

Abstract
Aims
The early repolarization syndrome (ERS) can cause ventricular fibrillation (VF) and sudden death in young, otherwise healthy individuals. There are limited data suggesting that ERS might be heritable. The aim of this study was to characterize the clinical phenotype and to identify a causal variant in an affected family using an exome-sequencing approach.
Methods and results
Early repolarization syndrome was diagnosed according to the recently proposed Shanghai ERS Score. After sequencing of known ERS candidate genes, whole-exome sequencing (WES) was performed. The index patient (23 years, female) showed a dynamic inferolateral early repolarization (ER) pattern and electrical storm with intractable VF. Isoproterenol enabled successful termination of electrical storm with no recurrence on hydroquinidine therapy during 33 months of follow-up. The index patient’s brother (25 years) had a persistent inferior ER pattern with malignant features and a history of syncope. Both parents were asymptomatic and showed no ER pattern. While there was no pathogenic variant in candidate genes, WES detected a novel missense variant affecting a highly conserved residue (p. H2245R) in the ANK3 gene encoding Ankyrin-G in the two siblings and the father.
Conclusion
We identified two siblings with a malignant ERS phenotype sharing a novel ANK3 variant. A potentially pathogenic role of the novel ANK3 variant is suggested by the direct interaction of Ankyrin-G with the cardiac sodium channel, however, more patients with ANK3 variants and ERS would be required to establish ANK3 as novel ERS susceptibility gene. Our study provides additional evidence that ERS might be a heritable condition.
Keywords: Early repolarization, Ventricular fibrillation, Electrical storm, Isoproterenol, Hydroquinidine, Exome sequencing, ANK3, Ankyrin-G, Sodium channel
What’s new?
The early repolarization syndrome (ERS) is an extremely rare and malignant primary arrhythmia syndrome. Early repolarization syndrome might be heritable, however, there are no co-segregation data of genetic variants and ERS in affected families.
We identified two siblings with symptomatic ERS and tried to identify a causal variant in the affected family by using a whole-exome sequencing (WES) approach.
Whole-exome sequencing detected a novel, ultra-rare missense variant affecting a highly conserved residue in the ANK3 gene encoding Ankyrin-G in three out of four family members.
There are clinical data supporting delayed depolarization as underlying mechanism in ERS and a potentially pathogenic role of the novel ANK3 variant in ERS is suggested by the direct interaction of Ankyrin-G with the cardiac sodium channel.
Introduction
The early repolarization (ER) pattern has been defined as J-point elevation of ≥0.1 mV in two continuous inferolateral leads on a standard 12-lead electrocardiogram (ECG) with either a distinct or partly buried J-wave.1 The malignant ER syndrome (ERS) is occurring in patients with an ER pattern that experience syncope , ventricular fibrillation (VF), or sudden cardiac death (SCD).2–5
About 15% of patients with ERS have a family history of sudden death and, conversely, relatives of sudden arrhythmic death victims are significantly more likely to display an ER pattern (31–23% of relatives vs. 13–11% of controls) indicating that ERS might be heritable.2,6–8 However, ERS-associated variants have been described only in unrelated individuals with ERS4,9 and there are no co-segregation data of genetic variants and ERS in affected families.6 We have identified two siblings with symptomatic ERS. This study aims to characterize the clinical phenotype and to identify a potentially causal variant in the affected family using a whole-exome sequencing (WES) approach.
Methods
The study was approved by the Institutional Ethics Committee on Human Research at the authors’ institution. All patients have given written informed consent for study participation, blood sampling, isolation of DNA, molecular genetic testing, and provision of their clinical data.
Clinical data analysis
Clinical characterization included medical history, analysis of 12-lead-, Holter-, and monitor ECGs, transthoracic echocardiography, coronary angiography, Ajmaline challenge (when deemed appropriate), exercise stress test, and Valsalva manoeuver. The peak of an end-QRS notch and/or onset of an end-QRS slur, J peak (Jp) was measured in standard 12-lead ECGs.1 A Shanghai ERS score of ≥5 points was required for a diagnosis of definitive ERS.10
Candidate gene analysis
DNA was extracted from peripheral blood lymphocytes of the two siblings and their parents and analysed by next-generation sequencing (NGS) using a customized (SureDesign, based on an Agilent SureSelectQXT) solution-capture enrichment strategy (Agilent, Santa Clara, CA, USA). Targeted sequencing of ERS- and Brugada syndrome (BrS)-related genes (GPD1L, SCN1B, KCNE3, SCN3B, KCNH2, RANGRF, KCNE1L, KCND3, HCN4, SLMAP, TRPM4, SCN2B, SCN10A, KCNJ8, CACNA1C, CACNB2, CACNA2D1, SCN5A, and ABCC9) was performed in the index patient. Target regions included coding exons (padding: ±25 bp). Sequencing was carried out on a MiSeq™- or a NextSeq™ 500-System (Illumina, San Diego, CA, USA). Bioinformatic analyses were performed using CLC Genomics Workbench (Qiagen, Venlo, the Netherlands) and variants were verified by conventional Sanger sequencing.
Whole-exome sequencing
Whole-exome sequencing was initially carried out on the proband, her affected brother and the father as we did not have enough high-quality DNA of the mother for WES. The coding exons were captured using the Agilent SureSelect Human All Exon v.5. The captured fragments were sequenced on the Hiseq2000 sequencer (Illumina San Diego, CA, USA) to an average depth of >50 reads per target base. The sequence reads were aligned to the human reference genome (UCSC NCBI37.1/hg19) using SOAPaligner (version 2.21), and functional annotation of high-quality variants was performed using SOAPsnp software for the single nucleotide variation. For insertion/deletion detection, sequence reads were aligned by BWA and annotated by GATK for break-point identification. Variants were annotated using ANNOVAR. Data analysis was performed in the statistical programming environment R version 3.2.1. Synonymous variations not located at splice sites and variants with a frequency >0.1% in the following publically available databases were excluded from further analysis: (i) the ExAC Browser, (ii) the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP), (iii), Phase 1 version 3 of the 1000 Genomes project (data release October 2012), (iv) the 69 genomes from Complete Genomics, and (v) the Genome Aggregation Database (gnomAD). Sequential analysis steps focused on (i) Candidate gene analysis: known genes associated with ERS- and BrS (BrS, cardiac arrhythmias and cardiomyopathies, see Supplementary material online, S3 for the gene lists); (ii) recessive inheritance: shared homozygous variants in both siblings, heterozygous in the father and the mother; (iii) reduced penetrance: novel variants (i.e. ultra-rare variants shared by both affected siblings and inherited from an unaffected parent but not found in any of the databases).
Results
Clinical phenotype
The index patient, individual II-2 in the pedigree (Figure 1A), experienced out-of-hospital-cardiac arrest at age 23 years in the early morning at home while sleeping. Basic life support was provided by her mother. Upon arrival of the emergency physician, the initially recorded rhythm was VF. After transfer to University Hospital Freiburg ischaemic and structural heart disease were ruled out by transthoracic echocardiography and coronary angiography. Due to recurrent VF, the patient underwent early percutaneous venoarterial extracorporeal membrane oxygenation (VA-ECMO) implantation. The initial 12-lead ECG showed normal sinus rhythm with a QTc of 421 ms, and an inferolateral ER pattern with Jp amplitudes ≥0.1 mV (Figure 1C). The telemetry monitor revealed a bradycardia-dependent inferior ER pattern with frequent premature ventricular complexes triggering polymorphic ventricular tachycardia (pVT) and recurrent VF (Figure 2). Conventional anti-arrhythmic drugs failed and the patient was eventually rescued from intractable VF with isoproterenol which enabled successful defibrillation and suppression of ER and VF—after more than 1 h of ongoing VF (Figure 3). During isoproterenol weaning, monomorphic couplets recurred and isoproterenol was replaced with hydroquinidine 300 mg bid, which again suppressed couplets and ER (Supplementary material online, S1). The patient recovered completely without any sequelae and was implanted with a dual-chamber implantable cardioverter-defibrillator (ICD) with atrial pacing set at 70 b.p.m. There has been no VF recurrence on hydroquinidine therapy during 33 months of follow-up. Ajmaline challenge including high precordial leads placed in 3rd and 2nd intercostal space in (para-)sternal positions was negative. The Shanghai ERS score of the index patient was consistent with a definitive diagnosis of ERS (cardiac arrest/documented VF: 3 points; ER ≥ 0.2 mV in ≥2 lateral ECG leads with horizontal ST-segment: 2 points).
Figure 1.

Pedigree and 12-lead electrocardiograms of the two siblings with early repolarization syndrome. (A) The index patient is marked with an arrow. Men are denoted by squares and women by circles. Solid symbols indicate individuals with early repolarization syndrome. Plus symbols next to each individual indicate carrier of the novel ANK3 H2245R variant; minus symbol indicates wild type. (B) Diagnostic inferior early repolarization pattern with high Jp amplitudes of ≥0.35 mV, slurred QRS and horizontal ST-segments in the index patient’s brother. (C) Diagnostic inferolaterlal early repolarization pattern in the index patient with maximal J peak (Jp) amplitudes of 0.20–0.25 mV and slurred QRS in lateral leads.
Figure 2.
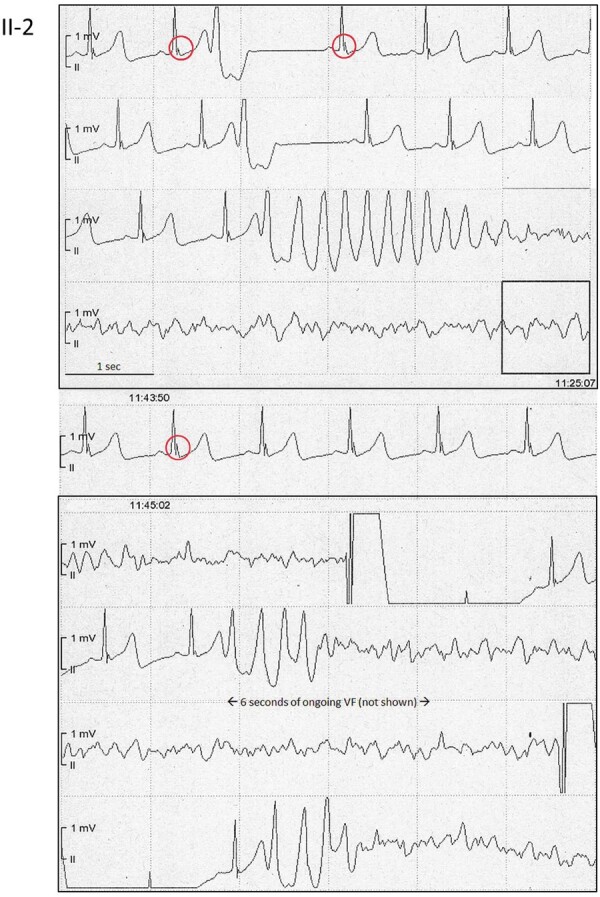
Electrical storm with dynamic early repolarization. Continuous electrocardiographic tracings from the telemetry monitor in the index patient: lead II demonstrates a distinct inferior J-wave with pause-dependent augmentation (red circles) and a monomorphic short-coupled premature ventricular complex triggering polymorphic ventricular tachycardia that rapidly degenerates into ventricular fibrillation. A further increase of the J-wave amplitude (red circles) precedes recurrent ventricular fibrillation. Following each defibrillation there is instantaneous ventricular fibrillation recurrence after four, respectively, two normal beats triggered by the very first monomorphic premature ventricular complex, resulting in three ventricular fibrillation episodes within 30 s. After this last defibrillation, the patient remained in ongoing VF for >1 h due to intractable ventricular fibrillation while being protected by venoarterial extracorporeal membrane oxygenation (VA-ECMO).
Figure 3.
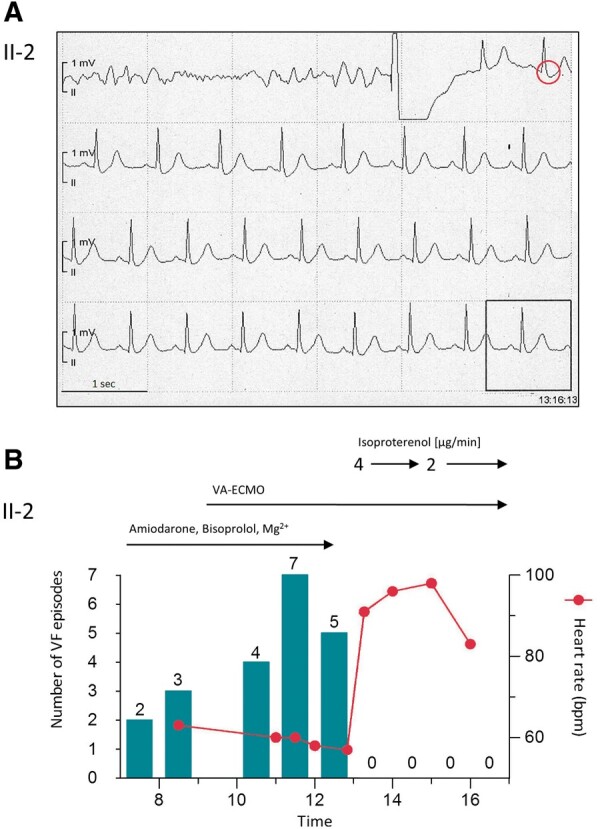
Termination of electrical storm and suppression of early repolarization. (A) Continuous electrocardiographic tracings from the telemetry monitor in the index patient: prior to the external shock shown isoproterenol was given for 2 min during ongoing ventricular fibrillation. Isoproterenol resulted in successful defibrillation, suppression of the early repolarization pattern (red circle) and of the ventricular fibrillation trigger (note absence of short-coupled premature beats). (B) Synopsis of electrical storm in the index patient: Conventional anti-arrhythmic drugs failed and, by slowing heart rate, retrospectively even increased the number of ventricular fibrillation episodes. Isoproterenol led to sinus tachycardia and suppression of any further ventricular fibrillation episode. VA-ECMO, venoarterial extracorporeal membrane oxygenation.
The index patient’s brother (25 years) demonstrated a persistent inferior ER pattern with high inferior Jp amplitudes ≥0.35 mV and horizontal ST-segments (Figure 1B). Ajmaline challenge with precordial leads placed as in the index was negative. The past medical history was significant for a witnessed arrhythmic syncope at rest without any prodrome or trigger at age 14 years. The Shanghai ERS score was calculated to be 6 points (suspected arrhythmic syncope: 2 points; ER ≥0.2 mV in ≥2 inferior ECG leads with horizontal ST-segment: 2 points; relative with definitive ERS: 2 points) consistent with a definitive diagnosis of ERS. In addition, the ER pattern showed a dynamic augmentation during Valsalva (Supplementary material online, S2). Based on a malignant ER pattern, a strong family history of ERS, and a history of arrhythmic syncope at rest the patient was deemed at increased risk of pVT/VF and a subcutaneous ICD was implanted. The patient has not received any shocks during follow-up.
Both parents were asymptomatic, they had a negative (extended) family history for unexplained syncope or SCD and showed no ER pattern on several 12-lead ECGs at baseline and during Valsalva (‘non-diagnostic’ ERS score of 2 points). We did not perform Ajmaline challenges in the parents due to the negative Ajmaline test results in both siblings.
Genetic analysis
A diagnostic screening by NGS showed no potentially causal variant in GPD1L, SCN1B, KCNE3, SCN3B, KCNH2, RANGRF, KCNE1L, KCND3, HCN4, SLMAP, TRPM4, SCN2B, SCN10A, KCNJ8, CACNA1C, CACNB2, CACNA2D1, SCN5A, and ABCC9 in the proband.
Whole-exome sequencing was performed in both affected siblings and the unaffected father. The analysis strategy for the WES data consisted of three steps (i) analysis of candidate gene panels, (ii) analysis for potential recessive inheritance; (iii) analysis of potentially dominant inheritance with reduced penetrance. No rare variants shared by both affected siblings were identified in genes associated with ERS in the literature, nor in genes associated with cardiac arrhythmias or cardiomyopathy (Supplementary material online, S3). Both siblings shared 1 rare homozygous variant in the gene FADS6 which was shared homozygously by the unaffected father as well and therefore did not fit a putative recessive pattern of inheritance with complete penetrance. No shared compound heterozygous variants were identified. We next investigated whether any ultra-rare variants (not present in all reference databases) were shared by both siblings. Thirteen variants fitted these criteria (Supplementary material online, S3). Among these was a novel variant in the gene ANK3 (NM_020987; exon37; c. A6734G; p. H2245R), shared by both siblings and inherited from the unaffected father which was absent in the mother. Histidine 2245 of ANK3 is located in a highly conserved domain of the protein, it is unchanged from Xenopus to fish, birds, and mammals (Figure 4) and the CADD-phred score of this variant is 14.77 indicating a strong conservation and large predicted effect on the protein. None of the other variants were in genes expressed in the heart (GTEX tpm >5), with a CADD-phred score >10, in genes with a significant constraint based on gnomAD pLoF (≤0.05).
Figure 4.

Alignment with ClustalW of the ANK3 protein in the region of the variant p. H2245R. The histidine at this position is conserved in fish, birds, mammals, marsupials and human.
Discussion
In this study, we made an attempt to investigate the genetic background of ERS in a small affected family by candidate gene analysis and a WES approach. The clinical presentation and dynamic ER pattern—aggravated by pauses, bradycardia, and changes in vagal tone (Valsalva) and resolved with sympathetic activation by isoproterenol—support the diagnosis of ERS in the two siblings, which is reflected by high ERS scores. Thus, our study provides additional evidence that ERS could be a heritable condition.
The underlying mechanism of ER is a matter of debate and currently, it is not clear whether ERS involves disturbed depolarization, repolarization, or both.11,12 The results of genetic and functional studies and pharmacological interventions with isoproterenol and hydroquinidine are compatible with the cellular model of ER, i.e. an imbalance between depolarizing currents (sodium and L-type calcium currents, INa and ICa, L) and repolarizing transient outward potassium current (Ito) during the early phase of the epicardial action potential. However, the magnitude of these currents and the resulting action potential morphology (i.e. the voltage-time integral ‘area under the curve’) also determine the safety of conduction and hence conduction velocity.13 As isoproterenol and hydroquinidine increase the area under the curve, such pharmacological interventions will also improve conduction which is compatible with disturbed depolarization in ERS similar to BrS.13 Indeed, high-density mapping and histological data obtained in patients with ERS have also demonstrated electrophysiological abnormalities (high-frequency electrograms with low amplitude and long duration) and structural abnormalities (interstitial fibrosis) in the inferolateral epicardium supporting delayed depolarization as the predominant underlying mechanism in ERS similar to BrS.11,12,14 Interestingly, the index patient’s brother showed a fragmented QRS in precordial and lateral leads compatible with abnormal depolarization (Supplementary material online, S4).
While we did not find any pathogenic variant in ERS candidate genes, WES detected a novel ultra-rare missense variant affecting a highly conserved residue (p. H2245R) in the ANK3 gene encoding Ankyrin-G in the two symptomatic siblings and the unaffected father. A potentially pathogenic role of the novel ANK3 variant is suggested by the direct interaction of Ankyrin-G with the cardiac sodium channel: ANK3 encodes Ankyrin-G which is required for targeting Nav1.5 to the intercalated disc.15–17 In animals, ankyrin-G expression is required for the targeting of Nav1.5 and CaMKII to the intercalated disc16 and Nav1.5 expression and INa are reduced in myocytes derived from mice with selective loss of cardiomyocyte ankyrin-G (cKO).16 Priori et al. identified an individual with BrS harbouring an SCN5A variant in the Nav1.5 motif that interacts with ankyrin-G. In vitro binding assays show that the p. E1053K variant disrupts the interaction between Nav1.5 and ankyrin-G resulting in abnormal targeting in myocytes.18 Interestingly, Ankyrin variants have also been found in two young, unrelated individuals with VF storm due to possible ERS, one of them with a family history of SCD.19 In summary, as Ankyrins are a critical part of the sodium channel complex, the novel ANK3 variant could impair conduction via loss-of-sodium channel function20 which may contribute to disturbed depolarization in ERS. However, more patients with variants in ANK3 and (familial) ERS would be required to establish ANK3 as a novel ERS susceptibility gene. In addition, the presence of the novel ANK3 variant in the unaffected father may support a more complex genetic architecture of ERS (e.g. additional genetic variants/modifiers), may be due to the fact that the novel variant is not disease causing but may also point to incomplete disease penetrance or variable disease expression which has been described in families with ERS.6
Limitations
The small number of affected individuals in our study does not allow to conclude a causal role of the novel variant in this family and in ERS in general. Functional studies, e.g. iPS cells, would be required to assess the effect on ionic currents, e.g. a reduction in INa. and could be of additional value to support the pathogenicity of the novel ANK3 variant.
Conclusion
Our study provides additional evidence that ERS could be a heritable condition with variable penetrance. This study is, however, limited by the small number of affected individuals which does not allow a conclusion regarding a pathogenic role of the novel ANK3 variant in this family and in ERS in general. Additional patients with variants in ANK3 and ERS as well as functional studies are required to establish a disease-modifying or pathogenic role of ANK3 in ERS.
Supplementary material
Supplementary material is available at Europace online.
Supplementary Material
Acknowledgements
The authors would like to thank the family for participation in this study.
Funding
The work of E.M.L. is partly financed by the Dutch Research Council (NWO) through the NWO Talent Scheme VIDI-91718361 and by the CVON RESCUED project.
Conflict of interest: none declared.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding authors.
References
- 1. Macfarlane PW, Antzelevitch C, Haïssaguerre M, Huikuri HV, Potse M, Rosso R. et al. The early repolarization pattern: a consensus paper. J Am Coll Cardiol 2015;66:470–7. [DOI] [PubMed] [Google Scholar]
- 2. Haïssaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L. et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med 2008;358:2016–23. [DOI] [PubMed] [Google Scholar]
- 3. Nam GB, Kim YH, Antzelevitch C.. Augmentation of J waves and electrical storms in patients with early repolarization. N Engl J Med 2008;358:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haïssaguerre M, Chatel S, Sacher F, Weerasooriya R, Probst V, Loussouarn G. et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol 2009;20:93–8. [DOI] [PubMed] [Google Scholar]
- 5. Nam GB, Ko KH, Kim J, Park KM, Rhee KS, Choi KJ. et al. Mode of onset of ventricular fibrillation in patients with early repolarization pattern vs. Brugada syndrome. Eur Heart J 2010;31:330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gourraud JB, Le Scouarnec S, Sacher F, Chatel S, Derval N, Portero V. et al. Identification of large families in early repolarization syndrome. J Am Coll Cardiol 2013;61:164–72. [DOI] [PubMed] [Google Scholar]
- 7. McCorquodale A, Poulton R, Hendry J, Norrish G, Field E, Mead-Regan S. et al. High prevalence of early repolarization in the paediatric relatives of sudden arrhythmic death syndrome victims and in normal controls. Europace 2017;19:1385–91. [DOI] [PubMed] [Google Scholar]
- 8. Nunn LM, Bhar-Amato J, Lowe MD, Macfarlane PW, Rogers P, McKenna WJ. et al. Prevalence of J-point elevation in sudden arrhythmic death syndrome families. J Am Coll Cardiol 2011;58:286–90. [DOI] [PubMed] [Google Scholar]
- 9. Mahida S, Sacher F, Berte B, Yamashita S, Lim H, Derval N. et al. Evaluation of patients with early repolarization syndrome. J Atr Fibrillation 2014;7:1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Antzelevitch C, Yan G-X, Ackerman MJ, Borggrefe M, Corrado D, Guo J. et al. J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Europace 2016;19:665–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nademanee K, Haïssaguerre M, Hocini M, Nogami A, Cheniti G, Duchateau J. et al. Mapping and ablation of ventricular fibrillation associated with early repolarization syndrome. Circulation 2019;140:1477–90. [DOI] [PubMed] [Google Scholar]
- 12. Haïssaguerre M, Nademanee K, Hocini M, Cheniti G, Duchateau J, Frontera A. et al. Depolarization versus repolarization abnormality underlying inferolateral J-wave syndromes: new concepts in sudden cardiac death with apparently normal hearts. Heart Rhythm 2019;16:781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoogendijk MG, Potse M, Coronel R.. Critical appraisal of the mechanism underlying J waves. J Electrocardiol 2013;46:390–4. [DOI] [PubMed] [Google Scholar]
- 14. Boukens BJ, Benjacholamas V, van Amersfoort S, Meijborg VM, Schumacher C, Jensen B. et al. Structurally abnormal myocardium underlies ventricular fibrillation storms in a patient diagnosed with the early repolarization pattern. JACC Clin Electrophysiol 2020;6:1395–404. [DOI] [PubMed] [Google Scholar]
- 15. Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K. et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A 2004;101:9137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Makara MA, Curran J, Little SC, Musa H, Polina I, Smith SA. et al. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res 2014;115:929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu HC, Yamankurt G, Luo J, Subramaniam J, Hashmi SS, Hu H. et al. Identification and characterization of two ankyrin-B isoforms in mammalian heart. Cardiovasc Res 2015;107:466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG. et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A 2004;101:17533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krogh Broendberg A, Pedersen LN, Nielsen JC, Jensen HK.. Ankyrin-2 variants associated with idiopathic ventricular fibrillation storm in patients with intermittent early repolarization pattern. Heart Rhythm Case Rep 2015;1:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. El Refaey MM, Mohler PJ.. Ankyrins and spectrins in cardiovascular biology and disease. Front Physiol 2017;8:852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding authors.