

The expression of epigenetic factors is often dysregulated in cancers or upon specific stress signals, which often results in a display of noncanonical functions of the epigenetic factors that are independent from their chromatin-modifying roles. We have previously demonstrated that KDM2B normally inhibits KSHV lytic cycle using its histone demethylase activity.
KEYWORDS: AP-1, KDM2B, Kaposi's sarcoma-associated herpesvirus, SCF E3 ubiquitin ligase complex, de novo infection
ABSTRACT
It is still largely unknown what host factors are involved in controlling the expression of the lytic viral gene RTA during primary Kaposi's sarcoma-associated herpesvirus (KSHV) infection, which determines if KSHV establishes latent or lytic infection. We have recently identified the histone demethylase KDM2B as a repressor of RTA expression during both de novo KSHV infection and latency based on an epigenetic factor short interfering RNA screen. Here, we report that, surprisingly, KDM2B overexpression can promote lytic de novo infection by using a mechanism that differs from what is needed for its repressor function. Our study revealed that while the DNA-binding and demethylase activities of KDM2B linked to its transcription repressive function are dispensable, its C-terminal F-box and LRR domains are required for the lytic infection-inducing function of KDM2B. We found that overexpressed KDM2B increases the half-life of the AP-1 subunit c-Jun protein and induces the AP-1 signaling pathway. This effect depends on the binding of KDM2B to the SKP1-CUL1-F-box (SCF) E3 ubiquitin ligase complex via its F-box domain. Importantly, the inhibition of AP-1 reduces KDM2B-mediated lytic de novo KSHV infection. Overall, our findings indicate that KDM2B overexpression induces the degradation of some host factors by using the SCF complex, resulting in the enrichment of c-Jun. This leads to increased AP-1 transcriptional activity, which facilitates lytic gene expression following de novo infection interfering with the establishment of viral latency.
IMPORTANCE The expression of epigenetic factors is often dysregulated in cancers or upon specific stress signals, which often results in a display of noncanonical functions of the epigenetic factors that are independent from their chromatin-modifying roles. We have previously demonstrated that KDM2B normally inhibits KSHV lytic cycle using its histone demethylase activity. Surprisingly, we found that KDM2B overexpression can promote lytic de novo infection, which does not require its histone demethylase or DNA-binding functions. Instead, KDM2B uses the SKP1-CUL1-F-box (SCF) E3 ubiquitin ligase complex to induce AP-1 transcriptional activity, which promotes lytic gene expression. This is the first report that demonstrates a functional link between SFCKDM2B and AP-1 in the regulation of the KSHV lytic cycle.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) (human herpesvirus 8) is the causative agent of several cancers, such as Kaposi’s sarcoma, primary effusion lymphoma, and some forms of multicentric Castleman’s disease (1–3). Following primary infection, KSHV establishes a persistent, latent infection in B cells and lymphatic endothelial cells that is characterized by the continuous expression of latent genes while the rest of the viral genes and lytic replication are repressed (4). Like other herpesviruses, KSHV can also enter a lytic replication cycle, resulting in the production of new viral particles, which is essential for viral transmission (5). The viral protein required for initiating and driving the lytic cycle is the replication and transcription activator (RTA), which induces the temporally ordered viral gene expression cascade, such as immediate-early (IE), early (E), and late (L) gene expression (6–8). Consequently, for KSHV to establish latency, RTA expression has to be suppressed following de novo viral infection (9, 10). However, it is still largely unknown what host factors are involved in controlling RTA expression, which determines if the virus establishes latency or goes into the lytic cycle after de novo infection.
Recently, we have identified a number of new host epigenetic factors that can play a role in the establishment and maintenance of KSHV latency (11, 12). Of these host factors, we showed that the histone demethylase KDM2B rapidly binds to the KSHV DNA during de novo infection and reduces the level of specific activating histone marks on lytic genes such as RTA. This ultimately promotes the downregulation of lytic viral gene expression, which is required for the establishment of viral latency (11). KDM2B (also known as FBXL10, NDY1, and JHDM1B) is a Jumonji (JmjC) domain-containing histone demethylase, which also has several other functional domains. The N-terminal catalytic JmjC domain is followed by a CXXC DNA-binding domain, a plant homeodomain zinc finger domain, an F-box, and seven leucine-rich repeats (LRR) (13). The JmjC domain of KDM2B preferentially catalyzes the demethylation of the activating histone marks H3K4me3, H3K36me2, and H3K79me2, thereby repressing active transcription (14–16). KDM2B can also bind to unmethylated CpG islands by its CXXC domain and can recruit polycomb repressive complex 1 (PRC1) to specific cellular genes by interacting with PRC1 via its LRR domain (17–19). Importantly, KDM2B and its paralog KDM2A are the only two histone demethylases that contain an F-box and an LRR domain whereby they can potentially function as F-box protein subunits of the SKP1-CUL1-F-box (SCF) E3 ubiquitin ligase complex (20, 21).
The SCF complex is composed of SKP1, CUL1, ROC1, and one F-box protein. The F-box protein binds to both SKP1 and the substrate protein to be ubiquitinated, while SKP1 serves as an adapter protein, which links the F-box protein to CUL1. CUL1 acts as the scaffold protein in the SCF complex recruiting ROC1, which is the E3 ubiquitin ligase component of SCF (22). Currently, there is only one report, which showed the role of KDM2B in SCF-dependent degradation of a cellular protein, while in another case the demethylase activity of KDM2B was required for KDM2B-mediated protein degradation (23, 24). It is still largely unknown what proteins can be targeted by KDM2B for degradation and whether the SCFKDM2B complex plays any role in the regulation of viral infections. There are a number of studies that have shown the deregulation of KDM2B in various diseases or upon specific physiological or stress stimuli. Overexpression of KDM2B can contribute to the progression of cancers by promoting cell proliferation, metastasis, and drug resistance (25–27). Interestingly, hypoxia, which can induce lytic reactivation of KSHV from latently infected B cell lymphoma cells, also increases the expression of KDM2B (28, 29). Since KDM2B is a potent repressor of the KSHV lytic cycle, the dysregulation of KDM2B expression can likely affect the outcome of KSHV infection.
Multiple host signaling pathways also regulate the lytic cycle of KSHV. Mitogenic stimuli or cellular stresses can initiate RTA expression through mitogen-activated protein kinases (MAPK) and stress-activated protein kinases (SAPK), respectively (30–32). These kinases lead to the activation of AP-1, which is a dimeric transcription factor composed of the Jun, Fos, and ATF proteins (32). The AP-1 signaling pathway has been shown to function as a positive regulator of the KSHV lytic cycle (30, 32, 33), which can be activated during both primary infection and lytic reactivation (34–36). However, it is still poorly understood how specific host signaling pathways cooperate with the epigenetic machinery to control KSHV infection.
In this study, we have discovered that the increased expression of KDM2B facilitates lytic de novo infection and lytic reactivation from latency. We found that this function of KDM2B is independent of its DNA-binding and demethylase activities but requires its C terminus, including the F-box and LRR domains. Our results show that KDM2B interacts with the SCF E3 ubiquitin ligase complex through its F-box domain. This results in increased AP-1 transcriptional activity, which can promote viral lytic gene expression during de novo KSHV infection. Based on our data, we propose that KDM2B can function as a double-edged sword, which can both repress and promote lytic KSHV infection by using distinct functional domains in a context-dependent manner.
RESULTS
Dysregulation of KDM2B expression facilitates lytic de novo KSHV infection and lytic reactivation.
We have previously demonstrated that the short hairpin RNA (shRNA) knockdown of KDM2B upregulates lytic KSHV genes both during de novo infection and in latently infected cells, indicating that KDM2B functions as a suppressor of the KSHV lytic cycle (11). Interestingly, hypoxia, which is known to reactivate KSHV from latently infected cells, has been shown to increase the expression of KDM2B (28, 29). We found that hypoxia stimulated by CoCl2 not only reactivates KSHV from latency but also can promote lytic de novo KSHV infection of SLK cells, and we confirmed that hypoxia indeed upregulates KDM2B expression (Fig. 1A). Next, we tested how the overexpression of KDM2B compared to KDM2B depletion impacts viral gene expression during de novo KSHV infection. We transduced SLK cells with lenti-GFP and lenti-3×FLAG-KDM2B or lenti-shControl and lenti-shKDM2B, and 3 days later the cells were infected with KSHV for 8, 24, 48, and 72 h (Fig. 1B to E). The overexpression and depletion of KDM2B was confirmed by immunoblotting (Fig. 1B and D). Importantly, the protein level of overexpressed KDM2B relative to that of the endogenous KDM2B in lentivirus-transduced cells was in the range of what can be observed in hypoxia-stimulated cells (Fig. 1A and B). In line with our previous study, shKDM2B resulted in significant upregulation of RTA expression as early as 8 h postinfection (hpi), which was further increased over time (Fig. 1C) (11). KDM2B overexpression (OE) reduced RTA expression at 8 hpi, in agreement with KDM2B acting as a repressor of KSHV lytic gene expression (Fig. 1C). Surprisingly, however, we found that KDM2B-OE increased RTA expression at 48 hpi and 72 hpi (Fig. 1C). Further analyses revealed that the expression of latent (LANA), E (ORF42 and ORF57), and L (ORF25 and ORF64) viral genes were also significantly increased at 72 hpi in KDM2B-OE cells (Fig. 1E). Figure 1F shows that the KSHV DNA level was increased by several folds in KSHV-infected KDM2B-OE cells compared to GFP-OE cells at 72 hpi. We confirmed that KDM2B overexpression also increased lytic viral gene expression in a KSHV-infected endothelial EA.hy926 cell line at 72 hpi, indicating that KDM2B-induced lytic de novo KSHV infection is not cell type specific (Fig. 1G and H). In addition, KDM2B-OE also induced KSHV gene expression in the latently infected KSHV+ B cell lymphoma cell line BCBL1 after 5 days of lenti-3×FLAG-KDM2B transduction (Fig. 1I and J).
FIG 1.

Increased lytic gene expression following de novo KSHV infection upon dysregulation of KDM2B expression. (A) SLK cells were treated with CoCl2 for 24 h to induce hypoxia followed by KSHV infection in the presence of CoCl2 for 72 h. Immunoblot analysis of host (HIF-1α and KDM2B) and viral proteins are shown. HIF-1α induction is an intrinsic marker of hypoxia. The numbers below the KDM2B blots represent the quantification of KDM2B bands in hypoxia samples relative to the normoxia sample (0 μM CoCl2). (B) Immunoblot analysis of KDM2B expression using anti-FLAG and anti-KDM2B antibodies. (C) SLK cells were transduced with lenti-GFP and lenti-3×FLAG-KDM2B or lenti-shControl and lenti-shKDM2B for 3 days and then infected with KSHV for various time periods. RT-qPCR analysis of RTA expression relative to 18S is shown. (D) Immunoblot analysis of KDM2B expression in the shRNA-treated SLK cells. (E) Viral gene expression measured by RT-qPCR relative to 18S at 72 hpi. (F) Viral DNA level at 72 hpi was measured by qPCR, normalized to the host DNA level, and then calculated relative to 2 hpi in GFP-OE and KDM2-OE cells. (G) EA.hy926 cells were transduced with lenti-GFP or lenti-3×FLAG-KDM2B for 3 days and then infected with KSHV for 3 days. Immunoblot analysis of KDM2B expression using anti-FLAG antibody is shown. (H) Viral gene expression measured by RT-qPCR relative to 18S at 72 hpi. (I) BCBL1 cells were transduced with lenti-GFP or lenti-3×FLAG-KDM2B for 1, 3, or 5 days. 3×FLAG-KDM2B expression was tested by anti-FLAG immunoblotting. Tubulin was used as a loading control. (J) RT-qPCR analysis of viral mRNAs in KDM2B-OE cells relative to GFP-OE cells at 1, 3, and 5 days. t test was performed between GFP-OE and KDM2B-OE or shControl and shKDM2B, and P < 0.05 (*) was considered statistically significant. The numbers on the left side of immunoblots in this and later figures represent the molecular weight in kilodaltons (kDa).
Next, we investigated whether KDM2A, a paralog of KDM2B, affects KSHV de novo infection similarly to KDM2B. We treated SLK cells with KDM2A short interfering RNAs (siRNAs) or overexpressed 3×FLAG-KDM2A for 3 days and then infected them with KSHV for 3 days (Fig. 2). We found that the inhibition of KDM2A expression induced (Fig. 2A and B), while KDM2A-OE repressed lytic viral gene expression during de novo KSHV infection (Fig. 2C and D). These data showed that KDM2A acts as a classical transcription repressor of KSHV genes, in contrast to KDM2B. Thus, despite KDM2A and KDM2B being structurally similar and their overexpression being comparable (Fig. 2E), they function differently in KSHV gene regulation. Taken together, these results show that the expression level of KDM2B is critical for controlling KSHV lytic gene transcription. We hypothesize that KDM2B-OE uses a mechanism to induce lytic viral genes that differs from what is normally used by KDM2B in the epigenetic control of gene transcription.
FIG 2.
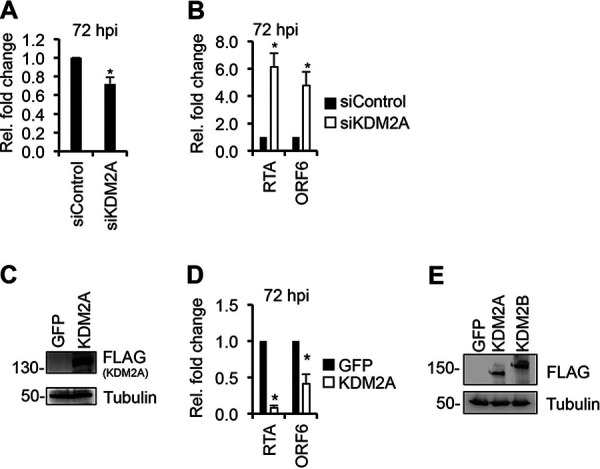
KDM2A functions as a transcriptional repressor of KSHV lytic cycle. (A and B) SLK cells were transfected with 100 nM siControl or siKDM2A for 3 days and then infected with KSHV for 3 days. (A) Fold change of KDM2A expression in siKDM2A cells relative to siControl cells at 72 hpi. (B) RT-qPCR testing of RTA and ORF6 gene expression relative to siControl at 72 hpi. (C and D) SLK cells were transduced with lenti-GFP or lenti-3×FLAG-KDM2A for 3 days and then infected with KSHV for 3 days. (C) Immunoblot analysis of 3×FLAG-KDM2A expression using anti-FLAG antibody. (D) RTA and ORF6 gene expression measured by RT-PCR in KDM2B-OE relative to that in GFP-OE at 72 hpi. t test was performed between siControl and siKDM2A and GFP-OE and KDM2A-OE (*, P < 0.05). (E) Comparison of the expression of 3×FLAG-KDM2A and 3×FLAG-KDM2B by FLAG immunoblot analysis.
KDM2B-induced lytic viral replication and viral gene expression during de novo infection depend on the expression of RTA.
To determine whether the KDM2B-induced viral DNA level increase during de novo KSHV infection is the result of increased latent or lytic viral DNA replication, KDM2B-OE SLK cells were infected with KSHV in the presence or the absence of PAA (phosphonoacetic acid). PAA is a potent viral DNA polymerase inhibitor that blocks lytic but not latent viral DNA replication as well as reduces viral DNA replication-dependent late gene expression. We found that PAA treatment abolished the increase of viral DNA level and reduced the expression of not only L genes (ORF25) but also IE (RTA) and E (ORF6 and ORF42) viral genes in KSHV-infected KDM2B-OE cells (Fig. 3A and B). We concluded that KDM2B-OE during de novo KSHV infection stimulates lytic viral DNA replication.
FIG 3.

RTA is required for KDM2B-induced lytic gene expression and viral DNA replication during de novo infection. (A and B) SLK cells were transduced with lenti-GFP or lenti-KDM2B for 3 days, followed by KSHV infection for 3 days in the presence or absence of 100 μM PAA. (A) Viral DNA load was measured by qPCR at 72 hpi relative to 2 hpi. (B) RT-qPCR analysis of viral gene expression at 72 hpi. (C to E) SLK cells transduced with lenti-GFP or lenti-3×FLAG-KDM2B for 3 days followed by infection with WT or RTA-KO KSHV for 3 days. (C) Viral DNA level at 72 hpi relative to 2 hpi was measured by qPCR. (D) RT-qPCR detection of viral gene expression in KDM2B-OE cells relative to GFP-OE cells at 72 hpi. (E) Immunoblot detection of lytic viral protein production in WT and RTA-KO KSHV-infected GFP-OE and KDM2B-OE cells at 72 hpi. Tubulin was used as a loading control. t test was performed between (−PAA) and (+PAA) samples (A and B) as well as between GFP-OE and KDM2B-OE samples (D), and P < 0.05 (*) was considered statistically significant. Asterisks at the immunoblots indicate nonspecific signal.
It is known that RTA expression is required for lytic replication and the induction of lytic genes, although some lytic genes can also be induced in an RTA-independent manner outside the canonical lytic cycle (37). Therefore, we tested whether KDM2B-induced lytic gene expression is RTA dependent or not. We infected GFP-OE and 3×FLAG-KDM2B-OE SLK cells with either wild-type (WT) or RTA knockout (RTA-KO) KSHV for 72 h and analyzed the viral DNA level and the expression of lytic viral genes. The results showed that viral DNA replication, lytic gene expression, and the production of lytic proteins were abolished in KDM2B-OE cells infected with RTA-KO KSHV (Fig. 3C to E), indicating that KDM2B-induced lytic viral DNA replication and lytic viral gene expression are dependent on RTA expression.
F-box and LRR domains of KDM2B are essential for KDM2B-induced lytic gene expression.
To investigate what domains of KDM2B are required for KDM2B-induced de novo lytic KSHV infection, we constructed a series of KDM2B mutants by introducing point mutations into the JmjC and CXXC domains or deleting the F-box or the LRR domain (Fig. 4A). SLK cells were first transduced with lenti-GFP or lentiviruses expressing the WT or different KDM2B mutants for 3 days, which was followed by KSHV infection for 2 and 72 h. Immunoblot analysis showed that the expression level of WT and KDM2B mutants was comparable, whereas viral DNA measurement by quantitative PCR (qPCR) at 2 hpi indicated that they did not affect KSHV infectivity (Fig. 4B and C). At 72 h KSHV postinfection, we analyzed how the mutations in KDM2B affected KDM2B-induced viral DNA replication and lytic viral gene expression. Intriguingly, we found that the deletion of the LRR (ΔLRR) or the F-box (ΔF-box) domain abolished KDM2B-induced lytic gene expression and lytic replication in de novo KSHV-infected cells (Fig. 4D to G). In contrast, the JmjC and CXXC domains were dispensable for KDM2B-induced lytic de novo infection. Strikingly, the CXXC mutant always resulted in greater lytic gene induction than the WT KDM2B. These results indicate that the F-box and the LRR domains are essential for KDM2B-driven lytic de novo KSHV infection while the histone demethylase (JmjC) and DNA binding (CXXC) activities of KDM2B are dispensable, supporting the notion that the upregulation of lytic viral genes is independent of the epigenetic and transcriptional role of KDM2B.
FIG 4.

Identifying the KDM2B domains required for KDM2B-mediated lytic gene expression. (A) Schematic representation of the domain structure of KDM2B. JmjC, Jumonji C histone demethylase domain; CXXC, DNA-binding domain; PHD, histone mark reader plant homeodomain; F-box, SKP1-binding domain; LRR, leucine-rich repeat. Point mutations in JmjCm and CXXCm are indicated. (B) FLAG immunoblot analysis of the expression of WT and mutant 3×FLAG-KDM2B in SLK cells transduced with lenti-3×FLAG-KDM2B. (C to G) SLK cells were transduced with lenti-GFP or lenti-3×FLAG-KDM2Bs for 3 days, followed by infection with KSHV for 3 days. (C) qPCR measurement of viral DNA at 2 hpi relative to GFP-OE cells and at 72 hpi relative to 2 hpi. (D) RT-qPCR analysis of viral gene expression in KDM2B-OE cells relative to GFP-OE cells at 72 hpi. (E) Immunoblot analysis of WT and mutant 3×FLAG-KDM2B expression in SLK cells. (F) qPCR analysis of viral DNA load at 72 hpi relative to 2 hpi. (G) RT-qPCR of viral mRNAs in KDM2B-OE cells relative to GFP-OE cells at 72 hpi. t tests were performed between GFP-OE and KDM2B-OE samples (C and D) and between WT and ΔF-box samples (F and G), and P < 0.05 (*) was considered statistically significant.
RING1B binding on the viral genome is reduced by KDM2B-OE, which depends on the C terminus of KDM2B and RTA expression.
It is well established that KDM2B can recruit RING1B, the catalytic subunit of Polycomb Repressive Complex 1 (PRC1), to unmethylated CpG islands in the mouse and human genomes, resulting in the inhibition of specific host genes (38, 39). It has also been shown that RING1B recruitment onto the KSHV genome is necessary for the downregulation of lytic genes following de novo infection so that KSHV can establish latency (10, 40). Thus, we tested how WT KDM2B or mutations in the functional domains of KDM2B affect the binding of RING1B on the viral genome in SLK cells during de novo KSHV infection. We found that while the overexpression of WT KDM2B or the CXXC domain mutant reduced RING1B binding on the viral genome, the ΔLRR mutant did not (Fig. 5). These results correlated with the WT and CXXC mutant being able to promote lytic de novo KSHV infection while the ΔLRR mutant could not (Fig. 4D).
FIG 5.

Overexpression of KDM2B reduces RING1B binding on the viral genome, which depends on the C terminus of KDM2B. SLK cells were first transduced with lenti-GFP or lenti-3×FLAG-KDM2B WT or mutants, which was followed by KSHV infection for 72 h. RING1B ChIP assay was performed to test the binding of RING1B to the indicated regions of the RTA promoter, RTA gene body +0.8 kb downstream of the transcription start site, and the promoter (pr) and the gene body (gb) of E (K2) and L (ORF25) genes. Cellular intergenic region Neg and the MYC gene promoter were used as controls. t tests were performed between GFP-OE and KDM2B-OE samples, and P < 0.05 (*) was considered statistically significant.
Next, we tested whether KDM2B directly reduces RING1B binding on the KSHV genome in KDM2B-OE cells during de novo infection or KDM2B-induced RTA expression is responsible for the reduced RING1B binding, given that RTA can antagonize the function of polycomb proteins on the viral genome (Fig. 6) (41). We infected GFP-OE and 3×FLAG-KDM2B-OE SLK cells with WT or RTA-KO KSHV for 72 h and analyzed the binding of RING1B on the RTA promoter. Immunoblot analysis showed that KDM2B expression in SLK cells infected with WT or RTA-KO KSHV was comparable (Fig. 6A). We confirmed that KDM2B overexpression enhances viral DNA replication only in WT but not in RTA-KO KSHV-infected cells (Fig. 6B). FLAG (KDM2B) chromatin immunoprecipitation (ChIP) analysis showed that KDM2B binding to the viral DNA was comparable in WT and RTA-KO KSHV-infected SLK cells (Fig. 6C). In contrast, RING1B ChIP analysis revealed that KDM2B-OE reduced RING1B binding on the promoter and the gene body of viral genes RTA, K2, and ORF25 in WT but not in RTA-KO KSHV-infected cells (Fig. 6D). We also confirmed that KDM2B overexpression did not change the expression of PRC1 factors (RING1B, RYBP, and BMI1), indicating that KDM2B-induced RING1B reduction on the viral genome in WT KSHV-infected cells is not due to decreased expression of PRC1 factors (Fig. 6E). Altogether, these results show that the reduced RING1B binding on the KSHV DNA in KDM2B-OE cells during de novo infection is due to KDM2B-OE-induced RTA expression and not because of a direct effect of KDM2B-OE on RING1B. These data are also in agreement with our previous study showing that KDM2B is not involved in PRC1 recruitment to the KSHV genome (11).
FIG 6.

RTA expression in KDM2B-OE cells is responsible for the reduced RING1B binding on the KSHV genome. SLK cells were transduced with lenti-GFP or lenti-3×FLAG-KDM2B for 3 days, followed by infection with WT or RTA-KO KSHV for 3 days. (A) Immunoblot analysis of 3×FLAG-KDM2B expression using anti-FLAG antibody. (B) qPCR measurement of the viral DNA level in KDM2B-OE cells relative to GFP-OE cells at 72 hpi. (C) FLAG ChIP for testing the binding of 3×FLAG-KDM2B on RTA promoter. (D) RING1B ChIP assay for analyzing RING1B-binding on the promoter (pr) and the gene body (gb) of lytic viral genes. (E) Immunoblot analysis of PRC1 factors at 72 hpi. t tests were performed between GFP-OE and KDM2B-OE samples, and P < 0.05 (*) was considered statistically significant; ns, nonsignificant.
KDM2B interacts with the E3 ubiquitin ligase SCF complex.
Since our results showed that the F-box and LRR domains of KDM2B are required for facilitating lytic de novo KSHV infection in KDM2B-OE cells, we tested the interaction of KDM2B with the endogenous components of the SCF complex (SKP1, CUL1, and ROC1). To this end, we transfected 293T cells with a plasmid expressing 3×FLAG-KDM2B and performed FLAG immunoprecipitation (IP). We could detect endogenous SKP1 but not CUL1 in the FLAG IP, indicating an interaction between endogenous SKP1 and the transfected 3×FLAG-KDM2B (Fig. 7B). We do not know why 3×FLAG-KDM2B does not pull down CUL1; however, our results are in line with previous studies, which showed SKP1, but not other SCF factors, in KDM2B complex purifications (17, 18). Next, we transfected 3×FLAG-KDM2B into 293T cells with epitope-tagged SCF components in different combinations, such as HA-ROC1 alone or HA-ROC1 and 3×Myc-CUL1 or HA-ROC1, 3×Myc-CUL1, and Myc-FLAG-SKP1. When we immunoprecipitated HA-ROC1, we could detect SKP1, CUL1, and KDM2B in the HA-ROC1 immunoprecipitation when all SCF components were coexpressed with 3×FLAG-KDM2B, indicating that KDM2B can interact with the entire SCF complex (Fig. 7C). To test if the F-box and/or LRR domain is required for the binding of KDM2B with SCF complex via SKP1, we transfected 293T cells with the WT or mutants of 3×FLAG-KDM2B (ΔF-box and ΔLRR) (Fig. 7D). Both FLAG and SKP1 immunoprecipitations showed that the deletion of the F-box or LRR domain of KDM2B completely disrupted its binding to SKP1 (Fig. 7E and F). These results indicate that the C terminus of KDM2B containing the F-box and LRR domains is essential for KDM2B to interact with the SCF complex.
FIG 7.

Interaction of KDM2B with the SCF complex. (A) Composition of the SCF E3 ubiquitin ligase complex: the substrate receptor F-box protein, the adaptor protein SKP1, the scaffold protein Cullin-1 (CUL1), and the RING finger E3 ubiquitin ligase ROC1. (B) FLAG IP using 3×FLAG-KDM2B-transfected HEK293T cells. KDM2B interacts with endogenous SKP1. (C) HEK293T cells were cotransfected with 3×FLAG-KDM2B along with different combinations of epitope-tagged CUL1, SKP1, and ROC1. HA-ROC1 IP was analyzed by immunoblotting. (D) Schematic representation of the full-length KDM2B (WT) and its mutants. (E) FLAG IP using HEK293T cells transfected with WT, the F-box mutant, or the LRR domain mutant of 3×FLAG-KDM2B. The IPs were analyzed by anti-FLAG antibody and for the presence of endogenous SKP1. (F) HEK293T cells were cotransfected with myc-FLAG-SKP1 and WT or the indicated mutants of 3×FLAG-KDM2B, followed by myc IP at 48 h posttransfection. KDM2B and SKP1 were detected by FLAG and myc antibodies, respectively.
The C terminus of KDM2B is sufficient for KDM2B-mediated KSHV lytic gene expression.
Because of the importance of KDM2B’s C terminus in SCF binding and in KDM2B-induced lytic KSHV infection, we tested if the KDM2B C terminus alone is sufficient for SCF binding and to increase KSHV lytic gene expression during KSHV infection. To test this, we generated 3×FLAG-tagged F-box–LRR and LRR domain expression constructs (Fig. 8A). Immunoprecipitation assays showed that the F-box–LRR fragment could, while LRR could not, interact with SKP1 (Fig. 8B). This indicates that the F-box domain is essential for KDM2B to interact with SKP1, which bridges KDM2B and the SCF complex as a whole, confirming our results shown in Fig. 7. In addition, we found that viral gene expression was increased in F-box–LRR-OE SLK cells similarly to WT KDM2B-OE cells during KSHV infection but not in LRR-OE cells (Fig. 8C and D). These results support our notion that KDM2B-mediated lytic gene expression of KSHV is mediated through the interaction of KDM2B with the E3 ubiquitin ligase SCF complex.
FIG 8.

Interaction of KDM2B with SKP1 is necessary for KDM2B-induced de novo lytic KSHV infection. (A) Schematic representation of the full-length KDM2B (WT) and its C-terminal fragments. (B) HEK293T cells were transfected with F-box–LRR and LRR domain expression constructs for 48 h followed by FLAG IPs, which were tested for the presence of endogenous SKP1. (C) SLK cells were transduced with lenti-GFP or lenti-3×FLAG-KDM2B (WT), 3×FLAG-F-box–LRR, or 3×FLAG-LRR for 3 days and then infected with KSHV for 3 days. At 72 hpi, viral gene expression was measured by RT-qPCR and calculated relative to viral gene expression in GFP-OE cells. (D) Immunoblot detection of viral proteins at 72 hpi. Tubulin was used as a loading control. t tests were performed between GFP-OE and KDM2B-OE samples, and P < 0.05 (*) was considered statistically significant.
KDM2B induces the transcriptional activity of AP-1 pathway through its F-box domain.
To gain insight into how KDM2B-OE induces lytic viral gene expression, we tested the effect of the WT and the DNA-binding mutant (CXXCm) of KDM2B on 10 different host signaling pathways, several of which have been shown to be critical in the regulation of the KSHV life cycle (Fig. 9A). We cotransfected 293T cells with WT or CXXCm 3×FLAG-KDM2B, along with luciferase reporter plasmids in which the promoters of luciferase gene were responsive to distinct signaling pathways, such as Wnt, p53, TGF-beta, c-Myc, Hypoxia, Hippo, AP-1, Notch, Oct4/Sox2, and NF-κB. We found that WT KDM2B induced Wnt, p53, Hippo, AP-1, Notch, and Oct4/Sox2 luciferase reporters by 2- to 4-fold. Strikingly, while the expression of WT and CXXCm KDM2B was comparable, CXXCm induced most of the luciferase reporter plasmids more strongly than WT KDM2B did (Fig. 9A and B). This observation correlated with CXXCm being more potent in the induction of lytic KSHV infection (Fig. 4). Based on these results, we speculated that KDM2B promotes lytic viral gene expression by inducing some of these signaling pathways in a way that does not require the DNA-binding domain of KDM2B linked to its transcriptional function.
FIG 9.

KDM2B induces AP-1 transcriptional activity through its F-box domain. (A) Identification of host signaling pathways that are regulated by KDM2B using luciferase reporter assay. HEK293T cells were cotransfected with 3×FLAG-KDM2B WT or CXXCm along with luciferase reporter plasmids whose promoters are responsive to the signaling pathways indicated on the graph. The fold change was calculated by comparing KDM2B-induced luciferase activity to the basal activity of the luciferase reporter plasmids alone. (B) Immunoblotting analysis of transfected WT and KDM2B mutants. Tubulin was used as a loading control. (C) KDM2B mutants were tested in the AP-1 luciferase reporter assay as described for panel A. (D) Expression of 3×FLAG-LRR reduced the KDM2B-induced AP-1 activity. HEK293T cells were cotransfected with AP-1 luciferase reporter plasmid and 3×FLAG-KDM2B WT along with an increasing amount of 3×FLAG-LRR plasmid (50 to 600 ng). The y axis shows luciferase light units. t tests were performed between samples with and without KDM2B or CXXCm (A) and between vector and KDM2B samples (C), and P < 0.05 (*) was considered statistically significant.
We noticed that while WT KDM2B only slightly (1.6-fold) induced the AP-1 luciferase reporter plasmid, CXXCm stimulated it much more strongly (6-fold), which correlated with CXXCm-OE being a stronger inducer of lytic de novo KSHV infection. Since AP-1 has been shown to facilitate the KSHV lytic cycle (30, 32, 33), we further assessed the effect of KDM2B-OE on AP-1-mediated transcription activation. To determine what domains of KDM2B are needed to promote the transcriptional activity of AP-1, we cotransfected the AP-1 luciferase reporter plasmid with vectors expressing the WT or KDM2B mutants (CXXCm, ΔF-box, ΔLRR, F-box–LRR, or LRR fragment) (Fig. 9B and C). Consistent with our earlier observation, CXXCm induced AP-1 activity more strongly than WT KDM2B did. In contrast, deletion of the F-box or LRR domain abrogated KDM2B-induced AP-1-mediated transcription activation. In addition, the expression of the F-box–LRR but not LRR domain significantly induced AP-1 transcriptional activity (Fig. 9C). These results suggest that KDM2B’s F-box domain, which is responsible for SCF interaction, is necessary for the KDM2B-mediated increase of AP-1 activity.
Based on our data, we hypothesized that SCFKDM2B degrades some suppressors of AP-1, which allows the upregulation of AP-1 activity. Importantly, the LRR domain of F-box proteins is involved in the targeting of proteins to SCF-mediated protein degradation. Thus, we tested if the overexpression of the LRR domain can function as a dominant-negative mutant by sequestering the target proteins from the SCFKDM2B complex and reverse KDM2B-mediated induction of AP-1 activity. Figure 9D shows that the increasing amount of LRR fragment gradually reduced KDM2B-induced AP-1 activity. This result supports the notion that, in addition to the F-box, the LRR domain is also crucial for KDM2B to induce AP-1 activity.
AP-1 is critical for KDM2B-induced KSHV lytic gene expression.
To determine whether KDM2B-induced KSHV lytic gene expression during de novo viral infection involves AP-1, we inhibited AP-1 activity in KDM2B-OE cells during KSHV infection using either an AP-1-specific inhibitor (SR11302) (Fig. 10) or AP-1-specific siRNAs (Fig. 11). Figure 10B shows that SR11302 significantly inhibited the expression of c-Fos, c-Jun, and IL-6, which are known to be regulated by AP-1, demonstrating that AP-1 inhibition worked. Importantly, viral lytic gene expression induced by KDM2B-OE or F-box–LRR-OE was also reduced by 2- to 3-fold when AP-1 was inhibited by SR11302 during KSHV infection (Fig. 10C to E). To further confirm the role of AP-1 in KDM2B-mediated lytic gene expression, the expression of the AP-1 subunit c-Fos or c-Jun was depleted by siRNAs in GFP-OE and KDM2B-OE cells during KSHV infection (Fig. 11A). At 72 hpi, we performed reverse transcription-qPCR (RT-qPCR) and immunoblotting, which showed that the expression of both c-Fos and c-Jun was reduced upon siRNA treatment (Fig. 11B and C). We confirmed that as a result of AP-1 inhibition, interleukin-6 (IL-6) expression was also reduced (Fig. 11B). Figure 11D shows that the siRNA knockdown of c-Fos or c-Jun significantly dampened KDM2B-mediated lytic gene expression compared to the siControl-treated cells (Fig. 11C and D). Collectively, these results indicate that KDM2B-OE-mediated KSHV lytic infection requires AP-1 transcription factor.
FIG 10.
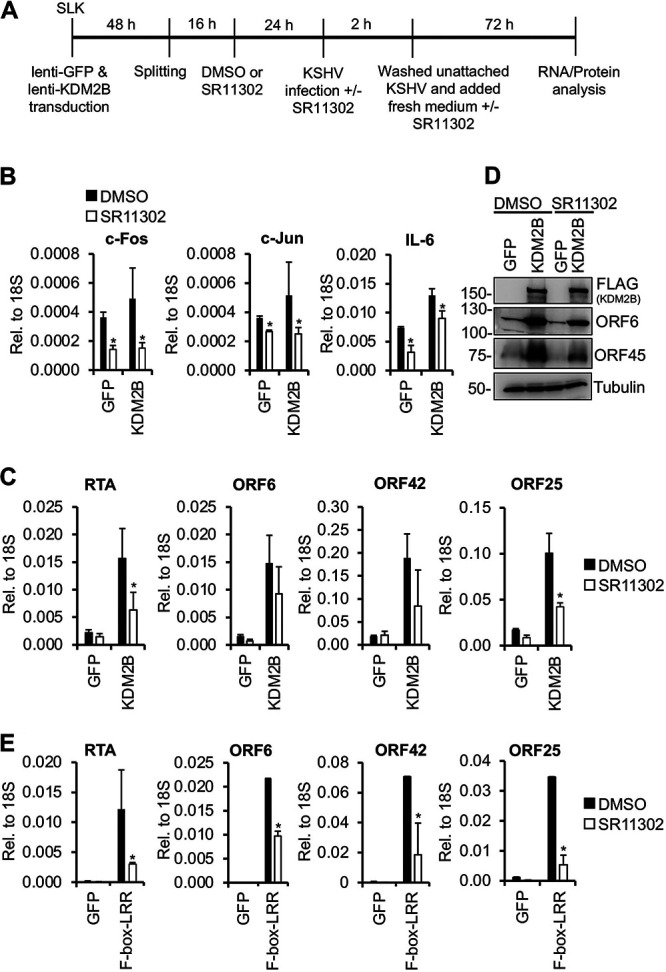
AP-1 inhibition reduces KDM2B-induced lytic viral gene expression. (A) Flowchart of the experiments. SLK cells were first transduced with lenti-GFP or lenti-3×FLAG-KDM2B for 48 h. Cells were split, and 16 h later the same number of cells was treated with DMSO or 50 μM SR11302. After 24 h, cells were infected with KSHV in the presence or absence of SR11302. Virus was removed at 2 hpi. At 72 hpi, cells were harvested for RNA and protein analysis. (B) The gene expression levels of c-Fos, c-Jun, and IL-6 were analyzed by RT-qPCR. (C) Viral gene expression was measured by RT-qPCR at 72 hpi. (D) Immunoblot analysis of viral ORF6 and ORF45 protein expression at 72 hpi. Tubulin was used as a loading control. (E) AP-1 inhibitor reduced lytic gene expression induced by 3×FLAG-F-box–LRR. The experiment was performed as described for panel B. The viral gene expression was analyzed by RT-qPCR at 72 hpi. t tests were performed between DMSO- and SR11302-treated cells, and P < 0.05 (*) was considered statistically significant.
FIG 11.

siRNA knockdown of AP-1 subunits reduces KDM2B-induced lytic KSHV gene expression. (A) Flowchart of the experiments similar to Fig. 10, except that SLK cells were transfected with 50 nM siRNAs prior to KSHV infection. (B) RT-qPCR analysis of gene expression in siRNA-treated cells. (C) Immunoblotting for c-Fos, c-Jun, and viral ORF6 protein at 72 hpi. Tubulin was used as a loading control. (D) Viral gene expression was measured by RT-qPCR at 72 hpi. t tests were performed between siRNA c-Fos or c-Jun and siControl; *, P < 0.05 was considered statistically significant.
KDM2B stabilizes c-Jun protein, which requires the F-box domain of KDM2B.
We noticed that KDM2B-OE in KSHV-infected cells was accompanied by increased expression of c-Jun (Fig. 11B and C). Since KSHV primary infection and the constitutively expressed KSHV factor LANA can induce c-Jun expression (34, 42), we could not tell if KDM2B-OE was responsible for the increased level of c-Jun in KSHV-infected cells. Hence, we tested the effect of KDM2B on c-Jun expression in KSHV-free SLK cells by expressing 3×FLAG-KDM2B (Fig. 12A to C). As a positive control, we used TPA treatment, which can induce c-Jun expression. We found that while TPA indeed induced c-Jun expression both at the mRNA and protein levels, KDM2B could also slightly increase c-Jun but not c-Fos expression (Fig. 12A to C).
FIG 12.

KDM2B prolongs the half-life of c-Jun protein, which requires KDM2B’s F-box domain. (A to C) Effect of KDM2B-OE on the mRNA and protein expression of AP-1 subunits. SLK cells were first transduced with lenti-GFP or lenti-3×FLAG-KDM2B for 72 h, followed by treatment with DMSO or 20 nM TPA. Seventy-two hours posttreatment, RT-qPCR and immunoblotting were performed. (A) Gene expression of c-Fos and c-Jun measured by RT-qPCR. t test was performed between GFP-OE and KDM2B-OE samples, and P < 0.05 (*) was considered statistically significant. (B) Immunoblot analysis of c-Jun expression. Tubulin was used as a loading control. (C) Quantification of c-Jun bands in panel B. c-Jun level was normalized by tubulin expression. (D) Effect of KDM2B on the half-life of c-Jun. HEK293T cells were transfected with empty vector or 3×FLAG-KDM2B WT, ΔF-box, or F-box–LRR mutant for 48 h, which was followed by treatment of the cells with 0.5 mg/ml CHX for the indicated length of time. Immunoblotting was performed for c-Jun, KDM2B, and tubulin at the indicated time points. (E) Quantification of c-Jun level by normalizing with tubulin expression. (F) Working model for illustrating the mechanism of how SCFKDM2B complex induces KSHV lytic gene expression through inducing AP-1 activity.
Since c-Jun transcription is positively autoregulated by c-Jun itself (43), the increased c-Jun protein level in KDM2B-OE cells could be the result of elevated c-Jun transcription. Based on our data that KDM2B mutants that lacked protein domains involved in transcriptional and epigenetic regulation induced AP-1 activity, we hypothesized that KDM2B increases c-Jun by stabilizing the rapidly degrading c-Jun at the protein level via interacting with SCF. To test this idea, we analyzed the half-life of c-Jun in the presence of WT KDM2B, the ΔF-box mutant, or the F-box–LRR domain alone (Fig. 12D and E). Forty-eight hours after transfection of 293T cells with the KDM2B-expressing plasmids, we treated the cells with cycloheximide (CHX) for 0.5, 1, 2, 4, and 6 h and then performed immunoblotting for c-Jun and KDM2B. The c-Jun protein level at each time point was normalized to tubulin. We found that while the steady-state level of c-Jun was reduced in the vector and ΔF-box mutant-expressing cells over time, c-Jun protein reduction was delayed in cells expressing either WT KDM2B or the F-box–LRR domain alone (Fig. 12D and E). These results suggest that KDM2B can increase the amount of c-Jun in cells by prolonging its half-life, which requires the F-box domain of KDM2B. Based on these results, we hypothesize that in KDM2B-OE cells, the SCFKDM2B complex degrades some c-Jun repressors (X target) that have yet to be identified, thereby increasing the c-Jun protein level (Fig. 12F).
DISCUSSION
KDM2B overexpression has been shown to enhance the development of several cancers (44). In this study, we demonstrated that KDM2B overexpression can also promote the replication of an oncogenic virus following de novo infection and lytic reactivation from latency. We propose that the overexpression of KDM2B leads to increased amounts of SCFKDM2B complexes in cells, which can induce the degradation of proteins that destabilize c-Jun. Consequently, the stabilized c-Jun protein enhances AP-1 activity, which facilitates lytic gene expression during de novo KSHV infection (Fig. 12F). This is the first report showing a functional connection between SCFKDM2B complex and AP-1 activity in the context of KSHV infection. However, further studies will be required to identify the target proteins of SCFKDM2B in KDM2B-OE cells to better understand how they can regulate de novo KSHV infection.
The repressive function of KDM2B in gene transcription has been linked to several of its functional domains, such as JmjC demethylase, CXXC DNA binding, and the C-terminal LRR domain, which binds to PRC1 (18, 38). Thus, KDM2B is well suited to act as a transcription repressor of KSHV gene expression. Accordingly, we have recently demonstrated that the shRNA knockdown of KDM2B during de novo KSHV infection or in latently infected cells results in increased lytic gene expression, indicating that KDM2B suppresses the lytic cycle of KSHV (11). Consequently, we would expect that KDM2B overexpression inhibits and does not induce lytic gene expression. However, a growing body of evidence indicates that epigenetic factors possessing multiple distinct functional domains can also display noncanonical functions independent from their chromatin-modifying roles, which is often manifested best when their expression is dysregulated. In fact, it was shown that when the histone demethylase LSD1, which normally demethylates mono- or dimethylated H3K4 and H3K9, is overexpressed, it can disrupt the dimerization of the F-box protein FBXW7 and promotes the self-ubiquitination and degradation of FBXW7. This noncanonical function of LSD1 is independent of its demethylase activity (45). Similarly, the histone demethylase LSD2 can also induce protein degradation, but in this case is uses its own E3 ubiquitin ligase activity (46). It was also shown that the H3K27 demethylases JMJD3 and UTX can play a role in general chromatin remodeling that does not require their histone demethylase activity (47). In addition, similarly to KDM2B, a number of transcription factors and epigenetic regulators (e.g., EZH2, p53, Brd4, RBP-Jκ, LSD1, and KAT5) have been shown to act both as activators and repressors of genes depending on what cofactors they interact with (48–54). Thus, it is not surprising that KDM2B, which has several distinct functional domains, can also show activities other than histone demethylation, some of which may become more pronounced when KDM2B expression is dysregulated.
In this study, we observed that the timing whereby shKDM2B and KDM2B-OE induce lytic genes following de novo KSHV infection is different. Notably, while shKDM2B induces significant lytic gene expression as early as 8 h after KSHV infection (corresponding to 72 h of shKDM2B expression), KDM2B-OE induces lytic genes significantly only after 48 hpi (corresponding to 5 days of KDM2B-OE) (Fig. 1). It takes 5 days for KDM2B-OE to induce lytic genes in latently infected BCBL1, while shKDM2B can induce lytic genes after 72 h of lenti-shKDM2B transduction (Fig. 2 and reference 11). These observations suggest that KDM2B-OE requires an extended period of time to switch from being a transcriptional repressor to a transcriptional activator. We speculate that this delay can be accounted for by the slow accumulation of c-Jun by KDM2B-OE over time, which ultimately turns on RTA expression. Interestingly, this functional switch did not occur with KDM2A, a paralog of KDM2B, which we have also previously identified as a transcriptional repressor of KSHV lytic genes during de novo infection (Fig. 2) (11). In addition, we found that while both JmjC and CXXC are dispensable, the F-box domain of KDM2B, which binds to SCF, is necessary for KDM2B-induced lytic viral gene expression during de novo infection (Fig. 4 and 8). In contrast, we previously showed that JmjC and CXXC domains are essential for KDM2B-mediated demethylation of viral chromatin, which is associated with viral gene repression (11). Altogether, our studies show that KDM2B utilizes different functional domains to repress and activate viral gene expression.
The C terminus of KDM2B, which we found to be essential for KDM2B-induced lytic gene expression, also includes an LRR domain that interacts with PRC1. We did not find any evidence of KDM2B affecting PRC1 binding to the KSHV genome in our previous or current study (Fig. 6) (11). Thus, we excluded the possibility of overexpressed KDM2B having a dominant-negative effect on PRC1 recruitment to lytic viral genes. Our data show that the lytic replication-inducing role of KDM2B-OE depends on the induction of RTA expression (Fig. 6). The CXXC mutant KDM2B and F-box–LRR fragment can induce RTA expression while the F-box and LRR mutants cannot, which strongly suggests that KDM2B-OE does not regulate RTA expression through directly binding to the RTA promoter but indirectly via interacting with the SCF complex.
One of the crucial roles of F-box proteins is to recruit substrate proteins to the SCF complex for degradation (55). Presently, there is only one report, which has shown that KDM2B can induce protein degradation, namely, c-Fos, using the SCF complex (23). This raises the question of how KDM2B-OE can promote the AP-1 transcriptional activity if c-Fos, one of the AP-1 subunits, is degraded by KDM2B. It is possible that KDM2B-mediated stabilization of c-Jun facilitates c-Jun homodimers that possess different transcriptional activities (56). In fact, c-Jun/c-Fos AP-1 heterodimers have been shown to have stronger DNA affinity and transcriptional activity than c-Jun homodimers (56). KDM2B-OE increasing c-Jun homodimer numbers can result in moderate AP-1 activity, which explains why the AP-1 luciferase reporter was only modestly induced in KDM2B-OE cells. We note, however, that it was demonstrated in KSHV-infected cells that the viral protein ORF45 can induce the accumulation of c-Fos during the progression of the KSHV lytic cycle (57). This may explain why we observed increased c-Fos protein levels in KSHV-infected KDM2B-OE cells (Fig. 11C). KSHV entry into cells during primary infection can also induce the AP-1 signaling pathway (34). Thus, the accumulation of c-Fos and c-Jun induced by KSHV infection and KDM2B-OE, respectively, can surge AP-1 activity, which can promote KSHV lytic gene expression during de novo infection.
F-box proteins other than KDM2B have also been reported to play a role in KSHV infection. A previous study demonstrated that viral interferon regulatory factor 3 (vIRF3) of KSHV interacts with the F-box protein SKP2 and uses it to activate cellular promoters regulated by c-Myc (58). They also showed that overexpressed vIRF3 can extend the half-life of c-Myc, which correlates with the increased expression of c-Myc target genes (58). Similarly, KSHV LANA can bind to the F-box protein FBXW7, resulting in increased protein stability of the activated intracellular domain of Notch (ICDN), which correlates with enhanced proliferation of KSHV-infected cells (59). In addition, the LANA-FBXW7 interaction can also enhance the stability of the antiapoptotic protein MCL-1 by preventing its proteasome-mediated degradation, which can contribute to the oncogenesis of KSHV+ PEL cells (60). However, none of these studies determined if these KSHV proteins utilize SCF to control the protein level of c-Myc, ICDN, or MCL-1 and whether SCF plays any role in viral gene regulation. Our study is the first report that provides evidence for the role of SCFKDM2B in controlling KSHV gene expression through upregulation of AP-1 activity. Additional studies will be required to identify the target proteins of SCFKDM2B that allow c-Jun accumulation and whether SCFKDM2B is also involved in the regulation of other signaling pathways critical in KSHV infection.
The dysregulation of KDM2B expression has been observed in several viral infections. While KDM2B gene expression is downregulated in Epstein-Barr virus-associated endemic Burkitt lymphoma (61), KDM2B is highly expressed in E6/E7-positive HPV16-infected keratinocytes, cervical cancer cell lines, and laryngeal squamous cell carcinomas (62). In contrast, we did not observe any KDM2B expression changes either during de novo KSHV infection of epithelial cells or upon lytic reactivation in PEL cells in our previous in vitro studies (11). However, this does not exclude the possibility that KSHV infects cells in vivo in which KDM2B is overexpressed upon some physiological or stress signals (e.g., hypoxia), which can result in increased lytic KSHV infection and viral transmission. It is still largely unknown what host factors determine if KSHV goes into latency or the lytic cycle following primary infection. Collectively, our data indicate that one such factor is KDM2B, whose expression level can play a critical role in determining the outcome of de novo KSHV infection.
MATERIALS AND METHODS
Cell lines, KSHV, and KSHV infection.
HEK293T (ATCC), EA.hy926 (ATCC), and SLK (NIH AIDS Reagent Program) cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (P/S). BCBL1 (NIH AIDS Reagent Program) was cultured in RPMI containing 10% FBS and P/S. KSHV (BAC16 clone) was received from Jae U. Jung (University of Southern California). KSHV BAC16 production and viral infection were performed as described previously (12). Hypoxia was induced by CoCl2 (Sigma) during de novo KSHV infection of SLK cells. RTA-KO KSHV was made by introducing a STOP codon and a frameshift in the second exon of RTA in BAC16. This mutant has been characterized and published previously (37). Cells were infected with 100 viral DNA copies/cell. The viral DNA copy number was calculated by qPCR using primers for KSHV ORF11 and a standard curve with BAC16 DNA.
Antibodies and oligonucleotides.
The list of antibodies used for ChIPs and immunoblot analyses, and the DNA sequences of oligonucleotides used for qPCR, are listed in Tables 1 and 2, respectively. We note that while all antibodies were diluted in phosphate-buffered saline-Tween 20 (PBST) containing 5% milk, the KDM2B antibody was diluted in Tris-buffered saline-Tween 20 (TBST) containing 5% milk for immunoblots.
TABLE 1.
Antibodies used in this study
| Antibody | Dilution (assay) | Source |
|---|---|---|
| RTA | 1/1,000 (WB) | Yoshihiro Izumiya, UC Davis |
| ORF6 | 1/1000 (WB) | Gary S. Hayward, Johns Hopkins University |
| ORF45 | 1/1,000 (WB) | Santa Cruz; sc-53883 |
| K8 | 1/1,000 (WB) | Santa Cruz; sc-57889 |
| K8.1 | 1/1,000 (WB) | Santa Cruz; sc-65446 |
| Tubulin | 1/10,000 (WB) | Sigma; T5326 |
| KDM2B | 1/1,000 (WB) | EMD Millipore; 09-864 |
| RING1B | 2 μg (ChIP) | Abcam; ab3832 |
| SKP1 | 1/1,000 (WB) | Santa Cruz; sc-5281 |
| CUL1 | 1/1,000 (WB) | Santa Cruz; sc-17775 |
| c-Jun | 1/1,000 (WB) | Cell Signaling; 9165 |
| c-Fos | 1/1,000 (WB) | Cell Signaling; 2250 |
| MYC | 1/1,000 (WB), 2 μg (IP) | BioLegend; 626802 |
| HA | 1/1,000 (WB), 2 μg (IP) | BioLegend; 901502 |
| FLAG | 1/1,000 (WB), 1 μg (ChIP) | Sigma; F1804 |
TABLE 2.
Primers used in this study
| Target | Forward (5′–3′) | Reverse (5′–3′) | Application |
|---|---|---|---|
| RTA (−1.7 kb) | GATCGGCGAAGTGGATAGAGT | CCCTATTGGTCACATCTCACG | ChIP-qPCR |
| RTA (−1.4 kb) | TGAGGTCTATTTCCCACGACA | ACAGCTCCGACGATGAGTATG | ChIP-qPCR |
| RTA (−1.0 kb) | CCCCAACACAAGGACCTTTA | GCTTTTGGATACCCTGGTGA | ChIP-qPCR |
| RTA (−0.6 kb) | AAGACACTGACCCACCAAGG | GGTGCCACCAATGTATGACC | ChIP-qPCR |
| RTA (−0.1 kb) | AAAGTCAACCTTACTCCGCAAG | GCTGCCTGGACAGTATTCTCAC | ChIP-qPCR |
| RTA (+0.8 kb) | TTGCCAAGTTTGTACAACTGCT | ACCTTGCAAAGACCATTCAGAT | ChIP/RT-qPCR |
| Neg | CAGGATCTCCGAGAATCAGC | GAGTTGGGAGAGCTGTCAGG | ChIP-qPCR |
| K2pr | CATACGCAGCCAAGCTATCA | GCTAGCACAGCAAATTGAGA | ChIP-qPCR |
| K2gb | TCACTGCGGGTTAATAGGATTT | CATGACGTCCACGTTTATCACT | ChIP/RT-qPCR |
| ORF25pr | AGTTGTCGGTGTCTATCTGT | TGCAGAGCGATACGCAGACT | ChIP-qPCR |
| ORF25gb | ACAGTTTATGGCACGCATAGTG | GGTTCTCTGAATCTCGTCGTGT | ChIP/RT-qPCR |
| ORF11 | GGCACCCATACAGCTTCTACGA | CGTTTACTACTGCACACTGCA | qPCR |
| HS1 | TTCCTATTTGCCAAGGCAGT | CTCTTCAGCCATCCCAAGAC | qPCR |
| ORF57 | AGGGATATCACCGCTCTCATAAGA | CTGCGGTTTCTCGACGGCAACTCA | RT-qPCR |
| ORF42 | CTGGTGGAGCACGAAGACAT | CGATAACAGGCAGCAGAGCT | RT-qPCR |
| ORF64 | CTTCCTCGAGGGCATCATATAC | TATACGGTGATGGACTTGATGG | RT-qPCR |
| LANA | GAGTCTGGTGACGACTTGGAG | AGGAAGGCCAGACTCTTCAAC | RT-qPCR |
| RTA | CAAGGTGTGCCGTGTAGAGAT | GGTCAAAGCCTTACGCTTCTT | RT-qPCR |
| KDM2B | AGTCCGAGACGTCAAACTCCTA | TAGTAACGCACAAACTGGGACA | RT-qPCR |
| SKP1 | GATGATGACCCAGTTCCTCTAC | CAAACAGGGATATCATCTGTTCG | RT-qPCR |
| CUL1 | CATTGGGTTCGCCGTGAATGTGAC | TACAACTCCACTAATCAATCTTGT | RT-qPCR |
| c-Fos | CATGGGCTCGCCTGTCAACGCGCA | GGTGAGGGGCTCTGGTCTGCGATG | RT-qPCR |
| c-Jun | AGCGCCTGATAATCCAGTCCAGCA | ACGTGACGCTGGGCAGCGTGTTCT | RT-qPCR |
| IL-6 | ATGTAACAAGAGTAACATGTGTGA | AGTGATGATTTTCACCAGGCAAGT | RT-qPCR |
| 18S | TTCGAACGTCTGCCCTATCAA | GATGTGGTAGCCGTTTCTCAGG | RT-qPCR |
shRNA knockdown and lentiviruses.
The shRNAs were expressed from the lentiviral pLKO.1 vector. The target sequence of shKDM2B is 5′-GCATGAAGCAGAGCTGCATCA-3′. We used pCDHCMV-MCS-EF1puro lentiviral vector to make KDM2B-expressing lentiviruses. The CXXC and JmjC domain KDM2B mutants were described in our previous study (11). The ΔF-box KDM2B mutant was made by the deletion of 48 residues containing the entire F-box domain (residues 1057 to 1105). The ΔLRR KDM2B mutant was generated by the deletion of the C-terminal leucine-rich repeat domain (residues 1083 to 1336). The KDM2B C-terminal fragments such as F-box–LRR (residues 1041 to 1336) and LRR (residues 1091 to 1336) were expressed as N-terminal 3×FLAG-tagged proteins from pCDHCMV-MCS-EF1puro vector. Lentivirus production and lentivirus infection were performed as described previously (12). After cells were transduced by lentiviruses for 2 days, the cells were split and the same number of different lentivirus-transduced cells was infected with KSHV on day 3 of the lentivirus transduction.
siRNA transfection.
The siRNAs targeting KDM2A (sc-96991), c-Fos (sc-29221), and c-Jun (sc-29223) were purchased from Santa Cruz Biotechnology. Lipofectamine RNAiMAX (Invitrogen) was used for siRNA transfections, which were performed according to the manufacturer’s instructions. The knockdown efficiency was examined by RT-qPCR and Western blot analysis.
Total RNA and DNA isolation and qPCR analysis.
Total RNA and DNA extraction from cells and qPCR analysis were performed as described in our previous publication (12). When GFP-OE and KDM2B-OE cells were infected with KSHV, viral gene expression was first normalized to 18S RNA expression, and then the viral gene expression in KDM2B-OE cells was compared to that of GFP-OE cells to calculate relative fold change (2−ΔΔCT method). Alternatively, the RT-qPCR data were presented as gene expression relative to 18S RNA expression (2−ΔCT method). The viral DNA was measured by qPCR using ORF11-specific primers. The amount of viral DNA was normalized for the cellular DNA level, which was measured by qPCR specific for the HS1 genomic region. The RT-qPCR and qPCR primers for KSHV genes and host genes, which were used in this study, are listed in Table 2. The RT-qPCR and qPCR results were calculated as an average from three independent experiments. For significance test, we used a two-tailed Student's t test where a P value of <0.05 was considered significant.
ChIP assay.
The ChIP assay was performed as we described previously (12). In the de novo KSHV infection ChIP experiments, SLK cells were first transduced with lenti-GFP or lenti-KDM2B for 2 days and then were split. On day 3, the same number of cells was infected with WT or RTA-KO KSHV for 72 h. The ChIP graphs show the averages from three independent ChIP experiments, which were calculated as the percentages of the immunoprecipitated DNA compared to input DNA. The ChIP antibodies are listed in Table 1, while the ChIP-qPCR primer sequences are shown in Table 2.
Coimmunoprecipitation assay.
The plasmid pCMV6-SKP1-myc-DDK is from OriGene (number RC206509), while pcDNA3-HA-ROC1 (number 19897) and pcDNA3-myc3-CUL1 (number 19896) are from Addgene. The transfected HEK293T cells were harvested at 48 h posttransfection for the immunoprecipitation experiments. Cells were washed with cold PBS once, lysed in NP-40 lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% NP-40) containing protease inhibitor cocktail (Roche), and passed through a 23-gauge needle 10 to 15 times, and then the cell lysates were incubated on ice for 15 min. After centrifugation, the cell lysates were subjected to preclearing using protein A Sepharose for 2 h at 4°C and then incubated with antibodies overnight. The next day, protein A/G Sepharose was added to the lysates, which were further incubated for 2 h at 4°C. Immunoprecipitates (IP) were washed three times with the lysis buffer, and the IPs were resuspended in 50 μl of 2× Laemmli buffer. The IP samples along with the input samples were analyzed by immunoblotting.
Luciferase reporter assay.
The luciferase reporter plasmids of signaling pathways were purchased from Addgene. The plasmids were the following: Wnt signaling (12456; M50 Super 8× TOPFlash), p53 signaling (16442; PG13-Luc WT p53 binding sites), TGF-beta signaling (16495; SBE4-Luc), c-Myc signaling (16564; pBV-Luc WT MBS1-4), Hypoxia signaling (26731; HRE-luciferase), Hippo signaling (34615; 8×GTIIC-luciferase), AP-1 signaling (40342; 3×AP1pGL3), Notch signaling (41726; 4×CSL-luciferase), OCT4/SOX2 (69445; 6×O/S luc), and NF-κB signaling (111216; 4×NF-kB Luc). HEK293T cells in 24-well plates were transfected with the luciferase reporter plasmids together with plasmids expressing 3×FLAG-KDM2B WT or CXXCm. Transfection was carried out by polyethylenimine (PEI). At 48 h posttransfection, cells were collected in 200 μl of lysis buffer (0.5% Triton X-100 diluted in 1× Dulbecco’s PBS). Twenty microliters of lysates was mixed with 20 μl of ONE-Glo luciferase substrate (Promega), and the luciferase activity was measured by a Promega GloMax-Multi detection system. All luciferase assays were carried out three times in triplicate.
Protein stability analysis.
To conduct translation shutoff (cycloheximide chase) experiments, HEK293T cells were transfected with empty vector or plasmids expressing 3×FLAG-KDM2B WT or mutants. Forty-eight hours after transfection, the cells were treated with 0.5 mg/ml cycloheximide (CHX) or dimethyl sulfoxide (DMSO) and chased for 0 to 6 h. At the indicated time points, cells were harvested for immunoblot analysis.
ACKNOWLEDGMENTS
This work was partly supported by an American Cancer Society Research Scholar Grant (RSG-18-221-01-MPC) and the National Institutes of Health (R01AI132554).
We thank Gary S. Hayward (Johns Hopkins University) for providing the ORF6 antibody and Yoshihiro Izumiya (University of California, Davis) for the RTA antibody. We also thank Bernadett Papp (University of Florida) and members of the Toth laboratory for helpful discussions during the project. We especially thank Lauren Roberts and Lauren McKenzie Spires for critical readings of the manuscript.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Hengge UR, Ruzicka T, Tyring SK, Stuschke M, Roggendorf M, Schwartz RA, Seeber S. 2002. Update on Kaposi's sarcoma and other HHV8 associated diseases. Part 1: epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect Dis 2:281–292. 10.1016/S1473-3099(02)00263-3. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz RA, Micali G, Nasca MR, Scuderi L. 2008. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol 59:179–206. 10.1016/j.jaad.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J Virol 72:8309–8315. 10.1128/JVI.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenner RG, Alba MM, Boshoff C, Kellam P. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J Virol 75:891–902. 10.1128/JVI.75.2.891-902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol 73:9348–9361. 10.1128/JVI.73.11.9348-9361.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J Virol 74:6207–6212. 10.1128/jvi.74.13.6207-6212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 95:10866–10871. 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, Pari GS. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J Virol 79:3479–3487. 10.1128/JVI.79.6.3479-3487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toth Z, Brulois K, Lee HR, Izumiya Y, Tepper C, Kung HJ, Jung JU. 2013. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog 9:e1003813. 10.1371/journal.ppat.1003813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naik NG, Nguyen TH, Roberts L, Fischer LT, Glickman K, Golas G, Papp B, Toth Z. 2020. Epigenetic factor siRNA screen during primary KSHV infection identifies novel host restriction factors for the lytic cycle of KSHV. PLoS Pathog 16:e1008268. 10.1371/journal.ppat.1008268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toth Z, Smindak RJ, Papp B. 2017. Inhibition of the lytic cycle of Kaposi's sarcoma-associated herpesvirus by cohesin factors following de novo infection. Virology 512:25–33. 10.1016/j.virol.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. 2006. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439:811–816. 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 14.Kang JY, Kim JY, Kim KB, Park JW, Cho H, Hahm JY, Chae YC, Kim D, Kook H, Rhee S, Ha NC, Seo SB. 2018. KDM2B is a histone H3K79 demethylase and induces transcriptional repression via sirtuin-1-mediated chromatin silencing. FASEB J 32:5737–5750. 10.1096/fj.201800242R. [DOI] [PubMed] [Google Scholar]
- 15.Janzer A, Stamm K, Becker A, Zimmer A, Buettner R, Kirfel J. 2012. The H3K4me3 histone demethylase Fbxl10 is a regulator of chemokine expression, cellular morphology, and the metabolome of fibroblasts. J Biol Chem 287:30984–30992. 10.1074/jbc.M112.341040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He J, Nguyen AT, Zhang Y. 2011. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 117:3869–3880. 10.1182/blood-2010-10-312736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farcas AM, Blackledge NP, Sudbery I, Long HK, McGouran JF, Rose NR, Lee S, Sims D, Cerase A, Sheahan TW, Koseki H, Brockdorff N, Ponting CP, Kessler BM, Klose RJ. 2012. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. Elife 1:e00205. 10.7554/eLife.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inagaki T, Iwasaki S, Matsumura Y, Kawamura T, Tanaka T, Abe Y, Yamasaki A, Tsurutani Y, Yoshida A, Chikaoka Y, Nakamura K, Magoori K, Nakaki R, Osborne TF, Fukami K, Aburatani H, Kodama T, Sakai J. 2015. The FBXL10/KDM2B scaffolding protein associates with novel polycomb repressive complex-1 to regulate adipogenesis. J Biol Chem 290:4163–4177. 10.1074/jbc.M114.626929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Z, Yang X, He J, Liu J, Wu F, Yu S, Liu Y, Lin R, Liu H, Cui Y, Zhou C, Wang X, Wu J, Cao S, Guo L, Lin L, Wang T, Peng X, Qiang B, Hutchins AP, Pei D, Chen J. 2017. Kdm2b regulates somatic reprogramming through variant PRC1 complex-dependent function. Cell Rep 21:2160–2170. 10.1016/j.celrep.2017.10.091. [DOI] [PubMed] [Google Scholar]
- 20.Shen J, Spruck C. 2017. F-box proteins in epigenetic regulation of cancer. Oncotarget 8:110650–110655. 10.18632/oncotarget.22469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorelik M, Manczyk N, Pavlenco A, Kurinov I, Sidhu SS, Sicheri F. 2018. A structure-based strategy for engineering selective ubiquitin variant inhibitors of Skp1-Cul1-F-box ubiquitin ligases. Structure 26:1226–1236. 10.1016/j.str.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP. 2002. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416:703–709. 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 23.Han XR, Zha Z, Yuan HX, Feng X, Xia YK, Lei QY, Guan KL, Xiong Y. 2016. KDM2B/FBXL10 targets c-Fos for ubiquitylation and degradation in response to mitogenic stimulation. Oncogene 35:4179–4190. 10.1038/onc.2015.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu L, Gao Y, Zhang Z, Cao Q, Zhang X, Zou J, Cao Y. 2015. Kdm2a/b lysine demethylases regulate canonical Wnt signaling by modulating the stability of nuclear beta-catenin. Dev Cell 33:660–674. 10.1016/j.devcel.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Ueda T, Nagamachi A, Takubo K, Yamasaki N, Matsui H, Kanai A, Nakata Y, Ikeda K, Konuma T, Oda H, Wolff L, Honda Z, Wu X, Helin K, Iwama A, Suda T, Inaba T, Honda H. 2015. Fbxl10 overexpression in murine hematopoietic stem cells induces leukemia involving metabolic activation and upregulation of Nsg2. Blood 125:3437–3446. 10.1182/blood-2014-03-562694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tzatsos A, Paskaleva P, Ferrari F, Deshpande V, Stoykova S, Contino G, Wong KK, Lan F, Trojer P, Park PJ, Bardeesy N. 2013. KDM2B promotes pancreatic cancer via Polycomb-dependent and -independent transcriptional programs. J Clin Invest 123:727–739. 10.1172/JCI64535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konuma T, Nakamura S, Miyagi S, Negishi M, Chiba T, Oguro H, Yuan J, Mochizuki-Kashio M, Ichikawa H, Miyoshi H, Vidal M, Iwama A. 2011. Forced expression of the histone demethylase Fbxl10 maintains, self-renewing hematopoietic stem cells. Exp Hematol 39:697–709. 10.1016/j.exphem.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Batie M, Druker J, D’Ignazio L, Rocha S. 2017. KDM2 family members are regulated by HIF-1 in hypoxia. Cells 6:8. 10.3390/cells6010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. 2001. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244–3250. 10.1182/blood.v97.10.3244. [DOI] [PubMed] [Google Scholar]
- 30.Wang SE, Wu FY, Chen H, Shamay M, Zheng Q, Hayward GS. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J Virol 78:4248–4267. 10.1128/jvi.78.8.4248-4267.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol 79:10308–10329. 10.1128/JVI.79.16.10308-10329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen A, Brodie C, Sarid R. 2006. An essential role of ERK signalling in TPA-induced reactivation of Kaposi's sarcoma-associated herpesvirus. J Gen Virol 87:795–802. 10.1099/vir.0.81619-0. [DOI] [PubMed] [Google Scholar]
- 33.Xie J, Ajibade AO, Ye F, Kuhne K, Gao SJ. 2008. Reactivation of Kaposi's sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 371:139–154. 10.1016/j.virol.2007.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie J, Pan H, Yoo S, Gao SJ. 2005. Kaposi's sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J Virol 79:15027–15037. 10.1128/JVI.79.24.15027-15037.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.An J, Lichtenstein AK, Brent G, Rettig MB. 2002. The Kaposi sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood 99:649–654. 10.1182/blood.v99.2.649. [DOI] [PubMed] [Google Scholar]
- 36.Ye FC, Blackbourn DJ, Mengel M, Xie JP, Qian LW, Greene W, Yeh IT, Graham D, Gao SJ. 2007. Kaposi's sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J Virol 81:3980–3991. 10.1128/JVI.02089-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toth Z, Brulois KF, Wong LY, Lee HR, Chung B, Jung JU. 2012. Negative elongation factor-mediated suppression of RNA polymerase II elongation of Kaposi's sarcoma-associated herpesvirus lytic gene expression. J Virol 86:9696–9707. 10.1128/JVI.01012-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu XD, Johansen JV, Helin K. 2013. Fbxl10/kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol Cell 49:1134–1146. 10.1016/j.molcel.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 39.Zhao X, Wang X, Li Q, Chen W, Zhang N, Kong Y, Lv J, Cao L, Lin D, Wang X, Xu G, Wu X. 2018. FBXL10 contributes to the development of diffuse large B-cell lymphoma by epigenetically enhancing ERK1/2 signaling pathway. Cell Death Dis 9:46. 10.1038/s41419-017-0066-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toth Z, Papp B, Brulois K, Choi YJ, Gao SJ, Jung JU. 2016. LANA-mediated recruitment of host polycomb repressive complexes onto the KSHV genome during de novo infection. PLoS Pathog 12:e1005878. 10.1371/journal.ppat.1005878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 6:e1001013. 10.1371/journal.ppat.1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.An J, Sun Y, Rettig MB. 2004. Transcriptional coactivation of c-Jun by the KSHV-encoded LANA. Blood 103:222–228. 10.1182/blood-2003-05-1538. [DOI] [PubMed] [Google Scholar]
- 43.Angel P, Hattori K, Smeal T, Karin M. 1988. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 55:875–885. 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 44.Yan M, Yang X, Wang H, Shao Q. 2018. The critical role of histone lysine demethylase KDM2B in cancer. Am J Transl Res 10:2222–2233. [PMC free article] [PubMed] [Google Scholar]
- 45.Lan H, Tan M, Zhang Q, Yang F, Wang S, Li H, Xiong X, Sun Y. 2019. LSD1 destabilizes FBXW7 and abrogates FBXW7 functions independent of its demethylase activity. Proc Natl Acad Sci U S A 116:12311–12320. 10.1073/pnas.1902012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y, Yin X, Yang H, Xu Y. 2015. Histone demethylase LSD2 acts as an E3 ubiquitin ligase and inhibits cancer cell growth through promoting proteasomal degradation of OGT. Mol Cell 58:47–59. 10.1016/j.molcel.2015.01.038. [DOI] [PubMed] [Google Scholar]
- 47.Miller SA, Mohn SE, Weinmann AS. 2010. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell 40:594–605. 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan J, Li B, Lin B, Lee PT, Chung TH, Tan J, Bi C, Lee XT, Selvarajan V, Ng SB, Yang H, Yu Q, Chng WJ. 2016. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 128:948–958. 10.1182/blood-2016-01-690701. [DOI] [PubMed] [Google Scholar]
- 49.Jiao L, Shubbar M, Yang X, Zhang Q, Chen S, Wu Q, Chen Z, Rizo J, Liu X. 2020. A partially disordered region connects gene repression and activation functions of EZH2. Proc Natl Acad Sci U S A 117:16992–17002. 10.1073/pnas.1914866117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laptenko O, Prives C. 2006. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ 13:951–961. 10.1038/sj.cdd.4401916. [DOI] [PubMed] [Google Scholar]
- 51.Sapountzi V, Logan IR, Robson CN. 2006. Cellular functions of TIP60. Int J Biochem Cell Biol 38:1496–1509. 10.1016/j.biocel.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 52.Wu SY, Chiang CM. 2007. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem 282:13141–13145. 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. 2007. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446:882–887. 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 54.Giaimo BD, Oswald F, Borggrefe T. 2017. Dynamic chromatin regulation at Notch target genes. Transcription 8:61–66. 10.1080/21541264.2016.1265702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skaar JR, Pagan JK, Pagano M. 2013. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol 14:369–381. 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smeal T, Angel P, Meek J, Karin M. 1989. Different requirements for formation of Jun: Jun and Jun: Fos complexes. Genes Dev 3:2091–2100. 10.1101/gad.3.12b.2091. [DOI] [PubMed] [Google Scholar]
- 57.Li X, Du S, Avey D, Li Y, Zhu F, Kuang E. 2015. ORF45-mediated prolonged c-Fos accumulation accelerates viral transcription during the late stage of lytic replication of Kaposi's sarcoma-associated herpesvirus. J Virol 89:6895–6906. 10.1128/JVI.00274-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baresova P, Pitha PM, Lubyova B. 2012. Kaposi sarcoma-associated herpesvirus vIRF-3 protein binds to F-box of Skp2 protein and acts as a regulator of c-Myc protein function and stability. J Biol Chem 287:16199–16208. 10.1074/jbc.M111.335216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lan K, Verma SC, Murakami M, Bajaj B, Kaul R, Robertson ES. 2007. Kaposi's sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein. Proc Natl Acad Sci U S A 104:16287–16292. 10.1073/pnas.0703508104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim YJ, Kim Y, Kumar A, Kim CW, Toth Z, Cho NH, Lee HR. 2021. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen dysregulates expression of MCL-1 by targeting FBW7. PLoS Pathog 17:e1009179. 10.1371/journal.ppat.1009179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vargas-Ayala RC, Jay A, Manara F, Maroui MA, Hernandez-Vargas H, Diederichs A, Robitaille A, Sirand C, Ceraolo MG, Romero-Medina MC, Cros MP, Cuenin C, Durand G, Le Calvez-Kelm F, Mundo L, Leoncini L, Manet E, Herceg Z, Gruffat H, Accardi R. 2019. Interplay between the epigenetic enzyme lysine (K)-specific demethylase 2B and Epstein-Barr virus infection. J Virol 93:e00273-19. 10.1128/JVI.00273-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peta E, Sinigaglia A, Masi G, Di Camillo B, Grassi A, Trevisan M, Messa L, Loregian A, Manfrin E, Brunelli M, Martignoni G, Palu G, Barzon L. 2018. HPV16 E6 and E7 upregulate the histone lysine demethylase KDM2B through the c-MYC/miR-146a-5p axys. Oncogene 37:1654–1668. 10.1038/s41388-017-0083-1. [DOI] [PubMed] [Google Scholar]