

Structures of the AVP-V2R-Gs protein complex provide molecular insights into the function of this clinically relevant target.
Abstract
The antidiuretic hormone arginine-vasopressin (AVP) forms a signaling complex with the V2 receptor (V2R) and the Gs protein, promoting kidney water reabsorption. Molecular mechanisms underlying activation of this critical G protein–coupled receptor (GPCR) signaling system are still unknown. To fill this gap of knowledge, we report here the cryo–electron microscopy structure of the AVP-V2R-Gs complex. Single-particle analysis revealed the presence of three different states. The two best maps were combined with computational and nuclear magnetic resonance spectroscopy constraints to reconstruct two structures of the ternary complex. These structures differ in AVP and Gs binding modes. They reveal an original receptor-Gs interface in which the Gαs subunit penetrates deep into the active V2R. The structures help to explain how V2R R137H or R137L/C variants can lead to two severe genetic diseases. Our study provides important structural insights into the function of this clinically relevant GPCR signaling complex.
INTRODUCTION
The biological actions of arginine-vasopressin (AVP), a cyclic nonapeptide, are mediated through three G protein–coupled receptor (GPCR) subtypes, V1a, V1b, and V2 (1). In addition, AVP is able to activate the related oxytocin (OT) receptor (OTR) (2). The V2 receptor (V2R) is mainly expressed at the basolateral membrane of principal cells of the kidney collecting ducts and governs the crucial physiological function of body water homeostasis (3). Binding of AVP to the V2R increases cyclic adenosine monophosphate (cAMP) intracellular level via coupling to the adenylyl cyclase stimulatory Gs protein, leading to activation of protein kinase A, phosphorylation of aquaporin 2 water channels (4), and, ultimately, to water reabsorption and urine concentration. Activation of the V2R also elicits arrestin-dependent pathways such as receptor internalization and mitogen-activated protein (MAP) kinase phosphorylation associated with cell growth and proliferation (5, 6). This GPCR is involved in many water balance disorders (hyponatremia consecutive to congestive heart failure, hypertension, or hepatic cirrhosis) and voiding disorders (incontinence and nocturia) and, hence, constitutes a major therapeutic target (7). Moreover, inactivating and constitutively active mutations in the V2R sequence are responsible for two rare X-linked genetic diseases with opposite clinical outcomes: (i) congenital nephrogenic diabetes insipidus (cNDI) characterized by excessive urine voiding (8) and (ii) nephrogenic syndrome of inappropriate antidiuresis (NSIAD) characterized by excessive water loading and hyponatremia (9). V2R is also a target for treating autosomal dominant polycystic kidney disease, the most frequent Mendelian inherited disorder affecting millions of people worldwide (10). This pathology results from increased cell proliferation, apoptosis, and dedifferentiation, in which cAMP- and MAP kinase–dependent signaling pathways are highly activated.
The structural biology of GPCRs has made substantial progress during the past decade with a wealth of information about ligand binding and G protein coupling that shed light on structural and dynamic aspects of their function (11, 12). V2R, similar to many GPCRs, has been refractory to high-resolution structure determination. Cryo–electron microscopy (cryo-EM) has emerged as a powerful method for the determination of challenging membrane protein structures (13), particularly when the intrinsic structural dynamics of the target prevents the use of crystallogenesis. A growing list of GPCR–G protein complex structures has thus been determined (14, 15), revealing key molecular mechanisms of agonist binding and G protein (Gi, Gs, Gq, and Go) coupling to class A and class B GPCRs. Here, we have developed an in vitro purification strategy to reconstitute the GPCR signaling complex comprising the AVP-bound V2R and the heterotrimeric Gs protein stabilized with the nanobody Nb35. Cryo-EM single-particle analysis revealed the presence of three distinct populations of the ternary complex with two best maps at a mean resolution of 4.0 and 4.1 Å. A novel hybrid approach was used to build both corresponding structures. Analyses of the structural features of the distinct conformations provide unprecedented molecular insights into the dynamic process of ternary complex formation between the hormone AVP, the V2R, and the Gs protein.
RESULTS
Determination of the AVP-V2R-Gs-Nb35 complex structure
To improve the expression of the human V2R and facilitate its purification, we constructed a receptor version with a hemagglutinin signal peptide followed by a flag tag at its N terminus and a twin strep tag at its C terminus (fig. S1A). In addition, N22 was substituted with a glutamine residue to avoid N-glycosylation, and C358 was mutated into an alanine to eliminate the possibility of intermolecular disulfide bridges. Apart from receptor engineering designed uniquely for expression and purification purpose and unlike many of the recently published GPCR structures, we did not modify the receptor sequence (the V2R is a wild-type from T31 to G345). Our aim was to avoid possible artifacts and irrelevant information due to the introduction of mutations in the transmembrane (TM) core domain of the receptor, even if this was at the expense of lower-resolution cryo-EM data. Moreover, and before the recombinant expression of the receptor in Sf9 insect cells, the pharmacological properties of the engineered V2R were verified in human embryonic kidney (HEK) mammalian cells (fig. S2, A to C). The cryo-EM version of the V2R bound a fluorescent nonpeptide antagonist and AVP with high affinity [dissociation constant (Kd) and inhibition constant (Ki) = 2.27 ± 0.24 nM (n = 3) and 1.12 ± 0.5 nM (n = 3), respectively], close to the values determined for a wild-type V2R (16). Moreover, the receptor was proven to be functional as it was able to stimulate cAMP accumulation upon AVP binding [Kact = 2.05 ± 0.11 nM (n = 4), similar to the wild-type V2R in transfected cells (17)].
Following infection of Sf9 cells with the V2R recombinant baculovirus, the receptor was purified through an orthogonal chromatography procedure (fig. S1B). It was then mixed with the heterotrimeric Gs protein and the Nb35 in the presence of an excess of AVP. The purified complex displayed a monodisperse peak on size exclusion chromatography (SEC) (fig. S1C), and SDS gel analyses confirmed the presence of all components of the complex [the V2R, the three subunits of the G protein (αs, β1, and γ2), and the Nb35; fig. S1D]. The complex was first characterized using negative stain electron microscopy (NS-EM), before the preparation of vitrified samples onto Quantifoil grids for cryo-EM single-particle analysis.
Images of the complex first recorded in NS-EM revealed a homogeneous distribution of the particles, as observed from two-dimensional (2D) class averages (fig. S3, A and B). More than 60% of the particles correspond to the complex. A reconstruction at 20 Å clearly showed the micelle of detergent and the G protein–Nb35 components. Fitting the 3D model of the crystal structure of the β2-adrenergic receptor (β2AR)–Gs-Nb35 complex (18) in this low-resolution reconstruction map confirms that V2R-Gs-Nb35 displays typical structural features of a TM signaling GPCR complex (fig. S3C). Moreover, the addition of the specific V2R nonpeptide antagonist SR121463 (19) and guanosine 5′-O-(3-thiotriphosphate) (GTPγS) to the purified complex led to the dissociation of the different components (fig. S3D), confirming the functionality of the signaling particle.
After validation of cryo-EM grid sample vitrification, a total number of 25,770 movies were recorded, with 3.5 million particles picked and sorted out for further data processing (figs. S4 and S5). After 3D classification of projections and 3D refinement, we identified three different conformational states of the complex, referred to as loose (L), tight-1 (T1), and tight-2 (T2). Reconstruction of each state was at 4.2, 4.5, and 4.7 Å, with a distribution of 16, 48, and 36%, respectively (fig. S4), the local resolution varying from 3.2 to 6.4 Å (fig. S5C). Using the recent algorithm developed to enhance cryo-EM maps by density modification (20), the resolution of density maps were improved to 4.0 Å (L state), 4.1 Å (T1 state), and 4.5 Å (T2 state), respectively (Table 1, and figs. S4 to S6). This step enhanced the visibility of many details for some V2R TM regions (fig. S6, A and B), for the hormone AVP (fig. S6C), for the Gαs subunit (fig. S6D), and for Gβ2 subunit (fig. S6E). The maps mainly differ in the angle of Gs-Nb35 with the receptor 7TM and may reflect an inherent high flexibility of the complex. A conformational heterogeneity analysis using multibody refinement revealed that more than 78% of the variance is accounted for by the four first eigenvectors related to rotations and translations between AVP-V2R and Gs-Nb35 (Fig. 1, A to C, and movie S1). The 4.5-Å map of the T2 state was not well enough resolved to compute a reliable structure. Therefore, only the L and T1 structures, referred to as L and T states, were used for further analysis (Table 1).
Table 1. Cryo-EM data collection, refinement, and validation statistics.
PDB, Protein Data Bank; RMSD, root mean square deviation.
|
AVP-V2R-Gαsβ1γ2-Nb35 (L state) (EMD-12128) (PDB code 7BB6) |
AVP-V2R-Gαsβ1γ2-Nb35 (T state) (EMD-12129) (PDB code 7BB7) |
|
| Data collection and processing | ||
| Magnification | 165,000 | 165,000 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e−/Å2) | 50.19*/41.19† | 50.19*/41.19† |
| Defocus range (μm) | −0.8 to −2.2 | −0.8 to −2.2 |
| Pixel size (Å) | 0.81 | 0.81 |
| Symmetry imposed | C1 | C1 |
| Initial particle images (no.) | 3,566,007 | 3,566,007 |
| Final particle images (no.) | 147,524 | 420,953 |
| Map resolution (Å) | 4.2 | 4.4 |
| FSC threshold | 0.143 | 0.143 |
| Map resolution range (Å) | 3.2–5.6 | 3.8–6 |
| Refinement | ||
| Model resolution (Å) | 4.2 | 4.4 |
| FSC threshold | 0.5 | 0.5 |
| Model resolution range (Å) | 3.2–5.6 | 3.8–6 |
| Map improved resolution (Å) (cryoresolve) | 4.0 | 4.1 |
| Map sharpening B factor (Å2) | −141.6 | −75 |
| Model composition | ||
| Nonhydrogen atoms | 7955 | 8040 |
| Protein residues | 1017 | 1022 |
| Ligands | 0 | 0 |
| B factors (Å2) | ||
| Protein | 87.61 | 72.14 |
| Ligand | 163.31 | 106.00 |
| RMSD | ||
| Bond lengths (Å) | 0.007 | 0.007 |
| Bond angles (°) | 0.947 | 1.027 |
| Validation | ||
| MolProbity score | 2.55 | 2.81 |
| Clashscore | 28.39 | 37.36 |
| Poor rotamers (%) | 0 | 0 |
| Ramachandran plot | ||
| Favored (%) | 87.46 | 78.74 |
| Allowed (%) | 12.54 | 21.26 |
| Disallowed (%) | 0 | 0 |
*Dataset 1.
†Dataset 2.
Fig. 1. Flexibility in the AVP-V2R-Gs-Nb35 complex.
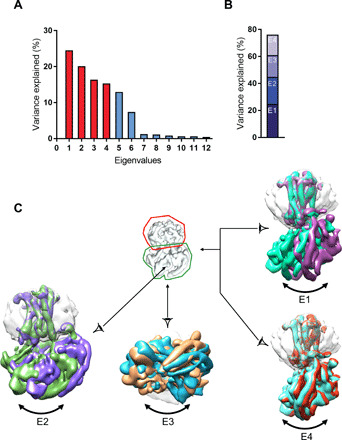
(A) The contribution of each of the 12 eigenvectors (numbered along x axis) to the variance of the overall final map is illustrated. (B) Eigenvectors 1 to 4 correspond to 78% of the variance. (C) Mask used for multibody refinement is shown in the middle, detergent micelle and V2R are surrounded by a red line, and Gs and Nb35 are surrounded by a green line. Maps corresponding to the four first vectors are illustrated, showing swing-like motion and tilting of Gs-Nb35 with respect to AVP-V2R. E4 is part of E1.
Because we could not unambiguously build the AVP in the calculated maps, we designed an original hybrid strategy based on a combination of cryo-EM maps, computational molecular dynamics simulations (MDSs), and experimental saturation transfer difference (STD) nuclear magnetic resonance (NMR) (Fig. 2 and figs. S7 to S10). First, the conformational sampling of the peptide-receptor complex was improved using the unbiased coarse-grained (CG) method coupled to replica exchange molecular dynamics (REMD) simulation protocol (Fig. 2Aand fig. S7). We successfully used this protocol to predict the binding modes of peptides in the class A GPCRs neurotensin receptor type 1 (NTSR1), C-X-C chemokine receptor type 4 (CXCR4), and growth hormone secretagogue receptor (GHSR) (21, 22). Three independent CG-REMD simulations were run, together representing about 3 ms of cumulated simulation time (fig. S8). Each of the three simulations led to, respectively, 288, 306, and 302 clusters of peptide:receptor conformations. The first 10 most populated clusters (Fig. 2B) were identically retrieved among the three independent simulations, as shown by the root mean square deviation (RMSD) matrix (fig. S8) and represented more than 60% of the whole explored conformations. After addition of the Gs heterotrimer and Nb35 proteins, refinement of each of these clusters was performed in the L cryo-EM density map (Fig. 2C). At this step, we used the correlation-driven molecular dynamics (CDMD) method (23) while keeping advantage of using a CG representation for sampling speed and better agreement with the resolution of the maps (fig. S9). Fitting of each cluster was repeated five times. Typical curves of cross-correlation coefficients as a function of time for each CDMD show that the used protocol reached a “plateau” in each case, indicating the convergence of the fit for all clusters (fig. S10). Small variability of the position of the peptide among the five obtained models for clusters 2 and 5 (mean RMSD of 3.0 and 2.2 Å, respectively) and in a lower manner for the clusters 6 and 8 (mean RMSD of 3.2 and 3.6 Å, respectively) was seen (Fig. 2, D and E). The higher values obtained for the other clusters (in the range 4.8 to 8.7 Å) were explained by the upper starting position of the peptide in the pocket, finding more easily the density located at the surface of the receptor during the fitting procedure (Fig. 2E). Last, the CG models obtained from the fitting procedure were back-mapped to an all-atom (AA) representation. Minimization, MDSs, iterative manual adjustment, and real-space refinement were carried out to finalize AVP docking.
Fig. 2. Overview of the hybrid strategy: A combination of cryo-EM, computational, and NMR.
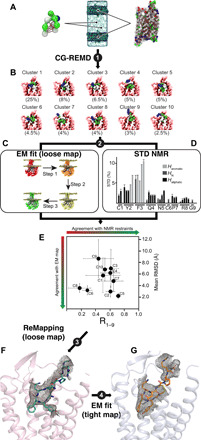
(A) Schematic representation of the unbiased CG ab initio approach. The internal elastic networks used for both AVP (left) and V2R (right) are shown. The full system (center) used for the CG-REMD simulations included two receptors and two peptides. (B) Ten most populated clusters (67.5% of the whole conformations) obtained for the AVP-V2R complex after three independent CG-REMD simulations. (C) Schematic representation of the successive steps using the CDMD method to fit the models resulting from the CG-REMD simulations into the L cryo-EM map. (D) Mapping AVP contact surface by experimental STD NMR. The STD effect profile (in percentage) is shown as a function of AVP protons [aliphatic, N (backbone), and aromatic]. (E) Cross-correlation between computational and STD NMR. Variability of the position of AVP was calculated as mean RMSD values (in angstroms) after cross-comparison of five models resulting from the fitting procedure of each of the 10 clusters in the L density map. Cluster 5 showed the smaller variability (2.2 Å). Experimental STD values were compared to the expected STD values from all-atom models issued from MDSs, and correlation coefficients were calculated for the whole peptide (R1–9). Cluster 5 appeared as the best cluster fitting to experimental STD values. (F) Building of the final L structure based on remapping cluster 5 into the L density map. (G) On the basis of the L structure, the T structure was built to match the T density map.
The AVP binding modes were further cross-validated using experimental STD NMR spectroscopy, which can efficiently monitor the binding and map the contact surface of a given ligand with its cognate GPCR (24, 25). 1D STD spectra were thus recorded either on a mixture of AVP with V2R or on AVP alone (fig. S11, A and B). Intense STD signals were only observed in the presence of V2R, mostly for the aromatic protons of Y2 and F3 residues of AVP (Fig. 2, D and E, and fig. S11). The addition of the orthosteric antagonist tolvaptan (TVP) significantly attenuated the STD signals, demonstrating specific binding of AVP to the V2R orthosteric site (fig. S11B). Calculation of normalized STD effects as ISTD − Iref/Iref showed that the most intense effects were observed for the N-terminal cyclic part of AVP, with a strong involvement of the aromatic side chains of Y2 and F3 (and to a lesser extend C1), whereas the residues in the C-terminal tripeptide (P7, R8, and G9 amide) were less affected upon V2R binding (Fig. 2D). In addition, we compared these experimental STD values to the expected STD values from AA models issued from MDSs and subsequently refined with the density maps. As explained in Materials and Methods, coefficient correlations between simulated and experimental STD values were calculated for the whole peptide (R1–9). Cluster 5 fitted on L density map appeared as the best cluster fitting to experimental STD values (Fig. 2E).
On the basis of this approach, the L and T models were then built in a more conventional manner to match the density maps as closely as possible (Fig. 2, E and F, and Table 1). In the final models, side chains of most residues are clearly identifiable in the 7TM and helix 8 of the V2R in both structures (fig. S12, A and B). Intracellular loop 1 (ICL1) was well defined in the maps, as well as the contacts between V2R and the Gs protein. The α-helical domain of Gαs subunit was subtracted during single-particle analysis for high-resolution map refinement. ICL2, ICL3, and the C terminus of V2R were not seen in the density maps and were not constructed in the final models.
Overall architecture of the ternary complex
Both L and T AVP-V2-Gs ternary complexes present a typical GPCR–G protein architecture with the receptor 7TM helix bundle engaging the peptide agonist on the extracellular side and the Gαs C-terminal domain (α5 helix) on the intracellular side (Fig. 3, A to D). However, the L and T states present large structural differences most notably in the position of the G protein heterotrimer relative to V2R (Fig. 3E). The α5 helix interacts more tightly in the T state than in the L state (Fig. 3), inducing a translation of the whole Gs heterotrimer (Fig. 3E). In particular, the α4 helix and the Ras-like domain of Gαs are translated from 4 and 5 Å between the L and T states, respectively. These movements position the αN helix 5 Å closer to the receptor in the T state in comparison to the L state (Fig. 3E). Those Gα movements are also accompanied by a 7-Å translation of the Gβ N-terminal helix, a translation of the γ subunit of 6 Å and a translation of Nb35 of 7 Å (Fig. 3E).
Fig. 3. Structures of AVP-V2R-Gs-Nb35 complexes in L and T conformations.
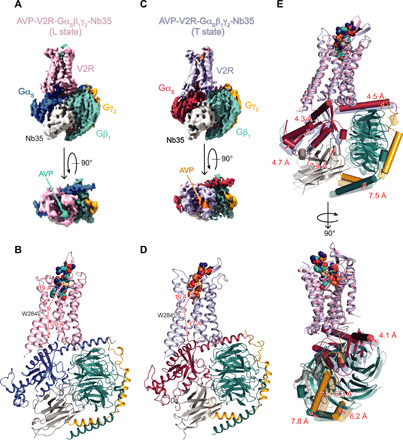
(A) Orthogonal views of the cryo-EM density maps of the L state of the AVP-V2R-Gs-Nb35 complex and (B) corresponding model as ribbon representation. V2R is colored in pink, Gαs in dark blue, Gβ1 in turquoise, Gγ2 in yellow, Nb35 in gray, and AVP in cyan. In (B), the distances between W2846.48 (at its Cα carbon) and the AVP center of mass (COM) and between W2846.48 and the C-terminal end of α5 helix of Gs (at the Cα carbon of the free carboxylic acid) are shown. (C and D) Corresponding maps and model for the T state. V2R is colored in blue gray, Gαs in raspberry, Gβ1 in turquoise, Gγ2 in yellow, Nb35 in gray, and AVP in orange. In (D), distances are measured as in (B). (E) L and T models are aligned on the V2R chains, and rotations/translations are shown by measuring displacement (in angstroms) of Gαs, Gβ1, Gγ2, and Nb35.
The presence of several conformational states and the multibody refinement analysis reflect the dynamics of V2-Gs complex formation. From the final L structure model, a principal components analysis obtained from classical MDSs revealed similar dynamics and suggests that the conformations captured by the cryo-EM 3D reconstructions represent averaged states that are part of a much larger conformational ensemble (fig. S13, A to D). Although those differences are less pronounced that the ones recently described for the neurotensin receptor NTSR1-Gi1 complexes (26), they further indicate that GPCR–G protein coupling is a dynamic process in which the G protein may explore different sets of conformations. The cryo-EM experimental structures appear to stem from sparsely populated regions of the conformational landscape of the signaling complex and at the junction between populated regions in principal component 1 space (fig. S13, A to D). The 3D reconstructions obtained from the maximum likelihood classification method (conformationally averaged substates), which select by repeated sifting clusters of homogeneous substates from the initial pool of particles, may not necessarily correspond to global energy minima conformations, highlighting the importance of using complementary approaches to determining functionally relevant structures such as MDSs and NMR. Similar problems have been documented for other systems (27) and remain an area of possible improvement in the field.
AVP binding pocket within V2R and comparison with OTR-binding site
Our hybrid approach allowed us to build convincing models of AVP binding poses in both L and T structures. The final calculated structures present a central position of AVP in the orthosteric pocket of the V2R along the axis of the helical bundle (Figs. 4, A to C, and 5). The extracellular domains of the V2R are widely opened in both L and T conformations, a feature consistent with the accommodation of a cyclic peptide such as AVP (Fig. 5), and in agreement with the recently reported inactive OTR structure (28). In the L and T structures, AVP contacts residues from both TM helices and extracellular loops (Figs. 4A and 5, A to F) in agreement with what was originally proposed on the basis of pharmacological data (29). Consistent with its amphipathic nature, AVP interacts with two chemically distinct interfaces in a 15-Å-deep binding pocket to form both polar and hydrophobic contacts (Fig. 4, B and C).
Fig. 4. AVP-binding site of the V2R, comparison with retosiban-binding site in OTR.
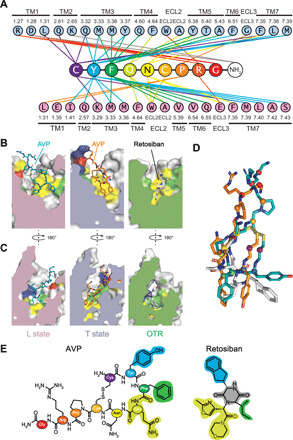
(A) Direct contacts between AVP and V2R in L and T structures. Interactions (within a maximum of 5 Å in distance) are shown between each AVP residue (and the C-terminal amide) with V2R residues in the L structure (pink) and in the T structure (blue). All TM helices, extracellular loop 2 (ECL2), and ECL3 interact with the hormone AVP. V2R residues are labeled according to the Ballesteros-Weinstein numbering. Each residue from AVP is colored differently for clarity. (B) Side views of the binding pocket in the L and T structures and in the inactive structure of OTR. AVP binding modes in the L (pink) and T (light blue) structures are compared to that of the small-molecule antagonist retosiban in the OTR structure (green), all viewed from TM3. Residues from receptors that interact with AVP or retosiban are depicted in different colors: yellow for hydrophobic, green for polar, and red and dark blue for negatively and positively charged, respectively. (C) Side views of the binding pockets after a 180° rotation. AVP and retosiban are viewed from TM6. The same color code is used. (D) Superimposition of AVP and retosiban. The peptide agonist and the nonpeptide antagonist are superimposed after alignment of V2R and OTR structures. The most hydrophobic parts of both ligands superimpose at the bottom of the orthosteric binding pocket. (E) Structure comparison of AVP and retosiban. AVP is shown using the same color code in (A). The retosiban indanyl moiety, the sec-butyl group, and the oxazol-morpholine amide moiety superimpose with AVP Y2, F3, and Q4, respectively. The retosiban 2,5-diketopiperazine core is positioned between AVP Y2 and F3.
Fig. 5. V2R and OTR binding pockets: Binding of AVP versus retosiban.
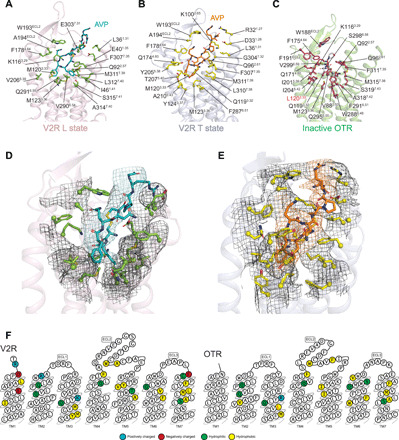
AVP binding poses are viewed from the side of the V2R helix bundle in L (A) or T (B) state and are compared with that of retosiban (in white sticks) in OTR (C). Receptor residues directly interacting with the ligands (at a maximum of 5 Å in distance) are indicated (Ballesteros-Weinstein numbering). In the OTR, L1203.33 (highlighted in red) is a mutation introduced in the sequence to increase thermostability and facilitate crystallogenesis (V120L). Densities of V2R residues in contact with AVP are shown in L (D) and T (E) states, respectively. (F) Residues of V2R and OTR involved in the binding of ligands are shown in receptor snake-like plot representations (https://gpcrdb.org).
While AVP conformations occupy a central position in both the L and T binding clefts, interesting changes are observed because of a translation of the Y2 residue side chain (from TM7 to TM3) and to a movement of the C-terminal tripeptide (inversion in R8 and G9-NH2 positions) at the V2R surface (Figs. 4, B to D, and 5, A and B). The cyclic part of AVP (C1 to C6) and the P7 are buried into the cleft defined by the seven-helix bundle of V2R, leaving only R8 residue and C-terminal glycinamide exposed to the solvent (Fig. 5). In both the L and T structures, the C1-Y2-F3 hydrophobic motif of AVP binds deeper in the binding site, creating key contacts with the receptor (Figs. 4 and 5), in agreement with STD spectroscopy data (Fig. 2, D and E, and fig. S11).
V2R and OTR belong to the same subfamily of peptide class A GPCRs and share a common orthosteric binding site (29, 30). Although V2R and OTR [Protein Data Bank (PDB) code 6TPK] structures (28) represent different GPCR conformations (active agonist-bound V2R versus inactive antagonist-bound OTR), it is interesting to compare the complete set of residues involved in the binding of the natural hormone AVP with the ones involved in retosiban binding to gain insights into ligand binding and efficacy in this receptor family (Figs. 4 and 5). Many OTR residues involved in the binding of retosiban are actually conserved among AVP/OTRs and also interact with AVP in the V2R (Figs. 4 and 5). The conserved W6.48 and F6.51 (Ballesteros-Weinstein numbering) in AVP/OTRs interact with the highly hydrophobic indanyl moiety of retosiban in the crystal structure of inactive OTR. AVP also makes contact with F6.51 through its Y2 but is not in contact with W6.48 in the V2R, probably because it is too bulky to bind deeper in the pocket. These data confirm that hydrophobic small-molecule nonpeptide antagonists and AVP partially superimpose at the bottom of the orthosteric binding pocket of AVP/OTRs (Figs. 4 and 5) (31–33).
Activation of the V2R and comparison with other class A GPCRs
The active-state structures of the V2R reveal key structural features of the activation process by comparison with the OTR inactive structure (Fig. 6, A to E, and fig. S14, A to C). Moreover, to get a more general view of V2R activation, it was also important to look at the canonical conformational changes of TMs and of conserved motifs involved in other ligand-activated GPCRs of class A (34, 35). Thus, compared to other active GPCR structures and to the inactive antagonist-bound OTR structure (Fig. 6, A to E, and fig. S14, A to C), the L and T structures of V2R present all the features of active conformations, i.e., a large-scale displacement of TM6 (Fig. 6A); conformational changes of W6.48 toggle switch (Fig. 6B); a rearrangement of the P5.50-S3.40-Y6.44 transmission switch, equivalent to the PIF motif in other GPCRs (Fig. 6C); a rotation of the conserved NPxxY7.53 motif (Fig. 6D); and a broken D1363.49-R1373.50 ionic lock (Fig. 6E).
Fig. 6. Active conformations of L and T V2R states, comparison with inactive structure of OTR and active/inactive structures of β2AR.
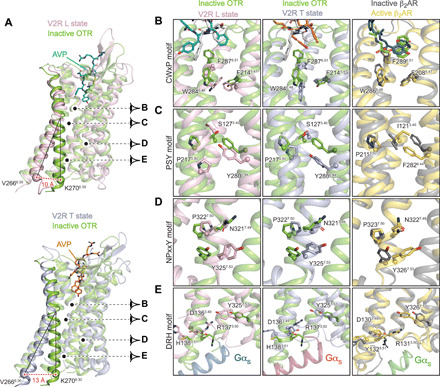
(A) Large-scale displacement of TM6. The V2R L (pink) and T (blue gray) active structures are aligned onto that of the inactive OTR (green) structure. Residue 6.30 (Ballesteros-Weinstein numbering) is chosen as a reference (V266 in V2R and K270 in OTR) for measuring the outward movement of TM6: 10 and 13 Å between OTR and V2R in the L and T states, respectively. Activation of molecular switches along the helix bundle of the V2R is viewed in (B) to (E). For comparison, rearrangements of those corresponding motifs in the b2AR are depicted. (B) Rotameric toggle switch in the conserved CWxP motif. Position of W6.48, F6.51, and F5.47 (284, 287, and 214 in V2R) are shown. (C) Rearrangement of the PSY transmission switch. The P5.50-S3.40-Y6.44 motif (217, 127, and 280 in V2R) is equivalent to the PIF motif in other GPCRs. (D) Rotation of the NPxxY conserved motif in TM7. The conserved Y7.53 (position 325 in V2R) is shown. (E) Breaking of the conserved ionic lock in TM3. Upon activation of V2R, the ionic bond between D1363.49 and R1373.50 is broken, and R1373.50 projects to Y3257.53.
By comparing the structures of the inactive antagonist-bound OTR with the active agonist-bound V2R, it appears that contacts between M1233.36 and F2876.51-W2846.48 motif (all in contact with Y2 of AVP) undergo large conformational rearrangements (Figs. 4 to 6). It is thus tempting to speculate that it is a key motif regulating the activity of this family of receptor.
As indicated above, the V2R R1373.50 participates in the ionic lock motif involved in the balance of active versus inactive states of class A GPCRs (34). Position of this R1373.50 in the V2R snake representation is shown in Fig. 7A. In the inactive structure of OTR (Figs. 6E and Fig. 7B), D1363.49 and R1373.50 interact with each other through this ionic lock (the distance between the two charged groups is 3 Å; Fig. 7B). For comparison, this salt bridge is broken in the L and T active conformations of the V2R-Gs complex (Figs. 6E and 7B). In that case, the distance between the two charges is 10 Å in the L state (Fig. 7B) and 8 Å in the T state. The observed constitutive activity toward Gs coupling for the missense mutations C1373.50 or L1373.50 responsible for NSIAD (9, 36, 37) can thus be explained from a structural point of view since these hydrophobic residues are not able to form such an ionic lock to stabilize the inactive state (Fig. 7C). On the contrary, the mutant H1373.50 causing cNDI (38, 39) might still be able to maintain the balance between active and inactive states of the V2R through its partial positive charge (Fig. 7C). Its loss of function rather reflects the loss of accessibility to AVP due to the constitutive internalization (37–39).
Fig. 7. Structural insights into V2R mutations associated with cNDI or NSIAD.
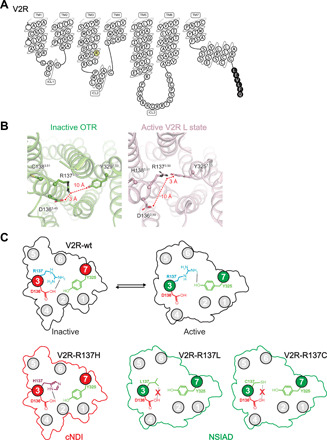
(A) Modified snake plot of the human V2R construct used in the study (https://gpcrdb.org). The R1373.50 residue involved in the ionic lock motif with D1363.49 is highlighted in yellow. The mutation of this residue into a histidine or a cysteine/leucine is responsible for two genetic diseases, cNDI and NSIAD, respectively. Part of the human rhinovirus 3C (HRV3C) protease cleavage site introduced in the C-terminal end is indicated in black. (B) Ionic lock motif in the inactive structure of OTR (left) and in the active V2R L state structure (right). In the inactive OTR, the distance between positively charged R1373.50 and negatively charged D1363.49 is 3 Å (locked conformation), whereas it is 10 Å in the active V2R (open conformation). In the active V2R L state, R1373.50 directly contacts Y3257.53, an interaction that is not seen in the inactive OTR. (C) Schematic representation of R1373.50 mutations responsible for either cNDI or NSIAD. In the top, equilibrium between inactive and active conformations of the wild-type V2R (V2R-wt) and the ionic lock motif are illustrated. Bottom: Mutations of R1373.50 that induce cNDI (V2R-R137H) or NSIAD (V2R-R137L and V2R-R137C) are compared. Breakage of the R1373.50-D1363.49 ionic lock is shown in the R137C and R137L mutants. TM3 and TM7 are depicted in red in the V2R-R137H mutant, whereas they are shown in green in the constitutive active mutants V2R-R137L/C.
V2R-Gs interactions
The cryo-EM maps of the ternary complex clearly establish the structural details of V2R-Gs coupling. As anticipated from the conserved mechanism of GPCR–G protein coupling (40, 41), both the L and T conformations show a similar overall architecture of the complex interface with the engagement of the Gαs C-terminal α5 helix in the core of the 7TM (Fig. 8 and figs. S14, D to F, S15). However, there are some interesting differences compared to other GPCR-Gs complex structures. Notably, in both the L and T structures, the V2R ICL1 makes many direct contacts with the Gβ subunit. In the T state, ICL1 residues L62-A63-R64-R65-G66 interact with Gβ R52, D312-N313, and D333-F335 (Fig. 8A). In the L state, ICL1 residues R65-G66-R67-R68 interact with Gβ R52, D312, and D333 (figs. S14 and S15). These contacts between V2R and Gβ are much more numerous than in the class A GPCR β2AR- or adenosine A2A receptor (A2AR)-Gs complexes (18, 42). Moreover, in the T conformation, there are some additional contacts between V2R ICL1 (R67-G69-H70) with the N-terminal α helix of Gαs (Q31, Q35, and R38), resulting in a more compact interaction (Fig. 8B). In the L state, V2R (W71) and N-terminal α helix of Gαs (Q35 and R38) contacts are more limited (fig. S15). Contacts between the N-terminal α helix of Gαs with GPCRs have only been seen in glucagon-like peptide-1 receptor (GLP1R) and calcitonin receptor (CTR) class B GPCR complexes (43, 44), not in class A GPCR–G protein complexes.
Fig. 8. Interface of the V2R T state with Gs.
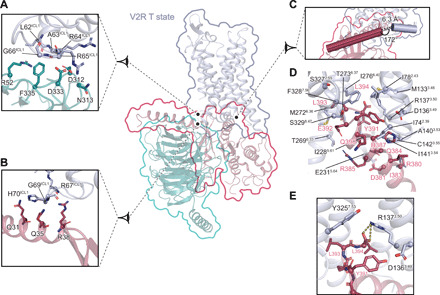
Interactions between V2R and Gs heterotrimer are shown. Specific interfaces are depicted, and residues in close proximity (within a maximum of 3.9 Å in distance) are highlighted (blue gray for V2R, turquoise for Gβ subunit, and raspberry for Gα subunit). (A) Interaction of V2R ICL1 with Gβ subunit. (B) Interaction of V2R ICL1 with the N-terminal helix of Gα subunit. (C) Position of C-terminal α5 helix of Gα subunit relative to V2R helix 8. The distance between Gα and helix 8 is indicated. Angle between these two domains is shown. (D) Interacting residues between the C-terminal α5 helix of Gα subunit and V2R. (E) Zoom on an ionic bridge between the C-terminal free carboxylic moiety of α5 helix of Gα subunit and V2R R1373.50.
In contrast to what was observed for the β2AR (18) and the mu-opioid receptor (μOR) (45), the Gαs C-terminal α5 helix appears to extend helix 8 (H8) of the V2R, lying almost parallel to the membrane plane (Fig. 8C and fig. S15, C and F to H). In addition, compared with the β2AR, the C terminus of Gαs is interacting deeper in the V2R 7TM core, making direct contact with the residues (L and T states, respectively) of V2R that are part or in close proximity to the conserved NPxxY (TM7) and DRH (TM3) activation motifs (Fig. 8, D and E, and fig. S15, D to H). In this respect, the V2-Gs interaction resembles more the interaction seen in the μOR-Gi complex (fig. S15). The V2R TM7-H8 hinge region also makes a strong contact with the Gαs ELL motif, particularly through hydrophobic contacts with the F3287.56 side chain (Fig. 8D). The T and L conformations differ here in the position of the Gαs L394 side chain originating from a distinct F3287.56 side-chain conformation (pointing toward I782.43 of the receptor in the T structure or toward Gαs L394 in the L structure) (Fig. 8D and fig. S15D). Most notably in the T state, the side chain of R1373.50, which is part of the ionic lock motif, forms an ionic interaction with the free carboxylic acid function of the Gαs C terminus (Fig. 8E), a direct contact that was not observed before between a GPCR and a G protein of any family (Gs, Gi, Go, or Gq) (14, 15, 46). Moreover, in the L state, the density map suggests that the R1373.50 side chain could adopt two conformations, one forming a similar ionic interaction with the carboxylic acid of Gαs L394 main chain and the other one pointing toward the Y3257.53 from the NPxxY motif (fig. S15E).
DISCUSSION
In this study, we identified three different states and solved two structures of the AVP hormone–bound V2R in complex with the Gs protein. They reveal distinct agonist and G protein binding modes and a more compact architecture compared to other class A GPCR–G protein complexes. Although this work provides structural insights into the mechanisms of G protein activation by V2R, additional data are needed to determine whether the different conformations represent distinct intermediates along the signaling activation pathway. However, their identification using single-particle analysis and all-atom MDSs reports a high intrinsic flexibility, in agreement with the concept that GPCRs can explore a wide range of conformations, adapting their shape in response to different ligands and/or intracellular signaling partners (47). We also consider that the characterization of three different populations of the AVP-V2R-Gs complex was made possible because of using a native receptor (the V2R is a wild-type from T31 to G345), which was not engineered with thermostabilizing mutations or fusion partners.
Despite their various physiological roles, the cyclic peptides AVP and OT share a common receptor family. The V1aR, V1bR, V2R, and OTR display a common binding pocket that accommodates peptide and nonpeptide orthosteric agonists and antagonist ligands (29, 30). Although V2R and OTR (28) structures represent different GPCR conformations (active agonist-bound V2R versus inactive antagonist-bound OTR), it is not unexpected to see that many residues involved in the binding of AVP (natural cyclic peptide agonist) are conserved among AVP/OTRs and also interact with retosiban (small nonpeptide antagonist) in the OTR. These data confirm that specific binding sites for nonpeptide antagonists and for AVP/OT peptides overlap at the bottom of the receptor binding pocket (31–33). Moreover, these are the most hydrophobic parts of AVP and retosiban that superimpose (AVP Y2 and F3 residues versus retosiban indanyl and sec-butyl moieties) in the binding pocket. The main pharmacophore responsible for activating V2R seems also to be the Y2-F3 AVP side chains (the message, i.e., efficacy), while the rest of the peptide rather seems to be responsible for the address (selectivity). In agreement, we demonstrated that the presence of the AVP F3 residue (L3 residue for OT) is responsible for partial agonist activity of AVP to the human OTR, whereas AVP hormone is a full agonist on V1aR (48), V1bR, and V2R. In addition, modification of residues at position 4 (glutamine for AVP and OT) and 8 (arginine for AVP and isoleucine for OT) has been shown to control selectivity of AVP analogs toward the different receptor subtypes in the AVP/OTR family (49).
The significance of our study also lies in the clinical relevance of the AVP receptor family, particularly for two rare X-linked genetic diseases involving mutations in the V2R, cNDI (8), and NSIAD (9), and our work provides a structural explanation on how those mutations can possibly affect the level of V2R activity and Gs protein coupling. These two pathologies are associated with V2R loss of function or constitutive activity, respectively. Substitution of R1373.50 of the V2R for histidine (H1373.50) leads to cNDI (38, 39), whereas substitution of the same residue to cysteine or leucine (C/L1373.50) causes NSIAD (9, 36, 37). Paradoxically, the three mutant receptors were shown to share common features, such as constitutive arrestin recruitment and endocytosis, resistance to AVP-stimulated cAMP accumulation and MAP kinase activation, and marked decrease in receptor cell surface expression (36–39). The unique difference observed between the H1373.50 mutant and the C/L1373.50 mutants resides in their basal constitutive activity toward the cAMP pathway (9). C/L1373.50 gain-of-function mutants promote a significant higher basal cAMP level as compared to the wild-type V2R or the H1373.50 loss-of-function mutant. In the present study, we proposed that the two hydrophobic cysteine or leucine residues are not able to form an ionic lock with D1363.49 to stabilize the inactive state, explaining their constitutive activity. That is, the conformation of these mutants may be comparable to that of active V2R in the L and T states of the AVP-V2R-Gs signaling complex, at least considering a broken D1363.49-C/L1373.50 ionic lock. We provided here a unique evaluation of these gain-of-function V2R mutations.
A patient bearing the V2R H1373.50 mutation was shown to increase his urine osmolality after a short-term therapeutic treatment with the V1a antagonist SR49059 (50). A structural knowledge about this ligand rescue is clinically important since this mutation is recurrent in independent cNDI families and also presents a phenotypic variability (51). SR49059 antagonist is used as a pharmacological chaperone (52). This lipophilic nonpeptide antagonist able to cross biological membranes is selective for V1aR subtype but still displays a measurable affinity for V2R. This ligand, which is a competitive analog of AVP, has been shown to be able to rescue the function of endoplasmic reticulum (ER)–trapped mutants of the V2R responsible for cNDI (38). Upon binding to the orthosteric site of the V2R mutants, SR49059 triggers targeting and stabilization of the mutated receptors to the plasma membrane of receptor-expressing cells, including R137H V2R. This mutant combines most of the properties of the wild-type receptor but is constitutively internalized (37, 38), leading to a reduced cell surface expression, thus explaining a cNDI phenotype. Treatment of the patient with the pharmacological chaperone probably allows us to stabilize the R137H mutant at the plasma membrane where it is displaced by endogenous circulating AVP hormone, eliciting an antidiuretic response (increase in the osmolality of urine from 150 to 300 mOsm/kg).
The use of cell-permeable pharmacological chaperones for rescuing function of misfolded V2R mutants responsible for cNDI is a very attractive therapeutic avenue, in particular, regarding those that are trapped in the ER but, otherwise, are functional once they are targeted to the cell plasma membrane (see above for the V2R H1373.50 mutation). It is thus tempting to interpret clinical observations (or in vitro pharmacological and cellular data) based on the present structures of the V2R. Importance of the structural data to help in understanding mutations is discussed here with two examples of cNDI loss-of-function mutations that can be rescued using pharmacological chaperones using the V2R-selective nonpeptide antagonist TVP, which is now used in thousands of patients with autosomal polycystic kidney disease with a reasonable safety profile (53). The V88M mutation is responsible for a mild phenotype, which is moderate polyuria and some degree of increased urine osmolality following treatment with desmopressin, an analog of AVP (54). Both the expression level and the hormone binding affinity are affected by this mutation. Structurally, V882.53 makes a direct contact with M1233.36, which belongs to the AVP-binding site. We hypothesize that V88M induces a local destabilization by a steric clash with M123, leading to the decreased AVP binding affinity observed in in vitro pharmacological experiments but to a substantial increase in urinary concentration after desmopressin treatment in vivo. The M272R mutation is responsible for a severe phenotype with polyuria and no response to desmopressin treatment (55). In Madin-Darby canine kidney cells, this mutant is trapped in the ER and is not accessible to AVP but can be rescued using the pharmacological chaperone TVP. Once it is at the cell surface, it is able to respond to desmopressin. M2726.36 is located at the bottom of TM6, a highly flexible region that moves outward the V2 core upon activation. On the basis of positioning of the corresponding conserved M2766.36 in the inactive structure of the related OTR (28), M2726.36 in the V2R is surrounded by an aromatic/hydrophobic residue cluster, made of I742.39, I782.43, V2756.39, I2766.40, Y3257.53, and F3287.56. Mutation of M272 into a positively charged arginine probably destabilizes this domain, induces misfolding of the receptor, and results in ER retention. TVP can rescue the receptor to the cell surface probably by stabilizing its unfolded structure.
While this manuscript was under review, another structure of AVP-bound V2R in complex with a modified Gs protein was published online (56). A major difference between this structure with L and T structures described herein comes from the use of a chimeric Gs/Gi protein in the first study, different from the wild-type Gs. This modification, together with the use of the ScFv16 antibody fragment further stabilizing the complex through interaction with the Gi domain, probably explains not only a better resolution but also why flexibility and dynamics of the signaling complex were not addressed. The use of a physiological Gs protein allowed us to probe the flexibility of the system and to characterize an original V2R-Gs interface. Nonetheless, positioning of AVP in the orthosteric binding pocket and superimposition of AVP with OTR-selective retosiban are comparable in the two studies. Hence, the different structures are complementary, help to have a complete view of this signaling system, and pave the way for future drug development to treat water balance disorders (7).
MATERIALS AND METHODS
Data analysis and figure preparation
Figures were created using the PyMOL 2.3.5 Molecular Graphics System (Schrödinger LLC) and the UCSF Chimera X 0.9 package. Data were plotted with GraphPad Prism 8.3.0.
V2R expression and purification
The optimized sequence of the human V2R was cloned into a pFastBac1 vector (Invitrogen) for insect cell expression. To facilitate expression and purification of the V2R construct used for cryo-EM, the hemagglutinin signal peptide (MKTIIALSYIFCLVFA) followed by a Flag tag (DYKDDDDA) was added at the N terminus, and a Twin-Strep-tag (WSHPQFEKGGGSGGGSGGGSWSHPQFEK) was inserted at the C terminus. In addition, N22 was substituted with a glutamine residue to avoid N-glycosylation, and C358 mutated into an alanine to eliminate potential intermolecular disulfide bridges during solubilization and purification. A Tobacco Etch Virus (TEV) protease cleavage site (following the Flag tag) and two Human Rhinovirus 3C (HRV3C) protease cleavage sites (one inserted in the N terminus between D30 and T31 and the other inserted in the C terminus between G345 and Q354 and replacing R346-TPPSLG-P353) were also added to remove N and C termini and facilitate structure determination. M1L2 residues were replaced by AS residues, and LE residues were added before the Twin-Strep-tag, during subcloning (introduction of Nhe I and Xho I restriction sites, respectively). Sequence modifications did not affect the receptor ligand binding or function. The V2R was expressed in Sf9 insect cells using the Bac-to-Bac baculovirus expression system (Thermo Fisher Scientific) according to the manufacturer’s instructions. Insect cells were grown in suspension in EX-CELL 420 medium (Sigma-Aldrich) to a density of 4 × 106 cells/ml and infected with the recombinant baculovirus at a multiplicity of infection of 2 to 3. The culture medium was supplemented with the V2R pharmacochaperone antagonist TVP (Sigma-Aldrich) at 1 μM to increase the receptor expression levels (52, 57). The cells were infected for 48 to 54 hours at 28°C, and expression of the V2R was checked by immunofluorescence using an anti-Flag M1 antibody coupled to Alexa Fluor 488. Cells were then harvested by centrifugation (two steps for 20 min at 3000g), and pellets were stored at −80°C until use.
The cell pellets were thawed and lysed by osmotic shock in 10 mM tris-HCl (pH 8), 1 mM EDTA buffer containing iodoacetamide (2 mg/ml), 1 μM TVP, and protease inhibitors [leupeptine (5 μg/ml), benzamidine (10 μg/ml), and phenylmethylsulfonyl fluoride (PMSF) (10 μg/ml)]. After centrifugation (15 min at 38,400g), the pellet containing crude membranes was solubilized using a glass dounce tissue grinder (15 and 20 strokes using A and B pestles, respectively) in a solubilization buffer containing 20 mM tris-HCl (pH 8), 500 mM NaCl, 0.5% (w/v) n-dodecyl-β-d-maltopyranoside (DDM; Anatrace), 0.2% (w/v) sodium cholate (Sigma-Aldrich), 0.03% (w/v) cholesteryl hemisuccinate (CHS; Sigma-Aldrich), 20% glycerol, iodoacetamide (2 mg/ml), biotin BioLock (0.75 ml/liter; IBA), 1 μM TVP, and protease inhibitors. The extraction mixture was stirred for 1 hour at 4°C and centrifuged (20 min at 38,400g). The cleared supernatant was poured onto an equilibrated Strep-Tactin resin (IBA) for a first affinity purification step. After 2 hours of incubation at 4°C under stirring, the resin was washed three times with 10 column volume (CV) of a buffer containing 20 mM tris-HCl (pH 8), 500 mM NaCl, 0.1% (w/v) DDM, 0.02% (w/v) sodium cholate, 0.03% (w/v) CHS, and 1 μM TVP. The bound receptor was eluted in the same buffer supplemented with 2.5 mM desthiobiotin (IBA).
The eluate was supplemented with 2 mM CaCl2 and loaded onto a M1 anti-Flag affinity resin (Sigma-Aldrich). The resin was washed with 10 CV of two successive buffers containing 20 mM Hepes (pH 7.5), 100 mM NaCl, 0.1% DDM, 0.01% CHS, 10 μM AVP, and 2 mM CaCl2 and then 20 mM Hepes (pH 7.5), 100 mM NaCl, 0.025% DDM, 0.005% CHS, 10 μM AVP, and 2 mM CaCl2, respectively. The receptor was eluted from the Flag resin using a buffer containing 20 mM Hepes (pH 7.5), 100 mM NaCl, 0.025% DDM, 0.005% CHS, 10 μM AVP, 2 mM EDTA, and Flag peptide (200 μg/ml) (Covalab).
After concentration using a 50-kDa molecular weight cutoff (MWCO) concentrator (Millipore), the V2R was purified by SEC using a Superdex 200 (10/300 column) connected to an ÄKTA purifier system (GE Healthcare). Fractions corresponding to the pure monomeric receptor were pooled (~2 ml) and concentrated to 50 to 100 μM with an excess of AVP (200 μM).
Gs expression and purification
Human Gαs, Gβ1 with an N-terminal Twin-Strep-tag, and Gγ2 were all expressed in Sf9 insect cells grown in EX-CELL 420 medium (Sigma-Aldrich). A recombinant baculovirus for Gαs subunit was prepared using the BestBac (Expression Systems) strategy, whereas a baculovirus for Gβ1 and Gγ2 was prepared using the Bac-to-Bac system. Gβ1 and Gγ2 were cloned in tandem into the pFastBac Dual vector (Thermo Fisher Scientific). Sf9 cells, at a density of 4 × 106 cells/ml, were coinfected with both viruses at a 1:2 Gαs:Gβ1γ2 ratio for 72 hours at 28°C. Cells were harvested and pellets were stored at −80°C.
Coinfected Sf9 cell pellets were thawed and lysed in a buffer containing 10 mM tris (pH 7.4), 1 mM EDTA, 5 mM β-mercaptoethanol, 10 μM guanosine diphosphate (GDP), and protease inhibitors [leupeptine (5 μg/ml), benzamidine (10 μg/ml), and PMSF (10 μg/ml)]. Lysed cells were centrifuged (20 min at 38,400g). The pellets containing the crude membranes were homogenized using a glass dounce tissue grinder (20 strokes with tight B pestle) in solubilization buffer containing 20 mM Hepes (pH 7.5), 100 mM NaCl, 1% DDM, 5 mM MgCl2 supplemented with 5 mM β-mercaptoethanol, 10 μM GDP, biotin BioLock (0.75 ml/liter), and protease inhibitors. The mixture was stirred for 40 min at 4°C and centrifuged (20 min at 38,400g). The supernatant was loaded onto a Strep-Tactin affinity resin equilibrated with the same buffer. The resin was washed three times, first with 5 CV of solubilization buffer, then with 5 CV of solubilization buffer supplemented with 100 μM tris(2-carboxyethyl)phosphine (TCEP) (instead of β-mercaptoethanol), and last with 10 CV of wash buffer containing 20 mM Hepes (pH 7.5), 50 mM NaCl, 0.1% DDM, 1 mM MgCl2, 100 μM TCEP, and 10 μM GDP. The Gs heterotrimer protein was eluted in the same buffer supplemented with 2.5 mM desthiobiotin. After a treatment with antarctic phosphatase (5 U; NEB Inc.) for 30 min at 4°C, the Gs protein was concentrated to 10 mg/ml using 50-kDa MWCO concentrators. Twenty percent of glycerol was added to the sample, and aliquots were flash-frozen in liquid nitrogen before storage at −80°C.
Nb35 expression and purification
The production and purification of Nb35 were performed following a protocol established by Kobilka and co-workers (18). Nb35 having a C-terminal 6His-tag was expressed in the periplasm of Escherichia coli strain BL21 following induction with 1 mM isopropyl-β-d-thiogalactopyranoside. Cultures of 2L were grown to an optical density at 600 nm of 0.6 at 37°C in LB medium containing 0.1% glucose and ampicillin (100 μg/ml). Induced cultures were grown overnight at 25°C. Cells were harvested by centrifugation and lysed in ice-cold buffer [50 mM tris-HCl (pH 8), 125 mM sucrose, and 2 mM EDTA]. Lysate was centrifuged to remove cell debris, and Nb35 was purified by nickel affinity chromatography. Eluate was concentrated to 5 mg/ml and loaded onto a Superdex 200 (16/600 column, GE Healthcare) at a 1 ml/min of flowrate. Fractions containing the monodisperse peak of Nb35 were pooled and dialyzed overnight against a buffer containing 10 mM Hepes (pH 7.5) and 100 mM NaCl at room temperature (RT). The dialyzed sample was concentrated to approximately 100 mg/ml using a 10-kDa MWCO concentrator (Millipore). Aliquots were stored at −80°C until use.
Purification of the AVP-V2R-Gs-Nb35 complex
Formation of a stable complex was performed by mixing the purified V2R with 1.2 molar excess of purified Gs heterotrimer, 250 μM AVP, and 2.5 mM MgCl2 (fig. S1). The coupling reaction was allowed to proceed at RT for 45 min and was followed by addition of apyrase (0.0125 U; NEB Inc.) to hydrolyze residual GDP and maintain the high-affinity nucleotide-free state of Gs. Fifteen minutes later, Nb35 was added at a twofold molar excess compared to Gs. After 15 more minutes at RT, the mix was incubated overnight at 4°C. In most reaction mixtures, the final concentration of V2R was 20 to 30 μM, that of Gs 30 to 40 μM, and the one of Nb35 around 80 μM. To remove excess of G protein heterotrimer and Nb35, the complex AVP-V2R-Gs-Nb35 was purified by a M1 anti-Flag affinity chromatography. After loading, the DDM detergent was then gradually exchanged with Lauryl Maltose Neopentyl Glycol (LMNG; Anatrace). The LMNG concentration was then decreased gradually from 0.5 to 0.01%. The complex and the unbound V2R were eluted in 20 mM Hepes (pH 7.5), 100 mM NaCl, 0.01% LMNG, 0.002% CHS, 2 mM EDTA, 10 μM AVP, and Flag peptide (0.2 mg/ml). The eluted AVP-V2R-Gs-Nb35 complex was separated from unbound V2R by SEC on a Superdex 200 (10/300 column) with a buffer containing 20 mM Hepes (pH 7.5), 100 mM NaCl, 0.002% LMNG, 0.0025% glyco-diosgenin (GDN; Anatrace), 0.002% CHS, and 10 μM AVP. The fractions corresponding to the complex were collected, concentrated with a 50-kDa MWCO concentrator, and subjected to a second SEC on a Superose 6 (10/300 GL, GE Healthcare) with a buffer containing 20 mM Hepes (pH 7.5), 100 mM NaCl, 0.0011% LMNG, 0.001% GDN, 0.002% CHS, and 10 μM AVP. Peak fractions were pooled and concentrated using a 50-kDa MWCO concentrator to concentrations ranging from ~1 to ~4 mg/ml for cryo-EM studies. The amphipol A8-35 (Anatrace) was added at 0.001% to help in the dispersion of the particles for cryo-EM grid preparation.
Negative stain microscopy observations
Before preparing cryo-EM grids, we first checked the quality and the homogeneity of the AVP-V2R-Gs-Nb35 sample by NS-EM. Three microliters of AVP-V2R-Gs-Nb35 complex at 0.04 mg/ml was applied for 2 min on glow-discharged carbon-coated grids and then negatively stained with 1% uranyl acetate for 1 min. Observation of EM grids was carried out on a JEOL 2200FS FEG operating at 200 kV under low-dose conditions (total dose of 20 electrons/Å2) in the zero–energy loss mode with a slit width of 20 eV. Images were recorded on a 4K × 4K slow-scan charge-coupled device camera (Gatan Inc.) at a nominal magnification of ×50,000 with defocus ranging from 0.5 to 1.5 μm. Magnifications were calibrated from cryo-images of tobacco mosaic viruses. In total, 37 micrographs were recorded, allowing us to pick 22,791 particles using e2boxer from Eman2 package (58). Further processing was performed with Relion 2.0 (13, 59). The particles were subjected to a 2D classification included to get rid of free micelles and dissociated components of the complex. From 2D classes, 14,545 particles corresponding to the V2R-Gs-Nb35 complexes were selected, representing 63% of all particles. This selection was used to calculate an ab initio low-resolution model. The sample was also subjected to NS-EM analysis after 5 days. At this point, after particle picking and 2D classification, 35% of particles were representing the complex. The fresh sample was also mixed with 100 μM GTPγS and 10 μM SR121463 V2R antagonist and visualized in negative stain to observe complete dissociation.
Cryo-EM sample preparation and image acquisition
In this study, two datasets have been recorded from two different preparations of AVP-V2R-Gs-Nb35. For the first dataset acquisition, 3 μl of purified AVP-V2R-Gs-Nb35 at a concentration of 0.75 mg/ml were applied on glow-discharged Quantifoil R 1.2/1.3 300-mesh copper holey carbon grids (Quantifoil Micro Tools GmbH, Germany), blotted for 4.5 s, and then flash-frozen in liquid ethane using the semiautomated plunge-freezing device Vitrobot Mark IV (Thermo Fisher Scientific) maintained at 100% relative humidity and 4°C. For the second dataset acquisition, cryo-EM grids were prepared as previously, but the purified V2R-Gs-Nb35 complex was at a concentration of 4 mg/ml, and the cryo-EM grids were prepared using an EM GP2 (Leica Microsystems) plunge freezer with a 4 s blotting time (100% humidity and 4°C).
Images were collected in two independent sessions on a TEI Titan Krios (Thermo Fisher Scientific) at the European Molecular Biology Laboratory (EMBL) of Heidelberg (Germany) at 300 keV through a Gatan Quantum 967 LS energy filter using a 20-eV slit width in zero-loss mode and equipped with a K2 Summit (Gatan Inc.) direct electron detector configured in counting mode. Movies were recorded at a nominal energy-filtered transmission electron microscope magnification of ×165,000 corresponding to a 0.81 Å calibrated pixel size. The movies were collected in 40 frames in defocus range between −0.8 and −2.2 μm with a total dose of 50.19 e−/Å2 (first dataset) and 41.19 e−/Å2 (second dataset). Data collection was fully automated using SerialEM (60).
Cryo-EM data processing
All data processing operations were performed with Relion-3.0.7 (61), unless otherwise specified. In total, 17,290 movies of the AVP-V2R-Gs-Nb35 sample at 0.75 mg/ml were collected. Dose-fractionated image stacks were subjected to beam-induced motion correction and dose weighting using Motioncorr own implementation. Gctf was used to determine the contrast transfer function (CTF) parameters (62) from non–dose-weighted images. After sorting, micrographs with maximum estimated resolution beyond 5 Å were discarded. Particle picking was carried out using Gautomatch [K. Zhang, Medical Research Council Laboratory of Molecular Biology (www.mrc-lmb.cam.ac.uk/kzhang/)], allowing us to pick out 2,291,432 particles. Particles were extracted in a box size of 240 Å, downscaled to 4 Å per pixel, and subjected to reference-free 2D classifications to discard false-positive particles or particles categorized in poorly defined classes. A subset of 1,109,475 particles was selected for further processing. This particle set was subjected to a 3D classification with four classes using the 30-Å low-pass filtered calcitonin receptor map as reference (44). Particles from the two classes representing 27% of total particles and showing a complete AVP-V2R-Gs-Nb35 complex were selected, reextracted with a pixel size of 1.62 Å, and subjected to a 3D refinement. This subset of 307,125 particles yielded a map with a global resolution [Fourier shell correlation (FSC) = 0.143] of 4.8-Å resolution. Particles were then subjected to a focused 3D classification without angular and translational alignments with a mask including the complex minus GαsAH (Gαs α-helical domain). The best class corresponding to 150,000 particles was reextracted without binning and submitted to a 3D refinement, allowing us to obtain a map at 4.4-Å resolution. All further processing including signal subtraction, using different types of masks, CTF refinement, and polishing did not improved the resolution of the map.
In total, 8490 movies of the AVP-V2R-Gs-Nb35 sample at 4.0 mg/ml were recorded. The image processing steps were the same as previously described, except that the picking was performed using boxnet from Warp software package (63), allowing us to extract 1,214,575 particles. After a 2D classification to clean the dataset, a subset of 917,990 particles was subjected to two successive rounds of 3D classification. A subset of 150,000 particles was used for further 3D refinements, yielding a final map at 4.4-Å resolution.
Both cleaned datasets were merged, corresponding to 1,109,475 particles from dataset 1 and 917,990 particles from dataset 2. Particles were subjected to 3D classification with three classes. One class displayed the expected structural features of the AVP-V2R-Gs-Nb35 complex corresponding to 877,003 particles and was selected for a new round of 3D classification with six classes. This classification revealed a structural variability in the ligand location and at the interface between the receptor and the Gs protein. Three subsets of particles were selected (L, T1, and T2 states), reextracted with a pixel size of 1.62 or 0.81 Å, and subjected to 3D refinements, yielding maps at 4.5, 4.7, and 5.5 Å, respectively. New rounds of 3D refinements were performed in applying a mask to exclude both the micelle and the GαsAH, yielding maps at 4.23, 4.4, and 4.7 Å. CTF refinement and polishing steps were applied on the three subsets of particles, allowing us to improve the resolution of the best map to 4.17 Å (FSC = 0.143). The T1 map (1.62 Å per pixel) was resampled at 0.81 Å per pixel for visualization purposes. Final refinements were processed with the option of masking individual particles with zero turned off. All our attempts to refine our final subsets in cisTEM (64) and cryoSPARC (65) using nonuniform refinement did not improve the resolution of final maps.
To investigate the conformational dynamics of the signaling complex, multibody refinement was performed on 877,003 particles, with two bodies corresponding to AVP-V2R and Gs-Nb35. Local resolution was estimated with the Bsoft 2.0.3 package (66, 67). Map sharpening was reevaluated with Phenix autosharpen tool (68). Phenix resolve_cryoEM tool (20) was used to improve the map interpretability and allowed to increase the estimated resolution to 4.04, 4.13, and 4.5 Å for L, T1, and T2 states, respectively.
Model building and refinement
Receptor and AVP initial models
The V2R was built by comparative modeling, using the MODELLER software (69) and the x-ray structure of the δ-opioid receptor at 3.4-Å resolution (PDB code 4EJ4) as a template (70), sharing a sequence similarity of about 44% with the V2R (on the modeled region). Because modeling loops or terminal regions is a very challenging task and their dynamical behavior is very poorly described in CG simulations, N and C termini of the receptor (residues 1 to 35 and 335 to 371, respectively) and part of the ICL3 loop (residues 237 to 262) were lacking in the used template. Thus, only residues 36 to 236 and 263 to 334 were modeled. Five hundred models were generated, and the one sharing the best objective function score was further selected as a starting point for the simulations. The disulfide bridge conserved among the class A GPCRs was included between residues 112 and 192 of the V2R.
The AVP peptide (NH3+-CYFQNCPRG-CONH2) was built from its x-ray structure available in the PDB (code 1JK4; 2.3-Å resolution), which describes the six-residue cycle of the peptide in interaction with neurophysin (71). This structure shows a cycle conformation equivalent to that found in bound (PDB code 1NPO) and unbound related peptide OT (PDB code 1XY2) (72, 73). It was thus preferred to the one describing the trypsin-vasopressin complex (PDB code 1YF4) (74) harboring a completely different conformation of the cycle. The three last residues of the peptide (7-PRG-9) were also built with the OT structure templates.
The obtained initial models of both receptor and peptide were then converted to a CG representation using the MARTINI force field (version 2.2; Elnedyn) (75). Using such a model, residues (backbone beads) closer than 9.0 Å are bound by a spring, displaying a force constant of 500 kJ/mol per nm2 (default value from the Elnedyn force field). Such a link is meant to maintain both the secondary and the tertiary structures of the polypeptides. For the peptide, only the springs involving two residues of the cycle were conserved for further calculations, the three last residues being free to move. The standard elastic network of the receptor was not modified and allowed the latter to open or close freely as no spring was bridging the extracellular loops.
Molecular dynamics simulations
The receptor was inserted in a 100 Å–by–100 Å lipid bilayer exclusively composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). To avoid the exploration by the peptide of the intracellular side of the membrane during molecular dynamics (because of periodic boundary conditions), the system was duplicated/rotated along the z axis (the two extracellular sides of the receptors were facing each other) to create an extracellular compartment. Two copies of the peptide were added to increase the interaction sampling with a 1:1 ratio. In a last step, water and chloride counterions were added to neutralize the system. The fully solvated system included 20,004 beads. After 10,000 steps of energy minimization using the conjugate gradient algorithm, the system was further equilibrated at 51 different temperatures (in the range 300:450 K by steps of 3 K) in the NVT (constant particle number, volume, and temperature) ensemble, using an integration step of 20 fs and over a period of 5 ns. The final production step was performed in the NPT (constant particle number, pressure, and temperature) ensemble, using an integration step of 20 fs and was stopped after 20 μs. During production, REMD was used to improve the sampling of all possible configurations of the peptide:receptor complex. The potential energy difference of adjacent replicas was computed every 1000 steps (20 ps), and their coordinates were exchanged according to a Boltzmann criterion. With the used parameters, the probability of exchange between adjacent replica was in the range 0.11 (300 K):0.23(450 K). Three independent CG-REMD simulations were run to verify the convergence of the obtained models, together representing a cumulated sampling time of ~3 ms. For each of these simulations, a clustering was performed on all conformations of the peptide:receptor complex obtained at the lowest temperature (300 K). To do so, we first concatenated the data corresponding to the four possible complexes (peptide1-receptor1, peptide1-receptor2, peptide2-receptor1, and peptide2-receptor2). For that step, only the conformations displaying at least one peptide:receptor contact were kept (a contact was defined using a cutoff distance of 7 Å). For clustering, we used the algorithm of Daura et al. (76) with an RMSD cutoff of 3.0 Å. The RMSD was computed only on the backbone beads of the peptide’s residues 1 to 6 after structural fit onto those of the V2R. The two cysteine side-chain beads were also included for RMSD calculations. All simulations and analyzes were performed with the GROMACS software (version 5) (77). Figures were produced with Visual Molecular Dynamics (78).
Refinement of the obtained CG models in the cryo-EM maps
The CDMD method (23) was used to refine the most populated clusters obtained in CG-REMD using the L-state cryo-EM map of the AVP-V2-Gs-Nb35 complex. The principle of the method is to use an accurate force field and thermodynamic sampling to improve the real-space correlation between the modeled structure and the cryo-EM maps. Before this refinement step, the Gs heterotrimer and the Nb35 were modeled using the structure of the β2AR-Gs-Nb35 complex (18) as a reference. The MARTINI force field restrained the internal conformations of the different partners with an internal elastic network. To increase significantly the conformational plasticity of the receptor and explore new conformations specific to the V2R, we modified its default elastic network. We automatically deleted the “long-range” springs involving two beads whose indexes differ by at least 15. This contributed to delete all interhelix springs. The standard elastic network was conserved for all other partners including the AVP peptide, the G protein, and the Nb35. No interchain springs were included for the G protein. After conversion of Gs and Nb35 into the CG model, the two proteins were placed at a rational position in respect to the V2R using the β2AR-Gs-Nb35 complex (18). The full system was inserted in a larger membrane (150 Å by 150 Å) and solvated on each side for further calculations.
The fit in each cryo-EM map was performed in four successive steps. First, a quick energy minimization (2000 steps of conjugate gradient) was performed on the full system without taking the map into account. This step was dedicated to the removal of bad contacts resulting from the addition of Gs and Nb35 proteins. Then, the second step consisted in a first equilibration of 5 ns (10-fs time step; NVT; 300 K) performed with CDMD and using a constant targeted low resolution of 5 Å together with a strength constant of 10,000 kJ/mol for the map potential. This bias was applied only to the backbone beads of the system. This step was useful to quickly optimize the alignment of the system with the targeted map. During this second step, an additional force of 50,000 kJ/mol per nm2 was added to keep the distance between the two centers of masses (COMs) of both the peptide and surrounding residues of the receptor close to its initial value. This force prevented a quick motion of the AVP peptide in the first steps of the simulation that resulted from large forces applying to the receptor. For the subsequent steps of the fitting procedure, this additional force on COMs was removed. During the step 3 (30 ns), the same molecular dynamics parameters were used but with a gradual increase in both the resolution (from 5 to 3 Å) and the strength constant (from 10,000 to 50,000 kJ/mol), over a period of 25 ns. During the last 5 ns, these values were kept constant. This step was the key step allowing the whole system to adapt and fit to the maps. Last, the last step (10 ns) consisted in keeping the resolution and the strength constant at their reached values (3 Å; 50,000 kJ/mol), but this time applying the force only to the backbone and side-chain beads of the peptide. All the other backbone beads of the system were restrained in positions during this step with a force constant of 5000 kJ/mol. This step was useful to refine the position of the peptide in the density, especially of its side chains. For every step of the fitting procedure, the fit of each cluster was performed five times to verify the convergence of the obtained models.
All-atom refinement of the models in the maps
The CG models obtained from the fitting procedure were back-mapped to a full-atom representation. We used the standard “initram” procedure provided by the developers of MARTINI (79) with subtle changes. These changes concerned restrains on ω angles and Cα positions for all chains (V2R, Gs, and Nb35) to keep ω angles in trans conformation and to avoid large backbone motions, which inevitably would lead to models out of cryo-EM maps. Those restrains were added during the minimization and the MDSs inherent to the default initram procedure. In practice, the initram procedure was as follows: (i) After the very raw guess of atomic positions, from CG beads, performed by the initram script, (ii) the Charmm36 force field (80) was used for 10,000 steps of steepest descent, disabling the nonbonded terms, (iii) followed by 5000 steps of steepest descent including all terms of the force field, and last, (iv) 300 steps of molecular dynamics were performed. Except the number of steps, the parameters for minimization and MDSs were set as default from the initram procedure. Minimization and MDSs were performed using the GROMACS package (77).
As a final step, iterative manual adjustments were carried out in Coot (81) and real-space refinement using Phenix programs (82). The model statistics were validated using MolProbity (83).
Classical all-atom MDSs
Following procedures previously described (84), the L-state cryo-EM structure was subjected to MDSs. The system was set up using the CHARMM-GUI micelle builder (85). The protein complex was inserted into a hydrated, equilibrated micelle composed of 60 molecules of LMNG after addition of missing protein loops in Coot. A total of 495 sodium and 511 chloride ions were added to neutralize the system, reaching a final concentration of approximately 150 mM. MDSs were performed in GROMACS 2020 using the CHARMM36m force field and the CHARMM TIP3P water model. The input systems were subjected to energy minimization, equilibration, and production simulation using the GROMACS input scripts generated by CHARMM-GUI (86). Briefly, the system was energy minimized using 5000 steps of steepest descent, followed by 375 ps of equilibration. NVT and NPT equilibrations were followed by NPT production runs. The van der Waals interactions were smoothly switched off at 10 to 12 Å by a force-switching function (87), whereas the long-range electrostatic interactions were calculated using the particle mesh Ewald method (88). The temperature and pressure were held at 310.15 K and 1 bar, respectively. The assembled system was equilibrated by the well-established protocol in Micelle Builder, in which various restraints were applied to the protein, detergents, and water molecules, and the restraint forces were gradually reduced during this process. During production simulations, an NPT ensemble was used with isotropic pressure coupling via the Parrinello-Rahman barostat method, while the Nose-Hoover thermostat was used to maintain a temperature of 310.15 K. A leapfrog integration scheme was used, all bonds were constrained, and hydrogen mass repartitioning was applied (89), allowing for a time step of 4 ps to be used during NPT equilibration and production MDSs. We performed 10 independent production runs starting from the highest-resolution L state model, for a total simulation time of ~2.6 μs. Production runs were subsequently pooled together, and the resulting trajectory was analyzed using GROMACS tools to yield principal components. The analysis was performed on the subset of Cα atoms common to the simulated and experimental structures using 1 frame/ns of trajectory. The experimental L, T1, and T2 states were included in the analysis for comparison.
NMR data analysis
The purified V2R was prepared either in neutral amphipol (90, 91) or in LMNG detergent. In both cases, the V2R was expressed in Sf9 insect cells and purified as described above, except it was cleaved overnight at 4°C using the HRV3C protease at a 1:20 weight ratio (HRV3C:V2R) before concentration and purification by SEC.
1D STD NMR spectra (92) were recorded either on a mixture of AVP with V2R (400:2 μM) or on AVP. Selective methyl resonance saturation was achieved by equally spaced 60-ms Gaussian 180° pulses separated by 1-ms delay at 0 parts per million (ppm) (−50 ppm for reference spectra) at 274 and 283 K. An irradiation test was performed on a free peptide sample (400 μM) to verify that only V2R resonances were irradiated. Subtraction of free induction decay with on- and off-resonance protein saturation was achieved by phase cycling. A relaxation delay of 2.6 s (Aq and D1) and 128 dummy scans were used to reduce subtraction artifacts. Investigation of the time dependence of the saturation transfer from 0.5 to 4 s with equally spaced 50-ms Gaussian-shaped pulses (separated by a 1-ms delay) showed that 2 s was needed for efficient transfer of saturation from V2R to the AVP. A T1ρ filter of 30 ms was applied to eliminate background resonances of V2R. The transient number was typically 4000. To determine the specificity of STD signals, similar samples were prepared with the antagonist TVP as competitor, using 3 μM V2R, 80 μM AVP, and 550 μM TVP. The STD effect was then calculated as (I0 − Isat)/I0, where I0 and Isat are the intensities of one signal in the reference NMR spectrum and in the on-resonance spectrum, respectively.
We discriminated the different molecular models issued from CG-REMD simulations by comparing the experimental STD values and the expected simulated STD from model structures. Back calculation of STD intensities were calculated with the 3.8 version of CORCEMA-ST software (93). An order parameter value of 0.85 for methyl groups and a Kon value of a 108 s−1 were used. The correlation times were set to 0.5 and 40 ns for the free and bound states, respectively. Calculations with different correlation time values exploring the 0.2 to 2 ns and 10 to 30 ns for the free and bound forms, respectively, showed that the simulated profiles, as well as, in particular, the correlation coefficient between calculated and experimental values, were much more dependent on the template model than on the correlation time values. Coefficient correlations between simulated and experimental values were calculated for the whole peptide (residues 1 to 9). Mean correlations factors R1–9 were calculated for five representative structures of each cluster.
Time-resolved fluorescence resonance energy transfer binding assays
V2R binding studies using TagLite assays (Cisbio Bioassays, Codolet, France) based on time-resolved fluorescence resonance energy transfer (FRET) measurements were previously described (16, 94). Briefly, HEK cells were plated in white-walled, flat-bottom, 96-well plates (Greiner CELLSTAR plate, Sigma-Aldrich) in Dulbecco’s minimum essential medium (DMEM) containing 10% fetal bovine serum (Lonza), 1% nonessential amino acids, and penicillin/streptomycin (GIBCO) at 15,000 cells per well. Cells were transfected 24 hours later with a plasmid coding for the V2R version used in cryo-EM studies fused at its N terminus to the SNAP-tag (SNAP-V2R) (Cisbio Bioassays, Codolet, France). Transfections were performed with X-tremeGENE 360 (Sigma-Aldrich), according to the manufacturer’s recommendations: 10 μl of a premix containing DMEM X-tremeGENE 360 (0.3 μl per well), SNAP-V2 coding plasmid (30 ng per well), and noncoding plasmid (70 ng per well) were added to the culture medium. After a 48-hour culture period, cells were rinsed once with Tag-lite medium (Cisbio Bioassays, Codolet, France) and incubated in the presence of Tag-lite medium containing 100 nM benzylguanine-Lumi4-Tb for at least 60 min at 37°C. Cells were then washed four times. For saturation studies, cells were incubated for at least 4 hours at 4°C in the presence of benzazepine-red nonpeptide vasopressin antagonist (BZ-DY647, Cisbio Bioassays, Codolet, France) at various concentrations ranging from 1 × 10−10 to 1 × 10−7 M. Nonspecific binding was determined in the presence of 10 μM vasopressin. For competition studies, cells were incubated for at least 4 hours at 4°C with benzazepine-red ligand (5 nM) and increasing concentrations of vasopressin ranging from 1 × 10−11 to 3.16 × 10−6 M. Fluorescent signals were measured at 620 nm (fluorescence of the donor) and at 665 nM (FRET signal) on a PHERAstar (BMG LABTECH, Champigny s/Marne, France). Results were expressed as the 665/620 ratio [10,000 × (665/620)]. Specific variation of the FRET ratio was plotted as a function of benzazepine-red concentration (saturation experiments) or competitor concentration (competition experiment). All binding data were analyzed with GraphPad 8.3.0 (GraphPad Software Inc.) using the one site-specific binding equation. All results are expressed as the means ± SEM of at least three independent experiments performed in triplicate. Ki values were calculated from median inhibitory concentration values with the Cheng-Prusoff equation.
cAMP accumulation assays
As for V2R binding studies, V2R functional studies based on time-resolved FRET measurements were described previously (36, 57). Briefly, Chinese hamster ovary cells were plated in six-well plates (Falcon) at 350,000 cells per well and transfected 24 hours later with jetPEI (Ozyme) with a pRK5 plasmid coding for the version of the V2R used in the cryo-EM studies. A mix of isotonic NaCl solution (200 μl per well) containing jetPEI (2 μl per well), V2R coding plasmid (1 ng per well), and noncoding plasmid (3000 ng per well) was added to the culture medium (2 ml). Twenty-four hours later, cells were harvested with trypsin and cultured in white-walled, flat-bottom, 96-well plates (Greiner CELLSTAR plate, Sigma-Aldrich) at a density of 30,000 cells per well in DMEM containing 10% fetal bovine serum (Lonza), 1% nonessential amino acids, and penicillin/streptomycin (GIBCO). After a 24-hour culture period, cells were treated for 30 min at 37°C in the cAMP buffer with or without increasing AVP concentrations (3.16 × 10−12 to 10−6 M) in the presence of 0.1 mM RO201724, a phosphodiesterase inhibitor (Sigma-Aldrich). The accumulated cAMP was quantified using the cAMP Dynamic 2 Kit (Cisbio Bioassays, Codolet, France) according to the manufacturer’s protocol. Fluorescent signals were measured at 620 and 665 nm on a Spark 20M multimode microplate reader (Tecan). Data were plotted as the FRET ratio [10,000 × (665/620)] as a function of AVP concentration [log(AVP)]. Data were analyzed with GraphPad Prism using the “dose-response stimulation” subroutine. Median effective concentrations were determined using the log(agonist) versus response variable slope (four parameters) fit procedure. Experiments were repeated at least three times on different cultures, each condition in triplicate. Data are presented as means ± SEM.
Acknowledgments
We thank the cryo-EM staff at EMBL of Heidelberg (Germany) and the IGF Arpege platform of Pharmacology. We thank R. Healey for critical reading of the manuscript and A. Sagar for helping in software installation. Funding: This work was supported by grants from FRM (grant DEQ20150331736) and ANR (grants ANR-19-CE11-0014 and ANR-17-CE11-0011) and core funding from CNRS, INSERM, ENSCM, and Université de Montpellier. We thank GENCI (Grand Équipement National de Calcul Intensif) and TGCC/IDRIS to have selected us for the “Great Challenge” phases of both the Irène JOLIOT-CURIE and Jean-ZAY supercomputers. The CBS is a member of the French Infrastructure for Integrated Structural Biology (FRISBI) supported by ANR (ANR-10-INBS-05). J.B. was supported by a doctoral fellowship from the Ministère de L’Enseignement Supérieur, de la Recherche et de l’Innovation. Author contributions: J.B. purified V2R and AVP-V2R-Gs-Nb35 complexes, screened samples by NS-EM and cryo-EM, prepared grids, collected and processed cryo-EM data, generated the cryo-EM maps, and built some extended data figures. H.O. managed the Sf9 cell culture and baculoviral infections, expressed and purified V2R, purified AVP-V2R-Gs-Nb35 complexes, and prepared grids for cryo-EM. N.F. developed the CG-REMD modeling approach, fitted the models onto the cryo-EM maps, and back-mapped the models to all-atom representation. C.L. built the final models of AVP-V2R-Gs-Nb35 complexes into cryo-EM and performed MDSs. J.L.-K.-H. and A.A. screened samples by NS-EM and prepared grids for cryo-EM. G.G. contributed to the expression and purification of V2R. J.S.-P. expressed and purified Gs protein and Nb35 nanobody. S.T. contributed to processing cryo-EM data for generating cryo-EM maps. M.L. contributed to CG-REMD MD modeling. S.G. established all the initial procedures for the ternary complex purification. R.S. designed Gs constructs, expressed and purified Gs protein, and built most of the figures. H.D. managed STD NMR experiments and generated the STD NMR data. B.M. designed the V2R construct and determined its pharmacological properties with the help of H.O. S.G., P.B., and B.M. wrote the paper with the input from J.B., N.F., and H.D. Last, S.G., P.B., and B.M. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The cryo-EM density maps for the AVP-V2R-Gs-Nb35 complex have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-12128 (L state) and EMD-12129 (T state). The coordinates for the models of AVP-V2R-Gs-Nb35 complex have been deposited in the PDB under accession numbers 7BB6 and 7BB7 (L and T states, respectively). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabg5628/DC1
REFERENCES AND NOTES
- 1.Barberis C., Mouillac B., Durroux T., Structural bases of vasopressin/oxytocin receptor function. J. Endocrinol. 156, 223–229 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Barberis C., Morin D., Durroux T., Mouillac B., Guillon G., Seyer R., Hibert M., Tribollet E., Manning M., Molecular pharmacology of AVP and OT receptors and therapeutic potential. Drug News Perspect. 12, 279–292 (1999). [Google Scholar]
- 3.Robben J. H., Knoers N. V. A. M., Deen P. M. T., Regulation of the vasopressin V2 receptor by vasopressin in polarized renal collecting duct cells. Mol. Biol. Cell 15, 5693–5699 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Treschan T. A., Peters J., Warltier D. C., The vasopressin system: Physiology and clinical strategies. Anesthesiology 105, 599–612 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Ren X.-R., Reiter E., Ahn S., Kim J., Chen W., Lefkowitz R. J., Different G protein-coupled receptor kinases govern G protein and β-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. U.S.A. 102, 1448–1453 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alonso G., Galibert E., Boulay V., Guillou A., Jean A., Compan V., Guillon G., Sustained elevated levels of circulating vasopressin selectively stimulate the proliferation of kidney tubular cells via the activation of V2 receptors. Endocrinology 150, 239–250 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Ball S. G., Vasopressin and disorders of water balance: The physiology and pathophysiology of vasopressin. Ann. Clin. Biochem. 44, 417–431 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Morello J.-P., Bichet D. G., Nephrogenic diabetes insipidus. Annu. Rev. Physiol. 63, 607–630 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Feldman B. J., Rosenthal S. M., Vargas G. A., Fenwick R. G., Huang E. A., Matsuda-Abedini M., Lustig R. H., Mathias R. S., Portale A. A., Miller W. L., Gitelman S. E., Nephrogenic syndrome of inappropriate antidiuresis. N. Engl. J. Med. 352, 1884–1890 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris P. C., Torres V. E., Polycystic kidney disease. Annu. Rev. Med. 60, 321–337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edward Zhou X., Melcher K., Eric Xu H., Structural biology of G protein-coupled receptor signaling complexes. Protein Sci. 28, 487–501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gusach A., Maslov I., Luginina A., Borshchevskiy V., Mishin A., Cherezov V., Beyond structure: Emerging approaches to study GPCR dynamics. Curr. Opin. Struct. Biol. 63, 18–25 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Fernandez-Leiro R., Scheres S. H. W., Unravelling biological macromolecules with cryo-electron microscopy. Nature 537, 339–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carpenter B., Tate C. G., Active state structures of G protein-coupled receptors highlight the similarities and differences in the G protein and arrestin coupling interfaces. Curr. Opin. Struct. Biol. 45, 124–132 (2017). [DOI] [PubMed] [Google Scholar]
- 15.García-Nafría J., Tate C. G., Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol. Cell. Endocrinol. 488, 1–13 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Loison S., Cottet M., Orcel H., Adihou H., Rahmeh R., Lamarque L., Trinquet E., Kellenberger E., Hibert M., Durroux T., Mouillac B., Bonnet D., Selective fluorescent nonpeptidic antagonists for vasopressin V2GPCR: Application to ligand screening and oligomerization assays. J. Med. Chem. 55, 8588–8602 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Ala Y., Morin D., Mouillac B., Sabatier N., Vargas R., Cotte N., Déchaux M., Antignac C., Arthus M. F., Lonergan M., Turner M. S., Balestre M. N., Alonso G., Hibert M., Barberis C., Hendy G. N., Bichet D. G., Jard S., Functional studies of twelve mutant V2 vasopressin receptors related to nephrogenic diabetes insipidus: Molecular basis of a mild clinical phenotype. J. Am. Soc. Nephrol. 9, 1861–1872 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Rasmussen S. G. F., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T. A., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K., Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serradeil-Le Gal C., Lacour C., Valette G., Garcia G., Foulon L., Galindo G., Bankir L., Pouzet B., Guillon G., Barberis C., Chicot D., Jard S., Vilain P., Garcia C., Marty E., Raufaste D., Brossard G., Nisato D., Maffrand J. P., Le Fur G., Characterization of SR 121463A, a highly potent and selective, orally active vasopressin V2 receptor antagonist. J. Clin. Invest. 98, 2729–2738 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terwilliger T. C., Ludtke S. J., Read R. J., Adams P. D., Afonine P. V., Improvement of cryo-EM maps by density modification. Nat. Methods 17, 923–927 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delort B., Renault P., Charlier L., Raussin F., Martinez J., Floquet N., Coarse-grained prediction of peptide binding to G-protein coupled receptors. J. Chem. Inf. Model. 57, 562–571 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Ferré G., Louet M., Saurel O., Delort B., Czaplicki G., M’Kadmi C., Damian M., Renault P., Cantel S., Gavara L., Demange P., Marie J., Fehrentz J.-A., Floquet N., Milon A., Banères J.-L., Structure and dynamics of G protein-coupled receptor–bound ghrelin reveal the critical role of the octanoyl chain. Proc. Natl. Acad. Sci. U.S.A. 116, 17525–17530 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Igaev M., Kutzner C., Bock L. V., Vaiana A. C., Grubmüller H., Automated cryo-EM structure refinement using correlation-driven molecular dynamics. eLife 8, e43542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Assadi-Porter F. M., Tonelli M., Maillet E., Hallenga K., Benard O., Max M., Markley J. L., Direct NMR detection of the binding of functional ligands to the human sweet receptor, a heterodimeric family 3 GPCR. J. Am. Chem. Soc. 130, 7212–7213 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartoschek S., Klabunde T., Defossa E., Dietrich V., Stengelin S., Griesinger C., Carlomagno T., Focken I., Wendt K. U., Drug design for G-protein-coupled receptors by a ligand-based NMR method. Angew. Chem. Int. Ed. Engl. 49, 1426–1429 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Kato H. E., Zhang Y., Hu H., Suomivuori C.-M., Kadji F. M. N., Aoki J., Krishna Kumar K., Fonseca R., Hilger D., Huang W., Latorraca N. R., Inoue A., Dror R. O., Kobilka B. K., Skiniotis G., Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 572, 80–85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dashti A., Mashayekhi G., Shekhar M., Hail D. B., Salah S., Schwander P., Georges A. D., Singharoy A., Frank J., Ourmazd A., Retrieving functional pathways of biomolecules from single-particle snapshots. Nat. Commun. 11, 4734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waltenspühl Y., Schöppe J., Ehrenmann J., Kummer L., Plückthun A., Crystal structure of the human oxytocin receptor. Sci. Adv. 6, eabb5419 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mouillac B., Chini B., Balestre M. N., Elands J., Trumpp-Kallmeyer S., Hoflack J., Hibert M., Jard S., Barberis C., The binding site of neuropeptide vasopressin V1a receptor. Evidence for a major localization within transmembrane regions. J. Biol. Chem. 270, 25771–25777 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Hibert M., Hoflack J., Trumpp-Kallmeyer S., Mouillac B., Chini B., Mahé E., Cotte N., Jard S., Manning M., Barberis C., Functional architecture of vasopressin/oxytocin receptors. J. Recept. Signal Transduct. Res. 19, 589–596 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Macion-Dazard R., Callahan N., Xu Z., Wu N., Thibonnier M., Shoham M., Mapping the binding site of six nonpeptide antagonists to the human V2-renal vasopressin receptor. J. Pharmacol. Exp. Ther. 316, 564–571 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Cotte N., Balestre M.-N., Aumelas A., Mahé E., Phalipou S., Morin D., Hibert M., Manning M., Durroux T., Barberis C., Mouillac B., Conserved aromatic residues in the transmembrane region VI of the V1a vasopressin receptor differentiate agonist vs. antagonist ligand binding. Eur. J. Biochem. 267, 4253–4263 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Tahtaoui C., Balestre M.-N., Klotz P., Rognan D., Barberis C., Mouillac B., Hibert M., Identification of the binding sites of the SR49059 nonpeptide antagonist into the V1a vasopressin receptor using sulfydryl-reactive ligands and cysteine mutants as chemical sensors. J. Biol. Chem. 278, 40010–40019 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Filipek S., Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 55, 114–120 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Deupi X., Relevance of rhodopsin studies for GPCR activation. Biochim. Biophys. Acta 1837, 674–682 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Tenenbaum J., Ayoub M. A., Perkovska S., Adra-Delenne A.-L., Mendre C., Ranchin B., Bricca G., Geelen G., Mouillac B., Durroux T., Morin D., The constitutively active V2 receptor mutants conferring NSIAD are weakly sensitive to agonist and antagonist regulation. PLOS ONE 4, e8383 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rochdi M. D., Vargas G. A., Carpentier E., Oligny-Longpré G., Chen S., Kovoor A., Gitelman S. E., Rosenthal S. M., von Zastrow M., Bouvier M., Functional characterization of vasopressin type 2 receptor substitutions (R137H/C/L) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: Implications for treatments. Mol. Pharmacol. 77, 836–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernier V., Lagacé M., Lonergan M., Arthus M.-F., Bichet D. G., Bouvier M., Functional rescue of the constitutively internalized V2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059. Mol. Endocrinol. 18, 2074–2084 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Barak L. S., Oakley R. H., Laporte S. A., Caron M. G., Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. U.S.A. 98, 93–98 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weis W. I., Kobilka B. K., The molecular basis of G protein-coupled receptor activation. Annu. Rev. Biochem. 87, 897–919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hilger D., Masureel M., Kobilka B. K., Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 25, 4–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.García-Nafría J., Lee Y., Bai X., Carpenter B., Tate C. G., Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 7, e35946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang Y.-L., Khoshouei M., Deganutti G., Glukhova A., Koole C., Peat T. S., Radjainia M., Plitzko J. M., Baumeister W., Miller L. J., Hay D. L., Christopoulos A., Reynolds C. A., Wootten D., Sexton P. M., Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang Y.-L., Khoshouei M., Radjainia M., Zhang Y., Glukhova A., Tarrasch J., Thal D. M., Furness S. G. B., Christopoulos G., Coudrat T., Danev R., Baumeister W., Miller L. J., Christopoulos A., Kobilka B. K., Wootten D., Skiniotis G., Sexton P. M., Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koehl A., Hu H., Maeda S., Zhang Y., Qu Q., Paggi J. M., Latorraca N. R., Hilger D., Dawson R., Matile H., Schertler G. F. X., Granier S., Weis W. I., Dror R. O., Manglik A., Skiniotis G., Kobilka B. K., Structure of the μ-opioid receptor–Gi protein complex. Nature 558, 547–552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim K., Che T., Panova O., DiBerto J. F., Lyu J., Krumm B. E., Wacker D., Robertson M. J., Seven A. B., Nichols D. E., Shoichet B. K., Skiniotis G., Roth B. L., Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 182, 1574–1588.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manglik A., Kobilka B., The role of protein dynamics in GPCR function: Insights from the β2AR and rhodopsin. Curr. Opin. Cell Biol. 27, 136–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chini B., Mouillac B., Balestre M. N., Trumpp-Kallmeyer S., Hoflack J., Hibert M., Andriolo M., Pupier S., Jard S., Barberis C., Two aromatic residues regulate the response of the human oxytocin receptor to the partial agonist arginine vasopressin. FEBS Lett. 397, 201–206 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Manning M., Misicka A., Olma A., Bankowski K., Stoev S., Chini B., Durroux T., Mouillac B., Corbani M., Guillon G., Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J. Neuroendocrinol. 24, 609–628 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernier V., Morello J.-P., Zarruk A., Debrand N., Salahpour A., Lonergan M., Arthus M.-F., Laperrière A., Brouard R., Bouvier M., Bichet D. G., Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 17, 232–243 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Kalenga K., Persu A., Goffin E., Lavenne-Pardonge E., van Cangh P. J., Bichet D. G., Devuyst O., Intrafamilial phenotype variability in nephrogenic diabetes insipidus. Am. J. Kidney Dis. 39, 737–743 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Morello J. P., Bouvier M., Petäjä-Repo U. E., Bichet D. G., Pharmacological chaperones: A new twist on receptor folding. Trends Pharmacol. Sci. 21, 466–469 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Torres V. E., Chapman A. B., Devuyst O., Gansevoort R. T., Perrone R. D., Lee J., Hoke M. E., Estilo A., Sergeyeva O., Multicenter study of long-term safety of tolvaptan in later-stage autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 16, 48–58 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bockenhauer D., Carpentier E., Rochdi D., van’t Hoff W., Breton B., Bernier V., Bouvier M., Bichet D. G., Vasopressin type 2 receptor V88M mutation: Molecular basis of partial and complete nephrogenic diabetes insipidus. Nephron Physiol. 114, p1–p10 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Prosperi F., Suzumoto Y., Marzuillo P., Costanzo V., Jelen S., Iervolino A., Guarino S., La Manna A., Miraglia Del Giudice E., Perna A. F., Zacchia M., Cordat E., Capasso G., Trepiccione F., Characterization of five novel vasopressin V2 receptor mutants causing nephrogenic diabetes insipidus reveals a role of tolvaptan for M272R-V2R mutation. Sci. Rep. 10, 16383 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang L., Xu J., Cao S., Sun D., Liu H., Lu Q., Liu Z., Du Y., Zhang C., Cryo-EM structure of the AVP-vasopressin receptor 2-Gs signaling complex. Cell Res., (2021); 10.1038/s41422-021-00483-z. [DOI] [PMC free article] [PubMed]
- 57.Jean-Alphonse F., Perkovska S., Frantz M.-C., Durroux T., Méjean C., Morin D., Loison S., Bonnet D., Hibert M., Mouillac B., Mendre C., Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 20, 2190–2203 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Kimanius D., Forsberg B. O., Scheres S. H. W., Lindahl E., Accelerated cryo-EM structure determination with parallelisation using GPUS in RELION-2. eLife 5, e18722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J. H., Lindahl E., Scheres S. H. W., New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang K., Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tegunov D., Cramer P., Real-time cryo-EM data pre-processing with Warp. bioRxiv, 338558 (2018). [Google Scholar]
- 64.Grant T., Rohou A., Grigorieff N., cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Heymann J. B., Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. 27, 159–171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cardone G., Heymann J. B., Steven A. C., One number does not fit all: Mapping local variations in resolution in cryo-EM reconstructions. J. Struct. Biol. 184, 226–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Terwilliger T. C., Sobolev O. V., Afonine P. V., Adams P. D., Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr. D Struct. Biol. 74, 545–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Webb B., Sali A., Protein structure modeling with MODELLER. Methods Mol. Biol. 1654, 39–54 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Granier S., Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Weis W. I., Kobilka B. K., Structure of the δ-opioid receptor bound to naltrindole. Nature 485, 400–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu C. K., Hu B., Rose J. P., Liu Z.-J., Nguyen T. L., Zheng C., Breslow E., Wang B.-C., Structures of an unliganded neurophysin and its vasopressin complex: Implications for binding and allosteric mechanisms. Protein Sci. 10, 1869–1880 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rose J. P., Wu C., Hsiao C.-D., Breslow E., Wang B.-C., Crystal structure of the neurophysin–oxytocin complex. Nat. Struct. Biol. 3, 163–169 (1996). [DOI] [PubMed] [Google Scholar]
- 73.Schmidt J. M., Ohlenschläger O., Rüterjans H., Grzonka Z., Kojro E., Pavo I., Fahrenholz F., Conformation of [8-arginine]vasopressin and V1 antagonists in dimethyl sulfoxide solution derived from two-dimensional NMR spectroscopy and molecular dynamics simulation. Eur. J. Biochem. 201, 355–371 (1991). [DOI] [PubMed] [Google Scholar]
- 74.Ibrahim B. S., Pattabhi V., Trypsin inhibition by a peptide hormone: Crystal structure of trypsin–vasopressin complex. J. Mol. Biol. 348, 1191–1198 (2005). [DOI] [PubMed] [Google Scholar]
- 75.Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., De Vries A. H., The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Daura X., Jaun B., Seebach D., van Gunsteren W. F., Mark A. E., Reversible peptide folding in solution by molecular dynamics simulation. J. Mol. Biol. 280, 925–932 (1998). [DOI] [PubMed] [Google Scholar]
- 77.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindah E., Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 1–2, 19–25 (2015). [Google Scholar]
- 78.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 79.Wassenaar T. A., Pluhackova K., Böckmann R. A., Marrink S. J., Tieleman D. P., Going backward: A flexible geometric approach to reverse transformation from coarse grained to atomistic models. J. Chem. Theory Comput. 10, 676–690 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Best R. B., Zhu X., Shim J., Lopes P. E. M., Mittal J., Feig M., Mackerell A. D. Jr., Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 82.Afonine P. V., Mustyakimov M., Grosse-Kunstleve R. W., Moriarty N. W., Langan P., Adams P. D., Joint x-ray and neutron refinement with phenix.refine. Acta Crystallogr. Sect. D 66, 1153–1163 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vasiliauskaité-Brooks I., Healey R. D., Rochaix P., Saint-Paul J., Sounier R., Grison C., Waltrich-Augusto T., Fortier M., Hoh F., Saied E. M., Arenz C., Basu S., Leyrat C., Granier S., Structure of a human intramembrane ceramidase explains enzymatic dysfunction found in leukodystrophy. Nat. Commun. 9, 5437 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu E. L., Cheng X., Jo S., Rui H., Song K. C., Dávila-Contreras E. M., Qi Y., Lee J., Monje-Galvan V., Venable R. M., Klauda J. B., Im W., CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee J., Cheng X., Swails J. M., Yeom M. S., Eastman P. K., Lemkul J. A., Wei S., Buckner J., Jeong J. C., Qi Y., Jo S., Pande V. S., Case D. A., Brooks C. L. III, Mac Kerell A. D. Jr., Klauda J. B., Im W., CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steinbach P. J., Brooks B. R., New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 15, 667–683 (1994). [Google Scholar]
- 88.Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G., A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
- 89.Balusek C., Hwang H., Lau C. H., Lundquist K., Hazel A., Pavlova A., Lynch D. L., Reggio P. H., Wang Y., Gumbart J. C., Accelerating membrane simulations with hydrogen mass repartitioning. J. Chem. Theory Comput. 15, 4673–4686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sharma K. S., Durand G., Gabel F., Bazzacco P., Le Bon C., Billon-Denis E., Catoire L. J., Popot J.-L., Ebel C., Pucci B., Non-ionic amphiphilic homopolymers: Synthesis, solution properties, and biochemical validation. Langmuir 28, 4625–4639 (2012). [DOI] [PubMed] [Google Scholar]
- 91.Rahmeh R., Damian M., Cottet M., Orcel H., Mendre C., Durroux T., Sharma K. S., Durand G., Pucci B., Trinquet E., Zwier J. M., Deupi X., Bron P., Banères J. L., Mouillac B., Granier S., Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 109, 6733–6738 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mayer M., Meyer B., Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 38, 1784–1788 (1999). [DOI] [PubMed] [Google Scholar]
- 93.Krishna N. R., Jayalakshmi V., Quantitative analysis of STD-NMR spectra of reversibly forming ligand-receptor complexes. Top. Curr. Chem. 273, 15–54 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Zwier J. M., Roux T., Cottet M., Durroux T., Douzon S., Bdioui S., Gregor N., Bourrier E., Oueslati N., Nicolas L., Tinel N., Boisseau C., Yverneau P., Charrier-Savournin F., Fink M., Trinquet E., A fluorescent ligand-binding alternative using Tag-lite® technology. J. Biomol. Screen. 15, 1248–1259 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabg5628/DC1