

Abstract
Peanut is one of the important oil and economic crops, among which the variegated testa peanut is a unique member. The molecular mechanisms underlying the pigment synthesis in variegated testa are still unclear. Differentially expressed genes (DEGs) in the flavonoid metabolism pathway in pigmented areas indicated that there were 27 DEGs highly related to the synthesis of variegated testa color among 1,050 DEGs. Of these 27, 13 were up-regulated and 14 were down-regulated, including 3 PALs, 1 C4H, 2 CHSs, 1 F3H, 1 F3'H, 2 DFRs, 2 LARs, 2 IAAs, 4 bHLHs, and 9 MYBs. GO (Gene Ontology) analysis indicated that DEGs were similarly enriched in three branches. KEGG (Kyoto Encyclopedia of Genes and Genomes) analysis suggested flavonoid biosynthesis is the most direct metabolic pathway for the synthesis of testa variegation. The liquid chromatography–tandem mass spectrometry (LC–MS/MS) results showed that cyanidin and delphinidin were the primary metabolites that caused the color differences between the pigmented and the non-pigmented areas. Through the verification of 20 DEGs via qPCR, the results were consistent with transcriptome sequencing in four comparison groups. The results in this study lay the foundation for revealing the molecular regulation mechanisms of flavonoid synthesis in variegated testa peanut.
Subject terms: Molecular biology, Transcriptomics, Plant sciences, Plant molecular biology, Secondary metabolism
Introduction
Peanut (Arachis hypogaea L.) is the primary oil and economic crop that occupies an important position in the world for grain, oil, and food. It is widely planted in developing countries in Asia, Africa, and South America, as well as some developed countries including the United States and Australia1. Peanut testa contains a variety of biologically active substances, which are important raw materials for cosmetics and medicinal health products2. The variegated testa peanut is a unique member of the peanut family. The variegated testa is composed of pigmented and non-pigmented areas, with the pigmented areas including purple, red, pink, and other colors. The variegated testa is a unique agronomic trait, which is combined with other traits such as high sugar and high oleic acid content in breeding for peanut quality. The distinctive phenotype of the trait facilitates planting and breeding. In addition, variegated testa peanuts enrich the peanut germplasm resources in China. Different types and content of anthocyanins will cause peanut testa to appear different colors. Anthocyanins also have many pharmacological functions in anti-oxidation, anti-inflammatory, eyesight protection, antibacterial, and other aspects3.
The development of different colors on the same variegated testa is an interesting natural phenomenon within the same genetic background. However, the molecular mechanism of pigment synthesis in the testa is still unclear. Plant anthocyanin metabolism is a complex process that included chalcone synthase (CHS), chalcone isomerase (CHI), dihydroflavonol-3-hydrogenase (F3H), dihydroflavonol-3′-hydrogenase (F3′H), dihydroflavonol-3′-5′hydrogena, dioxanonol-4-reductase (DFR), anthocyanidins synthase (ANS), and many other key enzymes4. Meanwhile, basic helix-loop-helix (bHLH), v-myb avian myeloblastosis viral oncogene homolog (MYB), WD40 protein, and other transcription factors can also regulate the expression of structural genes in the plant anthocyanin metabolic pathway5. The first enzyme (CHS) in anthocyanin synthesis can affect the shade of plant color6 and the flower color can change from purple to pink or even white if the CHS antisense gene is transformed in Petunia hybrida7. The color differentiation of tea tree (Camellia sinensis) buds and leaves are catalyzed by DFR and ANS from three main anthocyanidins8–11. MYB and bHLH are important regulatory genes in the anthocyanin synthesis pathway of Petunia hybrida12. Overexpression of Rs-MYB1 in petunia activates the expression of ANS and DFR in the anthocyanin synthesis process and increases the anthocyanin content13. The regulation of anthocyanin synthesis by bHLH transcription factors has been widely investigated in Arabidopsis, snapdragon, chrysanthemum, begonia, and other plants14–16. The degree of coloration often only requires the difference in the expression of one or several key anthocyanin genes between the pigmented and the non-spotted areas and does not require all of the anthocyanin genes to have an obvious difference in expression17. For example, the formation of white star-shaped spots in P. hybrida is caused by the inhibition of CHS-A expression18. The large purple spots in Phalaenopsis stuartiana are primarily due to the specific expression of DFR in the spot areas19, while other spots are caused by the expression difference of two or more anthocyanin genes. For example, the color develops if the expression of OgCHI and OgDFR is suppressed in Oncidium hybridum20. Xia et al. found that the up-regulation of DFR in Chinese peanut variety ZH9 is beneficial to the promotion of dihydroflavonol synthesis in the anthocyanin and flavonol synthesis pathways16. Li et al. studied four types of peanut testa with different colors 40 and 50 days after anthesis and found that CHS was expressed in large quantities, promoting anthocyanin synthesis. In addition, the expression of DFR was significantly related to the testa color21. Wan et al. adopted transcriptome sequencing to analyze the color accumulation of the testa of the pink peanut variety ZH16, showing that the expression of PAL, 4CL, CHS, CHI, and other enzymes increased, while the expression of ANR and LAR decreased22. Li et al. found that the relative expression level of F3H in purple peanut variety FH01 is positively correlated with anthocyanin content23. One reason for the lack of research on the pigment synthesis and molecular mechanism of the variegated peanut testa is related to the scarcity of the research materials.
Transcriptome sequencing is a powerful technique that can identify almost all transcripts and gene sequences of a specific cell or tissue of a certain species in a certain state and can be used to study gene expression, gene function, structure, alternative splicing, and forecasting new transcripts. Ye et al. adopted this technology to screen out 15 key structural genes involved in the metabolism of peach skin anthocyanins, including the upstream phenyl propionic acid metabolism pathway genes, such as PAL, C4H, and 4CL, and the late anthocyanin metabolism pathway genes, such as CHS, CHI, F3H, F3'H, DFR, LAR, and ANR24. Liu et al. found that the expression levels of anthocyanin biosynthesis genes PAL and C4H and transcription factor RsMYB1 and RsTT8 were significantly down-regulated during the development in a radish variety “Xin Li Mei”25. Jia et al. concluded that ANS is the key gene determining kiwifruit anthocyanin synthesis via transcriptome sequencing26. Jin et al. analyzed transcriptomes of differently pigmented Cineraria flowers (white and yellow), which were not able to accumulate anthocyanins due to mutations in the coding regions of specific genes that cause variations in color development27. Transcriptome sequencing has been widely used to study various species including apple28, grape29, kiwifruit30, and soybean31.
In this study, we used transcriptomics to quantitatively analyze the differential genes related to the flavonoid synthesis pathway. Different parts of the same peanut testa tissue were used as experimental materials. The differences in metabolites were compared between the pigmented and non-pigmented areas at the same developmental stage, and the same testa areas (pigmented and non-pigmented) at different developmental stages were also compared. The results show different relative contents of metabolites, and also demonstrate the changes in metabolites between the pigmented and non-pigmented areas. The relative content of flavonoids in different parts of the variegated testa at different developmental stages was determined with metabonomics by screening out key genes involved in flavonoid synthesis in peanut. This paper initially revealed the molecular regulation mechanism of flavonoid synthesis in the variegated testa of peanut and facilitate further in-depth studies in peanut functional genomics.
Results
Characterization of variegated testa by stereomicroscope
Pigmentation in the pigmented areas (F) of the variegated peanut testa was 30 days after flowering and needling (DAF30), which was the active period of related gene expression. The “red, green” (a) and “yellow, blue” (b) values of the non-pigmented areas (B) were lower than in the pigmented areas during testa development. The “lightness” value (L) in the non-pigmented areas was higher than in the pigmented areas, which was possibly due to the white color of the testa in non-pigmented areas. As the pod matured 45 days after flowering and needling, the color of the variegated testa stabilized. The values of “a” and “b” in the pigmented areas changed slightly and “L” increased to 12.85% (Fig. 1, Table S1). The pigmented and non-pigmented areas sampled were located on the same testa with identical genetic backgrounds, which is ideal for the study of DEGs in variegated testa pigmentation.
Figure 1.

Phenotype and chromatic changes in the synthesis of color in the variegated peanut testa: Numbers 1–6: day at flowering and needling (DAF) 30, 35, 40, 45, 50, and 55 days. F1 and F2 represent the sample names of the pigmented areas at DAF30 and DAF45, respectively. B1 and B2 represent the sample names of the non-pigmented areas at DAF30 and DAF45, respectively. Bar colors represent different values of “a”, “b”, and “L”. The absorbance value “a” (“a” value) represents red and green; the absorbance value “b” (“b” value) represents yellow and blue; and the “L” value represents lightness.
Measurement of flavonoids
The pigmented (F1 and F2) and non-pigmented (B1 and B2) areas were compared at DAF30 and DAF45. In the comparison of F1–B1, the total content of the ten different metabolites measured in the pigmented areas was 7.24 times higher than that in the non-pigmented areas. The content of procyanidin A1, A2, B2, B3, delphinidin, cyanidin, cyanidin 3-O-galactoside, and rosinidin O-hexoside in the pigmented areas increased by 185.40–895.58%, while the relative content of petunidin 3-O-glucoside and cyanidin O-syringic acid in the pigmented areas were lower than in the non-pigmented areas, by 62.70–76.92% (Fig. 2a). In the comparison of F2–B2, the total content of the 11 different metabolites measured in the pigmented areas was 9.95 times higher than that in the non-pigmented areas. The relative content of procyanidin A1, A2, B2, B3, delphinidin 3-O glucoside, delphinidin, cyanidin, cyanidin 3-O-glueoside, rosinidin O-hexoside in the pigmented areas increased by 106.54–1759.77%. However, compared with the pigmented areas, the relative content of cyanidin 3-O-galactoside and cyanidin O-syringic acid in the non-pigmented areas was increased by 35.25–80.35% (Fig. 2b). The relative content of cyanidin and delphinidin are highest in colored metabolites in the comparison F1–B1, which is same as F2–B2. In the comparison of B1–B2, at DAF45, the total content of the seven different metabolites measured was 1.33 times higher than that at DAF30 in the non-pigmented areas. The relative content of petunidin 3-O-glucoside, delphinidin, cyanidin 3-O-galactoside at DAF45 increased by 43.73–81.62%, while the relative content of cyanidin O-syringic acid, cyanidin 3-O-glucoside, procyanidin A1, rosinidin O-hexoside at DAF45 decreased by 152.39–313.82% compared with DAF30 (Fig. 2c). The metabolites between F1 and F2 were almost identical. At DAF45, the total content of the four different metabolites measured was 3.35 times higher than that at DAF30 in the pigmented areas. The relative content of delphinidin 3-O-glucoside, petunidin 3-O-glucoside, and rosinidin O-hexoside at DAF45 was 48.44–92.25% higher than at DAF30, while the relative content of cyanidin O-syringic acid at DAF45 was lower by 386.24% compared to DAF30 (Fig. 2d, Table S2).
Figure 2.

Types and relative contents of flavonoids in different areas of peanut testa at different developmental stages. (a, b) represent the type and content differences of metabolized flavonoids between the pigmented and non-pigmented areas after DAF30 and DAF45, respectively. (c) represents the different metabolized flavonoid types and content in the non-pigmented areas after DAF30 and DAF45. (d) represents the different metabolized flavonoid types and content in the pigmented areas at DAF30 and DAF45. The horizontal axis represents the types of flavonoids in the variegated testa and the vertical axis represents the peak areas value determined by the LC–MS/MS detection platform.
RNA sequencing
Transcriptome sequencing was performed on F1, B1, F2, and B2 of the variegated peanut (accession VG-01). The adapter sequences and low-quality read sequences were all removed from the sequencing data. A total of 64.33 Gb clean data were obtained, and the clean data of each sample reached 6.53 Gb. The base recognition error rate was about 0.02%, the Q20 value of the obtained sequence was above 97.45%, the Q30 base percentage was above 93.23%, and the GC content was above 43.90% (Table S3).
Detection of DEGs
With reference to the tetraploid reference genome of peanut and hierarchical clustering analysis, a total of 214 differential genes in F1–B1 (F1 is the control group and B1 is the comparison group) were identified, of which 53 were up-regulated and 161 were down-regulated. There were 348 differential genes identified in F2–B2 (F2 is the control group and B2 is the comparison group), including 97 up-regulated and 251 down-regulated. There was a total of 152 differential genes in B1–B2 (B1 is the control group and B2 is the comparison group), including 82 up-regulated and 70 down-regulated genes, and 213 differential genes in F1–F2 (F1 is the control group and F2 is the comparison group), with 169 up-regulated and 44 down-regulated (Fig. 3, Table S4).
Figure 3.

Analysis of the volcano map of different genes in various parts of the peanut testa at different developmental stages. The abscissa indicates the multiple of difference, and the ordinate indicates − log10 (q-value). Red dots indicate up-regulation of gene expression, green dots indicate down-regulation of gene expression, and black dots indicate that there is no significant difference in gene expression.
GO enrichment analysis of DEGs
1050 DEGs (accounting for 82.48% of all differential genes) were compared with GO public databases of tetraploid cultivated peanut and diploid wild peanut, respectively. The comparison results of the tetraploid reference genome showed that F1–B1, F2–B2, F1–F2, and B1–B2 comparison groups were all enriched in 51 GO entries and the diploid genome comparison showed that four comparison groups were all enriched in 41 GO entries (Table S5). Following comparative analysis, the enrichment items in three branches of four comparison groups were horizontally similar. Analysis of the tetraploid cultivar transcriptome data revealed that the enriched items in the cellular components category were membrane, and membrane part, the molecular function were enriched in catalytic activity, binding, and transporter activity. In addition, biological processes showed that DEGs enriched in metabolic, single-organism, and cellular processes (Fig. 4).
Figure 4.

GO functional enrichment classification of variegated peanut testa transcriptome. Differently colore bars represent four comparison groups. The y-axis represents the number of genes mapped to the indicated GO term and the x-axis represents each GO term. GO terms with an adjusted p-value of < 0.05 were considered significantly enriched.
COG enrichment analysis of DEGs
The annotated genes were compared with the COG database and it was found that F1–B1 contained the largest DEGs proportion (R-General functional prediction only), accounting for 12.61%, followed by Transcription, accounting for 6.54%. Amino acid transport and metabolism accounted for 5.61%. Energy production and conversion, and Defense mechanisms accounted for 4.67%. F2–B2 had the largest DEGs proportion (R-General functional prediction only), accounting for 10.63%, followed by Transcription, accounting for 6.32%. Amino acid transport and metabolism accounted for 5.61%. Energy production and conversion, and Defense mechanisms accounted for 4.89%. DEGs in B1–B2 contained the largest DEGs proportion (R-General functional prediction only), accounting for 5.92%, followed by Transcription, accounting for 4.61%. The synthesis, transport, and metabolism of Secondary metabolites biosynthesis accounted for 3.29%. F1–F2 had the largest DEGs proportion (R-General functional prediction only), accounting for 7.51%, followed by Modification and transport after transcription and protein translation, accounting for 5.63% (Fig. 5).
Figure 5.

COG functional classification compared with the tetraploid peanut reference genome. A: RNA processing and modification; B: Chromatin structure and dynamics; C: Energy production and conversion; D: Cell cycle control, cell division, chromosome partitioning: E: Amino acid transport and metabolism; F: Nucleotide transport and metabolism; G: Carbohydrate transport and metabolism; H: Coenzyme transport and metabolism; I; Lipid transport and metabolism; J: Translation, ribosomal structure and biogenesis; K: Transcription; L: Replication, recombination and repair: M: Cell wall/membrane/envelope biogenesis; N: Cell motility; O: Posttranslational modification protein turnover, chaperones; P: Inorganic ions transport and metabolism; Q: Secondary metabolites biosynthesis, transport and catabolism; R: General functional prediction only; S: Function unknown; T: Signal transduction mechanism; U: Intracellular trafficking, secretion, and vesicular transport; V: Defense mechanisms; W: Extracellular structures; Y: nuclear structure; Z: Cytoskeleton.
KEGG pathways enrichment analysis of DEGs
In order to understand the biological functions of DEGs discussed above, the transcriptome sequencing results were compared with the KEGG public database of tetraploid cultivated peanut and diploid wild peanut. The results showed that 363 DEGs (28.52% of all differential genes) were annotated in the KEGG database. Among them, the metabolic pathways associated with testa color enrichment in F1–B1 included phenylalanine metabolism, phenylpropanol biosynthesis, flavonoid biosynthesis, and plant circadian rhythm. Testa color-related enrichment metabolic pathways in F2–B2 included phenylpropanol biosynthesis, flavonoids and flavonol biosynthesis, and flavonoid biosynthesis. Testa color-related enrichment metabolic pathways in B1–B2 included plant hormone signals transduction and biosynthesis of phenylalanine, tyrosine and tryptophan. Testa color-related enrichment pathways in F1–F2 included phenylalanine, tyrosine, and tryptophan biosynthesis. The diploid and tetraploid reference genomes were compared based on different parts of the same period and different periods of the same part and six metabolic pathways related to anthocyanin biosynthesis were screened out, including phenylpropane acid metabolism, phenylpropanol biosynthesis, flavonoids and flavonol biosynthesis, flavonoid biosynthesis, plant hormone signal transduction, and circadian rhythm in plants. (Fig. 6, Table S6).
Figure 6.
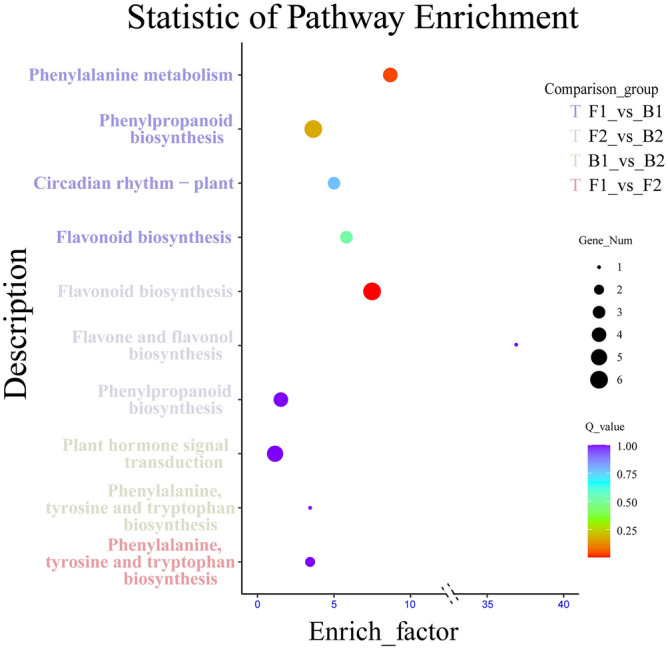
Pathway enrichment analysis of DEGs. The ordinate represents the pathway entry, and the abscissa represents the rich factor. The dots represent the number of significant DEGs, and the significance increases as the dots increase in size. The dots with different colors indicate different Q-values. (The figures were created by R version 4.0.3 based on the KEGG pathway database www.kegg.jp/kegg/kegg1.html).
Structural genes related to testa pigment synthesis
The Venn diagram analysis showed that 71 DEGs were co-expressed between F1–B1 and F2–B2, 143 DEGs presented specifically in F1–B1, and 277 DEGs were particularly expressed in F2–B2 (Fig. 7c, Table S7). Fifteen DEGs were co-expressed between B1–B2 and F1–F2, 137 DEGs presented specifically in B1–B2, and 198 DEGs were primarily expressed in F1–F2 (Fig. 7c, Table S7). KEGG analysis showed that both F1–B1 and F2–B2 involved differential genes related to peanut testa color and the expression of DEGs was closely related to testa development. Genes involved in the pigment synthesis included 3 PAL, 1 C4H, 2 CHS, 1 F3H, 1 F3'H, 2 DFR, 2 LAR, and 2 IAA (Table 1). Three PAL, 1 C4H, 2 CHS, and 1 F3'H in the non-pigmented areas were down-regulated compared with the pigmented areas. Two DFR, 2 LAR, and 2 IAA in non-pigmented areas were up-regulated compared with the pigmented areas (Fig. 7a), involving the entire anthocyanin metabolism pathway key enzyme gene metabolic pathway.
Figure 7.

(a, b) Heat map of the DEGs related to anthocyanin synthesis in the variegated peanut testa among different comparison groups of structural genes and transcription factors. Gene expression was scaled in this analysis using FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) Z-scores based on the mean value of two biological replicates in the heatmap. The key is located on the right-hand side in each case with FPKM values increasing from blue to orange. (c) The Venn diagram of the DEGs among different comparison groups.
Table 1.
Genes related to anthocyanin synthesis in the variegated peanut testa among different comparison groups.
| Annotation | Gene ID F1–B1 | F1 FPKM | B1 FPKM | FDR | log2FC | Regulated |
|---|---|---|---|---|---|---|
| PAL | arahy.Tifrunner.gnm1.ann1.0B4MFB | 9.05 | 1.45 | 7.31E − 04 | − 2.72 | down |
| PAL | arahy.Tifrunner.gnm1.ann1.NB2RRK | 9.63 | 0.92 | 8.56E − 11 | − 3.47 | Down |
| PAL | arahy.Tifrunner.gnm1.ann1.PUMP6L | 12.60 | 3.96 | 4.95E − 03 | − 1.74 | Down |
| C4H | arahy.Tifrunner.gnm1.ann1.AYA1A5 | 2.32 | 0.26 | 5.48E − 03 | − 3.07 | Down |
| DHS2 | arahy.Tifrunner.gnm1.ann1.CY60UM | 22.02 | 6.74 | 3.17E − 03 | − 1.77 | Down |
| CHS | arahy.Tifrunner.gnm1.ann1.6J3HHE | 10.01 | 0.68 | 5.04E − 10 | − 3.92 | Down |
| CHS | arahy.Tifrunner.gnm1.ann1.UDJX6I | 91.99 | 20.08 | 1.65E − 05 | − 2.26 | Down |
| IAA14 | arahy.Tifrunner.gnm1.ann1.CB6084 | 0.55 | 9.01 | 4.54E − 09 | 3.96 | Up |
| IAA14 | arahy.Tifrunner.gnm1.ann1.QUY0YV | 0.68 | 5.80 | 1.56E − 04 | 3.02 | up |
| CSNK2B | arahy.Tifrunner.gnm1.ann1.X2EPYD | 0.00 | 14.94 | 8.91E − 43 | Inf | Up |
| Annotation | Gene ID F2–B2 | F2 FPKM | B2 FPKM | FDR | Log2FC | Regulated |
|---|---|---|---|---|---|---|
| PAL | arahy.Tifrunner.gnm1.ann1.NB2RRK | 5.90 | 0.92 | 8.56E − 11 | − 3.47 | Down |
| F3'H | arahy.Tifrunner.gnm1.ann1.K8H9R8 | 11.11 | 26.27 | 5.59E − 03 | 1.20 | Up |
| F3H | arahy.Tifrunner.gnm1.ann1.HE8J5U | 27.03 | 62.10 | 9.35E − 03 | 1.16 | Up |
| DFR | arahy.Tifrunner.gnm1.ann1.X8EVF3 | 29.29 | 152.10 | 1.81E − 05 | 2.33 | Up |
| DFR | arahy.Tifrunner.gnm1.ann1.7JZ58T | 11.70 | 144.98 | 4.15E − 08 | 3.60 | Up |
| LAR | arahy.Tifrunner.gnm1.ann1.M05HM0 | 0.81 | 8.85 | 8.14E − 13 | 3.27 | Up |
| LAR | arahy.Tifrunner.gnm1.ann1.T1J2UZ | 1.27 | 18.35 | 2.87E − 08 | 3.85 | Up |
| BG | arahy.Tifrunner.gnm1.ann1.D1ITL5 | 1.70 | 0.19 | 9.04E − 04 | − 3.16 | Down |
| EC | arahy.Tifrunner.gnm1.ann1.13Q8F8 | 30.51 | 0.00 | 2.99E − 32 | #NAME? | Down |
| POD | arahy.Tifrunner.gnm1.ann1.49I11A | 12.86 | 3.04 | 2.45E − 07 | − 2.12 | Down |
| ORR10 X1 | arahy.Tifrunner.gnm1.ann1.D687JL | 0.44 | 5.66 | 8.33E − 08 | 3.52 | Up |
| IAA14 | arahy.Tifrunner.gnm1.ann1.CB6084 | 0.55 | 9.01 | 4.54E − 09 | 3.96 | Up |
| IAA14 | arahy.Tifrunner.gnm1.ann1.QUY0YV | 0.68 | 5.80 | 1.56E − 04 | 3.02 | Up |
Analysis of transcription factors differentially expressed in the transcriptome
The transcription factors in the sequencing results were analyzed and it was found that nine differentially expressed MYBs were screened from 410 MYBs transcription factors, four differentially expressed bHLHs were screened from 278 bHLH transcription factors, and there were no differentially expressed WD40 family transcription factors (Fig. 7b). All MYB regulatory factors were enriched in the two metabolic pathways of splicing and plant circadian transduction. All bHLH regulatory factors were also enriched in the two metabolic pathways of plant hormone signal transduction and plant circadian rhythm. However, the differentially expressed MYB and bHLH transcription factors were not annotated clearly among the metabolic pathways, and the regulatory role of these in the metabolic pathway was unclear (Table S8).
Joint analysis of transcriptome and metabolome
The combined analysis of metabolome and transcriptome showed that the differential genes in the flavonoid biosynthetic pathway are directly related to the synthesis of delphinidin and cyanidin. The F1–B1 correlation results showed a higher delphinidin and cyanidin content in the pigmented areas compared with the non-pigmented areas. Concomitantly, FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) of 2 CHSs and 1 C4H increased in the pigmented areas. The F2–B2 correlation results showed that the FPKM of 2 DFRs, 1 F3'H, 1 F3H, and 2 LARs in non-pigmented areas increased, and different metabolites resulted in the highest procyanidin content in the non-pigmented areas (Fig. 8).
Figure 8.

Combined analysis of transcriptome and metabolome of flavonoid biosynthesis. The red graphic represents the down-regulation of gene expression, the blue graphic represents the up-regulation of gene expression, and the gray graphic indicates that there is no significant difference in gene expression.
qPCR analysis of differential gene expression levels
Fluorescence quantitative qPCR verification was performed on 20 genes related to anthocyanin metabolism. The results showed that 11 genes were verified in F1–B1 and F2–B2 (Fig. 9a,b), 12 genes were verified in B1–B2 (Fig. 9c), and ten genes were verified in F1–F2 (Fig. 9d), consistent with transcriptome results. The 20 selected differential genes in the four comparison groups showed similar qPCR expression trends to the transcriptome detection results (Fig. 9).
Figure 9.

DEGs qRT-PCR verified results. (a–d) represent F1–B1, F2–B2, B1–B2, and F1–F2, respectively. The x-axis shows the relative gene expression levels analyzed by qRT-PCR. *p < 0.05; **p < 0.01; and ***p < 0.001. ↑ means that the gene is up-regulated in the transcriptome, ↓ the gene is down-regulated in the transcriptome.
Discussion
Peanut is one of the five largest oil crops in the world, containing testa that is enriched with biologically active substances. Peanut testae have significant differences in color, with white, red, purple, pink, and variegated colors, of which the variegated testa is unique. Anthocyanin is one of the key substances responsible for color variation in plants32. The ingenious sampling is where this research differs from previous studies. In this study, accession VG-01 material with a variegated testa was used, allowing for direct comparisons of mechanisms underlying the color variation within the same genetic background. Because the genetic background of the samples is identical, measurement of the relative flavonoid metabolites can also represent the difference between the pigments in the pigmented and non-pigmented areas. At the same time, the sequencing results were compared and analyzed to both diploid wild and tetraploid cultivated peanuts. Moreover, the molecular regulation mechanism of anthocyanin synthesis has not been previously found in the variegated peanut testa, so this research is of significance to the field of research.
CHS of the circadian pathway determines the early differentiation of peanut testa color
The synthesis of pigments in the pigmented and non-pigmented areas is comprehensively regulated by multiple structural genes in the variegated peanut testa. The analysis results showed that two key CHS genes are the first to regulate pigment synthesis. The up-regulated expression of CHS provides a large amount of substrate for the metabolism of anthocyanins downstream of the pigmented areas of variegated testa. Deng et al. identified 10 CHS genes that were significantly up-regulated, using materials of different colors and different parts on the same wild banana peel33. Gao et al. adopted transcriptome sequencing to screen RsCHS- B2 and found that RsCHS is up-regulated in red radishes when they formed fleshy roots, which were important genes for red color34. Wan et al. studied the increase in CHS expression during the growth process from DAF20d to DAF60d, which promoted the color accumulation in the peanut testa22. The significant difference in our study was that two CHS enriched in plant circadian metabolic pathways were identified. We hypothesize that the two CHS may be key genes for the color differentiation between the peanut mottling and non-pigmented areas in the early testa development. The F1–B1 correlation results showed that higher delphinidin and cyanidin content in the pigmented areas compared with the non-pigmented areas resulted in the red color of the variegated testa due to the up-regulation of the two CHS genes.
Up-regulated genes in the flavonoid synthesis pathway jointly promote the accumulation of pigments
In the flavonoid synthesis pathway at DAF30, three PAL, one C4H, and two CHS genes are all up-regulated in the pigmented areas, which provides a preliminary product for pigment accumulation in the pigmented areas. Wan et al. analyzed the pink peanut testa ZH16 and the results showed that the expression of PAL, 4CL, CHS, CHI, and other enzymes increased with the peanut testa growth22. Previous studies reported that F3H, F3'H, and F3′5'H could catalyze the synthesis of three dihydroflavonols (DHK, DHQ, and DHM), where DHK eventually produced brick red geranium pigment, DHQ produced red cyanidin glycosides, and DHM produced blue to purple delphinidin glycosides35. In the process of flower color synthesis in dahlia, the expression of F3H is significantly down-regulated, changing the color from orange to light red, or even white36. Interestingly, we observed up-regulation of F3H and F3'H in the non-pigmented areas at DAF45. This may be due to the up-regulation of downstream genes in the flavonoid synthesis pathway and that the reverse regulation promotes the increase of F3H and F3′H expression. Previous studies have shown that silencing the endogenous gene F3′H with RNA interference technology could increase the delphinidin content and make the petals of chrysanthemums appear a new type of blue37. Therefore, the pigment accumulation pattern of variegated peanut testa may be similar to the studies cited above.
Procyanidin expressing high content as pre-products accumulated in pigmented areas
DFR is the first key downstream gene that affects the anthocyanin synthesis branch in the variegated testa. High expression of DFR is advantageous to the accumulation of plant anthocyanins. For example, brick red petunidin 3-O-glucoside can be obtained if DFR is introduced into white gerbera38,39. Zhang et al. studied the up-regulation of DFR gene expression in purple leaves in mustard, which contributes to the accumulation of anthocyanins31. Different from previous studies, the expression levels of two DFR selected by the transcriptome in our study were higher in the non-pigmented areas than in the pigmented areas. The metabolomic analysis determined that the content of Delphinidin and Cyanidin in the pigmented areas was significantly higher than that in non-pigmented areas. There are two possible explanations for the up-regulation of DFR in the non-pigmented areas. One is that there is no significant correlation between DFR enzyme activity and testa anthocyanin accumulation during pigment synthesis. Wang et al. have reported that DFR enzyme activity in the pericarp was not closely related to anthocyanin synthesis in litchi40. Furthermore, there is no significant correlation detected between anthocyanin content and DFR enzyme activity in sweet cherry41. The second reason for the significant up-regulation of DFR expression in the non-pigmented areas may be that the up-regulated expression of LAR causes the leuco anthocyanins to accumulate to procyanidin in the testa. The leuco anthocyanidins are synthesized and accumulated in vacuoles under the catalysis of LAR42.
Hormone inhibition of pigment accumulation in the pigmented areas of variegated peanut testa
Hormones have an antagonistic effect on the synthesis of pigments. The transcription factors bHLH and MYB indirectly regulate the synthesis of anthocyanins by regulating the synthesis of hormones. The results showed that four differentially expressed bHLH and nine MYB regulatory genes were not clearly annotated in the metabolic pathway. It is speculated that bHLH and MYB regulatory genes in the plant circadian rhythm pathway may promote the synthesis of CHS to facilitate the accumulation of metabolites in the early stage and make the pigmented areas darker. The synthesis of testa pigment was indirectly regulated by hormones in the plant signal transduction pathway. Wan et al. identified 16 DEGs related to plant hormone signal transduction, four of which were down-regulated by the AUX signal and concluded that hormones had an antagonistic effect on pigment synthesis22. In this study, we found that the expression levels of 2 IAA14 hormone-regulated genes in the non-pigmented areas at DAF30 and DAF45 were all up-regulated, which is consistent with previous studies.
Joint analysis and mutual verification of pigment accumulation
The results of the combined analysis of the transcriptome and metabolome data were corroborated. We found that the metabolic pathway was the most closely integrated pathway of flavonoid biosynthesis, and the nine most closely integrated genes are all key genes in the anthocyanin metabolic pathway. This suggested that the selected 2 CHS, 1 C4H, 2 DFR, 1 F3'H, 1 F3H, and 2 LAR were the key genes for anthocyanin synthesis in peanut. Two different metabolites, Delphinidin and Cyanidin, were enriched in this pathway. Wan et al., in a metabolomic study, showed that flavonoids were redirected in WSC, while multi-omics analyses of WSC mutant seeds and testae demonstrated the influence of WSC on flavonoid biosynthesis in the testa43. Kovinich et al. found that UGT78K1 and 19 other anthocyanin, (iso)flavonoid, and phenylpropanoid isogenes were differentially expressed, resulting in different testa colors of black (iRT) and brown (irT) soybean (Glycine max)44, which is consistent with the results of our study. In this study, the transcriptome and metabolome of the pigmented and non-pigmented variegated testa were closely integrated. Following analysis, it was determined that delphinidin and cyanidin were the key pigments that make the pigmented areas color different from that in the non-pigmented areas.
In summary, the identified DEGs in this study were involved in 96 metabolic pathways. Phenylalanine metabolism, phenylpropanol biosynthesis, flavonoids and flavonol biosynthesis, flavonoid biosynthesis, plant hormone signal transduction, and circadian rhythm plants were related pigment synthesis in variegated testa. During the testa development, 14 main structural enzyme genes and 13 regulatory genes affected the pigment synthesis, including phenylalanine ammonia-lyase, cinnamic 4-hydroxylase enzyme, chalcone synthase, dihydroflavonol-3-hydrogenase, dihydroflavonol-3′-hydrogenase, dioxyflavonol-4-reductase, leuco anthocyanins, four bHLHs, and nine MYB regulatory genes. Cyanidin and delphinidin were the primary metabolites that caused the color differences between the pigmented and the non-pigmented areas.The results in this study reveal the molecular regulation mechanism of pigment synthesis in peanut testa, which is of great significance in breeding and planting resource protection.
Materials and methods
Materials
The variegated testa (accession VG-01) is composed of two parts, the non-pigmented areas (white) and the pigmented areas (red). The seeds were provided by the Peanut Breeding Laboratory of Hebei Agricultural University in P. R. China. VG-01 was flowering and needling on May 6, 2018, and was marked by the hanging thread method at 30, 35, 40, 45, 50, and 55 days after flowering and needling (DAF). 0.6 g variegated testa were used for RNA extraction which was repeated twice.
Characterization by stereomicroscope and chromatic aberration value
The peanut testa after needling for different days was sliced and the color changes were observed using the stereomicroscope (Olympus SZ61, Japan). The chromatic aberration value measurements result were repeated in triplicate. Different testa parts at different periods were taken, and “L”, “a”, and “b” values were measured using a colorimeter (Konica Minolta Colorimeter CR-10Plus), where “L” represents the lightness value, ”a” represents red and green values, and “b” represents yellow and blue value. F1 and F2 represent the sample names of the pigmented areas at DAF30 and DAF45, respectively. B1 and B2 represent the sample names of the non-pigmented areas at DAF30 and DAF45, respectively.
Flavonoids extraction from variegated testa
Peanut testa in different pigmented areas at DAF30 and DAF45 were stored at − 80 ℃. Samples were crushed using a mixer mill (MM 400, Retsch) with one zirconia bead (1.5 min, 30 Hz). One-hundred mg of powder was weighed and extracted overnight at 4 ℃ with 1.0 mL of 70% aqueous methanol. Following centrifugation at 10,000g for 10 min, the extracts were absorbed (CNWBOND Carbon-GCB SPE Cartridge, 250 mg, 3 mL; ANPEL, Shanghai, China) and filtrated (SCAA-104, 0.22 μm pore size; ANPEL, Shanghai, China) before liquid chromatography-tandem mass spectrometry (LC–MS) analysis.HPLC Conditions: HPLC: column, Waters ACQUITY UPLC HSS T3 C18 (1.8 µm, 2.1 mm*100 mm); solvent system, water (0.04% acetic acid): acetonitrile (0.04% acetic acid); gradient program,100:0 V/V at 0 min, 5:95 V/V at 11.0 min, 5:95 V/V at 12.0 min, 95:5 V/V at 12.1 min, 95:5 V/V at 15.0 min; flow rate, 0.40 mL/min; temperature, 40 ℃; injection volume: 5 μL.
Species and quantitative analysis of flavonoids in variegated peanut testa
LC–MS/MS was adopted to qualitatively and quantitatively detect the different areas of the variegated testa. The testa was dissolved in dimethyl sulfoxide or methanol and stored at − 20 ℃, which was diluted with 70% methanol to different gradient concentrations before mass spectrometry analysis with ultra-high-performance liquid chromatography and tandem mass spectrometry (MS/MS) (Applied Biosystems 4500 QTRAP45,46, were used to qualitatively analyze the primary and secondary spectral data detected by mass spectrometry. Analyst 1.6.3 software was used to analyze the detected data. MultiaQuant software was used to integrate and calibrate the mass spectrometry results to finally obtain the different metabolites.The size of the peak area represented the relative content of metabolites.Two biological replicates and three technical replicates were adopted for each sample.
Transcriptome sequencing and DEGs analysis
The samples were sequenced and analyzed using the Illumina sequencing platform. The original data sequencing results were compared and analyzed with the diploid wild (Araip. K30076. a1. m1&&Aradu. V14167. a1. M1, Peanutbase) and Tifrunner genomes (http://peanutbase.org/datahypogaea/Tifrunner.gnm1.ann1.CCJH/) and functional annotations were performed. The fragments per kilobase of transcript sequence per million base pairs (FPKM) values in four samples were calculated and DEseq software was used to perform DEGs analysis. Fold Change ≥ 2 and FDR < 0.01 were chosen as DEGs screening criteria. The Benjamini–Hochberg was used to correct the significance p-value obtained from the original hypothesis test to reduce the false positive rate, and FDR was used as the key indicator for DEGs screening.
GO and KEGG analysis of DEGs
GO and KEGG pathway enrichment analysis of the DEGs was based on the standard of p-value < 0.05 and FDR ≤ 0.05. The GO predicted protein information was obtained when the similarity > 30%, E-value < Ie − 5 using the BLAST comparison tool. The metabolic synthesis pathway was annotated by KEGG47 (http://www.genome.jp/kegg/pathway.html), which was obtained when compared with the KEGG database under the condition of E-value < le3.
Joint analysis of transcriptome and metabolome
The differential genes and metabolites screened from the metabolome and transcriptome were jointly analyzed according to the Pearson correlation analysis method. The joint analysis was carried out on the metabolome and transcriptome data based on the standard of the correlation coefficient threshold ≥ 0.8 and correlation P < 0.05.
Verification by qRT-PCR
The selected 20 DEGs from the pigmented areas were all verified by real-time quantitative PCR (qRT-PCR). Total RNA was extracted by the TRIZOL method, and the corresponding cDNA was synthesized. According to the sequence library required for sequencing, the fluorescent real-time quantitative PCR primers were designed (Table S9) by using Primer Premier5.0 software, in which the peanut actin ACT7 gene was used as the internal reference gene. The ΔΔCt data analysis method was used for gene expression analysis and 2−ΔΔCt was used to calculate the relative gene expression48. The expression level in the target gene of the control part (B1, B2 non-pigmented areas) was 1, and two replicates were set in this experiment.
Ethical statement
All experimental research and field studies on this paper complied with relevant guidelines and legislation of Hebei Agricultural University P. R. China. The experimental methods in this study were performed in accordance with relevant guidelines and legislation of the University Committee.
Supplementary Information
Acknowledgements
We thank Key project of science and technology research in colleges and universities of the department of education in Hebei Province (ZD2019051) and Key project of science and technology research of modern seed industry of the department of S&T in Hebei Province (19226363D) for providing funds.This research was supported by North China Key Laboratory for Crop Germplasm Resources of Education Ministry, College of Agronomy, Hebei Agricultural University and Hebei Yiyuan Ecological Agriculture Technology Co, Ltd.
Author contributions
G.M. the corresponding author. G.M. and L.L. developed the concept, planned, coordinated and executed the research. Me.H. performed most of the experiments, analyzed the data, and prepared the figures and tables; Me.H. wrote the manuscript; J.L., Mi.H. and X.L. performed some analyses and prepared Figs. 1, 2, 3, 4, 5, 6, 7, 8 and 9; S.C., X.Y. and X.J. supervised the experiments; G.M., Mi.H. and X.Y. modifed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-90141-6.
References
- 1.Wang Y, et al. Research progress of peanut aflatoxin control and detection methods. Liaoning Agric. Sci. 2019;05:66–68. [Google Scholar]
- 2.Bulgakov VP, Avramenko TV, Tsitsiashvili GS. Critical analysis of protein signaling networks involved in the regulation of plant secondary metabolism: focus on anthocyanins. Crit. Rev. Biotechnol. 2017;37:685–700. doi: 10.3109/07388551.2016.1141391. [DOI] [PubMed] [Google Scholar]
- 3.Sun Q, et al. Color change and pigment deposition of colored peanut testa. Acta Arachis. 2015;44:1–6. [Google Scholar]
- 4.Hu J, et al. Ultraviolet B-induced MdWRKY72 expression promotes anthocyanin synthesis in apple. Plant Sci. 2020;292:110377. doi: 10.1016/j.plantsci.2019.110377. [DOI] [PubMed] [Google Scholar]
- 5.Fukusaki E, et al. Flower color modulations of Torenia hybrida by downregulation of chalcone synthase genes with RNA interference. J. Biotechnol. 2004;111:229–240. doi: 10.1016/j.jbiotec.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Chen Q, et al. Research progress of MYB transcription factors in plants. Genom. Appl. Biol. 2009;28:365–372. [Google Scholar]
- 7.van der Krol AR, Mur LA, de Lange P, Mol JN, Stuitje AR. Inhibition of flower pigmentation by antisense CHS genes: Promoter and minimal sequence requirements for the antisense effect. Plant Mol. Biol. 1990;14:457–466. doi: 10.1007/BF00027492. [DOI] [PubMed] [Google Scholar]
- 8.Yamazaki M, et al. Metabolomics and differential gene expression in anthocyanin chemo-varietal forms of Perilla frutescens. Phytochemistry. 2003;62:987–995. doi: 10.1016/S0031-9422(02)00721-5. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Z, Lu Y. Anthocyanin metabolic pathway and plant color variation. Acta Botanica Sinica. 2016;51:107–119. [Google Scholar]
- 10.Zhang X. Regulation of ANS, ANR and LAR Genes in Anthocyanin Metabolism Of Strawberry. Sichuan Agricultural University; 2013. [Google Scholar]
- 11.Chen Y. Cloning and Functional Verification of CsUGGT Gene from Tea Plant. Hunan Agricultural University; 2017. [Google Scholar]
- 12.Elomaa P, et al. Activation of anthocyanin biosynthesis in Gerbera hybrida (Asteraceae) suggests conserved protein–protein and protein–promoter interactions between the anciently diverged monocots and eudicots. Plant Physiol. 2003;133:1831–1842. doi: 10.1104/pp.103.026039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baudry A, et al. TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 2004;39:366–380. doi: 10.1111/j.1365-313X.2004.02138.x. [DOI] [PubMed] [Google Scholar]
- 14.Almeida J, Carpenter R, Robbins TP, Martin C, Coen ES. Genetic interactions underlying flower color patterns in Antirrhinum majus. Genes Dev. 1989;3:1758–1767. doi: 10.1101/gad.3.11.1758. [DOI] [PubMed] [Google Scholar]
- 15.Xiang LL, et al. A novel bHLH transcription factor involved in regulating anthocyanin biosynthesis in Chrysanthemums (Chrysanthemum morifolium Ramat.) PLoS ONE. 2015;10:e0143892. doi: 10.1371/journal.pone.0143892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia H, et al. Comparative transcriptome analysis of anthocyanin synthesis in black and pink peanut. Plant Signal Behav. 2020;15:1721044. doi: 10.1080/15592324.2020.1721044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shang X, et al. Advances in molecular mechanism of plant spot synthesis. J. Horticult. 2014;41:1485–1494. [Google Scholar]
- 18.Koseki M, Goto K, Masuta C, Kanazawa A. The star-type color pattern in Petunia hybrida 'red Star' flowers is induced by sequence-specific degradation of chalcone synthase RNA. Plant Cell Physiol. 2005;46:1879–1883. doi: 10.1093/pcp/pci192. [DOI] [PubMed] [Google Scholar]
- 19.Ma D, Constabel CP. MYB repressors as regulators of phenylpropanoid metabolism in plants. Trends Plant Sci. 2019;24:275–289. doi: 10.1016/j.tplants.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Chiou CY, Yeh KW. Differential expression of MYB gene (OgMYB1) determines color patterning in floral tissue of Oncidium Gower Ramsey. Plant Mol. Biol. 2008;66:379–388. doi: 10.1007/s11103-007-9275-3. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Qiu J, Sun X, Liang X. Cloning and sequence analysis of genes related to anthocyanin synthesis in Peanut Cultivars with different testa colors. Journal of Tropical Crops. 2017;39:600–605. [Google Scholar]
- 22.Wan L, et al. Transcriptomic profiling reveals pigment regulation during peanut testa development. Plant Physiol. Biochem. 2018;125:116–125. doi: 10.1016/j.plaphy.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 23.Li M, et al. Cloning and expression analysis of peanut Flavanone-3-hydroxylase Gene ahf3h. Shandong Agric. Sci. 2013;45:1–6. [Google Scholar]
- 24.Ye J, et al. Identification of candidate genes involved in anthocyanin accumulation using Illmuina-based RNA-seq in peach skin. Sci. Horticult. 2019;250:184–198. doi: 10.1016/j.scienta.2019.02.047. [DOI] [Google Scholar]
- 25.Liu T, et al. Transcriptome analyses reveal key genes involved in skin color changes of 'Xinlimei' radish taproot. Plant Physiol. Biochem. 2019;139:528–539. doi: 10.1016/j.plaphy.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Jia Z, et al. Transcriptome and Anthocyanin Accumulation Mechanism in Fruit Synthesis of 'Hongyang' Kiwifruit. School of Chinese Academy of Sciences (Wuhan Botanical Garden); 2017. [Google Scholar]
- 27.Jin X. Synthesis of Anthocyanin from Chrysanthemum morifolium Based on High Throughput Sequencing. Beijing Forestry University; 2013. [Google Scholar]
- 28.Bai Y, Dougherty L, Xu K. Towards an improved apple reference transcriptome using RNA-seq. Mol. Genet. Genom. 2014;289:427–438. doi: 10.1007/s00438-014-0819-3. [DOI] [PubMed] [Google Scholar]
- 29.Venturini L, et al. De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity. BMC Genom. 2013;14:41. doi: 10.1186/1471-2164-14-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, et al. MicroRNA858 negatively regulates anthocyanin biosynthesis by repressing AaMYBC1 expression in kiwifruit (Actinidia arguta) Plant Sci. 2020;296:110476. doi: 10.1016/j.plantsci.2020.110476. [DOI] [PubMed] [Google Scholar]
- 31.Zhang D, et al. Genome-wide identification and expression analysis of anthocyanin biosynthetic genes in Brassica juncea. J. Integr. Agric. 2020;19:1250–1260. doi: 10.1016/S2095-3119(20)63172-0. [DOI] [Google Scholar]
- 32.Jin X, Huang H, Wang L, Sun Y, Dai S. Transcriptomics and metabolite analysis reveals the molecular mechanism of anthocyanin biosynthesis branch pathway in different Senecio cruentus cultivars. Front. Plant Sci. 2016;7:1307. doi: 10.3389/fpls.2016.01307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng S. Study on the Molecular Mechanism of Color Difference Synthesis of Wild Banana Peel Based on RNA Seq. Fujian Agriculture and Forestry University; 2018. [Google Scholar]
- 34.Gao J, Li WB, Liu HF, Chen FB. De novo transcriptome sequencing of radish (Raphanus sativus L.) fleshy roots: Analysis of major genes involved in the anthocyanin synthesis pathway. BMC Mol. Cell Biol. 2019;20:45. doi: 10.1186/s12860-019-0228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohno S, Hori W, Hosokawa M, Tatsuzawa F, Doi M. Post-transcriptional silencing of chalcone synthase is involved in phenotypic lability in petals and leaves of bicolor dahlia (Dahlia variabilis) 'Yuino'. Planta. 2018;247:413–428. doi: 10.1007/s00425-017-2796-3. [DOI] [PubMed] [Google Scholar]
- 36.Enrique GA, et al. Elicitation with Bacillus QV15 reveals a pivotal role of F3H on flavonoid metabolism improving adaptation to biotic stress in blackberry. PloS One. 2018;15:e0232626. doi: 10.1371/journal.pone.0232626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brugliera F, et al. Violet/blue chrysanthemums–metabolic engineering of the anthocyanin biosynthetic pathway results in novel petal colors. Plant Cell Physiol. 2013;54:1696–1710. doi: 10.1093/pcp/pct110. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Gao Z, Zhang C, Li N, Liu C. Research progress of molecular regulation mechanism in anthocyanin synthesis pathway. J. Ecol. 2015;34:2937–2942. [Google Scholar]
- 39.Johnson ET, et al. Alteration of a single amino acid changes the substrate specificity of dihydroflavonol 4-reductase. Plant J. 2001;25:325–333. doi: 10.1046/j.1365-313x.2001.00962.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, Huang X, Hu G, Huang H. Study on the relationship Between Anthocyanin Synthesis and Related Enzymes in Litchi Pericarp. Agricultural Science in China; 2004. pp. 2028–2032. [Google Scholar]
- 41.Shen Y. Study on Nutritional Components and Anthocyanins of Cherry Fruit. Nanjing Forestry University; 2014. [Google Scholar]
- 42.Gargouri M, Gallois B, Chaudière J. Binding-equilibrium and kinetic studies of anthocyanidin reductase from Vitis vinifera. Arch Biochem. Biophys. 2009;491:61–68. doi: 10.1016/j.abb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 43.Liyun W, et al. Transcriptome and metabolome reveal redirection of flavonoids in a white testa peanut mutant. BMC Plant Biol. 2009;20:161. doi: 10.1186/s12870-020-02383-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovinich N, Saleem A, Arnason JT, Miki B. Combined analysis of transcriptome and metabolite data reveals extensive differences between black and brown nearly-isogenic soybean (Glycine max) seed coats enabling the identification of pigment isogenes. BMC Genom. 2011;12:381. doi: 10.1186/1471-2164-12-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wishart DS, et al. HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu ZJ, et al. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013;8:451–460. doi: 10.1038/nprot.2013.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Methods. 2017;25:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(− Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.