

Fluorescent sensors targeted to endogenous proteins measure native signaling dynamics within signaling compartments.
Abstract
Growing evidence suggests that many essential intracellular signaling events are compartmentalized within kinetically distinct microdomains in cells. Genetically encoded fluorescent biosensors are powerful tools to dissect compartmentalized signaling, but current approaches to probe these microdomains typically rely on biosensor fusion and overexpression of critical regulatory elements. Here, we present a novel class of biosensors named FluoSTEPs (fluorescent sensors targeted to endogenous proteins) that combine self-complementing split green fluorescent protein, CRISPR-mediated knock-in, and fluorescence resonance energy transfer biosensor technology to probe compartmentalized signaling dynamics in situ. We designed FluoSTEPs for simultaneously highlighting endogenous microdomains and reporting domain-specific, real-time signaling events including kinase activities, guanosine triphosphatase activation, and second messenger dynamics in live cells. A FluoSTEP for 3′,5′-cyclic adenosine monophosphate (cAMP) revealed distinct cAMP dynamics within clathrin microdomains in response to stimulation of G protein–coupled receptors, showcasing the utility of FluoSTEPs in probing spatiotemporal regulation within endogenous signaling architectures.
INTRODUCTION
Compartmentalization of intracellular signals by macromolecular complexes can reshape the kinetics of cellular processes and provide diversity and specificity in signaling. Our understanding of this microdomain architecture of signaling networks has greatly benefited from the design and utilization of genetically encoded fluorescent biosensors (1). By attaching these sensors to proteins of interest (POIs) and introducing these fusions into living cells, researchers can monitor compartmentalized signals in real time (2–5). Despite the utility of these biosensors, this fusion strategy has drawbacks primarily stemming from unintended effects from the concomitant overexpression of the POIs. Overexpression of enzymes or scaffolds can disrupt native signaling pathways by causing mislocalization, artificially enhancing/weakening certain biochemical reactions, and imbalancing the stoichiometry of macromolecular interactions. In addition, some biosensors themselves contain enzymatic components known to affect global signaling within the cell. For example, the dimerization optimized reporter for activation (DORA) RhoA sensor for measuring activity of the guanosine triphosphatase (GTPase) RhoA contains active RhoA within a conformational switch (6); however, the overexpression of RhoA is a hallmark of several cancers with associated, downstream signaling effects (7, 8). Strategies have been developed to address the overexpression concerns such as using nanobodies for highlighting endogenous, active receptors and using intrabodies to recruit biosensors to endogenous compartments (9, 10). However, perturbations in trafficking and signaling due to nanobody binding, as well as issues of compartment specificity, are still ever-present (9, 11, 12). Ideally, an approach that combined the strength of quantitative biosensing, specificity of genetic fusions, and minimal perturbation of endogenous POIs would be valuable for dissecting compartmentalized signaling within living cells.
In a recent study of cyclic adenosine 3′,5′-monophosphate (cAMP) signaling (13), we introduced a pair of fluorescent biosensors engineered based on a new strategy for probing endogenous microdomains. Here, we present the design and characterization of a suite of novel fluorescent biosensors based on this strategy. By using a self-complementing split green fluorescent protein (GFP) as a fluorescence resonance energy transfer (FRET) donor, we designed ratiometric sensors that can be recruited and reconstituted at a tagged POI (14, 15), giving rise to fluorescent sensors targeted to endogenous proteins (FluoSTEPs). The generation of the functional biosensors only at a POI ensures compartment specificity, and the self-complementing split GFP donor facilitates endogenous protein tagging when combined with CRISPR-Cas9 gene editing. We demonstrate the generalizability of FluoSTEPs by applying the modular design to measure kinase activities, GTPase activation, and second messenger dynamics. We showcase the applicability of FluoSTEPs by deploying the new sensors to uncover mechanisms governing sustained cAMP dynamics at clathrin membrane microdomains after G protein–coupled receptor (GPCR) stimulation.
RESULTS
FluoSTEP–A-kinase activity reporter is reconstituted and functional at microdomains of interest
Our goal with the FluoSTEP design was to construct a logic-gated FRET sensor that exists predominantly in a nonfunctional (i.e., FRET-incapable) state except when localized to the desired protein target. To install a FRET-based sensor with this control logic, we adopted the robust and bright split super-folder GFP (sfGFP) as the FRET donor and a red fluorescent protein (RFP) as the FRET acceptor. Split sfGFP is divided between the 10th and 11th β strands into a pair of nonfluorescent components (GFP1–10 and GFP11) capable of undergoing spontaneous fragment complementation to reconstitute intact, fluorescent sfGFP (15). Thus, when the small GFP11 fragment (16 amino acids) is fused to a POI and expressed in the presence of GFP1–10, the reconstitution of the donor fluorophore should occur and give rise to a functional FRET-based sensor only at the POI (Fig. 1A). When GFP11 is introduced into a specific gene locus using CRISPR-Cas9 technology, this domain-specific logic control further enables ratiometric FRET measurements of compartmentalized signaling activities at endogenous protein loci.
Fig. 1. FluoSTEP-AKAR is reconstituted and functional at microdomains of interest.
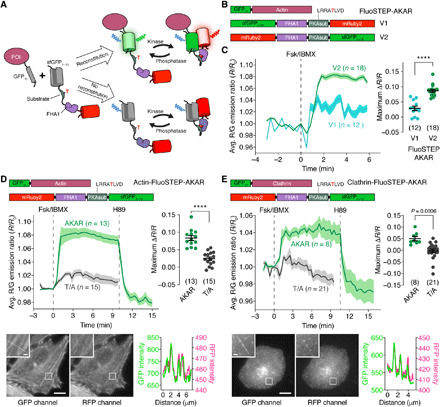
(A) FluoSTEPs use the spontaneous fragment complementation of split sfGFP to reconstitute a functional FRET-based biosensor at a POI, as shown here with a kinase activity reporter. Probe species that fail to reconstitute contain a nonfluorescent donor and thus do not contribute to the FRET signal. (B) Domain structures of GFP11-actin, G1–10-R-FluoSTEP-AKAR (V1), and R-G1–10–FluoSTEP-AKAR (V2). (C) Average red/green (R/G) emission ratio time courses (left) and maximum emission ratio changes (right) in HEK293T cells expressing GFP11-actin and either G1–10-R–FluoSTEP-AKAR (V1, teal, n = 12 cells) or R-G1–10–FluoSTEP-AKAR (V2, green, n = 18 cells) following stimulation with 50 μM Fsk and 100 μM IBMX (Fsk/IBMX). (D and E) Top: Average R/G emission ratio time courses (left) or maximum emission ratio change (right) in HeLa cells expressing GFP11-actin (D) or GFP11-clathrin (E) and either FluoSTEP-AKAR (green; actin: n = 13 cells; clathrin: n = 8 cells) or FluoSTEP-AKAR (T/A) negative control (gray; actin: n = 15 cells; clathrin: n = 21 cells) upon stimulation with Fsk/IBMX followed by 10 μM H89. Bottom: Representative confocal fluorescence images depicting the localization of biosensor fluorescence in the GFP (left) and RFP (middle) channels. Insets are enlarged from the outlined regions. Line-profile intensity plots (right) highlight colocalization of the fluorescence signals along the indicated region. ****P < 0.0001, unpaired two-tailed Student’s t test. Solid lines in time courses indicate average responses; shaded areas denote SEM. Horizontal lines in scatter plots indicate means ± SEM. Scale bars, 10 μm (inset, 1 μm).
We initially applied this concept to generate a FluoSTEP A-kinase activity reporter (FluoSTEP-AKAR) in which protein kinase A (PKA)–mediated phosphorylation of a kinase-specific substrate domain causes intramolecular binding to the Forkhead-associated 1 (FHA1) phospho-amino acid–binding domain (PAABD), resulting in a conformational change that increases FRET between the donor and acceptor fluorescent proteins (FPs) (Fig. 1A). Specifically, we exchanged the Cerulean (cyan) donor and cpVenus (yellow) acceptor FPs in AKAR4 (16) with GFP1–10 and mRuby2 (17), respectively, to make a complementation-dependent green-red FRET probe (G1–10-R–FluoSTEP-AKAR; Fig. 1B). Given that FRET efficiency is determined not only by the distance between the donor and acceptor fluorescence transition dipole moments but also by their relative orientation (18) and because the repositioned C terminus of sfGFP1–10 may alter the orientation of the donor analogous to the effect of circular permutation (19), we also tested a variant in which the GFP1–10 donor and mRuby2 acceptor were swapped (R-G1–10–FluoSTEP-AKAR; Fig. 1B).
In human embryonic kidney (HEK) 293T cells transiently expressing actin N-terminally tagged with GFP11 (GFP11-actin), both sensor variants demonstrated the spontaneous reconstitution of GFP fluorescence. However, only the R-G1–10–FluoSTEP-AKAR sensor produced a robust increase in the red/green emission ratio (sensitized acceptor RFP emission due to FRET divided by direct donor GFP emission) after the addition of the transmembrane adenylyl cyclase (AC) activator forskolin (Fsk; 50 μM) and the phosphodiesterase (PDE) inhibitor 3-isobutyl-1-methylxanthine (IBMX; 100 μM) [normalized ratio change (ΔR/R) = 8.7 ± 0.44% (means ± SEM), n = 18 cells] (Fig. 1C), with a time–to–half-maximum response (t1/2) of 0.51 ± 0.04 min (table S1). Intracellular FRET efficiencies for this sensor were measured by acceptor photobleaching to be 7 ± 2% and 11 ± 1% (n = 34 cells) before and after Fsk/IBMX stimulation, respectively. The G1–10-R–FluoSTEP-AKAR sensor generated a smaller Fsk/IBMX-induced response compared to its counterpart (ΔR/R = 2.9 ± 0.74%, n = 12 cells, P < 0.0001). This observation is consistent with the critical role of the relative orientations of the donor/acceptor fluorescence transition dipole moments in the performance of FRET-based biosensors (20).
To further characterize R-G1–10–FluoSTEP-AKAR, which we renamed FluoSTEP-AKAR, in another cell type, we coexpressed the sensor with GFP11-actin in HeLa cells. Confocal fluorescence imaging revealed proper targeting of the biosensor to actin, as shown by the actin cytoskeletal structure visible in both the GFP and RFP channels (Fig. 1D). After Fsk/IBMX stimulation, FluoSTEP-AKAR also produced a robust increase in red/green emission ratio in these cells (ΔR/R = 8.3 ± 0.70%, t1/2 = 0.99 ± 0.05 min, n = 13 cells) (Fig. 1D and table S1). Subsequent addition of the PKA inhibitor H89 (10 μM) acutely reversed the emission ratio change, while mutating the phospho-acceptor threonine residue in FluoSTEP-AKAR to alanine (T/A) largely abolished the emission ratio change induced by Fsk/IBMX stimulation (ΔR/R = 2.4 ± 0.50%, n = 15 cells, P < 0.0001), highlighting the specificity of the response (Fig. 1D). To test the recruitment and functioning at a different POI, we coexpressed FluoSTEP-AKAR with GFP11-tagged clathrin (GFP11-clathrin) in HeLa cells and observed GFP reconstitution via confocal fluorescence microscopy, which highlighted clathrin microdomains along the plasma membrane (PM) (Fig. 1E). Similar to the actin-targeted sensor, Fsk/IBMX stimulation triggered a robust increase in the FluoSTEP-AKAR red/green emission ratio (ΔR/R = 5.1 ± 0.92%, t1/2 = 1.2 ± 0.12 min, n = 8 cells), which was acutely reversed upon H89 addition, while the FluoSTEP-AKAR (T/A) negative control construct showed no response to Fsk/IBMX treatment (ΔR/R = −0.24 ± 0.76%, n = 21 cells) (Fig. 1E). Our complementation-dependent biosensor design thus enables robust activity measurements around specific POIs with minimal disruption of molecular organization.
The FluoSTEP design can be generalized to multiple targets
Many FRET-based biosensors are generated on the basis of a modular design where a signal-specific conformational switch is sandwiched between a pair of FPs. This modular architecture facilitates the straightforward assembly of a suite of sensors for detecting different biological activities by simply swapping out the signal-specific switch domain (21). We therefore took advantage of this feature to expand the FluoSTEP arsenal. For instance, most sensors designed to probe cAMP, the upstream regulator of PKA and a second messenger with diverse regulatory roles (22), use the conformational change induced by the binding of cAMP to a single-protein domain (23, 24) to modulate FRET between flanking FPs. Thus, to construct a FluoSTEP cAMP sensor, we tested cAMP-binding switches derived from the cAMP-binding domains of two different isoforms of exchange protein activated by cAMP (Epac), Epac2B (285 to 443) (25) and Epac1 (149 to 881) (25, 26), inserted between mRuby2 and GFP1–10 (fig. S1). Only the sensor containing Epac1 (149 to 881) produced an increase in the green/red emission ratio (ΔR/R = 4.1 ± 0.63%, t1/2 = 4.8 ± 0.44 min, n = 15 cells; FRET efficiency change from 17 ± 1% to 9 ± 1%, n = 33 cells) upon Fsk/IBMX addition in HEK293T cells coexpressing GFP11-actin and was thus named FluoSTEP-ICUE (indicator of cAMP using Epac) (Fig. 2A; fig. S1, A and B; and table S1). FluoSTEP-ICUE targeted to the PM via GFP11 fused to Lyn-tagged FK506-binding protein (FKBP)–rapamycin-binding domain (FRB) (Lyn-FRB-GFP11) also responded to β-adrenergic receptor (β-AR) stimulation, producing an increase in the green/red emission ratio upon treatment with the β-AR agonist isoproterenol and showing identical kinetics to a previously developed PM-targeted cAMP indicator that uses the same switch domain as FluoSTEP-ICUE (PM-ICUE3) (fig. S1C) (26). As a negative control, the cAMP-binding site in Epac1 was mutated (R279E). When coexpressed with GFP11-actin, FluoSTEP-ICUE (R279E) produced no change in emission ratio after Fsk/IBMX stimulation (ΔR/R = 0.52 ± 0.46%, n = 22 cells, P < 0.0001) (Fig. 2A).
Fig. 2. The FluoSTEP design can be generalized to probe multiple targets.
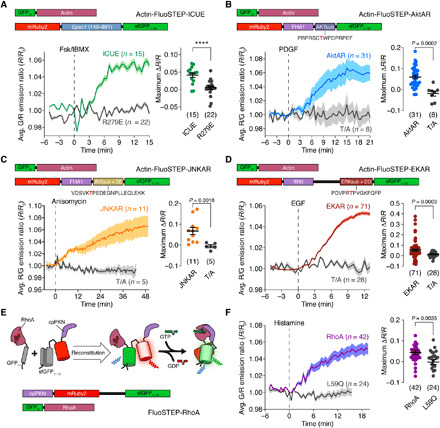
(A) Top: Domain structure of actin-targeted FluoSTEP-ICUE. Bottom: HEK293T cells expressing GFP11-actin and FluoSTEP-ICUE (green; n = 15 cells) or FluoSTEP-ICUE (R279E) (gray; n = 22 cells) stimulated with 50 μM Fsk and 100 μM IBMX (Fsk/IBMX). Left: Average green/red (G/R) emission ratio time courses and (right) maximum ratio changes. (B) Top: Domain structure of actin-targeted FluoSTEP-AktAR. Bottom: NIH3T3 cells expressing GFP11-actin and FluoSTEP-AktAR (blue; n = 31 cells) or FluoSTEP-AktAR (T/A) (gray; n = 8 cells) stimulated with PDGF (50 ng/ml). Left: Average red/green (R/G) emission ratio time courses and (right) maximum ratio changes. (C) Top: Domain structure of actin-targeted FluoSTEP-JNKAR. Bottom: HeLa cells expressing GFP11-actin and FluoSTEP-JNKAR (orange; n = 11 cells) or FluoSTEP-JNKAR (T/A) (gray; n = 5 cells) stimulated with 5 μM anisomycin. Left: Average R/G emission ratio time courses and (right) maximum ratio changes. (D) Top: Domain structure of actin-targeted FluoSTEP-EKAR. Bottom: HEK293T cells expressing GFP11-actin and FluoSTEP-EKAR (red; n = 71 cells) or FluoSTEP-EKAR (T/A) (gray; n = 28 cells) stimulated with epidermal growth factor (EGF; 100 ng/ml). Left: Average R/G emission ratio time courses and (right) maximum ratio changes. (E) Binding of GFP11-RhoA to cpPKN-mRuby2-GFP1–10 reconstitutes FluoSTEP-RhoA. (F) (left) Average G/R emission ratio time courses and (right) maximum ratio change in HEK293T cells expressing p63-P2A-mTagBFP2 and FluoSTEP-RhoA (purple curve; n = 42 cells) or FluoSTEP-RhoA (L59Q) (gray curve; n = 24 cells) stimulated with 100 μM histamine. ****P < 0.0001, unpaired two-tailed Student’s t test without (A, B, and F) or with (C and D) Welch’s correction. Solid lines in time courses indicate average responses; shaded areas denote SEM. Horizontal lines in scatter plots indicate means ± SEM.
In addition to PKA, many kinases are organized in macromolecular complexes and subcellularly targeted to tune signaling kinetics and target specificity (27). For example, compartmentalization of Akt and C-jun N-terminal kinase (JNK), two kinases important within cellular survival and stress pathways, has been uncovered using FRET-based biosensors (28–30). By simply swapping out the PKA substrate domain for the substrate sequences corresponding to Akt and JNK, we created FluoSTEP versions of the previously published Akt activity reporter (AktAR) (28) and JNK activity reporter (JNKAR) (29), respectively. Upon activation of Akt via platelet-derived growth factor (PDGF; 50 ng/ml) in NIH3T3 fibroblasts expressing GFP11-actin, FluoSTEP-AktAR produced a 6.0 ± 0.90% increase in the red/green emission ratio (ΔR/R, t1/2 = 5.3 ± 0.1 min, n = 31 cells; FRET efficiency change from 10 ± 4% to 16 ± 3%, n = 11 cells), while the nonphosphorylatable T/A mutant was nonresponsive (ΔR/R = −1.9 ± 1.2%, n = 8 cells, P = 0.0002) (Fig. 2B and table S1). Similarly, upon stimulation of JNK activity with anisomycin (5 μM), HeLa cells expressing GFP11-actin and FluoSTEP-JNKAR showed a 6.8 ± 1.7% increase in the red/green emission ratio (ΔR/R, t/12 = 23 ± 1.8 min, n = 11 cells; FRET efficiency change from 12 ± 5% to 15 ± 4%, n = 10 cells), in contrast to cells expressing GFP11-actin and the nonphosphorylatable T/A mutant (ΔR/R = −0.54 ± 0.73%, n = 5 cells, P = 0.0018) (Fig. 2C and table S1). Other FRET-based kinase sensors use different substrate and PAABD pairs for the conformational switch. A FluoSTEP-EKAR (Erk kinase activity reporter) for Erk, a kinase essential in cell growth and differentiation, was also created by using an Erk substrate/docking domain sequence, phospho-amino acid–binding WW domain, and extended linker (EV) (31). In HEK293T cells coexpressing GFP11-actin and stimulated with epidermal growth factor (EGF; 100 ng/ml) to activate Erk, FluoSTEP-EKAR produced a 5.6 ± 0.99% increase in the red/green emission ratio (ΔR/R, t1/2 = 6.7 ± 0.18 min, n = 71 cells; FRET efficiency change from 10 ± 3% to 13 ± 2%, n = 18 cells) (Fig. 2D and table S1). As a negative control, the nonphosphorylatable FluoSTEP-EKAR (T/A) mutant showed no response (ΔR/R = 1.5 ± 0.49%, n = 28 cells, P = 0.0003).
Enzyme activation biosensors often incorporate the targets of interest within their design architecture (32), which can cause side effects from overexpression (33). Thus, we hypothesized that FluoSTEPs could help untangle this dependence by uncoupling the expression of the target of interest from the rest of the sensor. As a prototype, we used the DORA RhoA sensor that measures the activation of RhoA, a small GTPase important in cytoskeletal regulation, by transducing a binding event between guanosine 5′-triphosphate (GTP)–bound RhoA and a circularly permuted RhoA interactor domain from Protein kinase C-related kinase 1 (cpPKN) into an increase in FRET (6). We created a FluoSTEP version of this RhoA activation sensor by splitting the sensor into two parts: one part containing the cpPKN interactor domain and the two FPs (split sfGFP and mRuby2), with an EV linker separating the FPs, and the other containing full-length RhoA tagged with GFP11 at its N terminus, similar to the organization of DORA RhoA (Fig. 2E). Expression of both parts reconstitutes the donor GFP to engage in FRET (Fig. 2E). The FluoSTEP architecture for this sensor reversed the activity-induced FRET change; therefore, we plotted the FluoSTEP-RhoA response as the increase in green/red emission ratio. Upon stimulation of RhoA activity via activation of the Gαq-coupled histamine-1 receptor through histamine (100 μM) addition in HeLa cells expressing Gαq-activatable p63 (6, 34), FluoSTEP-RhoA exhibited a 4.4 ± 0.95% increase in the green/red emission ratio (ΔR/R, t1/2 = 6.1 ± 0.29 min, n = 42 cells; FRET efficiency change from 14 ± 4% to 5 ± 2%, n = 5 cells) (Fig. 2F and table S1). In comparison, a FluoSTEP-RhoA (L59Q) negative control in which the cpPKN interactor domain was mutated to prevent binding to RhoA-GTP showed no response to histamine stimulation (ΔR/R = −0.20 ± 1.2%, n = 24 cells, P = 0.0035) (Fig. 2F). The design of FluoSTEP-RhoA could potentially be adapted for endogenous tagging of RhoA with GFP11 and thus offer a strategy for monitoring RhoA activation at the endogenous level.
Variants of FluoSTEP to increase dynamic range and brightness
Having established the FluoSTEP design as a general approach for probing localized signaling activities, we next explored various strategies to improve biosensor performance, using FluoSTEP-AKAR as a template. We first exchanged the split sfGFP donor for a brighter, split version of mNeonGreen with an orthogonal FP11 tag attached to clathrin (35) and tested the variant in HEK293T cells; however, we observed a smaller dynamic range (mNeonGreen: 4.1 ± 0.30%, n = 9 cells; sfGFP: 11 ± 1.4%, n = 6 cells; P < 0.0001) (fig. S2A). We also replaced the mRuby2 acceptor in FluoSTEP-AKAR with brighter RFPs such as mRuby3 (36) or mScarlet-I (37) and tested these variants with GFP11-actin. While the mRuby3-containing construct failed to produce a discernable response (ΔR/R = 2.2 ± 0.98%, n = 7 cells) (fig. S2A), mScarlet-I did yield a sensor with an approximately twofold increased dynamic range versus the mRuby2 version (ΔR/R = 19 ± 0.38%, n = 27 cells, P < 0.0001) (Fig. 3A and fig. S2A). However, we observed that the mScarlet-I sensor exhibited a strong, diffuse green signal even in the absence of GFP donor reconstitution (fig. S2, B and C), possibly due to direct excitation of the bright mScarlet-I FP or incomplete fluorophore maturation (37), which could be mitigated by applying calibration or choosing alternative imaging conditions.
Fig. 3. Variants of FluoSTEP to improve dynamic range.

(A) Top: Domain structures of GFP11-actin and FluoSTEP-AKAR containing either mRuby2 (Ruby) or mScarlet-I (Scarlet) as the FRET acceptor. Average R/G emission ratio time courses (left) and maximum emission ratio changes (right) in HEK293T cells coexpressing GFP11-actin and either FluoSTEP-AKAR “Ruby” (red; n = 14 cells) or FluoSTEP-AKAR “Scarlet” (dark red; n = 27 cells) upon Fsk/IBMX stimulation. Solid lines in time courses indicate average responses; shaded areas denote SEM. Horizontal lines in scatter plots indicate means ± SEM. (B) Tagging a POI with multiple tandem copies of GFP11 to recruit multiple copies of GFP1–10. (C) Comparison of normalized GFP intensity in HeLa cells coexpressing FluoSTEP-AKAR (left) or FluoSTEP-ICUE (right) and either GFP11(×1)-actin (teal) or GFP11(×7)-actin (green). AKAR: n = 25 cells each; ICUE: n = 10 (×1) and n = 17 (×7). Solid and dashed lines indicate the median and quartiles, respectively. Raw fluorescence intensity values were normalized to the median intensity of the GFP11(×1) group. (D) Top: Domain structures of GFP11(×n)-actin and FluoSTEP-AKAR. Average R/G emission ratio time courses (left) and maximum emission ratio changes (right) in HeLa cells coexpressing FluoSTEP-AKAR and either GFP11(×1)-actin (teal; n = 13 cells) or GFP11(×7)-actin (green; n = 5 cells) after stimulation with 50 μM Fsk and 100 μM IBMX (Fsk/IBMX). (E) Top: Domain structures of GFP11(×n)-actin and FluoSTEP-ICUE. Average G/R emission ratio time courses (left) and maximum emission ratio changes (right) in HEK293T cells coexpressing FluoSTEP-ICUE and either GFP11(×1)-actin (teal; n = 10 cells) or GFP11(×7)-actin (green; n = 17 cells) upon Fsk/IBMX stimulation. ****P < 0.0001, unpaired two-tailed Student’s t test (A, D, and E) or Mann-Whitney U test (C).
We then tested whether different FluoSTEP color variants can be developed for multiplexed applications. The donor GFP1–10 was exchanged for two previously described sfGFP color variants (38): yellow fluorescent protein (YFP1–10) and cyan fluorescent protein (CFP1–10). We created a yellow-red version of FluoSTEP-AKAR by using YFP1–10 as the donor and mRuby2 as the acceptor and a cyan-yellow version of FluoSTEP-AKAR by using CFP1–10 as the donor and cpVenus from AKAR4 as the acceptor. In HEK293T cells coexpressing GFP11-actin and stimulated with Fsk/IBMX, cyan-yellow FluoSTEP-AKAR yielded a robust response (ΔR/R = 9.1 ± 1.2%, n = 17 cells), although the reconstituted CFP exhibited dim fluorescence in cells, while the response from the yellow-red FluoSTEP-AKAR variant was weaker (ΔR/R = 3.2 ± 0.51%, n = 26 cells) (fig. S2A).
Fluorescence intensity is particularly important for targeted biosensing at endogenous proteins expressed at low levels; thus, we set out to increase the brightness of our sensors. The split GFP system offers a unique approach for boosting the fluorescent signal by fusing multiple copies of the small GFP11 tag in tandem (38). We hypothesized that a similar strategy could be used to recruit multiple FluoSTEP copies and thus amplify the fluorescence intensity (Fig. 3B) (24). We found that coexpressing actin fused to a seven-copy array of GFP11 [GFP11(×7)-actin] along with FluoSTEP-AKAR in HeLa cells yielded an over fivefold increase in the brightness of the GFP channel versus GFP11(×1)-actin (P < 0.0001, n = 25 cells) (Fig. 3C), without affecting the Fsk/IBMX-induced response [GFP11(×1)-actin: ΔR/R = 8.3 ± 0.70%, n = 13 cells; GFP11(×7)-actin: ΔR/R = 9.4 ± 0.90%, n = 5 cells, P = 0.4217; Fig. 3D). The same strategy was applied to FluoSTEP-ICUE. By using an array of GFP11 tags, we achieved a 3.6-fold enhancement in the brightness of the GFP channel (P < 0.0001, n = 17) (Fig. 3C). We also observed a 1.9-fold enhancement in the dynamic range of FluoSTEP-ICUE when coexpressed with the GFP11 array [GFP11(×1)-actin: ΔR/R = 6.1 ± 0.54%, n = 10 cells; GFP11(×7)-actin: ΔR/R = 10 ± 0.64%, n = 17 cells; P < 0.0001] (Fig. 3E). Together, these data indicate that recruiting an array of biosensors to a POI can be used to enhance the brightness of FluoSTEPs without degrading probe sensitivity.
Endogenous signaling compartments are accessible by FluoSTEPs
To test our compartment-specific FluoSTEPs in an endogenous context, we sought to knock-in GFP11 at a specific genomic locus in HEK293T cells. Because of the small size of GFP11, knock-in via CRISPR and homology-directed repair (HDR) with a single-stranded oligonucleotide donor is efficient and versatile and can theoretically be extended to multiple genomic loci of interest (38, 39). We previously used FluoSTEP-AKAR and FluoSTEP-ICUE to measure the PKA activities and cAMP levels in endogenous type I-alpha regulatory subunit of PKA (RIα) phase-separated bodies (13), highlighting the utility of FluoSTEPs. To showcase the versatility of FluoSTEPs at endogenous POIs, we chose to measure cAMP and PKA dynamics around clathrin, which has a role in regulating cAMP/PKA signaling, as this scaffold protein is important in the early steps of endocytosis of receptors such as GPCRs. During endocytosis, clathrin-coated pits form at the PM (40), which appear visually as discrete puncta (41). After stabilization and maturation, these pits can bud off from the PM and traffic to early endosomes, with clathrin promptly dissociating from the vesicle and recycling back to the cytoplasm for subsequent endocytosis events (42, 43). When we expressed either FluoSTEP-AKAR or FluoSTEP-ICUE in clathrin light chain A (CLTA)–FP11 cells (38), a HEK293T cell line in which the GFP11 tag is knocked-in to the CLTA gene, the GFP donor was reconstituted and distinct clathrin-containing microdomains were observed, indicating correct probe localization (Fig. 4, A and E). Localization to endogenously expressed clathrin did not negatively affect FluoSTEP-AKAR dynamic range compared to GPF11-clathrin overexpression, as Fsk/IBMX treatment reliably induced a rapid 16 ± 0.91% increase in the red/green emission ratio (ΔR/R, n = 33 cells), which completely returned to baseline levels upon subsequent addition of H89 (Fig. 4, B and D). Similarly, treating FluoSTEP-ICUE–expressing CLTA-FP11 cells with Fsk/IBMX successfully triggered a rapid 12 ± 1.1% increase in the green/red emission ratio (ΔR/R, n = 19 cells), demonstrating the utility of FluoSTEPs to monitor cAMP levels at clathrin microdomains (Fig. 4, C and D, and fig. S3A). Together, these results confirm the utility of FluoSTEPs as a platform for monitoring compartmentalized signaling dynamics near specific target proteins expressed at endogenous levels from their native loci.
Fig. 4. FluoSTEPs deployed at endogenously expressed clathrin.
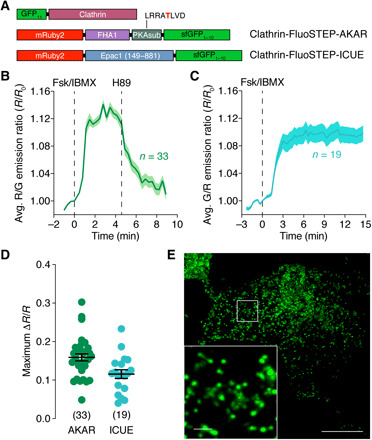
(A) Domain structures of clathrin-targeted FluoSTEP-AKAR and FluoSTEP-ICUE. (B and C) Average time course of (B) the R/G emission ratio from FluoSTEP-AKAR (n = 33 cells) or (C) the G/R emission ratio from FluoSTEP-ICUE (n = 19 cells) expressed in CLTA-FP11 cells and stimulated either with (B) 50 μM Fsk and 100 μM IBMX (Fsk/IBMX) followed by 10 μM H89 or (C) Fsk/IBMX alone. Solid lines in time courses indicate average responses; shaded areas denote SEM. (D) Summary of the maximum emission ratio changes from FluoSTEP-AKAR and FluoSTEP-ICUE in CLTA-FP11 cells following Fsk/IBMX stimulation. Horizontal lines indicate means ± SEM. (E) Representative GFP fluorescence image depicting the localization of FluoSTEP-AKAR in CLTA-FP11 cells, which are HEK293T cells in which the GFP11 tag is stably expressed at the clathrin N terminus via CRISPR-mediated knock-in at the endogenous CLTA gene locus. Punctate clathrin structures are seen in zoomed-in inset. Scale bars, 10 μm (inset, 1 μm).
Transmembrane ACs regulate sustained cAMP production at long-lived clathrin microdomains following β-AR stimulation
After activation, GPCRs at the PM undergo desensitization, endocytosis and trafficking to endosomes, and eventual recycling back to the PM (40). Evidence suggests that this process of GPCR endocytosis governs prolonged cAMP signaling from endosomes (44). Clathrin plays a central role in the spatiotemporal regulation of cAMP signaling by promoting GPCR internalization and trafficking (45). However, little is known about the receptor-mediated signaling within clathrin microdomains.
To probe clathrin-specific cAMP dynamics after GPCR stimulation, we expressed FluoSTEP-ICUE in CLTA-FP11 cells and stimulated them with the β-AR agonist isoproterenol (10 μM), followed by Fsk/IBMX to maximally induce cAMP production. Tagging with FluoSTEP-ICUE had little effect on endogenous clathrin dynamics, as assessed via total internal reflection fluorescence (TIRF) imaging of CLTA-FP11 cells transfected with either GFP1–10 or FluoSTEP-ICUE (movie S1; fig. S3, B and C). While the median puncta lifetime observed via TIRF imaging was <1 min (fig. S3, B and C), the isoproterenol-induced response from clathrin-targeted FluoSTEP-ICUE was largely sustained over 20 min [sustained activity metric (46) 20 min after stimulation (SAM20) = 0.90 ± 0.019, n = 55 cells; see Materials and Methods] (Fig. 5A). Clathrin microdomains are known to exhibit heterogeneous dynamics and can be roughly divided among smaller structures that are only transiently present (1 to 2 min) at the membrane surface and other longer-lived structures known as clathrin plaques (47, 48). Our analysis of the cAMP dynamics within these different types of clathrin structures suggests that the sustained cAMP increases occur primarily in the longer-lived clathrin structures (fig. S3, D and E).
Fig. 5. Transmembrane ACs regulate sustained cAMP production at clathrin after β-AR stimulation.
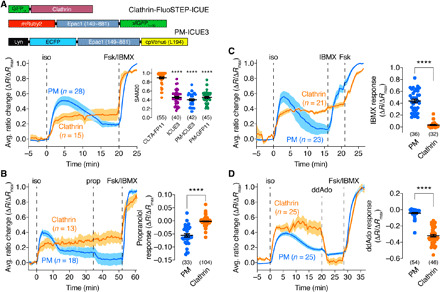
(A) Top: GFP11-clathrin (from CLTA-FP11 cells), FluoSTEP-ICUE, and PM-targeted ICUE3 (PM-ICUE3). Bottom: CLTA-FP11 cells expressing FluoSTEP-ICUE (clathrin) or PM-ICUE3 (PM) were stimulated with 10 μM isoproterenol (iso), 50 μM Fsk, and 100 μM IBMX (Fsk/IBMX). Representative average time courses (left) showing G/R (clathrin) or cyan/yellow (PM) emission ratio change (ΔR/ΔRmax; see Materials Methods). Right: Sustained activity metric at 20 min (SAM20) following iso stimulation in CLTA-FP11 cells expressing FluoSTEP-ICUE (CLTA-FP11), diffusible ICUE3 (ICUE3), or PM-ICUE3 or in HEK293T cells expressing Lyn-FRB-GFP11/FluoSTEP-ICUE (PM-GFP11). ****P < 0.0001 versus CLTA-FP11; Kruskal-Wallis test followed by Dunn’s multiple comparisons test. (B) CLTA-FP11 cells expressing FluoSTEP-ICUE or PM-ICUE3 were stimulated with 10 μM iso, 10 μM propranolol (prop), and Fsk/IBMX. Left: Representative average time courses showing G/R (clathrin) or cyan/yellow (PM) emission ratio changes (ΔR/ΔRmax). Right: Summary of prop-induced ratio change (ΔR/ΔRmax) for FluoSTEP-ICUE (clathrin) and PM-ICUE3 (PM). (C) CLTA-FP11 cells expressing FluoSTEP-ICUE or PM-ICUE3 were stimulated with 10 μM iso, 100 μM IBMX, and 50 μM Fsk. Left: Representative average time courses showing G/R (clathrin) or cyan/yellow (PM) emission ratio changes (ΔR/ΔRmax). Right: Summary of IBMX-induced ratio change (ΔR/ΔRmax) for FluoSTEP-ICUE (clathrin) and PM-ICUE3 (PM). (D) CLTA-FP11 cells expressing FluoSTEP ICUE or PM-ICUE3 were stimulated with 10 μM iso, 100 μM ddAdo, and Fsk/IBMX. Left: Representative average time courses showing G/R (clathrin) or cyan/yellow (PM) emission ratio changes (ΔR/ΔRmax). Right: Summary of ddAdo-induced ratio change (ΔR/ΔRmax) for FluoSTEP-ICUE (clathrin) and PM-ICUE3 (PM). ****P < 0.0001, unpaired two-tailed Student’s t test with Welch’s correction [in (B) and (D)]. Solid lines in time courses indicate average responses; shaded areas denote SEM. Horizontal lines in scatter plots indicate means ± SEM. Values shown in parentheses indicate number of cells.
To compare the dynamics of clathrin microdomain-specific cAMP signaling with those of bulk cAMP signals, we transfected CLTA-FP11 cells with either untargeted or PM-targeted ICUE3. Both ICUE3 and PM-ICUE3 reported transient cAMP accumulation upon isoproterenol stimulation, exhibiting sharp increases in the normalized ratio that gradually decayed to a submaximum steady state (ICUE3: SAM20 = 0.46 ± 0.026, n = 40 cells; PM-ICUE3: SAM20 = 0.40 ± 0.022, n = 42 cells) (Fig. 5A and fig. S4A), consistent with previous reports of cAMP clearance due to desensitization and internalization during canonical β-AR signaling (23). Reconstituting FluoSTEP-ICUE at the PM via Lyn-FRB-GFP11 similarly revealed a transient response upon isoproterenol treatment (SAM20 = 0.46 ± 0.21, n = 45 cells) (fig. S4B), indicating that the sustained cAMP responses observed with clathrin-targeted FluoSTEP-ICUE were not an artifact of the complementation-based targeting strategy.
We next set out to investigate the molecular mechanisms responsible for driving the unique compartmentalized cAMP dynamics that we observed within these endogenous clathrin microdomains. We hypothesized that the presence of sustained cAMP signaling might be driven by continuous signaling by active β-ARs (49). To test this, we treated isoproterenol-stimulated CLTA-FP11 cells expressing either FluoSTEP-ICUE or PM-ICUE3 with the β-AR antagonist 2-propranolol (10 μM). Whereas propranolol treatment yielded an acute decrease in the normalized ratio of PM-ICUE3 (ΔR/ΔRmax = −5.6 ± 0.64%, n = 33 cells) to almost basal levels, propranolol addition had no effect on the isoproterenol-induced response of clathrin-targeted FluoSTEP-ICUE (ΔR/ΔRmax = −0.18 ± 0.14%, n = 104 cells, P < 0.0001), indicating that this sustained cAMP response does not require active receptors (Fig. 5B).
Local variations in cAMP accumulation within the cell can be controlled by the spatial organization of ACs and PDEs, which synthesize and degrade cAMP, respectively (22), and the compartmentalized cAMP dynamics observed using clathrin-targeted FluoSTEP-ICUE may be due to the differential distribution of these enzymes. For instance, AC3 and AC9 have been shown to undergo internalization after GPCR stimulation and traffic to endosomes containing the receptors (44, 50). Thus, to test the role of these enzymes in regulating the sustained cAMP accumulation detected by clathrin-targeted FluoSTEP-ICUE, we treated isoproterenol-stimulated CLTA-FP11 cells expressing FluoSTEP-ICUE or PM-ICUE3 with IBMX (100 μM) to acutely inhibit PDE activity or 2′,3′-dideoxyadenosine (ddAdo; 100 μM) to acutely inhibit transmembrane AC activity. Whereas pan-PDE inhibition induced a large increase in the normalized ratio of PM-ICUE (ΔR/ΔRmax = 44 ± 3.0%, n = 36 cells), the response from clathrin-targeted FluoSTEP-ICUE was largely unaffected (ΔR/ΔRmax = 3.1 ± 0.89%, n = 32 cells, P < 0.0001) (Fig. 5C). A similar trend was seen upon selective inhibition of PDE4 (fig. S5). On the other hand, transmembrane AC inhibition using ddAdo induced a sharp reversal of the normalized ratio of clathrin-targeted FluoSTEP-ICUE back to baseline levels (ΔR/ΔRmax = −32 ± 1.5%, n = 46 cells) but had a minimal effect on the PM-ICUE3 response (ΔR/ΔRmax = −4.2 ± 0.80%, n = 54 cells, P < 0.0001) (Fig. 5D). Together, these data suggest that the compartmentalized cAMP dynamics associated with long-lived clathrin microdomains result from a combination of low PDE activity and high transmembrane AC activity leading to sustained cAMP accumulation following β-AR stimulation.
DISCUSSION
Here, we present FluoSTEPs, an adaptable biosensor framework for monitoring compartmentalized signaling at endogenous cellular locations, and deploy them to study a specific cAMP signaling microdomain. The complementation and functional reconstitution of the biosensors at endogenously tagged proteins rely on a logic gate and thus confer domain specificity. Efficient knock-in of GFP11 at a specific genomic locus via CRISPR-mediated HDR (38) precludes overexpression of the POI while also bypassing the need for knocking in an entire FRET-based sensor. In addition, the ratiometric readout further strengthens the utility of this biosensor collection by allowing quantitative comparisons of localized signaling.
Despite the demonstrated advantages of this platform, potential obstacles such as low-sensor dynamic range and the dependence on the level of endogenous protein expression must be considered. While we have demonstrated the ability to enhance FP reconstitution at an endogenous POI and amplify biosensor dynamic range using an array of GFP11 tags, future development and engineering of FluoSTEPs will include linker optimization and additional FP screening. For example, mScarlet-I incorporation into FluoSTEP-AKAR increased the dynamic range. Certain applications, where detecting subtle changes in signaling activities is required, might benefit from the enhanced dynamic range of the mScarlet-I–based sensor variant. While the cyan-yellow FluoSTEP-AKAR sensor (fig. S2A) produced comparable responses to green-red FluoSTEP-AKAR, the dim fluorescence of the reconstituted CFP currently limits the utility of this sensor. Future studies will focus on generating brighter split CFP variants. Furthermore, by extending the FluoSTEP toolkit to use orthogonal, multicolored FP variants, several sensors may be multiplexed to simultaneously report differential compartmentalized signaling in the same cell.
GPCR endocytosis starts a cascade of intricate signaling events such as activating a second wave of cAMP signaling (51) and is implicated in pathological processes such as opioid addiction (52). While the kinetics of bulk cAMP accumulation during GPCR endocytosis have been characterized (53), the dynamics of cAMP accumulation within the specific microdomains important in this process are elusive. Here, by deploying FluoSTEP-ICUE to probe cAMP dynamics at an endogenous signaling compartment, we were able to detect sustained cAMP accumulation within clathrin microdomains following β-AR activation. We show that this sustained cAMP accumulation primarily arises from a subset of long-lived clathrin structures known as plaques, a recently found class of clathrin structures that persist in the membrane for longer periods than clathrin-coated pits (47, 49). Initially, clathrin plaques were thought to be biologically inert structures (48, 54); however, recent work has shed light on their functional importance (55–57). Clathrin plaques can recruit various GPCRs such as the β2-AR (49) and also undergo typical scission events (47, 49, 58). Furthermore, these longer-lasting clathrin structures are implicated in various cellular processes such as mechanotransduction (57, 59) and sarcomere organization and function (55, 56). Our results shed more light onto the signaling role of long-lived clathrin structures such as plaques by suggesting that they play an important role in regulating local cAMP dynamics. The sustained cAMP elevations within clathrin plaques are likely achieved by the exclusion or inhibition of PDEs and by the recruitment of ACs with the endocytosed receptor, consistent with previous reports of Gαs and AC cointernalization with GPCRs (44, 50, 60). Notably, our studies were performed in HEK293T cells, and the unique cAMP dynamics we observed within these clathrin structures may differ in other cell types depending on the expression of specific ACs isoforms and other cAMP effectors in this microdomain (61, 62). Compared with canonical PM GPCR signaling, other sources of GPCR signaling, including from endosomes, produce distinct signaling profiles and dictate different cellular processes, such as cAMP-dependent transcription (63–65). Future studies of the cAMP signaling originating within the clathrin microdomains versus the general PM (Fig. 5 and figs. S3 and S4) may thus reveal unique downstream signaling effects and cellular functions regulated by these closely juxtaposed membrane compartments. We envision that FluoSTEPs can be further used to probe many specific GPCR compartments and habitats, thus adding to the existing toolkit (66, 67) to enable a better understanding of the intricate spatiotemporal organization of GPCR signaling.
In summary, FluoSTEPs provide a strategy for observing microdomain-specific signaling at endogenous protein expression levels. Using FluoSTEPs to elucidate a unique cAMP compartment associated with clathrin microdomains uncovers new aspects of cAMP regulation during GPCR internalization and highlights the utility of FluoSTEPs to advance our understanding of the spatiotemporal regulation of biochemical networks in endogenous biological contexts.
MATERIALS AND METHODS
Biosensor construction
All assembly of constructs was performed using Gibson Assembly (NEB 2× High Fidelity Master Mix). To construct FluoSTEP-AKAR, mRuby2 was polymerase chain reaction (PCR)–amplified from pcDNA3 AKAR-CR (gift of M. Lin, Stanford University, Palo Alto, CA) using the primers (lowercase is Gibson overlap region, and uppercase is priming region) 5′-cccaagctggctagcgtttaaacttaagcttggATGGTGTCTAAGGGCGAAGAGCTGATC-3′ and 5′-gatctgttcttgagaaaacttatgcatgcgCTTGTACAGCTCGTCCATCCCACC-3′ and the FHA1 and PKA substrate from AKAR4 (16) using primers 5′- ggtgggatggacgagctgtacaagCGCATGCATAAGTTTTCTCAAGAACAGATC-3′ and 5′- tcctttggacatagatctgttaacgaattcGAGCTCGCTGCCGCCGGTGCCGCCGTCC-3′. The resulting PCR fragments were Gibson-assembled into Hind III– and Eco RI–digested pcDNA3.1 GFP1–10. GFP1–10-FHA1-PKAsub-mRuby2 (version 1 of FluoSTEP-AKAR in Fig. 1B) was constructed similarly by Gibson Assembly using primers 5′-agcaaagatccaaatgaaaaaCGCATGCATAAGTTTTCTCAAGAACAGATCGGCGAAAAC-3′ and 5′-ccagtgtgatggatatctgcaGAATTCTTACTTGTACAGCTCGTCCATCCCACC-3′ to amplify FHA1-PKAsub-mRuby2 from AKAR-CR and primers 5′-gttttcgccgatctgttcttgagaaaacttatgcatgcgTTTTTCATTTGGATCTTTGCT-3′ and 5′-GAATTCTGCAGATATCCATCACACTGGCGG-3′ to amplify the GFP1–10 pcDNA3.1 backbone. FluoSTEP-AKAR (T/A) was constructed by Gibson Assembly of PCR products amplified from FluoSTEP-AKAR using primers 5′-CTGCGTCGCGCCGCCCTGGTTGAC-3′ and 5′-GTCAACCAGGGCGGCGCGACGCAG-3′. FluoSTEP-ICUE was constructed similarly, except that the Epac1 (149 to 881) fragment from ICUE3 (26) was PCR-amplified using primers 5′-ggtggtgggatggacgagctgtacaagGAGGAGAAGAAGGAGTGTGATGAAGAA-3′ and 5′-ggtaaacagttcttctcctttggacatCTCAACGTCCCTCAAAATCCGATTGAA-3′. FluoSTEP ICUE R279E was constructed by Gibson Assembly of PCR products amplified from FluoSTEP-ICUE using primers 5′-gatgcaccccggGCAGCCACCATCATCCTG-3′ and 5′-ggtggctgcccgGGGTGCATCATTCACCAGAG-3′. FluoSTEP-Epac2B (285 to 443) was constructed via Gibson Assembly of the Epac2B (285 to 443) fragment PCR-amplified from the Epac2camps biosensor (gift from M. Lohse) using the forward primer 5′-ggtggtgggatggacgagctgtacaagGAGGAGAAGAAGGAGTGTGATGAAGAA-3′ and the reverse primer 5′-ggtaaacagttcttctcctttggacatCTCAACGTCCCTCAAAATCCGATTGAA-3′. FluoSTEP-AktAR was constructed similarly to FluoSTEP-AKAR, except that the FHA1 and Akt substrate domains were amplified from pcDNA3 AktAR (28) using the forward primer 5′-ggatggacgagctgtacaagCGCATGCATAAGTTTTCTCAA-3′ and the reverse primer 5′-agttcttctcctttggacatAAGTTCACTGCCGCCGGTACCTC-3′. FluoSTEP-AktAR (T/A) was made by Gibson Assembly of a PCR product amplified from FluoSTEP-AktAR using the forward primer 5′-gtgcgcatggcctgatCCCAGGCCGGAGTTTGG-3′ and the reverse primer 5′-tgggatcaggccatgcGCACGAGCGCGGACGA-3′. FluoSTEP-JNKAR was constructed similarly to FluoSTEP-AKAR, except that the FHA1 and JNK substrate domains were amplified from pcDNA3-JNKAR1 (29) using the forward primer 5′-ggatggacgagctgtacaagCGCATGCATAAGTTTTCTCAA-3′ and the reverse primer 5′-agttcttctcctttggacataagttctgaacctcctgtacctccCTTCTTCTCGAGCTGCTC-3′. FluoSTEP-JNKAR (T/A) was made by Gibson Assembly of a PCR fragment amplified from FluoSTEP-JNKAR using the forward primer 5′-agtgtcaaggcCCCCGAGGATGAAGGCAAC-3′, and the reverse primer 5′-tcctcgggggcCTTGACACTGTCGACcaggc-3′. FluoSTEP-EKAR was constructed by Gibson Assembly of an Sph I/Sac I–digested fragment of pcDNA3-Rab-EKARev (68) encoding the WW domain, EV linker, and Erk substrate sequence with a PCR fragment amplified from FluoSTEP-AKAR using the forward primer 5′-tcccccgcgcacgggagctcATGTCCAAAGGAGAAGAACTGTTTACCGGTGTT-3′ and the reverse primer 5′-tcgtccgccatgtgcatgcgCTTGTACAGCTCGTCCATCCCACCACC-3′. FluoSTEP-EKAR (T/A) was constructed similarly to FluoSTEP-EKAR, except that an Sph I/Sac I–digested fragment from pcDNA3-Rab-EKARev (T/A) (68) was used. GFP11-RhoA was constructed by Gibson Assembly of the PCR-amplified fragment of DORA RhoA (6) to add GFP11 onto the N terminus of RhoA using the forward primers 5′-AGACCCAAGCTGGCTAGCGTTTAAACTTAAGCTTGGGCCACCATGCGTGACCACAT-3′, 5′-CACCATGCGTGACCACATGGTCCTTCATGAGTATGTAAATGCTGCTGGGATTA-3′, 5′-GAGTATGTAAATGCTGCTGG GATTACAGGTGGAACAGGAGGTTCA-3′, and 5′-AGGTGGAACAGGAGGTTCAATGGCTGCCATCCGGAAGA-3′ and the reverse primer 5′-ggactagtggatccgagctcggtaTCACAAGACAAGGCAACCAG-3′ into pcDNA3.1 backbone (Invitrogen). cpPKN-mRuby2-EV-GFP1–10 was constructed by Gibson Assembly of cpPKN PCR-amplified from DORA RhoA (6) using the forward primer 5′-agacccaagctggctagcgtttaaacttaagcttggatgAGCCTGGGCCCCGTAG-3′ and the reverse primer 5’-tcttcgcccttagacaccattgaacctcctgttccaccGCGGCCCAGGTCAGT-3′; mRuby2 PCR-amplified from FluoSTEP-AKAR using the forward primer 5′-ATGGTGTCTAAGGGCGAAGA-3′ and the reverse primer 5′-gcactggttcctccggagccCTTGTACAGCTCGTCCATCC-3′; and a Kpn2 I/Kpn I–digested fragment of FluoSTEP EKAR encoding the EV linker and GFP1–10 PCR-amplified from FluoSTEP-AKAR using the forward primer 5′-gtggtagtgctggtggtaccATGTCCAAAGGAGAAGAACTGTTT-3′ and the reverse primer 5′-gtttaaacgggccctctagaCTATTTTTCATTTGGATCTTTGCTC-3′. p63-P2A-mTagBFP2 was constructed by Gibson Assembly of p63 PCR-amplified from p63-mCherry (6) using the forward primer 5′-AGACCCAAGCTGGCTAGCGTTTAAACTTAAGCTTGGGCCACCatgcggggggggcacaaag-′3 and 5′-CCAACAGGTGGATCTCGTGCTCGTGGcagctcatcttcatccagcttg-3′ and mTagBFP2 PCR-amplified from mTagBFP2-pBAD [gift from M. Davidson (Addgene plasmid #54572)] using the forward primers 5′-GCTGCCACGAGCACGAGATCCACCTGTTGGATCAGGTGCTACTAACTTCAGCCTGTTAAAGC-3′, 5′-GCTACTAACTTCAGCCTGTTAAAGCAAGCTGGAGATGTAGAGGAGAACCCTGGACCT-3′, and 5′-AGAACCCTGGACCTGGGTCCatgagcgagctgattaaggag-3′ and the reverse primer 5′-TAAACGGGCCCTCTAGACTAttaattaagcttgtgccccagt-3′.
FluoSTEP-AKAR color variants were made similarly by PCR amplification of the FP to swap in and the remainder of the FluoSTEP-AKAR minus FP to swap out. ICUE3 and PM-ICUE3 were described previously (26). Lyn-FRB-FP11 was generated via Gibson Assembly of an Nhe I/Bam HI–digested fragment encoding the N-terminal targeting sequence from PM-ICUE3 (26), a PCR fragment encoding FRB amplified from AKAP95-FRB (3) using the forward primer 5′-aagcgcaaggacaaggatccATCCTCTGGCATGAGATGTG-3′ and the reverse primer 5′-ACTAGTCTTTGAGATTCGTC-3′, and a PCR fragment encoding GFP11 along with the pcDNA3.1 backbone amplified from FluoSTEP-AKAR using the forward primer 5′-aactggggcacaagcttaatGGTGGAACAGGAGGTTCACG-3′ and the reverse primer 5′-ACGCTAGCCAGCTTGGGTCT-3′.
Cell culture and transfection
HeLa and HEK293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco) containing glucose (1 g/liter) and supplemented with 10% (v/v) fetal bovine serum (Sigma-Aldrich) and 1% (v/v) penicillin-streptomycin (Pen-Strep; Sigma-Aldrich). NIH3T3 cells were cultured in DMEM (Gibco) containing glucose (1 g/liter) and supplemented with 10% (v/v) fetal calf serum and 1% (v/v) Pen-Strep (Sigma-Aldrich). All cells were maintained in a humidified incubator at 37°C with a 5% CO2 atmosphere. Before transfection, cells were plated onto sterile 35-mm glass-bottomed dishes and grown to 50 to 70% confluence. Cells were then transfected using Lipofectamine 2000 (Invitrogen) or PolyJet (SignaGen Laboratories) and grown an additional 24 hours (HeLa, HEK293T) before imaging. NIH3T3 cells were changed to serum-free DMEM immediately before transfection and serum-starved for 24 hours before imaging.
Generation of stable GFP11 cell lines via CRISPR-mediated knock-in
For knock-in experiments, 200 ng of Cas9 + single guide RNA vector (designed with px330) and 400 ng of an oligonucleotide donor DNA were transfected to HEK293FT cells (50% confluence) per well in a 24-well plate (Eppendorf). For CLTA, transient transfection of GFP1–10 and fluorescence-activated cell sorting enrichment for GFP+ cells were performed, followed by a negative sort 2 weeks later to select against stable incorporation of GFP1–10. Genomic DNA was extracted from cells and sequenced to confirm knock-in.
Time-lapse fluorescence imaging
Cells were washed twice with Hank’s balanced salt solution (HBSS; Gibco) and subsequently imaged in HBSS in the dark at 37°C. Fsk (Calbiochem), IBMX (Sigma-Aldrich), PDGF (Sigma-Aldrich), anisomycin (Sigma-Aldrich), EGF (Sigma-Aldrich), histamine (Sigma-Aldrich), isoproterenol (Sigma), 2-propranolol (Sigma-Aldrich), and ddAdo (Cayman Chemical) were added as indicated. Epifluorescence imaging was performed on a Zeiss Axiovert 200 M microscope (Carl Zeiss) equipped with a xenon lamp, a 40×/1.3 numerical aperture objective and an Orca-FLASH 4.0LT digital CMOS camera (Hammamatsu) and controlled by METAFLUOR 7.7 software (Molecular Devices). The following excitation/emission filter combinations (center/bandwidth in nanometer) were used: CFP, EX420/20 and EM475/25; GFP, EX480/30 and EM535/45; YFP, EX495/10 and EM535/25; RFP, EX568/55 and EM653/95; C-Y FRET, EX420/20 and EM535/25; G-R FRET, EX480/30 and EM653/95. All filter sets were alternated using a Lambda 10-2 filter-changer (Sutter Instruments). Exposure times were 50 ms (for acceptor direct channel) and 500 ms (for all other channels), and images were acquired every 30 s. All epifluorescence experiments were subsequently analyzed using METAFLUOR 7.7 software.
Raw fluorescence images were corrected by subtracting the background fluorescence intensity of a cell-free region from the emission intensities of biosensor-expressing cells at each time point. Emission ratios were then calculated at each time point. Biosensor response time courses shown in Figs. 1 to 4 and fig. S1 were subsequently plotted as the normalized emission ratio with respect to time zero (e.g., R/R0), where R is the ratio value at a given time point, and R0 is the initial ratio value at the time point immediately preceding drug addition or the average emission ratio (−5 to 0 min) before drug addition. Biosensor responses shown in Fig. 5 and figs. S3D and S4 were plotted as the normalized-to-max emission ratio change (ΔR/ΔRmax), calculated as (R − R0) / (Rmax − R0), where R and R0 are defined as above, and Rmax is the maximum ratio value recorded after Fsk/IBMX stimulation. Maximum ratio (ΔR/R) changes shown in Figs. 1 to 4 and figs. S1 and S2 were calculated as (Rmax − Rmin) / Rmin, where Rmax and Rmin are the maximum and minimum ratio value recorded after stimulation, respectively. Sustained activity levels in Fig. 5A and fig. S3D were assessed using the SAM20 or at 15 min (SAM15), calculated as (Rt − R0) / (Rmax,t − R0), where Rt is the ratio value recorded either 20 min after stimulation for SAM20 or 15 min after stimulation for SAM15, Rmax,t is the maximum ratio value recorded within either the 20-min window for SAM20 or 15-min window for SAM15, and R0 is the ratio value at t = 0. Graphs were plotted using GraphPad Prism 8 (GraphPad Software).
Acceptor photobleaching to measure FRET efficiencies
Epifluorescence time course imaging of cells expressing GFP11-actin and the different FluoSTEPs were performed using the GFP/RFP FRET, GFP, and RFP channels and the 0.3 neutral density (ND) filter. At the end of the experiment, acceptor photobleaching was performed by illuminating RFP without any ND filters for 10 min. After confirming that most of the RFP is photobleached, images were taken with the 0.3 ND filter. FRET efficiency was calculated as 1 − (GFPbefore / GFPafter), where GFPbefore and GFPafter are the GFP intensities before and after acceptor photobleaching, respectively. For calculating the FRET efficiency before drug stimulation, the GFP intensity right before drug stimulation was used as GFPbefore; for calculating the FRET efficiency after drug stimulation, the GFP intensity right before acceptor photobleaching was used as GFPbefore.
TIRF imaging and analysis
CLTA-FP11 cells were plated onto glass-bottom 35-mm dishes coated with poly-d-lysine (100 μg/ml; Sigma-Aldrich). Cells were transfected 24 hours after plating and then incubated an additional 24 hours after transfection. Cells were imaged using a 488-nm laser on a Nikon A1R TIRF microscope using a 100× objective at a 1840 TIRF angle with 20% laser power, gain multiplier of 300, and 2-s interval time. TIRF image for single-particle tracking of the clathrin-coated structures was analyzed via Fiji plugin TrackMate (69). Single clathrin-coated structures in each frame were detected using LoGdetector with 3 pixels of estimated blob diameter. Detected clathrin-coated structures were linked to get the trajectory by linear assignment problem (LAP) tracker, with the gap closing after arbitrary thresholding of dot detection for each sample. The lifetimes of single clathrin-coated structures were estimated by the durations of their trajectories.
Statistics and reproducibility
Statistical analyses were performed in GraphPad Prism 8 (GraphPad). All data were assessed for normality. For normally distributed data, pairwise comparisons were performed using Student’s t tests or Welch’s unequal variance test as indicated, and comparisons among three or more groups were performed using ordinary one-way analysis of variance (ANOVA) followed by Dunnett’s test for multiple comparisons. Non-Gaussian data were analyzed using the Mann-Whitney U test for pairwise comparisons or the Kruskal-Wallis test followed by Dunn’s multiple comparisons test for analyses of three or more groups. Statistical significance was set at P < 0.05. Unless stated otherwise, average time courses and summary bar graphs shown in Figs. 1 to 5 (bar graphs only) and figs. S1 (C and D), S2, and S3 (A, B, and D) (bar graphs only) are pooled from at least three independent experiments, and average time courses shown in Fig. 5 (time courses only) and figs. S1 (A and B), S3D (time courses only), S4, and S5 are representative of at least three independent experiments.
Acknowledgments
The authors are grateful to W. Lin for help in TIRF imaging. Funding: This work was supported by R01 DK073368 and R35 CA197622 (to J.Z.); R21 EB022798, R01 GM124334, and R01 GM131641 (to B.H.); and a National Science Foundation predoctoral fellowship DGE-1650112 (to J.Z.Z.). Author contributions: S.M. conceived of FluoSTEPs. B.T. developed FluoSTEPs. B.T., J.Z.Z., S.M., and J.Z. designed experiments. V.P., S.F., and B.H. provided crucial reagents and cell lines. B.T., J.Z.Z., and Y.K. performed experiments and analyzed the data. J.Z. supervised the project. S.M. generated final figures. B.T., J.Z.Z., S.M., and J.Z. wrote the manuscript. Competing financial interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabe4091/DC1
REFERENCES AND NOTES
- 1.Greenwald E. C., Mehta S., Zhang J., Genetically encoded fluorescent biosensors illuminate the spatiotemporal regulation of signaling networks. Chem. Rev. 118, 11707–11794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Surdo N. C., Berrera M., Koschinski A., Brescia M., MacHado M. R., Carr C., Wright P., Gorelik J., Morotti S., Grandi E., Bers D. M., Pantano S., Zaccolo M., FRET biosensor uncovers cAMP nano-domains at b-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 8, 15031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clister T., Greenwald E. C., Baillie G. S., Zhang J., AKAP95 organizes a nuclear microdomain to control local cAMP for regulating nuclear PKA. Cell Chem. Biol. 26, 885–891.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohse C., Bock A., Maiellaro I., Hannawacker A., Schad L. R., Lohse M. J., Bauer W. R., Experimental and mathematical analysis of cAMP nanodomains. PLOS ONE 12, e0174856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tenner B., Mehta S., Zhang J., Optical sensors to gain mechanistic insights into signaling assemblies. Curr. Opin. Struct. Biol. 41, 203–210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Unen J., Reinhard N. R., Yin T., Wu Y. I., Postma M., Gadella T. W. J., Goedhart J., Plasma membrane restricted RhoGEF activity is sufficient for RhoA-mediated actin polymerization. Sci. Rep. 5, 14693 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamai T., Yamanishi T., Shirataki H., Takagi K., Asami H., Ito Y., Yoshida K.-I., Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin. Cancer Res. 10, 4799–4805 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Liu X., Chen D., Liu G., Overexpression of RhoA promotes the proliferation and migration of cervical cancer cells. Biosci. Biotechnol. Biochem. 78, 1895–1901 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Irannejad R., Tomshine J. C., Tomshine J. R., Chevalier M., Mahoney J. P., Steyaert J., Rasmussen S. G. F., Sunahara R. K., El-Samad H., Huang B., Von Zastrow M., Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez-Alvarez A., Fearey B. C., O’Toole R. J., Yang W., Arganda-Carreras I., Lamothe-Molina P. J., Moeyaert B., Mohr M. A., Panzera L. C., Schulze C., Schreiter E. R., Wiegert J. S., Gee C. E., Hoppa M. B., Oertner T. G., Freeze-frame imaging of synaptic activity using SynTagMA. Nat. Commun. 11, 2464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menzel S., Schwarz N., Haag F., Koch-Nolte F., Nanobody-based biologics for modulating purinergic signaling in inflammation and immunity. Front. Pharmacol. 9, 266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jank L., Pinto-Espinoza C., Duan Y., Koch-Nolte F., Magnus T., Rissiek B., Current approaches and future perspectives for nanobodies in stroke diagnostic and therapy. Antibodies 8, 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J. Z., Lu T.-W., Stolerman L. M., Tenner B., Yang J., Zhang J.-F., Falcke M., Rangamani P., Taylor S. S., Mehta S., Zhang J., Phase separation of a PKA regulatory subunit controls cAMP compartmentation and oncogenic signaling. Cell 182, 1531–1544.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabantous S., Terwilliger T. C., Waldo G. S., Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23, 102–107 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Pédelacq J.-D., Cabantous S., Tran T., Terwilliger T. C., Waldo G. S., Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Depry C., Allen M. D., Zhang J., Visualization of PKA activity in plasma membrane microdomains. Mol. Biosyst. 7, 52–58 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Lam A. J., St-Pierre F., Gong Y., Marshall J. D., Cranfill P. J., Baird M. A., McKeown M. R., Wiedenmann J., Davidson M. W., Schnitzer M. J., Tsien R. Y., Lin M. Z., Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 9, 1005–1012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritz R. D., Letzelter M., Reimann A., Martin K., Fusco L., Ritsma L., Ponsioen B., Fluri E., Schulte-Merker S., Van Rheenen J., Pertz O., A versatile toolkit to produce sensitive FRET biosensors to visualize signaling in time and space. Sci. Signal. 6, rs12 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Baird G. S., Zacharias D. A., Tsien R. Y., Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 11241–11246 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.J. R. Lakowicz, Principles of fluorescence spectroscopy (Second edition. New York : Kluwer Academic/Plenum, [1999] ©1999; https://search.library.wisc.edu/catalog/999882542202121).
- 21.Zhang J., Allen M. D., FRET-based biosensors for protein kinases: Illuminating the kinome. Mol. Biosyst. 3, 759–765 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Calebiro D., Maiellaro I., cAMP signaling microdomains and their observation by optical methods. Front. Cell. Neurosci. 8, 350 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Violin J. D., DiPilato L. M., Yildirim N., Elston T. C., Zhang J., Lefkowitz R. J., β2-Adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J. Biol. Chem. 283, 2949–2961 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Klarenbeek J., Goedhart J., van Batenburg A., Groenewald D., Jalink K., Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLOS ONE 10, e0122513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolaev V. O., Bünemann M., Hein L., Hannawacker A., Lohse M. J., Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 279, 37215–37218 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Dipilato L. M., Zhang J., The role of membrane microdomains in shaping β2-adrenergic receptor-mediated cAMP dynamics. Mol. Biosyst. 5, 832–837 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Wang Y., Yang F., Fu Y., Huang X., Wang W., Jiang X., Gritsenko M. A., Zhao R., Monore M. E., Pertz O. C., Purvine S. O., Orton D. J., Jacobs J. M., Camp D. G. II, Smith R. D., Klemke R. L., Spatial phosphoprotein profiling reveals a compartmentalized extracellular signal-regulated kinase switch governing neurite growth and retraction. J. Biol. Chem. 286, 18190–18201 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao X., Zhang J., Spatiotemporal analysis of differential Akt regulation in plasma membrane microdomains. Mol. Biol. Cell 19, 4366–4373 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fosbrink M., Aye-Han N.-N., Cheong R., Levchenko A., Zhang J., Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc. Natl. Acad. Sci. U.S.A. 107, 5459–5464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X., Herbst-Robinson K. J., Zhang J., Visualizing dynamic activities of signaling enzymes using genetically encodable FRET-based biosensors from designs to applications. Methods Enzymol. 504, 317–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu N., Aoki K., Yamada M., Yukinaga H., Fujita Y., Kamioka Y., Matsuda M., Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 22, 4647–4656 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donnelly S. K., Miskolci V., Garrastegui A. M., Cox D., Hodgson L., Characterization of genetically encoded FRET biosensors for Rho-family GTPases. Methods Mol. Biol. 1821, 87–106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pertz O., Hodgson L., Klemke R. L., Hahn K. M., Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 440, 1069–1072 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Zhang J. Z., Nguyen A. H., Miyamoto S., Brown J. H., McCulloch A. D., Zhang J., Histamine-induced biphasic activation of RhoA allows for persistent RhoA signaling. PLOS Biol. 18, e3000866 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng S., Sekine S., Pessino V., Li H., Leonetti M. D., Huang B., Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun. 8, 370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bajar B. T., Wang E. S., Lam A. J., Kim B. B., Jacobs C. L., Howe E. S., Davidson M. W., Lin M. Z., Chu J., Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 6, 20889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bindels D. S., Haarbosch L., van Weeren L., Postma M., Wiese K. E., Mastop M., Aumonier S., Gotthard G., Royant A., Hink M. A., Gadella T. W. J. Jr., MScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53–56 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Kamiyama D., Sekine S., Barsi-Rhyne B., Hu J., Chen B., Gilbert L. A., Ishikawa H., Leonetti M. D., Marshall W. F., Weissman J. S., Huang B., Versatile protein tagging in cells with split fluorescent protein. Nat. Commun. 7, 11046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M., RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., Lefkowitz R. J., Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eichel K., Jullié D., Barsi-Rhyne B., Latorraca N. R., Masureel M., Sibarita J.-B., Dror R. O., von Zastrow M., Catalytic activation of β-arrestin by GPCRs. Nature 557, 381–386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mettlen M., Chen P.-H., Srinivasan S., Danuser G., Schmid S. L., Regulation of clathrin-mediated endocytosis. Annu. Rev. Biochem. 87, 871–896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirchhausen T., Owen D., Harrison S. C., Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 6, a016725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Calebiro D., Nikolaev V. O., Persani L., Lohse M. J., Signaling by internalized G-protein-coupled receptors. Trends Pharmacol. Sci. 31, 221–228 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Kaksonen M., Roux A., Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 19, 313–326 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Keyes J., Ganesan A., Molinar-Inglis O., Hamidzadeh A., Zhang J., Ling M., Trejo J., Levchenko A., Zhang J., Signaling diversity enabled by Rap1-regulated plasma membrane ERK with distinct temporal dynamics. eLife 9, e57410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grove J., Metcalf D. J., Knight A. E., Wavre-Shapton S. T., Sun T., Protonotarios E. D., Griffin L. D., Lippincott-Schwartz J., Marsh M., Flat clathrin lattices: Stable features of the plasma membrane. Mol. Biol. Cell 25, 3581–3594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saffarian S., Cocucci E., Kirchhausen T., Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLOS Biol. 7, e1000191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lampe M., Pierre F., Al-Sabah S., Krasel C., Merrifield C. J., Dual single-scission event analysis of constitutive transferrin receptor (TfR) endocytosis and ligand-triggered β2-adrenergic receptor (β2AR) or Mu-opioid receptor (MOR) endocytosis. Mol. Biol. Cell 25, 3070–3080 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lazar A. M., Irannejad R., Baldwin T. A., Sundaram A. B., Gutkind J. S., Inoue A., Dessauer C. W., Von Zastrow M., G protein-regulated endocytic trafficking of adenylyl cyclase type 9. eLife 9, e58039 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pavlos N. J., Friedman P. A., GPCR signaling and trafficking: The long and short of it. Trends Endocrinol. Metab. 28, 213–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koch T., Höllt V., Role of receptor internalization in opioid tolerance and dependence. Pharmacol. Ther. 117, 199–206 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Ferrandon S., Feinstein T. N., Castro M., Wang B., Bouley R., Potts J. T., Gardella T. J., Vilardaga J.-P., Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol. 5, 734–742 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Batchelder E. M., Yarar D., Differential requirements for clathrin-dependent endocytosis at sites of cell-substrate adhesion. Mol. Biol. Cell 21, 3070–3079 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franck A., Lainé J., Moulay G., Lemerle E., Trichet M., Gentil C., Benkhelifa-Ziyyat S., Lacène E., Bui M. T., Brochier G., Guicheney P., Romero N., Bitoun M., Vassilopoulos S., Clathrin plaques and associated actin anchor intermediate filaments in skeletal muscle. Mol. Biol. Cell 30, 579–590 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vassilopoulos S., Gentil C., Lainé J., Buclez P.-O., Franck A., Ferry A., Précigout G., Roth R., Heuser J. E., Brodsky F. M., Garcia L., Bonne G., Voit T., Piétri-Rouxel F., Bitoun M., Actin scaffolding by clathrin heavy chain is required for skeletal muscle sarcomere organization. J. Cell Biol. 205, 377–393 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baschieri F., Dayot S., Elkhatib N., Ly N., Capmany A., Schauer K., Betz T., Vignjevic D. M., Poincloux R., Montagnac G., Frustrated endocytosis controls contractility-independent mechanotransduction at clathrin-coated structures. Nat. Commun. 9, 3825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leyton-Puig D., Isogai T., Argenzio E., van den Broek B., Klarenbeek J., Janssen H., Jalink K., Innocenti M., Flat clathrin lattices are dynamic actin-controlled hubs for clathrin-mediated endocytosis and signalling of specific receptors. Nat. Commun. 8, 16068 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franck A., Lainé J., Moulay G., Trichet M., Gentil C., Fongy A., Bigot A., Benkhelifa-Ziyyat S., Lacène E., Thao Bui M., Brochier G., Guicheney P., Mouly V., Romero N., Coirault C., Bitoun M., Vassilopoulos S., Clathrin plaques form mechanotransducing platforms. bioRxiv, 321885 (2018). [Google Scholar]
- 60.Calebiro D., Nikolaev V. O., Gagliani M. C., de Filippis T., Dees C., Tacchetti C., Persani L., Lohse M. J., Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLOS Biol. 7, e1000172 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Merriam L. A., Baran C. N., Girard B. M., Hardwick J. C., May V., Parsons R. L., Pituitary adenylate cyclase 1 receptor internalization and endosomal signaling mediate the pituitary adenylate cyclase activating polypeptide-induced increase in guinea pig cardiac neuron excitability. J. Neurosci. 33, 4614–4622 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cai W., Du A., Feng K., Zhao X., Qian L., Ostrom R. S., Xu C., Adenylyl cyclase 6 activation negatively regulates TLR4 signaling through lipid raft-mediated endocytosis. J. Immunol. 191, 6093–6100 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Bowman S. L., Shiwarski D. J., Puthenveedu M. A., Distinct G protein-coupled receptor recycling pathways allow spatial control of downstream G protein signaling. J. Cell Biol. 214, 797–806 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eichel K., von Zastrow M., Subcellular organization of GPCR signaling. Trends Pharmacol. Sci. 39, 200–208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsvetanova N. G., von Zastrow M., Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol. 10, 1061–1065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ni Q., Mehta S., Zhang J., Live-cell imaging of cell signaling using genetically encoded fluorescent reporters. FEBS J. 285, 203–219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clister T., Mehta S., Zhang J., Single-cell analysis of G-protein signal transduction. J. Biol. Chem. 290, 6681–6688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mehta S., Zhang Y., Roth R. H., Zhang J.-f., Mo A., Tenner B., Huganir R. L., Zhang J., Single-fluorophore biosensors for sensitive and multiplexed detection of signalling activities. Nat. Cell Biol. 20, 1215–1225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tinevez J.-Y., Perry N., Schindelin J., Hoopes G. M., Reynolds G. D., Laplantine E., Bednarek S. Y., Shorte S. L., Eliceiri K. W., TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/21/eabe4091/DC1