

Japanese encephalitis virus (JEV) is the leading cause of viral encephalitis epidemics in Southeast Asia, affecting mostly children, with high morbidity and mortality. Identification of host factors is thus essential for the rational design of antivirals that are urgently need as therapeutics.
KEYWORDS: dengue virus, Japanese encephalitis virus, UFD-1, West Nile virus, clathrin-coated vesicles, flavivirus, valosin-containing protein, viral replication, virus entry
ABSTRACT
Host factors provide critical support for every aspect of the virus life cycle. We recently identified the valosin-containing protein (VCP)/p97, an abundant cellular ATPase with diverse cellular functions, as a host factor important for Japanese encephalitis virus (JEV) replication. In cultured cells, using small interfering RNA (siRNA)-mediated protein depletion and pharmacological inhibitors, we show that VCP is crucial for replication of three flaviviruses, JEV, dengue, and West Nile viruses. An FDA-approved VCP inhibitor, CB-5083, extended survival of mice in the animal model of JEV infection. While VCP depletion did not inhibit JEV attachment on cells, it delayed capsid degradation, potentially through the entrapment of the endocytosed virus in clathrin-coated vesicles (CCVs). Early during infection, VCP-depleted cells showed an increased colocalization of JEV capsid with clathrin and also higher viral RNA levels in purified CCVs. We show that VCP interacts with the JEV nonstructural protein NS5 and is an essential component of the virus replication complex. The depletion of the major VCP cofactor UFD-1 also significantly inhibited JEV replication. Thus, mechanistically, VCP affected two crucial steps of the JEV life cycle—nucleocapsid release and RNA replication. Our study establishes VCP as a common host factor with a broad antiviral potential against flaviviruses.
IMPORTANCE Japanese encephalitis virus (JEV) is the leading cause of viral encephalitis epidemics in Southeast Asia, affecting mostly children, with high morbidity and mortality. Identification of host factors is thus essential for the rational design of antivirals that are urgently need as therapeutics. Here, we have identified the valosin-containing protein (VCP) as one such host-factor. This protein is highly abundant in cells and engages in diverse functions and cellular pathways by its ability to interact with different cofactors. Using small interfering RNA (siRNA)-mediated protein knockdown, we show that this protein is essential for release of the viral RNA into the cell so that it can initiate replication. The protein plays a second crucial role for the formation of the JEV replication complex. FDA-approved drugs targeting VCP show enhanced mouse survival in JE model of disease, suggesting that this could be a druggable target for flavivirus infections.
INTRODUCTION
The genus Flavivirus comprises 53 virus species with nearly 30 viruses of medical importance (1). Its members, such as Japanese encephalitis virus (JEV), dengue virus (DENV), yellow fever virus (YFV), West Nile virus (WNV), and Zika virus, pose a significant threat to human health across the world (2). Urbanization, transportation, and climate change have led to an expansion of the flavivirus horizon, which makes them viruses of global relevance. No antivirals are currently available against any of the flaviviruses. Understanding the virus life cycle and the role of the host proteins in virus replication will significantly assist in this direction.
All flaviviruses share common features of a 50-nm enveloped particle with an ∼11-kb-long, positive-sense single-stranded RNA genome that is packaged with the help of a capsid protein. Once the virus enters a host cell by receptor-mediated endocytosis, the genome is released as the RNA-protein complex (RNP). The viral RNA is translated into a single polypeptide with the help of the host machinery and is processed by the host and viral proteases to make three structural (envelope, E; precursor membrane protein, prM; capsid, C) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) (3, 4). Viruses are known to hijack the cell machinery for their life cycle (3). The flaviviral RNA replication takes place on endoplasmic reticulum (ER)-derived vesicles and is accomplished with the help of various viral (NS1, NS4A, NS3, NS5, etc.) and host factors (for example, DDOST, hnRNPk, RPL19, RPS3, IPO9, GRP78, LC3I, PTB, etc.) (5–9). The virus life cycle is heavily dependent on the host membrane trafficking network for entry and egress. It also requires extensive membrane rearrangements, such as the formation of replication factories at the ER and transportation in a membranous structure for egress (10, 11).
To identify the host factors involved in the JEV life cycle, we previously carried out a small interfering RNA (siRNA) screen for the membrane trafficking genes (12, 13). Valosin-containing protein (VCP)/p97 was identified as a crucial host factor involved in JEV replication. VCP is a member of the AAA+ family of ATPase proteins with diverse cellular activities. The VCP monomer has three domains; the N domain interacts with the various adaptors and substrates, and D1 and D2 are the ATPase domains. The protein functions in a homohexameric state wherein six monomers come together to form a barrel-like structure (14). VCP is a known segregase protein that recognizes ubiquitin signature on the target protein directly or with the help of adaptor proteins. The target proteins can either be degraded through the ubiquitin proteasomal system or be extracted from their aggregates to perform their specific function (15). VCP thus plays essential and diverse roles in the ER-associated degradation (ERAD), the ubiquitin-proteasome system (UPS), cell cycle regulation, endosome trafficking, autophagy, mitophagy, and DNA repair (16–19). This protein has also been studied in the context of several viruses, such as Yellow fever virus (YFV), poliovirus (PV), hepatitis C virus (HCV), coronavirus, human cytomegalovirus (HCMV), Sindbis virus, chikungunya virus, and is involved in different steps of the virus life cycle (20–27).
In this report, we show that VCP plays a crucial role in the life cycle of flaviviruses. We have further characterized the role of VCP in JEV entry and replication. We show that VCP governs both the viral nucleocapsid release from the clathrin-coated vesicle (CCV) and the virus replication as a part of the virus replication complex. Pharmacological inhibitors of the VCP show a significant antiviral effect in the cell culture and the mouse model of infection. Our study establishes VCP as a crucial host factor with the potential to aid in the development of a broad antiviral against the flaviviruses.
RESULTS
VCP has a crucial role in flavivirus replication.
A recent RNA interference-based study in our lab targeting membrane-trafficking genes established VCP as a key host factor for JEV replication (12). An earlier study has shown VCP’s role in WNV replication (28). To further characterize the role of VCP in the flavivirus life cycle, replication of three flaviviruses—JEV, DENV-2, and WNV—was studied in HeLa and Huh7 cells where endogenous VCP levels were depleted by siRNA treatment. The siRNA-treatment did not affect the cell viability at 48, 72, and 96 h posttransfection (hpt), whereas a significant VCP knockdown was observed at both the transcript and protein levels (Fig. 1A). A 70 to 80% reduction in the VCP levels was observed in cells at 72 hpt (Fig. 1A). The siRNA-treated HeLa cells (for JEV) and Huh7 cells (for DENV-2 and WNV) were infected with the virus at 72 hpt, and the viral RNA levels and titers were compared at 24 h postinfection (hpi). VCP depletion resulted in a significant reduction in the viral RNA levels and titers at 24 hpi. A reduction in the viral RNA levels of ∼80% for JEV, and ∼60% for DENV-2 and WNV, was seen (Fig. 1B). Importantly, the JEV titers were reduced by ∼90%, and a reduction of ∼50% was seen in DENV-2 and WNV titers (Fig. 1B), thus indicating a crucial role of VCP in flavivirus replication. VCP levels in JEV-infected cells did not show any notable change (Fig. 1C). In accordance with reduced JEV RNA levels and titers under VCP depletion, a significant reduction was seen in viral protein translation (Fig. 1C) and in the number of infected cells (Fig. 1D) at 24 hpi.
FIG 1.
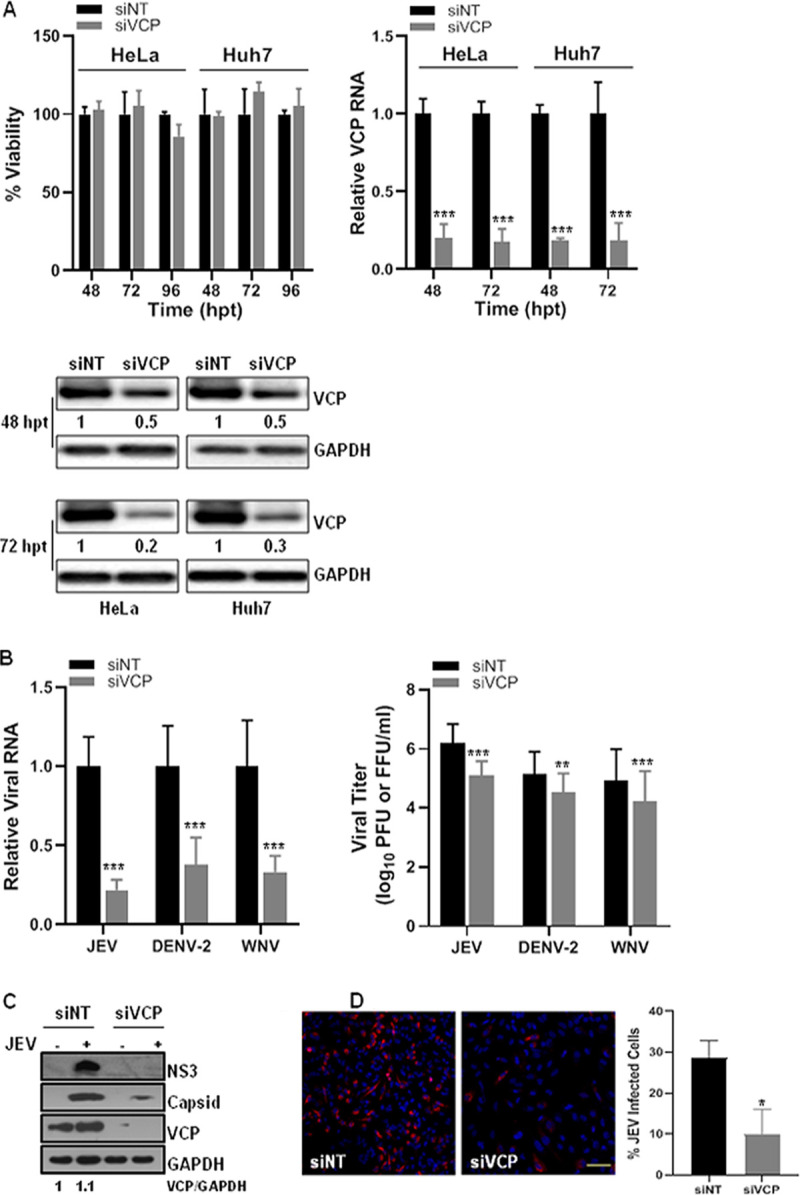
VCP knockdown reduces flavivirus replication. (A) HeLa and Huh7 cells were transfected with the nontargeting siRNA (siNT) or VCP siRNA (siVCP) and processed at different time points posttransfection (p.t.). The bar graph in the top-left panel shows the viability of the siRNA-treated cells. The level of VCP mRNA relative to the GAPDH transcript is shown in the top-right panel. The bottom panel shows the Western blots depicting VCP levels, and GAPDH as the loading control. The values below the VCP bands indicate VCP protein levels normalized to GAPDH. (B) The siRNA-treated HeLa cells were infected with JEV, and siRNA-treated Huh7 cells were infected with DENV-2 or WNV at 1 MOI. The left panel has the bar graph showing intracellular viral RNA levels at 24 hpi relative to GAPDH transcript, normalized to siNT treatment. The right panel shows extracellular virus titer at 24 hpi. Each experiment had biological triplicates, and the data shown are from three independent experiments. (C) Representative Western blots showing levels of JEV NS3 and capsid proteins at 24 hpi in siNT- and siVCP-treated HeLa cells infected with JEV (3 MOI). The GAPDH blot served as the loading control. (D) HeLa cells were treated with siNT or siVCP and at 72 hpt were infected with JEV (1 MOI). The cells were stained at 24 hpi with JEV NS1 antibody for immunofluorescence-based quantification of JEV infection. The left panel shows NS1 staining in siNT- and siVCP-treated HeLa cells (scale bar = 100 μm). The right panel has data from three independent coverslips. Student’s t test was used for comparing the data from the siVCP-treated cells with that from siNT-treated control cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Pharmacological inhibitors of VCP show strong antiviral effects.
VCP is an established drug target for cancer therapy, and some small molecule inhibitors of the VCP have been tested as drugs (29, 30). We evaluated three VCP inhibitors for their potential antiviral effects. CB-5083 {1-[7,8-dihydro-4-[(phenylmethyl)amino]-5H-pyrano[4,3-d]pyrimidin-2-yl]-2-methyl-1H-indole-4-carboxamide} is a reversible, competitive inhibitor of VCP ATPase activity. DBeQ (N2, N4-dibenzylquinazoline-2, 4-diamine) is a selective, reversible, ATP-competitive inhibitor that targets the two ATPase domains of VCP and blocks both autophagy and proteasomal degradation (31). Eeyarestatin 1 (Eer1) irreversibly binds to VCP and blocks the associated deubiquitinase ataxin-3, preventing deubiquitination of substrates and resulting in accumulation of misfolded proteins and unfolded protein response (UPR) induction (30). Nontoxic concentrations of these drugs were identified by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assays (Fig. 2A) and tested in the virus replication experiments. Drugs were added at 2 hpi, and the virus replication at 24 hpi was analyzed with quantitative real-time PCR (qRT-PCR) for the viral RNA synthesis and with plaque assays for the viral titer. While the JEV RNA synthesis and viral titers were significantly suppressed by all three drugs (Fig. 2B), the DENV-2 RNA synthesis and titers were inhibited predominantly by Eer1 (Fig. 2B). CB-5083 and Eer1 inhibited WNV RNA synthesis; however, only Eer1 showed some suppressive effect on virus titers (Fig. 2B). That Eer1 exerted a profound negative effect on the replication of all three flaviviruses suggested that the deubiquitinase activities of VCP might be crucial for the flavivirus life cycle.
FIG 2.

Pharmacological inhibition of the VCP suppresses flavivirus replication. (A) HeLa and Huh7 cells were treated with the indicated concentrations of the VCP inhibitors for 24 h. The viability of cells, as determined by MTT assay, is shown normalized to DMSO-treated cells. (B) HeLa cells were infected with JEV, and Huh7 cells were infected with DENV-2 or WNV at 1 MOI. VCP inhibitors were added to cells 2 hpi at the indicated concentrations. Cells and supernatants were harvested at 24 hpi to analyze the intracellular viral RNA levels by qRT-PCR (left panels) and extracellular viral titers by plaque assays (right panels). Viral RNA and titers in the drug-treated cells were normalized to the DMSO-treated cells. Each experiment had biological triplicates, and the data shown are from three independent experiments. One-way ANOVA followed by Tukey’s post hoc test was used for comparing the data from drug-treated cells with those treated with DMSO alone. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
CB-5083 is an orally bioavailable inhibitor of VCP, which is a potent inducer of the unfolded protein response and leads to retention of ERAD substrates and cancer cell death (31–34). Since the drug displayed strong anti-JEV effects in cultured cells, we tested it in the mouse model of virus infection. JEV-infected C57BL/6 mice were administered CB-5083 by oral gavage once every day, starting at 2 hpi (Fig. 3A). In the virus-infected group, typical JE symptoms such as movement restriction, hind-limb paralysis, and piloerection were observed in all mice by day 5 postinfection (p.i.). An average survival time of 10.8 days in the virus-infected group was recorded. In the drug-treated mice, there was a delay in the onset of JE symptoms; the symptoms first appeared on day 7 p.i., with an average survival time of 12.3 days. While the survival data were not significant, the drug-treated mice consistently showed lower virus titers in the brain on all days, and these were significantly decreased on day 7 p.i., suggestive of a protective effect of this drug in the mouse model (Fig. 3B). These observations are in line with the antiviral effect of CB-5083 seen in the tissue-cultured cells.
FIG 3.

VCP inhibitor CB-5083 enhances the survival of JEV-infected mice. Three-week-old C57BL/6 mice were mock-infected or injected with 107 PFU of JEV intraperitoneally. CB-5083 was dissolved in DMSO and freshly mixed with corn oil in a 1:9 ratio just before the treatment. The drug was administered to mock- and virus-infected mice at a dose of 30 mg/kg body weight by oral gavage at 2 hpi. The drug dose was repeated every 24 h until day 12 p.i. Mice were monitored for the visible symptoms of JEV infection and death. (A) The survival curve of mock- and JEV-infected mice with or without the drug treatment is shown. The number of mice in each group is indicated. The data are pooled from two independent experiments. (B) Virus titers in brains from the JEV-infected and the JEV-infected and treated with CB-5083 mouse groups are shown. Mean titers in each of the groups on different days are shown with a line. Statistical analysis was performed using Student’s t test. *, P < 0.05.
VCP depletion does not affect JEV attachment to the host cell.
We examined flavivirus entry in VCP-depleted cells by assaying the intracellular viral RNA levels at 1 hpi, which provided a direct estimate of the internalized virus (5). A significant increase in JEV RNA levels was observed in VCP-depleted cells, suggesting that JEV entry in the cell was not inhibited by VCP knockdown, but rather, was enhanced (Fig. 4A).
FIG 4.

VCP depletion does not affect JEV attachment to the host cell. (A) Virus binding was performed on HeLa cells with JEV (5 MOI) and on Huh7 cells with DENV-2 (10 MOI) or WNV (5 MOI) on ice for 1 h. After washing, the cells were incubated at 37°C in a complete cell culture medium for 1 h. The cells were trypsinized to remove the bound virus, and the levels of internalized virus were determined as viral RNA by qRT-PCR relative to the GAPDH transcript levels. The viral RNA levels in the siVCP-treated samples were normalized to their respective siNT-treated cell controls. (B) HeLa cells were transfected with siNT or siVCP, and 72 hpt were incubated with JEV (50 MOI) on ice for 1 h. Cells were washed with phosphate-buffered saline (PBS) and treated with trypsin as indicated to remove the extracellular attached virus. Western blotting was performed to detect the level of the attached virus using the JEV capsid antibody. The VCP Western blot shows the protein knockdown where GAPDH was used as the loading control. The bar graph shows the levels of JEV capsid protein bound to siNT- and siVCP-treated HeLa cells from three independent experiments. The protein quantitation was done by measuring the band intensities using ImageJ software. (C) The siNT- and siVCP-treated HeLa cells were incubated 72 hpt with JEV (1 MOI) for 1 h. Cells were washed, and the level of bound virus was determined by qRT-PCR relative to GAPDH levels, normalized to siNT treatment. The bar graph shows the relative RNA levels of bound JEV virions from three independent experiments. (D) HeLa cells were incubated with JEV (1 MOI) on ice for 1 h, followed by incubation at 37°C. Cells were harvested at the indicated time points to analyze the viral RNA levels. Relative JEV RNA levels were normalized to siNT treatment at 1 hpi. Each experiment had biological triplicates, and the data are plotted from two independent experiments. The data for the siVCP-treated cells were compared with those for the siNT-treated cells using Student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We next dissected the role of VCP in the JEV life cycle by examining different steps of the virus infection process. We first analyzed the JEV attachment on cells by Western blotting for the capsid protein in nontargeting siRNA (siNT)- and VCP siRNA (siVCP)-treated HeLa cells at 1 h post-virus binding on ice (Fig. 4B). A significant amount of virus (as seen by the capsid protein band intensity) was found adhered to cells, which could be efficiently removed by trypsin treatment. The capsid signal here represents the bound but uninternalized virus, as virus binding on ice does not permit virus entry. The data also showed that all bound virus could be removed from the cells by trypsin treatment (Fig. 4B). Quantification of bound capsid protein levels (Fig. 4B, bar graph) and bound viral RNA levels (Fig. 4C) showed no significant difference between the two conditions, suggesting that virus binding to cells is not affected by VCP depletion. These data suggest that virus receptor levels are unlikely to be altered by VCP deficiency.
We next studied the time kinetics of viral RNA levels during the JEV replication and observed that the siNT-treated cells showed a previously described pattern of JEV RNA levels (7), wherein the viral RNA levels showed an ∼50% decrease at 6 hpi compared to 1 hpi, suggestive of some viral RNA degradation upon nucleocapsid uncoating (Fig. 4D). In these cells, the viral RNA replication proceeded rapidly after 6 hpi. Compared to the siNT-treated cells, the VCP-depleted cells showed higher levels of viral RNA during the early phase of virus life cycle (both at 1 h and at 6 hpi), although at the later time points (12 and 24 hpi), the viral RNA levels were significantly reduced (Fig. 4D). Collectively, these data point to two effects of VCP depletion on the JEV replication cycle, at the early time points that correspond to the nucleocapsid release and genomic RNA uncoating and later during the genomic RNA replication.
VCP depletion delays internalized virus capsid degradation.
Acidification occurring along the endocytic pathway is essential for the uncoating of the flavivirus particle and release of the RNP complex. After the RNP release, the capsid protein is degraded, and the viral RNA is free to initiate translation. Earlier studies have shown that VCP has a vital role in endosome maturation and membrane trafficking pathways (24, 35–37). To check if VCP is affecting an early step of the virus life cycle, levels of the internalized viral RNA were determined at early time points p.i. (Fig. 5A). We observed significantly higher levels of JEV RNA in VCP-depleted cells at 3 hpi, which could be indicative of a defect in virus uncoating and viral RNA release. To check for this possibility, virus capsid protein levels were analyzed during the early hours of the virus replication cycle (Fig. 5B). HeLa cells were infected with JEV at a multiplicity of infection (MOI) of 50, and trypsinization was done before the lysate preparation to ensure that only the internalized virus was detected. The early time points (1, 2, and 3 hpi) were chosen, as viral RNA translation does not initiate by this time, and the capsid detected will be indicative of the internalized virus only (5). In the siNT-treated JEV-infected cells, the capsid protein was detectable at 1 hpi, and its levels progressively decreased, with protein being undetectable by 3 hpi, indicative of the virion disassembly and genome release. However, in the VCP-depleted cells, the internalized capsid protein could be detected until 3 hpi (Fig. 5B). We also quantified the endocytosed JEV by immunofluorescence analysis of the capsid protein at 3 hpi in the siNT- and siVCP-treated cells. Numerous capsid puncta were specifically detected in the JEV-infected cells (Fig. 5C). Quantification of the data revealed a significantly higher number of capsid puncta per cell (Fig. 5C), and higher capsid protein levels (Fig. 5D), in the VCP-depleted cells than in the siNT-treated cells. We also tested capsid protein levels under conditions of protein synthesis and degradation block and observed an enhancement in the band intensity in siNT-treated cells, suggesting that capsid protein starts undergoing degradation by 3 hpi under control conditions (Fig. 5D). This experiment could not be done in siVCP-treated cells due to the loss of cell viability on bortezomib and cycloheximide treatment. Collectively, these observations are indicative of two possibilities; either the VCP depletion is leading to entrapment of the internalized virus in an endocytic compartment, or the VCP is involved in the degradation of the capsid protein in the RNP complex.
FIG 5.

VCP depletion delays the internalized virus capsid degradation. (A) The siNT- and siVCP-treated HeLa cells were incubated 72 hpt with JEV (1 MOI) for 1 h on ice. After washing, cells were incubated at 37°C in a complete cell culture medium. At the indicated time points, cells were trypsinized to remove the bound virus, and levels of internalized virus were determined by qRT-PCR relative to the GAPDH transcript levels. The relative JEV RNA levels were normalized to siNT treatment at 1 hpi. The data are derived from three independent experiments. (B) HeLa cells were transfected with siNT or siVCP, and 72 h later were incubated on ice for 1 h with JEV (50 MOI). After a PBS wash, cells were incubated at 37°C in complete cell culture medium. At the indicated time points, cells were treated with trypsin, and cell lysates were prepared. The internalized virus was quantified by Western blotting for JEV capsid (left panel). The VCP blot shows the level of protein knockdown, and GAPDH blots are loading controls. The bar graph in the right panel shows the densitometric analysis of capsid protein bands normalized to GAPDH of the respective sample, performed using ImageJ software. All time points were plotted relative to the value for 1 hpi of the respective treatment groups. Data are presented as the mean and standard deviation of the values obtained from 3 independent experiments. (C) HeLa cells were treated with siNT or siVCP, and 72 hpt were incubated with JEV (100 MOI) for 1 h on ice and then incubated at 37°C for 3 h. The cells were fixed and immunostained for the JEV capsid. Representative images are shown. Scale bar, 10 μm. The number of capsid puncta per cell were counted using Fiji particle analysis. Each experiment had biological triplicates, and the weighted mean ± standard error (SE) are plotted in the right panel using two independent experiments. The data for the siVCP-treated cells were compared with those for the siNT-treated cells using Student’s t test. *, P < 0.05; **, P < 0.01. (D) HeLa cells were transfected with siNT or siVCP, and 72 h later, siNT cells were treated with a combination of bortezomib (BTZ, 1 μM) and cyclohexamide (CHX, 100 μg/ml) for 1 h followed by infection with JEV (50 MOI) for 1 h on ice. After a PBS wash, cells were incubated at 37°C in complete cell culture medium in the presence of BTX+CHX until 3 hpi. Cells were treated with trypsin, and cell lysates were prepared. The internalized virus was quantified by Western blotting for JEV capsid, as shown in the left panel. The VCP blot shows the level of protein knockdown, and GAPDH blots are loading controls. SE is the short exposure of the blot (upper panel), while LE is the long exposure (lower panel) of the same blot. The bar graph in the right panel shows the densitometric analysis of capsid protein bands normalized to GAPDH of the respective sample. Data are presented as the mean and standard deviation of the values obtained from 2 independent experiments. Statistics were performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
VCP depletion traps JEV in an early endocytic compartment.
Earlier studies have shown that VCP associates with clathrin and Hsc 70 (38) and also with the early endosome autoantigen EEA1 (36). VCP depletion by siRNA leads to clustering and enlargement of early endosomes and altered trafficking of transferrin (Tf) (36, 37). We analyzed the kinetics of the Tf receptor cycle by quantification of labeled Tf uptake at different times under siNT- and siVCP-treated conditions. In accordance with the published studies, we observed that VCP depletion moderately impacted Tf kinetics (Fig. 6A). Higher levels of internalized Tf were observed in VCP-depleted cells at 10 min onward, suggesting a delay in Tf recycling (Fig. 6A). We next performed endosome fractionation of the siNT- and siVCP-treated HeLa cells that were infected with JEV and treated with trypsin at 3 hpi, followed by qRT-PCR to determine viral RNA levels in different fractions (Fig. 6B). For the siNT-treated cells, EEA1 was detected in fractions 5 and 6, while clathrin heavy chain (CHC) was seen in fractions 6 to 9 (Fig. 6B, left panel). The VCP-depleted cells showed a broader distribution for EEA1 (fractions 6 to 8) which could reflect enlarged and immature early endosomes. While similar RNA levels were seen across fractions 5 and 6, fraction 7 showed significantly higher JEV RNA in the VCP-depleted cells (Fig. 6B, right panel). This fraction was positive for CHC but negative for EEA1 under the VCP-depleted conditions, suggesting that the viral RNA could be entrapped in clathrin-coated vesicles (CCVs). We further purified CCVs from both siNT- and siVCP-depleted cells and checked for capsid protein and JEV RNA in the purified fraction (Fig. 6C). Higher capsid protein and JEV RNA levels were observed in the purified CCV from VCP-depleted cells, suggesting that the virus particles could be trapped in the CCV, restricting the virus genome release during the early phase of virus infection.
FIG 6.

VCP depletion traps JEV in an early endocytic compartment. (A) Trafficking kinetics of Tf were measured in siNT- and siVCP-treated cells. Cells were given a pulse of Alexa 488 Tf for the indicated time points. The graph in the left panel shows the mean fluorescence intensity of Tf plotted against time. The solid line represents the logarithmic trendline. The right panel has the bar graph showing the levels of internalized Tf. Each experiment was performed using duplicate coverslips for each time point, and intensity measurements were made from ∼100 cells per coverslip. The data are from two independent experiments and are plotted as the weighted mean ± SE. Statistical analysis was performed using Student’s t test. *, P < 0.05. (B) HeLa cells were treated with siNT or siVCP, and 72 hpt were infected with JEV (50 MOI). The cells were harvested at 3 hpi by treating them with trypsin. Cells were homogenized, and the nuclei were removed by centrifugation. The supernatant was subjected to density gradient endosome fractionation as described in Materials and Methods. The left panel has the Western blots showing the presence of EEA1 and CHC in the different fractions. The total RNA was isolated from the equal volume of each of the fractions, and the JEV RNA levels were quantified by qRT-PCR. The bar graph in the right panel shows the absolute threshold cycle (CT) values in different fractions. The data shown are representative of three independent experiments. (C) Clathrin-coated vesicles (CCV) were isolated from siNT- and siVCP-treated and JEV-infected (50 MOI) HeLa cells. Representative Western blots showing levels of different early and late endosome marker proteins and JEV capsid (SE, short exposure; LE, long exposure) in the total cell lysate and purified CCV are presented. The bar graph on the right shows relative JEV RNA in CCV isolated from siNT- and siVCP-treated HeLa cell CCV fractions. The relative JEV RNA levels were normalized to siNT treatment. Data are presented as the mean and standard deviation of the values obtained from 2 independent experiments. Significance was established using Student’s t test. *, P < 0.05.
We further performed immunofluorescence experiments to test if the trapped virus particles could be detected in the EEA1- or clathrin-positive endosomes in the VCP-depleted cells (Fig. 7A). The siNT- and siVCP-treated cells were infected with JEV (100 MOI), and at 3 hpi were checked for the localization of the capsid with EEA1 or clathrin light chain (CLC). The VCP-depleted cells showed significantly more capsid puncta, and these showed significantly higher colocalization with the CLC- but not with the EEA1-positive structures (Fig. 7A and B). The higher colocalization of the endocytosed virus particles with the clathrin-positive structures in the VCP-depleted cells suggested a longer residence time for the virus in the CCV, which could potentially delay the progression of the virus through the early endocytic compartments. As reported earlier (36), we observed larger EEA1 endosomes in VCP-depleted cells (Fig. 7C). The total cellular levels of EEA1 and CLC were also increased in VCP-depleted cells (Fig. 7D), suggesting that VCP plays a critical role in maintaining the levels of these proteins.
FIG 7.

VCP depletion results in the viral arrest in clathrin-coated vesicles. HeLa cells were transfected with siNT or siVCP, and 72 hpt were infected with JEV (100 MOI). The cells were fixed at 3 hpi. (A) Cells were immunostained for JEV capsid and EEA1 or clathrin light-chain (CLC). Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole; blue). Insets in the upper two rows of the second panel (EEA1 staining) represent a magnified view of the region corresponding to the white box. Imaging was performed on the FV3000 Olympus confocal microscope. Scale bar, 10 μm. (B) Plot showing Pearson’s correlation coefficient between capsid and EEA1 or CLC in control and VCP-depleted cells. Each experiment had biological triplicates. The data are from two independent experiments and plotted as the weighted mean ± SE. (C) The bar graph shows mean ± standard deviation (SD) of early endosome size calculated from 8 fields (1 cell/field) using Fiji (particle analysis). (D) Representative Western blot showing levels of EEA1 and CLC in siNT- and siVCP-treated HeLa cells. The GAPDH blot is the loading control. The statistical significance was established using Student’s t test. *, P < 0.05; **, P < 0.01.
Role of VCP adaptors UBXD1 and UFD1 on JEV infection kinetics.
VCP has been shown to function as an interaction hub and interacts with nearly 30 different cofactors to carry out its diverse functions (14). In an attempt to identify potential VCP cofactors that could be playing a role in the context of JEV infection, we examined two major VCP adaptors, UBXD1 and UFD1, through siRNA depletion (Fig. 8A). UBXD1 is the only known VCP adaptor that has been implicated in endocytic trafficking, and since movement through the endosomal system is crucial for flavivirus infection, we checked if UBXD1 depletion would impact JEV infection. The VCP-UBXD1 complex has been shown to bind monoubiquitinated caveolin1 (Cav1) and is essential for Cav1 trafficking and degradation in lysosomes. UBXD1 depletion has been shown to block Cav1 in Rab5-positive enlarged endosomes (35). UBXD1 depletion had no effect on JEV entry (Fig. 8B) or any effect on JEV RNA replication and egress (Fig. 8C), indicating that the described roles of VCP-UBXD1 in the context of Cav1 are not involved in the JEV infection process.
FIG 8.
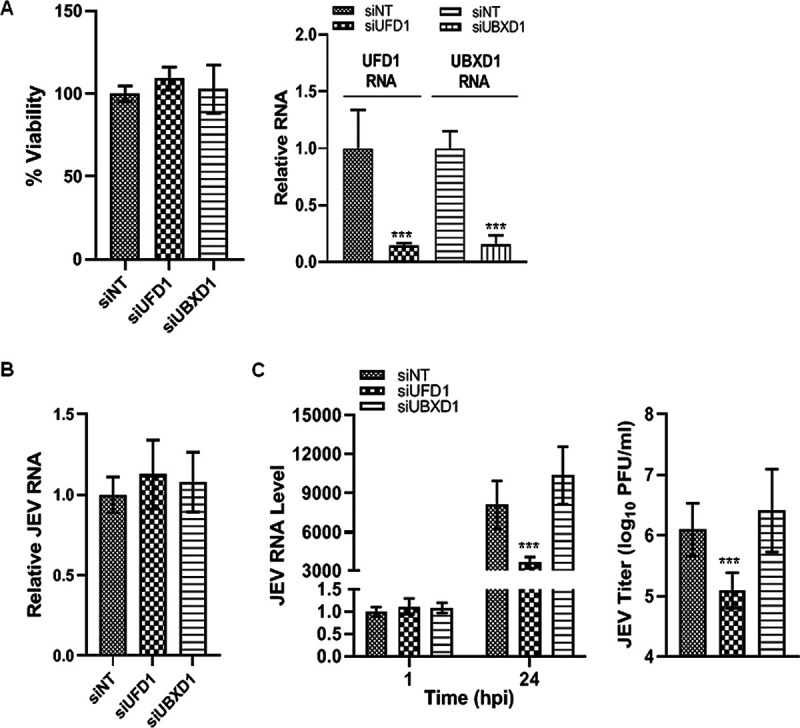
The VCP adaptor UFD1 complex has a vital role in the JEV life cycle. (A) HeLa cells were transfected with siNT, siUFD1, or siUBXD1. An MTT assay of transfected cells was performed at 48 hpt to establish the siRNA cytotoxicity (left panel). The bar diagram in the right panel shows relative RNA levels in siRNA-treated cells at 48 hpt normalized to that in the siNT-treated cells. (B) The JEV entry experiment was performed as described previously. Intracellular viral RNA levels in siRNA-treated cells were analyzed by qRT-PCR and normalized to that in the siNT-treated cells. (C) HeLa cells were infected with JEV at 1 MOI. Samples were harvested at 1 and 24 hpi. The qRT-PCR was performed to analyze the intracellular RNA levels relative to GAPDH. The left panel shows the RNA levels at 1 hpi normalized to that seen in the siNT-treated cells, and at 24 hpi normalized to that seen at 1 hpi for the respective siRNA treatment. The right panel shows the virus titers in the culture supernatants at 24 hpi. Each experiment had biological triplicates. The data are from three independent experiments. The statistical significance was established using Student’s t test. *, P < 0.05; **, P < 0.01.
Another well-characterized VCP cofactor is UFD1. VCP forms a major core complex with the cofactor UFD1 (ubiquitin fusion degradation 1)-NPL4 (nuclear protein localization homolog 4) heterodimer, which is essential for ERAD through the dislocation of polyubiquitinated substrates from the ER to the cytosol (39). Flavivirus replication is closely associated with ER-derived membranes (11, 40), and we thus hypothesized that UFD1 could be involved in the JEV replication process. The depletion of UFD1 did not affect JEV entry (Fig. 8B); however, it significantly inhibited JEV replication and infectious virus titer in the culture supernatant (Fig. 8C), indicating its crucial role at a later time point of infection. Since JEV replication is intricately linked to ERAD (7), it is likely that the inhibition of virus replication through UFD1 depletion is due to its impact on ERAD.
VCP is an essential host factor for JEV RNA replication.
VCP is a chaperone performing a variety of distinct and unrelated functions in the cell. To study if VCP is also involved at the later steps of the virus life cycle, we bypassed the virus entry by transfecting the siNT- and siVCP-treated cells with the viral RNA (Fig. 9). Similar levels of transfected viral RNA were detected under the two conditions (Fig. 9A). Viral replication as seen by immunofluorescence staining for viral NS1 protein was detected in siNT-treated cells at 12 hpt (∼3%), and the number of infected cells did not increase until 24 hpt, indicating just the first round of virus replication (Fig. 9C). The intensity of NS1 signal in the infected cells, however, increased from 12 to 24 hpt, suggestive of viral protein translation and accumulation (Fig. 9D). In comparison, infected cells were virtually undetectable under the siVCP-treated conditions (Fig. 9B). No detectable virus titers were observed in either condition at 12 hpt. At 24 hpt, while the viral RNA levels and titers increased in the control siNT-treated cells, indicating RNA replication, these remained almost stalled in the VCP-depleted cells, establishing a crucial role of the protein to achieve the viral RNA replication (Fig. 9E).
FIG 9.

VCP plays a critical role in JEV RNA replication. JEV viral RNA was isolated from viral supernatant using RNA extraction columns. HeLa cells were transfected with siNT or siVCP, and 72 hpt were transfected with 250 ng viral RNA. (A) Relative levels of JEV RNA in siNT- and siVCP-treated HeLa cells at 6 hpt normalized to siNT treatment. (B) Immunofluorescence staining for JEV-infected cells at 6, 12, and 24 hpt. (C) Bar graph showing quantification of NS1-positive cells under the siNT conditions at 12 and 24 hpt. (D) Fluorescence intensity distribution of NS1 in positive cells under the siNT conditions at 12 (n = 28) and 24 hpt (n = 45). (E) Cells were harvested at 24 hpi. The left panel shows the relative JEV intracellular RNA levels determined by qRT-PCR normalized to those seen in the siNT-treated cells. The right panel shows the virus titer in the culture supernatant at 24 hpi. Each experiment had biological triplicates, and the data are from two independent experiments. The data in siVCP-treated cells were compared with those from the siNT-treated controls using Student’s t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To check if VCP had a role in the formation of the virus replication complex, we checked for the colocalization of VCP with the JEV proteins NS1 and NS5 at 24 hpi (Fig. 10A). Our earlier studies have established that the JEV replication complex can be detected by immunofluorescence staining of the NS1 and NS5 proteins starting from 10 hpi (5, 7). VCP distribution in JEV-infected cells showed as larger puncta around the nucleus. VCP showed strong colocalization with NS5 and NS1, suggesting its potential role in virus replication. In contrast, very little overlap was seen between VCP and capsid, which is known to be in close proximity, but distinct from the replication complex (41). Further, the specificity of the replication complex localization was confirmed by staining with the ER marker calnexin (Fig. 10A).
FIG 10.

VCP colocalizes to the JEV RNA replication complex. (A) Mock- or JEV-infected (3 MOI) HeLa cells were fixed at 24 hpi and immunostained for VCP and calnexin along with JEV NS1, NS5, or capsid. Images were acquired on a confocal microscope using a ×60 objective. The bar graph shows Pearson’s correlation coefficient of colocalization between NS1/VCP, NS5/VCP, capsid/VCP, and NS1/calnexin. Scale bar, 10 μm.
We next performed immunoprecipitation studies to confirm this interaction. We observed that VCP could immunoprecipitate the JEV NS5 from the virus-infected cells and vice-versa (Fig. 11A). We also performed coimmunoprecipitation experiments in cells overexpressing NS5-HA and NS3-HA. VCP could not be detected in the immunoprecipitates from the overexpressing cells, suggesting that the NS5 and VCP interaction requires additional host factors/adaptors or viral proteins available during the virus replication (Fig. 11B). Interestingly, overexpressed NS5-HA could immunoprecipitate VCP and NS3 from the JEV-infected cells but not from the mock-infected cells (Fig. 11C), further validating the role of the virus replication for VCP interaction with the JEV NS5.
FIG 11.
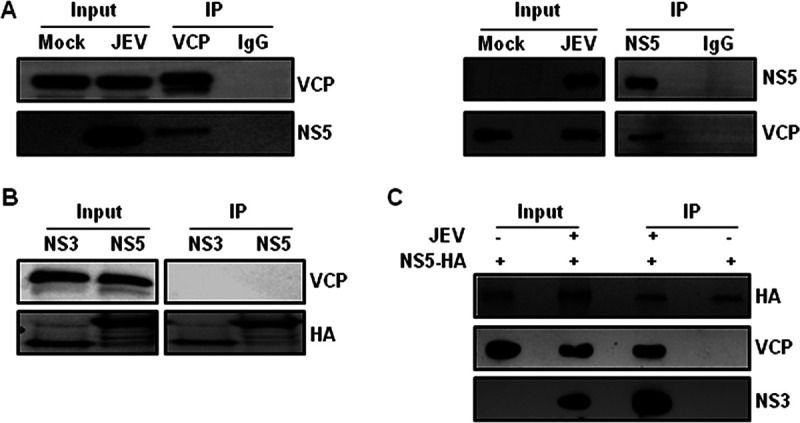
VCP interacts with JEV NS5 in infected cells. (A) HeLa cells were infected with JEV (3 MOI), and the cell lysates prepared at 24 hpi were immunoprecipitated (IP) using the VCP antibody or rabbit IgG as the isotype control (left panel), and NS5 antibody or rabbit IgG (right panel). The input and the IP samples were Western blotted with the VCP and NS5 antibodies, as indicated on the right side of the blots. (B) HEK 293T cells were transfected with 1 μg plasmid expressing NS5-HA or NS3-HA. The cell lysates were prepared at 24 hpt, and immunoprecipitation was performed using the HA antibody. The input and the IP samples were Western blotted with the VCP and HA antibodies, as indicated at the right side of the blot. (C) NS5-HA transfected HEK 293T cells were either mock- or JEV-infected (10 MOI), and lysates prepared at 24 hpi were immunoprecipitated using the HA antibody. Western blotting was done using the HA, VCP, or NS3 antibodies, as indicated at the right side of the blot.
DISCUSSION
VCP/p97 belongs to the AAA+ family of proteins and is one of the most abundant and complex proteins found in the cell (16, 18, 19). The protein is conserved across eukaryotes and has a nonredundant function during development (42). In the case of humans, mutations in VCP lead to late adulthood development of inclusion body myopathy associated with Paget’s disease of bone and frontotemporal dementia (IBMPFD) (43). The protein was first characterized as a “segregase,” as it extracts ubiquitinated proteins from cellular structures, membranes, and aggregates and leads to their degradation by the proteasome. It ensures the degradation of terminally misfolded/damaged proteins in different cellular compartments (ER, mitochondria, nucleus) and thus plays a crucial role in maintaining cellular homeostasis. However, VCP can also bind to proteins tagged with monoubiquitin/nondegradative ubiquitin chains and can perform other cellular functions, such as disassembly of RNA-protein complexes and separation of transcription factors from chromatin (44, 45). VCP regulates and drives multiple cellular functions based upon specific combinatorial interactions with diverse cofactors, 30 of which are known to date (15). Various VCP cofactor proteins are known and characterized, such as UFD1-NPL4, p47, UBXD1, UBXD2, gp78, HRD1, VCIP135, and many more (19). These cofactors can bind to the VCP N or C terminus and provide spatiotemporal specificity for its diverse functions in the cell.
Given its abundance and multiple roles in several cellular processes, it is perhaps not surprising that VCP is an important host factor for virus replication/life cycle. VCP has been studied in the context of several viruses, such as poliovirus, enterovirus (EV) 71, coronavirus, HCMV, Sindbis virus (SINV), vesicular stomatitis virus (VSV), Rift Valley fever virus (RVFV), chikungunya virus, HCV, and WNV (20–26, 28, 46, 47). In this study, we have examined the role of VCP in the context of the flavivirus life cycle and dissected its role in detail for JEV. Below, we analyze the role that VCP plays in the flavivirus life cycle and, in particular, JEV.
VCP depletion does not affect JEV attachment.
Our study shows that JEV attachment on cells was not affected by VCP deficiency. Flavivirus entry is primarily mediated through clathrin-mediated endocytosis (CME) (12, 48, 49). While an earlier study has shown that VCP depletion delays transferrin receptor (TfR) recycling, (36) it was observed not to have any effect on the plasma membrane levels of TfR or other adhesion molecules (cadherin, JAM1), ion transporters (ATP1A1, ATP7A), and poliovirus receptor. The entry of the alphaviruses chikungunya virus (CHIKV) and Ross River virus (RRV) through CME and Junin pseudotyped virus through the TfR, was also shown to be unaffected by VCP depletion (24). However, the same study showed that VCP depletion altered the expression of the major cellular iron transporter NRAMP2 and β1 integrin and blocked SINV entry through NRAMP2. These effects were independent of ERAD and were suggested to be caused by mislocalization of NRAMP2 to lysosomes, suggesting cargo-specific regulation of membrane trafficking by VCP (24). Similar levels of virus attachment observed in our study indicate that virus receptor levels are not significantly altered under VCP-deficient conditions.
VCP depletion traps the virus nucleocapsid in CCV.
VCP has been shown to interact with clathrin and EEA1 and regulate early endosomal dynamics and trafficking of the TfR and caveolin 1 (35). Interestingly, we also observed higher CLC and EEA1 levels in VCP depleted cells. Accumulation of coronaviruses in the early endosomes of VCP-depleted cells has been reported (23). Since VCP inactivation leads to the formation of large immature early endosomes (36, 37), we hypothesized that these conditions might lead to the entrapment of the internalized virion particles in the early endocytic compartments. This arrest of virus particles in the early endosomes could lead to reduced and/or delayed release of the virus-RNP complex. We observed higher capsid protein and viral RNA levels in purified CCV from VCP-depleted cells. There was also enhanced internalized virus capsid colocalization with the clathrin light chain, but not with EEA1. Collectively, these data are indicative of virus entrapment in CCVs under VCP-deficient conditions.
Vaccinia virus genome release was shown to be ubiquitination-independent, but it required the proteasomal activity with the core protein existing in a ubiquitinated state inside the virion (50). Ubiquitination was described to be essential for uncoating of the influenza A virus (IAV) also, wherein the capsid protein was associated with ubiquitin chains noncovalently in the virion (51). The E3 ubiquitin ligase Itch was shown to be essential for the IAV release from endosomes (52). Ubiquitination, but not a proteasomal activity, was shown to be essential for the DENV genome uncoating and release (53). Though direct ubiquitination of the DENV capsid was not seen, the internalized capsid protein was degraded by the host ubiquitin-proteasomal system; however, proteasomal activity and capsid degradation were not required for the initiation of the first round of viral translation. The study concluded that capsid protein degradation takes place independently of the engagement of the viral genome in viral translation. Using a yellow fever reporter virus, a recent study showed an essential role for ubiquitination in an early prereplication step of the virus life cycle that was also sensitive to the VCP pharmacological inhibitors DBeQ and NMS-873 (27). In our study, we did not detect ubiquitination on internalized JEV virion capsid protein (data not shown). However, since overexpressed JEV capsid protein undergoes K48 ubiquitination (our unpublished observations), it is possible that the ubiquitination of the capsid occurs at the level of nucleocapsid disassembly.
VCP in complex with the UBXD1 cofactor was essential for sorting mono-ubiquitinated Cav1 to the lysosome for degradation (35); however, UBXD1 depletion did not impact JEV replication at any step, ruling out its involvement.
VCP is critical for JEV replication and is a part of the replication complex.
We established an essential role for VCP in JEV replication through the transfection of viral RNA, thus bypassing the JEV endocytosis and nucleocapsid uncoating. Our study also showed that VCP displayed a strong colocalization with JEV NS5 and NS1. NS5 is the RNA-dependent RNA polymerase (RdRP), and NS1 also plays a critical role in flavivirus RNA replication (54, 55). Thus, VCP localizes with components of the virus replication complex and is likely to play an important role in virus replication. The VCP-NS5 interaction was also validated by immunoprecipitation experiments. Published literature has shown VCP to be associated with the HCV viral replicase, and its ATPase activity was essential for virus replication (21). In this context, it may be of interest to note that the VCP pharmacological inhibitor CB-5083, which interferes with its ATPase activity, caused significant inhibition of JEV RNA synthesis and viral titers, suggesting that the ATPase activity of the VCP may have a role in the JEV RNA replication. Besides the role as the ATPase in the virus replication, studies on the HCV replication complex suggest that VCP might have a role in the disaggregation of aggregation-prone replicase components to maintain its functionality (22). Additionally, VCP might have a role in the Alphavirus replication, as it was shown to colocalize with the replication proteins and compartments of these viruses (20, 21, 25, 46).
The UFD1-NPL4 heterodimer is a well-characterized cofactor of VCP. It is essential for ERAD through the dislocation of polyubiquitinated substrates from the ER (15, 19). Depletion of UFD1 did not affect virus entry but resulted in significant inhibition of virus replication and egress, validating its role as an essential host factor for JEV replication. A role of UFD1 for EV71 entry has been described in an siRNA-based screen of 31 VCP cofactors (56). The VCP-UFD-NPL4 complex has also been shown to negatively regulate type I interferon signaling through proteasomal degradation of RIG-I in the context of VSV infection (57).
In summary, our study demonstrates a role of the VCP in the flavivirus replication and presents a detailed analysis of its role in the JEV life cycle ranging from the nucleocapsid release to the genome replication. Further mechanistic studies with the VCP core complexes will enhance our understanding of how the VCP cofactors modulate the virus replication. Because of its important role in the replication of the flaviviruses, VCP presents a pan-flavivirus target for the novel antiviral development.
MATERIALS AND METHODS
Cell lines and viruses.
The HeLa cell line (CCL-2) was obtained from ATCC. The Huh7, HEK-293T, Vero, and C6/36 cell lines were obtained from the cell repository at the National Centre for Cell Sciences, Pune, India. Dulbecco’s modified Eagle medium (DMEM) (HyClone) supplemented with 10% fetal bovine serum (FBS) was used to culture HeLa, Huh7, and HEK-293T cells. Leibovitz’s L-15 medium (HyClone) with 10% FBS was used for culturing C6/36 cells. Minimum essential medium Eagle (MEM) (HyClone) was used for culturing Vero cells. All the media were supplemented with 1× penicillin-streptomycin-glutamine (PSG) (Invitrogen). The following virus strains were used for the study: JEV (strain P20778, GenBank accession no. AF080251), DENV-2 (strain P23085 INDI-60, GenBank accession no. KJ918750), and WNV (strain 68856, GenBank accession no. EU249803). The C6/36 cell line was used for culturing and generating JEV and DENV-2. The Vero cell line was used to culturing WNV, and for plaque assays and foci forming assays to determine the titer of all three viruses.
Reagents, antibodies, and primers.
CB-5083 (1542705-92-9), Eeyarestatin 1 (4129650-54-4), and DBeQ (177355-84-9) were purchased from Cayman Chemical. The following primary antibodies were used in the study: VCP (ab11433; CST-2648S), GAPDH (GTX100118), pan-flavi antibody 4G2 harvested from HB-112 (ATCC) culture supernatant, clathrin light chain (CLC; Santa Cruz; SC-32815), clathrin heavy chain (CHC; CST-P1663), EEA1 (CST-2411S), JEV capsid (GTX131368); JEV NS1 (ab41651), JEV NS5 (GTX131359), HA (GTX), and rabbit IgG (CST-2729). A polyclonal JEV NS5 rabbit antibody with high specificity and sensitivity was generated in the lab and used for immunoprecipitation studies. Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Jackson Immunochemicals. Alexa 488 Tf (T13342) and fluorophore-coupled secondary antibodies were from Invitrogen, Thermo Fisher Scientific. Human ON-TARGETplus- VCP-SMARTpool (L-008727-00-0020), UBXD1-SMARTpool (L-008785-01-0005), UFD1 (L-017918-00-0005), ON-TARGETplus control siRNA nontargeting pool (D-001810-10-20), and DharmaFECT 1 transfection reagent (T-2001-02) were purchased from Dharmacon. The primers (5′-3′) used in the study were as follows: JEV: F-AGAGCACCAAGGGAATGAAATAGT, R-AATAAGTTG TAGTTGGGCACTCTG; JEV TaqMan probe CCACGCCACTCGACCCATAGACTG (5′ end, 6-carboxyfluorescein [FAM]; 3′ end, 6-carb-oxytetramethylrhodamine [TAMRA]): DENV-2: F-TCAATATGCTGAAACGCGAGAAAACCG, R-CGCCACAAGGGCCATGAACAGTTT; WNV: F-GCTCCGCTGTCCCTGTGA, R-CACTCTCCTCCTGCATGGATG; GAPDH: F-TGCACCACCAACTGCTTAGC; R-GGCATGGACTGTGGTCATGAG; VCP: F-AAACCGTGGTAGAGGTGCCA, R-CTTGGAAGGTGTCATGCCAA; UFD1: F-CCCATGCTGTTCAAACTGACC, R-AAGTGCGTTTTCTAATACGGCT; UBXN6: F-GGAGCGCATTAACTGCCTG, R-GCTCA GCACGTAGAACTCCTC.
Cell treatment and virus assays.
Cells were transfected with siRNA for 48 h (UBXD1, UBA1; 30 nM) or 72 h (VCP; 12.5 nM), and cell viability, protein, and/or RNA levels of the target gene were checked. For the VCP inhibitor experiments, cells were treated with the indicated concentration of the drug 1 h before infection (virus entry experiments) or 2 h postinfection (virus replication experiments). siRNA/drug-treated cells were infected with the virus at 1 MOI, and infection was monitored by harvesting cells for qRT-PCR and supernatant for virus titers at different time points. For virus entry assays, cells were incubated with the infection medium (virus plus incomplete medium; JEV and WNV, 5 MOI; DENV-2, 10 MOI) on ice for 1 h (JEV and WNV) or 2 h (DENV-2). After incubation, cells were washed with 1× PBS, complete medium was added, and cells were shifted to 37°C for 1 h. Cells were harvested by washing, and trypsin treatment was given to remove any extracellular attached virus. qRT-PCR was done to measure the levels of internalized viral RNA relative to the GAPDH transcript. To monitor the levels of capsid protein at early time points of virus infection, HeLa cells were infected with JEV at 50 MOI 72 h post-siRNA treatment. Cells were harvested by trypsin treatment at 1 h intervals, and lysates were processed for Western blotting. To block protein synthesis and proteasomal degradation, siNT-transfected cells were treated with 100 μg/ml cycloheximide and 1 μM bortezomib 1 h prior to infection and replenished after infection. Cells were infected with JEV at 50 MOI on ice for 1 h. Cells were harvested at 3 hpi by trypsin treatment and processed for Western blotting. Alternatively, the cells were infected with JEV at 100 MOI and processed at 3 hpi for immunofluorescence staining of the capsid protein for confocal analysis.
RNA isolation and quantitative real-time PCR (qRT-PCR).
Total cellular RNA was isolated with the phenol-chloroform method using RNAiso reagent (TaKaRa Bio). The cDNA was prepared using the Prime-script transcription kit (Promega). SyBr mix and TaqMan mix (TaKaRa Bio) were used to set up real-time PCR on QuantStudio 6 (Applied Biosystems). Each experiment had biological triplicates, and qPCR for each sample was done in technical triplicates.
Western blot analysis.
Cells were lysed in buffer (1% TritonX-100 in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and protease inhibitor cocktail; Sigma-Aldrich, Merck). Then, 5× loading buffer was added to the lysates and heated at 95°C for 10 min. Different percentages of SDS-PAGE were run depending on the target protein’s molecular weight. Proteins from the gel were transferred onto a polyvinylidene difluoride (PVDF) membrane. Posttransfer blocking was done using 5% nonfat milk in 1× PBS. Membranes were incubated at 4°C overnight with primary antibodies (mentioned above) diluted in milk. Three washes with 1× PBST (1% Tween in PBS) were given. HRP-conjugated secondary antibodies were diluted in milk, and membranes were incubated for 1 h at room temperature (RT). Three washes with 1× PBST were given before subjecting the membrane to the HRP substrate, Luminol (Santa Cruz).
Immunofluorescence staining.
Cells were plated on glass coverslips for all immunofluorescence experiments. After the indicated treatment and/or time point postinfection, cells were fixed with 2% paraformaldehyde for 15 min and permeabilized using 0.3% Tween in 1× PBS for another 15 min at RT. After blocking, staining was performed using primary (JEV-capsid, NS1, NS5, EEA1, CLC, VCP) and appropriate fluorescence-labeled secondary antibodies. Imaging was performed on an Olympus FV3000 confocal microscope using a ×60 1.4-numerical-aperature (NA) objective.
Transferrin kinetics.
HeLa cells were plated on glass coverslips. At 72 hpt (siNT and siVCP), cells were incubated in a starvation medium (incomplete DMEM plus HEPES buffer, pH 7.4, plus 0.5% bovine serum albumin [BSA]) for 30 min. Cells were moved to ice and were incubated with Alexa 488 conjugated Tf for 10 min. After washing, complete medium was added, and cells were shifted to 37°C and incubated for 2, 5, 10, 15, 30, and 120 min. After the incubation, cells were washed with chilled PBS and acid buffer (50 mM NaCl, 150 mM glycine, pH 3) for 3 min before fixing with 2% paraformaldehyde. Imaging was done using a FV3000 microscope (Olympus). The integrated fluorescence intensity of Alexa 488 Tf was calculated per cell using Fiji software (NIH) (https://imagej.net/Fiji/Downloads). Two coverslips were used for each time point. Fluorescence intensity was calculated from 12 or more fields (∼100 cells/coverslip). Fluorescence associated with internalized Tf was plotted against time and fitted using a logarithmic trendline.
Estimation of EEA1 endosome diameter.
The images were converted to 8 bit. The image calibration was done by drawing a straight line on the scale bar, and the scale was set. The images were zoomed, and a straight line was drawn across the diameter of the EEA1-positive structures, and particle analysis was performed to obtain the length of the line drawn.
Endosome fractionation.
HeLa cells treated with siRNA were infected with JEV at 50 MOI for 3 h, as described above. Cells were trypsinized to remove any extracellular attached virus and were harvested. Endosome isolation was done using the published protocols (58–60). Then, 1-ml aliquots were collected starting from the top of the gradient. Western blotting was performed to analyze the proteins present in different fractions. RNA was isolated from every fraction, and JEV RNA levels were analyzed by qRT-PCR.
Clathrin-coated vesicle purification and isolation of associated viral RNA.
Cells were infected and harvested as described for endosome fractionation. Cells were washed with buffer A [100 mM 2-(N-morpholino)ethanesulfonic acid (MES), 0.2 mM EGTA, 0.5 mM MgCl2; pH 6.5] and homogenized in buffer A using a 26-gauge syringe. Centrifugation at 4,100 × g was done to remove the debris and unlysed cells. Ultracentrifugation was used to remove the heavy membrane fractions from the lysates. Buffer A-FS (buffer A with 12.5% wt/vol OptiPrep and 12.5% wt/vol sucrose) was diluted using buffer A in a 1:4 ratio to precipitate clathrin-coated vesicles (61). The final pellet was resuspended in 80 μl buffer A, and 20 μl of it was processed for RNA isolation.
Transfection of JEV RNA.
JEV RNA was isolated from purified JEV virions using the Qiagen RNeasy kit, 74104. siNT- and siVCP-transfected HeLa cells were transfected 72 h later with 250 ng viral RNA. To confirm the internalization of viral RNA, cells were harvested at 6 hpt, and levels of viral RNA were analyzed by qRT-PCR. To determine the role of VCP in the first round of replication, cells were fixed with 2% paraformaldehyde at 6, 12, and 24 hpt, and NS1-positive cells were observed by confocal microscopy. To access the role of VCP in virus RNA replication, cells were harvested 24 h later. Relative JEV intracellular RNA levels were determined by qRT-PCR, and supernatants were used to perform the plaque assay to measure the virus titer.
Immunoprecipitation.
Cells were infected with JEV at 3 MOI and harvested at 24 hpi. For overexpression studies, 1 μg of the plasmid expressing NS5-HA and NS3-HA was transfected into HEK-293T using Lipofectamine 2000, and cell lysates were made 24 hpt. A coimmunoprecipitation (Co-IP) kit (Pierce) was used and as per the protocol; 10 μg antibody was used for 1 mg of total cell lysate. The results of the co-IP were analyzed by Western blotting.
Virus titration.
Vero cells were infected with the serial dilutions of the viral supernatant at 90% to 95% confluence. Cells were incubated with infection medium for 1 h (for JEV and WNV) or 2 h (for DENV-2). After washing, cells were overlaid with an agarose plug (1:1, agarose VIII: 2× MEM; for JEV and WNV) or with CMC medium (0.5% carboxymethyl cellulose in MEM; for DENV-2). For titration of JEV and WNV, cells were fixed with 3.7% formaldehyde overnight and stained with crystal violet after removing the agarose plug for visualizing the plaques. For DENV-2 titration, CMC was removed, and cells were washed and fixed with 2% paraformaldehyde. The focus-forming assay was done by immunostaining with 4G2 primary antibody, followed by the TrueBlue substrate for the peroxidase-conjugated secondary antibody.
Cytotoxicity assay.
Cell viability assays were performed by using the MTT assay (TOX1; Sigma-Aldrich, Merck) as per the product manual. Cell viability was measured relative to the controls.
Survival assay in the JEV mouse model.
Experiments using the JEV mouse model were conducted at the Regional Centre for Biotechnology after approval from the animal ethics committee (RCB/IAEC/2018/040). Animals were handled as per the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India. C57BL/6 mice were maintained and provided by the Small Animal Facility in the NCR Biotech Science Cluster. For virus generation, 2- to 3-day-old mice pups were given an intracranial injection of JEV. The pups’ brains were harvested at the onset of symptoms such as movement impairment and constant shivering/body tremors. Brain tissue was homogenized in incomplete MEM medium, and the supernatant was titrated by plaque assay. For the survival assay, 3-week-old mice were weighed, and grouping was done by matching weight. Mice were given an intraperitoneal injection with 107 PFU of JEV. The drug was administered by the oral route at a concentration of 30 mg/kg body weight starting from 2 hpi until day 12 at intervals of 24 h. The drug dosage was based on observable toxicity in mice and published literature (33, 34). Mice were observed for visual symptoms and mortality. To analyze the effect of CB-5083 on JEV titer, mice were segregated in two groups, JEV and JEV+CB-5083 (12 mice each). Both the groups received 107 PFU of JEV via intraperitoneal injection. The drug regime was followed as described above. Three mice from each group were sacrificed, and their brains were harvested for virus titration by plaque assay.
Statistical analysis.
Statistical analysis of the data was done using the paired Student’s t test or one-way ANOVA followed by Tukey’s post hoc test. Differences were considered significant at P values of <0.05.
ACKNOWLEDGMENTS
The work benefitted from the DIACENTER grant (BT/BI/14/042/2017) from the Department of Biotechnology, Government of India, to S.V. This work was supported by THSTI & RCB intramural research funds. The Small Animal Facility is supported by DBT grant BT/PR5480/INF/22/158/2012. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. S.S. was supported by a research fellowship from the CSIR and the DIACENTER grant (BT/BI/14/042/2017) to S.V. R.K. was supported by ICMR, A.D. by CSIR, and S.K.P. by DBT fellowships.
We thank Padmakar Tambare and the staff at the Small Animal Facility for their help with animal experiments. We thank Arup Banerjee, Sankar Bhattacharyya, Krishnan H. Harshan, Vishal Gupta, Manish Sharma, and Riya Sarkar for input and help with reagents. All virology lab members are acknowledged for their support and encouragement.
We have no conflict of interest to declare.
REFERENCES
- 1.Thomas SJ, Martinez LJ, Endy TP. 2014. Flaviviruses: yellow fever, Japanese B, West Nile, and others, p 383–415. In Kaslow RA, Stanberry LR, Le Duc JW (ed), Viral infections of humans: epidemiology and control. Springer US, Boston, MA. [Google Scholar]
- 2.Gould EA, Solomon T. 2008. Pathogenic flaviviruses. Lancet (London, England) 371:500–509. 10.1016/S0140-6736(08)60238-X. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez-Garcia M-D, Mazzon M, Jacobs M, Amara A. 2009. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe 5:318–328. 10.1016/j.chom.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Mukhopadhyay S, Kuhn RJ, Rossmann MG. 2005. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol 3:13–22. 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 5.Nain M, Mukherjee S, Karmakar SP, Paton AW, Paton JC, Abdin MZ, Basu A, Kalia M, Vrati S. 2017. GRP78 Is an Important host factor for Japanese encephalitis virus entry and replication in mammalian cells. J Virol 91:e02274-16. 10.1128/JVI.02274-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrova E, Gracias S, Beauclair G, Tangy F, Jouvenet N. 2019. Uncovering flavivirus host dependency factors through a genome-wide gain-of-function screen. Viruses 11:68. 10.3390/v11010068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma M, Bhattacharyya S, Nain M, Kaur M, Sood V, Gupta V, Khasa R, Abdin MZ, Vrati S, Kalia M. 2014. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 10:1637–1651. 10.4161/auto.29455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhullar D, Jalodia R, Kalia M, Vrati S. 2014. Cytoplasmic translocation of polypyrimidine tract-binding protein and its binding to viral RNA during Japanese encephalitis virus infection inhibits virus replication. PLoS One 9:e114931. 10.1371/journal.pone.0114931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brunetti JE, Scolaro LA, Castilla V. 2015. The heterogeneous nuclear ribonucleoprotein K (hnRNP K) is a host factor required for dengue virus and Junín virus multiplication. Virus Res 203:84–91. 10.1016/j.virusres.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 10.de Armas-Rillo L, Valera M-S, Marrero-Hernández S, Valenzuela-Fernández A. 2016. Membrane dynamics associated with viral infection. Rev Med Virol 26:146–160. 10.1002/rmv.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 6:363–374. 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khasa R, Vaidya A, Vrati S, Kalia M. 2019. Membrane trafficking RNA interference screen identifies a crucial role of the clathrin endocytic pathway and ARP2/3 complex for Japanese encephalitis virus infection in HeLa cells. J Gen Virol 100:176–186. 10.1099/jgv.0.001182. [DOI] [PubMed] [Google Scholar]
- 13.Khasa R, Sharma P, Vaidya A, Vrati S, Kalia M. 2020. Proteins involved in actin filament organization are key host factors for Japanese encephalitis virus life-cycle in human neuronal cells. Microb Pathog 149:104565. 10.1016/j.micpath.2020.104565. [DOI] [PubMed] [Google Scholar]
- 14.Meyer H, Weihl CC. 2014. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127:3877–3883. 10.1242/jcs.093831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stach L, Freemont PS. 2017. The AAA+ ATPase p97, a cellular multitool. Biochem J 474:2953–2976. 10.1042/BCJ20160783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Boom J, Meyer H. 2018. VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol Cell 69:182–194. 10.1016/j.molcel.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 17.Dargemont C, Ossareh-Nazari B. 2012. Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim Biophys Acta 1823:138–144. 10.1016/j.bbamcr.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Bug M, Meyer H. 2012. Expanding into new markets: VCP/p97 in endocytosis and autophagy. J Struct Biol 179:78–82. 10.1016/j.jsb.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Ye Y, Tang WK, Zhang T, Xia D. 2017. A mighty “protein extractor” of the cell: structure and function of the p97/CDC48 ATPase. Front Mol Biosci 4:39. 10.3389/fmolb.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arita M, Wakita T, Shimizu H. 2012. Valosin-containing protein (VCP/p97) is required for poliovirus replication and is involved in cellular protein secretion pathway in poliovirus infection. J Virol 86:5541–5553. 10.1128/JVI.00114-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi Z, Fang C, Zou J, Xu J, Song W, Du X, Pan T, Lu H, Yuan Z. 2016. Affinity purification of the hepatitis C virus replicase identifies valosin-containing protein, a member of the ATPases associated with diverse cellular activities family, as an active virus replication modulator. J Virol 90:9953–9966. 10.1128/JVI.01140-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi Z, Yuan Z. 2017. Aggregation of a hepatitis C virus replicase module induced by ablation of p97/VCP. J Gen Virol 98:1667–1678. 10.1099/jgv.0.000828. [DOI] [PubMed] [Google Scholar]
- 23.Wong HH, Kumar P, Tay FPL, Moreau D, Liu DX, Bard F. 2015. Genome-wide screen reveals valosin-containing protein requirement for coronavirus exit from endosomes. J Virol 89:11116–11128. 10.1128/JVI.01360-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panda D, Rose PP, Hanna SL, Gold B, Hopkins KC, Lyde RB, Marks MS, Cherry S. 2013. Genome-wide RNAi screen identifies SEC61A and VCP as conserved regulators of Sindbis virus entry. Cell Rep 5:1737–1748. 10.1016/j.celrep.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carissimo G, Chan Y-H, Utt A, Chua T-K, Bakar FA, Merits A, Ng LFP. 2019. VCP/p97 is a proviral host factor for replication of chikungunya virus and other alphaviruses. Front Microbiol 10:236. 10.3389/fmicb.2019.02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin Y-T, Prendergast J, Grey F. 2017. The host ubiquitin-dependent segregase VCP/p97 is required for the onset of human cytomegalovirus replication. PLoS Pathog 13:e1006329. 10.1371/journal.ppat.1006329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramanathan HN, Zhang S, Douam F, Mar KB, Chang J, Yang PL, Schoggins JW, Ploss A, Lindenbach BD. 2020. A sensitive yellow fever virus entry reporter identifies valosin-containing protein (VCP/p97) as an essential host factor for flavivirus uncoating. mBio 11:e00467-20. 10.1128/mBio.00467-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phongphaew W, Kobayashi S, Sasaki M, Carr M, Hall WW, Orba Y, Sawa H. 2017. Valosin-containing protein (VCP/p97) plays a role in the replication of West Nile virus. Virus Res 228:114–123. 10.1016/j.virusres.2016.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lan B, Chai S, Wang P, Wang K. 2017. VCP/p97/Cdc48, a linking of protein homeostasis and cancer therapy. Curr Mol Med 17:608–618. 10.2174/1566524018666180308111238. [DOI] [PubMed] [Google Scholar]
- 30.Chapman E, Maksim N, de la Cruz F, La Clair JJ. 2015. Inhibitors of the AAA+ chaperone p97. Molecules 20:3027–3049. 10.3390/molecules20023027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bastola P, Neums L, Schoenen FJ, Chien J. 2016. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with Salubrinal. Mol Oncol 10:1559–1574. 10.1016/j.molonc.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson KG, Stromnes IM, Greenberg PD. 2017. Obstacles posed by the tumor microenvironment to t cell activity: a case for synergistic therapies. Cancer Cell 31:311–325. 10.1016/j.ccell.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou H-J, Wang J, Yao B, Wong S, Djakovic S, Kumar B, Rice J, Valle E, Soriano F, Menon M-K, Madriaga A, Kiss von Soly S, Kumar A, Parlati F, Yakes FM, Shawver L, Le Moigne R, Anderson DJ, Rolfe M, Wustrow D. 2015. Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083). J Med Chem 58:9480–9497. 10.1021/acs.jmedchem.5b01346. [DOI] [PubMed] [Google Scholar]
- 34.Le Moigne R, Aftab BT, Djakovic S, Dhimolea E, Valle E, Murnane M, King EM, Soriano F, Menon M-K, Wu ZY, Wong ST, Lee GJ, Yao B, Wiita AP, Lam C, Rice J, Wang J, Chesi M, Bergsagel PL, Kraus M, Driessen C, Von Soly SK, Yakes FM, Wustrow D, Shawver L, Zhou H-J, Martin TG, Wolf JL, Mitsiades CS, Anderson DJ, Rolfe M.2017. The p97 inhibitor CB-5083 is a unique disrupter of protein homeostasis in models of multiple myeloma. Mol Cancer Ther 16:2375–2386. 10.1158/1535-7163.MCT-17-0233. [DOI] [PubMed] [Google Scholar]
- 35.Ritz D, Vuk M, Kirchner P, Bug M, Schütz S, Hayer A, Bremer S, Lusk C, Baloh RH, Lee H, Glatter T, Gstaiger M, Aebersold R, Weihl CC, Meyer H. 2011. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol 13:1116–1123. 10.1038/ncb2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramanathan HN, Ye Y. 2012. The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res 22:346–359. 10.1038/cr.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramanathan HN, Zhang G, Ye Y. 2013. Monoubiquitination of EEA1 regulates endosome fusion and trafficking. Cell Biosci 3:24. 10.1186/2045-3701-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pleasure IT, Black MM, Keen JH. 1993. Valosin-containing protein, VCP, is a ubiquitous clathrin-binding protein. Nature 365:459–462. 10.1038/365459a0. [DOI] [PubMed] [Google Scholar]
- 39.Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. 2000. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J 19:2181–2192. 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neufeldt CJ, Cortese M, Acosta EG, Bartenschlager R. 2018. Rewiring cellular networks by members of the Flaviviridae family. Nat Rev Microbiol 16:125–142. 10.1038/nrmicro.2017.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarkar R, Sharma KB, Kumari A, Asthana S, Kalia M. 2020. Japanese encephalitis virus capsid protein interacts with non-lipidated MAP1LC3 on replication membranes and lipid droplets. J Gen Virol 10.1099/jgv.0.001508. [DOI] [PubMed] [Google Scholar]
- 42.Müller JMM, Deinhardt K, Rosewell I, Warren G, Shima DT. 2007. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Commun 354:459–465. 10.1016/j.bbrc.2006.12.206. [DOI] [PubMed] [Google Scholar]
- 43.Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. 2004. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36:377–381. 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 44.Ndoja A, Cohen RE, Yao T. 2014. Ubiquitin signals proteolysis-independent stripping of transcription factors. Mol Cell 53:893–903. 10.1016/j.molcel.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stolz A, Hilt W, Buchberger A, Wolf DH. 2011. Cdc48: a power machine in protein degradation. Trends Biochem Sci 36:515–523. 10.1016/j.tibs.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 46.Wu KX, Phuektes P, Kumar P, Goh GYL, Moreau D, Chow VTK, Bard F, Chu JJH. 2016. Human genome-wide RNAi screen reveals host factors required for enterovirus 71 replication. Nat Commun 7:13150. 10.1038/ncomms13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brahms A, Mudhasani R, Pinkham C, Kota K, Nasar F, Zamani R, Bavari S, Kehn-Hall K. 2017. Sorafenib impedes Rift Valley fever virus egress by inhibiting valosin-containing protein function in the cellular secretory pathway. J Virol 91:e00968-17. 10.1128/JVI.00968-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helenius A. 2018. Virus entry: looking back and moving forward. J Mol Biol 430:1853–1862. 10.1016/j.jmb.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smit JM, Moesker B, Rodenhuis-Zybert I, Wilschut J. 2011. Flavivirus cell entry and membrane fusion. Viruses 3:160–171. 10.3390/v3020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mercer J, Snijder B, Sacher R, Burkard C, Bleck CKE, Stahlberg H, Pelkmans L, Helenius A. 2012. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep 2:1036–1047. 10.1016/j.celrep.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 51.Banerjee I, Miyake Y, Nobs SP, Schneider C, Horvath P, Kopf M, Matthias P, Helenius A, Yamauchi Y. 2014. Influenza A virus uses the aggresome processing machinery for host cell entry. Science 346:473–477. 10.1126/science.1257037. [DOI] [PubMed] [Google Scholar]
- 52.Su W-C, Chen Y-C, Tseng C-H, Hsu PW-C, Tung K-F, Jeng K-S, Lai MMC. 2013. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc Natl Acad Sci U S A 110:17516–17521. 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Byk LA, Iglesias NG, De Maio FA, Gebhard LG, Rossi M, Gamarnik AV. 2016. Dengue virus genome uncoating requires ubiquitination. mBio 7:e00804-16. 10.1128/mBio.00804-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rastogi M, Sharma N, Singh SK. 2016. Flavivirus NS1: a multifaceted enigmatic viral protein. Virol J 13:131. 10.1186/s12985-016-0590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edward Z, Takegami T. 1993. Localization and functions of Japanese encephalitis virus nonstructural proteins NS3 and NS5 for viral RNA synthesis in the infected cells. Microbiol Immunol 37:239–243. 10.1111/j.1348-0421.1993.tb03206.x. [DOI] [PubMed] [Google Scholar]
- 56.Yan J, Wang M, Wang M, Dun Y, Zhu L, Yi Z, Zhang S. 2020. Involvement of VCP/UFD1/nucleolin in the viral entry of enterovirus A species. Virus Res 283:197974. 10.1016/j.virusres.2020.197974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hao Q, Jiao S, Shi Z, Li C, Meng X, Zhang Z, Wang Y, Song X, Wang W, Zhang R, Zhao Y, Wong CCL, Zhou Z. 2015. A non-canonical role of the p97 complex in RIG-I antiviral signaling. EMBO J 34:2903–2920. 10.15252/embj.201591888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Araújo MEG, Lamberti G, Huber LA. 2015. Purification of early and late endosomes. Cold Spring Harb Protoc 2015:pdb.top074443. 10.1101/pdb.top074443. [DOI] [PubMed] [Google Scholar]
- 59.de Araújo MEG, Lamberti G, Huber LA. 2015. Homogenization of mammalian cells. Cold Spring Harb Protoc 2015:1009–1012. 10.1101/pdb.prot083436. [DOI] [PubMed] [Google Scholar]
- 60.de Araújo MEG, Lamberti G, Huber LA. 2015. Isolation of early and late endosomes by density gradient centrifugation. Cold Spring Harb Protoc 2015:1013–1016. 10.1101/pdb.prot083444. [DOI] [PubMed] [Google Scholar]
- 61.Borner GHH, Fielding AB. 2014. Isolating HeLa cell fractions enriched for clathrin-coated vesicles. Cold Spring Harb Protoc 2014:1184–1187. 10.1101/pdb.prot083147. [DOI] [PubMed] [Google Scholar]