

Respiratory syncytial virus (RSV) has an enormous impact on infants and the elderly, including increased fatality rates and potential for causing lifelong lung problems. Humans become infected with RSV through the inhalation of viral particles exhaled from an infected individual.
KEYWORDS: RSV, CX3CR1, CX3CL1, cilia, interferon, leukotrienes, cytokines, virus, respiratory
ABSTRACT
Respiratory syncytial virus (RSV) contains a conserved CX3C motif on the ectodomain of the G protein. The motif has been indicated as facilitating attachment of the virus to the host, initiating infection via the human CX3CR1 receptor. The natural CX3CR1 ligand, CX3CL1, has been shown to induce signaling pathways resulting in transcriptional changes in the host cells. We hypothesize that binding of RSV to CX3CR1 via CX3C leads to transcriptional changes in host epithelial cells. Using transcriptomic analysis, the effect of CX3CR1 engagement by RSV was investigated. Normal human bronchial epithelial (NHBE) cells were infected with RSV virus containing either wild-type (WT) G protein or a mutant virus containing a CX4C mutation in the G protein. RNA sequencing was performed on mock-infected and 4-days-postinfected cultures. NHBE cultures were also treated with purified recombinant wild-type A2 G protein. Here, we report that RSV infection resulted in significant changes in the levels of 766 transcripts. Many nuclear-associated proteins were upregulated in the WT group, including nucleolin. In contrast, cilium-associated genes, including CC2D2A and CFAP221 (PCDP1), were downregulated. The addition of recombinant G protein to the culture led to the suppression of cilium-related genes while also inducing nucleolin. Mutation of the CX3C motif (CX4C) reversed these effects on transcription, decreasing nucleolin induction and lessening the suppression of cilium-related transcripts in culture. Furthermore, immunohistochemical staining demonstrated decreases in ciliated cells and altered morphology. Therefore, it appears that engagement of CX3CR1 leads to induction of genes necessary for RSV entry as well as dysregulation of genes associated with the function of cilia.
IMPORTANCE Respiratory syncytial virus (RSV) has an enormous impact on infants and the elderly, including increased fatality rates and potential for causing lifelong lung problems. Humans become infected with RSV through the inhalation of viral particles exhaled from an infected individual. These virus particles contain specific proteins that the virus uses to attach to human ciliated lung epithelial cells, initiating infection. Two viral proteins, G protein and F protein, have been shown to bind to human CX3CR1and nucleolin, respectively. Here, we show that the G protein induces nucleolin and suppresses gene transcripts specific to ciliated cells. Furthermore, we show that mutation of the CX3C-motif on the G protein, CX4C, reverses these transcriptional changes.
INTRODUCTION
Respiratory syncytial virus causes respiratory disease in humans (1). Initial infections occur usually within the first year of life and continually throughout childhood and adulthood (2–5). RSV infection initiates in the upper airways and can be found in the lower airways during severe disease (6). Symptoms are generally mild, manifesting in stages similar to the common cold (e.g., rhinorrhea, cough, sneezing, fever, and wheezing) but can manifest as viral pneumonia during severe disease, which occurs mostly in young children and elderly adults (7). The annual economic impact of RSV in the United States is estimated at over a half a billion dollars and the World Health Organization has listed RSV as a public health priority (8, 9).
RSV contains a negative-sense, single-stranded genome. The RSV genome encodes 11 separate proteins using host translation machinery (1). Two proteins, G and F, facilitate attachment and viral penetration into the host cell (10). The RSV G protein contains a CX3C motif that has been implicated as an attachment motif for RSV (11–16). The CX3C motif is similar to that found in fractalkine (CX3CL1) and has been shown to bind the host fractalkine receptor, CX3CR1, although perhaps in a manner unique from fractalkine (11). RSV F protein has been implicated in both attachment and entry into the host epithelial cell. The attachment motif of F is still unknown, but attachment to the host protein via nucleolin has been well reported (12).
RSV primarily targets host epithelial cells in the lungs (13), although many cell types are infectible (14–16). Multiple host proteins have been indicated in RSV attachment and binding may be strain specific (17). The RSV G protein has been shown to bind heparan sulfate or chondroitin sulfate proteoglycans, although the presence of these proteoglycans on the apical surface of the human lung has not been shown. More recently, the G protein has been shown to bind human CX3CR1, which is expressed on epithelial cells at the apical surface of human lung (18). The RSV F protein has also been shown to bind heparan sulfate proteoglycans and human nucleolin, which has been shown to be expressed apically in vitro and in vivo. Both G protein and F protein are thought to bind TLR4, aiding in cellular entry potentially through outside-in signaling (19). Other attachment receptors have included annexin II, epidermal growth factor receptor, lectins, and ICAM-1 (20).
Human CX3CR1 has been shown to exist on the apical surface of human lung tissue and lung epithelial cell air-liquid interface models (12–15). CX3CR1 has been shown to be a coreceptor for other viruses, including HIV (21), and variations in the CX3CR1 allele have been shown to increase susceptibility to HIV-1 infection and progression to AIDS (22). Although the exact mechanism behind attachment and fusion of RSV to the host is still being investigated, host CX3CR1 appears to play a role in RSV infection.
CX3CR1 engagement by its ligand CX3CL1 (fractalkine) has been shown to regulate cellular transcription via its heterotrimeric Gαi protein. Specifically, the binding of CX3CR1 by CX3CL1 has been shown to increase numerous signaling molecules, including several different secondary messengers and transcription factors in neuronal, endothelial, and epithelial cells (20, 23, 24). Whether attachment of RSV to CX3CR1 results in host epithelial cell transcriptional changes is unknown. Here, we use primary human differentiated epithelial cell cultures to evaluate the effect of CX3CR1 engagement by RSV.
RESULTS
Transcriptional changes after RSV infection.
The transcriptional profile of normal human bronchial epithelial cells after infection with RSV was determined. Infection by RSV both increased and decreased gene transcripts relative to mock infected (Table 1, Table S1 in the supplemental material). RSV infection significantly increased 558 gene transcripts. Infection resulted in the induction of transcripts encoding antiviral enzymes (OAS1, OAS2, CMPK2, HERC5, and HERC6), antiviral peptides (RSAD2), interferon response genes (RIG-I, STAT1, STAT2, MX1, IFIT1, IFIT2, IFI44L, IRF1, and OASL), mucin genes (MUC5AC, MUC5B, MUC15, MUC21, and MUC13), stat inhibitor genes (SOCS1 and SOCS53), chemokines/cytokines (CX3CL1, CCL5, CXCL9, CXCL8, CXCL10, and TGFA), interleukins and their receptors (IL1A, IL-15, IL15R IL22RA1, IL2RG, and IL18R1), major histocompatibility complex genes (HLA-A, HLA-B, HLA-C, HLA-E, HLA-F, HLA-DMA, and HLA-DRB1), type-II helper T cell-associated genes (PSENEN), Toll-like receptors (TLR2), and innate immune cell differentiation/survival genes (BATF2 and JAGN1). Both proapoptotic (CASP1, CASP4, CASP7, and CASP8) and antiapoptotic (Birc3, BCL2L13, BCL2L14, and BCL2L15) transcripts were increased during infection. The 2′-O-ribose cap methyltransferase (CMTR1), a protein required for viral RNA cap snatching and shown to decrease interferon type I, was also increased after infection. Taken together, these results demonstrate many genes were upregulated in bronchial epithelial cells in response to RSV.
TABLE 1.
Mock infection versus wild-type RSV infection differentially expressed genes
| Expression relative to mock | Gene symbol | Gene description | NCBI GeneID | Regression coefficient B | P value |
|---|---|---|---|---|---|
| Up | BATF2 | Basic leucine zipper ATF-like transcription factor 2 | 116071 | −2.219807223 | 0.00000841 |
| Up | STAT1 | Signal transducer and activator of transcription 1 | 6772 | −1.932894088 | 0.00000841 |
| Up | MX1 | MX dynamin like GTPase 1 | 4599 | −3.026455262 | 0.00000841 |
| Up | TRIM14 | Tripartite motif containing 14 | 9830 | −1.373708618 | 0.00000887 |
| Up | MX2 | MX dynamin like GTPase 2 | 4600 | −3.352521625 | 0.00000887 |
| Up | OASL | 2′-5′-oligoadenylate synthetase like | 8638 | −3.527345499 | 0.00000887 |
| Up | HSH2D | Hematopoietic SH2 domain containing | 84941 | −1.924851569 | 0.00000887 |
| Up | IFI27 | Interferon alpha inducible protein 27 | 3429 | −2.936030535 | 0.00000887 |
| Up | HERC6 | HECT and RLD domain containing E3 ubiquitin protein ligase family member 6 | 55008 | −2.27262453 | 0.00000887 |
| Up | HELZ2 | Helicase with zinc finger 2 | 85441 | −2.022967637 | 0.00000887 |
| Down | MTURN | Maturin, neural progenitor differentiation regulator homolog | 222166 | 0.663532629 | 0.000362109 |
| Down | EOGT | EGF domain specific O-linked N-acetylglucosamine transferase | 285203 | 0.199675192 | 0.001184083 |
| Down | GXYLT2 | Glucoside xylosyltransferase 2 | 727936 | 0.494404491 | 0.001387687 |
| Down | MYLK | Myosin light chain kinase | 4638 | 0.637636967 | 0.001431977 |
| Down | DHX40 | DEAH-box helicase 40 | 79665 | 0.31391556 | 0.001549972 |
| Down | RP11-410E4.1 | lincRNA | - | 0.139630264 | 0.001792911 |
| Down | ACO1 | Aconitase 1 | 48 | 1.139317636 | 0.001839785 |
| Down | GALNT11 | Polypeptide N-acetylgalactosaminyltransferase 11 | 63917 | 0.273330797 | 0.002265838 |
| Down | PDE5A | Phosphodiesterase 5A | 8654 | 0.743763137 | 0.00297151 |
| Down | HS3ST4 | Heparan sulfate-glucosamine 3-sulfotransferase 4 | 9951 | 0.225923605 | 0.003275347 |
Pathway analysis of significantly upregulated genes (mock versus RSV) demonstrated 1,033 significantly enriched biological processes (Fig. 1A, Table S2). The most significantly enriched biological processes included defense processes, responses to foreign objects, innate immune responses, and cytokine-associated responses. Taken together, we found that cellular defense and immune system biological processes were associated with increased viral transcripts after RSV infection.
FIG 1.

Changes in biological processes after RSV infection. (A) Top 10 biological processes and the associated genes that are significantly enriched in transcripts increased after RSV infection. (B) Top 10 biological processes and associated genes that are significantly enriched in transcripts decreased after RSV infection. Colors represent the biological processes described in the legend.
RSV infection also resulted in the reduction of 208 transcripts relative to mock infection, with many of these transcripts associated with cell morphology (Table 1, Table S1). This included microtubule-associated protein transcript levels (MAP6, MAPT, and MAST1), and myosin transcript levels (MYO6, MYO9A, MYO3A, and MY05A) were decreased after infection. We found many cilium-associated genes (CC2D2A and PCDP1), lysosomal genes (LAMP1), leukotriene B4 hydrolase (LTA4H), WNT signaling transcripts (MCC, WNT2B), and RNA helicases (DHX40) to also be decreased during infection. Interestingly, the heparan sulfate sulfotransferase (HS3ST4) was among one of the most significantly associated downregulated transcripts.
Pathway analysis of differentially expressed downregulated genes identified 110 enriched biological processes (Fig. 1B, Table S3). Down-regulated genes were associated with biological process related to cellular morphology, including cell projection. Microtubule and cellular transplant-related biological processes were also enriched for downregulated genes, including microtubule bundle formation, cytoskeletal transport, and protein localization.
CX3C-specific transcriptional changes.
We found that transcriptional changes for both wild-type RSV-infected and mutant RSV-infected cells were in the same direction compared to mock-treated cells, but to a lesser magnitude. Infection with an A2 RSV virus containing a mutation in the CX3C motif (CX4C) resulted in five host transcripts that were significantly increased relative to wild type. After mutant virus infection, these transcripts were still higher than mock, but significantly lower than wild-type virus infections (Table 2, Table S4). We found that NCL (nucleolin), the eukaryotic nucleolar phosphoprotein, was the most significantly decreased gene in the mutant virus infection compared to wild type. Other transcripts also decreased after infection with the mutated versus the wild-type virus included those for adenylate kinase AK2 (an apoptotic activator), the Golgi homeostasis protein TMEM199 (recently identified to be essential in influenza A virus infection), and the transcriptional coactivator SNW1, which can bind the vitamin D receptor-binding domain and retinoid receptors to enhance many gene expression pathways.
TABLE 2.
Wild type versus CX4C mutant differentially expressed genes
| Expression relative to WT | Gene symbol | Gene description | NCBI GeneID | Regression coefficient B | P value | P value (BH) |
|---|---|---|---|---|---|---|
| Down | NCL | Nucleolin | 4691 | −0.1118883 | 5.84E−06 | 0.032346124 |
| Down | AK2 | Adenylate kinase 2 | 204 | −0.1604704 | 7.28E−06 | 0.032346124 |
| Down | TMEM199 | Transmembrane protein 199 | 147007 | −0.1412261 | 8.51E−06 | 0.032346124 |
| Down | SNW1 | SNW domain containing 1 | 22938 | −0.1587004 | 1.13E−05 | 0.035780973 |
| Down | CDKAL1 | CDK5 regulatory subunit associated protein 1 like 1 | 54901 | −0.2689998 | 1.81E−05 | 0.049244299 |
| Down | GPKOW | G-patch domain and KOW motifs | 27238 | −0.2037808 | 3.54E−05 | 0.070297745 |
| Down | TRAFD1 | TRAF-type zinc finger domain containing 1 | 10906 | −0.1964691 | 3.76E−05 | 0.070297745 |
| Down | PELI3 | Pellino E3 ubiquitin protein ligase family member 3 | 246330 | −0.1745717 | 3.77E−05 | 0.070297745 |
| Down | CALCOCO2 | Calcium binding and coiled-coil domain 2 | 10241 | −0.2155026 | 4.07E−05 | 0.070297745 |
| Down | ZMYND8 | Zinc finger MYND-type containing 8 | 23613 | −0.2766429 | 4.65E−05 | 0.073595815 |
| Up | C2orf81 | Chromosome 2 open reading frame 81 | 388963 | 0.34626864 | 7.19E−06 | 0.032346124 |
| Up | CC2D2A | Coiled-coil and C2 domain containing 2A | 57545 | 0.59449472 | 7.39E−06 | 0.032346124 |
| Up | CFAP221 | Cilia and flagella associated protein 221 | 200373 | 0.77283464 | 6.46E−05 | 0.076430535 |
| Up | CERKL | Ceramide kinase like | 375298 | 0.52666645 | 7.28E−05 | 0.076780874 |
| Up | BBOF1 | Basal body orientation factor 1 | 80127 | 0.48543271 | 0.00016933 | 0.104057284 |
| Up | PPIL6 | Peptidylprolyl isomerase like 6 | 285755 | 0.73103569 | 0.0001922 | 0.104057284 |
| Up | CCDC60 | Coiled-coil domain containing 60 | 160777 | 0.33676074 | 0.00020953 | 0.104057284 |
| Up | AKAP14 | A-kinase anchoring protein 14 | 158798 | 0.78099765 | 0.00021646 | 0.104057284 |
| Up | PSENEN | Presenilin enhancer, gamma-secretase subunit | 55851 | 0.24667032 | 0.00023446 | 0.104057284 |
| Up | DNAH6 | Dynein axonemal heavy chain 6 | 1768 | 0.5313297 | 0.00023723 | 0.104057284 |
There were 354 biological processes enriched for gene transcripts that were decreased in the mutant CX4C virus compared to wild-type infection (Fig. 2A, Table S5). Biological processes enriched for decreased genes included response to cytokines, response to type I interferon, and RNA processing. Innate immune response biological processes, including nucleolin and many virus-signaling genes (e.g., BCL6, BATF, and IRF2) were also decreased after mutant compared to wild-type infection.
FIG 2.

Changes in biological processes between wild-type and mutant RSV Infection. (A) Top 10 biological processes and associated genes that are significantly enriched in transcripts increased after wild-type RSV infection compared to mutant (CX4C) infection. (B) Top 10 biological processes and associated genes that are significantly enriched in transcripts decreased after wild-type RSV infection compared to mutant (CX4C) infection. Colors represent the biological processes described in the legend.
After multiple comparison corrections, two gene transcripts were significantly increased with mutant virus infection relative to wild type (Table 2). The most significantly associated transcript, C2orf81, has no known function and the other transcript, CC2D2A, encodes a protein that forms a complex localized at the transition zone of primary cilia.
The 1,067 transcripts increased (uncorrected P value < 0.05) after mutant virus infection relative to wild-type infection resulted in 78 biological pathways that were significantly enriched (Fig. 2B, Table S6). The biological processes were all associated with cilia and microtubule function and formation. These included transport, organization, and assembly. The genes associated with these pathways included many cilium-related genes (CC2D2A, CFAP221, CFAP43, and CFAP52). Dynein-related genes (DNAH3, DNAH6, DNAH9, DNAH10, and DNAH12) were also associated with enriched pathways. Taken together, mutating the CX3C domain resulted in a significant increase in cilium-related genes and biological processes.
Transcriptional changes with CX3CR1 ligand.
To focus on differences specific to the G protein, we exposed cells to soluble wild-type G protein. Recombinant soluble wild-type G protein-exposed cells had significant changes in transcript levels (Fig. 3). Cilium-related genes (CC2D2A and CFAP221) were both significantly decreased, while nucleolin was increased compared to mock-treated cells after addition of soluble G protein. Alternatively, nucleolin was increased after treatment with G protein ligand, although the relative fold change was minor and the difference did not reach the significance threshold (P = 0.065). Taken together, soluble G protein affected transcript levels similarly to the differences seen between wild type and CX4C mutant infections.
FIG 3.

Effect of G protein on epithelial cell transcripts. (A to C) Transcript levels of expression after treatment of epithelial cell cultures with recombinant G protein for CC2D2A (A), CFAP331 (B), and nucleolin (C). Error bars represent standard error of the mean from averages of three subjects, with three replicates per subject per condition.
Ciliated cells after RSV infection.
In order to provide support for evidence of decreased cilium-related gene transcripts after RSV infection, epithelial cell cultures were stained with acetylated-tubulin antibody, a cilium marker, after either mock or RSV infection. The number of ciliated cells per nuclei was counted. Ciliated cells were found in both infected and noninfected cultures (Fig. 4A). Mock-infected cultures contained on average 26% ciliated cells. Epithelial cells infected with wild-type RSV contained 13% ciliated cells on average (Fig. 4B). Taken together, both mock-infected and RSV-infected cells contained ciliated cells, but RSV infection decreased the percentage of ciliated cells relative to mock infection, supporting the changes in transcription of cilium-related genes seen after RSV infection.
FIG 4.
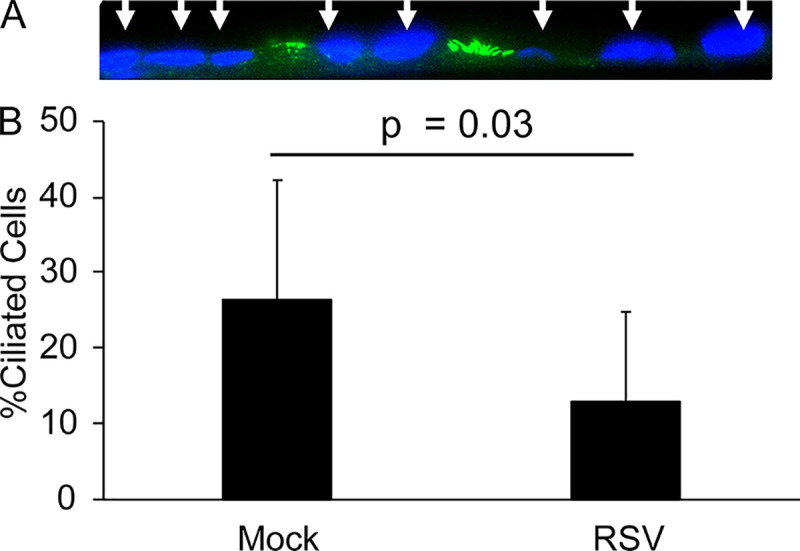
Ciliated cells after RSV infection. Quantification of the number of ciliated cells staining positive for acetylated tubulin per nucleus (DAPI, blue) in mock-infected or RSV-infected cultures. Averages represent three subjects, with three replicates per subject.
DISCUSSION
Respiratory epithelial cells act as both the primary target for RSV and the primary defense against the virus. Therefore, it is unsurprising that so many genes and biological processes were altered during RSV infection. For successful replication, the virus must first attach to the cell and then enter the cell, use host machinery to make new virus, and release from the cell (1). During this process, the host epithelium cell recognizes the presence of foreign proteins and RNA or DNA, produces antiviral peptides, and signals to neighboring cells (including immune cells) of the infection (25). Therefore, in order to successfully reproduce, the virus must both avoid inducing and becoming susceptible to host responses while simultaneously hijacking cellular processes.
Here, we demonstrate that these underlying biological processes occur during virus infection of adult primary epithelial cells grown at a physiological air-liquid interface. Given our experimental design, it is impossible to distinguish the transcriptional effects due to the host reaction to the virus and those caused directly by the virus. Both viral proteins and viral genetic material can interfere with biological processes by directly acting on host cellular machinery, resulting in cellular signaling cascades that lead to changes in transcriptional regulation. It is of no surprise that these changes have to do with mechanistic cilium function that removes foreign objects from the respiratory tract and the induction of nucleolin, an RSV F protein receptor (12), which would presumably provide a fitness advantage for the virus.
These introductory studies demonstrate a unique relationship between two host genes, CX3CR1 and nucleolin, and two virus genes, G protein and F protein. These studies suggest that CX3CR1 engagement by the RSV G protein CX3C motif results in intercellular signaling impacting nucleolin expression. The role of nucleolin in RSV infection is still being determined, although it has been suggested it has a role in virus attachment and fusion (12). Although our experimental design did not allow us to discern any specifics in the signaling cascade, manipulating the cytokine pathway in order to provoke nucleolin production would likely prove advantageous for the virus. Furthermore, these finding are consistent with the other viruses that manipulate host transcription in order to increase host proteins necessary to the virus (26).
Disruption of ciliary function has been described for many pathogens, although no morphology changes were seen after 24 h postinfection (27). A primary function of host immunity is the physical removal of foreign objects, including dead/infected cells, during an infection. It is important to note that the decrease in ciliated cells may be due to instability of the cilia that were disrupted during processing of the cultures, since the transcriptomics analysis support the likelihood of altered cilia function, including possibility of structural defects. We found, as has been noted in other reports of the upper-airway epithelial cells, that less than half of the airway epithelial cells express cilia (28–30), however, other models, including nare-derived epithelial cell models, show higher levels of airway epithelial cells expressing cilia; whether the effect of RSV infection might explain lower levels of cilia expressing cells is unknown (29). Regardless, it is clear that cilia, or the cells that express cilia, are affected by RSV infection. Furthermore, our pathway analysis suggests that a functional dysregulation is occurring. Given the role of cilia in the removal of foreign bodies, future studies will be needed to further characterize cilium function after RSV infection.
The CX4C mutant virus does not replicate at the level of the wild-type virus. Therefore, our statistical analysis required correction for variable levels of viral transcript. Although mathematical correction for viral transcripts levels using linear modeling have been continually shown to be accurate for these types of studies, it is not possible to completely distinguish nucleolin induction or cilia dysregulation between wild type and mutant based on CX3CR1 engagement from virus replication differences.
Furthermore, what effect disruption of the CX3CL1/CX3CR1 axis is having is unknown. It is entirely possible that the effect we see here is due to interference with autocrine signaling of CX3CL1 on CX3CR1. Crystal structures (31) suggest that engagement of CX3CR1 by RSV G protein may be fundamentally different than fractalkine. Future studies will be needed to determine if transcriptional changes occur due to the G protein blocking CX3CL1 from binding the binding pocket of CX3CR1 or from direct activation of the CX3CR1 receptor by G protein. Moreover, our results do not rule out contributions from other parts of the G protein to our findings. Future studies will be needed to establish the mechanism by which the specific contribution of G protein binding to CX3CR1 has an effect on increased nucleolin and decreased cilium-related transcripts.
It is important to note that these studies involved only a single strain of the A subtype (A2). Given the large differences between subtype A and subtype B G protein genes, and the fact that G protein has undergone significant genetic changes since the isolation of A2, future studies will need to determine if these transcriptional changes are associated with other RSV strains, including those with different recently circulating genotypes.
Taken together, RSV infection changes host gene expression and CX3CR1 appears to play a role in facilitating these changes. Our results suggest that RSV disrupts defense mechanisms while also increasing the expression of proviral proteins.
MATERIALS AND METHODS
Virus propagation.
Four RSV strains were used for the NHBE infection: (i) A2; (ii) A2CX4C mutant with an alanine A186 insertion in the CX3C motif (182CWAIAC187) of G protein; (iii) recombinant rA2 line 19F (r19F); and (iv) r19FCX4C mutant (provided by M.L. Moore, formerly at Emory University and presently with Meissa Vaccines, Inc., South San Francisco, CA). The prototype A2 strain of RSV (ATCC, Manassas, VA) was propagated in HEp-2 cells after serial plaque purification to reduce defective interfering particles. A stock virus designated RSV/A2 (lot number 092215 SSM) containing approximately 5.0 × 108 PFU/ml was grown in sucrose-stabilizing medium and stored at −80°C for inoculation. This stock of virus has been characterized and validated for upper and lower respiratory tract replication in the cotton rat model. The A2CX4C, r19F, and r19FCX4C viruses were grown in HEp-2 cells at 0.1 or 0.01 multiplicity of infection (MOI) to reduce defective interfering particles, purified through a sucrose cushion, and stored at −80°C. For inoculation, the virus was thawed, diluted to a final concentration with phosphate-buffered saline (PBS) (pH 7.4, without Ca2+ or Mg2+), kept on ice, and used within an hour.
NHBE cells.
Normal human bronchial epithelial (NHBE) cells derived from the upper airways of healthy adult patients were kindly provided by C.U. Cotton (Case Western Reserve University), expanded, and cultured as previously described (32–34). Briefly, cells were plated on a layer of mitomycin C (Sigma) treated 3T3 mouse fibroblast feeder cells and grown until 70% confluence in the 3:1 mixture of Ham’s F12/DMEM medium (HyClone) supplemented with 5% fetal bovine serum (FBS) (Sigma), 24 μg/ml adenine (Sigma), 0.4 μg/ml hydrocortisone (Sigma), 5 μg/ml insulin (Sigma), 10 ng/ml epidermal growth factor (EGF) (Sigma), 8.4 ng/ml cholera toxin, and 10 μM ROCK1 inhibitor Y-2763 (Selleck Chemical LLC). After expansion, cells (passage 3 or 4) were plated on Costar Transwell inserts (Corning Inc., Corning, NY), grown until confluence, then transferred to air-liquid interface where cells were maintained in a 1:1 mixture of Ham’s F12/DMEM medium supplemented with 2% Ultroser G (Pall Biosepra, SA, Cergy-Sainte-Christophe, France) for 4 weeks until they were differentiated. Differentiation was confirmed by transepithelial resistance measurements of >500 Ω and flow cytometry staining for FoxJ1 (eBioscience) and acetylated α-tubulin (Life Technologies).
NHBE inoculation.
The differentiated NHBE cultures were inoculated with RSV at an MOI of 0.1 or 0.3 as determined by infectivity titration in HEp-2 cells. The NHBE were washed with PBS and then virus in PBS or PBS alone was added to the apical surface of the cells and incubated for 1 h at 37°C. The virus inoculum was then aspirated, the apical surface washed with PBS, and fresh medium was added to the basolateral compartment. The RSV-infected NHBE cells were incubated for 4 days at 37°C and 5% CO2, and then cell lysates were collected by washing cells twice with PBS and adding 100 μl of RNA lysis buffer (Qiagen) to each well.
RNA isolation.
RNA was extracted, DNase treated, and purified using a Qiagen RNeasy kit (Qiagen). The RNA was reverse transcribed into cDNA using an iScript cDNA synthesis kit (Bio-Rad) following the manufacturer’s instructions.
Real-time PCR.
Semiquantitative PCR was carried out on a 7500 Fast real-time PCR system (Applied Biosystems) using Power SYBR Green PCR master mix (Applied Biosystems). The threshold cycle (CT) values were normalized using control PPIA CT values from the same samples. Top genes were validated using qPCR performed on samples from a separate experiment. Primer sequences were as follows: NCL primer FWD GAACCGACTACGGCTTTCAAT and NCL primer REV AGCAAAAACATCGCTGATACCA; CFAP221 primer FWD GGGTTGTTCGCAATCAAGAAGA and CFAP221 primer REV GTTGAAACCGTTTTTGTGCCC; CC2D2A primer FWD CAAGCAGCGAGGTCCAAAG and CC2D2A primer REV GGCTCTGTGCCAAATTCAGTC.
RNA sequencing.
Purified RNA was sequenced using the HiSeq 2500. Genes with low reads counts were filtered as previously described (35). Filtered reads were aligned to the GRCh38 reference genome. The RSV A2 virus genome (GenBank KT992094.1) was used for virus transcript alignment. Gene counts were normalized and log transformed (R 3.4.4, rLog package). Multiple comparisons were corrected using the Benjamini-Hochberg method (R 3.4.4, “p.adjust()” command).
Statistical models.
Significant association between transcript levels and condition, both marginal association (mock infection versus RSV infection) and viral-transcript-level-adjusted models (wild type and CX4C mutant), was determined by linear regression (R 3.4.4. lm function). Regression coefficients (B) and associated P values are reported in Tables 1 and 2 and in Tables S1 and S4 in the supplemental material. Transcript fold change (mock/infected) are listed in Tables S1 and S4 under the header “Fold change (mock/infected).” To account for any differences in virus replication between cultures and viruses, we obtained viral gene levels by mapping the transcriptome sequencing (RNAseq) data to the RSV genome. We performed principal-component analysis (FractalMineR) of 11 RSV gene transcript levels. The eigenvalue of the first principal component was used as a pseudogene to represent the level of virus in each culture. For wild type and CX4C mutant comparisons, the virus pseudogene level was used to adjust the statistical model for any differences in the virus level. Virus pseudogene-associated regression coefficient (B [vgPC1]) and P value (P value [vgPC1]) are located in Table S4. Resulting P values were adjusted for multiple testing using the Benjamini-Hochberg method (R 3.4.4. p.adjust function) and are listed in Table 2 under the header “P value (BH)” and in Table S4 under the header “P value (BH) (Mock/Infected).” P values of less than 0.05 were considered significant.
Pathway analysis.
Gene symbols with unadjusted P values of less than 0.05 were used for pathway analysis. Pathway analysis of gene sets was performed using ToppGene functional analysis software (36) and genes were referenced against the gene ontology (GO) biological process GO terms as are referenced at Gene Ontology (geneontology.org). Complete results are located in Tables S2, S3, S5, and S6 in the supplemental material. Pathway annotation (ID, Name), raw P values (P value), adjusted P values (q-value Bonferroni, q-value FDR B&H, q-value FDR B&Y), number of differentially expressed genes in pathway (Hit Count in Query List) or in genome (Hit Count in Genome), and genes included in the query list (Hit in Query List) are included in the supplemental documents.
Cilium staining.
Mock-infected or wild-type RSV-infected epithelial cell transwell cultures were stained for cilia. Transwell membranes were cut off from the plate, formalin fixed, and paraffin embedded. Cross-sectional slices were taken from each well. Mouse anti-acetylated tubulin monoclonal antibody (Sigma-Aldrich, clone 6-11B-1) was diluted 1:5,000 in Tris-buffered saline (TBS) with 1% BSA. Two hundred microliters of diluted antibody mixture was added to each slide and incubated overnight at 4°C. Normal mouse immunoglobulin G (IgG) and goat IgG served as negative controls. After overnight incubation, slides were rinsed twice for 5 min with TBS with 0.025% Triton, with gentle agitation. Fluorophore-conjugated anti-mouse secondary antibody was diluted 1:1,000 in TBS with 1% BSA. Two hundred microliters of diluted secondary antibody mixture was added to each slide and incubated for 1 h at room temperature (RT) in the dark. After incubation, slides were rinsed three times for 5 min with TBS. Slides were mounted using ProLong Gold Antifade mounting medium (Life Technologies). Slides were incubated overnight at RT to allow slides to cure. Images were taken by first locating pseudostratified columnar structured cells on the section using bright field, then taking images with fluorescent filters. Immunohistochemistry was performed and images were taken on three subjects with three slides per subject. Ciliated cells and nuclei were counted by an observer blinded to the condition (mock versus infected) of the slide.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Jason Myers for his help in RNA sequencing and data processing. We also thank M. L. Moore for providing the viruses used in this study.
This original work was supported by the NIH/NIAID HHSN272201200005C, the University of Rochester Pulmonary Training Fellowship NIH/NHLBI T32-HL066988, the University of Rochester HSCCI OP211341, the University of Rochester SAC Incubator Award, and the Department of Pediatrics, Emory University School of Medicine.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Fields BN, Knipe DM, Howley PM. 2013. Fields virology, 6th edition. Lippincott Williams & Wilkins, Philadephia, PA. [Google Scholar]
- 2.Piedimonte G, Perez MK. 2014. Respiratory syncytial virus infection and bronchiolitis. Pediatr Rev 35:519–530. 10.1542/pir.35-12-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi T, McAllister DA, O'Brien KL, Simoes EAF, Madhi SA, Gessner BD, Polack FP, Balsells E, Acacio S, Aguayo C, Alassani I, Ali A, Antonio M, Awasthi S, Awori JO, Azziz-Baumgartner E, Baggett HC, Baillie VL, Balmaseda A, Barahona A, Basnet S, Bassat Q, Basualdo W, Bigogo G, Bont L, Breiman RF, Brooks WA, Broor S, Bruce N, Bruden D, Buchy P, Campbell S, Carosone-Link P, Chadha M, Chipeta J, Chou M, Clara W, Cohen C, de Cuellar E, Dang D-A, Dash-Yandag B, Deloria-Knoll M, Dherani M, Eap T, Ebruke BE, Echavarria M, de Freitas Lázaro Emediato CC, Fasce RA, Feikin DR, Feng L, RSV Global Epidemiology Network, et al. 2017. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390:946–958. 10.1016/S0140-6736(17)30938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali A, Lopardo G, Scarpellini B, Stein RT, Ribeiro D. 2020. Systematic review on respiratory syncytial virus epidemiology in adults and the elderly in Latin America. Int J Infect Dis 90:170–180. 10.1016/j.ijid.2019.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han LL, Alexander JP, Anderson LJ. 1999. Respiratory syncytial virus pneumonia among the elderly: an assessment of disease burden. J Infect Dis 179:25–30. 10.1086/314567. [DOI] [PubMed] [Google Scholar]
- 6.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759. 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 7.Bennett JE, Dolin R, Blaser MJ. 2015. Mandell, Douglas, and Bennetts’s principles and practice of infectious diseases, 8th edition. Elsevier Saunders, Philadelphia, PA. [Google Scholar]
- 8.Paramore LC, Ciuryla V, Ciesla G, Liu L. 2004. Economic impact of respiratory syncytial virus-related illness in the US: an analysis of national databases. Pharmacoeconomics 22:275–284. 10.2165/00019053-200422050-00001. [DOI] [PubMed] [Google Scholar]
- 9.Forum of International Respiratory Societies. 2017. The global impact of respiratory disease, 2nd ed. https://www.who.int/gard/publications/The_Global_Impact_of_Respiratory_Disease.pdf?ua=1.
- 10.Meng J, Hotard AL, Currier MG, Lee S, Stobart CC, Moore ML. 2016. Respiratory syncytial virus attachment glycoprotein contribution to infection depends on the specific fusion protein. J Virol 90:245–253. 10.1128/JVI.02140-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones HG, Ritschel T, Pascual G, Brakenhoff JPJ, Keogh E, Furmanova-Hollenstein P, Lanckacker E, Wadia JS, Gilman MSA, Williamson RA, Roymans D, van T Wout AB, Langedijk JP, McLellan JS. 2018. Structural basis for recognition of the central conserved region of RSV G by neutralizing human antibodies. PLoS Pathog 14:e1006935. 10.1371/journal.ppat.1006935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. 2011. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med 17:1132–1135. 10.1038/nm.2444. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol 76:5654–5666. 10.1128/jvi.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raiden S, Sananez I, Remes-Lenicov F, Pandolfi J, Romero C, De Lillo L, Ceballos A, Geffner J, Arruvito L. 2017. Respiratory syncytial virus (RSV) infects CD4+ T cells: frequency of circulating CD4+ RSV+ T cells as a marker of disease severity in young children. J Infect Dis 215:1049–1058. 10.1093/infdis/jix070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhivaki D, Lemoine S, Lim A, Morva A, Vidalain P-O, Schandene L, Casartelli N, Rameix-Welti M-A, Hervé P-L, Dériaud E, Beitz B, Ripaux-Lefevre M, Miatello J, Lemercier B, Lorin V, Descamps D, Fix J, Eléouët J-F, Riffault S, Schwartz O, Porcheray F, Mascart F, Mouquet H, Zhang X, Tissières P, Lo-Man R. 2017. Respiratory syncytial virus infects regulatory B cells in human neonates via chemokine receptor CX3CR1 and promotes lung disease severity. Immunity 46:301–314. 10.1016/j.immuni.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cirino NM, Panuska JR, Villani A, Taraf H, Rebert NA, Merolla R, Tsivitse P, Gilbert IA. 1993. Restricted replication of respiratory syncytial virus in human alveolar macrophages. J General Virology 74:1527–1537. 10.1099/0022-1317-74-8-1527. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths C, Drews SJ, Marchant DJ. 2017. Respiratory syncytial virus: infection, detection, and new options for prevention and treatment. Clin Microbiol Rev 30:277–319. 10.1128/CMR.00010-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson CS, Chu C-Y, Wang Q, Mereness JA, Ren Y, Donlon K, Bhattacharya S, Misra RS, Walsh EE, Pryhuber GS, Mariani TJ. 2020. CX3CR1 as a respiratory syncytial virus receptor in pediatric human lung. Pediatr Res 87:862–867. 10.1038/s41390-019-0677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchant D, Singhera GK, Utokaparch S, Hackett TL, Boyd JH, Luo Z, Si X, Dorscheid DR, McManus BM, Hegele RG. 2010. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J Virol 84:11359–11373. 10.1128/JVI.00804-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imaizumi T, Yoshida H, Satoh K. 2004. Regulation of CX3CL1/fractalkine expression in endothelial cells. J Atheroscler Thromb 11:15–21. 10.5551/jat.11.15. [DOI] [PubMed] [Google Scholar]
- 21.Combadiere C, Salzwedel K, Smith ED, Tiffany HL, Berger EA, Murphy PM. 1998. Identification of CX3CR1. A chemotactic receptor for the human CX3C chemokine fractalkine and a fusion coreceptor for HIV-1. J Biol Chem 273:23799–23804. 10.1074/jbc.273.37.23799. [DOI] [PubMed] [Google Scholar]
- 22.Faure S, Meyer L, Costagliola D, Vaneensberghe C, Genin E, Autran B, Delfraissy JF, McDermott DH, Murphy PM, Debré P, Théodorou I, Combadiere C. 2000. Rapid progression to AIDS in HIV+ individuals with a structural variant of the chemokine receptor CX3CR1. Science 287:2274–2277. 10.1126/science.287.5461.2274. [DOI] [PubMed] [Google Scholar]
- 23.Brand S, Sakaguchi T, Gu X, Colgan SP, Reinecker H-C. 2002. Fractalkine-mediated signals regulate cell-survival and immune-modulatory responses in intestinal epithelial cells. Gastroenterology 122:166–177. 10.1053/gast.2002.30329. [DOI] [PubMed] [Google Scholar]
- 24.Poniatowski ŁA, Wojdasiewicz P, Krawczyk M, Szukiewicz D, Gasik R, Kubaszewski Ł, Kurkowska-Jastrzębska I. 2017. Analysis of the role of CX3CL1 (fractalkine) and its receptor CX3CR1 in traumatic brain and spinal cord injury: insight into recent advances in actions of neurochemokine agents. Mol Neurobiol 54:2167–2188. 10.1007/s12035-016-9787-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janeway C. 1996. Immunobiology. Taylor & Francis Group, New York, NY. [Google Scholar]
- 26.Seitz C, Frensing T, Hoper D, Kochs G, Reichl U. 2010. High yields of influenza A virus in Madin-Darby canine kidney cells are promoted by an insufficient interferon-induced antiviral state. J Gen Virol 91:1754–1763. 10.1099/vir.0.020370-0. [DOI] [PubMed] [Google Scholar]
- 27.Jumat MR, Yan Y, Ravi LI, Wong P, Huong TN, Li C, Tan BH, Wang DY, Sugrue RJ. 2015. Morphogenesis of respiratory syncytial virus in human primary nasal ciliated epithelial cells occurs at surface membrane microdomains that are distinct from cilia. Virology 484:395–411. 10.1016/j.virol.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 28.Gras D, Petit A, Charriot J, Knabe L, Alagha K, Gamez AS, Garulli C, Bourdin A, Chanez P, Molinari N, Vachier I. 2017. Epithelial ciliated beating cells essential for ex vivo ALI culture growth. BMC Pulm Med 17:80. 10.1186/s12890-017-0423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antunes MB, Woodworth BA, Bhargave G, Xiong G, Aguilar JL, Ratner AJ, Kreindler JL, Rubenstein RC, Cohen NA. 2007. Murine nasal septa for respiratory epithelial air-liquid interface cultures. Biotechniques 43:195–204. 10.2144/000112531. [DOI] [PubMed] [Google Scholar]
- 30.Chapelin C, Coste A, Gilain L, Poron F, Verra F, Escudier E. 1996. Modified epithelial cell distribution in chronic airways inflammation. Eur Respir J 9:2474–2478. 10.1183/09031936.96.09122474. [DOI] [PubMed] [Google Scholar]
- 31.McLellan JS, Ray WC, Peeples ME. 2013. Structure and function of respiratory syncytial virus surface glycoproteins. Curr Top Microbiol Immunol 372:83–104. 10.1007/978-3-642-38919-1_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, Kinter M, Shank S, Cotton C, Kelley TJ, Ziady AG. 2008. Dysfunction of Nrf-2 in CF epithelia leads to excess intracellular H2O2 and inflammatory cytokine production. PLoS One 3:e3367. 10.1371/journal.pone.0003367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ziady AG, Sokolow A, Shank S, Corey D, Myers R, Plafker S, Kelley TJ. 2012. Interaction with CREB binding protein modulates the activities of Nrf2 and NF-κB in cystic fibrosis airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 302:L1221–31. 10.1152/ajplung.00156.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ha B, Chirkova T, Boukhvalova MS, Sun HY, Walsh EE, Anderson CS, Mariani TJ, Anderson LJ. 2019. Mutation of respiratory syncytial virus G protein's CX3C motif attenuates infection in cotton rats and primary human airway epithelial cells. Vaccines (Basel) 7:69. 10.3390/vaccines7030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chu C-Y, Qiu X, Wang L, Bhattacharya S, Lofthus G, Corbett A, Holden-Wiltse J, Grier A, Tesini B, Gill SR, Falsey AR, Caserta MT, Walsh EE, Mariani TJ. 2016. The healthy infant nasal transcriptome: a benchmark study. Sci Rep 6:33994. 10.1038/srep33994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, Bardes EE, Aronow BJ, Jegga AG. 2009. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37:W305–W311. 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.