

With the emergence of new variants, PEDV remains a major problem in the global swine industry. Efficient PEDV infection usually requires trypsin, but the mechanism of trypsin dependency is complex.
KEYWORDS: porcine epidemic diarrhea virus, spike protein, trypsin dependency, S2′ site, reverse genetic system
ABSTRACT
Porcine epidemic diarrhea virus (PEDV) is an enteric pathogen of importance to the swine industry, causing high mortality in neonatal piglets. Efficient PEDV infection usually relies on the presence of trypsin, yet the mechanism of trypsin dependency is ambiguous. Here, we identified two PEDV strains, the trypsin-enhanced strain YN200 and the trypsin-independent strain DR13; the spike (S) protein of YN200 exhibits a stronger ability to induce syncytium formation and to be cleaved by trypsin than that of DR13. Using a full-length infectious YN200 cDNA clone, we confirmed that the S protein is a trypsin dependency determinant by comparison of rYN200 and rYN200-SDR13. To explore the trypsin-associated sites of the YN200 S protein, we then constructed a series of mutations adjacent to the fusion peptide. The results show that the putative S2′ cleavage site (R892G) is not the determinant for virus trypsin dependency. Hence, we generated viruses carrying chimeric S proteins: the S1 subunit, the S2 subunit, and the S2720∼892 domain (NS2′) were individually replaced by the corresponding DR13 sequences. Intriguingly, only the S2 substitution, not the S1 or NS2′ substitution, provides trypsin-independent growth of YN200. Additionally, the NS2′ recombinant virus significantly abrogated effective infection, indicating a vital role for NS2′ in viral entry. These findings suggest that the trypsin dependency of PEDV is controlled mainly by mutations in the S2 subunit rather than directly by a trypsin cleavage site.
IMPORTANCE With the emergence of new variants, PEDV remains a major problem in the global swine industry. Efficient PEDV infection usually requires trypsin, but the mechanism of trypsin dependency is complex. Here, we used two PEDV strains, the trypsin-enhanced strain YN200 and the trypsin-independent strain DR13. By using a YN200 reverse genetic system, we showed that the S protein determined PEDV trypsin dependency. The S2 subunit was verified as the main portion of PEDV trypsin dependency, although the putative S2′ site mutation cannot provide trypsin-independent growth of YN200. Finally, these results provide new insight into PEDV trypsin dependency and might inspire vaccine development.
INTRODUCTION
Porcine epidemic diarrhea virus (PEDV) is an enveloped, positive-sense, single-stranded RNA virus belonging to the Alphacoronavirus genus within the Coronaviridae family. PEDV causes acute watery diarrhea, vomiting, and dehydration in piglets. Classical PEDV was first identified in the United Kingdom in the 1970s (1) and then spread throughout Europe and to Asia (1–3). In the 2010s, a highly pathogenic PEDV strain suddenly emerged in China, resulting in almost 100% mortality for suckling piglets (4–6). Subsequently, PEDV variants caused outbreaks of swine disease in the United States, resulting in ∼$1.8 billion in economic losses from 2013 to 2014 (7, 8). On the basis of phylogenetic analyses of the whole genome, PEDV is divided into two subtypes, G1 and G2 (9, 10). G1 is represented by the prototype PEDV CV777, which is commonly used as a vaccine strain. However, the currently circulating strains with high virulence (belonging to subtype G2) are genetically different from CV777, especially with regard to the spike (S) gene, leading to different immunogenicity (11).
The S gene encodes a highly glycosylated homotrimeric protein anchored on the virus envelope, and the S protein exerts membrane fusion activity during virus entry (12, 13). For coronaviruses, the conversion of the S protein from the prefusion to the postfusion conformation requires multiple host factors, such as receptor binding, protease cleavage, or a low-pH environment (14). To establish an efficient infection in host cells, coronaviruses have evolved the ability to utilize multiple proteases, which have different binding motifs and cleavage efficiencies, including trypsin, chymotrypsin, transmembrane serine proteases (TMPRSSs), and cathepsin (15). Proteolytic cleavage usually occurs near the viral fusion peptide (FP) of the S fusion protein, exposing the hydrophobic FP, which can be inserted into the host cell membrane to initiate membrane fusion. The S proteins of betacoronaviruses, such as severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and mouse hepatitis virus (MHV), require two steps of protease cleavage, at S1–S2 and S2′, to activate fusion (16–18); however, a cleavage motif is rarely reported for alphacoronaviruses. The latest report on the 229E-CoV S protein concluded that it is not cleaved at S1–S2; in contrast, the cleavage of the S2′ site is particularly important (19).
The PEDV S protein can also be cleaved by a variety of proteases to facilitate entry, especially by trypsin, which plays a critical role in PEDV isolation (20–22). Previous studies have shown that trypsin cleavage of the S protein occurs only after PEDV binds to a receptor, which can greatly promote virus entry (23), and the addition of trypsin is also conducive to viral particle release (24). Wicht et al. suggested that the key factor determining the conversion of a PEDV strain from trypsin dependency to trypsin independence is the amino acid mutation of S2′ from arginine to glycine (25). Sequence alignment also showed an arginine at the S2′ site of clinical PEDV; as a result, trypsin is required for virus isolation. Another group discovered that during serial passage of PEDV, when the virus was passaged 30 to 40 generations, the S protein had only one mutation at the S2′ site (R895G), leading to trypsin-independent growth of the virus and a different cytopathic effect (CPE) (26). These results underline the importance of the S2′ site and suggest that further validation with infectious clones is needed.
In this study, we investigated the difference in trypsin dependency between two PEDV strains, the G2 strain YN200 and the G1 strain DR13. Then we constructed a series of chimeric recombinant viruses with mutations of the S gene introduced via bacterial artificial chromosomes (BACs) by a CRISPR-Cas9 system in vitro and studied their characteristics. Intriguingly, mutations at the S2′ site showed that the putative S2′ site had no impact on the trypsin dependency of YN200. The key regions affecting the trypsin dependency of the virus were further explored by S protein subdomain substitution. In addition, two basic amino acid residues, R887 and K891 of the PEDV S protein, were identified as significant factors for virus entry.
RESULTS
YN200, but not DR13, facilitates infection by utilizing trypsin in vitro.
To examine the differences in trypsin dependency between PEDVs, two strains belonging to different PEDV types were selected: YN200 and DR13. The former is a highly cell-adapted strain belonging to G2 (27); the latter is a classic G1 attenuated vaccine strain (28). Vero cells were infected with YN200 or DR13 and were subsequently cultured in the presence or absence of trypsin. Immunofluorescence and hematoxylin-and-eosin (HE) staining were performed, and multistep growth curves were drawn to examine the expansion of the viral infection. Only in the presence of trypsin did YN200 establish a highly efficient infection and form large syncytia with many more nuclei than those for DR13 (Fig. 1A to C). While YN200 spread was limited in the absence of trypsin, the replication of DR13 was more efficient without trypsin. At the same time, although the growth of DR13 in Vero cells was significantly inhibited in the presence of trypsin (Fig. 1E), the growth kinetics of YN200 were highly promoted by trypsin (Fig. 1D). After 24 h postinfection, cells infected with YN200 in the presence of trypsin were quickly detached and died, causing a rapid drop in virus titers. These results showed that PEDVs have different trypsin dependencies and that trypsin is able to promote virus infection and induce cell-to-cell fusion for strains such as YN200 but not for strains such as DR13.
FIG 1.

Growth characteristics of YN200 and DR13 with or without trypsin. (A) Vero cells were infected with YN200 or DR13 at an MOI of 0.01 in the presence or absence of trypsin. Infected cells were fixed at 12 h or 24 h postinfection and were then detected by immunofluorescence staining against the nucleocapsid protein (green). Nuclei were stained with DAPI (blue). Bars, 200 μm. (B) The numbers of nuclei per syncytium were determined and are displayed in the scatter plot. Statistical significance was assessed by an unpaired one-tailed Student t test. (C) Cells were stained with hematoxylin and eosin at 18 h postinfection. Bars, 20 μm. (D and E) Growth kinetics of YN200 (D) and DR13 (E) in Vero cells at an MOI of 0.01. The supernatant was harvested at 6, 12, 18, 24, 36, 48, 60, and 72 h postinfection and was titrated on Vero cells. Three replicates were performed. Error bars represent means ± standard deviations. Asterisks indicate significant differences (*, P < 0.05; ***, P < 0.001); ns, not significant.
The S protein determines the trypsin dependency of the virus and mediates cell-cell fusion.
According to previous reports, the S gene is considered a key trypsin-enhanced gene (23, 25). To further validate this concept, eukaryotic expression plasmids carrying the S proteins of YN200 and DR13 were transfected into Vero cells. As shown in Fig. 2A to C, only YN200-S mediated cell-to-cell fusion in the presence of trypsin, indicating that the different CPEs induced by the viruses were determined by the S protein. To confirm the trypsin cleavage of the S protein, 293T cells transiently expressing the YN200 or DR13 S protein were treated with trypsin. The Western blot results show that a small cleavage product of the YN200 S protein (below 100 kDa, at S2*) is absent for the DR13 S protein, indicating that only the YN200 S protein can be activated by trypsin (Fig. 2D).
FIG 2.

The S protein induced syncytium formation and trypsin sensitivity. (A) Vero cells were transiently transfected with PCAGGS expressing YN200 S or DR13 S for 24 h. Cells were either left untreated or treated with trypsin for another 18 h and were subsequently monitored by immunofluorescence staining against the S protein (green). Nuclei were stained with DAPI (blue). (B) The numbers of nuclei per syncytium were determined and are displayed in the scatter plot. Asterisks indicate significant differences (**, P < 0.01; ***, P < 0.001); ns, not significant. (C) 293T cells were transfected with PCAGGS encoding the S protein. After 24 h, cells were removed from the plate and collected. Then the samples were lysed by ultrasonication before treatment for 1 h with 0, 5, 10, or 20 μg/ml of trypsin at 37°C. Cleaved S proteins were detected by Western blotting with a C-terminal His tag antibody. S0, full-length S protein; S2*, a cleavage product.
We subsequently constructed a full-length infectious YN200 cDNA clone and replaced the S gene with the DR13 S gene (Fig. 3A). Through virus recovery, we obtained two recombinant PEDVs containing YN200-S or DR13-S. Immunofluorescence, HE staining, and kinetic growth analysis showed that recombinant YN200 (rYN200) exhibited characteristics similar to those of wild-type YN200, and rapid infection and syncytial formation occurred only in the presence of trypsin (Fig. 3B to E). As expected, the replication of recombinant YN200 with the DR13 S protein (rYN200-SDR13) was trypsin independent, and higher virus titers were observed without trypsin addition. Thus, the S protein is indeed a crucial factor affecting PEDV trypsin dependency.
FIG 3.

The trypsin dependency of PEDV is determined by the S protein. (A) Schematic diagram depicting the construction of a full-length cDNA clone of YN200. The YN200 genome was divided into eight contiguous cDNAs (A to H) by restriction enzymes for construction. The structures of the recombinant virus genomes carrying the YN200 or DR13 S gene (rYN200 and rYN200-SDR13, respectively) are also shown. (B) Vero cells were infected with rYN200 or rYN200-SDR13 with or without trypsin for 24 h. Cells were examined by immunostaining against the nucleocapsid protein (green), and nuclei were stained with DAPI (blue). (C) Cells were stained with hematoxylin and eosin under the same conditions as those for panel B. (D and E) Vero cells were inoculated with rYN200 or rYN200-SDR13 in the presence or absence of trypsin at an MOI of 0.01. The supernatant was harvested at 12, 24, 36, 48, 60, and 72 h postinfection and was titrated. Asterisks indicate significant differences (*, P < 0.05).
R892 is not a prerequisite for trypsin dependency, and R885 is related to YN200 cell adaptation.
Comparison of the amino acids of the S proteins in the region from S1–S2 to the FP showed that 9 amino acids (aa) differed between YN200 and DR13, including in particular two arginines in YN200, R885 and R892 (Fig. 4A). Notably, R892 is the putative S2′ site for trypsin cleavage in PEDV (25). To investigate the effects of R885 and R892 on the trypsin dependency of YN200, we constructed and rescued single- and double-recombinant mutant viruses. Surprisingly, all three recombinant viruses exhibited extensive syncytial formation in Vero cells (Fig. 4B). In the presence of trypsin, the growth curve of rYN200-R892G was slightly greater than that of rYN200 (Fig. 4C), with comparable plaque sizes. However, the plaques formed by rYN200-R885S,R892G were slightly smaller, and the growth titer was lower than that of rYN200 without trypsin (Fig. 4D). These results showed that R892 is not a prerequisite site for viral trypsin dependency, but the double mutations of R885 and R892 reduced virus infection in vitro, indicating that there might be an interaction between the two sites to affect trypsin cleavage.
FIG 4.

Effects of R885S and R892G substitutions in YN200 during viral infection. (A) Amino acid alignment between YN200 S and DR13 S in the region from the S1–S2 cleavage site to the FP. There are nine amino acid differences, including R885 and R892. (B) Comparison of rYN200 infection with infections with the recombinant viruses (rYN200-R885S, rYN200-R892G, and rYN200-R885S,R892G) in the presence of trypsin. Vero cells were inoculated with viruses at an MOI of 0.01 in the presence of trypsin for 24 h. HE, hematoxylin-and-eosin staining; IF, immunofluorescence; Plaque, plaque formation. (C and D) Growth kinetics of the four viruses with (C) or without (D) trypsin at an MOI of 0.01. Asterisks indicate significant differences (*, P < 0.05). (E) Effects of the R885S and R892G mutations on viral fitness as determined by a competition assay. Vero cells were coinfected with rYN200 and one of the mutants at a 1:1 ratio in the presence of trypsin, and supernatants were passaged serially 3 times every 24 h. Three replicates were performed.
Notably, wild-type YN200 evolved two arginines during passage. To further evaluate the fitness of recombinant viruses in a competition assay, we mixed the rYN200 and mutant viruses at a 50:50 ratio in Vero cells. As shown in Fig. 4E, nearly 10% of rYN200-R885S and 40% of rYN200-R892G were retained after three passages. However, the double-mutant virus, rYN200-R885S,R892G, is almost undetectable. This result indicates that the mutation at position 885 (S to R) of the S protein provides a fitness advantage in Vero cells and a novel cell adaptation.
Mutations at two conserved sites, R887 and K891, seriously damage virus recovery.
The S protein of YN200 was analyzed structurally using a cryo-electron microscopy (cryo-EM) structure of the PEDV S as a model (29). In agreement with a previous study (30), the putative FP at aa 893 to 900 exhibited the typical alpha-helix (Fig. 5A). For most coronaviruses, the cleavage of S2′ occurs at the N terminus of the FP (17, 30). However, the PEDV S protein model lacks a spatial structure at aa 882 to 891, indicating that these residues may form a flexible loop. Therefore, we investigated two additional conserved sites in this region, R887 and K891, along with R885 and R892 (Fig. 5B). As shown in Table 1, we constructed multiple mutations at those four sites and tried to recover the recombinant viruses. While mutants with single mutations of R887A or K891A were successfully recovered with no impact on trypsin dependency (Fig. 5C), the rest of the recombinant viruses could not be recovered. Moreover, these S proteins were expressed normally by transfection in 293T cells (Fig. 5E). These findings indicate that the failure to rescue the viruses may be due to the inability of the mutated S protein to utilize proteases.
FIG 5.

Structure and sequence analysis of the PEDV S protein. (A) Three-dimensional structure of the PEDV S protein monomer from the Protein Data Bank (PDB accession number 6U7K). S1, S2, and FP are color-coded cyan, green, and red, respectively. (Inset) Amino acids 882 to 891 are represented by a dashed green line. (B) Multisequence alignment of PEDV S sequences in the vicinity of the proposed S2′ position. Arrows indicate the amino acids of four critical sites. (C) Vero cells were infected with either rYN200-R887A, rYN200-K891A, rYN200-SDR13-R887A, or rYN200-SDR13-K891A with or without trypsin for 18 h (MOI, 0.01). Cells were examined by immunostaining against the nucleocapsid protein (green), and nuclei were stained with DAPI (blue). (D) rYN200-F672V was characterized by the same procedure as that used for panel C. (E) 293T cells were transfected with PCAGGS encoding the S protein. After 24 h, the S proteins were detected by Western blotting using the anti-S1 antibody or the anti-GAPDH antibody. CON, empty vector control.
TABLE 1.
Mutations of putative cleavage sites in the PEDV S protein
| Virus | S protein backbone | Amino acids adjacent to FPa | Recoveryb | Trypsin dependencyc |
|---|---|---|---|---|
| rYN200-R887A | YN200 S | IGVSVYDPARGAVVQKR | √ | + |
| rYN200-K891A | IGVSVYDPARGRVVQAR | √ | + | |
| rYN200- R887A,R892G | IGVSVYDPARGAVVQKG | × | ND | |
| rYN200-R885S,R887A,R892G | IGVSVYDPASGAVVQKG | × | ND | |
| rYN200-R885S,K891A,R892G | IGVSVYDPASGRVVQAG | × | ND | |
| rYN200-R885S,R887A,K891A,R892G | IGVSVYDPASGAVVQAG | × | ND | |
| rYN200-SDR13-R887A | DR13 S | IGVSVYDPASGAVVQKG | √ | − |
| rYN200-SDR13-K891A | IGVSVYDPASGRVVQAG | √ | − | |
| rYN200-SDR13-R887A,K891A | IGVSVYDPASGAVVQAG | × | ND |
Underlined letters represent mutant amino acids.
√, virus successfully recovered; ×, virus could not be recovered.
+, trypsin-enhanced strain; −, trypsin-independent strain; ND, not detected.
The S2 subdomain is responsible for the trypsin dependency of chimeric recombinant viruses.
According to the results presented above, a single point mutation may not be enough to alter the trypsin dependency of the virus; therefore, we constructed chimeras of the S protein. The YN200 S1 domain, S2 domain, and S2 domain from aa 720 to aa 892 (S2720–892 domain) (NS2′) were replaced with the corresponding domains of DR13 (Fig. 6A), and all three viruses were successfully recovered and characterized. In agreement with the findings presented above, both rYN200-S1DR13 and rYN200-NS2′DR13 formed typical syncytia, whereas rYN200-S2DR13 generated trypsin-independent infection without syncytium formation (Fig. 6B). Additionally, there was no obvious band of the trypsin cleavage product of rYN200-S2DR13 S protein, as there was for the others (Fig. 6C). These results showed that the overall structure of the S2 domain is more likely than the S1 domain to affect trypsin cleavage. Moreover, the infection of rYN200-NS2’DR13 was significantly inhibited, indicating that the NS2’ domain has an important role during virus entry.
FIG 6.
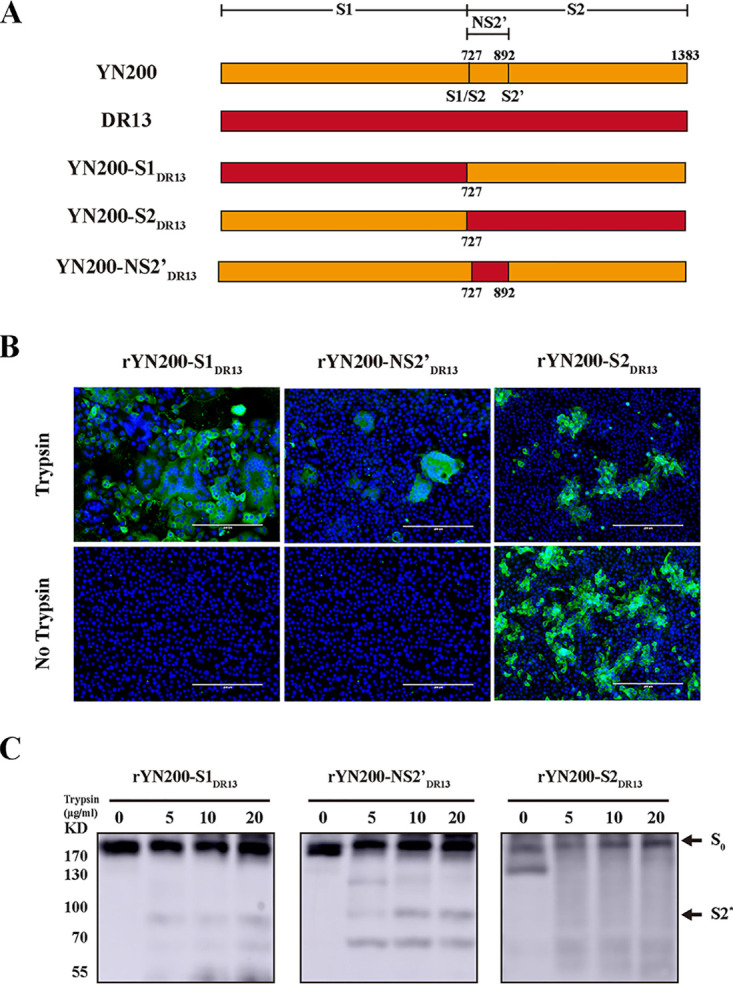
The critical region of the S protein subdomains for PEDV trypsin dependency. (A) Schematic map of the chimeric S proteins of the recombinant viruses. Orange and red regions represent YN200 S and DR13 S, respectively. (B) Vero cells were infected with rYN200-S1DR13 or rYN200-S2DR13 with or without trypsin at an MOI of 0.01 for 24 h, while rYN200-NS2′DR13-infected cells were incubated for another 48 h before monitoring. (C) Trypsin sensitivities of chimeric S proteins, determined as described in the legend to Fig. 2C. S0, full-length S protein; S2*, a cleavage product.
DISCUSSION
Coronavirus entry into host cells relies on the receptor binding and proteolytic priming of the S protein by a variety of host cell proteases, such as furin, trypsin, TMPRSS2, and cathepsin L (31). For both the current global pandemic virus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the newly emerging porcine delta coronavirus (PDCoV) in the pig industry, trypsin can promote the replication of wild-type strains and is therefore widely used in virus isolation (32, 33). Especially for PEDV, trypsin is indispensable in virus isolation (34). In this study, we analyzed two PEDV strains: DR13, belonging to the G1 genotype, and YN200, belonging to the G2 genotype. YN200 is a trypsin-enhanced strain, exhibiting rapid replication and typical syncytium formation in the presence of trypsin, while YN200 infection is limited without trypsin addition. Trypsin-independent infection with YN200 is strongly inhibited by a serine protease inhibitor (data not shown), a finding consistent with previous studies (21, 22), indicating the evolutionary progress of PEDV from trypsin dependency to trypsin independence. In contrast, the presence of trypsin reduced DR13 virus titers according to a previous report (25), so that the growth curve of DR13 (with trypsin) shows a lower rate in the early stage.
In this study, we constructed a novel PEDV infectious clone of YN200 in a BAC system, which has been used for many other coronaviruses (35, 36). To quickly manipulate the full-length PEDV genome, the CRISPR-Cas9 system was used as described previously (37). Through multiple virus recovery assays (n > 10), the recombinant viruses were successfully obtained in a short time, indicating a highly efficient and stable reverse genetic operating system. The trypsin dependency of the recombinant virus carrying the DR13 S protein was successfully changed, confirming that the S protein is the key factor.
To explore the mechanism by which trypsin activates the PEDV S protein, we focused on the S protein cleavage site. Studies have shown that the number of arginine residues contained in S1–S2 and S2′ determines the enzyme cleavage efficiency (38). Notably, the cleavage site sequence can determine the zoonotic potential of coronaviruses (39); for instance, a multibasic cleavage site is present in SARS-CoV-2 but not in bat coronavirus RaTG13 (40). Here, we compared the cleavage sites of the two S proteins and found that there are two extra arginines at the S2′ site of YN200 (R885 and R892). R892 is considered to be located at the N terminus of the FP, while arginine at the same position in SARS-CoV is the main trypsin cleavage site (16). However, we found that the in vitro infection characteristics of the R892G recombinant virus were similar to those of the parent trypsin-enhanced strain, in which R885 may act as an extra cleavage site. Sequence analysis of PEDV S in the GenBank database showed that some other PEDV strains also evolved the R885 site (Fig. 6B), indicating, along with the virus competition assay, that R885 is likely to be a cell-adaptive mutation. Notably, double mutation only slightly affects virus infection compared with wild-type infection, indicating that there must be another trypsin cleavage site.
To further explore the possible trypsin cleavage sites, we focused on the structures of the S1–S2 and S2′ sites. According to reports, the trypsin cleavage site of 229E alphacoronavirus is located at the S2′ arginine instead of S1–S2 (19), indicating that the S2′ site of PEDV is more likely to be the putative cleavage site. Through trypsin cleavage of the YN200 S protein, a cleavage band was observed between 100 and 70 kDa, suggesting that the S protein is more likely cleaved at the S2′ site rather than the S1–S2 site. Since trypsin cleaves only at arginine and lysine residues, we then focused on R887 and K891. Similarly, single mutations of R887A or K891A on rYN200 or rYN200-SDR13 did not affect the trypsin dependency of the recombinant viruses. However, after the R887A and K891A mutations were combined with R885S and R892G, neither rYN200 nor rYN200-SDR13 was successfully recovered. According to GenBank, R887 and K891 on the PEDV S protein are extremely highly conserved, indicating that there may be a conservative cleavage mechanism for the PEDV S protein. On this point, further research is needed for verification.
Based on our results, one may speculate that the substitution of multiple amino acids in the S protein is necessary for changing the trypsin dependency of PEDV. Thus, chimeric S proteins of recombinant viruses were generated, including S1, S2, and NS2′ chimeras. The results showed that S2, not S1 or NS2′, provided trypsin-independent growth of YN200, suggesting a vital impact for the S2 subdomain. Moreover, we also tried to mutate F672 to V in the S1 subdomain of YN200, as mentioned previously (41). Although the cell-cell fusion of the F672V recombinant virus was reduced (Fig. 5D), it was still a trypsin-enhanced strain, indicating that S1 is not the critical region that affects trypsin dependency. Although the NS2′ recombinant virus was still a trypsin-enhanced strain, it exhibited a reduced infection rate and restricted syncytia. In conclusion, the non-cleavage site regions of the S2 subunit are critical for PEDV trypsin dependency. Overall, PEDV trypsin dependency involves a complex and variable mechanism, including the interaction between S1 and S2 as well as the possibility of multiple cleavage sites.
MATERIALS AND METHODS
Cell lines, viruses, and plasmid construction.
Vero cells (CCL-81) and HEK293T cells were obtained from the American Type Culture Collection (ATCC) and were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Natocor, Argentina), 100 U/ml penicillin, and 100 μg/ml streptomycin. PEDV strain YN200 (GenBank accession no. KT021233) was propagated in DMEM supplemented with 5 μg/ml trypsin (Gibco) in Vero cells. Another strain used was the attenuated DR13 strain (GenBank accession no. JQ023162.1), which was propagated under the same conditions except for trypsin addition. The recombinant viruses used in this research were also propagated in Vero cells with or without 5 μg/ml trypsin, as described in this article. The S genes of YN200, DR13, and other recombinant viruses were amplified from the virus genomes and were designed to be flanked by EcoRI and XhoI restriction sites for cloning into the pCAGGS(+) vector. Recombinant plasmids were sequenced to authenticate the subject sequences.
Virus infection assay.
Vero cell monolayers were seeded in 24-well plates and were subsequently inoculated with YN200 and DR13 at a multiplicity of infection (MOI) of 0.01 in the presence or absence of 5 μg/ml trypsin for 1 h. After virus adsorption, cells were washed twice with DMEM and then cultured for 24 h under the conditions described above. For recombinant viruses, the process of cell infection was similar to that for parental viruses at an MOI of 0.01, except for rYN200-NS2′DR13, which was inoculated with an equivalent volume and then cultured for 72 h. Virus-infected Vero cells were visualized by immunofluorescence microscopy.
Viral growth kinetics.
Vero cells were seeded in 12-well plates and then inoculated with YN200, DR13, or recombinant viruses at an MOI of 0.01 in the presence or absence of 5 μg/ml trypsin. After 1 h of incubation, cells were washed twice with DMEM, followed by the addition of maintenance medium (DMEM with or without 5 μg/ml trypsin), and the supernatants of cells were collected at 6, 12, 18, 24, 36, 48, 60, and 72 h postinfection. The viral titers were determined by a 50% tissue culture infective dose (TCID50) assay. Briefly, Vero cells were prepared in 96-well plates at 100% confluence and were washed three times with DMEM. Serial 10-fold dilutions of virus samples were inoculated in eight replicates per dilution. Viral CPE was monitored for 3 to 5 days, and virus titers were calculated by using the Reed-Muench method (42).
S protein-mediated cell-cell fusion.
Vero cells were transfected with pCAGGS expression plasmids encoding the S protein of YN200 or DR13 by using jetPRIME (Polyplus, France) for 24 h. Then the cells were washed twice with DMEM and cultured in maintenance medium with or without 5 μg/ml trypsin for another 18 h. Cell-cell fusion was monitored by immunofluorescence staining using an anti-S1 mouse monoclonal antibody. Plates were photographed to quantify syncytium formation. The nuclei in each syncytium were counted.
S protein cleavage by trypsin.
293T cells were seeded into 6-well plates and transfected with 2 μg/well of an empty plasmid or a plasmid encoding PEDV S, including wild-type and mutant forms, by using ExFect2000 (Vazyme, China) according to the manufacturer’s protocol. At 24 h posttransfection, cells were scraped into the culture medium and pelleted. Cell samples were washed twice with cold phosphate-buffered saline (PBS) to remove FBS residue and were then lysed by ultrasonication for 1 min on ice. Cell lysates were mixed with equal volumes of trypsin at different concentrations (to final concentrations of 0, 5, 10, or 20 μg/ml) at 37°C for 30 min before denaturation. Proteins were separated by SDS-PAGE and were transferred to a polyvinylidene fluoride membrane (Merck Millipore, USA). The PEDV S protein was detected by staining with a mouse monoclonal antibody specific for S1, followed by incubation with a horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (Thermo Fisher Scientific, USA).
Construction and recovery of recombinant viruses.
The YN200 genome was divided into eight continuous fragments (A to H), and each fragment was amplified from the total cDNA. Fragments B to G were first cloned into the pMD18-T vector (TaKaRa, Kusatsu, Japan), while A and H were cloned into a BAC plasmid (kindly provided by Cao Gang). Fragments A and H were cloned with the SfiI and Van91I sites, and other fragments were cloned with the Van91I site. A natural Van91I site was constructed by introducing the A11107C mutation, which was maintained as the rescue marker. All the fragments were then sequenced for verification. Subsequently, all subclones were digested with Van91I or SfiI and then ligated for 2 h at 16°C before being transformed into chemically competent DH10B cells (Biomed, Beijing, China). After determination of all the fragments by bacterial PCR, the positive clones were further determined by restriction fragment length polymorphism with KpnI, and the correct clone was designated pBAC-PEDV-YN200 after sequencing (GenScript, Nanjing, China).
To construct the recombinant viruses, a method reported by our laboratory previously was used (37). The design of the relevant primers is shown in Table 2. Each corresponding mutant virus was efficiently constructed by CRISPR-Cas9 technology. Briefly, two specific enzyme sites encompassing the sequence of the S gene were selected. Two types of single-stranded DNA forward primers (sgS-F or sgS-R) were used, and a constant reverse primer (single guide RNA [sgRNA]) corresponding to the two enzyme cutting sites was used. After annealing PCR using the forward and reverse primers, the purified PCR products of short DNA fragments were transcribed by T7 RNA polymerase. The transcribed products corresponding to two sites were incubated with the nuclease Cas9 to digest the YN200 BAC in vitro, and the digestion yielded a linearized BAC and a 4-kb DNA fragment that included the sequence of the S gene. Subsequently, pBAC-PEDV-YN200 was digested in a 50-μl reaction mixture consisting of 5 μg of pBAC-PEDV-YN200, 5 μl of Cas9 (New England Biolabs [NEB]), 10 μg of sgRNA, and 5 μl of nuclease reaction buffer by incubation at 37°C overnight.
TABLE 2.
Primers used to produce the specific sgRNA for pBAC-PEDV-YN200 digestion
| Primer | Sequence |
|---|---|
| sgS-F | TTAATACGACTCACTATAGGTAAGACATAGTCATAATTGGTTTTAGAGCTAGA |
| sgS-R | TTAATACGACTCACTATAGGCCATGTTCTTTTTCAGAGCGTTTTAGAGCTAGA |
| sgRNA | AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC |
For virus recovery, 293T cells seeded in 6-well plates (1 × 106 cells per well) were transfected with 2 μg of purified BAC DNA and 4 μl of the ExFect2000 transfection reagent (Vazyme) according to the manufacturer’s instructions. Transfected cultures were infected with Vero cells at 24 h after transfection. Virus recovery was monitored by CPE, and the newly reconstituted viruses were then plaque-purified and propagated in Vero cells.
Virus competition assays.
Vero cells were coinfected with rYN200 and one of the three recombinant viruses at an MOI of 0.01, and the initial proportion was 1:1. At 24 h postinfection, the supernatants were collected and serially passaged three times. The relative abundances of the wild type and the mutants at the third passage were determined by gene sequencing of the mutation at R885 or R892.
Immunofluorescence and HE staining assay.
Cell samples were washed with PBS and fixed with 4% paraformaldehyde (PFA) for 20 min, followed by membrane permeabilization with 0.1% Triton X-100 in PBS for 15 min at room temperature. Cells were then washed, blocked with PBS containing 2% bovine serum albumin (BSA) for 1 h at 37°C, and then incubated with a rabbit polyclonal antibody against PEDV nucleocapsid protein (dilution, 1:300) or a mouse monoclonal antibody against PEDV S1 (dilution, 1:1,000) in 2% BSA in PBS for 1 h at 37°C. After the cells were washed three times, Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, USA) or Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) in PBS at a dilution of 1:1,000 was added and further incubated for 45 min. 4′,6-Diamidino-2-phenylindole (DAPI) was used for nuclear staining at a dilution of 1:1,000 for 1 min. Samples were observed using a fluorescence microscope (Thermo Fisher Scientific). For HE staining, Vero cells, which were preseeded onto the cell slide, were fixed in acetone and stained with Harris hematoxylin (Sigma-Aldrich) and 1% eosin (Sigma-Aldrich). Images were captured with a microscope.
Statistical analysis.
Statistical analysis of the data was performed with Prism software (version 6.01) using an unpaired one-tailed Student t test. Error bars in figures indicate standard deviations. Asterisks indicate levels of significance (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (grants 31873020 and 317220656), the National Key Research and Development Plan of China (grant 2018YFD0500102), and the Huazhong Agricultural University Scientific and Technological Self-Innovation Foundation Program (grant 2662017PY028).
REFERENCES
- 1.Chasey D, Cartwright SF. 1978. Virus-like particles associated with porcine epidemic diarrhoea. Res Vet Sci 25:255–256. 10.1016/S0034-5288(18)32994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kweon C-H, Kwon B-J, Jung T-S, Kee Y-J, Hur D-H, Hwang E-K, Rhee J-C, An S-H. 1993. Isolation of porcine epidemic diarrhea virus (PEDV) in Korea. Korean J Vet Res 33:249–254. [Google Scholar]
- 3.Chen J-F, Sun D-B, Wang C-B, Shi H-Y, Cui X-C, Liu S-W, Qiu H-J, Feng L. 2008. Molecular characterization and phylogenetic analysis of membrane protein genes of porcine epidemic diarrhea virus isolates in China. Virus Genes 36:355–364. 10.1007/s11262-007-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, Tang X, He Q. 2012. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg Infect Dis 18:1350–1353. 10.3201/eid1808.120002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun R-Q, Cai R-J, Chen Y-Q, Liang P-S, Chen D-K, Song C-X. 2012. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg Infect Dis 18:161–163. 10.3201/eid1801.111259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X, Huo J-Y, Chen L, Zheng F-M, Chang H-T, Zhao J, Wang X-W, Wang C-Q. 2013. Genetic variation analysis of reemerging porcine epidemic diarrhea virus prevailing in central China from 2010 to 2011. Virus Genes 46:337–344. 10.1007/s11262-012-0867-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ. 2013. Emergence of porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Invest 25:649–654. 10.1177/1040638713501675. [DOI] [PubMed] [Google Scholar]
- 8.Vlasova AN, Marthaler D, Wang Q, Culhane MR, Rossow KD, Rovira A, Collins J, Saif LJ. 2014. Distinct characteristics and complex evolution of PEDV strains, North America, May 2013–February 2014. Emerg Infect Dis 20:1620–1628. 10.3201/eid2010.140491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin C-M, Saif LJ, Marthaler D, Wang Q. 2016. Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Res 226:20–39. 10.1016/j.virusres.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarvis MC, Lam HC, Zhang Y, Wang L, Hesse RA, Hause BM, Vlasova A, Wang Q, Zhang J, Nelson MI, Murtaugh MP, Marthaler D. 2016. Genomic and evolutionary inferences between American and global strains of porcine epidemic diarrhea virus. Prev Vet Med 123:175–184. 10.1016/j.prevetmed.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, Zhang L, Zhang Q, Zhou P, Fang Y, Zhao D, Feng J, Li W, Zhang Y, Wang Y. 2019. Evaluation and comparison of immunogenicity and cross-protective efficacy of two inactivated cell culture-derived GIIa- and GIIb-genotype porcine epidemic diarrhea virus vaccines in suckling piglets. Vet Microbiol 230:278–282. 10.1016/j.vetmic.2019.02.018. [DOI] [PubMed] [Google Scholar]
- 12.Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77:8801–8811. 10.1128/jvi.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heald-Sargent T, Gallagher T. 2012. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4:557–580. 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belouzard S, Millet JK, Licitra BN, Whittaker GR. 2012. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4:1011–1033. 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belouzard S, Chu VC, Whittaker GR. 2009. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A 106:5871–5876. 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millet JK, Whittaker GR. 2014. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A 111:15214–15219. 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuyama S, Taguchi F. 2009. Two-step conformational changes in a coronavirus envelope glycoprotein mediated by receptor binding and proteolysis. J Virol 83:11133–11141. 10.1128/JVI.00959-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonnin A, Danneels A, Dubuisson J, Goffard A, Belouzard S. 2018. HCoV-229E spike protein fusion activation by trypsin-like serine proteases is mediated by proteolytic processing in the S2′ region. J Gen Virol 99:908–912. 10.1099/jgv.0.001074. [DOI] [PubMed] [Google Scholar]
- 20.Park J-E, Cruz DJM, Shin H-J. 2014. Clathrin- and serine proteases-dependent uptake of porcine epidemic diarrhea virus into Vero cells. Virus Res 191:21–29. 10.1016/j.virusres.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C, Ma Y, Yang Y, Zheng Y, Shang J, Zhou Y, Jiang S, Du L, Li J, Li F. 2016. Cell entry of porcine epidemic diarrhea coronavirus is activated by lysosomal proteases. J Biol Chem 291:24779–24786. 10.1074/jbc.M116.740746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi W, Fan W, Bai J, Tang Y, Wang L, Jiang Y, Tang L, Liu M, Cui W, Xu Y, Li Y. 2017. TMPRSS2 and MSPL facilitate trypsin-independent porcine epidemic diarrhea virus replication in Vero cells. Viruses 9:114. 10.3390/v9050114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park J-E, Cruz DJM, Shin H-J. 2011. Receptor-bound porcine epidemic diarrhea virus spike protein cleaved by trypsin induces membrane fusion. Arch Virol 156:1749–1756. 10.1007/s00705-011-1044-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirato K, Matsuyama S, Ujike M, Taguchi F. 2011. Role of proteases in the release of porcine epidemic diarrhea virus from infected cells. J Virol 85:7872–7880. 10.1128/JVI.00464-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wicht O, Li W, Willems L, Meuleman TJ, Wubbolts RW, van Kuppeveld FJ, Rottier PJ, Bosch BJ. 2014. Proteolytic activation of the porcine epidemic diarrhea coronavirus spike fusion protein by trypsin in cell culture. J Virol 88:7952–7961. 10.1128/JVI.00297-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun M, Ma J, Yu Z, Pan Z, Lu C, Yao H. 2017. Identification of two mutation sites in spike and envelope proteins mediating optimal cellular infection of porcine epidemic diarrhea virus from different pathways. Vet Res 48:44. 10.1186/s13567-017-0449-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen F, Zhu Y, Wu M, Ku X, Ye S, Li Z, Guo X, He Q. 2015. Comparative genomic analysis of classical and variant virulent parental/attenuated strains of porcine epidemic diarrhea virus. Viruses 7:5525–5538. 10.3390/v7102891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song D, Oh J, Kang B, Yang JS, Moon H, Yoo HS, Jang Y, Park B. 2007. Oral efficacy of Vero cell attenuated porcine epidemic diarrhea virus DR13 strain. Res Vet Sci 82:134–140. 10.1016/j.rvsc.2006.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wrapp D, McLellan JS. 2019. The 3.1-angstrom cryo-electron microscopy structure of the porcine epidemic diarrhea virus spike protein in the prefusion conformation. J Virol 93:e00923-19. 10.1128/JVI.00923-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madu IG, Roth SL, Belouzard S, Whittaker GR. 2009. Characterization of a highly conserved domain within the severe acute respiratory syndrome coronavirus spike protein S2 domain with characteristics of a viral fusion peptide. J Virol 83:7411–7421. 10.1128/JVI.00079-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millet JK, Whittaker GR. 2015. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res 202:120–134. 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G, Shi Z-L. 2020. Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. BioRxiv 10.1101/2020.01.22.914952. [DOI]
- 33.Hu H, Jung K, Vlasova AN, Chepngeno J, Lu Z, Wang Q, Saif LJ. 2015. Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J Clin Microbiol 53:1537–1548. 10.1128/JCM.00031-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hofmann M, Wyler R. 1988. Propagation of the virus of porcine epidemic diarrhea in cell culture. J Clin Microbiol 26:2235–2239. 10.1128/JCM.26.11.2235-2239.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.St-Jean JR, Desforges M, Almazán F, Jacomy H, Enjuanes L, Talbot PJ. 2006. Recovery of a neurovirulent human coronavirus OC43 from an infectious cDNA clone. J Virol 80:3670–3674. 10.1128/JVI.80.7.3670-3674.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye C, Chiem K, Park J-G, Oladunni F, Platt RN, Anderson T, Almazan F, de la Torre JC, Martinez-Sobrido L. 2020. Rescue of SARS-CoV-2 from a single bacterial artificial chromosome. mBio 11:e02168-20. 10.1128/mBio.02168-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang G, Liang R, Liu Z, Shen Z, Shi J, Shi Y, Deng F, Xiao S, Fu ZF, Peng G. 2019. The N-terminal domain of spike protein is not the enteric tropism determinant for transmissible gastroenteritis virus in piglets. Viruses 11:313. 10.3390/v11040313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Follis KE, York J, Nunberg JH. 2006. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 350:358–369. 10.1016/j.virol.2006.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menachery VD, Dinnon KH, Yount BL, McAnarney ET, Gralinski LE, Hale A, Graham RL, Scobey T, Anthony SJ, Wang L, Graham B, Randell SH, Lipkin WI, Baric RS. 2020. Trypsin treatment unlocks barrier for zoonotic bat coronavirus infection. J Virol 94:e01774-19. 10.1128/JVI.01774-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, Müller MA, Drosten C, Pöhlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280.e8. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wanitchang A, Saenboonrueng J, Kaewborisuth C, Srisutthisamphan K, Jongkaewwattana A. 2019. A single V672F substitution in the spike protein of field-isolated PEDV promotes cell-cell fusion and replication in Vero E6 cells. Viruses 11:282. 10.3390/v11030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]