

CTCF insulators are important for transcriptional control, and increasing evidence suggests that CTCF insulators, together with the cohesin complex, regulate viral transcription in DNA viruses. The CTCF-cohesin interaction is important for the formation of chromatin loops, structures that orient distance-separated elements in close spatial proximity for transcriptional control.
KEYWORDS: insulator, CTCF, cohesin, HSV-1 latency, epigenetics, chromatin, mouse ocular, HSV-1, Rad21, latency
ABSTRACT
Herpes simplex virus 1 (HSV-1) is a human pathogen that can establish a lifelong infection in the host. During latency, HSV-1 genomes are chromatinized and are abundantly associated with histones in sensory neurons, yet the mechanisms that govern the latent-lytic transition remain unclear. We hypothesize that the latent-lytic switch is controlled by CTCF insulators, positioned within the HSV-1 latent genome. CTCF insulators, together with the cohesin complex, can establish and maintain chromatin loops that allow distance-separated gene regions to be spatially oriented for transcriptional control. In the current study, we demonstrated that the cohesin subunit Rad21 was recruited to latent HSV-1 genomes near four of the CTCF insulators during latency. We showed that the CTCF insulator known as CTRS1/2, positioned downstream from the essential transactivating immediate-early (IE) region of ICP4, was only enriched in Rad21 prior to but not during latency, suggesting that the CTRS1/2 insulator is not required for the maintenance of latency. Further, deletion of the CTRL2 insulator, positioned downstream from the latency-associated transcript (LAT) enhancer, resulted in a loss of Rad21 enrichment at insulators flanking the ICP4 region at early times postinfection in mouse ganglia, suggesting that these insulators are interdependent. Finally, deletion of the CTRL2 insulator resulted in a loss of Rad21 enrichment at the CTRL2 insulator in a cell type-specific manner, and this loss of Rad21 enrichment was correlated with decreased LAT expression, suggesting that Rad21 recruitment to viral genomes is important for efficient gene expression.
IMPORTANCE CTCF insulators are important for transcriptional control, and increasing evidence suggests that CTCF insulators, together with the cohesin complex, regulate viral transcription in DNA viruses. The CTCF-cohesin interaction is important for the formation of chromatin loops, structures that orient distance-separated elements in close spatial proximity for transcriptional control. Herpes simplex virus 1 (HSV-1) has seven putative CTCF insulators that flank the LAT and the IE, indicating that CTCF insulators play a role in the transition from latency to reactivation. Contributions from the work presented here include the finding that CTCF insulators in HSV-1 genomes are differentially enriched in the cohesin subunit Rad21, suggesting that CTCF-cohesin interactions are establishing and anchoring chromatin loop structures to control viral transcription.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a human pathogen that can establish a lifelong infection in the host. During acute infection, the virus infects mucosal or corneal epithelial cells, where it replicates, produces progeny virus, and spreads. Following acute infection, HSV-1 is transported by retrograde axonal transport to sensory neurons in the peripheral nervous system, where it establishes a lifelong latent infection. Latent viral genomes can periodically reactivate in response to a number of external stimuli, and, following reactivation, the virus can travel back to the periphery and cause clinical disease, including diseases of the cornea that can ultimately lead to blindness (1, 2).
During acute infection, the HSV-1 genome is organized into dynamic nucleosomes that rapidly associate with histones to complete a series of sequential gene activation, reversal of epigenetic silencing, and nucleosome destabilization steps that subsequently combat host chromatinization mechanisms aimed at silencing lytic gene expression (3–11). In contrast, during latency, HSV-1 genomes are regularly chromatinized and are abundantly associated with histones in sensory neurons (12–14). During latency, the HSV-1 genome is essentially silenced with the exception of the latency-associated transcript (LAT). LAT is a multifunctional RNA that is abundantly expressed in a fraction of sensory neurons (15). During latency, the LAT is populated with permissive euchromatin marks, including acetyl-H3K9K14 and H3K4me2 (13, 16), while the nearby immediate-early (IE) lytic regions bear repressive histone marks, including H3K27me3 and H3K9me3 (17–20). At early times following the application of reactivation stimuli that include epinephrine iontophoresis in rabbits or sodium butyrate and ganglion explant in mice, the histone marks associated with IE regions ICP0 and ICP4 rapidly undergo dynamic changes and become more euchromatic in nature (21–23). However, the epigenetic mechanisms that govern the initial transcription of lytic genes appear to be distinct from the epigenetic mechanisms that are involved in reactivation from latency, the latter being linked to changes in neuronal stress response pathways (24, 25). Understanding the key differences between epigenetic regulation of HSV-1 during the acute infection and those mechanisms involved in reactivation are critical to understanding the latent-lytic switch in vivo. Nonetheless, these distinct epigenetic mechanisms involved in controlling the latent-lytic switch remain elusive.
One intriguing possibility is that lytic and latent viral genomes and their transcriptional abilities are controlled, in part, by chromatin insulators. Classically defined, chromatin insulators are the regulatory domains that separate distinct gene clusters and prevent inappropriate signaling across transcriptional domains (26). CTCF insulators are well-characterized vertebrate chromatin insulators that function when the 11-zinc finger DNA binding protein CTCF (CCCTC-binding factor) binds to a conserved DNA binding motif (26). These insulator elements can act as barriers and/or enhancer-blockers and can recruit corepressive or coactivating proteins to maintain transcriptional control over gene regions. CTCF insulators can also self-dimerize to establish long-range chromosomal interactions that facilitate the formation of chromatin loops (26). In eukaryotic cells, host genomes are folded into CTCF-mediated spatially oriented domains that are organized into compartmentalized structures, known as topologically associated domains (TADs), to control transcription (27, 28). In TAD organization, CTCF insulators can colocalize with the cohesin complex, a 4-membered protein complex that is essential for chromatid cohesion in cell division and the establishment and maintenance of long-range chromosomal interactions (chromatin loops) (29–33). The cohesin complex is composed of four core proteins (SMC1, SMC3, SCC1 [Rad21], and SA1/2 subunits) that form a ring-shaped structure (34). Cohesin colocalizes with CTCF insulators and contributes to the organization and maintenance of chromatin loops, in part by anchoring the loop structures formed (27, 34–36). The Rad21 subunit of cohesin was originally identified for its role in DNA double-strand break repair (37), and this subunit interacts with the SMC1 and SMC3 protein subunits of the cohesin complex to form a tripartite ring-shaped structure that interacts with the SA subunit of the cohesin complex for stabilization (38, 39). The C-terminal domain of the Rad21 subunit has also been described as a participant in loading and holding of DNA in the cohesin complex (40–42). Consequently, knockdown of cohesin subunits, including Rad21, results in the reorganization of chromatin loops (30, 33, 43–51).
Interestingly, these CTCF-mediated long-range spatial interactions have also been identified in a number of DNA viruses, including Epstein-Barr virus (EBV), Kaposi’s sarcoma-associated herpesvirus (KSHV), and human papillomavirus (HPV), where they play roles in gene silencing and gene activation, chromatin barrier formation, nucleosome positioning, and RNA pol II progression and viral genome persistence (52–69). Most recently, cohesin subunits Rad21 and SMC1 were shown to be recruited to HSV-1 lytic replication compartments, and knockdown of Rad21 and SMC1 resulted in reduced RNA pol II occupancy and increased H3K27me3 enrichment, further suggesting that cohesion recruitment facilitates HSV-1 lytic transcription (70).
In latent HSV-1 genomes, 7 putative CTCF insulators were previously identified and cluster around the LAT and IE genes, likely to facilitate the formation of distinct chromatin domains during latency (Fig. 1) (64). We have characterized 4 of the 7 putative insulators as enhancer-blocking insulators, some acting as enhancer-blocking insulators in a cell type-specific manner (62), and demonstrated that (i) CTCF protein was differentially enriched at these sites during latency, (ii) that the protein Suz12, part of the Polycomb Repressive Complex 2, differentially colocalized to CTCF insulators flanking the IE genes of HSV-1 during latency, and (iii) that CTCF was differentially evicted from the sites flanking IE genes at very early times following the application of reactivation stimuli in vivo (60, 62). Further, we showed that the global depletion of CTCF using recombinant adeno-associated virus (rAAV) vectors resulted in long-term persistent shedding of virus in the absence of reactivation stimuli in rabbits, suggesting that CTCF depletion facilitated HSV-1 reactivation (61). These results indicated that CTCF insulators played a key role in the maintenance of latency and that eviction of CTCF protein from the viral genome was important for reactivation.
FIG 1.

Schematic diagram of the nucleotide locations of the putative CTCF insulator sites in latent HSV-1 genomes. Genomic positions of CTCF binding motifs in the HSV-1 genome. CTRS1/2 is 2 distinct CTCF binding clusters separated by fewer than 100 nucleotides. The nucleotide position of the core CTCF binding cluster, known as CTRL2, and the depiction of the CTRL2 deletion site are shown.
In the current study, we explored the hypothesis that cohesin is recruited to latent HSV-1 genomes at or near CTCF insulators, potentially enabling chromatin loop formation. To test this, we utilized chromatin immunoprecipitation (ChIP) assays using an antibody to the cohesin complex protein subunit Rad21 and combined those assays with quantitative PCR (qPCR) at CTCF insulator sites to show that Rad21 was enriched near 4 of the CTCF insulators in latent HSV-1 genomes in vivo. Interestingly, a CTCF insulator downstream from the essential transactivating IE region of ICP4 (CTRS1/2) was not significantly enriched in Rad21 during latency, but in ganglia harvested from mice at 7 and 14 days postinfection (dpi), this site was significantly enriched in Rad21, suggesting that the CTRS1/2 insulator is important for the establishment of latency but not the maintenance of latency. Deletion of one of the CTCF insulators, the CTRL2 insulator located downstream from the LAT enhancer (Fig. 1), did not impact the overall enrichment of Rad21 during latency but significantly decreased Rad21 accumulation at all CTCF insulators at 7 and 14 dpi in mouse ganglia, suggesting that Rad21 localization to CTCF insulators prior to the establishment of latency is dependent on the CTRL2 insulator. Finally, during productive infection, Rad21 was abundant near all CTCF insulators in HeLa cells infected with wild-type (wt) virus, but deletion of the CTRL2 insulator resulted in a loss of Rad21 localization to insulators near LAT and ICP4. In productively infected differentiated Lund human mesencephalic (LUHMES) cells, Rad21 localized to only two sites, CTRL2 and CTRS3, near the LAT and ICP4 regions, independent of the presence of the CTRL2 insulator, suggesting that the CTRL2 insulator maintained distinct cell type-specific roles during productive infection and that CTRL2 and CTRS3 insulator function was interdependent.
RESULTS
The mouse cellular control Tsix Site A was enriched in the cohesin subunit Rad21.
Chromatin immunoprecipitation assays (ChIP) using the Rad21 antibody were combined with qPCR of near CTCF insulators in the HSV-1 genome using mouse trigeminal ganglion (TG) tissues harvested at 7, 14, and 28 days postinfection (dpi) with HSV-1 strain 17Syn+. Since there were 5 individual CTCF insulator sites being evaluated for potential Rad21 enrichment, we first validated ChIP assays using cellular controls prior to analyses of the individual insulators. As a positive control, we selected the mouse cellular control Tsix Site A, a site that has a known CTCF insulator that is enriched in CTCF in the mouse genome (71). In contrast, the negative cellular control MT498 does not contain a CTCF binding site. Both Tsix and MT498 have been used by us and others to successfully validate in vivo ChIP assays with the CTCF antibody (60, 62). To validate the Tsix site as a control for cohesin binding using the Rad21 antibody, we measured both specific and nonspecific binding of Rad21 to cellular controls. After sonication, samples were divided in two parts and incubated with either the Rad21 antibody or IgG as a control for nonspecific binding. Following DNA purification for each of the ChIP replicates, we performed qPCR with primers specific for Tsix Site A and MT498 (Table 1). Relative copies for the bound (B), input (I), and IgG fractions were determined based on standard curves for each specific primer set. Relative bound/input ratios and IgG/input ratios were calculated for each of the individual replicates for comparison and statistical analyses. Here, we found significant enrichment (greater than 10-fold) of the positive Tsix Site A compared to IgG, indicating that the pulldowns were efficient and specific for the Rad21 protein and not due to nonspecific binding (data not shown). The relative bound/input fractions for the cellular genes were plotted for each of the ChIP assay time points, and one-way analysis of variance (ANOVA) was done on the individual data sets to confirm that Tsix Site A was significantly enriched in Rad21 relative to the negative cellular control MT498 (Fig. 2A to C). Experiments that did not validate (no enrichment of Rad21 over IgG or less than a 2-fold enrichment of Rad21 at Tsix Site A compared to MT498) were excluded from further analyses.
TABLE 1.
Primers and probes
| Target (accession no.) | Primer or probe | Sequence (5′–3′) |
|---|---|---|
| CTRL1 | Forward primer | AGCCAAGTTAACGGGCTACG |
| Reverse primer | ATGCCAGCCCAACAAAATC | |
| Probe | CCTTCGGGAATGGGACTGG | |
| CTRL2 | Forward primer | CCGCGGCTCTGTGGTTA |
| Reverse primer | GGATGCGTGGGAGTGGG | |
| Probe | ACACCAGAGCCTGCCCAACATGGCA | |
| CTam | Forward primer | GCTGCCACAGGTGAAACC |
| Reverse primer | TGTAGCAGGAGCGGTGTG | |
| Probe | ACCTGCCCAACAACACAACT | |
| CTRS1/2 | Forward primer | GCCCCTCGAATAAACAACGCTA |
| Reverse primer | GTTGTGGACTGGGAAGGCGC | |
| Probe | GGTTGTTGCCGTTTATTGCG | |
| CTRS3 | Forward primer | ATCGCATCGGAAAGGGACACG |
| Reverse primer | CCAAGGTGCTTACCCGTGCAAA | |
| Probe | ACAGAAACCCACCGGTCCGCCTTT | |
| Tsix Site A (AJ421479) | Forward primer | CGCAGGGCAGCCAGAA |
| Reverse primer | TCTGGTGTTATCCCTTCCTGTCTT | |
| Probe | CAGCCATTCACAATCC | |
| MT498 (NT_039554) | Forward primer | TACAAGATGCAAGTCTAGATATTTTAAGTCTATGTAT |
| Reverse primer | ACACACACACACACACACACA | |
| Probe | CACACACACACAAACAC | |
| LAT | Forward primer | ACCCACGTACTCCAAGAAGGC |
| Reverse primer | TAAGACCCAAGCATAGACAGCCA | |
| Probe | TCCCACCCCGCCTGTGTTTTT | |
| DNA Pol | Forward primer | AGAGGGACATCCAGGACTTTGT |
| Reverse primer | CAGGCGCTTGTTGGTGTA | |
| Probe | ACCGCCGAACTGAGCG |
FIG 2.

Validation of ChIP assays. ChIP assays were done using trigeminal ganglia harvested from mice infected with wt 17Syn+ using the Rad21 antibody (n = 5). Assays were validated using the positive cellular control mouse Tsix Site A and the negative cellular control mouse MT498 (a cellular region with no known CTCF insulators present). Following ChIP with Rad21 antibody on ganglia harvested at 7, 14, or 28 dpi, qPCR was performed using primers specific for the cellular controls (Table 1). Rad21 enrichment at each site was calculated as the relative bound/input (B/I) ratio following qPCR. The bar graph presented is the average B/I for all 5 biological replicates, and the individual experiment values are represented by the dot plots. Error bars are measured for the standard deviations from the means for the 5 replicates. One-way ANOVA on independent samples was used to determine statistical significance for each time point.
Rad21 enrichment was observed at four CTCF insulators during latency in vivo.
To determine if the cohesin subunit Rad21 could be detected at or near the individual CTCF insulators in latently infected mouse TG, we harvested ganglia at 28 dpi and performed ChIP assays using the antibody for the cohesin complex subunit Rad21 and combined that with qPCR at unique nucleotide sequences immediately adjacent to each individual CTCF site using primers and probes specific for that nucleotide region (Table 1). Validation of the experiments was done prior to qPCR as described above, and only replicates that showed greater than 2-fold enrichment of the positive cellular control (Tsix Site A) over the negative cellular control (MT498) were used for further analyses. Our result showed significant Rad21 enrichment relative to IgG at four of the CTCF insulator sites, namely, the CTRL1 and CTRL2 insulators that flank the LAT 5′ exon region, the CTam upstream from ICP0, and the CTRS3 insulator, upstream from ICP4 (Fig. 3), suggesting that these sites are involved in the formation of chromatin loops in latent viral genomes. Strikingly, one insulator downstream from the IE ICP4, the CTRS1/2 insulator, was not enriched in Rad21 relative to the IgG control, suggesting that this insulator does not contribute to the maintenance of latency.
FIG 3.
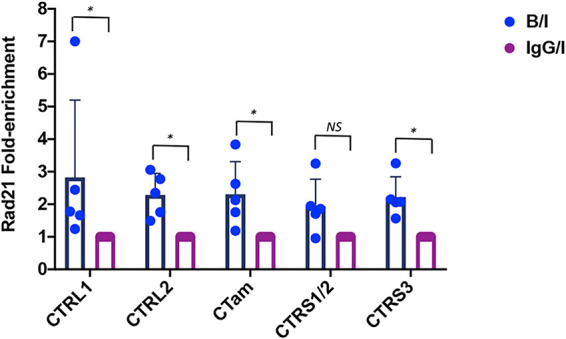
Rad21 enrichment at CTCF insulators during latency in the mouse TG. Following ocular infection of mice, TGs were harvested on 28 dpi, homogenized, and immediately fixed and processed for ChIP assays using the Rad21 antibody or the IgG control. Subsequent qPCR using primers and custom probes specific for nucleotide regions within 100 to 150 bp of the CTCF insulator sites was performed on both the antibody aliquot and the IgG control. Relative copy numbers in the B, I, or IgG fractions were determined from the equation for the standard curve specific to the primer/probe set used. Each ChIP assay was done using a pooled sample of 3 mice (6 TG pooled); n = 5. All relative B/I values were normalized to IgG (set to 1). The bar graph is presented as the average fold enrichment of 5 biological replicates, and the individual experiment fold enrichment value is represented by the dot plot. Error bars are measured for the standard deviations from the means for the 5 replicates. One-way ANOVA on correlated samples was used to determine statistical significance (*, P < 0.05).
Rad21 accumulation was dynamic prior to the establishment of HSV-1 latency in vivo.
Previous evidence from our laboratory showed that individual CTCF sites were enhancer-blocking insulators in a cell type-specific manner, suggesting that individual CTCF insulators have different roles in lytic versus latent infections (59, 62). To determine whether binding of Rad21 at the viral insulator sites was differential, as the virus established latency in mouse sensory neurons, we performed a series of ChIP assays on mice infected with wild-type virus at 7 and 14 dpi. Assays were validated using cellular controls as described above for the 28-dpi samples (Fig. 2A and B). The relative bound/input ratios were normalized to the positive cellular control Tsix Site A in all samples to normalize ChIP samples across the time points. Here, we found that overall Rad21 enrichment was very low in the 7-dpi samples compared to the 14- and 28-dpi samples, suggesting that cohesin loading on the genome was progressive over time and could play a role in the establishment of latency in vivo (Fig. 4). Nonetheless, the direct comparison of the individual insulators over the 3 infection time points showed that the CTRL1 (insulator upstream from the LAT promoter) and CTRS3 (upstream from the ICP4 region) insulators were significantly enriched in Rad21 relative to IgG at all time points (Fig. 4A and E). The CTRL2 insulator downstream from the LAT enhancer was not significantly enriched in Rad21 relative to IgG until 14 dpi, but this could be due to limits of detection in these earlier samples (Fig. 4B). A similar observation was made for the CTam insulator upstream ICP0 promoter (Fig. 4C). Interestingly, the CTRS1/2 insulator located downstream from the ICP4 region was significantly enriched in Rad21 at early times in the infection (7 and 14 dpi), but by 28 dpi no significant enrichment of Rad21 was observed (Fig. 4D). These data suggest that Rad21 localization to HSV-1 genomes in neurons is dynamic, as latency is established, and that site-specific Rad21 enrichment is necessary for the establishment of latency (CTRS1/2), the maintenance of latency (CTRL2 and CTam), or both (CTRL1 and CTRS3).
FIG 4.

Rad21 accumulation was dynamic in vivo. Following ocular infection of mice with wt 17Syn+, TGs were harvested on 7 and 14 dpi for comparison to 28-dpi samples. Five biological replicates containing 3 mice (6 TG) were processed for ChIP assay with the Rad21 antibody or the IgG control. qPCR using primers listed in Table 1 were was for both antibody and IgG samples. The relative B/I ratios were normalized to the Tsix B/I ratio for comparison across time points. The bar graph is presented as the average B/I for all 5 biological replicates, and the individual experiment value is represented by the dot plot. Error bars are measured for the standard deviations from the means for the 5 replicates. Insets are presented to show 7 and/or 14 dpi for scale differences. One-way ANOVA on correlated samples was used to determine statistical significance (*, P < 0.05). (A) Normalized B/I and IgG/I for CTRL1 insulator upstream LAT. (B) Normalized B/I and IgG/I for CTRL2 insulator downstream LAT. (C) Normalized B/I and IgG/I for CTam insulator upstream ICP0. (D) Normalized B/I and IgG/I for CTRS1/2 insulator downstream ICP4. (E) Normalized B/I and IgG/I for CTRS3 insulator upstream ICP4.
Deletion of the CTRL2 insulator resulted in a significant decrease in genomes during latency but not at early times postinfection.
We previously reported that a recombinant virus with a 135-bp deletion of the CTRL2 insulator (ΔCTRL2) (Fig. 1) had significantly higher mortality in infected mice and the mice that survived to 28 dpi harbored significantly lower viral genomes in the mouse TG than those infected with the wt 17Syn+ virus. Further, deletion of the CTRL2 insulator altered H3K27me3 enrichment and increased lytic gene expression in vivo (63). Here, we sought to establish whether viral genome loads were significantly different in mouse TG prior to the establishment of latency in ΔCTRL2-infected mice compared to the wt-infected mice. Mice were sacrificed at 7 and 14 dpi, and genome loads were quantified using the viral cellular gene CTRS3 and the host cellular gene Tsix Site A. Relative copy numbers were determined using qPCR as described for each of the biological replicates and were plotted as a ratio between viral/host genes for both viruses (Fig. 5A and B). We detected no significant difference in genome loads at 7 and 14 dpi between the two viruses (Fig. 5C). However, both wt- and ΔCTRL2-infected mouse genome loads decreased significantly by 28 dpi, and we found a significant (∼5-fold) decrease in viral genomes per ganglion in the ΔCTRL2- compared to wt-infected mice, consistent with our previous report, suggesting that latency is not efficiently established in the absence of the CTRL2 insulator (63). The fact that equivalent numbers of viral genomes were found at 7 and 14 dpi in ganglia infected with both the wt and recombinant viruses suggested that transport of viral genomes to neurons was not affected by the deletion of the CTRL2 insulator, confirming that the inability for the mutant ΔCTRL2 to establish latency in the neurons was not due to fewer genomes being delivered to TG neurons.
FIG 5.
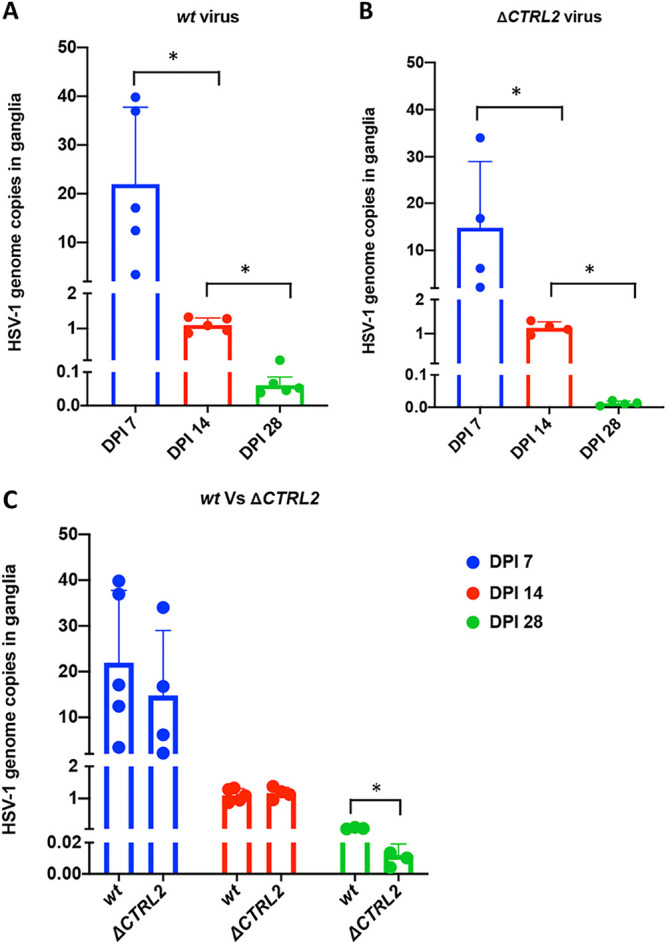
HSV-1 genome copies in mice ganglia in wt 17Syn+ and the ΔCTRL2 recombinant following infection. The number of HSV-1 genomes per sample was determined between wt 17Syn+ and ΔCTRL2 viral infections. Relative copies of HSV-1 CTRS3 and mouse Tsix Site A were determined by PCR for 7, 14, and 28 dpi. qPCR data are presented as the CTRS3/Tsix Site A ratio. (A) Genome copies in wt 17Syn+. One-way ANOVA on correlated samples was used to determine statistical significance between the individual dpi (*, P < 0.05). (B) Genome copies in ΔCTRL2. One-way ANOVA on correlated samples was used to determine statistical significance between the individual dpi (*, P < 0.05). (C) Comparison between genome copies of wt and mutant viruses. One-way ANOVA on independent samples was used to determine statistical significance between the mutant and wt at each individual dpi (*, P < 0.05).
Deletion of the CTRL2 insulator does not affect viral replication in differentiated LUHMES cells.
We previously reported that the ΔCTRL2 recombinant virus had attenuated replication in epithelial cell lines but increased replication in the undifferentiated mouse neuronal cell line (Neuro2A) (63). To determine what, if any, effect the deletion of the CTRL2 insulator had on replication in terminally differentiated LUHMES cells, a human neuronal cell line capable of supporting HSV-1 quiescence (72), we infected differentiated LUHMES with wt and mutant viruses at an MOI of 1 and monitored HSV-1 replication through 48 hpi. HeLa cells were also infected with both viruses to serve as a human epithelial cell line comparison. We found no significant difference in replication of the wt and mutant viruses though 48 hpi in LUHMES cells, consistent with our findings in mouse ganglia at 7 and 14 days postinfection (Fig. 6A). In contrast, there was a significant attenuation of replication of the ΔCTRL2 recombinant at earlier times, but by 48 hpi, the replication defect was no longer observed in the mutant, suggesting that at later times postinfection in HeLa cells the replication defect can be overcome, likely due to the higher number of genomes present at later times (Fig. 6B). Nevertheless, these data support that there are distinct differences in epithelial and neuronal infections with the ΔCTRL2 virus.
FIG 6.
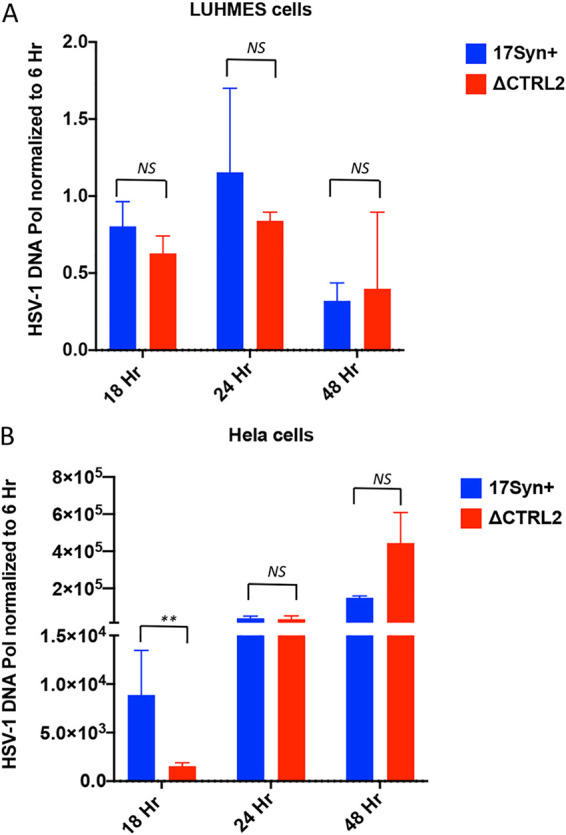
Replication was attenuated in HeLa cells, but not LUHMES cells, when the CTRL2 insulator was deleted. (A and B) Differentiated LUHMES cells (A) or HeLa cells (B) were infected with either wt or ΔCTRL2 at an MOI of 1 in triplicate. The cells were harvested at 6, 18, 24, and 48 h postinfection (hpi). HSV-1 genomes were quantified by qPCR using HSV-1 DNA polymerase primers (Table 1). At 18, 24, and 48 h, samples were normalized to the 6-h genome copies for an input normalization. Horizontal bars represent the averages from 3 biological replicates with standard deviations from the means. Statistical analyses were done by paired Student's t test (**, P < 0.001).
Deletion of the CTRL2 insulator did not affect Rad21 enrichment during latency.
To determine whether deletion of the CTRL2 insulator resulted in an altered enrichment of Rad21 near the CTCF insulators, we performed ChIP assays using Rad21 antibody on mice infected with the ΔCTRL2 recombinant at 28 dpi and compared relative enrichments over IgG to wt virus for each of the corresponding CTCF insulators. All ChIP assays were done using ganglia harvested at 28 dpi from ΔCTRL2-infected mice and were validated using the positive and negative cellular controls Tsix and MT498, as previously described. To compare the two viruses, B/I ratios were normalized to IgG for each individual experimental replicate, and the fold enrichments of Rad21 at each individual site were compared between the wt and mutant viruses (Fig. 7). We found no significant difference in Rad21 enrichment at any of the CTCF insulator sites in the ΔCTRL2 mutant relative to wt virus. It should also be noted that in both the wt and mutant viruses, there was no significant enrichment of Rad21 near the CTRS1/2 site relative to IgG (fold enrichment, <1.6) (Fig. 7). These findings showed that the deletion of the CTRL2 insulator did not significantly change Rad21 enrichment near CTCF insulators during latency, suggesting that the CTRL2 insulator is responsible for efficient latency establishment but not the maintenance of latency.
FIG 7.
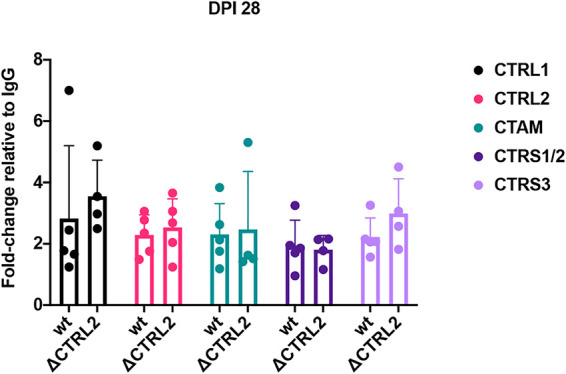
Deletion of the CTRL2 insulator did not affect Rad21 enrichment at 28 dpi. ChIP assays using the Rad21 antibody or the IgG control were done on mouse TG infected with the ΔCTRL2 virus at 28 dpi. Subsequent qPCR using primers and custom probes specific for nucleotide regions within 100 to 150 bp of the CTCF insulator sites was performed on both the antibody aliquot and the IgG control. Relative copy numbers in the B, I, or IgG fraction were determined from the equation for the standard curve specific to the primer/probe set and then normalized to IgG. B/I ratios for each site were divided by the IgG/I normalized value so that comparisons between the ΔCTRL2 and wt 17Syn+ virus are graphed as fold enrichment relative to IgG. The bar graph is presented as the average fold enrichment for all 5 biological replicates at 28 dpi for the wt and mutant viruses. One-way ANOVA on independent samples was used to determine statistical significance between fold changes relative to IgG between the two viruses.
Deletion of the CTRL2 insulator in HSV-1 decreased Rad21 enrichment near CTCF insulators at early time points following infection in vivo.
To explore the possibility that Rad21 deposition to viral genomes was linked to the CTRL2 insulator at early times postinfection in ganglia, we performed ChIP assays using Rad21 antibody on mice infected with the ΔCTRL2 recombinant at 7 and 14 dpi (Fig. 8). Following ChIP of TG from ΔCTRL2-infected mice, several key differences were noted between the ΔCTRL2- and wt-infected animals at early times following infection. First, in the ΔCTRL2 mutant the CTRL1 insulator was not enriched in Rad21 at 7 and 14 dpi (Fig. 8A). For comparison, in the wt-infected animals, the CTRL1 insulator was significantly enriched in Rad21 at all infection time points (Fig. 4A). Rad21 enrichment was not observed near the CTRL2 site in the ΔCTRL2 mutant virus through 14 dpi (Fig. 8B), in contrast to the wt, where the site is enriched in Rad21 at 14 dpi. Finally, no Rad21 enrichment was observed in the ΔCTRL2 mutant at the CTRS1/2 or CTRS3 site that flanks ICP4 (Fig. 8D and E), suggesting that the deposition of Rad21 on these sites is at least in part dependent on the presence of the CTRL2 insulator.
FIG 8.

Deletion of the CTRL2 insulator in HSV-1 decreased Rad21 enrichment 7 and 14 dpi. ChIP assays using the Rad21 antibody or the IgG control were done on mouse TG infected with the ΔCTRL2 virus at 7 and 14 dpi (n = 4 to 5). Subsequent qPCR using primers and custom probes listed in Table 1 were done on both the antibody aliquot and the IgG control. Relative copy numbers in the B, I, or IgG fractions were determined from the equation for the standard curve specific to the primer/probe set and then normalized to the cellular control Tsix Site A B/I for that ChIP assay for comparison across time points. Bar graphs are presented for B/I and IgG/I, and one-way ANOVA on correlated samples was used to determine statistical significance for the enrichment of Rad21 relative to IgG for the individual dpi (*, P < 0.05). (A) Normalized B/I and IgG/I for CTRL1 insulator upstream LAT. (B) Normalized B/I and IgG/I for CTRL2 insulator downstream LAT. (C) Normalized B/I and IgG/I for CTam insulator upstream ICP0. (D) Normalized B/I and IgG/I for CTRS1/2 insulator downstream ICP4. (E) Normalized B/I and IgG/I for CTRS3 insulator upstream ICP4.
Deletion of the CTRL2 insulator eliminated Rad21 enrichment at the CTRL2 and CTRS3 insulators in HeLa cells but not LUHMES cells.
Our experiments in mouse TG suggested that Rad21 enrichment at CTRL2 and CTRS3 is interdependent, as lytic replication is occurring. To explore this during the productive HSV-1 infection, we performed ChIP assays using Rad21 antibody and qPCR at the CTCF insulator sites following infection of differentiated LUHMES cells, a human neuronal model that supports quiescent HSV-1 infections (72, 73). For comparison purposes, identical ChIP assays were done in HeLa cells as a representative human epithelial cell line. The ChIP assays were validated by comparing the bound/input values for the positive control for Rad21 enrichment in human cell lines (ERE2) to IgG/input. Experiments with a significant enrichment of ERE2 to IgG (over 5-fold) were considered validated and used for qPCR. During productive infection in HeLa cells, Rad21 was significantly enriched over IgG at all of the CTCF insulators analyzed (Fig. 9A). Deletion of the CTRL2 insulator resulted in a loss of Rad21 enrichment at the CTRL2 and CTRS3 insulators in HeLa cells (Fig. 9B). However, in differentiated LUHMES cells, Rad21 enrichment was only observed at the CTRL2 and CTRS3 insulators in wt virus, and deletion of the CTRL2 insulator had no effect on Rad21 enrichment of viral genomes in neuronal cells during the productive infection (Fig. 9C and D). These findings were exciting, again indicating the CTCF-Rad21 interactions are cell type specific and that the CTRL2 and CTRS3 insulators are interdependent.
FIG 9.

Deletion of the CTRL2 insulator eliminated Rad21 enrichment at the CTRL2 and CTRS3 insulators in HeLa cells but not LUHMES cells. ChIP assays using a Rad21 antibody were done in HeLa and differentiated LUHMES cells at 24 hpi with either the wt or ΔCTRL2 virus (n = 4). ChIP assays were validated using the positive cellular control ERE2, as described in Materials and Methods. All graphs represent the average bound/input (B/I) values normalized to IgG and are presented as fold enrichments with standard deviations from the means. gC represents the negative viral control (not associated with either CTCF or Rad21). Statistical analyses were performed with a Student's t test for each of the CTCF sites B/I and IgG/I (*, P < 0.01, ***, P < 0.0001).
LAT expression decreased in the absence of Rad21 enrichment at the CTRL2 insulator.
A recent study by Li et al. showed that cohesin subunits Rad21 and SMC1 were recruited to HSV-1 lytic replication compartments, and knockdown of Rad21 and SMC1 resulted in reduced RNA pol II occupancy, suggesting that cohesin recruitment is important for HSV-1 lytic transcription (70). To determine if Rad21 enrichment at the CTRL2 insulator could be correlated with LAT expression, we infected HeLa and differentiated LUHMES cells at two multiplicities of infection (MOI; 0.1 and 1) and performed quantitative reverse transcription-PCR (qRT-PCR) to quantify LAT expression in the two cell types for the wt and the ΔCTRL2 mutant. We found strikingly different LAT expression profiles between these two viruses depending on cell type. First, in HeLa cells, LAT expression was significantly lower in the ΔCTRL2 mutant (Fig. 10A). These data suggest that LAT expression is dependent on Rad21 enrichment at or near the CTRL2 insulator, since deletion of the CTRL2 insulator resulted in a loss of Rad21 enrichment at the CTRL2 and CTRS3 insulators in HeLa cells. In contrast, in LUHMES cells, Rad21 enrichment at both the CTRL2 and CTRS3 insulators is consistent between the wt and mutant viruses, and LAT expression is significantly increased in the ΔCTRL2 mutant compared to the wt, supporting that Rad21 enrichment at the CTRL2 and CTRS3 insulators is required for efficient LAT expression (Fig. 10B).
FIG 10.

LAT expression decreased when Rad21 enrichment at the CTRL2 insulator decreased. (A and B) HeLa cells (A) and differentiated LUHMES cells (B) were infected with wt or ΔCTRL2 virus at 0.1 and 1.0 MOI in triplicate. After 6 h, cells were harvested and RNA was extracted. qRT-PCR was performed for LAT intron primer and probe (Table 1). Threshold values used for PCR analyses were set within the linear range of PCR target amplification based on a standard curve generated for each plate. Viral transcripts were normalized to GAPDH. Statistical analyses were performed with a Student's t test (***, P < 0.0001).
DISCUSSION
CTCF insulators are important regulatory domains that have multiple functions in the context of DNA viral genomes, including establishing chromatin barriers, recruiting coactivating and corepressing complexes, and maintaining the integrity of gene expression (74, 75). CTCF insulators can self-dimerize in an orientation-specific manner to establish chromatin loops to control gene expression over large genomic distances, and other DNA viruses, such as EBV and KSHV, control lytic transcription in a CTCF cohesin-dependent manner (65–67). To date, 7 putative CTCF insulators have been identified in latent HSV-1 genomes, and the location of each of these sites within the context of HSV-1 are oriented to flank the LAT and IE gene regions of the genome (64). The locations of CTCF insulators in the context of the latent HSV-1 genome have long suggested that CTCF insulators play a key role in not only the establishment of latency but also the maintenance of latency. The disruption of CTCF binding to latent genomes precedes reactivation, a finding that reinforces the hypothesis that CTCF insulators maintain transcriptional integrity of the LAT and IE genes during latency.
We previously showed that the protein CTCF was differentially bound to individual insulator sites, that the enhancer-blocking functions of key CTCF insulators in HSV-1 were cell type specific, and that CTCF was differentially evicted at early times postreactivation, suggesting that the individual CTCF insulators in HSV-1 have independent roles in the regulation of transcription during the lytic infection and during latency that are likely location dependent (60, 62). Orientation-specific self-dimerization of CTCF insulators results in the formation of higher-order chromatin structures known as chromatin loops. In eukaryotic cells, genomes are folded into three-dimensional structures that are further organized into epigenomic compartments known as topologically associated domains (TADs). TADs serve as interaction hubs where regulatory elements, such as enhancers, can interact with genes to activate or silence transcription (76–78). Recent studies have shown that the cohesin complex is an integral part of not only the establishment of chromatin loops but also the organization and maintenance of TADs. CTCF-cohesin interactions are codependent; the knockdown of cohesin results in the disruption of TADs, the loss of loop formation, and the disruption of gene expression in eukaryotic cells. Finally, recent evidence from the gammaherpesvirus field has indicated that cohesin is colocalized with CTCF and facilitates the formation of DNA loops that are both CTCF and cohesin dependent (65, 67). Considering this, we hypothesized that HSV-1 would utilize similar mechanisms for transcriptional regulation during latency. Our key finding that one of the cohesin complex subunits, Rad21, colocalized within ∼600 bp of 4 of the CTCF insulators during latency suggests that these four insulators are involved in the formation of chromatin loops to regulate transcription. The location of the CTCF insulators that maintain Rad21 enrichment during latency, namely, the CTRL1 and CTRL2 insulators that flank the LAT and the CTRS3 insulator upstream from the ICP4 promoter (Fig. 1), suggests the 3-dimensional structure of the latent viral genome is oriented in such a way that the LAT enhancer could be spatially oriented near the ICP4 promoter. This is further supported by our findings from the HeLa and LUHMES ChIP assays that indicated that the CTRL2 and CTRS3 insulators were interdependent. Genomes with a spatial orientation of the LAT and ICP4 regions could allow for the LAT enhancer to activate ICP4 following instances where CTCF binding is disrupted at these sites. Earlier work done in our laboratory has already shown that CTCF binding is abolished at the CTRS3 insulator at early times postreactivation (60).
Interestingly, not all of the CTCF insulators that have been identified were enriched in Rad21 during latency. Specifically, the CTRS1/2 insulator, positioned downstream of the IE transactivator ICP4, notably was not enriched in Rad21 during latency in wt virus-infected mice. This was an intriguing finding, considering that we had previously characterized the site as an enhancer-blocking insulator in both epithelial and neuronal cell types, suggesting that it plays a role in the regulation of gene expression by blocking the LAT enhancer (60, 62). One possibility is that the CTRS1/2 insulator functions in a linear manner during latency rather than as part of a chromatin loop. Another, perhaps more intriguing, possibility is that the CTRS1/2 insulator is involved in the formation of a chromatin loop when the virus is lytically replicating. This possibility is supported by our findings that in wt virus at 7 and 14 dpi, the CTRS1/2 insulator is enriched in Rad21 when the viral genome loads are very high in TG, but by 28 dpi, the CTRS1/2 insulator is no longer enriched in Rad21. One possibility is that genomes enriched in Rad21 at the CTRS1/2 insulator target a different population of neurons that are either permissive for lytic expression (79) or maintain different LAT expression levels (73). Our ChIP data from wt-infected HeLa cells showing that CTRS1/2 is enriched in Rad21 and this enrichment is correlated with LAT expression support this possibility.
Finally, we knew from previous studies that deletion of the CTRL2 insulator resulted in a loss of barrier insulator function (59), decreased latency establishment, decreased H3K27me3 occupation on the viral genome, and increased lytic transcription in neurons (63). In the current study, Rad21 enrichment during latency was not affected when the CTRL2 insulator was deleted. However, we found significantly decreased enrichment in Rad21 at the insulators flanking ICP4 and the CTRL2 site prior to latency in the CTRL2 mutant virus. These findings suggest that Rad21 enrichment of CTRL2 during a productive infection is involved in the establishment of latency. This is consistent with our data showing all of the CTCF insulators are enriched in Rad21 in HeLa cells infected with wt virus, but in the CTRL2 deletion mutant, Rad21 enrichment is lost at the CTRL2 and CTRS3 insulators. The fact that both the CTRL2 and CTRS3 insulators remain enriched in Rad21 in LUHMES cells in both viruses highlights the significant cell type differences in insulator function. We speculate that these cell type-specific differences are due to different spatial orientations of genomes in epithelial cells versus neuronal cells during productive versus latent infections, and work to address this important question is underway.
MATERIALS AND METHODS
Viruses and cells.
Vero cells (ATCC CCL-81) and HeLa cells (ATCC-CCL-2) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (catalog no. 15-013; Corning) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic solution (catalog no. 15240062; Gibco) in a humidified atmosphere with 5% CO2 at 37°C and passaged at regular intervals when the cells were 70 to 80% confluent. The wild-type HSV-1 strain, 17Syn+ (GenBank accession number NC_001806), and the recombinant ΔCTRL2 viruses were both obtained from D. Bloom (University of Florida), and the mutant virus was sequenced prior to use in animals (63). Both viruses were grown in Vero cells supplemented with 1% FBS and 1% antibiotic-antimycotic solution at 37°C. The titers of viruses were determined on Vero cells in triplicate.
LUHMES cells.
The LUHMES neuronal cell line (CRL-2927; ATCC) was cultured and differentiated as described previously (72, 80). Briefly, for CHIP assay, 1 × 105 LUHMES cells were seeded in T-75 flasks precoated with 50 μg/ml poly-l-ornithine (catalog no. P3655; Sigma) and 1 μg/ml fibronectin (catalog no. F2006; Sigma) in DMEM-F12 medium supplemented with 1% N2 supplement, fibroblast growth factor, and 1% antibiotics in humidified atmosphere with 5% CO2 at 37°C. After 48 h, cells were differentiated for 5 days in DMEM-F12, 1% N2 supplement, 1% antibiotics, 10 mg/ml tetracycline, 100 μg/ml rhDGNF, 204 mM cyclic AMP (cAMP). An extra flask was used for differentiated LUHMES cell counting before infection. Cells were infected with wt or recombinant HSV-1 virus at 1 MOI in the differentiation media. After 1 h, medium was removed and replaced with fresh differentiation media. Cells were harvested and processed for ChIP, followed by qPCR as described below. For LAT expression experiments, 50,000 cells were seeded in precoated 6-well plates, and cells were differentiated for 5 days. Cells were infected with wt or recombinant HSV-1 virus at 0.1 and 1 MOI in differentiation media. Cells were harvested for RNA at 6 hpi. qRT-PCR was performed using LAT primers and probe (Table 1).
Mouse ocular infections.
Four- to 6-week-old female BALB/c mice (Taconic) were anesthetized by isoflurane in accordance with University of Wisconsin veterinary guidelines. Using a 27-gauge needle, a light 2 by 2 crosshatch pattern was made on the corneal epithelium. Equal MOIs of virus (150,000 PFU/eye), as determined by standard plaque assay, were applied to both eyes of each mouse. Mice were monitored daily through postinfection day 28 for signs of morbidity and mortality. Mice were monitored through the duration of the experiment for signs of morbidity and mortality. Animals were considered latent at >28 days postinfection. Mice were sacrificed at 7, 14, or 28 dpi and processed immediately for ChIP assays. Each ChIP assay replicate contained 3 mice (6 TG) per sample.
HSV-1 DNA replication in cell culture.
HeLa cells and differentiated LUHMES cells were infected with the wt and recombinant HSV-1 strains described above at an MOI of 1 in triplicate. The media were replenished (DMEM plus 1% FBS plus 1% antibiotics) for HeLa and differentiation media for LUHMES (DMEM-F12, 1% N2, 1% antibiotics, 10 mg/ml tetracycline, 100 μg/ml rhDGNF, 204mM cAMP) after an hour following infection. The cells were harvested at 6 h, 18 h, 24 h, and 48 h. The genomic DNA was extracted by phenol-chloroform extraction. The HSV-1 DNA was quantified by qPCR using HSV-1 DNA polymerase primers (Table 1). The genome copy numbers at 18, 24, and 48 h were normalized to the 6-h time as an input measure.
qPCR for genome copies/ganglion.
Genome copies per ganglion were measured by quantitative real-time PCR using primers and a probe specific for CTRS3 (Table 1). All real-time PCR experiments were performed using TaqMan universal PCR master mix and No AmpErase uracil N-glycosylase on Agilent AriaMx real-time PCR using custom-designed primer-probe mixes (Life Technologies) with the general protocol of 95°C for 10 min (1×) and then 95°C for 15 s, followed by 60°C for 1 min (45×). Threshold values used for PCR analyses were set within the linear range of PCR target amplification based on a standard curve generated for each plate. The relative HSV-1 copy numbers for each sample were normalized to the relative copy numbers of the host control Tsix Site A (Table 1).
RNA expression.
HeLa cells and differentiated LUHMES cells were infected with wt and recombinant HSV-1 virus at 0.1 and 1.0 MOI in triplicate. The viruses were replaced with their respective fresh low-serum media after an hour of infection. After 6 h, cells were harvested in TRIzol reagent (T9424; Sigma), and RNA was extracted according to the manufacturer’s protocol. The qRT-PCR was performed using a Superscript III platinum one-step qRT-PCR system (11732-088; Invitrogen) for LAT intron primer and probe (Table 1) with a general protocol of 50°C for 15 min (1×) followed by 95°C for 3 min (1×) and then 95°C for 15 s followed by 60°C for 1 min (40×). Threshold values used for PCR analyses were set within the linear range of PCR target amplification based on a standard curve generated for each plate. Viral transcripts were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 4333764; TaqMan gene expression assay; ThermoFisher).
ChIP assays.
ChIP assays were performed as previously described for mice using the specific antibody anti-Rad21 (Millipore). Each ChIP assay contained three mice pooled (6 TG). The TG were rapidly removed following euthanasia, the ganglia were homogenized, and the chromatin was cross-linked in formaldehyde (Sigma-Aldrich). The cross-linked cell lysates were sonicated to shear the chromatin into fragments between 300 and 800 bp. Fragment size following sonication was confirmed by agarose gel electrophoresis using a 1.5% gel. The sheared chromatin was precleared with salmon sperm DNA protein A-agarose beads (Millipore) prior to antibody incubation. An aliquot representing 1/5 of the total sample volume was removed as a sample input (I). The remaining sample was incubated in a cold room overnight with shaking with 2 μg of antibody per 1 ml of sample or with IgG as a nonspecific antibody binding control. The chromatin-antibody complexes were collected with salmon sperm DNA protein A-agarose beads and eluted to represent the bound (B) fraction. Both bound and input fractions were treated with 5 M NaCl, RNase A, and proteinase K, and the DNA was purified using a QIAquick PCR purification kit (Qiagen).
qPCR analysis of ChIP experiments.
All qPCR experiments were performed using TaqMan universal PCR master mix and No AmpErase uracil N-glycosylase on an Agilent AriaMx real-time PCR system using custom-designed primer-probe mixes (Applied Biosystems) (Table 1) with the general protocol of 95°C for 10 min (1×) and then 95°C for 15 s, followed by 60°C for 1 min (45×). Threshold values used for PCR analyses were set within the linear range of PCR target amplification based on a standard curve generated for each plate.
Determination of B/I ratios from ChIP experiments.
ChIP data are presented as a ratio of the relative copies of the PCR target in two fractions (bound and input), where the bound fractions are representative of aliquots incubated overnight with the antibody or IgG and the input fractions are precleared chromatin only (no specific antibody incubation). Standard curves were generated for each PCR run for each gene region analyzed using serial dilutions of purified HSV-1 DNA or purified mouse DNA using primers/probes specific for that region (Table 1). Each ChIP assay was analyzed as follows. First, qPCR was performed using DNA purified from either the B, I, or IgG fraction as the target. The average cycle threshold (CT) for the bound fraction and the average CT for the input fraction were then used to determine the relative quantity of target DNA in each fraction by using the equation for the standard curve specific to the primer and probe set used. For IgG incubated samples, the ratios were calculated as IgG/I. The quantity was expressed as a ratio of the relative bound quantity to the relative input quantity (B/I ratio) and then compared to IgG/I ratios for graphs. For some figures, the relative B/I ratios from each gene region were normalized to the B/I ratio for the cellular control Tsix Site A B/I so that comparisons between different treatment groups could be done.
ChIP validation.
Prior to real-time PCR analyses of viral targets, all ChIP assays were validated by determining the B/I ratios of the cellular controls Tsix imprinting/choice center CTCF site A (positive control) and MT498 (negative control) (81). Only assays having a greater than 2-fold abundance of Rad21 bound to Tsix imprinting/choice center CTCF site A relative to MT498 were used in further analyses. For ChIP assays done in the human cell lines (HeLa and LUHMES), validations were performed using the positive-control ERE2 compared to IgG as a negative control, as previously described (82) (Table 1).
Statistical analysis.
One-way analysis of variance (ANOVA) and Student’s t test were performed using SigmaPlot 12.5 by analyzing the individual normalized B/I ratios for a specific gene region to the IgG/I values as correlated samples. For comparisons between samples at different time points or between the two viruses, one-way ANOVA with independent samples was used. The P values are indicated on the graphs above the time point at which a significant change was determined statistically.
ACKNOWLEDGMENTS
This work was supported in part by grant R01AI134807 (D.M.N.) from NIH-NIAID, the Core Grant for Vision Research from the NIH to the University of Wisconsin-Madison (P30 EY016665), and an unrestricted grant from Research to Prevent Blindness (Department of Ophthalmology, University of Wisconsin).
We also thank David C. Bloom, University of Florida, for providing reagents, protocols for LUHMES cells, and the viruses used in this study.
REFERENCES
- 1.Austin A, Lietman T, Rose-Nussbaumer J. 2017. Update on the management of infectious keratitis. Ophthalmology 124:1678–1689. 10.1016/j.ophtha.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bloom DC. 2016. Alphaherpesvirus latency: a dynamic state of transcription and reactivation. Adv Virus Res 94:53–80. 10.1016/bs.aivir.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Gibeault RL, Conn KL, Bildersheim MD, Schang LM. 2016. An essential viral transcription activator modulates chromatin dynamics. PLoS Pathog 12:e1005842. 10.1371/journal.ppat.1005842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu M, Depledge DP, Flores Cortes E, Breuer J, Schang LM. 2019. Chromatin dynamics and the transcriptional competence of HSV-1 genomes during lytic infections. PLoS Pathog 15:e1008076. 10.1371/journal.ppat.1008076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conn KL, Hendzel MJ, Schang LM. 2013. The differential mobilization of histones H3.1 and H3.3 by herpes simplex virus 1 relates histone dynamics to the assembly of viral chromatin. PLoS Pathog 9:e1003695. 10.1371/journal.ppat.1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh J, Sanders IF, Chen EZ, Li H, Tobias JW, Isett RB, Penubarthi S, Sun H, Baldwin DA, Fraser NW. 2015. Genome wide nucleosome mapping for HSV-1 shows nucleosomes are deposited at preferred positions during lytic infection. PLoS One 10:e0117471. 10.1371/journal.pone.0117471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JS, Raja P, Knipe DM. 2016. Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic infection. mBio 7:e02007-15. 10.1128/mBio.02007-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oh HS, Neuhausser WM, Eggan P, Angelova M, Kirchner R, Eggan KC, Knipe DM. 2019. Herpesviral lytic gene functions render the viral genome susceptible to novel editing by CRISPR/Cas9. Elife 8:e51662. 10.7554/eLife.51662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, Berger SL. 2004. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol 78:10178–10186. 10.1128/JVI.78.18.10178-10186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacasse JJ, Schang LM. 2010. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J Virol 84:1920–1933. 10.1128/JVI.01934-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lacasse JJ, Schang LM. 2012. Herpes simplex virus 1 DNA is in unstable nucleosomes throughout the lytic infection cycle, and the instability of the nucleosomes is independent of DNA replication. J Virol 86:11287–11300. 10.1128/JVI.01468-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen C, Corpet A, Roubille S, Maroui MA, Poccardi N, Rousseau A, Kleijwegt C, Binda O, Texier P, Sawtell N, Labetoulle M, Lomonte P. 2018. Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV-1 genomes chromatinization through a PML NB/histone H3.3/H3.3 chaperone axis. PLoS Pathog 14:e1007313. 10.1371/journal.ppat.1007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubat NJ, Tran RK, McAnany P, Bloom DC. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J Virol 78:1139–1149. 10.1128/jvi.78.3.1139-1149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kristie TM. 2016. Chromatin modulation of herpesvirus lytic gene expression: managing nucleosome density and heterochromatic histone modifications. mBio 7:e00098-16. 10.1128/mBio.00098-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cabrera JR, Charron AJ, Leib DA. 2018. Neuronal subtype determines herpes simplex virus 1 latency-associated-transcript promoter activity during latency. J Virol 92:e00430-18. 10.1128/JVI.00430-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubat NJ, Amelio AL, Giordani NV, Bloom DC. 2004. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol 78:12508–12518. 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cliffe AR, Coen DM, Knipe DM. 2013. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. mBio 4:e00590-12. 10.1128/mBio.00590-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181. 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arbuckle JH, Gardina PJ, Gordon DN, Hickman HD, Yewdell JW, Pierson TC, Myers TG, Kristie TM. 2017. Inhibitors of the histone methyltransferases EZH2/1 induce a potent antiviral state and suppress infection by diverse viral pathogens. mBio 8:e01141-17. 10.1128/mBio.01141-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM. 2007. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J Virol 81:13248–13253. 10.1128/JVI.01569-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Creech CC, Neumann DM. 2010. Changes to euchromatin on LAT and ICP4 following reactivation are more prevalent in an efficiently reactivating strain of HSV-1. PLoS One 5:e15416. 10.1371/journal.pone.0015416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amelio AL, Giordani NV, Kubat NJ, O'Neil JE, Bloom DC. 2006. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J Virol 80:2063–2068. 10.1128/JVI.80.4.2063-2068.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avgousti DC, Weitzman MD. 2015. Stress flips a chromatin switch to wake up latent virus. Cell Host Microbe 18:639–641. 10.1016/j.chom.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cliffe AR, Wilson AC. 2017. Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J Virol 91:e01419-16. 10.1128/JVI.01419-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips JE, Corces VG. 2009. CTCF: master weaver of the genome. Cell 137:1194–1211. 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380. 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159:1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gassler J, Brandao HB, Imakaev M, Flyamer IM, Ladstatter S, Bickmore WA, Peters JM, Mirny LA, Tachibana K. 2017. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J 36:3600–3618. 10.15252/embj.201798083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, Huang X, Shamim MS, Shin J, Turner D, Ye Z, Omer AD, Robinson JT, Schlick T, Bernstein BE, Casellas R, Lander ES, Aiden EL. 2017. Cohesin loss eliminates all loop domains. Cell 171:305–320. 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Haarhuis JHI, Sedeño Cacciatore Á, Oldenkamp R, van Ruiten MS, Willems L, Teunissen H, Muir KW, de Wit E, Rowland BD, Panne D. 2020. The structural basis for cohesin–CTCF-anchored loops. Nature 578:472–476. 10.1038/s41586-019-1910-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maya-Miles D, Andujar E, Perez-Alegre M, Murillo-Pineda M, Barrientos-Moreno M, Cabello-Lobato MJ, Gomez-Marin E, Morillo-Huesca M, Prado F. 2019. Crosstalk between chromatin structure, cohesin activity and transcription. Epigenet Chromatin 12:47. 10.1186/s13072-019-0293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen AS, Pustova I, Cattoglio C, Tjian R, Darzacq X. 2017. CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6:e25776. 10.7554/eLife.25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng H, Zhang N, Pati D. 2020. Cohesin subunit RAD21: from biology to disease. Gene 758:144966. 10.1016/j.gene.2020.144966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang LH, Ghosh S, Noordermeer D. 2020. TADs and their borders: free movement or building a wall? J Mol Biol 432:643–652. 10.1016/j.jmb.2019.11.025. [DOI] [PubMed] [Google Scholar]
- 36.Rowley MJ, Corces VG. 2018. Organizational principles of 3D genome architecture. Nat Rev Genet 19:789–800. 10.1038/s41576-018-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKay MJ, Troelstra C, van der Spek P, Kanaar R, Smit B, Hagemeijer A, Bootsma D, Hoeijmakers JH. 1996. Sequence conservation of the rad21 Schizosaccharomyces pombe DNA double-strand break repair gene in human and mouse. Genomics 36:305–315. 10.1006/geno.1996.0466. [DOI] [PubMed] [Google Scholar]
- 38.Zhang N, Jiang Y, Mao Q, Demeler B, Tao YJ, Pati D. 2013. Characterization of the interaction between the cohesin subunits Rad21 and SA1/2. PLoS One 8:e69458. 10.1371/journal.pone.0069458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birot A, Eguienta K, Vazquez S, Claverol S, Bonneu M, Ekwall K, Javerzat JP, Vaur S. 2017. A second Wpl1 anti-cohesion pathway requires dephosphorylation of fission yeast kleisin Rad21 by PP4. EMBO J 36:1364–1378. 10.15252/embj.201696050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcos-Alcalde I, Mendieta-Moreno JI, Puisac B, Gil-Rodriguez MC, Hernandez-Marcos M, Soler-Polo D, Ramos FJ, Ortega J, Pie J, Mendieta J, Gomez-Puertas P. 2017. Two-step ATP-driven opening of cohesin head. Sci Rep 7:3266. 10.1038/s41598-017-03118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu X, Kanai R, Nakazawa N, Wang L, Toyoshima C, Yanagida M. 2018. Suppressor mutation analysis combined with 3D modeling explains cohesin's capacity to hold and release DNA. Proc Natl Acad Sci U S A 115:E4833–E4842. 10.1073/pnas.1803564115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higashi TL, Eickhoff P, Sousa JS, Locke J, Nans A, Flynn HR, Snijders AP, Papageorgiou G, O'Reilly N, Chen ZA, O'Reilly FJ, Rappsilber J, Costa A, Uhlmann F. 2020. A structure-based mechanism for DNA entry into the cohesin ring. Mol Cell 79:917–933. 10.1016/j.molcel.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ba Z, Lou J, Ye AY, Dai HQ, Dring EW, Lin SG, Jain S, Kyritsis N, Kieffer-Kwon KR, Casellas R, Alt FW. 2020. CTCF orchestrates long-range cohesin-driven V(D)J recombinational scanning. Nature 586:305–310. 10.1038/s41586-020-2578-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng G, Kanchwala M, Xing C, Yu H. 2018. MCM2-7-dependent cohesin loading during S phase promotes sister-chromatid cohesion. Elife 7:e33920. 10.7554/eLife.33920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Senaratne TN, Joyce EF, Nguyen SC, Wu CT. 2016. Investigating the interplay between sister chromatid cohesion and homolog pairing in Drosophila nuclei. PLoS Genet 12:e1006169. 10.1371/journal.pgen.1006169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fisher JB, Peterson J, Reimer M, Stelloh C, Pulakanti K, Gerbec ZJ, Abel AM, Strouse JM, Strouse C, McNulty M, Malarkannan S, Crispino JD, Milanovich S, Rao S. 2017. The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia 31:712–719. 10.1038/leu.2016.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia L, Wang M, Li H, Tang X, Chen F, Cui J. 2018. The effect of aberrant expression and genetic polymorphisms of Rad21 on cervical cancer biology. Cancer Med 7:3393–3405. doi:10.1002/cam4.1592. 10.1002/cam4.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pugacheva EM, Kubo N, Loukinov D, Tajmul M, Kang S, Kovalchuk AL, Strunnikov AV, Zentner GE, Ren B, Lobanenkov VV. 2020. CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention. Proc Natl Acad Sci U S A 117:2020–2031. 10.1073/pnas.1911708117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wutz G, Ladurner R, St Hilaire BG, Stocsits RR, Nagasaka K, Pignard B, Sanborn A, Tang W, Varnai C, Ivanov MP, Schoenfelder S, van der Lelij P, Huang X, Durnberger G, Roitinger E, Mechtler K, Davidson IF, Fraser P, Lieberman-Aiden E, Peters JM. 2020. ESCO1 and CTCF enable formation of long chromatin loops by protecting cohesin(STAG1) from WAPL. Elife 9:e52091. 10.7554/eLife.52091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wutz G, Varnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, Walther N, Koch B, Kueblbeck M, Ellenberg J, Zuber J, Fraser P, Peters JM. 2017. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J 36:3573–3599. 10.15252/embj.201798004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davidson IF, Bauer B, Goetz D, Tang W, Wutz G, Peters JM. 2019. DNA loop extrusion by human cohesin. Science 366:1338–1345. 10.1126/science.aaz3418. [DOI] [PubMed] [Google Scholar]
- 52.Li DJ, Verma D, Mosbruger T, Swaminathan S. 2014. CTCF and Rad21 act as host cell restriction factors for Kaposi's sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog 10:e1003880. 10.1371/journal.ppat.1003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li D, Mosbruger T, Verma D, Swaminathan S. 2019. Complex interactions between cohesin and CTCF in regulation of Kaposi's sarcoma-associated herpesvirus lytic transcription. J Virol 94:e01279-19. 10.1128/JVI.01279-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lupey-Green LN, Caruso LB, Madzo J, Martin KA, Tan Y, Hulse M, Tempera I. 2018. PARP1 stabilizes CTCF binding and chromatin structure to maintain Epstein-Barr virus latency type. J Virol 92:e00755-18. 10.1128/JVI.00755-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mehta K, Gunasekharan V, Satsuka A, Laimins LA. 2015. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog 11:e1004763. 10.1371/journal.ppat.1004763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pentland I, Campos-Leon K, Cotic M, Davies KJ, Wood CD, Groves IJ, Burley M, Coleman N, Stockton JD, Noyvert B, Beggs AD, West MJ, Roberts S, Parish JL. 2018. Disruption of CTCF-YY1-dependent looping of the human papillomavirus genome activates differentiation-induced viral oncogene transcription. PLoS Biol 16:e2005752. 10.1371/journal.pbio.2005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang H, Cho H, Sung GH, Lieberman PM. 2013. CTCF regulates Kaposi's sarcoma-associated herpesvirus latency transcription by nucleosome displacement and RNA polymerase programming. J Virol 87:1789–1799. 10.1128/JVI.02283-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lang F, Li X, Vladimirova O, Hu B, Chen G, Xiao Y, Singh V, Lu D, Li L, Han H, Wickramasinghe JM, Smith ST, Zheng C, Li Q, Lieberman PM, Fraser NW, Zhou J. 2017. CTCF interacts with the lytic HSV-1 genome to promote viral transcription. Sci Rep 7:39861. 10.1038/srep39861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee JS, Raja P, Pan D, Pesola JM, Coen DM, Knipe DM. 2018. CCCTC-binding factor acts as a heterochromatin barrier on herpes simplex viral latent chromatin and contributes to poised latent infection. mBio 9:e02372-17. 10.1128/mBio.02372-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ertel MK, Cammarata AL, Hron RJ, Neumann DM. 2012. CTCF occupation of the herpes simplex virus 1 genome is disrupted at early times postreactivation in a transcription-dependent manner. J Virol 86:12741–12759. 10.1128/JVI.01655-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Washington SD, Edenfield SI, Lieux C, Watson ZL, Taasan SM, Dhummakupt A, Bloom DC, Neumann DM. 2018. Depletion of the insulator protein CTCF results in herpes simplex virus 1 reactivation in vivo. J Virol 92:e00173-18. 10.1128/JVI.00173-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Washington SD, Musarrat F, Ertel MK, Backes GL, Neumann DM. 2018. CTCF binding sites in the herpes simplex virus 1 genome display site-specific CTCF occupation, protein recruitment, and insulator function. J Virol 92:e00156-18. 10.1128/JVI.00156-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Washington SD, Singh P, Johns RN, Edwards TG, Mariani M, Frietze S, Bloom DC, Neumann DM. 2019. The CCCTC binding factor, CTRL2, modulates heterochromatin deposition and the establishment of herpes simplex virus 1 latency in vivo. J Virol 93:e00415-19. 10.1128/JVI.00415-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amelio AL, McAnany PK, Bloom DC. 2006. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J Virol 80:2358–2368. 10.1128/JVI.80.5.2358-2368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen HS, Martin KA, Lu F, Lupey LN, Mueller JM, Lieberman PM, Tempera I. 2014. Epigenetic deregulation of the LMP1/LMP2 locus of Epstein-Barr virus by mutation of a single CTCF-cohesin binding site. J Virol 88:1703–1713. 10.1128/JVI.02209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen HS, De Leo A, Wang Z, Kerekovic A, Hills R, Lieberman PM. 2017. BET-inhibitors disrupt Rad21-dependent conformational control of KSHV latency. PLoS Pathog 13:e1006100. 10.1371/journal.ppat.1006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. 2008. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J 27:654–666. 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM. 2011. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog 7:e1002140. 10.1371/journal.ppat.1002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen HS, Wikramasinghe P, Showe L, Lieberman PM. 2012. Cohesins repress Kaposi's sarcoma-associated herpesvirus immediate early gene transcription during latency. J Virol 86:9454–9464. 10.1128/JVI.00787-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li X, Yu Y, Lang F, Chen G, Wang E, Li L, Li Z, Yang L, Cao X, Fraser NW, Zhou J. 2021. Cohesin promotes HSV-1 lytic transcription by facilitating the binding of RNA Pol II on viral genes. Virol J 18:26. 10.1186/s12985-021-01495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giorgetti L, Lajoie BR, Carter AC, Attia M, Zhan Y, Xu J, Chen CJ, Kaplan N, Chang HY, Heard E, Dekker J. 2016. Structural organization of the inactive X chromosome in the mouse. Nature 535:575–579. 10.1038/nature18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edwards TG, Bloom DC. 2019. Lund human mesencephalic (LUHMES) neuronal cell line supports herpes simplex virus 1 latency in vitro. J Virol 93:e02210-18. 10.1128/JVI.02210-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grams TR, Edwards TG, Bloom DC. 2020. Herpes simplex virus 1 strains 17syn (+) and KOS(M) differ greatly in their ability to reactivate from human neurons in vitro. J Virol 94:e00796-20. 10.1128/JVI.00796-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Golan-Mashiach M, Grunspan M, Emmanuel R, Gibbs-Bar L, Dikstein R, Shapiro E. 2012. Identification of CTCF as a master regulator of the clustered protocadherin genes. Nucleic Acids Res 40:3378–3391. 10.1093/nar/gkr1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pentland I, Parish JL. 2015. Targeting CTCF to control virus gene expression: a common theme amongst diverse DNA viruses. Viruses 7:3574–3585. 10.3390/v7072791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang DC, Wang W, Zhang L, Wang X. 2019. A tour of 3D genome with a focus on CTCF. Semin Cell Dev Biol 90:4–11. 10.1016/j.semcdb.2018.07.020. [DOI] [PubMed] [Google Scholar]
- 77.Hansen AS, Cattoglio C, Darzacq X, Tjian R. 2018. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 9:20–32. 10.1080/19491034.2017.1389365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chung IM, Ketharnathan S, Kim SH, Thiruvengadam M, Rani MK, Rajakumar G. 2016. Making sense of the tangle: insights into chromatin folding and gene regulation. Genes 7:71. 10.3390/genes7100071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bertke AS, Ma A, Margolis MS, Margolis TP. 2013. Different mechanisms regulate productive herpes simplex virus 1 (HSV-1) and HSV-2 infections in adult trigeminal neurons. J Virol 87:6512–6516. 10.1128/JVI.00383-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scholz D, Poltl D, Genewsky A, Weng M, Waldmann T, Schildknecht S, Leist M. 2011. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem 119:957–971. 10.1111/j.1471-4159.2011.07255.x. [DOI] [PubMed] [Google Scholar]
- 81.Chao W, Huynh KD, Spencer RJ, Davidow LS, Lee JT. 2002. CTCF, a candidate trans-acting factor for X-inactivation choice. Science 295:345–347. 10.1126/science.1065982. [DOI] [PubMed] [Google Scholar]
- 82.McEwan MV, Eccles MR, Horsfield JA. 2012. Cohesin is required for activation of MYC by estradiol. PLoS One 7:e49160. 10.1371/journal.pone.0049160. [DOI] [PMC free article] [PubMed] [Google Scholar]