

Abstract
Introduction
Lewy body–related pathology is commonly observed at autopsy in individuals with dementia, but in vivo biomarkers for α‐synucleinopathy are lacking.
Methods
Baseline cerebrospinal fluid (CSF) biomarkers, polygenic risk score (PRS) for Parkinson's disease (PRS‐PD) and Alzheimer's disease (PRS‐AD), longitudinal cognitive scores, and magnetic resonance imaging were measured in 217 participants from the Alzheimer's Disease Neuroimaging Initiative. Linear mixed models were used to find the relationship of CSF biomarkers and the PRS with cognition and cortical atrophy.
Results
Higher PRS‐PD and PRS‐AD were associated with lower CSF α‐synuclein and amyloid beta (Aβ), respectively. Lower CSF α‐synuclein and the interaction of CSF α‐synuclein and Aβ were associated with lower cognitive scores and global cortical atrophy most prominently in the occipital cortex.
Discussion
Lower CSF α‐synuclein could be a biomarker for α‐synucleinopathy, and the simultaneous evaluation of CSF biomarkers for AD and CSF α‐synuclein could reveal the independent and interactive effects on cognition and cortical atrophy.
Keywords: α‐synuclein, Alzheimer's disease, cerebrospinal fluid biomarkers, Lewy body disease, polygenic risk score
1. INTRODUCTION
Dementia with Lewy bodies (DLB) is the second most common neurodegenerative cause of dementia after Alzheimer's disease (AD). Concomitant Lewy body (LB)‐related pathology and Lewy body disease (LBD) is observed in up to 60% of neuropathologic‐confirmed AD cases, 1 , 2 and is associated with more severe symptoms. 3 , 4 , 5 Diagnostic sensitivity for LBD or LB‐related pathology in AD patients is suboptimal due to the absence of direct biomarkers for α‐synuclein (α‐syn). Cerebrospinal fluid (CSF) biomarkers provide useful information for AD and LBD. 6 , 7 However, there are complex relationships between CSF biomarkers for AD and CSF α‐syn. Decreased CSF α‐syn has been reported in patients with Parkinson's disease (PD) and DLB, 7 but increased in AD patients compared to controls and LBD patients. 8 Therefore, CSF AD biomarkers need to be considered to find the clinical implication of CSF α‐syn. 9 , 10
Recent genome‐wide association studies (GWAS) in AD and LBD revealed multiple risk alleles with small effect sizes. 11 , 12 , 13 , 14 The polygenic risk score (PRS) summarizes these genetic factors into one score. PRS for AD was used to predict the age of symptom onset, amyloid beta (Aβ) deposition, and neuropathology. 15 , 16 PRS for PD was associated with the progression 17 , 18 and striatal dopaminergic degeneration in PD. 19 Although there is high genetic pleiotropy between AD and DLB, 20 , 21 and the prevalence of mixed AD and LBD pathology is high, the implication of the PRS for α‐synucleinopathy has not been evaluated simultaneously considering AD.
We evaluated the implication of CSF biomarkers for AD and CSF α‐syn by investigating their relationship with PRS for AD (PRS‐AD) and PD (PRS‐PD). Then, we investigated the effects of CSF α‐syn on cognitive scores and cortical thickness using the Alzheimer's Disease Neuroimaging Initiative (ADNI) cohort, using subjects with normal cognition (NC), mild cognitive impairment (MCI), and dementia. We hypothesized that CSF α‐syn would be related to PRS‐PD, and cognitive decline and longitudinal cortical atrophy with LBD‐specific patterns after controlling for the effects of AD.
2. METHODS
2.1. Participants and CSF biomarkers
Data used in this article were obtained from the ADNI database. Detailed methods for ADNI protocol for the diagnosis of NC, MCI, and dementia are in Text S1 in supporting information.
Subjects with baseline CSF α‐syn (n = 368) were included, from which 149 with hemoglobin contamination (>200 ng/mL) were excluded. 9 , 10 Baseline AD CSF biomarkers, including tau, phosphorylated tau (p‐tau), and Aβ1‐42, were retrieved and if subjects had a CSF Aβ1‐42 level <192 pg/mL, they were considered Aβ positive. 22 Pre‐calculated PRS‐AD was available from the ADNI. 15 PRS‐PD was calculated, based on 48 single nucleotide polymorphisms (SNPs) of previous GWAS, 11 , 12 in 187 subjects (Figure 1).
FIGURE 1.
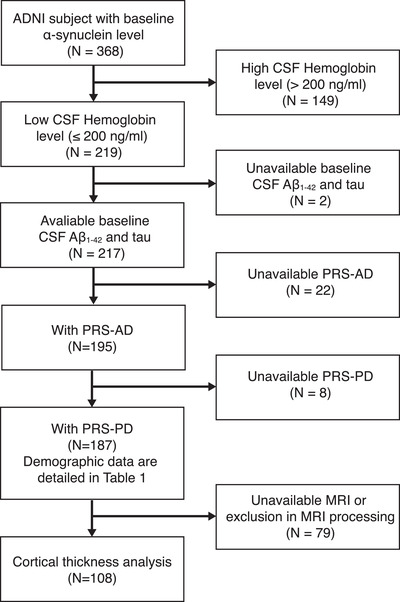
Flowchart of the study participants. Aβ, amyloid beta; AD, Alzheimer's disease; ADNI, Alzheimer's Disease Neuroimaging Initiative; CSF, cerebrospinal fluid; PD, Parkinson's disease; PRS, polygenic risk score
2.2. Processing of genotyping data and calculation of PRS
Genotyping of the study participants was conducted using an Illumina Human610‐Quad BeadChip array. The genetic relationship between individuals was calculated using PLINK 23 and those with first‐ or second‐degree relatives were removed. Quality control was performed with the following exclusion criteria: SNPs on non‐autosome, low minor allele frequencies (< 0.01), deviations from Hardy‐Weinberg equilibrium (P < 1E‐6), and high rates of missing genotypes (> 0.05). Population structures were calculated using principal component analysis after pruning steps and ancestry outliers were excluded.
HIGHLIGHTS
Lower cerebrospinal fluid (CSF) α‐synuclein (α‐syn) was associated with higher polygenic risk score (PRS) of Parkinson's disease (PD).
Lower CSF α‐syn and CSF α‐syn × amyloid beta (Aβ) were associated with cognitive dysfunction.
Lower CSF α‐syn and CSF α‐syn × Aβ were associated with widespread cortical atrophy.
Higher PRS of PD was associated with thickening of the cingulum.
RESEARCH IN CONTEXT
Systemic review: The authors reviewed the literature using PubMed. Although Lewy body‐related pathology is commonly observed at autopsy in individuals with dementia, in vivo biomarkers for α‐synucleinopathy are lacking.
Interpretation: Decreased cerebrospinal fluid (CSF) α‐synuclein levels were associated with higher polygenic risk scores designed for Parkinson's disease after controlling the CSF tau and amyloid beta (Aβ) levels. After considering the effects of Alzheimer's disease (AD) biomarkers, decreased α‐synuclein was associated with cognitive decline and cortical atrophy with Lewy body disease‐specific pattern, which had detrimental interactions with Aβ.
Future directions: CSF α‐synuclein could reflect α‐synucleinopathy after considering the effects of AD. Future research with pathologic confirmation will be needed.
Risk SNPs of PD were selected from two previous GWAS for PD. 11 , 12 Meta odds ratio of the risk alleles were obtained from the PDgene database (https://www.pdgene.org). Genotyping data were phased using SHAPEIT2 24 and missing SNPs were imputed using IMPUTE2. 25 After post‐imputation filtering and clumping based on linkage disequilibrium structures of samples, the final 48 SNPs were selected (Table S1 in supporting information). PRS was calculated by the summation of the number of risk alleles weighted by log‐transformed odds ratio from each individual. PRS for AD was calculated based on 31 risk SNPs from survival analysis for age of symptom onset using log hazard ratios as effect size. 15
2.3. Neuropsychological assessment
Composite scores of attention, language, visuospatial function, memory, and executive function were calculated using z‐transformed scores of the Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐Cog) and a neuropsychological battery described in Text S2 in supporting information. Mini‐Mental State Examination (MMSE) was included to evaluate general cognition.
2.4. Magnetic resonance imagin acquisition and image processing
3D MPRAGE scans were acquired at 1.5 Tesla MRI. Details of the magnetic resonance imaging (MRI) protocols are listed on the ADNI website (http://adni.loni.usc.edu/methods/documents/mri‐protocols/). We selected minimally pre‐processed T1‐weighted MRIs (n = 384) that underwent post‐acquisition correction such as gradient warping, B1 calibration, intensity non‐uniformity correction, and scaling for better image analysis results. Low‐quality scans identified in the csv files were removed before the image processing.
We processed the T1‐weighted image using the CIVET pipeline (version 2.1.1, http://mcin.ca/civet). Each subject's image was linearly registered to the Montreal Neurological Institute standard space. 26 The images were tissue‐classified and the inner and outer cortical surfaces were extracted resulting in 40,962 vertex points per hemisphere. 27 To obtain intersubject correspondence, the surfaces were registered to an unbiased group template by matching the sulcal folding pattern. 28 Cortical thickness was calculated using three‐dimensional Laplace's equation between the inner and outer surfaces and smoothed using a 30 mm full width at half maximum surface‐based diffusion smoothing kernel.
All results from the anatomical pipeline were visually inspected by three researchers (Y. Lee, S. Jeon, and B.S. Ye) who were blinded to subject information for quality assurance. We excluded 36 subjects due to image processing errors in brain masking, tissue classification, and surface reconstruction. Only subjects with scans of at least two timepoints were included. Finally, 332 scans of 108 subjects were used in the statistical analysis (Figure 1).
2.5. Statistical analysis
Statistical analyses were performed using IBM SPSS version 25.0 (SPSS Inc.). An analysis of variance and chi‐square test were used to compare the demographic variables. We selected CSF tau rather than CSF p‐tau as previous studies have shown that CSF tau positively correlates with CSF α‐syn 8 , 9 and that CSF tau and CSF p‐tau are closely correlated (r > 0.7) in our analysis. General linear models for each CSF biomarker were used with the remaining CSF biomarkers as predictors to evaluate the relationship between PRS and CSF biomarkers. Covariates included age, sex, and education.
To evaluate the effects of PRS and CSF biomarkers on cognitive composite scores, we used linear mixed models for the composite scores using the baseline CSF biomarkers, PRS‐AD, PRS‐PD, and year (follow‐up years from baseline) as predictors. Covariates included age, sex, and education. Interaction terms were included in the final statistical models if they were significantly associated with outcome variables. To avoid multicollinearity, if a pair of variables had an r‐value of > 0.7 and variance inflation factor of > 2.5 (Table S2 in supporting information), those with higher statistical significance were selected. Accordingly, Aβ1‐42 × α‐syn and tau × year were included as predictors in the final model. P‐values were corrected for multiple comparisons using the false discovery rate method.
We used the SurfStat toolbox (http://www.math.mcgill.ca/keith/surfstat/) for vertex‐wise cortical thickness analyses. Linear mixed models for cortical thickness were performed to evaluate the effects of the predictors after controlling for age, sex, education, and intracranial volume. Aforementioned criteria for multicollinearity were also applied. We used the random field theory method (RFT, corrected P < .05) to correct for multiple comparisons. 29
3. RESULTS
3.1. Demographic and clinical characteristics
There were no significant differences in terms of baseline age, sex, and education years among the NC, MCI, and dementia groups (Table 1). The MCI and dementia groups had lower CSF Aβ1‐42 and higher CSF tau and p‐tau levels than the NC group. The proportion of Aβ‐positive subjects was significantly higher in the MCI and dementia groups than the NC group (Figure S1 in supporting information). CSF α‐syn levels were not different between the groups. The MCI group had better and worse scores than the dementia and NC groups, respectively, in MMSE, and neuropsychological composite scores for executive, language, memory, and visuospatial domains. For the attention domain, the dementia group had a worse score than the MCI and NC groups. The MCI and dementia groups had higher PRS‐AD than the NC group. The three groups had comparable PRS‐PD. Proportion of individuals with available MRI was not different among three groups.
TABLE 1.
Demographics, CSF biomarkers, and clinical characteristics of study participants
| NC | MCI | Dementia | P | |
|---|---|---|---|---|
| Number | 55 | 87 | 45 | |
| Age, year | 75.8 (4.9) | 73.8 (7.4) | 74.6 (8.8) | .302 |
| Sex, female | 25 (45.5) | 27 (31.0) | 20 (44.4) | .146 |
| Education, year | 16.2 (2.3) | 15.8 (3.1) | 15.0 (3.2) | .159 |
| CSF biomarkers | ||||
| Aβ1‐42, pg/mL | 213.0 (50.9) a,b | 159.8 (51.2) a | 152.2 (42.4) b | < .001 |
| tau, pg/mL | 71.8 (29.4) a,b | 96.8 (46.9) a,c | 122.0 (66.4) b,c | < .001 |
| P‐tau181p, pg/mL | 22.8 (10.4) a,b | 33.4 (15.7) a | 37.6 (18.8) b | < .001 |
| α‐syn, ng/mL | 0.52 (0.14) | 0.54 (0.14) | 0.56 (0.15) | .288 |
| Aβ‐positivity | 18 (32.7) a,b | 66 (75.9) a,c | 39 (86.7) b,c | < .001 |
| Baseline cognitive scores | ||||
| Attention | –0.18 (0.88) b | –0.40 (0.87) c | ‐0.83 (0.61) b,c | < .001 |
| Executive function | 0.05 (0.63) a,b | –0.69 (1.00) a,c | ‐2.16 (1.51) b,c | < .001 |
| Language | –0.15 (0.69) a,b | –0.78 (1.00) a,c | ‐1.42 (1.07) b,c | < .001 |
| Memory | –0.08 (0.59) a,b | –1.28 (0.83) a,c | ‐1.84 (0.57) b,c | < .001 |
| Visuospatial function | –0.03 (0.66) a,b | –0.63 (1.05) a,c | ‐1.34 (1.07) b,c | < .001 |
| MMSE score | 29.2 (0.9) a,b | 27.0 (1.7) a,c | 23.7 (1.9) b,c | < .001 |
| Genetic risk scores | ||||
| PRS‐PD | 1.97 (0.17) | 1.95 (0.23) | 1.91 (0.20) | .369 |
| PRS‐AD | ‐0.10 (0.62) a,b | 0.68 (0.78) a | 0.86 (0.95) b | < .001 |
| MRI analysis | 34 (61.8) | 50 (57.5) | 24 (53.3) | .696 |
Abbreviations: α‐syn, α‐synuclein; Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; MRI, magnetic resonance imaging; NC, normal cognition; PD, Parkinson's disease; PRS, polygenic risk score; p‐tau, phosphorylated tau.
Notes: Results from the analysis of variance or chi‐square test as appropriate. Data are expressed as the mean (standard deviation) or number (%).
A significant difference between NC and MCIa, NC and dmentiab, and MCI and dementia groupsc.
3.2. Association between PRS and CSF biomarkers
CSF Aβ1‐42 was independently associated with PRS‐AD, CSF tau, and CSF α‐syn, while CSF α‐syn was independently associated with PRS‐PD and CSF tau (Table S3 in supporting information). Lower CSF α‐syn was associated with higher PRS‐PD but not with PRS‐AD (Figure 2A and 2B), while lower CSF Aβ1‐42 was associated with higher PRS‐AD but not with PRS‐PD (Figure 2C and 2D). CSF tau was not associated with PRS‐AD or PRS‐PD (Figure 2E and 2F). CSF α‐syn positively correlated with CSF Aβ1‐42 and CSF tau. CSF tau negatively correlated with CSF Aβ1‐42.
FIGURE 2.
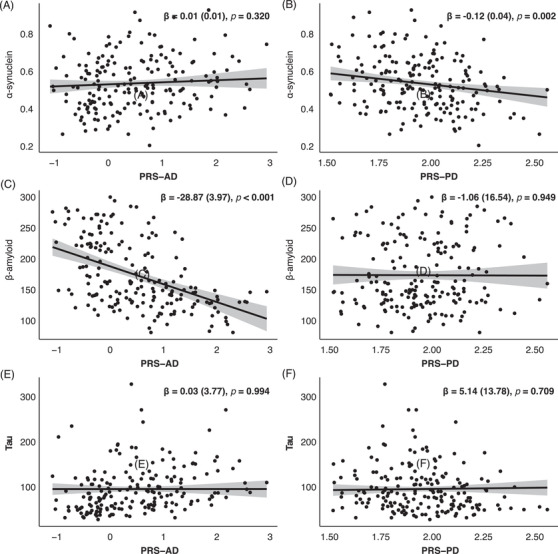
Association between cerebrospinal fluid (CSF) biomarkers and polygenic risk scores. Data are results of general linear models for CSF α‐synuclein (A and B), CSF β‐amyloid (C and D), and CSF tau (E and F) using polygenic risk score‐Alzheimer's disease (PRS‐AD), polygenic risk score‐Parkinson's disease (PRS‐PD), and other CSF biomarkers as predictors. Covariates included age, sex, and education. Each dot represents individuals and predicted regression lines (black line) with confidence intervals (gray shade) are displayed
3.3. Effect of CSF biomarkers and genetic risk scores on cognitive composite scores
Lower baseline CSF α‐syn, lower CSF Aβ1‐42, and higher CSF tau were independently associated with lower composite scores for language, memory, visuospatial, and executive domains (Table 2). Higher PRS‐AD was associated with lower composite scores for language and memory domains, while PRS‐PD was not independently associated with cognitive composite scores. Baseline CSF Aβ1‐42 × CSF α‐syn and CSF tau × year had significant detrimental effects on composite scores for language, memory, visuospatial, and executive domains.
TABLE 2.
Effects of CSF biomarkers and genetic risk scores on longitudinal cognitive scores
| Attention | Language | Memory | Visuospatial | Executive | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| β (SE) | P | β (SE) | P | β (SE) | P | β (SE) | P | β (SE) | P | |
| Aβ1‐42 | 0.01 (0.003) | .032 | 0.02 (0.01) | .007 a | 0.02 (0.003) | < .001 a | 0.02 (0.005) | < .001 a | 0.02 (0.01) | < .001 a |
| α‐syn | 1.90 (1.25) | .130 | 7.36 (2.01) | < .001 a | 6.15 (1.29) | < .001 a | 7.69 (1.90) | < .001 a | 9.01 (2.22) | < .001 a |
| Tau | −0.002 (0.001) | .107 | −0.01 (0.002) | < .001 a | −0.01 (0.002) | < .001 a | −0.01 (0.002) | < .001 a | −0.01 (0.003) | < .001 a |
| PRS‐AD | −0.07 (0.07) | .349 | −0.32 (0.12) | .006 a | −0.31 (0.07) | < .001 a | ‐0.11 (0.11) | .301 | −0.14 (0.13) | .288 |
| PRS‐PD | 0.08 (0.26) | .749 | −0.18 (0.42) | .667 | 0.13 (0.27) | .623 | 0.55 (0.39) | .162 | 0.93 (0.47) | .048 |
| Aβ1‐42 × α‐syn | −0.01 (0.01) | .158 | −0.03 (0.01) | .010 a | −0.02 (0.01) | .001 a | −0.03 (0.01) | .003 a | −0.03 (0.01) | .004 a |
| Tau × Year | −0.0004 (0.0002) | .092 | −0.002 (0.0003) | < .001 a | −0.001 (0.0001) | < .001 a | −0.001 (0.0002) | < .001 a | −0.002 (0.0003) | < .001* |
Abbreviations: α‐syn, α‐synuclein; Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; PD, Parkinson's disease; PRS, polygenic risk score; SE, standard error.
Notes: Data are the results of linear mixed models for longitudinal cognitive scores using CSF Aβ1‐42, α‐syn, and tau, PRS‐AD, PRS‐PD, Aβ1‐42 × α‐Syn and tau × year as predictors. Covariates included age, sex, and education.
Significant after correction for multiple comparisons using the false discovery method.
Because CSF Aβ1‐42 and CSF α‐syn had detrimental interaction effects on the composite scores, separate analyses were performed on the Aβ‐positive and Aβ‐negative subgroups (Table S4 in supporting information). In the Aβ‐positive subgroup, lower CSF α‐syn was associated with lower composite scores for language, memory, visuospatial, and executive domains. Higher tau was associated with lower composite scores for all cognitive domains. CSF tau and year had detrimental interaction effects on the composite scores for all cognitive domains. Higher PRS‐AD was associated with a lower composite score for the memory domain, while PRS‐PD was not independently associated with composite scores. In the Aβ‐negative subgroup, only CSF tau and year had a detrimental interaction effect on the memory domain score.
3.4. Effect of CSF biomarkers and genetic risk scores on vertex‐wise cortical thickness
Vertex‐wise cortical thickness analyses showed that lower baseline CSF α‐syn and lower CSF Aβ1‐42 were associated with decreased cortical thickness in the widespread cortical regions (Figure 3A). Higher baseline CSF tau was associated with decreased cortical thickness in the lateral parietal and inferior temporal and occipital cortices. CSF α‐syn × CSF Aβ1‐42 and CSF tau × year had detrimental effects on cortical thickness in the widespread cortical regions. Higher PRS‐AD was associated with decreased cortical thickness in the bilateral medial temporal cortices, while higher PRS‐PD was associated with increased cortical thickness in the cingulate cortex.
FIGURE 3.
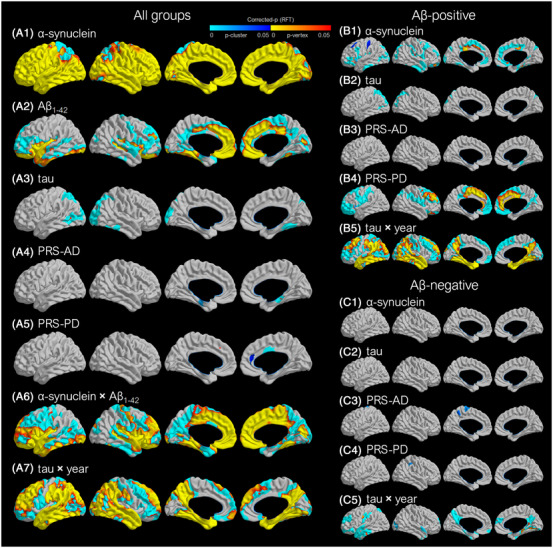
Effect of CSF biomarkers and the genetic risk score on regional cortical thickness. A, Results are based on a linear mixed model for voxel‐wise cortical thickness using baseline CSF α‐synuclein, CSF Aβ1‐42, CSF tau, PRS‐AD, PRS‐PD, α‐synuclein × Aβ1‐42, and tau × year as predictors after controlling for age, sex, education, and intracranial volume. B,C, Results for subgroups differing by Aβ positivity are based on a linear mixed model without CSF Aβ1‐42 and α‐synuclein × Aβ1‐42. Positive correlations were tested for Aβ1‐42 and PRS‐PD, while negative correlations were tested for α‐synuclein, tau, and PRS‐AD. The RFT was used to correct for multiple comparisons over the whole cortical mantle. In the p‐maps, blue color indicates the brain regions where the RFT‐corrected cluster P‐values were significant; yellow to red indicates the brain regions where the RFT‐corrected vertex P‐values were significant. Brain images are displayed in the neurologic convention. Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; PD, Parkinson's disease; PRS, polygenic risk score; RFT, random field theory
Subgroup analyses performed in the Aβ‐positive subgroup showed that the effects of lower baseline CSF α‐syn, higher CSF tau, the interaction term of CSF tau × year, and PRS‐AD were similar to those observed in the analysis for overall participants (Figure 3B). The effect of PRS‐PD was more prominent and widespread, in that higher PRS‐PD was associated with increased cortical thickness, mainly involving the cingulate cortex. Subgroup analyses performed in the Aβ‐negative subgroup showed that the effects of baseline CSF α‐syn and tau were not significant (Figure 3C). CSF tau × year had detrimental effects on cortical thickness in the temporal, parietal, and occipital cortices.
3.5. Effect of CSF biomarkers and genetic risk scores on mean lobar cortical thickness
Lower baseline CSF α‐syn level was associated with lower mean cortical thickness in all lobar regions (Table S5 in supporting information). Lower baseline CSF Aβ1‐42 was associated with lower mean occipital cortical thickness, while higher baseline CSF tau was associated with lower mean lobar cortical thickness in all lobar regions. CSF α‐syn × CSF Aβ1‐42 and CSF tau × year had detrimental effects on the mean lobar cortical thickness in all lobar regions. PRS‐AD and PRS‐PD were not associated with the mean lobar cortical thickness.
Subgroup analyses of the mean lobar cortical thickness performed in the Aβ‐positive subgroup showed that higher baseline CSF tau and CSF tau × year were associated with decreased cortical thickness in all lobar regions (Table S6 in supporting information). The effects of baseline CSF α‐syn, PRS‐AD, and PRS‐PD were not significant. In the Aβ‐negative subgroup, only CSF tau × year had significant detrimental effects on the mean cortical thickness in all lobar regions.
3.6. Effect of PRS‐PD on the mean cortical thickness in the cingulate cortex
Because PRS‐PD was associated with increased cortical thickness in the cingulate cortex (Figure 3), the correlation of PRS‐PD with mean cingulate cortical thickness was evaluated using Pearson's correlation analyses (Figure 4). The positive correlation was significant in the dementia (r = 0.548, P < .001) and Aβ‐positive subgroups (r = 0.193, P = .004), but not significant in the MCI, NC, and Aβ‐negative subgroups.
FIGURE 4.
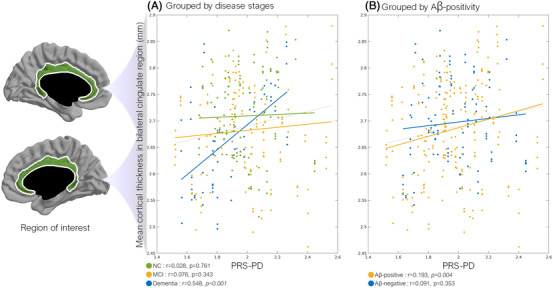
Correlation of PRS‐PD with mean cingulate cortical thickness. Scatter plots of PRS‐PD against mean cingulate cortical thickness in separate subgroups according to (A) cognitive status and (B) Aβ positivity. Results are based on a mixed model showing correlation between mean cingulate cortical thickness and PRS‐PD in each subgroup. Age, sex, years of education, and intracranial volume were corrected. Gray dashed line indicates the relationship without the group separation as a reference. The cingulate region was defined by a surface parcellation map provided in the anatomical pipeline (green area). Aβ, β amyloid beta; MCI, mild cognitive impairment; NC, normal cognition; PD, Parkinson's disease; PRS, polygenic risk score
4. DISCUSSION
In this study, we evaluated the implication of CSF α‐syn as a biomarker for α‐synucleinopathy by evaluating its relationship with PRS for PD, cognitive dysfunction, and cortical atrophy, simultaneously considering the effects of AD. The major findings of our study are as follows. First, lower CSF α‐syn and CSF Aβ1‐42 were associated with higher PRS‐PD and PRS‐AD, respectively. Second, lower CSF α‐syn, lower CSF Aβ1‐42, and CSF α‐syn × CSF Aβ1‐42 were independently associated with widespread cognitive dysfunction. Third, lower CSF α‐syn and CSF α‐syn × CSF Aβ1‐42 were associated with widespread cortical atrophy most prominently in the occipital cortex. Fourth, higher PRS‐PD was associated with increased cortical thickness in the cingulate cortex.
Lower CSF α‐syn was associated with higher PRS‐PD. The significant correlation between the PRS‐PD and lower CSF α‐syn suggests that decreased CSF α‐syn could be a biomarker reflecting α‐synucleinopathy. Previous studies found that CSF α‐syn is lower in patients with LBD compared to AD and control subjects. 7 Similar to the reduction in CSF Aβ1‐42, which reflects increased Aβ deposition in the AD brain, the aggregation and sequestration of α‐syn in LB could lead to a reduction of CSF α‐syn in LBD. However, a previous study showed that PRS for PD is not associated with CSF α‐syn levels. 30 This discrepancy could be related to different alleles being used to calculate the PRS, exclusion of samples with CSF hemoglobin contamination, and further adjustment for CSF tau and Aβ1‐42 in our study.
Simultaneous evaluation of CSF AD biomarkers and CSF α‐syn was important to find their relationship with neurodegeneration. Without consideration for CSF α‐syn × CSF Aβ1‐42, CSF α‐syn nor CSF Aβ1‐42 was significantly associated with cognitive dysfunction and cortical atrophy. Also, without consideration for CSF tau, the effects of CSF α‐syn × CSF Aβ1‐42 on cognitive dysfunction and cortical atrophy were not significant. CSF α‐syn positively correlated with CSF Aβ1‐42 and CSF tau, while CSF tau negatively correlated with CSF Aβ1‐42. The positive correlation of CSF Aβ1‐42 and CSF α‐syn could reflect a synergistic relationship between α‐synuclein and Aβ accumulation. 31 , 32 The negative correlation between CSF tau and CSF Aβ1‐42 suggests the abundance of AD pathology in the ADNI cohort. 33 The positive correlation between CSF α‐syn and CSF tau, which was independent of the AD‐related CSF changes, has also been reported previously. 7 , 9 Simultaneously increased CSF tau and α‐syn levels could be CSF biomarkers for neuronal damage causing a release of neuronal tau and α‐syn into the CSF. Because various etiologies including AD could cause neuronal damage or elevation of CSF tau, our results suggest that the consideration of CSF tau is needed for CSF α‐syn to reflect α‐synucleinopathy. Supporting this point of view, a previous study also proposed that the mismatch of high CSF p‐tau and low CSF α‐syn indicates the presence of additional LB pathology in AD patients. 9
We found that lower CSF α‐syn, lower CSF Aβ1‐42, and CSF α‐syn × CSF Aβ1‐42 were independently associated with cognitive dysfunction in language, memory, visuospatial, and executive domains. This finding is in agreement with previous autopsy‐confirmed studies that showed faster cognitive decline in patients with both AD and LBD pathologies than those with pure AD pathology. 3 , 5 , 34 However, to our knowledge, this is the first study to report the significant synergistic interaction effects of AD and α‐synucleinopathy biomarkers on cognitive dysfunction. In our previous imaging–clinical correlation study, significant negative interaction was observed between AD and LBD on cognitive dysfunction. 35 In a previous autopsy–clinical correlation study, patients with both AD and LBD pathologies had a lower rate of progression than would be expected if each pathology independently and additively contributed to progression. 5 These differences could be due to the lack of quantitative biomarkers for LB pathology in imaging–clinical correlation studies, and much‐advanced AD pathology in autopsy–clinical correlation studies that could overwhelm the effect of co‐occurring LB pathology. Considering the significant synergistic interaction of α‐syn and Aβ on the acceleration of neuropathology and cognitive decline in a preclinical study, 36 our results suggest the possibility of CSF α‐syn as a quantitative biomarker for α‐synucleinopathy.
We also found that lower CSF α‐syn and CSF α‐syn × CSF Aβ1‐42 were associated with diffuse cortical atrophy. Lower CSF α‐syn was associated with decreased cortical thickness only in the Aβ‐positive subgroup. Contrastingly, lower CSF α‐syn was associated with increased cortical thickness in the Aβ‐negative subgroup, although statistical significance was not observed (Table S6). This interaction of α‐syn and Aβ1‐42 on cortical thickness could explain previous discrepancies in the distribution of cortical atrophy in LBD, 37 which could be related to participants’ Aβ burden. Notably, the effects of CSF α‐syn and CSF Aβ1‐42 on cortical thickness were not significant without CSF α‐syn × CSF Aβ1‐42 (Table S7 in supporting information). Therefore, the interaction of Aβ1‐42 and α‐syn on neurodegeneration must be considered.
Cortical thinning related to CSF α‐syn and CSF α‐syn × CSF Aβ1‐42 was pronounced in parieto‐occipital and temoporo‐occipital cortices. This posterior emphasis on cortical thinning is consistent with previous clinical–imaging correlation studies showing that occipital atrophy is associated with dementia 37 and visual hallucinations 38 in PD patients. Furthermore, occipital LB pathology is associated with faster dementia progression in PD patients, and non‐AD type clinical diagnosis in demented subjects with AD and LB pathology. 39 A previous study also showed a positive correlation between occipital Aβ deposition and striatal dopamine depletion in LBD patients. 40 Our results suggest a possibility that the spreading of α‐synucleinopathy and the interaction of Aβ and α‐syn could converge in the occipital cortex.
Higher PRS‐PD was associated with increased cortical thickness in the cingulate cortex. Our result may seem counterintuitive, as the concept of neurodegeneration manifesting as cortical atrophy is widely accepted. However, increased metabolism is observed in patients with DLB involving the cingulate cortex, 41 and in brain regions including the somatomotor cortex, posterior putamen, and cerebellar vermis, 42 which negatively correlates with dopaminergic loss. 43 Because the correlation was significant only in the dementia subgroup, we can infer that the relationship is not compensatory but pathologic. Although several putative mechanisms, including LB‐induced axonal swelling, 44 neuroinflammation, 45 and disinhibited synaptic synchrony 46 could explain the link between PRS‐PD and increased cortical thickness, future studies are warranted.
Higher CSF tau and its interaction with year showed detrimental effects on cognitive dysfunction and cortical atrophy. The topography of CSF tau‐related cortical thinning was similar to the cortical thinning patterns of AD. 47 Combined with a significant negative correlation between CSF Aβ1‐42 and CSF tau, our result supports the previous suggestion that CSF tau levels could reflect AD‐related neurodegeneration. 48 , 49 However, even in Aβ‐negative subjects, the interaction of CSF tau and year was associated with memory dysfunction (Table S4) and cortical thinning in the temporal and parietal cortices (Table S6). This suggests that the dichotomization of Aβ status by CSF Aβ1‐42 could be problematic, especially when the effects of CSF α‐syn are considered. There is a possibility that subjects with concomitant α‐synucleinopathy could have symptom onset with subthreshold Aβ deposition.
Our study has several strengths. First, we considered the effect of AD and α‐synucleinopathy simultaneously on cognition and cortical thickness. As mixed pathologies are common in dementia, our results could help us understand the independent and interaction effects of each pathology. Second, our data included protein‐level CSF biomarkers and genetic‐level PRS of AD and PD. Our study also has several limitations. First, we could not perform pathologic confirmation. Although we showed a significant correlation between lower CSF α‐syn and higher PRS‐PD, causal relationships among PRS‐PD, lower CSF α‐syn, and LBD are not guaranteed. Second, PRS‐PD was based on the summary statistics from two previous GWAS of PD, 11 , 12 rather than the currently updated study from which the full summary statistics are not publicly available. 14 Third, we did not consider the effects of vascular pathology. 5 Further quantification of vascular burden could enhance the understanding of mixed pathologies in dementia.
Overall, our findings suggest that simultaneous evaluation of CSF biomarkers for AD and CSF α‐syn was important to find their relationship with neurodegeneration, and CSF α‐syn could be a biomarker for α‐synucleinopathy related with the genetic risk for PD. Significant effect of CSF α‐syn and its interaction with CSF Aβ1‐42 on cognitive dysfunction and cortical atrophy emphasize the importance of α‐synucleinopathy with or without AD in pathologic aging.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors are grateful to all the participants who have taken part in this study. This research was conducted in collaboration with Biostatistics Collaboration Unit, Department of Biomedical Systems Informatics, Yonsei University College of Medicine. Data collection and sharing for this project was funded by ADNI (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research has provided funding to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org/). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. This research was supported by a National Research Foundation of Korea Grant funded by the Korean Government (NRF‐2019R1I1A1A01059454), new faculty research seed money grant of Yonsei University College of Medicine (2020‐32‐0034), and by grants from the Canadian Institute of Health Research awarded to Professor Alan C. Evans (201085 & 247003).
Lee Y‐g, Jeon S, Kang SW, et al. Interaction of CSF α‐synuclein and amyloid beta in cognition and cortical atrophy. Alzheimer's Dement. 2021;13:e12177. 10.1002/dad2.12177
Young‐gun Lee and Seun Jeon contributed equally to this study.
REFERENCES
- 1. Lippa CF, Fujiwara H, Mann DM, et al. Lewy bodies contain altered alpha‐synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 1998;153:1365‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha‐synuclein immunohistochemistry. Brain Pathol 2000;10:378‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olichney JM, Galasko D, Salmon DP, et al. Cognitive decline is faster in Lewy body variant than in Alzheimer's disease. Neurology 1998;51:351‐357. [DOI] [PubMed] [Google Scholar]
- 4. Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC. Clinical features of Alzheimer disease with and without lewy bodies. JAMA Neurol 2015;72:789‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brenowitz WD, Hubbard RA, Keene CD, et al. Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimers Dement 2017;13:654‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimer's Dement 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Steenoven I, Majbour NK, Vaikath NN, et al. α‐Synuclein species as potential cerebrospinal fluid biomarkers for dementia with lewy bodies. Mov Disord 2018;33:1724‐1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Slaets S, Vanmechelen E, Le Bastard N, et al. Increased CSF α‐synuclein levels in Alzheimer's disease: correlation with tau levels. Alzheimers Dement 2014;10:S290‐S298. [DOI] [PubMed] [Google Scholar]
- 9. Toledo JB, Korff A, Shaw LM, Trojanowski JQ, Zhang J. CSF α‐synuclein improves diagnostic and prognostic performance of CSF tau and Aβ in Alzheimer's disease. Acta Neuropathol 2013;126:683‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang H, Stewart T, Toledo JB, et al. A longitudinal study of total and phosphorylated α‐synuclein with other biomarkers in cerebrospinal fluid of Alzheimer's disease and mild cognitive impairment. J Alzheimers Dis 2018;61:1541‐1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nalls MA, Pankratz N, Lill CM, et al. Large‐scale meta‐analysis of genome‐wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 2014;46:989‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang D, Nalls MA, Hallgrimsdottir IB, et al. A meta‐analysis of genome‐wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet 2017;49:1511‐1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol 2019;18:1091‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age‐associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med 2017;14:e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tan CH, Bonham LW, Fan CC, et al. Polygenic hazard score, amyloid deposition and Alzheimer's neurodegeneration. Brain 2019;142:460‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pihlstrøm L, Morset KR, Grimstad E, Vitelli V, Toft M. A cumulative genetic risk score predicts progression in Parkinson's disease. Mov Disord 2016;31:487‐490. [DOI] [PubMed] [Google Scholar]
- 18. Paul KC, Schulz J, Bronstein JM, Lill CM, Ritz BR. Association of polygenic risk score with cognitive decline and motor progression in parkinson disease. JAMA Neurol 2018;75:360‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee MJ, Pak K, Kim JH, et al. Effect of polygenic load on striatal dopaminergic deterioration in Parkinson disease. Neurology 2019;93:e665‐e674. [DOI] [PubMed] [Google Scholar]
- 20. Guerreiro R, Escott‐Price V, Darwent L, et al. Genome‐wide analysis of genetic correlation in dementia with Lewy bodies, Parkinson's and Alzheimer's diseases. Neurobiol Aging 2016;38:214.e7‐.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bellou E, Stevenson‐Hoare J, Escott‐Price V. Polygenic risk and pleiotropy in neurodegenerative diseases. Neurobiol Dis 2020;142:104953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delaneau O, Zagury JF, Marchini J. Improved whole‐chromosome phasing for disease and population genetic studies. Nat Methods 2013;10:5‐6. [DOI] [PubMed] [Google Scholar]
- 25. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Collins DL, Neelin P, Peters TM, Evans AC. Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J Comput Assist Tomogr 1994;18:192‐205. [PubMed] [Google Scholar]
- 27. Kim JS, Singh V, Lee JK, et al. Automated 3‐D extraction and evaluation of the inner and outer cortical surfaces using a Laplacian map and partial volume effect classification. Neuroimage 2005;27:210‐221. [DOI] [PubMed] [Google Scholar]
- 28. Lyttelton O, Boucher M, Robbins S, Evans A. An unbiased iterative group registration template for cortical surface analysis. Neuroimage 2007;34:1535‐1544. [DOI] [PubMed] [Google Scholar]
- 29. Worsley KJ, Taylor JE, Tomaiuolo F, Lerch J. Unified univariate and multivariate random field theory. Neuroimage 2004;23(Suppl 1):S189‐S195. [DOI] [PubMed] [Google Scholar]
- 30. Ibanez L, Dube U, Saef B, et al. Parkinson disease polygenic risk score is associated with Parkinson disease status and age at onset but not with alpha‐synuclein cerebrospinal fluid levels. BMC Neurol 2017;17:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lashley T, Holton JL, Gray E, et al. Cortical alpha‐synuclein load is associated with amyloid‐beta plaque burden in a subset of Parkinson's disease patients. Acta Neuropathol 2008;115:417‐425. [DOI] [PubMed] [Google Scholar]
- 32. Colom‐Cadena M, Gelpi E, Charif S, et al. Confluence of alpha‐synuclein, tau, and beta‐amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 2013;72:1203‐1212. [DOI] [PubMed] [Google Scholar]
- 33. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kraybill ML, Larson EB, Tsuang DW, et al. Cognitive differences in dementia patients with autopsy‐verified AD, Lewy body pathology, or both. Neurology 2005;64:2069‐2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kang SW, Jeon S, Yoo HS, et al. Effects of Lewy body disease and Alzheimer disease on brain atrophy and cognitive dysfunction. Neurology 2019;92:e2015‐e2026. [DOI] [PubMed] [Google Scholar]
- 36. Clinton LK, Blurton‐Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha‐synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 2010;30:7281‐7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burton EJ, McKeith IG, Burn DJ, Williams ED, O'Brien JT. Cerebral atrophy in Parkinson's disease with and without dementia: a comparison with Alzheimer's disease, dementia with Lewy bodies and controls. Brain 2004;127:791‐800. [DOI] [PubMed] [Google Scholar]
- 38. Goldman JG, Stebbins GT, Dinh V, et al. Visuoperceptive region atrophy independent of cognitive status in patients with Parkinson's disease with hallucinations. Brain 2014;137:849‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Toledo JB, Gopal P, Raible K, et al. Pathological alpha‐synuclein distribution in subjects with coincident Alzheimer's and Lewy body pathology. Acta Neuropathol 2016;131:393‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoo HS, Lee S, Chung SJ, et al. Dopaminergic depletion, β‐amyloid burden, and cognition in Lewy body disease. Ann Neurol 2020;87:739‐750. [DOI] [PubMed] [Google Scholar]
- 41. Graff‐Radford J, Murray ME, Lowe VJ, et al. Dementia with Lewy bodies: basis of cingulate island sign. Neurology 2014;83:801‐809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ye BS, Lee S, Yoo H, et al. Distinguishing between dementia with Lewy bodies and Alzheimer's disease using metabolic patterns. Neurobiol Aging 2020;87:11‐17. [DOI] [PubMed] [Google Scholar]
- 43. Huber M, Beyer L, Prix C, et al. Metabolic correlates of dopaminergic loss in dementia with lewy bodies. Mov Disord 2020;35:595‐605. [DOI] [PubMed] [Google Scholar]
- 44. Mezey E, Dehejia AM, Harta G, et al. Alpha synuclein is present in Lewy bodies in sporadic Parkinson's disease. Mol Psychiatry 1998;3:493‐499. [DOI] [PubMed] [Google Scholar]
- 45. Lin Y, Wen L. Inflammatory response following diffuse axonal injury. Int J Med Sci 2013;10:515‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burkhardt JM, Jin X, Costa RM. Dissociable effects of dopamine on neuronal firing rate and synchrony in the dorsal striatum. Front Integr Neurosci 2009;3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid‐positive individuals. Cereb Cortex 2009;19:497‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tosun D, Schuff N, Shaw LM, Trojanowski JQ, Weiner MW. Relationship between CSF biomarkers of Alzheimer's disease and rates of regional cortical thinning in ADNI data. J Alzheimers Dis 2011;26(Suppl 3):77‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mattsson N, Schöll M, Strandberg O, et al. (18)F‐AV‐1451 and CSF T‐tau and P‐tau as biomarkers in Alzheimer's disease. EMBO Mol Med 2017;9:1212‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information