

Single-molecule and cryo-EM studies reveal molecular acrobatics of signal recognition particle that initiates protein translocation.
Abstract
The conserved signal recognition particle (SRP) cotranslationally delivers ~30% of the proteome to the eukaryotic endoplasmic reticulum (ER). The molecular mechanism by which eukaryotic SRP transitions from cargo recognition in the cytosol to protein translocation at the ER is not understood. Here, structural, biochemical, and single-molecule studies show that this transition requires multiple sequential conformational rearrangements in the targeting complex initiated by guanosine triphosphatase (GTPase)–driven compaction of the SRP receptor (SR). Disruption of these rearrangements, particularly in mutant SRP54G226E linked to severe congenital neutropenia, uncouples the SRP/SR GTPase cycle from protein translocation. Structures of targeting intermediates reveal the molecular basis of early SRP-SR recognition and emphasize the role of eukaryote-specific elements in regulating targeting. Our results provide a molecular model for the structural and functional transitions of SRP throughout the targeting cycle and show that these transitions provide important points for biological regulation that can be perturbed in genetic diseases.
INTRODUCTION
Proper biogenesis of nascent proteins is a prerequisite for the generation and maintenance of protein homeostasis in all cells (1). The signal recognition particle (SRP) pathway is an ancient and universally conserved pathway that cotranslationally delivers most membrane and secretory proteins, which comprise nearly 30% of the proteome, to the eukaryotic endoplasmic reticulum (ER) or the bacterial plasma membrane (2, 3). SRP recognizes translating ribosomes bearing an exposed N-terminal hydrophobic signal sequence or transmembrane domain (TMD) on the nascent polypeptide. The interaction of SRP with the SRP receptor (SR) recruits the ribosome-nascent chain complex (RNC) to the Sec61p translocase at the ER (or SecYEG in bacteria), where translation of the nascent protein continues and is coupled to their insertion into or translocation across the membrane (2, 3).
The most conserved components of SRP and SR are found in bacteria, where SRP is composed of a 4.5S SRP RNA tightly bound to the SRP54 protein (named Ffh in bacteria). SRP54 contains a methionine-rich, M domain that binds the SRP RNA and recognizes the signal sequence or TMD on the nascent polypeptide emerging from the ribosome exit tunnel (Fig. 1A). A special guanosine triphosphatase (GTPase) domain in SRP54, termed NG, forms a guanosine 5′-triphosphate (GTP)–dependent complex with a homologous NG domain in SR (Fig. 1A). The two NG domains undergo cooperative rearrangements upon their assembly, which culminate in the reciprocal activation of the GTPase activity of one another (4, 5). The essential 4.5S RNA accelerates SRP-SR assembly 103-fold to activate protein targeting, via a conserved GNRA tetraloop that makes transient contacts with the SR NG domain before the formation of the stable NG heterodimer (6–8). Compared to the bacterial homologs, eukaryotic SRP and SR underwent an extensive expansion in size and complexity. Eukaryotic SRP contains a larger 7SL RNA on which five additional protein subunits (SRP19, SRP68/72, and SRP9/14) are assembled (9). While bacterial SR is a single protein in which the NG domain is preceded by two lipid-binding helices (10, 11), eukaryotic SR is an α/β heterodimer anchored at the ER via association of the SRα X domain with SRβ, an integral membrane protein (Fig. 1A) (12, 13). The role of many eukaryote-specific SRP subunits is not well understood. For example, a recent work identified a molecular recognition feature (MoRF), a short interaction motif (residues 242 to 261) in the disordered SR linker connecting the SRα X domain to its NG domain (Fig. 1A) (14). The SR MoRF specifically accelerates SRP•SR assembly in response to the ribosome and was proposed to functionally replace the role of the 4.5S SRP RNA in bacterial SRP (7, 8, 15), but how it carries out these functions is unclear.
Fig. 1. smFRET detects multiple conformational changes upon SRP-SR assembly.
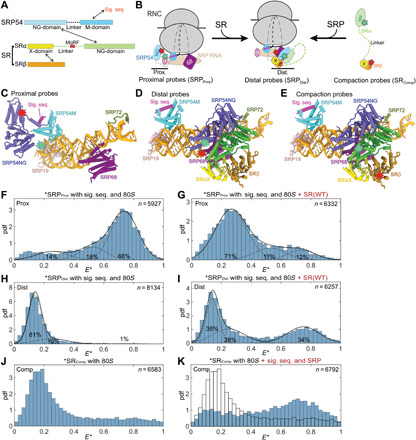
(A) Domain structures of SRP54 and SR. (B) Förster resonance energy transfer (FRET) donor (green stars) and acceptor (red stars) pairs to measure conformational changes in SRP and SR. Left: The “proximal” conformation with SRP54-NG near the ribosome exit. Middle: The “distal” conformation with SRP54-NG at SRP68/72. Right: FRET pair to measure the end-to-end distance of SR. “19,” SRP19. (C to E) Location of the FRET dyes in the structures of RNC-bound SRP [(C); Protein Data Bank (PDB): 3JAJ] and RNC•SRP•SR in the distal state [(D and E); PDB: 6FRK]. The ribosome is not shown for simplicity. (F to I) FRET at single-molecule (sm) resolution (smFRET) histograms of SRP labeled with SRPProx (F and G) or SRPDistal (H and I) probes without (F and H) and with (G and I) SR present. pdf, probability density function; E*, uncorrected FRET efficiency. n is the number of bursts in each histogram, obtained from at least three independent measurements. The data were fit to the sum (solid line) of three Gaussian functions with low, medium, and high FRET (dashed lines). WT, wild type. (J and K) smFRET histograms of SR labeled with SRComp without (J) and with (K) SRP present. These histograms were not fit as the intermediate E* arises from dynamic sampling of SR (fig. S5A).
How eukaryotic SRP transitions from cargo recognition to later stages of the targeting pathway is a long-standing question. Cross-linking, cryo–electron microscopy (cryo-EM), and single-molecule (sm) studies of the RNC•SRP complex showed that SRP is in a “proximal” conformation during cargo recognition, in which the signal sequence binds in a hydrophobic groove in the SRP54 M domain, and SRP54-NG docks at ribosomal proteins uL23/uL29 near the exit tunnel (Fig. 1B, left, and Fig. 1C) (5, 16, 17). However, the density for SRP54-NG at the exit site was not observed upon SR addition in early cryo-EM analyses (17, 18). A recent cryo-EM structure of the RNC•SRP•SR complex resolved SRP in a distinct conformation, in which the SRP•SR NG complex docks at a “distal” site of 7SL RNA where SRP68/72 is located (Fig. 1B, middle, and Fig. 1D) (19). This structure is proposed to represent a late, “prehandover” conformation of the targeting complex, in which the ribosome exit site is vacated for interaction with the Sec61p translocase (18, 19). These observations highlight the conformational dynamics of SRP and raised further questions as to the mechanisms by which SRP transitions between different functional states in the targeting pathway. It was unclear where SRP assembles with SR, given the distinct locations of SRP-NG in the different structures. Although kinetic analyses showed that SRP preorganized in the proximal conformation is optimized for assembly with SR and suggested the ribosome exit site as the site of initial SR recruitment (5), why this conformation is conducive to SR binding was not understood, nor the mechanism that drives SRP from the proximal to the prehandover structure. Furthermore, the mechanism of initiation of protein translocation remains unresolved as there are still a number of open questions. For example, a ~200–amino acid disordered linker separates SRαNG from the membrane proximal X/β domains in the SR complex (14, 20), posing another potential barrier for cargo loading onto the membrane translocase. In addition, GTPase activation in the SRP•SR complex acts as a double-edged sword: While GTP hydrolysis drives disassembly of SRP from SR to enable their turnover (21, 22) and is not required for protein translocation per se (21, 23), premature GTP hydrolysis could abort targeting before the RNC engages Sec61p and/or other translocases (24, 25). Whether and how the timing of GTP hydrolysis is regulated is unclear. Last, the functional and physiological roles of the conformational dynamics in SRP have not been demonstrated.
SRP is an essential targeting machine in bacteria and higher eukaryotic organisms (26, 27), and SRP54 deficiency is embryonically lethal in zebrafish models (28). Recently, mutations in SRP54 (T115A, T117Δ, or G226E) were linked to severe syndromic neutropenia with Shwachman-Diamond–like features, a rare recessive autosomal disease associated with neurodevelopmental defects or pancreatic dysfunction (29, 30). In zebrafish models, overexpression of SRP54G226E in SRP54+/− is dominant negative and recapitulated multiple disease phenotypes (28, 29). Intriguingly, part of the phenotype was attributed to reduced expression of the XBP1 transcription factor in SRP54G226E expressing cells (28, 29) as a result of the failure of SRPG226E to localize the XBP1u mRNA to the ER for unconventional splicing by the ER stress sensor Ire1 (28). However, the precise molecular basis of the defect associated with SRP54G226E remains unknown. A recent study suggested that the SRP54 NG domain bearing G226E did not form a stable complex with SRαNG (31), leading to the hypothesis that SRPG226E fails to target ribosomes to the ER (28). However, as SRP•SR complex formation is strongly regulated by additional domains and subunits in mammalian SRP and SR and further by the ribosome and signal sequence (5), the molecular basis of the defect associated with SRPG226E remains unclear.
To address these questions, we studied the global conformational changes in the human SRP•SR complex bound to the ribosome and signal sequence using Förster resonance energy transfer (FRET) at sm resolution. Three pairs of smFRET probes detected multiple, sequential conformational rearrangements in SRP and SR that prime the translating ribosome for engagement with translocases at the ER. We isolated multiple mutations that disrupt these conformational rearrangements at distinct stages, including SRP54G226E linked to congenital neutropenia. These mutant analyses elucidate the sequential molecular movements that occur after SRP•SR binding and demonstrate the essential roles of these rearrangements in the initiation of protein translocation. Cryo-EM structures of the targeting complex with SRPG226E reveal the architectural features of the targeting complex at early stages. Our results provide a molecular model to explain how the SRP pathway transitions from the cargo recognition to the pretranslocation stage and could inform on the mechanism of analogous transitions in other protein targeting pathways.
RESULTS
SRP and SR undergo multiple large-scale rearrangements during targeting
We developed three FRET pairs that monitor distinct molecular movements in the targeting complex. On the basis of the cryo-EM structure of SRP bound to signal sequence–bearing ribosome (16, 17), detachment of SRP54-NG from the ribosome exit was detected using a donor dye (Atto550) labeled at SRP19(C64) and an acceptor dye (Atto647N) labeled at SRP54(C12) (Fig. 1, B and C, proximal probes or SRPProx) (5, 16). On the basis of the structure of RNC•SRP•SR distal state complex (18, 19), docking of SRP54-NG at the distal site was detected using a donor dye (Cy3B) labeled at SRP54(C47) and Atto647N labeled near SRP68(P149) (Fig. 1, B and D, distal probes or SRPDist). Last, we monitored the end-to-end distance of SR using Atto550 labeled at the C terminus of SRαNG and Atto647N labeled at the N terminus of SRβ (Fig. 1, B and E, compaction probes or SRComp). The estimated distances between the dye pairs are ~44 Å for SRPProx in the proximal conformation and ~39 and ~36 Å for SRPDist and SRComp, respectively, in the distal conformation.
We detected FRET between all three pairs of probes using a diffusion-based sm technique with microsecond time-scale alternating laser excitation (μs-ALEX) (fig. S1, D to F) (32–34). This method allows optical purification of doubly labeled particles (fig. S1, E and F) and thus minimizes contributions from singly labeled species in the analysis (Supplementary Methods). Unless otherwise specified, all the measurements were made in the presence of a saturating concentration of ribosome (150 nM or ~1000-fold molar excess over labeled SRP), so that the FRET measurements report on the conformations in the complete targeting complex in which SRP•SR is also bound to the ribosome and signal sequence. We used a soluble SR complex in which the single SRβ TMD is removed. SRβ TMD is dispensable for function in yeast (35), while the GTPase domain was found to be essential and sufficient for targeting and translocation (36). Moreover, soluble SR show comparable stimulated GTPase activity in complex with SRP compared to wild-type SR in proteoliposome (4). Thus, the soluble SR is a reasonable mimic of wild-type SR to probe its interaction and allostery with SRP. Fluorescently labeled SRP and SR retained the ability to target preproteins to the ER [fig. S1, A to C; (5)]. We confirmed that the tested reaction conditions did not alter the photophysical properties of fluorophores (fig. S2), so that the observed FRET changes can be ascribed solely to conformational changes in SRP and SR.
Signal sequence– and ribosome-bound SRPProx mainly displayed a high FRET population (Fig. 1F), indicating that SRP54-NG initially docks near the ribosome exit tunnel as previously reported (5). In contrast, ~70% of SRPProx displayed low FRET upon SR addition (Fig. 1G), suggesting that interaction with SR induces SRP54-NG to move away from the ribosome exit. The opposite was observed with the distal probes: The smFRET histogram of SRPDist was dominated by low FRET populations (Fig. 1H), whereas approximately 34% of the complex acquired high FRET upon addition of SR (Fig. 1I), indicating acquirement of the distal state in this population. These results corroborate the recent structural analysis (18, 19) and together demonstrate that the SRP NG domain detaches from the ribosome exit site and docks at the distal site upon assembly with SR. Intriguingly, the fraction of the targeting complex that reached the distal state is substantially lower than those that detached from the ribosome exit as detected by the proximal probes, suggesting that the targeting complex samples additional conformations in which the NG complex is not stably docked at either the ribosome exit or the distal site.
Last, we monitored the global conformational changes of SR using the compaction probes that report on the proximity of its folded NG and X/β domains (Fig. 1, B and E). As SR was implicated in ribosome binding (14, 20), we first measured the conformation of free SR with the 80S ribosome present. The smFRET histogram of SRcomp exhibited a main peak at FRET ~0.15 (Fig. 1J), indicating that the NG and X/β domains are separated by ≥90 Å in free SR. When signal sequence– and ribosome-bound SRP were present, however, the FRET distribution of SRComp became broader and shifted to higher FRET with a major peak at FRET ~0.7 (Fig. 1K). These results show that SR undergoes a global compaction upon binding with cargo-loaded SRP, bringing its NG domain much closer to the membrane-proximal X/β domains.
Sequential conformational rearrangements in the targeting complex
To understand the molecular mechanisms that drive these conformational changes, we introduced mutations that disrupt the interaction surfaces of SRαNG with SRβ, SRX, or SRP68/72 (Fig. 2A and fig. S3, A to C). In addition, we characterized one of the SRP54 mutations (G226E) that cause congenital neutropenia with Shwachman-Diamond syndrome (Fig. 2A and fig. S3, A and C) (29, 30). None of the mutations impaired SRP•SR complex assembly or their reciprocal GTPase activation (fig. S3, D to G). Contrary to previous reports that suggested defects in the GTPase activity and SR interaction of SRP54G226E (28, 29), we found that SRPG226E displays basal GTPase activity and stimulated GTPase reactions with SR as efficiently as wild-type SRP (fig. S3, E and G). Using a pair of FRET probes incorporated in the NG domains of SRP and SR (5), kinetic measurements further showed that SRPG226E associates with and dissociates from SR three- and fivefold faster, respectively, than wild-type SRP (Fig. 3, A to C). The FRET efficiency between the SRP54 and SRα NG domains was 57% lower for the targeting complex formed by SRPG226E (Fig. 3D), suggesting that this mutation alters the conformation or dynamics in the NG heterodimer. The more dynamic nature of the SRPG226E•SR complex and/or its stabilization by the ribosome and the SR MoRF element (see the next section) may explain why an earlier size exclusion chromatography study using isolated SRP54-NG and SR failed to detect this complex (31). Nevertheless, the equilibrium dissociation constant (Kd) of the targeting complex formed with SRPG226E (23 nM) is within twofold of that of the wild-type complex. As efficient SRP•SR interaction requires both the ribosome and signal sequence (5), these results also rule out defects of these mutants in ribosome binding or signal sequence interaction. Thus, all of the defects observed in the following analyses can be attributed to conformational defects that occur after SRP•SR assembly.
Fig. 2. SR compaction drives GTPase movements in the targeting complex.
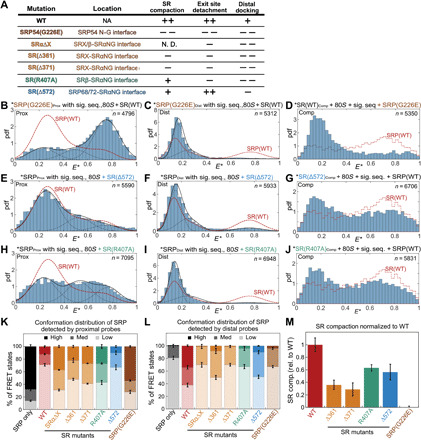
(A) Summary of the mutations characterized in this study and their phenotypes derived from the data in (B) to (M). Mutations are colored on the basis of the interactions disrupted. Details of each mutation are shown in fig. S3 (A to C). (B to J) smFRET histograms of targeting complex containing mutants SRPG226E (B to D), SR(Δ572) (E to G), or SR(R407A) (H to J) detected by the proximal (B, E, and H), distal (C, F, and I), and compaction (D, G, and J) probes. The data were analyzed as in Fig. 1. The red dashed lines outline the corresponding histograms of the wild-type targeting complex. (K and L) Quantification of the populations in low ( ), median (
), median ( ), and high (
), and high ( ) FRET states detected by the proximal (K) or distal (L) probes. (M) Quantification of SR compaction, calculated from the fraction of targeting complex displaying high FRET (E* = 0.6 to 0.8) and subtracting the corresponding value in the histogram of ribosome-bound SR. All values are normalized to that of the wild-type targeting complex. Error bars in (K) to (M) denote SD from at least three independent experiments.
) FRET states detected by the proximal (K) or distal (L) probes. (M) Quantification of SR compaction, calculated from the fraction of targeting complex displaying high FRET (E* = 0.6 to 0.8) and subtracting the corresponding value in the histogram of ribosome-bound SR. All values are normalized to that of the wild-type targeting complex. Error bars in (K) to (M) denote SD from at least three independent experiments.
Fig. 3. Biochemical characterization and cryo-EM maps of RNC•SRPG226E•SR.
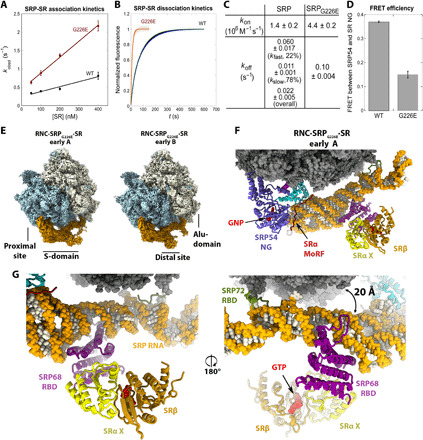
(A to C) Association (A) and dissociation (B) kinetics of SRP and SRPG226E with SR. The data in (A) were fit to eq. S5. The data in (B) were fit to eq. S6 for SRPG226E and eq. S7 for SRP. The obtained kon (mean ± fitting error, n = 2) and koff (means ± SD, n = 3 to 5) values are summarized in (C). kfast and kslow for SRP are the rate constants of the fast and slow phases (eq. S7), and the overall koff is the weighted sum of the two phases. (D) FRET efficiency between donor and acceptor dyes on SRP54-NG and SRαNG in RNC•SRP•SR and RNC•SRPG226E•SR complexes. Values are means ± SD (n = 2 to 3). (E) Cryo-EM maps (filtered to 4 Å resolution) of RNC•SRPG226E•SR in the early A and B states. Large and small ribosomal subunits are colored blue and beige, respectively. SRP•SR is colored orange. (F) Coordinates of the early SRPG226E•SR in state A, with SRP RNA in orange, ribosome in gray, signal sequence in magenta, and GTP/GNP in red. The position of GTP was based on PDB: 2FH5. (G) Close-up view of the distal site. The arrow indicates displacement of the SRP68 loop.
ALEX measurements suggest that these mutants block conformational rearrangements in the targeting complex at distinct steps. SRP54G226E severely impaired all three rearrangements in the targeting complex (Fig. 2, B to D; summarized in Fig. 2, K to M, brown). Similar albeit less pronounced defects were observed with mutants SR(Δ361) and SR(Δ371) that disrupt the intramolecular interactions between SRX and SRαNG: These mutants compromised SR compaction, as expected from their location at protein-interaction interfaces (fig. S4, C and F; summarized in Fig. 2, A and M, orange), and also impaired the detachment of the NG-domain complex from the ribosome exit site and its docking at the distal site (fig. S4, A, B, D and E; summarized in Fig. 2, K and L, orange). These results suggest that the intramolecular interactions within SR are crucial for the movement of the NG complex from the proximal to the distal site. Reciprocally, all the mutations that disrupted distal docking also reduced SR compaction to varying degrees (Fig. 2M), suggesting that interaction with the distal site helps stabilize a highly compact SR. Nevertheless, several mutants showed specific defects. SR(Δ572), which disrupts the contact of SRαNG with SRP68/72 (Fig. 2A and fig. S3, A and B, blue), specifically destabilized distal site docking but did not affect the removal of the NG complex from the ribosome exit and only modestly reduced SR compaction (Fig. 2, E to G; summarized in Fig. 2, K to M, blue). This shows that distal docking is not required for, and probably occurs after, the other rearrangements. Last, SR(R407A) disrupted the interaction of SRαNG with SRβ (Fig. 2A and fig. S3, A, B, and G, green). This mutant was impaired in both of the lateral movements of the NG-domain complex as strongly as SR(Δ361) and SR(Δ371) (Fig. 2, H and I; summarized in Fig. 2, K and L, green) but undergoes substantial compaction (Fig. 2, K and M, green), suggesting that SR can sample the compact conformation before the other rearrangements. The distinct mutational phenotypes (qualitatively summarized in Fig. 2A) suggest a sequential model in which SR compaction precedes and potentially drives the movement of the NG complex from the ribosome exit to the distal site of SRP.
Analysis of the dynamics of the rearrangements supported this sequential model. We first performed burst variance analysis, which detects dynamics by comparing the SD of E* (σE*) for individual molecules to the static limit defined by photon statistics (Supplementary Methods) (34, 37–39). If multiple conformations interconvert on the submillisecond or faster time scale, then the observed σE* would be higher than the static limit, whereas σE* would lie on the static limit curve if conformational interconversions are slower compared to molecular diffusion through the observation volume (1 to 5 ms) (34, 37–39). SRcomp displayed σE* values significantly higher than the static limit (fig. S5A, triangles versus dashed curve). This indicates that SR samples the compact state on the sub-millisecond time scale and is consistent with the disordered nature of the SR linker (14, 20). In contrast, σE* for SRPProx and SRPDist is much closer to the static limit (fig. S5, B to D), suggesting that these rearrangements occur on a time scale slower than the diffusion through the observation volume.
To further test this model, we measured the kinetics of the GTPase movements. Using the SRPProx and SRPDist probes, we carried out stopped-flow measurements to determine the rates of NG-domain departure from the ribosome exit site and its docking at the distal site, respectively. The observed rate constants of the SR-induced fluorescence change for both FRET pairs saturated at SR concentrations above 0.5 μM (fig. S5E), indicating that the observed reactions are rate-limited by a unimolecular conformational change at saturating SR concentrations. These measurements gave rate constants of 0.21 and 0.07 s−1 for exit site detachment and distal site docking, respectively (fig. S5F). Together, the mutational and kinetic analyses show that SR can rapidly undergo compaction upon binding of cargo-loaded SRP, followed by detachment of the NG complex from the ribosome exit site and its subsequent docking at the distal site.
Structures of targeting intermediates reveal the molecular basis of early SRP-SR recognition
The smFRET analyses strongly suggest the presence of early targeting intermediates before the prehandover conformation. To better understand these early events during protein targeting, we used SRPG226E, which accumulates early targeting intermediates in which both SRP and SR fail to attain the conformation observed in the prehandover structure (Fig. 2). We assembled SRPG226E with RNC and SR in the presence of 5′-guanylyl imidodiphosphate and determined the structures of the assembled complexes using cryo-EM, which resolved two SRPG226E•SR early targeting complexes referred to as “early A” and “early B” states (Fig. 3, E and F, and figs. S6 and S7). In agreement with the results of smFRET studies, the observed early targeting complexes assembled with SRPG226E were abundant and comprised most of the particle images, in contrast to wild-type SRP for which the early conformational states were not observed (19). In addition, we observed an SRPG226E•SR class from a small subset in the prehandover state (19), although the occupancy of the class is much lower compared with wild-type SRP and was not further analyzed here (fig. S6).
In all the complexes, we observed the SRP and SR proteins and the SRP RNA in its entirety, including the S and Alu domains docked at the ribosome tunnel and subunit interface, respectively (Figs. 3E and 4). The main difference between the two early SRPG226E•SR structures is in the conformation of the SRP RNA, which is resolved at 4 Å resolution at the proximal site and 7 to 10 Å resolution the distal site (fig. S7). The M domain is resolved at the ribosomal tunnel exit with a bound signal sequence found inside a hydrophobic pocket formed by the M domain and the GM linker of SRP54. We did not resolve a density of SR NG domain at the proximal site of the SRP RNA; this suggests a more dynamic NG heterodimer and is consistent with the lower FRET efficiency of the NG heterodimer and the faster association/dissociation kinetics of the SRPG226E•SR complex compared to the wild-type complex (Fig. 3, A to D). Both early A and B states resolve the SRX/β heterodimer bound at the distal site where it interacts with the SRP68 and SRP RNA, burying a surface area of almost 300 Å2 (Fig. 3, E and F, and figs. S7 and S8). In the early A state, the SRP RNA is lifted such that the loop of SRP68 RBD that binds the ribosome in the prehandover state (19) is now rotated and displaced away from the ribosome by 20 Å (Fig. 3G).
Fig. 4. Conformational changes in the SRP RNA in the RNC•SRPG226E•SR intermediates.
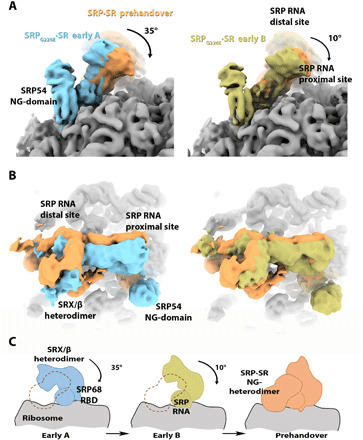
(A and B) Close-ups of the SRP RNA proximal and distal sites, respectively, depicting the conformational changes in the SRP RNA distal site are shown for the early A (cyan), early B (light green), and SRPG226E•SR states compared to the SRP•SR prehandover state (light orange, EMDB-4300). Cryo-EM maps were overlaid using the ribosome as a reference. The ribosome is colored gray. Cryo-EM maps are filtered to 8 Å resolution. (C) Schematic of the conformational change in SRP RNA, SRP68 RBD, and SRX/β heterodimer as it proceeds from early to prehandover states. The rotation of the SRP RNA moves the SRX/β heterodimer away from the ribosome surface to allow the docking of the SRP-SR NG heterodimer as observed in the prehandover states. The dashed orange line indicates that the position of the SRP-SR NG heterodimer would clash with the conformation SRX/β heterodimer, as observed in the early A and B states.
The conformation of the SRP RNA observed in the early A state is similar to a previously solved cryo-EM structure of the RNC•SRP complex (16). To reach the conformation observed in the prehandover complex, the SRP RNA needs to rotate by 35° and 10° compared to the early A and B complexes (Fig. 4, A and B). The observed conformational change is a prerequisite for the formation of the prehandover complex because the SRP RNA in the early A state, as well as in state B, would sterically prevent binding of the NG heterodimer to the distal site. Thus, the observed structures can be considered as snapshots of successive conformational states in the targeting pathway. We hypothesize that state A precedes state B and that the SRP RNA has to rotate toward state B to allow docking of the NG heterodimer at the distal site as seen in the prehandover conformation (Fig. 4C).
A MoRF in the SRα linker was recently shown to functionally replace the tetraloop of the bacterial 4.5S RNA in accelerating SRP•SR assembly, suggesting that the MoRF makes a key contact with SRP in an early intermediate before the rate-limiting transition state of SRP•SR complex formation (14). The structure of the early A SRPG226E•SR complex revealed that the SR MoRF binds to the SRP RNA in close proximity to the highly conserved GNRA tetraloop, where it further extends and bridges the M and NG domains of SRP54 (Fig. 5A and fig. S8D). Because of the lower resolution in this region of the cryo-EM map, we tested the effects of MoRF mutation on the stability of the targeting complexes formed by SRP and SRPG226E. We used the SRΔMoRF mutant, in which the MoRF region (residues 242 to 261) is replaced by a GS (gly-ser) linker and which specifically disrupts early stages of SRP•SR complex formation (14). As reported previously (14), the equilibrium dissociation constant (Kd) of the wild-type RNC•SRP•SR complex was only modestly sensitive to the MoRF deletion (Fig. 5C and fig. S9). In contrast, the deletion of MoRF destabilized the SRPG226E•SR complex over 30-fold, increasing the Kd value from 31 nM to 1.1 μM (Fig. 5, B and C). These results demonstrate that the SR MoRF provides a key contact to initiate the assembly between the NG domains of SRP and SR, whereas these interactions are lost after the NG heterodimer translocates to the SRP RNA distal site at later stages of targeting. They also provide strong evidence that the targeting complex formed with SRPG226E captures critical on-pathway interactions at early stages of protein targeting.
Fig. 5. The SR MoRF modulates SRP•SR assembly by binding SRP54 in close proximity to the SRP RNA tetraloop.

(A) Close-up of the proximal site in the early A SRPG226E•SR complex highlighting the interactions of SR MoRF with the SRP RNA and SRP54. The GNRA tetraloop of the SRP RNA is colored red. (B) The binding of wild-type SR (red) and mutant SRΔMoRF (black) to SRPG226E was measured using inhibition methods as described in Supplementary Methods. The data were fit to eq. S4, and the obtained binding constants (Ki) are summarized in (C). (C) Summary of the equilibrium dissociation constants (Kd) for the binding of SRP and SRPG226E to SR and the effect of the SR MoRF on the stability of the SRP•SR complex. Values of Kd were from the data in (B) and fig. S9 and are reported as means ± fitting error.
The contact of SRXβ with SRP68 in the SRPG226E•SR structures raised the question of whether SRXβ can interact autonomously with the SRP distal site before the NG-domain complex is formed. To address this question, we purified SRXβ and SRXLβ, in which the SR NG domain was deleted. However, a combination of FRET and competition experiments failed to detect an autonomous interaction of either construct with SRP (figs. S10 and S11). Furthermore, SRαΔX, in which the Xβ domain is removed, binds SRP with the same rate constant as SR (5, 40), indicating that the SRXβ interaction is not required before and in the transition state of SRP-SR assembly. The SRP•SR complex is 2.5-fold more stable with SR (32 nM; Fig. 5C) than with SRαΔX (78 nM; (5)), indicating that the Xβ interaction forms in the final SRP•SR complex as observed in the structure of the prehandover complex. These observations are in contrast to the interaction of the SR MoRF, which specifically affects the rate of SRP-SR assembly (14). We therefore conclude that the SRXβ interaction occurs late, after the rate-limiting transition state of SRP•SR assembly. This interaction was observed in the SRPG226E•SR structure, likely because the G226E mutation blocked the normal complex assembly pathway, and avidity effects allowed an otherwise late interaction to be favored (fig. S12).
These structural and biochemical results provide mechanistic insights into early stages of protein targeting. They explain how the conserved MoRF in the SR linker substitutes and extends the function of the SRP RNA tetraloop to activate targeting and illustrate the eukaryotic-specific sequential structural transitions in the targeting complex en route to the prehandover conformation.
Post-targeting conformational rearrangements in SRP•SR are essential for protein translocation
Last, we tested the role of the conformational dynamics of SRP and SR in their function. As stimulated GTP hydrolysis in the SRP•SR complex drives their irreversible disassembly and is an important regulatory point in the bacterial SRP pathway (41, 42), we first tested how the conformational rearrangements in the SRP•SR complex regulate the stimulated GTPase activity of the targeting complex (kcat) using mutants that bias the conformational equilibria (Fig. 2). The targeting complexes assembled with all the SR conformational mutants displayed higher GTPase rates (kcat) than the wild-type complex (Fig. 6A), strongly suggesting that docking at the distal site inhibits GTP hydrolysis and thus increases the lifetime of the targeting complex at the ER membrane. This is consistent with our previous observation that mutations in the SRP72 C terminus, which is positioned near the GTPase active site in the distal state structure, hyperactivated the GTPase reaction (19). On the other hand, the targeting complex bearing SRPG226E, in which the NG-domain complex is trapped at the ribosome exit site, hydrolyzed GTP at ~5 min−1. As SRPG226E alters the conformation within the NG heterodimer as well as its global movements, this observed GTPase rate may reflect a combination of these defects. Nevertheless, the observations with SR mutants, which specifically disrupt the global conformational rearrangements in SRP and SR, strongly suggest that these rearrangements tune the timing of GTP hydrolysis, possibly providing a balance between efficient SRP turnover and cargo handover.
Fig. 6. Conformational rearrangements in SRP and SR are essential for protein translocation.
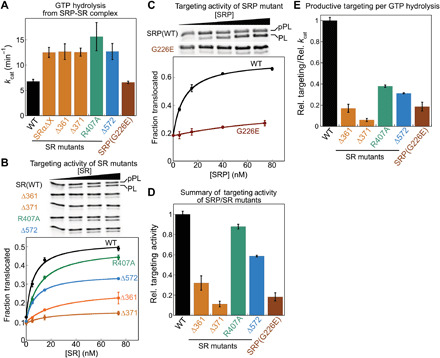
(A) Summary of the rate constants of stimulated GTP hydrolysis (kcat) in targeting complexes assembled with wild-type protein or the indicated SRP and SR mutants. Values are reported as means ± SD, with n = 3. (B and C) Cotranslational translocation of preprolactin (pPL) mediated by wild type or the indicated SRP (C) and SR (B) mutants. (D) Summary of the translocation efficiency of each mutant relative to wild-type SRP/SR at saturating protein concentrations. Translocation efficiencies were quantified from the data in (B) and (C) and in fig. S13 and are reported as means ± SD, with n = 2 to 3 technical replicates. (E) Coupling of GTP hydrolysis to translocation for each mutant relative to wild-type SRP/SR. Relative translocation efficiency (D) was divided by relative kcat (A) to obtain the relative coupling efficiency.
To test whether the conformational rearrangements in the targeting complex are important in cotranslational protein translocation, we used a reconstituted assay that measures the ability of purified SRP and SR to insert a model substrate, preprolactin (pPL), into ER microsomes depleted of endogenous SRP and SR (fig. S1A) (5, 43). Most of the SR conformational mutants are defective in pPL translocation (Fig. 6, B to D, and fig. S13). The largest defects were observed with SR(Δ361) and SR(Δ371), which blocked the rearrangements at the earliest stage (Figs. 2 and 6, B and D, orange). SR(Δ572), which specifically blocks distal site docking, also reduced translocation efficiency (Figs. 2 and 6, B and D, blue), supporting a role of the distal conformation in ensuring efficient protein translocation. The only exception was SR(R407A), which did not substantially affect pPL translocation despite impairments in the lateral movements of the NG heterodimer; this might reflect contributions from additional factors in the cell lysate and ER microsomes during translocation that were not present in smFRET measurements of the purified targeting complex. Notably, mutant SRPG226E, which causes severe congenital neutropenia (29, 30), strongly impaired the cotranslational translocation of pPL (Fig. 6, C and D, brown) despite its ability to assemble a stable early targeting complex and undergo reciprocal GTPase activation with SRP (Figs. 3 and 6A). Furthermore, given the faster GTPase rate in the targeting complexes assembled with these conformational mutants compared to wild-type SRP•SR, the energetic coupling of the GTPase cycle to productive work (translocation) was strongly reduced in all of the mutants (Fig. 6E).
Together, the results in this section demonstrate that the post-targeting conformational rearrangements in SRP and SR are essential for coupling the SRP/SR GTPase cycle to protein translocation. Disruption of these rearrangements leads to futile GTPase cycles and abortive targeting reactions and can lead to devastating pathology.
DISCUSSION
Cotranslational targeting of nascent proteins by the SRP pathway is essential for the generation and maintenance of compartmentalization in all cells (3, 44) and has served as a paradigm for understanding the molecular basis of protein localization. Despite decades of research, how the mammalian SRP pathway transitions from the cargo recognition phase, in which SRP captures translating ribosomes in the cytosol, to the targeting and translocation phases, in which the ribosomes are delivered to the ER membrane and handed off to the Sec61p translocase, remains unclear. In this work, smFRET measurements detected a sequential series of conformational rearrangements upon SRP•SR assembly that culminates in the prehandover conformation observed in a recent structure (19), in which the RNC is primed for unloading onto translocation machineries at the ER. Structural and biochemical analyses of early targeting intermediates revealed the molecular basis of early SRP•SR recognition and the structural transitions en route to the prehandover state. Biochemical measurements further demonstrate an essential role of these rearrangements in cotranslational protein translocation. Our results provide a molecular model for how the components of this universally conserved pathway transition from cargo recognition to the targeting and pretranslocation stages.
The evolution to higher eukaryotes is accompanied by an expansion of disordered regions in the proteome (45). This was also the case with SR. While the interaction of bacterial SR with the plasma membrane is primarily mediated by an amphipathic helix that immediately precedes its NG domain (46, 47), eukaryotic SR contains a ~200–amino acid disordered linker sequence between its NG domain and the X/β domain that anchors this receptor at the ER (48). The smFRET measurements here show that SR assumes an extended conformation and that interaction with cargo-loaded SRP is required to induce a global compaction of SR that brings the NG- and X/β domains into close proximity (Fig. 1). These findings suggest an attractive model in which the disordered SR linker initially acts as a “leash” that increases the search space in the cytosol and, upon recognition of cargo-bound SRP, compacts to bring the ribosome and nascent polypeptide close to the ER membrane. As SR is membrane-anchored, this compaction step as well as the subsequent GTPase movements could be further regulated by Sec61p or other membrane components (20), allowing SR to sense both cytosolic (ribosome-bound SRP) and membrane components (Sec61p and potentially other translocases) to determine the optimal timing of compaction. Moreover, membrane-anchored SR may have spatial restrictions and different physicochemical properties compared to the soluble SR used in this study, which could further tune the efficiency of the pathway. These possibilities remain to be explored in the future.
Molecular genetics and kinetic measurements (Fig. 2) further show that SR compaction precedes and drives the subsequent movements of the SRP•SR NG heterodimer from the ribosome exit site to the distal site. This model is also supported by the observation that the mutations that disrupt interactions within the SR complex (Δ361, Δ371, and R407A) also impair the detachment of NG complex from the ribosome. Intriguingly, a single-point mutation in SRP54 linked to congenital neutropenia, G226E (29, 30), abolishes all the conformational changes studied here. G226 is located at the interface between the N and G domains of SRP54 (fig. S3C, brown). Comparison of the structures of free SRP and SR with the SRP•SR complex in both bacterial and human systems showed that the N-G interface acts as a fulcrum that undergoes cooperative adjustments in both the SRP54 and SR NG domains upon their GTP-dependent assembly (31, 49–52). Conceivably, these rearrangements within the SRP•SR NG-domain complex are coupled to restructuring of the SR linker and thus amplified into the subsequent large-scale molecular motions. The extensive defect of mutants SRΔ361, SRΔ371, and SRPG226E in mediating cotranslational protein translocation further emphasized the central role of SR compaction. Intriguingly, SRPG226E expressed in zebrafish disease models also disrupts the expression of XBP1 (28), whose mRNA is targeted to the ER via the SRP pathway for splicing by the ER-anchored Ire1 (53, 54), suggesting that compaction of the targeting complex at the ER membrane is also important for unconventional SRP substrates that do not require translocation.
The cryo-EM structures of the early targeting complexes assembled with SRPG226E further reveal the structural basis of early SRP•SR recognition in the mammalian SRP pathway. We observed that the early complex is stabilized by bidentate interactions at the proximal site. In addition to the initial dynamic interaction between the SRP and SR NG domains, the MoRF in the SRα linker physically connects the M and NG domains of SRP54 in proximity to the GNRA tetraloop of the SRP RNA and is critical for the stability of the early targeting complex. Our analyses rationalize how the MoRF specifically activates early stages of NG heterodimer assembly analogously to the role of SRP RNA tetraloop in bacteria (14). In addition, the multiple specific interactions of SR MoRF at the proximal site of SRP explain why SRP preorganized in the proximal conformation is optimized for rapid assembly with SR (5). Furthermore, the comparison of the early and prehandover structures reveals extensive rotation in the SRP RNA, which is required to allow docking of the SRP•SR NG heterodimer at the distal site and anchoring of SRP to the ribosome via the SRP68 RBD. Together, the structural and biochemical analyses emphasize that critical events in SRP targeting in mammals are heavily regulated by eukaryotic-specific protein elements, which replace and extend the roles of the bacterial SRP RNA.
The movements of the SRP•SR NG heterodimer from the ribosome exit site to the distal site are conserved between bacterial and eukaryotic SRP pathways. In both cases, these rearrangements displace SRP-NG from uL23 and free up this universal docking site for subsequent interaction with Sec61p and/or other translocases at the ER (19, 55, 56). Nevertheless, distal docking in the mammalian SRP pathway displays distinct features compared to its bacterial homologs. While distal docking of the bacterial NG heterodimer is mediated solely by the SRP RNA that also activates GTP hydrolysis (41, 42, 55), this event is primarily mediated by eukaryote-specific protein elements, the SRP68/72 subunits and SR X/β domain, and requires extensive rotations of the SRP RNA to expose the NG-heterdimer docking site. (19). In addition, while distal docking of the bacterial NG heterodimer activates GTP hydrolysis, this event inhibits the GTPase activity of the mammalian targeting complex (Fig. 6A). This delays the turnover of mammalian SRP and may explain why a substantial fraction of mammalian SRP is found at the ER membrane (9, 57), whereas most of the bacterial SRP is cytosolic (58, 59). A longer lifetime of the SRP•SR complex at the ER could serve multiple roles. Measurements in yeast suggested that gating of Sec61p is slow (60), and therefore, delayed GTP hydrolysis may be necessary to provide sufficient time for Sec61p to engage RNCs with a functional signal or signal anchor sequence. The presence of additional translocases at the ER, such as the ER-associated membrane complex (61, 62), raises the possibility that RNCs with different nascent chains are triaged to distinct translocases, and a long-lived SRP•SR complex could provide a mechanism to improve both the efficiency and accuracy of the triaging process. Last, mammalian SRP evolved to mediate unconventional functions, such as delivering the XBP1u mRNA to the ER (53, 63, 64), which could benefit from an increased lifetime of the targeting complex at the ER.
Failures in SRP function have mostly been attributed to impaired interaction with the RNC and/or binding with the SR. Our results demonstrate a previously unidentified class of mutations that disrupt later stages of the SRP pathway, by interfering with conformational rearrangements of the targeting complex at the ER. This is most notable with mutant SRP54G226E linked to severe congenital neutropenia (29, 30), which undergoes ribosome- and signal sequence–activated assembly with SR at efficiencies comparable to wild-type SRP. Both the smFRET and cryo-EM studies of the targeting complex assembled with SRPG226E show that its dysfunction is due to the accumulation of trapped RNC•SRP•SR intermediates. The severe defects of SRPG226E in cotranslational protein translocation (this work) and the rescue of SRPG226E-expressing zebrafish by expression of XBP1 (28), a key branch of the integrated stress response (54), strongly suggest that a defect in protein homeostasis in the endomembrane system underlies the onset of diseases associated with this mutation.
In summary, our results provide a mechanistic model for the mammalian SRP pathway. During cargo recognition, signal sequence–bearing ribosomes promote SRP binding in the proximal conformation in which the SRP54 NG domain docks at uL23/uL29 near the ribosome exit site (Fig. 7) (5). At the same time, SR anchored at the membrane is in an extended conformation, potentially increasing the search space for SRPs in the cytosol. In these conformations, the SRP and SR NG domains first assemble a loose complex that is stabilized by interactions of the SR MoRF with the SRP proximal site (step 1). Cooperative rearrangements in the NG complex upon its assembly, especially those at the interface between the N and G domains, generate a tighter NG heterodimer and are further transmitted to the SR linker, amplifying into a global compaction of SR (step 2). Both the NG-complex rearrangements and SR compaction could drive the detachment of the GTPase complex from the ribosome exit (step 2), exposing universal docking sites at the ribosome exit tunnel for subsequent interaction with the Sec61p translocase. Throughout this process, the SRP RNA is remodeled to expose the NG-heterodimer docking site at the SRP distal end, to which the GTPase complex docks (step 3). This generates a prehandover conformation in which the ribosome is primed for unloading, and delayed GTP hydrolysis could generate an extended time window during which the targeting complex searches for and allows the ribosome to engage the appropriate translocase. These post-targeting molecular movements resolve multiple mechanistic challenges during the initiation of protein translocation and could serve as points for biological regulation as well as disease intervention.
Fig. 7. New model for cotranslational protein targeting and translocation.
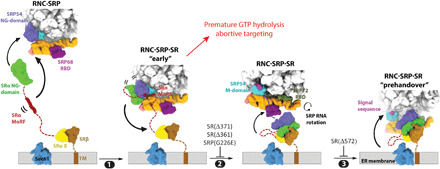
SRP is preorganized into the proximal conformation on signal sequence–bearing ribosomes and initially recruits SR via dynamic interaction between their NG domains, which is stabilized by additional interaction of the SR MoRF with the 7SL RNA (step 1). A conformational rearrangement within the NG domains generates a more stable NG complex and induces SR compaction, which together drive the detachment of the NG complex from the ribosome exit (step 2). Docking of the GTPase complex at the distal site primes the RNC for handover to Sec61p (step 3).
MATERIALS AND METHODS
Vectors
The vectors for expression of SRP and SR subunits and for fluorescence labeling of SRP19, SRP54, and SRα C terminus have been described (5). To fluorescently label SRP68, an Sfp recognition motif (ybbr6, DSLEFI) (65) was inserted after Pro149 using FastCloning. To fluorescently label SRβΔTM, a longer Sfp recognition motif (ybbr11, DSLEFIASKLA) (65) was inserted at the SRβ N terminus using FastCloning. Expression vectors for mutant SRP and SRs were generated using the QuikChange mutagenesis protocol (StrataGene).
Biochemical preparations
Wild-type and mutant SRP and SR proteins were expressed and purified as described (5). Mammalian SRP was prepared as described (5). Briefly, SRP protein subunits were expressed and purified in bacteria or yeast. A circularly permutated 7SL RNA variant was in vitro transcribed and purified on a denaturing polyacrylamide gel. SRP was assembled by first refolding 7SL RNA and sequentially adding SRP19, SRP68/72, SRP9/14, and SRP54. Holo-SRP was purified using a DEAE ion-exchange column. Unless otherwise specified, the C terminus of human SRP54 was fused to the 4A10L signal sequence (hSRP54-4A10L) to generate signal sequence–bound SRP, as described (5). Ribosome from rabbit reticulocyte lysate was purified by ultracentrifugation through a sucrose gradient, as described (5). The use of ribosome and signal sequence fusion to SRP54 reproduced the effects of signal sequence–bearing RNCs on the conformation and activity of SRP (5).
Fluorescence labeling
SRP54(C12), SRP54(C47), and SRP19(C64) were labeled with Atto550, Atto647N, or Cy3B using maleimide chemistry as described (5). Labeling efficiency of these proteins was 70 to 80%. SRα was labeled with Atto647N at the C terminus using sortase-mediated ligation, as described (5).
SRP68/72 was labeled via Sfp-mediated conjugation of coenzyme A (CoA)–Atto647N, CoA-Cy3B, or CoA-BODIPY FL at Ser2 in ybbr6-tagged SRP68 following the procedure described in (65). The labeling reaction contained 0.4 molar ratio of protein to Sfp enzyme and a threefold excess of CoA-dye conjugate and was carried out for 20 min at room temperature in Sfp-labeling buffer [50 mM KHepes (pH 7.5), 10 mM MgCl2, 150 mM NaCl, and 20% glycerol]. Labeling efficiency was close to 100%. Labeled SRP68/72 was immediately used for SRP assembly. SRXβ and SRXLβ were labeled via Sfp-mediated conjugation of CoA-TMR at a ybbr11 tag inserted between T155 and R156 C terminus to the SR X domain. The labeling reaction contained 0.4 molar ratio of SR to Hisless Sfp enzyme and twofold excess of CoA-conjugated TMR and was carried out for 1 hour and 30 min at room temperature by rotation in Sfp-labeling buffer. The labeling efficiency was close to 100%. Labeled SRXβ and SRXLβ were purified using Ni-HP Sepharose resin and stored in SRP assay buffer.
SRαβΔTM was doubly labeled via sortase-mediated ligation at the SRα C terminus and Sfp-mediated conjugation of CoA-dye at the N-terminal ybbr11 tag on SRβ. The labeling reaction contained 0.4 molar ratio of Sfp to protein and a twofold excess of CoA-Atto647N (or CoA-Atto550) and was carried out for 30 min at room temperature in Sfp-labeling buffer. A 4-fold molar excess of sortase,10-fold excess of GGGC-Atto550 (or GGGC-Atto647N), and 0.1 volume of 10× sortase buffer [500 mM tris-HCl (pH 7.5), 1.5 M NaCl, and 100 mM CaCl2) were then added, and the labeling reaction was carried out for an additional 3 hours at room temperature. Labeled SRαβΔTM was purified using Ni-Sepharose resin. Labeling efficiency was close to 100% for the Sfp reaction and ~60 to 70% for the sortase reaction.
Biochemical assays
All proteins except for SRP were ultracentrifuged at 4°C, 100,000 rpm in a TLA100 rotor for 30 to 60 min to remove aggregates before all assays. GTPase reactions were performed in SRP assay buffer [50 mM KHepes (pH 7.5), 150 mM KOAc, 5 mM Mg(OAc)2, 10% glycerol, 2 mM dithiothreitol, and 0.04% Nikkol] at 25°C and were followed and analyzed as described (5, 66). Details for the determination of the GTPase rate constants are described in Supplementary Methods. Cotranslational targeting and translocation of pPL into salt-washed and trypsin-digested rough ER microsomes were performed and analyzed as described in Supplementary Methods. Steady-state fluorescence measurements were carried out on a Fluorolog 3-22 spectrofluorometer (Jobin Yvon) at 25°C in SRP assay buffer supplemented with bovine serum albumin (0.6 mg/ml). Acquisition and analyses of fluorescence data are described in Supplementary Methods.
Small-molecule Förster resonance energy transfer
Measurements were performed as described (5, 32, 33). Labeled SRP was diluted to 100 to 200 pM in SRP assay buffer containing 200 μM nonhydrolysable GTP (guanosine-5′-[β,γ-imido]triphosphate) (GppNHp), 150 nM 80S, and 1.5 μM SRαβΔTM where indicated. To measure the conformation of SR, doubly labeled SRαβΔTM was diluted to 100 to 200 pM in SRP assay buffer containing 200 μM GppNHp, 400 nM 80S, and 400 nM SRP or SRP-4A10L where indicated. Data were collected over 30 to 60 min using an ALEX-FAMS setup with two single-photon Avalanche photodiodes (PerkinElmer) and 532 nm (CNI laser) and 638 nm (Opto Engine LLC) continuous wave lasers operating at 150 and 70 μW, respectively. Analysis of μs-ALEX data is described in Supplementary Methods.
Cryo–electron microscopy
For the protein targeting complexes containing human SRPG226E, 80 nM RNCs prepared as previously described (19) were mixed with human recombinant SRP54G226E and SR with a molar ratio of 1:1.5:5.8 in buffer C [50 mM Hepes-KOH, (pH 7.6), 100 mM KOAc, and 5 mM Mg(OAc)2] in the presence of 2 mM GppNHp and 0.02% Nikkol (Sigma-Aldrich). The reaction was incubated at 30°C for 20 min after adding SRP and then another 20 min after adding SR. The reaction was lastly chilled for 15 min on ice. Sample (5 μl) was applied on Quantifoil R2/2 grid holey carbon grids, which has been coated freshly with an extra layer of carbon and glow discharged with the Pelco EasyGlow system for the 15 s. The sample (5 μl) was incubated on the grid at 4°C with 95% relative humidity for 1 min before being blotted and then plunged into liquid ethane/propane mix cooled to liquid nitrogen temperature using a Thermo Fisher Scientific Vitrobot. Details of the purification of RNC, SRP, and SR for cryo-EM are described under Supplementary Methods.
Data collection was performed on a Titan Krios electron microscope (Thermo Fisher Scientific) operated at 300 kV, using the EPU software (Thermo Fisher Scientific) for automated data acquisition in counting mode using the Gatan K3 direct electron detector with an energy filter slit width of 20 eV. Data were collected at a defocus of −1.2- to −2.5-μm range with a step size of 0.1 μm and at a nominal magnification of ×81,000, which resulted into a calibrated pixel size of 1.062 Å per pixel (0.529 Å per pixel in superresolution mode). Illumination conditions were adjusted to an exposure rate of 8 e− per pixel per second. Micrographs were recorded as movie stacks with an exposure time for each movie stack of 1.4 s, corresponding to an electron dose of ~50 electrons/Å2 fractionated into total of 40 frames. Drift and bright gain reference corrections in addition to dose weighting were performed with MotionCor2 (67). Details of data processing are described under Supplementary Methods.
Acknowledgments
We thank the members of the Shan and Ban laboratory for comments on the manuscript. We thank A. Scaiola for the support with EM data processing and M. Leibundgut for the support with model building. Cryo-EM data were collected at the Scientific Center for Optical and Electron Microscopy at the ETH Zurich (ScopeM). Funding: This work was supported by National Institutes of Health grants GM078024 and R35 GM136321, National Science Foundation grant MCB-1929452, and the Gordon and Betty Moore Foundation grant GBMF2939 to S.S.; by the Swiss National Science Foundation (SNSF) (grant number 310030B_163478) and National Center of Excellence in Research (NCCR) RNA and Disease Program of the SNSF (grant number 51NF40_141735) to N.B.; and by National Institutes of Health grant GM130942 and Dean Willard Chair funds to S.W. We gratefully acknowledge the support of NVIDIA Corporation for the Titan Xp GPU used in this research through a GPU Grant program awarded to A.J. Author contributions: J.H.L., A.J., Y.-H.H.F., N.B., and S.S. designed research. J.H.L., Y.-H.H.F., R.Q., X.S., H.-H.H., X.B., and S.C. performed biochemical experiments and analyzed data. A.J. and S.M. purified RNCs for cryo-EM data collection. A.J., S.M., and D.B. collected EM data. A.J. processed cryo-EM data and built atomic models. J.H.L., R.Q., and S.Y.C. performed μs-ALEX experiments and analyzed data. S.W. provided guidance for μs-ALEX analysis. J.H.L., S.S., A.J., and N.B. wrote the manuscript with input from S.C., H.-H.H., S.Y.C. and S.W. Competing interests: S.W. is a consultant to Bio-Rad. The authors declare that they have no other competing interests. Data and materials availability: All the data and associated procedures are described in the manuscript and/or in Supplementary Materials. Cryo-EM maps and model coordinates are deposited in the EMDB as EMD-12303, EMD-12304, and EMD-12305 and in the PDB as PDB ID 7NFX.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabg0942/DC1
REFERENCES AND NOTES
- 1.Hartl F. U., Bracher A., Hayer-Hartl M., Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Akopian D., Shen K., Zhang X., Shan S., Signal recognition particle: An essential protein-targeting machine. Annu. Rev. Biochem. 82, 693–721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang X., Shan S., Fidelity of cotranslational protein targeting by the signal recognition particle. Annu. Rev. Biophys. 43, 381–408 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandon E. C., Jiang Y., Gilmore R., Dual recognition of the ribosome and the signal recognition particle by the SRP receptor during protein targeting to the endoplasmic reticulum. J. Cell Biol. 162, 575–585 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J. H., Chandrasekar S., Chung S., Hwang Fu Y.-H., Liu D., Weiss S., Shan S., Sequential activation of human signal recognition particle by the ribosome and signal sequence drives efficient protein targeting. Proc. Natl. Acad. Sci. U.S.A. 115, E5487–E5496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jomaa A., Boehringer D., Leibundgut M., Ban N., Structures of the E. coli translating ribosome with SRP and its receptor and with the translocon. Nat. Commun. 7, 10417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen K., Zhang X., Shan S.-o., Synergistic actions between the SRP RNA and translating ribosome allow efficient delivery of the correct cargos during cotranslational protein targeting. RNA 17, 892–902 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X., Lam V. Q., Mou Y., Kimura T., Chung J., Chandrasekar S., Winkler J. R., Mayo S. L., Shan S.-O., Direct visualization reveals dynamics of a transient intermediate during protein assembly. Proc. Natl. Acad. Sci. U.S.A. 108, 6450–6455 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter P., Blobel G., [53] Signal recognition particle: A ribonucleoprotein required for cotranslational translocation of proteins, isolation and properties. Methods Enzymol. 96, 682–691 (1983). [DOI] [PubMed] [Google Scholar]
- 10.Angelini S., Boy D., Schiltz E., Koch H.-G., Membrane binding of the bacterial signal recognition particle receptor involves two distinct binding sites. J. Cell Biol. 174, 715–724 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Leeuw E., te Kaat K., Moser C., Menestrina G., Demel R., de Kruijff B., Oudega B., Luirink J., Sinning I., Anionic phospholipids are involved in membrane association of FtsY and stimulate its GTPase activity. EMBO J. 19, 531–541 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tajima S., Lauffer L., Rath V. L., Walter P., The signal recognition particle receptor is a complex that contains two distinct polypeptide chains. J. Cell Biol. 103, 1167–1178 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz T., Blobel G., Structural basis for the function of the β subunit of the eukaryotic signal recognition particle receptor. Cell 112, 793–803 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Hwang Fu Y.-H., Chandrasekar S., Lee J. H., Shan S., A molecular recognition feature mediates ribosome-induced SRP-receptor assembly during protein targeting. J. Cell Biol. 218, 3307–3319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estrozi L. F., Boehringer D., Shan S., Ban N., Schaffitzel C., Cryo-EM structure of the E. coli translating ribosome in complex with SRP and its receptor. Nat. Struct. Mol. Biol. 18, 88–90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voorhees R. M., Hegde R. S., Structures of the scanning and engaged states of the mammalian SRP-ribosome complex. eLife 4, e07975 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halic M., Becker T., Pool M. R., Spahn C. M. T., Grassucci R. A., Frank J., Beckmann R., Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature 427, 808–814 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Halic M., Gartmann M., Schlenker O., Mielke T., Pool M. R., Sinning I., Beckmann R., Signal recognition particle receptor exposes the ribosomal translocon binding site. Science 312, 745–747 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi K., Jomaa A., Lee J. H., Chandrasekar S., Boehringer D., Shan S., Ban N., Structure of a prehandover mammalian ribosomal SRP·SRP receptor targeting complex. Science 360, 323–327 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jadhav B., McKenna M., Johnson N., High S., Sinning I., Pool M. R., Mammalian SRP receptor switches the Sec61 translocase from Sec62 to SRP-dependent translocation. Nat. Commun. 6, 10133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Connolly T., Rapiejko P., Gilmore R., Requirement of GTP hydrolysis for dissociation of the signal recognition particle from its receptor. Science 252, 1171–1173 (1991). [DOI] [PubMed] [Google Scholar]
- 22.Rapiejko P. J., Gilmore R., Empty site forms of the SRP54 and SRα GTPases mediate targeting of ribosome-nascent chain complexes to the endoplasmic reticulum. Cell 89, 703–713 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Wilson C., Connolly T., Morrison T., Gilmore R., Integration of membrane proteins into the endoplasmic reticulum requires GTP. J. Cell Biol. 107, 69–77 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chitwood P. J., Juszkiewicz S., Guna A., Shao S., Hegde R. S., EMC is required to initiate accurate membrane protein topogenesis. Cell 175, 1507–1519.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shurtleff M. J., Itzhak D. N., Hussmann J. A., Schirle Oakdale N. T., Costa E. A., Jonikas M., Weibezahn J., Popova K. D., Jan C. H., Sinitcyn P., Vembar S. S., Hernandez H., Cox J., Burlingame A. L., Brodsky J. L., Frost A., Borner G. H., Weissma J. S., The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. eLife 7, e37018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips G. J., Silhavy T. J., The E. coli ffh gene is necessary for viability and efficient protein export. Nature 359, 744–746 (1992). [DOI] [PubMed] [Google Scholar]
- 27.Althoff S. M., Stevens S. W., Wise J. A., The Srp54 GTPase is essential for protein export in the fission yeast Schizosaccharomyces pombe. Mol. Cell. Biol. 14, 7839–7854 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schürch C., Schaefer T., Müller J. S., Hanns P., Arnone M., Dumlin A., Schärer J., Sinning I., Wild K., Skokowa J., Welte K., Carapito R., Bahram S., Konantz M., Lengerke C., SRP54 mutations induce Congenital Neutropenia via dominant-negative effects on XBP1 splicing. Blood 137, 1340–1352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carapito R., Konantz M., Paillard C., Miao Z., Pichot A., Leduc M. S., Yang Y., Bergstrom K. L., Mahoney D. H., Shardy D. L., Alsaleh G., Naegely L., Kolmer A., Paul N., Hanauer A., Rolli V., Müller J. S., Alghisi E., Sauteur L., Macquin C., Morlon A., Sancho C. S., Amati-Bonneau P., Procaccio V., Mosca-Boidron A.-L., Marle N., Osmani N., Lefebvre O., Goetz J. G., Unal S., Akarsu N. A., Radosavljevic M., Chenard M.-P., Rialland F., Grain A., Béné M.-C., Eveillard M., Vincent M., Guy J., Faivre L., Thauvin-Robinet C., Thevenon J., Myers K., Fleming M. D., Shimamura A., Bottollier-Lemallaz E., Westhof E., Lengerke C., Isidor B., Bahram S., Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond–like features. J. Clin. Invest. 127, 4090–4103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bellanné-Chantelot C., Schmaltz-Panneau B., Marty C., Fenneteau O., Callebaut I., Clauin S., Docet A., Damaj G. L., Leblanc T., Pellier I., Stoven C., Souquere S., Antony-Debré I., Beaupain B., Aladjidi N., Barlogis V., Bauduer F., Bensaid P., Boespflug-Tanguy O., Berger C., Bertrand Y., Carausu L., Fieschi C., Galambrun C., Schmidt A., Journel H., Mazingue F., Nelken B., Quah T. C., Oksenhendler E., Ouachée M., Pasquet M., Saada V., Suarez F., Pierron G., Vainchenker W., Plo I., Donadieu J., Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond – Like syndrome. Blood 132, 1318–1331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juaire K. D., Lapouge K., Becker M. M. M., Kotova I., Michelhans M., Carapito R., Wild K., Bahram S., Sinning I., Structural and functional impact of SRP54 mutations causing severe congenital neutropenia. Structure 29, 15–28.e7 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Kapanidis A. N., Lee N. K., Laurence T. A., Doose S., Margeat E., Weiss S., Fluorescence-aided molecule sorting: Analysis of structure and interactions by alternating-laser excitation of single molecules. Proc. Natl. Acad. Sci. U.S.A. 101, 8936–8941 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kapanidis A. N., Laurence T. A., Lee N. K., Margeat E., Kong X., Weiss S., Alternating-laser excitation of single molecules. Acc. Chem. Res. 38, 523–533 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Ingargiola A., Lerner E., Chung S., Weiss S., Michalet X., FRETBursts: An open source toolkit for analysis of freely-diffusing single-molecule FRET. PLOS ONE 11, e0160716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogg S. C., Barz W. P., Walter P., A FUnctional GTPase domain, but not its transmembrane domain, is required for function of the SRP receptor β-subunit. J. Cell Biol. 142, 341–354 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fulga T. A., Sinning I., Dobberstein B., Pool M. R., SRβ coordinates signal sequence release from SRP with ribosome binding to the translocon. EMBO J. 20, 2338–2347 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torella J. P., Holden S. J., Santoso Y., Hohlbein J., Kapanidis A. N., Identifying molecular dynamics in single-molecule FRET experiments with burst variance analysis. Biophys. J. 100, 1568–1577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robb N. C., Cordes T., Hwang L. C., Gryte K., Duchi D., Craggs T. D., Santoso Y., Weiss S., Ebright R. H., Kapanidis A. N., The transcription bubble of the RNA polymerase-promoter open complex exhibits conformational heterogeneity and millisecond-scale dynamics: Implications for transcription start-site selection. J. Mol. Biol. 425, 875–885 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chio U. S., Chung S., Weiss S., Shan S., A protean clamp guides membrane targeting of tail-anchored proteins. Proc. Natl. Acad. Sci. 114, E8585–E8594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsieh H.-H., Lee J. H., Chandrasekar S., Shan S., A ribosome-associated chaperone enables substrate triage in a cotranslational protein targeting complex. Nat. Commun. 11, 5840 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen K., Wang Y., Hwang Fu Y.-H., Zhang Q., Feigon J., Shan S., Molecular mechanism of GTPase activation at the signal recognition particle (SRP) RNA distal end. J. Biol. Chem. 288, 36385–36397 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voigts-Hoffmann F., Schmitz N., Shen K., Shan S., Ataide S. F., Ban N., The structural basis of FtsY recruitment and GTPase activation by SRP RNA. Mol. Cell 52, 643–654 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shan S., Chandrasekar S., Walter P., Conformational changes in the GTPase modules of the signal reception particle and its receptor drive initiation of protein translocation. J. Cell Biol. 178, 611–620 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X., Rashid R., Wang K., Shan S.-o., Sequential checkpoints govern substrate selection during cotranslational protein targeting. Science 328, 757–760 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Light S., Sagit R., Sachenkova O., Ekman D., Elofsson A., Protein expansion is primarily due to indels in intrinsically disordered regions. Mol. Biol. Evol. 30, 2645–2653 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Parlitz R., Eitan A., Stjepanovic G., Bahari L., Bange G., Bibi E., Sinning I., Escherichia coli signal recognition particle receptor FtsY contains an essential and autonomous membrane-binding amphipathic helix. J. Biol. Chem. 282, 32176–32184 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Lam V. Q., Akopian D., Rome M., Henningsen D., Shan S., Lipid activation of the signal recognition particle receptor provides spatial coordination of protein targeting. J. Cell Biol. 190, 623–635 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nyathi Y., Wilkinson B. M., Pool M. R., Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 1833, 2392–2402 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Shan S., Stroud R. M., Walter P., Mechanism of association and reciprocal activation of two GTPases. PLOS Biol. 2, e320 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Egea P. F., Shan S. O., Napetschnig J., Savage D. F., Walter P., Stroud R. M., Substrate twinning activates the signal recognition particle and its receptor. Nature 427, 215–221 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Focia P. J., Shepotinovskaya I. V., Seidler J. A., Freymann D. M., Heterodimeric GTPase core of the SRP targeting complex. Science 303, 373–377 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wild K., Bange G., Motiejunas D., Kribelbauer J., Hendricks A., Segnitz B., Wade R. C., Sinning I., Structural basis for conserved regulation and adaptation of the signal recognition particle targeting complex. J. Mol. Biol. 428, 2880–2897 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Kanda S., Yanagitani K., Yokota Y., Esaki Y., Kohno K., Autonomous translational pausing is required for XBP1u mRNA recruitment to the ER via the SRP pathway. Proc. Natl. Acad. Sci. 113, E5886–E5895 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karagöz G. E., Acosta-Alvear D., Walter P., The unfolded protein response: Detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 11, a033886 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen K., Arslan S., Akopian D., Ha T., Shan S., Activated GTPase movement on an RNA scaffold drives co-translational protein targeting. Nature 492, 271–275 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jomaa A., Fu Y.-H. H., Boehringer D., Leibundgut M., Shan S., Ban N., Structure of the quaternary complex between SRP, SR, and translocon bound to the translating ribosome. Nat. Commun. 8, 15470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walter P., Blobel G., Subcellular distribution of signal recognition particle and 7SL-RNA determined with polypeptide-specific antibodies and complementary DNA probe. J. Cell Biol. 97, 1693–1699 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yosef I., Bochkareva E. S., Adler J., Bibi E., Membrane protein biogenesis in Ffh- or FtsY-depleted Escherichia coli. PLOS ONE 5, e9130 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishihama Y., Schmidt T., Rappsilber J., Mann M., Hartl F. U., Kerner M. J., Frishman D., Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics 9, 102 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng Z., Gilmore R., Slow translocon gating causes cytosolic exposure of transmembrane and lumenal domains during membrane protein integration. Nat. Struct. Mol. Biol. 13, 930–936 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Aviram N., Schuldiner M., Targeting and translocation of proteins to the endoplasmic reticulum at a glance. J. Cell Sci. 130, 4079–4085 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Chitwood P. J., Hegde R. S., The role of EMC during membrane protein biogenesis. Trends Cell Biol. 29, 371–384 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Yanagitani K., Imagawa Y., Iwawaki T., Hosoda A., Saito M., Kimata Y., Kohno K., Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell 34, 191–200 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Yanagitani K., Kimata Y., Kadokura H., Kohno K., Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science 331, 586–589 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Yin J., Lin A. J., Golan D. E., Walsh C. T., Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 1, 280–285 (2006). [DOI] [PubMed] [Google Scholar]
- 66.Peluso P., Shan S., Nock S., Herschlag D., Walter P., Role of SRP RNA in the GTPase cycles of Ffh and FtsY. Biochemistry 40, 15224–15233 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilmore R., Walter P., Blobel G., Protein translocation across the endoplasmic reticulum. II. Isolation and characterization of the signal recognition particle receptor. J. Cell Biol. 95, 470–477 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nir E., Michalet X., Hamadani K. M., Laurence T. A., Neuhauser D., Kovchegov Y., Weiss S., Shot-noise limited single-molecule FRET histograms: Comparison between theory and experiments. J. Phys. Chem. B 110, 22103–22124 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang K., Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J. H., Lindahl E., Scheres S. H. W., New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown A., Shao S., Murray J., Hegde R. S., Ramakrishnan V., Structural basis for stop codon recognition in eukaryotes. Nature 524, 493–496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.W. L. DeLano, The PyMOL Molecular Graphics System, Version 2.3. Schrödinger LLC (2020); http://www.pymol.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabg0942/DC1