (1:1000; CST, #3039), p65 (1:1000; CST, #8242), phosphorylated IκBα (Ser32) (1:1000; CST, #2859), IκBα (1:1000; CST, #9242), phosphorylated p38MAPK (Thr180/Tyr182) (1:2000; CST, #9216), p38MAPK (1:1000; CST, #8690), phosphorylated ERK (Thr202/Tyr204) (1:1000; CST, #4376), ERK (1:1000; CST, #4695), Lamin A/C (1:3000; CST, #5174), phosphorylated PKCθ (Ser643/676) (1:1000; CST, #4376), and PKCθ (1:1000; CST, #13643), phospho-PKCα (Thr638) (1:1000; CST, #9375), PKCα (1:1000; CST, #2056), phospho-PKCβ (Ser660) (1:1000; CST, #9371), PKCβ (1:1000; CST, #46809), phospho-PKCλ (Thr403) (1:1000; CST, #9378), PKCλ (1:1000; CST, #2998), and GAPDH (1:3000; CST, #4777).
Clinical samples
From July 2018 to December 2019, a total of 60 successive newly diagnosed Chinese male CAS from Wuhan area (aged 35 to 65 years; mean, 48.60 ± 0.65), who referred to our hospital for health examination, were selected randomly in this study. (i) Inclusion criteria: Newly diagnosed male CAS, older than 18 years, Chinese Han people from Wuhan area, body mass index of 18.5 to 30 kg/m2. (ii) Exclusion criteria: Individuals without CAS; diabetes; hypertension; thyroid diseases; autoimmune disease, such as rheumatoid arthritis; malignant tumors; other endocrinological diseases, such as Cushing syndrome; taking any drugs, such as aspirin, statin, antioxidative agents (coenzyme Q10 and vitamins E and C); smokers; alcohol drinkers; renal failure; heart failure; and liver failure. (iii) Diagnosis criteria: CAS was measured using B-mode ultrasonography and defined as stenosis 25% and/or intima-media thickness of 1.2 mm (32). The diagnosis of hypertension was made according to the WHO/ISH (World Health Organization/International Society of Hypertension) (33). Cigarette smoker was defined as subjects who had smoked at least one cigarette daily for 1 year. Alcohol drinker was defined as a man who was current or in the past 6 months with alcohol consumption ≧ 140 g/week. During the same period, 60 male healthy individuals (aged 35 to 65 years; mean, 47.45 ± 0.66) were selected as control subjects. The vascular studies of the brachial artery were performed by high-resolution ultrasound (128XP/10 with a 7.0-MHz linear array transducer; Acuson, Mountain View, CA) as described by us previously (22, 34). All subjects enrolled in the study signed informed consent. The study protocol agreed with the guidelines of the ethics committee of our hospital and approved by the ethics committee of General Hospital of Central Theater Command (Wuhan, China).
Human biochemical measurements
Blood samples were obtained from participants after a 12-hour fast. Plasma samples were stored at −80°C until further analysis. Plasma MYDGF level was detected by LC-MS assay. TC, TG, LDL-C, HDL-C, HbA1c, and creatinine were determined by colorimetric assays using the commercially available kit (Jiancheng Bioengineering Institute, Nanjing, China). FBG and postprandial 2-hour blood glucose (2-hour BG) (after 75-g glucose loading) were measured by a glucose oxidase procedure. Complement-reactive protein (CRP) was measured by particle-enhanced immunoturbidimetric assay. Serum fasting insulin concentration was measured by electrochemiluminescence immunoassay. FFA was determined using a kit from Roche Diagnostics (Mannheim, Germany) by enzymatic colorimetric assay according to the manufacturer’s instructions. Homeostasis model assessment of IR was calculated by fasting serum insulin (mU/liter) × FBG (mM)/22.5. Serum levels of TNF-α, IL-1β, IL-6, ICAM-1, and VCAM-1, as well as E-selectin, were measured using ELISA kits (R&D Systems Inc., Minneapolis, USA). Coefficients of variation for these assays were 1 to 2% (HbA1c and HDL-C), 2 to 3% (blood glucose, creatinine, TNF-α, IL1-β, and MYDGF), and 3 to 6% (insulin, LDL-C, FFA, TG, IL-6, and CRP).
Statistical analysis
All data are expressed as the means ± SEM. Comparisons between groups were analyzed using Student’s t test or one-way analysis of variance (ANOVA) with a least significant difference test. Pearson correlations were used to identify correlations between variables. A P value < 0.05 was considered significant. All data analyses were performed using SPSS 22.0 software (IBM Corp., Armonk, NY).
Acknowledgments
We authors thank Z. Gu for editing the manuscript and the General Hospital of Central Theater Command (Wuhan, China). Funding: This work was supported by grants from the National Natural Science Foundation of China (NSFC 81870573, 81370896, and 81570730), National Key Research and Development Program of China (2016YFC1305601), Research Project of Health Commission of Hubei Province (WJ2017H0031), and Hubei Province’s Outstanding Medical Academic Leader program (2014-9). Author contributions: B.M., Y.D., X.X., and J.Z. conducted the animal experiments. Y.L., L.X., J.D., and M.L. performed the in vitro experiments. Y.D., B.Z., and B.G. wrote the manuscript. L.X. and L.W. conducted the study design and data analysis. G.X. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabe6903/DC1
REFERENCES AND NOTES
- 1.Bos D., Van Der Rijk M. J. M., Geeraedts T. E. A., Hofman A., Krestin G. P., Witteman J. C. M., Van Der Lugt A., Ikram M. A., Vernooij M. W., Intracranial carotid artery atherosclerosis: Prevalence and risk factors in the general population. Stroke 43, 1878–1884 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Tanaka H., Gouskova N., Folsom A. R., Evenson K., Windham G., Aguilar D., Matsushita K., Heiss G., Habitual physical activity and arterial stiffness in older adults: The Atherosclerosis Risk in Community (ARIC) study. Circulation 135, 1–2 (2014). [Google Scholar]
- 3.Libby P., Inflammation in atherosclerosis. Nature 420, 868–874 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Flach R. J. R., Skoura A., Matevossian A., Danai L. V., Zheng W., Cortes C., Bhattacharya S. K., Aouadi M., Hagan N., Yawe J. C., Vangala P., Menendez L. G., Cooper M. P., Fitzgibbons T. P., Buckbinder L., Czech M. P., Endothelial protein kinase MAP4K4 promotes vascular inflammation and atherosclerosis. Nat. Commun. 6, 8995 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pithova P. P. P., Pitha J. P. J., Stechova K. S. K., Kvapil M. K. M., Preclinical atherosclerosis is associated with insulin resistance in type 1 diabetes women. Atherosclerosis 263, e54 (2017). [Google Scholar]
- 6.Libby P., Tabas I., Fredman G., Fisher E. A., Inflammation and its resolution as determinants of acute coronary syndromes. Circ. Res. 114, 1867–1879 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin Z., Pan X., Wu F., Ye D., Zhang Y., Wang Y., Jin L., Lian Q., Huang Y., Ding H., Triggle C., Wang K., Li X., Xu A., Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation 131, 1861–1871 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng Y., Guan M., Li C., Xu L., Zheng Z., Li J., Xue Y., Bitter melon (Momordica charantia) attenuates atherosclerosis in apo-E knock-out mice possibly through reducing triglyceride and anti-inflammation. Lipids Health Dis. 17, 251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korf-Klingebiel M., Reboll M. R., Klede S., Brod T., Pich A., Polten F., Napp L. C., Bauersachs J., Ganser A., Brinkmann E., Reimann I., Kempf T., Niessen H. W., Mizrahi J., Schönfeld H. J., Iglesias A., Bobadilla M., Wang Y., Wollert K. C., Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat. Med. 21, 140–149 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Wang L., Li Y., Guo B., Zhang J., Zhu B., Li H., Ding Y., Meng B., Zhao H., Xiang L., Dong J., Liu M., Zhang J., Xiang L., Xiang G., Myeloid-derived growth factor promotes intestinal glucagon-like peptide-1 production in male mice with type 2 diabetes. Endocrinology 161, bqaa003 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Mei W., Xiang G., Li Y., Li H., Xiang L., Lu J., Xiang L., Dong J., Liu M., GDF11 protects against endothelial injury and reduces atherosclerotic lesion formation in apolipoprotein E-null mice. Mol. Ther. 24, 1926–1938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M., Xiang G., Lu J., Xiang L., Dong J., Mei W., TRAIL protects against endothelium injury in diabetes via Akt-eNOS signaling. Atherosclerosis 237, 718–724 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Lu J., Xiang G., Liu M., Mei W., Xiang L., Dong J., Irisin protects against endothelial injury and ameliorates atherosclerosis in apolipoprotein E-Null diabetic mice. Atherosclerosis 243, 438–448 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Finney A. C., Funk S. D., Green J. M. Jr., Yurdagul A., Rana M. A., Pistorius R., Henry M., Yurochko A., Pattillo C. B., Traylor J. G., Chen J., Woolard M. D., Kevil C. G., Orr A. W., EphA2 expression regulates inflammation and fibroproliferative remodeling in atherosclerosis. Circulation 136, 566–582 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu L., Xu R., Wang S., Li S., Sheng H., Wu J., Qu Y., Honokiol ameliorates endothelial dysfunction through suppression of PTX3 expression, a key mediator of IKK/IκB/NF-κB, in atherosclerotic cell model. Exp. Mol. Med. 47, e171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rahman K., Vengrenyuk Y., Ramsey S. A., Vila N. R., Girgis N. M., Liu J., Gusarova V., Gromada J., Weinstock A., Moore K. J., Loke P., Fisher E. A., Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Invest. 127, 2904–2915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia Z., Nallasamy P., Liu D., Shah H., Li J. Z., Chitrakar R., Si H., McCormick J., Zhu H., Zhen W., Li Y., Luteolin protects against vascular inflammation in mice and TNF-alpha-induced monocyte adhesion to endothelial cells via suppressing IkBα/NF-κB signaling pathway. J. Nutr. Biochem. 26, 293–302 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gareus R., Kotsaki E., Xanthoulea S., van der Made I., Gijbels M. J., Kardakaris R., Polykratis A., Kollias G., de Winther M. P., Pasparakis M., Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 8, 372–383 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Kopp E., Ghosh S., Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265, 956–959 (1994). [DOI] [PubMed] [Google Scholar]
- 20.Ma X. F., Zhang J., Shuai H. L., Guan B. Z., Luo X., Yan R. L., IKKβ/NF-κB mediated the low doses of bisphenol A induced migration of cervical cancer cells. Arch. Biochem. Biophys. 573, 52–58 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Liu X., Chen W., Liu Q., Dai J., Abietic acid suppresses non-small-cell lung cancer cell growth via blocking IKKβ/NF-κB signaling. Onco. Targets. Ther. 12, 4825–4837 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang G. D., Xiang L., He H. L., Zhang J. X., Dong J., Impact of cardiac magnetic resonance on endothelial function in type 2 diabetic patients. Atherosclerosis 239, 131–136 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Sehnert B., Burkhardt H., Wessels J. T., Schroeder A., May M. J., Vestweber D., Zwerina J., Warnatz K., Nimmerjahn F., Schett G., Duebel S., Voll R. E., NF-κB inhibitor targeted to activated endothelium demonstrates a critical role of endothelial NF-κB in immune-mediated diseases. Proc. Natl. Acad. Sci. U.S.A. 110, 16556–16561 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu Y., Zhang C., Zhang G., Tao J., Endothelial progenitor cells in cardiovascular diseases. Aging Med. 1, 204–208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briasoulis A., Tousoulis D., Antoniades C., Papageorgiou N., Stefanadis C., The role of endothelial progenitor cells in vascular repair after arterial injury and atherosclerotic plaque development. Cardiovasc. Ther. 29, 125–139 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Ventura A., Meissner A., Dillon C. P., McManus M., Sharp P. A., Van Parijs L., Jaenisch R., Jacks T., Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl. Acad. Sci. U.S.A. 101, 10380–10385 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu B., Li Y., Xiang L., Zhang J., Wang L., Guo B., Liang M., Chen L., Xiang L., Dong J., Liu M., Mei W., Li H., Xiang G., Alogliptin improves survival and health of mice on a high-fat diet. Aging Cell 18, e12883 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu G., Gao X., Hu Q., Kang T., Liu Z., Jiang M., Miao D., Song Q., Yao L., Tu Y., Pang Z., Chen H., Jiang X., Chen J., The influence of the penetrating peptide iRGD on the effect of paclitaxel-loaded MT1-AF7p-conjugated nanoparticles on glioma cells. Biomaterials 34, 5138–5148 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Song Y., Huang Z., Xu J., Ren D., Wang Y., Zheng X., Shen Y., Wang L., Gao H., Hou J., Pang Z., Qian J., Ge J., Multimodal SPION-CREKA peptide based agents for molecular imaging of microthrombus in a rat myocardial ischemia-reperfusion model. Biomaterials 35, 2961–2970 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Li C.-J., Xiao Y., Yang M., Su T., Sun X., Guo Q., Huang Y., Luo X.-D., Long noncoding RNA Bmncr regulates mesenchymal stem cell fate during skeletal aging. J. Clin. Invest. 128, 5251–5266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J., Li Y., Li H., Zhu B., Wang L., Guo B., Xiang L., Dong J., Liu M., Xiang G., GDF11 improves angiogenic function of EPCs in diabetic limb ischemia. Diabetes 67, 2084–2095 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Jae S. Y., Carnethon M. R., Heffernan K. S., Choi Y.-H., Lee M.-K., Park W. H., Fernhall B., Slow heart rate recovery after exercise is associated with carotid atherosclerosis. Atherosclerosis 196, 256–261 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Whitworth J. A., 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J. Hypertens. 21, 1983–1992 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Xiang G. D., Xu L., Zhao L. S., Yue L., Hou J., The relationship between plasma osteoprotegerin and endothelium-dependent arterial dilation in type 2 diabetes. Diabetes 55, 2126–2131 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabe6903/DC1