

SUMMARY
The importance of pancreatic versus intestinal-derived GLP-1 for glucose homeostasis is controversial. We detected active GLP-1 in the mouse and human pancreas, albeit at extremely low levels relative to glucagon. Accordingly, to elucidate the metabolic importance of intestinal proglucagon-derived peptides (PGDPs), we generated mice with reduction of Gcg expression within the distal (GcgDistalGut−/−) or entire (GcgGut−/−) gut. Substantial reduction of gut Gcg expression markedly reduced circulating levels of GLP-1, and impaired glucose homeostasis, associated with increased levels of GIP, and accelerated gastric emptying. GcgDistalGut−/− mice similarly exhibited lower circulating GLP-1 and impaired oral glucose tolerance. Nevertheless, plasma levels of insulin remained normal following glucose administration in the absence of gut-derived GLP-1. Collectively, our findings identify the essential importance of gut-derived PGDPs for maintaining levels of circulating GLP-1, control of gastric emptying, and glucose homeostasis.
INTRODUCTION
Glucagon, a 29-amino-acid peptide, was originally identified as a hormone isolated from the pancreas with hyperglycemic activity. The molecular cloning of cDNAs and genes encoding proglucagon expanded the complexity of the proglucagon-derived peptides (PGDPs) (Drucker, 2006). Mammalian species transcribe a single proglucagon mRNA transcript yet generate a different profile of PGDPs in brain, pancreas, and intestine through tissue-specific proteolytic cleavage, mediated by members of the prohormone convertase enzyme family (Sandoval and D’Alessio, 2015). Within the pancreas, glucagon is the major liberated PGDP, and the glucagon-like peptides are contained within a larger, unprocessed major proglucagon fragment (Patzelt and Schiltz, 1984). In contrast, enteroendocrine L cells process proglucagon to liberate glicentin, oxyntomodulin, and two glucagon-like peptides: glucagon-like peptide-1 (GLP-1) and glucagon-like peptide-2 (GLP-2) (Mojsov et al., 1986).
Within the gut, two bioactive GLP-1 moieties, GLP-1(7–37), and GLP-1(7–36) amide are derived from proglucagon (Orskov et al., 1994). Both peptides circulate at low basal levels in the fasting state, are secreted rapidly following meal ingestion, and are equipotent stimulators of insulin secretion (Vahl et al., 2003). Indeed, pharmacological blockade of GLP-1 action in animals and humans, or genetic interruption of GLP-1 receptor signaling in mice, impairs the insulinotropic action of GLP-1, establishing the physiological importance of GLP-1 as an incretin hormone (Drucker, 2007).
Despite the preponderance of evidence demonstrating the synthesis and secretion of bioactive GLP-1 in the small and large intestine (Kreymann et al., 1988; Mojsov et al., 1986; Orskov et al., 1994), studies have also implicated pancreatic islet α cells as a source of bioactive GLP-1. Rodent and human islet cells cultured ex vivo secrete GLP-1 with insulinotropic activity (Ellingsgaard et al., 2011; Hansen et al., 2011; Heller and Aponte, 1995; Marchetti et al., 2012; Timper et al., 2016). Moreover, experimental pancreatic injury, including surgical manipulation, chemical damage, aging, or the prolonged nutritional stress of energy-dense diets, is associated with increased pancreatic production of GLP-1 (Kilimnik et al., 2010; Nie et al., 2000; Vasu et al., 2014).
We recently used mouse genetics and the GLP-1R antagonist exendin(9–39) to examine the consequences of selective re-induction of endogenous Gcg gene expression in the intestine versus the pancreas of Gcg−/− mice (Chambers et al., 2017). Reactivation of intestinal Gcg expression using the Villin-Cre system (GcgRAΔVilCre) restored intestinal Gcg and GLP-1 production, in the setting of complete loss of pancreatic Gcg expression (Chambers et al., 2017). Surprisingly, the GLP-1R antagonist exendin(9–39) failed to impair glycemic excursions to both oral and intraperitoneal (i.p.) glucose loads in GcgRAΔVilCre mice. In contrast, selective reactivation of pancreatic Gcg expression in GcgRAΔPDX1Cre mice and concomitant administration of exendin(9–39) led to the impairment of glycemic excursions in both oral and i.p. glucose tolerance tests. Collectively, these observations elevated the putative importance of the pancreas as a key source for GLP-1 action (Habener and Stanojevic, 2017).
Interpretation of these findings requires careful consideration of the experimental context, namely reactivation of Gcg expression in Gcg−/− mice known to exhibit α cell hyperplasia (Hayashi et al., 2009). These conditions favor the generation of pancreatic islet GLP-1 production in dedifferentiated α cells following the reintroduction of pancreatic Gcg expression (Hayashi et al., 2009). Moreover, Gcg−/− mice may exhibit compensatory induction of islet β cell GIP expression (Fukami et al., 2013), further complicating interpretation of incretin action. Critically, the importance of pancreatic versus intestinal GLP-1 activity was inferred indirectly via the glycemic response to exendin(9–39) (Chambers et al., 2017). Since the publication of these provocative findings, several studies have demonstrated that exendin(9–39) also blocks the insulinotropic actions of glucagon at the islet GLP-1 receptor (Capozzi et al., 2019a; Svendsen et al., 2018; Zhu et al., 2019), complicating precise mechanistic attribution of glycemic responses following use of exendin(9–39).
Accordingly, we have now re-examined the metabolic importance of endogenous gut-derived PGDPs in the absence of exendin(9–39) using two new lines of mice with the elimination of Gcg expression in the distal gut, or throughout the small and large bowel. Our findings re-establish gut-derived PGDPs as important metabolically active peptides, highlighting the potential for both enteroendocrine cells and pancreatic islet cells to serve as sources of PGDPs with glucoregulatory action.
RESULTS
Assessment of GLP-1 and Glucagon Content in Mouse and Human Pancreas
Although GLP-1 may be detected in the pancreas in the context of pancreatic injury and inflammation (Drucker, 2013; Habener and Stanojevic, 2017), active GLP-1 was not detected in mouse pancreatic perfusate (Svendsen et al., 2018) and no extractable active GLP-1 was detectable in the normal mouse pancreas (Galsgaard et al., 2018). Hence, we re-examined levels of active GLP-1 in mouse and human pancreas. Active GLP-1 content in the pancreas of wild-type (WT) Gcg+/+ mice, maintained on normal chow (NC) or fed a 60% high-fat diet (HFD), was extremely low but detectable (Figure 1A), several orders of magnitude lower than active GLP-1 content in the colon (Figure 1A). Moreover, the levels of active GLP-1 (liberated from a common proglucagon precursor; Figure S1) in the mouse pancreas were 20-to 30-fold lower than that of glucagon and were not different after 7 weeks of HFD feeding (Figures 1B and 1C). Similar findings were observed in Cre control and mice with Gcg reactivation in the pancreas (GcgRAΔPDX1Cre) (Chambers et al., 2017) (Figure 1D), where glucagon content greatly exceeded that of active GLP-1 in pancreas-tissue extracts (Figure 1E). In contrast, neither glucagon nor active GLP-1 was detectable in the pancreas of Gcg-null or intestinal Gcg-reactivated (GcgRAΔVilCre) mice despite abundant levels of insulin (Figure 1D).
Figure 1. Assessment of Peptide Content in Mouse and Human Pancreas.
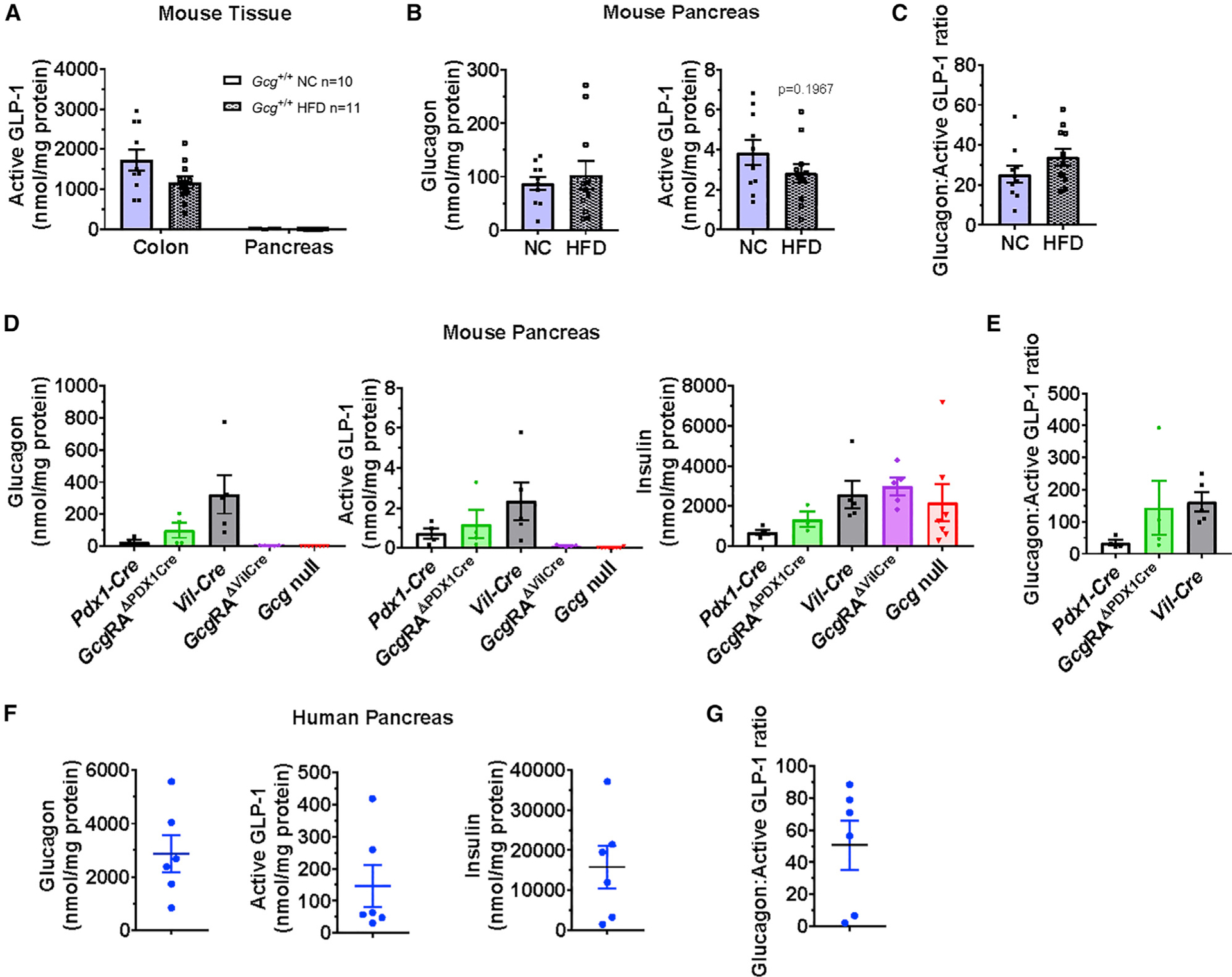
(A and B) Comparison of active GLP-1 tissue levels in colon versus pancreas (A), or glucagon (left panel) and active GLP-1 tissue levels (right panel) in pancreas
(B), normalized to protein content, in tissue extracts from 18-to 21-week-old WT (Gcg+/+) male mice fed normal chow (NC) or a 60% high-fat diet (HFD) for 7 weeks.
(C) Abundance of glucagon relative to active GLP-1 (glucagon-active GLP-1 ratio) in pancreas of NC or HFD-fed mice (n = 10–11).
(D and E) Tissue levels of (D) glucagon, active GLP-1, and insulin in extracts from pancreas, normalized to protein content, and (E) relative glucagon to active GLP-1 tissue levels in pancreas of 28-week-old male Cre controls, Gcg-null, and pancreas-(GcgRAΔPDX1Cre) or intestinal-reactivated (GcgRAΔVilCre) mice (n = 4–5/group). For (E), glucagon:active GLP-1 ratios are not reported for GcgRAΔVilCre or Gcg-null mice, as glucagon and/or active GLP-1 levels were below detectable limits of the assays.
(F and G) Tissue levels of (F) glucagon, active GLP-1, and insulin in extracts from normal human pancreas, normalized to protein content, and (G) glucagon-active GLP-1 ratio in human pancreas samples (n = 6).
To assess the potential for species-specific differences in pancreatic GLP-1 production, we examined the human pancreas. Active GLP-1 was also detectable in normal human pancreas extracts at levels higher than in the mouse pancreas (Figure 1F, middle panel). Levels were somewhat variable, as were both glucagon and insulin content (Figure 1F, left and right panels, respectively), most likely reflecting different numbers of islets in each sample and/or age of the respective donor (Table S1). Nevertheless, glucagon content greatly exceeded that of active GLP-1 in the human pancreas (Figures 1F and 1G). Taken together, active GLP-1 is very low, but detectable, in tissue extracts from both mouse and human pancreas, albeit at much lower levels relative to glucagon.
Generation and Characterization of GcgGut–/– and GcgDistalGut–/– Mice
Our previous studies addressed the glucoregulatory activity of GLP-1 in mice with reactivation of intestinal or pancreatic proglucagon (Gcg) expression in Gcg−/− mice (Chambers et al., 2017). Interpretation of the biological importance of GLP-1 in these studies relied on exendin(9–39), a GLP-1 receptor antagonist subsequently shown to block the activity of both GLP-1 and glucagon at the pancreatic GLP-1 receptor (Capozzi et al., 2019a; Svendsen et al., 2018; Zhu et al., 2019). Accordingly, to elucidate the importance of intestinal PGDP action in the absence of exendin(9–39), we generated two new lines of mice enabling the selective loss of Gcg expression in the intestine. Gcgflox/flox mice were crossed with Vil-Cre mice to generate GcgGut−/− mice (Figure 2A). GcgGut−/− mice exhibited a >95% reduction in Gcg mRNA levels throughout the entire small intestine and colon, whereas Gcg mRNA transcripts in the brainstem were normal (Figure 2A). Islet histology was similar (Figure S2A) and pancreatic glucagon mRNA transcripts levels were higher (Figure 2A), yet pancreatic levels of active GLP-1 were very low and not different in GcgGut−/− mice (Figure 2B). Moreover, plasma levels of glucagon were similar in GcgGut+/+ versus GcgGut−/− mice (Figure S2B). Consistent with the marked reduction of Gcg mRNA transcripts, active and total GLP-1 content was markedly reduced in all regions of the small intestine and colon from GcgGut−/− mice (Figures 2B and S2C), whereas intestinal GIP levels were not different (Figure 2C).
Figure 2. Characterization of GcgGut–/– and GcgDistalGut–/– Mice.
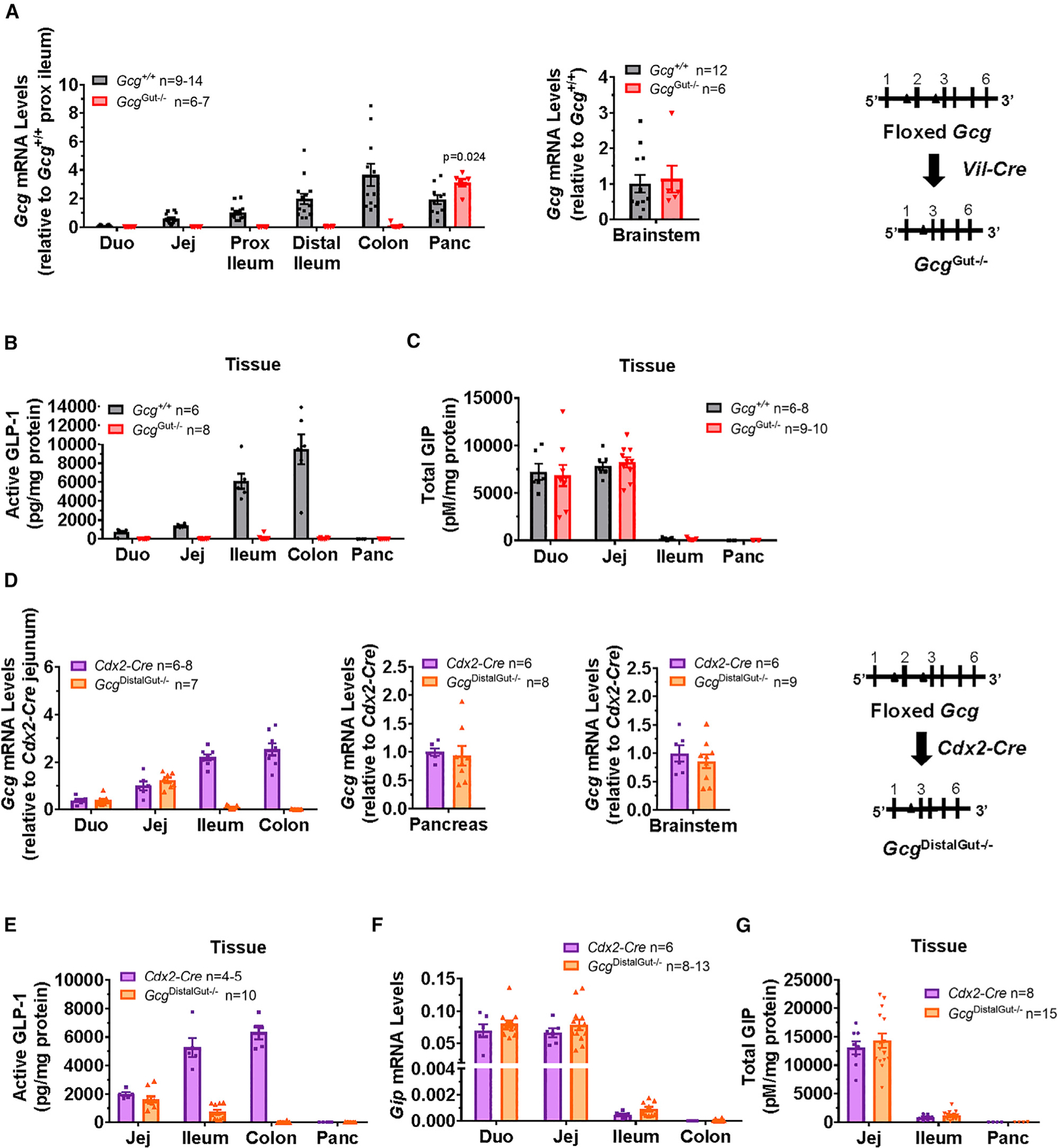
(A) Proglucagon (Gcg) mRNA abundance normalized to levels of mRNA for ribosomal protein L32 (Rpl32) in different regions of the small intestine, colon, and pancreas (left panel) or brainstem (right panel) of 16- to 18-week-old female mice by qPCR using a primer probe set against exons 1–2 of the Gcg gene (n = 6–14/group). Gene expression was expressed relative to values for Gcg mRNA transcripts in proximal ileum (prox ileum; left panel), or brainstem (right panel) of control Gcg+/+ mice.
(B and C) Active GLP-1 (B) and total GIP levels (C), normalized to total protein content, in whole-tissue extracts from different regions of the intestine and pancreas of 16- to 18-week-old female mice (n = 6–8/group for GLP-1, n = 6–10 for GIP). Duo, duodenum; Jej, jejunum; Prox ileum, proximal ileum; Panc, pancreas. Gcg+/+ mice represent combined data from WT, Gcgflox/flox, and Vil-Cre control mice.
(D) Gcg mRNA abundance normalized to cyclophilin (Ppia) in different regions of the small intestine and colon (left panel), pancreas (middle panel), or brainstem (right panel) of 20- to 23-week-old male mice by qPCR (n = 6–9/group). Gene expression was expressed relative to Gcg mRNA levels in control (Cdx2-Cre) mice.
(E) Active GLP-1 levels, normalized to total protein content, in whole-tissue extracts from different regions of the intestine and pancreas of 20- to 23-week-old male mice (n = 4–10/group).
(F) Gip mRNA abundance normalized to cyclophillin (Ppia) from different regions of the small intestine and colon of 20- to 23-week-old male mice (n = 6–13/group).
(G) Total GIP levels, normalized to total protein content, in whole-tissue extracts from the small intestine of 20- to 23-week-old male mice (n = 8–15/group). See also Figures S2 and S3.
Despite detection of some active GLP-1 in the pancreas of both GcgGut−/− and Gcg+/+ mice, levels were markedly lower than in the small intestine or colon of Gcg+/+ mice (pancreas, ~6.7 pg/mg protein versus jejunum, 1,389 pg/mg protein; colon, 9,470 pg/mg protein) (Figure 2B). Similarly, relative levels of active GLP-1 remained lower in the pancreas versus the small intestine and colon of GcgGut−/− mice (pancreas, ~9.4 pg/mg protein versus jejunum, 35.5 pg/mg protein; colon, 57.7 pg/mg protein) (Figure 2B), whereas active GLP-1 levels in the pancreas of Gcg+/+ versus GcgGut−/− mice were not different (6.7 ± 1.8 pg/mg protein versus 9.4 ± 3.2 pg/mg protein, p = 0.09). Although increased pancreatic GLP-1 production and activity have been demonstrated in the presence of metabolic stress and experimental diabetes (Kilimnik et al., 2010; Traub et al., 2017), active GLP-1 levels from pancreatic extracts of Vil-Cre and GcgGut−/− mice remained exceptionally low and did not increase after 7 weeks of HFD feeding (Figure S2D). In contrast to the low pancreatic levels of active GLP-1, assessment of total immunoreactive GLP-1 in the pancreas using an assay that recognizes all molecular forms of GLP-1 (Figure S1) revealed much higher levels (Figure S2C), consistent with the immunodetection of the GLP-1 sequence contained within the major proglucagon fragment (MPGF) (Patzelt and Schiltz, 1984).
We next established a second mouse model of intestinal PGDP deficiency within the distal, but not proximal gut, by crossing Gcgflox/flox mice with Cdx2-Cre mice to generate GcgDistalGut−/− mice. Gcg mRNA transcripts were markedly reduced in the ileum and colon of GcgDistalGut−/− mice, without perturbation of Gcg mRNA levels in the proximal small intestine, pancreas, or brainstem (Figure 2D). Both active and total GLP-1 levels were markedly reduced in the ileum and colon of GcgDistalGut−/− mice; however, GLP-1 content was not reduced in the jejunum (Figures 2E and S3A). Similarly, Gip mRNA levels and tissue GIP content were not different in GcgDistalGut−/− mice (Figures 2F and 2G).
GcgGut–/– Mice Exhibit Impaired Oral but Normal i.p. Glucose Tolerance
Despite the preservation of pancreatic Gcg expression in GcgGut−/− mice (Figure 2A), fasting plasma levels of total immunoreactive GLP-1 were lower in GcgGut−/− mice (Figure 3A). Moreover, fasting plasma active GLP-1 levels were markedly reduced in GcgGut−/− mice (Figure 3B), highlighting the predominant contribution of the intestine to the circulating pool of bioactive GLP-1. Conversely, although intestinal GIP levels were not different in GcgGut−/− versus Gcg+/+ mice (Figure 2C), fasting plasma levels of GIP were higher in GcgGut−/− mice (Figure S2B) without differences in fasting levels of insulin and glucagon (Figure S2B). Remarkably, despite the importance of basal GLP-1R signaling for control of food intake and body weight (Campbell and Drucker, 2013; Patterson et al., 2011; Sandoval and D’Alessio, 2015), both incremental and cumulative food intake and body weight on regular chow and HFD were similar in GcgGut−/− versus control mice (Figures S2E and S2F). Thus, marked reductions in GLP-1 of intestinal origin have no meaningful impact on food intake and body weight.
Figure 3. Loss of Gut Gcg Expression Produces Glucose Intolerance Following Oral, but not i.p., Glucose Administration.
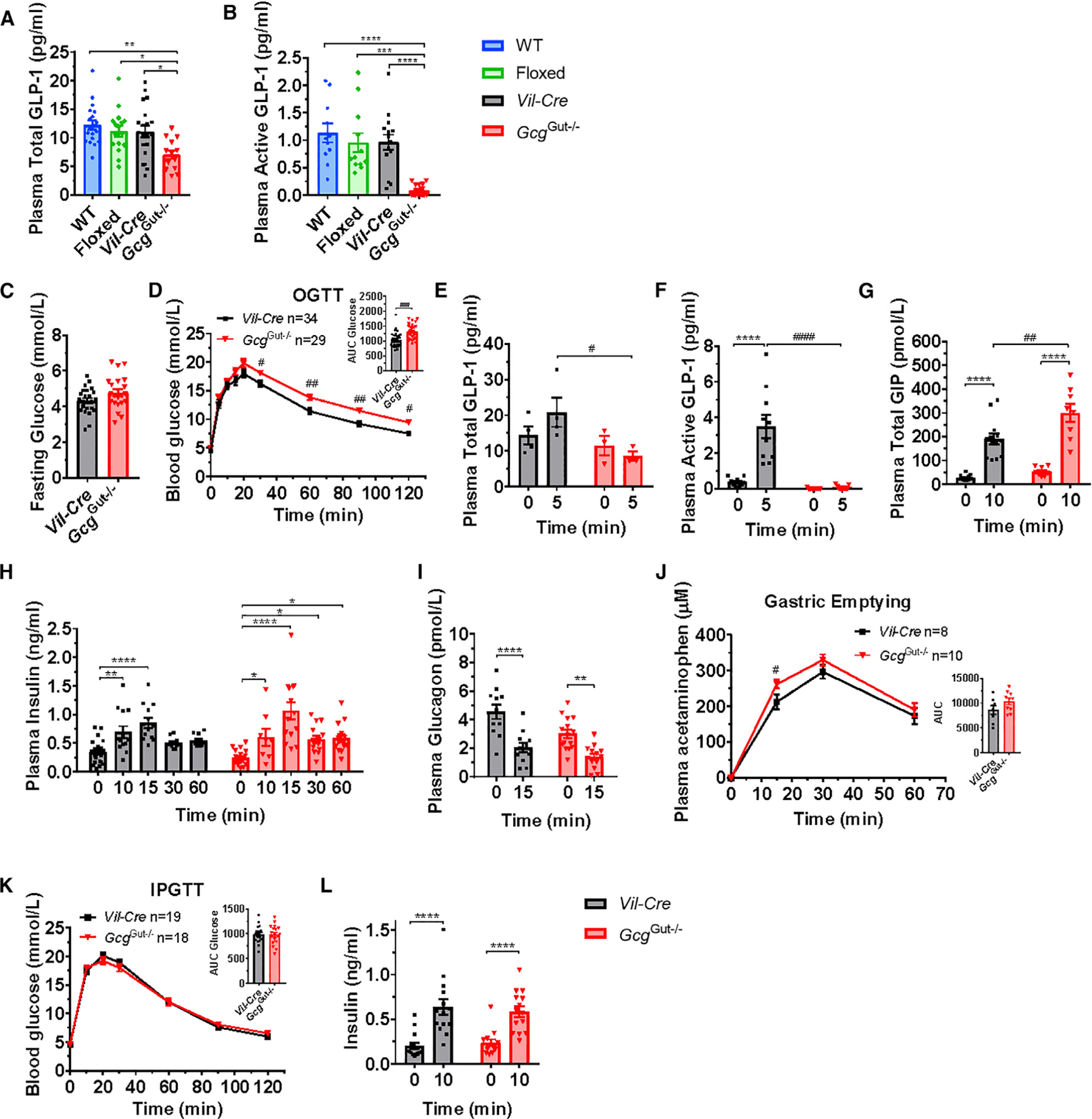
(A and B) Fasting plasma levels of (A) total GLP-1 or (B) active GLP-1 in overnight fasted 10- to 17-week-old male GcgGut−/− and control mice (n = 11–19/group).
(C and D) Fasting blood glucose levels (C) and blood glucose levels (D) 0–120 min after oral glucose (OGTT, 1.5g/kg) and area under the curve (AUC, inset) for glucose excursions in overnight fasted 12- to 17-week-old male GcgGut−/− and Vil-Cre control mice (n = 29–34/group).
(E–I) Plasma total GLP-1 (E), active GLP-1 (F), total GIP (G), insulin (H), and glucagon (I) measured before and 5–60 min after oral glucose challenge, as indicated (n = 4–15/group).
(J) Plasma acetaminophen levels, as a measurement of gastric emptying, 0–60 min after co-administration of oral acetaminophen and glucose (n = 8–10/group).
(K) Blood glucose levels before and after i.p. administration of glucose (IPGTT, 1.5 g/kg) and AUC (inset) for i.p. glucose excursions in overnight fasted 14- to 19-week-old male GcgGut−/− and Vil-Cre control mice (n = 19–18/group).
(L) Plasma insulin measured before and 10 min after an intraperitoneal glucose challenge (n = 14/group).
Data are presented as the mean ± SEM. (A and B) *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, control versus GcgGut−/− mice. (D–L) *p < 0.05, **p < 0.01, ****p < 0.0001, 0 versus 5–60 min. #p < 0.05, ##p<0.01, ###p < 0.001, ####p < 0.0001, GcgGut−/− versus Vil-Cre mice. See also Figures S2 and S4.
Following an oral glucose challenge, GcgGut−/− mice exhibited higher glycemic excursions compared to Vil-Cre controls without differences in fasting glucose (Figures 3C and 3D). Consistent with the marked reduction of Gcg mRNA transcripts in the intestine, both total and active plasma GLP-1 levels did not increase following enteral glucose loading in GcgGut−/− mice but increased in GcgGut+/+ mice (Figures 3E and 3F). In contrast, plasma GIP levels were higher following glucose challenge in GcgGut−/− mice than Vil-Cre controls (Figure 3G). Surprisingly, glucose-stimulated insulin levels and reductions in plasma glucagon were similar between GcgGut−/− and Vil-Cre control mice (Figures 3H and 3I). However, gastric emptying was accelerated in GcgGut−/− mice (Figure 3J). Accordingly, we bypassed the gut and administered i.p. glucose. GcgGut−/− mice exhibited glycemic excursions comparable to controls following i.p. glucose challenge (Figure 3K), without differences in plasma insulin levels (Figure 3L).
GcgDistalGut–/– Mice Exhibit Impaired Oral and i.p. Glucose Tolerance
Although levels of Gcg mRNA transcripts and tissue content of PGDPs are higher in the distal gut (Roberts et al., 2019; van der Wielen et al., 2016), the relative importance and contribution of the proximal versus distal gut to GLP-1 secretion and action remains uncertain and controversial (Svendsen et al., 2015). In contrast to findings in GcgGut−/− mice, fasting plasma total GLP-1 levels were comparable between GcgDistalGut−/− mice and controls (Figure 4A), suggesting that proximal gut Gcg expression contributes to circulating immunoreactive GLP-1. Strikingly, plasma levels of active GLP-1 were markedly reduced in fasted GcgDistalGut−/− mice (Figure 4B), revealing the predominant contribution of distal gut Gcg expression to circulating bioactive GLP-1. In contrast, fasting plasma GIP, insulin, and glucagon were similar between GcgDistalGut−/− mice and controls (Figure S3B), and food intake and body weight were not different in GcgDistalGut−/− mice on regular chow or after several weeks of HFD feeding (Figures S3C and S3D).
Figure 4. GcgDistalGut–/– Mice Exhibit Impaired Glucose Tolerance.
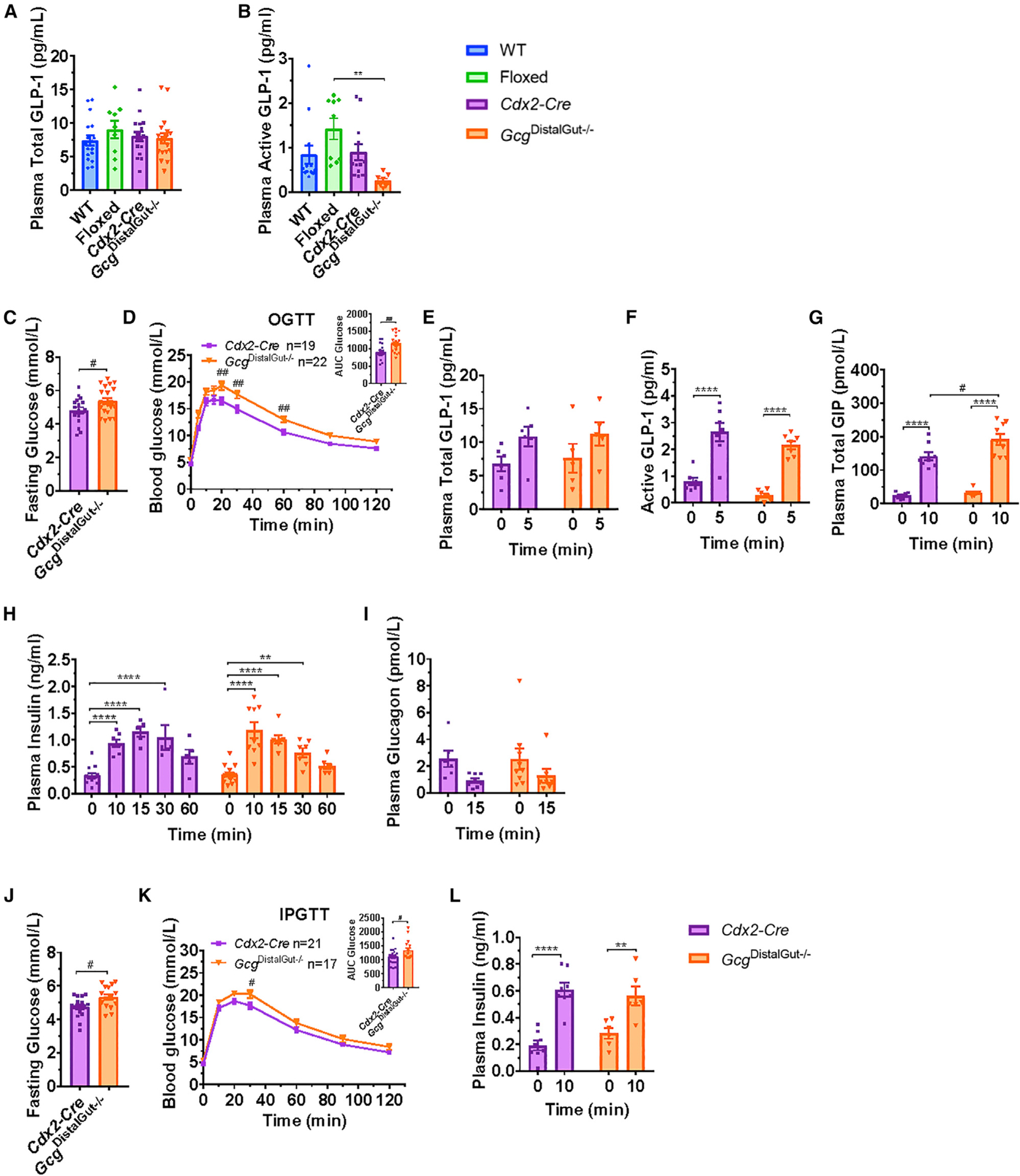
(A and B) Fasting plasma levels of (A) total GLP-1 or (B) active GLP-1 in overnight fasted 10- to 14-week-old male GcgDistalGut−/− and control mice (n = 7–17/group).
(C and D) Fasting glucose (C) blood glucose levels (D) after oral glucose (OGTT, 1.5 g/kg) and area under the curve (AUC, inset) for glucose excursions in overnight fasted 12- to 15-week-old male GcgDistalGut−/− and Cdx2-Cre control mice (n = 19–22/group).
(E–I) Plasma total GLP-1 (E), active GLP-1 (F), total GIP (G), insulin (H), and glucagon (I) measured before and 5–60 min after oral glucose challenge as indicated (n = 5–9/group).
(J and K) Fasting glucose levels (J) and blood glucose levels (K) following i.p. administration of glucose (IPGTT, 1.5 g/kg) and AUC (inset) for i.p. glucose excursions in overnight fasted 14- to 17-week-old male GcgDistalGut−/− and Cdx2-Cre control mice.
(L) Plasma insulin before and 10 min after an i.p. glucose challenge.
For (J) and (K), n = 17–21/group and for (L), n = 6–8/group. Data are presented as the mean ± SEM. (B) **p < 0.01, control versus GcgDistalGut−/− mice. (C–L)
**p < 0.01, ****p < 0.0001, 0 versus 5–60 min. #p < 0.05, ##p < 0.01, GcgDistalGut−/− versus Cdx2-Cre mice. See also Figures S3 and S4.
Consistent with the reductions in plasma active GLP-1, GcgDistalGut−/− mice exhibited oral glucose intolerance, associated with slightly higher fasting glucose levels (Figures 4C and 4D). Surprisingly, both total and active GLP-1 plasma levels increased equivalently following oral glucose challenge in GcgDistalGut−/− versus Cdx2-Cre control mice, despite the elimination of distal gut Gcg expression (Figures 4E and 4F), unmasking the importance of proximal gut Gcg expression for the rapid rise in plasma GLP-1 levels following oral glucose. Consistent with findings in GcgGut−/− mice, GIP levels were higher after ral glucose challenge in GcgDistalGut−/− versus Cdx2-Cre mice (Figure 4G). Despite elevated glucose excursions, glucose-stimulated insulin levels and reductions in plasma glucagon were similar between GcgDistalGut−/− and Cdx2-Cre control mice after oral glucose challenge (Figures 4H and 4I). GcgDistalGut−/− mice also exhibited modestly higher glucose excursions following an intraperitoneal glucose load; however, this was partially driven by higher fasting glucose levels (Figures 4J and 4K), and glucose-stimulated insulin levels were not different in GcgDistalGut−/− versus Cdx2-Cre control mice (Figure 4L)
DISCUSSION
Initial concepts of GLP-1 action as a gut-derived incretin hormone stemmed from its isolation from the small and large bowel, whereas pancreatic extracts contained little bioactive intact GLP-1 (Orskov et al., 1987), and studies with perfused pancreas preparations did not detect GLP-1 in pancreatic effluents (Orskov et al., 1986; Svendsen et al., 2018). Moreover, circulating GLP-1 levels increased briskly following oral glucose or meal ingestion, consistent with a gut-derived source for the peptide. Nevertheless, some (Mojsov et al., 1990), but not all (Galsgaard et al., 2018; Larraufie et al., 2019), studies report small amounts of GLP-1(7–36)amide in the normal rat or mouse pancreas and both pancreatic GLP-1 and Pcsk1 expression were increased in the rat pancreas following induction of experimental diabetes with streptozotocin (Nie et al., 2000). A considerable body of subsequent evidence is now consistent with the production of islet GLP-1 in the context of experimental pancreatic inflammation, diabetes, and aging (Chen et al., 2018; Ellingsgaard et al., 2011; Habener and Stanojevic, 2017). Our current findings further support the concept of pancreatic GLP-1 production by demonstrating low but detectable levels of active GLP-1 in the mouse and human pancreas.
The studies described herein stemmed from our provocative findings revealing the importance of pancreatic PGDPs and GLP-1 (Chambers et al., 2017), motivated in part by new data confirming exendin(9–39) as a dual glucagon and GLP-1 antagonist at the GLP-1R (Capozzi et al., 2019a; Svendsen et al., 2018; Zhu et al., 2019). Using a different experimental design, Chambers et al. (2017) assessed the consequences of reactivating pancreatic or intestinal Gcg expression in Gcg−/− mice using the glycemic response to exendin(9–39) to infer the relative importance of pancreatic versus intestinal GLP-1 production. Nevertheless, multiple studies (Capozzi et al., 2019a, 2019b; Svendsen et al., 2018; Zhu et al., 2019) have recently demonstrated that (1) glucagon acts as a potential insulinotropic factor via the islet GLP-1 receptor and (2) the insulinotropic actions of glucagon are blocked by exendin(9–39).
Notably, Gcg−/− mice exhibit resistance to the development of glucose intolerance (Fukami et al., 2013), reflecting the absence of pancreatic glucagon, which in turn lowers hepatic glucose production. Moreover, Gcg−/− mice exhibit substantial pancreatic enlargement and both islet and α cell hyperplasia, further magnifying the potential contribution of islet-derived GLP-1 or glucagon produced following the re-establishment of Gcg expression within hyperplastic islets (Chambers et al., 2017; Hayashi et al., 2009). Furthermore, some lines of Gcg−/− mice exhibit upregulation of islet GIP expression (Fukami et al., 2013), additionally complicating interpretation of incretin action, insulin levels, and islet function in vivo.
Given these limitations, we re-examined the importance of intestinal PGDPs and GLP-1 for glucose homeostasis without the concomitant use of exendin(9–39). We generated two new mouse models using Cre driver lines targeting the epithelium, including enteroendocrine cells, in the distal or the entire gut. These two new mouse lines enabled physiological analysis of the metabolic consequences of distal (GcgDistalGut−/−) or virtually complete (GcgGut−/− mice) elimination of intestinal Gcg expression, while preserving normal pancreatic islet structure, function, and PGDP expression. Our current studies reveal the importance of gut Gcg expression for normal circulating levels of active GLP-1 and glucoregulation. First, plasma active GLP-1 levels were clearly reduced in both GcgGut−/− and in GcgDistalGut−/− mice. Second, plasma GLP-1 levels failed to increase following oral glucose challenge in GcgGut−/− mice, providing clear proof that the gut is a key source of circulating GLP-1 following enteral nutrient ingestion. Third, loss of gut-derived PGDPs was associated with impaired oral glucose tolerance in GcgGut−/− mice and GcgDistalGut−/− mice, highlighting the importance of gut PGDPs for glucose homeostasis. Collectively, these findings are consistent with substantial evidence that intestinal-derived GLP-1 acts as a circulating hormone, or locally through a gut-brain axis (Grasset et al., 2017), to control gut motility, islet hormone secretion, and glucose homeostasis.
Nevertheless, despite the loss of gut GLP-1 production and reduced glucose-stimulated levels of GLP-1 in GcgGut−/− mice, we did not detect any changes in food intake and body weight or any reduction of glucose-stimulated insulin levels following the oral glucose challenge. Notably, however, GcgGut−/− mice and GcgDistalGut−/− mice are unable to fully augment insulin secretion to prevent hyperglycemia in the face of an enteral glucose challenge. Another explanation for the relatively normal insulin response resides in the increased levels of GIP following oral glucose challenge in both GcgGut−/− and GcgDistalGut−/− mice. It also seems likely that both pancreatic GLP-1 and glucagon may act as local glucoincretins to enhance insulin secretion in the face of reduced intestinal GLP-1 production (Capozzi et al., 2019a; Svendsen et al., 2018; Zhu et al., 2019). A notable phenotype of gut GLP-1 deficiency demonstrated herein is the acceleration of gastric emptying. This observation is consistent with an essential role for GLP-1 in the inhibitory control of basal and meal-regulated antroduodenal motility (Schirra et al., 2006; Witte et al., 2011). Collectively, these findings emphasize the essential importance of gut-derived PGDPs for control of gastric emptying and glucose homeostasis.
Considerable historical debate and controversy surround the relative abundance and importance of proximal versus distal gut L cells for the control of glucose homeostasis. Some studies have highlighted a key role for proximal-distal gut communication, wherein distal gut L cells are rapidly activated via neural and hormonal amplification signals (Lim and Brubaker, 2006). Alternatively, direct perfusion of the proximal versus distal human gut with glucose revealed a relatively greater capacity of the distal small bowel for GLP-1 secretion and acute glucose disposal (Zhang et al., 2019). Recent studies have suggested that the proximal gut may be a more important source of functionally competent L cells than originally postulated (Glass et al., 2017; Jorsal et al., 2018; Svendsen et al., 2015). Surprisingly, levels of active GLP-1 rose briskly following oral glucose administration and were not different in control versus GcgDistalGut−/− mice. Hence, the proximal gut is indeed capable of mounting a brisk GLP-1 response to oral glucose despite the marked reduction of distal gut Gcg expression. Nevertheless, GcgDistalGut−/− mice exhibit reduced fasting levels of circulating active GLP-1, together with impaired oral glucose tolerance. These findings buttress the importance of the distal gut as a critical source of circulating PGDPs with glucoregulatory activity.
In normal healthy mice, the relevance of pancreatic production of GLP-1 for the control of glucose homeostasis remains uncertain, in that pancreatic levels of active GLP-1 are at the lower limit of detection. In contrast, pancreatic levels of glucagon are substantially higher than those of GLP-1, and recent studies highlight the activity of islet glucagon, acting predominantly through the GLP-1 receptor, as a local regulator of insulin secretion (Capozzi et al., 2019a; Svendsen et al., 2018; Zhu et al., 2019). Taken together, our new findings integrated together with previous studies of pancreatic PGDP activity (Capozzi et al., 2019a; Chambers et al., 2017; Svendsen et al., 2018; Traub et al., 2017) emphasize the potential contributions of both the pancreas and the intestine to the generation of glucoregulatory PGDPs, and the regulation of GLP-1R-dependent insulin secretion and glucose homeostasis.
Limitations of Study
Our experimental design and findings have a number of limitations. We studied predominantly healthy mice and did not examine mice with experimental diabetes or prolonged HFD feeding and severe obesity. The new mouse lines studied here exhibit germline elimination of the Gcg gene; hence, we cannot rule out the possibility that one or both lines have adapted physiologically to compensate for developmental loss of gut PGDPs. Indeed, plasma levels of GIP were increased in both GcgGut−/− and GcgDistalGut−/− mice. Hence, the absence of gut GLP-1 and its impact on the incretin effect were potentially modified by compensatory upregulation of GIP action, as described previously for Glp1r−/− and Gcg−/− mice (Fukami et al., 2013; Pederson et al., 1998). Notably, we detected a very small amount of active GLP-1 in mouse pancreatic extracts. Hence, we cannot rule out a role for local islet GLP-1 or glucagon action in the context of the distal or complete elimination of intestinal Gcg expression. Importantly, we also report variable yet consistently detectable levels of active GLP-1 in the human pancreas. Furthermore, the current study was not designed to distinguish the extent of pancreatic GLP-1 receptor activation arising from locally produced glucagon and GLP-1, versus circulating GLP-1 or oxyntomodulin produced in the gut. Nevertheless, our new findings highlight the unequivocal importance of intestinal PGDPs in the control of gut motility and the regulation of glucose homeostasis.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel Drucker. Drucker@lunenfeld.ca
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mouse experiments were approved by the Animal Care and Use Subcommittee at the Toronto Centre for Phenogenomics (TCP), Mt. Sinai Hospital and the University of Michigan animal care committee. Mice were housed up to five per cage, kept under a 12-h light/12h-dark cycle in the TCP facility, and maintained on regular chow (RC; 18% kcal from fat, 2018, Harlan Teklad, Mississauga, ON), or High Fat Diet (HFD; 60% kcal from fat, D12492i, Research Diets, New Brunswick, NJ). All mice were given free access to food and water unless otherwise indicated. All experiments were performed using age- and sex-matched littermates. B6.Cg-Tg(Vil1-cre) 997Gum/J (Vil-Cre) (Cat No: 004586) and B6.Cg-Tg(CDX2-cre)101Erf/J (Cdx2-Cre) (Cat No: 009350) mice (Hinoi et al., 2007) were obtained from Jackson Laboratories (Bar Harbor ME). Gcgtm1Rsy (Gcgflox/flox) mice, in which exon 2 of the Gcg gene is flanked by loxP sites, was provided by Darleen Sandoval (Chambers et al., 2017). To generate GcgGut−/− and GcgDistalGut−/− mice, Vil-Cre and Cdx2-Cre mice were bred with Gcgflox/flox mice (Chambers et al., 2017), respectively. Germline deletion was minimized by restricting Cre expression to female breeders for the Vil-Cre line, and male breeders for the Cdx2-Cre line, and genotypes were verified by isolating RNA and determining length of Gcg transcripts in relevant tissues. To control for gene dosage, breeders were heterozygous for the Cre gene. Intercrossing Cre-positive and Cre-negative Gcg loxP heterozygotes from these 2 lines resulted in 6 genotypes: wildtype mice with no Cre (WT), mice homozygous for the LoxP Gcg gene (Gcgflox/flox)(Floxed), wildtype mice expressing Cre recombinase (Vil-Cre or Cdx2-Cre) and Gcgflox/flox mice expressing Cre recombinase (GcgGut−/− and GcgDistalGut−/− mice). All mice were born at the expected Mendelian ratios and appeared healthy. Whenever possible, we carried out experiments in all groups of mice; no major phenotypic differences in glucose homeostasis were observed between any of the control lines (Figure S4). For the purposes of clarity, data was presented as comparisons between Vil-Cre or Cdx2-Cre control mice to GcgGut−/− and GcgDistalGut−/− mice, respectively.
METHOD DETAILS
Assessment of Gcg Knockdown
Following euthanasia, the small intestine was removed, flushed with PBS and divided into four equal sections: the first quarter was defined as duodenum, the second quarter, jejunum and the final quarter defined as ileum. Gcg expression was also assessed in brainstem, pancreas and colon. Total RNA was isolated from the tail of the pancreas, the first 1 cm piece of each intestinal section (for duodenum, jejunum and proximal ileum), the last 1 cm piece (ileum/distal ileum), or from the proximal half of the colon. Total RNA was extracted from tissues using Tri Reagent (Molecular Research Center, Cincinnati, OH). cDNA was synthesized from DNase I-treated (Thermo-Fisher Scientific, Markham, ON) total RNA (2 μg) using random hexamers and Superscript III (Thermo-Fisher Scientific, Markham, ON). Real-time PCR was carried out using a QuantStudio 5 System and TaqMan Gene Expression Assays (Thermo-Fisher Scientific, Markham, ON). Primer-probe sets were manufactured by Taqman Assays-on-Demand (Applied Biosystems) to measure Exons 1–2 of proglucagon (Gcg: Mm01269053_m1), Exons 3–4 of Gastric Inhibitory Polypeptide (Gip: Mm00433601_m1). Relative RNA expression was normalized to levels of ribosomal protein L32 (Rpl32), or cyclophilin (Ppia) mRNA.
Glucose Tolerance Testing
Mice were fasted overnight (~16 h) and oral or intraperitoneal glucose tolerance tests (OGTT or IPGTT, respectively) were carried out using 1.5 g/kg body weight of glucose (15% solution). Blood glucose levels were assessed in tail vein blood using a hand-held glucometer (Contour glucometer, Bayer Healthcare, Toronto, ON). In accordance with animal protocol guidelines regarding blood volumes collected per mouse in a given experiment, blood was collected at either 0, and 5, or 10 minutes (for GLP-1, insulin and GIP), or 0, 15, 30 and 60 minutes (for insulin and glucagon) after glucose administration in heparin-coated capillary microvette tubes. For measurement of total or active GLP-1, insulin, total GIP, and glucagon, blood was mixed with 10% TED (vol/vol) (5,000 KIU/ml Trasylol, 1.2 mg/ml EDTA and 0.1 nmol/l Diprotin A) and plasma was isolated after centrifugation (13,000 rpm 5 min 4°C) and stored at −80°C until further analysis.
Metabolic Measurements
Insulin (#80-INSMSU-E01, Alpco, Salem, NH), glucagon (#10-1281-01, Mercodia, Winston Salem, NC), total GIP (#81517, Crystal Chem, Elk Grove Village, IL), active and total GLP-1 (#K150JWC-1 and #K150JVC-1, Mesoscale, Rockville, MD) were measured in plasma samples obtained before and 5–60 minutes after glucose administration.
Pancreas Samples
Pancreata samples from six non-diabetic human donors (male donors:18, 18, 20, 35 years of age; female donors: 45, 55 years of age) were obtained through a partnership of Vanderbilt University Medical Center) and Dr. Rita Bottino (Institute of Cellular Therapeutics, Allegheny Health Network, Pittsburgh, PA, USA) working with the International Institute for Advancement of Medicine (IIAM) and the National Disease Research Interchange (NDRI), as previously described (Brissova et al., 2018). Small (< 1 cm) human pancreatic samples were snap-frozen and stored at −80°C until analysis. The Vanderbilt University Institutional Review Board declared that studies on de-identified human pancreatic specimens do not qualify as human subject research.
Pancreas-reactivated (GcgRAΔPDX1Cre), intestinal-reactivated (GcgRAΔVilCre), Gcg-null, Pdx-1-Cre and Vil-Cre control pancreas samples from 28-week-old male mice were kindly provided by Dr. Darleen Sandoval (University of Michigan).
Tissue Peptide Content
To measure tissue GLP-1, glucagon, insulin or GIP content, a 0.5 cm segment of intestine, 20–35 mg murine pancreas tissue, or 6–37 mg human pancreas tissue were homogenized in 500 μl (or 250 μl for human pancreas tissue) of lysis buffer (50 mM Tris HCl (pH 8 at 4°C), 1 mM EDTA, 10% glycerol (wt/vol), 0.02% Brij-35 (wt/vol)) supplemented with protease inhibitors. Peptide levels were measured in homogenates using the above mentioned assays and normalized to protein content (Bradford assay), or tissue weight.
Gastric Emptying
Male mice were subjected to an acetaminophen absorption test to assess the rate of gastric emptying. Following an overnight fast (~16 h), oral administration of a glucose solution (1.5 g/kg BW) containing 1% (w/v) acetaminophen (#A7085, Sigma, Oakville, ON) was administered at a dose of 100 mg/kg. Blood was collected from the tail vein into heparin-coated tubes before and 15, 30 and 60 minutes after acetaminophen administration as described (Maida et al., 2008). Acetaminophen levels were measured in plasma using an enzymatic-spectrophotometric assay (Acetaminophen-L3K, #506–30, Sekisui Diagnostics, Lexington, MA).
Food Intake
After an overnight fast (~16 h), mice were weighed, singly housed and given a pre-weighed amount of food (RC; 18% kcal from fat, 2018, Harlan Teklad, Mississauga, ON) with free access to water. Food was then weighed after 2, 4, 8 and 24 hours.
Immunohistochemistry
The pancreas was fixed in 10% neutral-buffered formalin and paraffin embedded. Immunohistochemistry was done on 5-μm histological sections, and serial sections were stained for insulin (#ab181547, Abcam, Toronto, ON), or glucagon (#2760S, Cell Signaling, Danvers, MA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are expressed as the mean ± SEM. Statistical comparisons were made by ANOVA, followed by a Dunnet or Tukey post-hoc, or by a 2-way ANOVA followed by a Sidak post-hoc, or by Student t test (when only 2 conditions) using GraphPad Prism 7. A p value <0.05 was considered to be statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| insulin | Abcam | Cat# ab181547; RRID: AB_2716761 |
| glucagon | Cell Signalling | Cat# 2760S; RRID: AB_659831 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| acetaminophen | Sigma | Cat# A7085 |
| Critical Commercial Assays | ||
| Ultrasensitive Mouse Insulin ELISA | ALPCO | Cat# 80-INSMSU-E01; RRID: AB_2792981 |
| Active GLP-1 (ver. 2) | Mesoscale | Cat# K150JWC-1 |
| Total GLP-1 (ver. 2) | Mesoscale | Cat# K150JVC-1; RRID: AB_2801383 |
| Total GIP | Crystal Chem | Cat# 81517 |
| Glucagon | Mercodia | Cat# 10-1281-01; RRID: AB_2783839 |
| Acetaminophen-L3K | Sekisui Diagnostics | Cat# 506–30 |
| Insulin ELISA (human) | ALPCO | Cat# 80-INSHU-E01.1; RRID: AB_2801438 |
| Experimental Models: Organisms/Strains | ||
| B6.Cg-Tg(Vil1-cre)997Gum/J (Vil-Cre) | Jackson Laboratories | Cat# JAX:004586; RRID: IMSR_JAX:004586 |
| B6.Cg-Tg(CDX2-cre)101Erf/J (Cdx2-Cre) | Jackson Laboratories | Cat# JAX009350; RRID: IMSR_JAX:009350 |
| Gcgtm1Rsy (Gcgflox/flox) | Darleen Sandoval, University of Michigan | N/A |
| Oligonucleotides | ||
| Proglucagon (Gcg) | Applied Biosystems | Cat# Mm01269053_m1 |
| Gastric Inhibitory Polypeptide (Gip) | Applied Biosystems | Cat# Mm00433601_m1 |
| Cyclophilin (Ppia) | Applied Biosystems | Cat# Mm02342430_g1 |
| Ribosomal protein L32 (Rpl32) | Applied Biosystems | Cat# Mm02528467_g1 |
| Software and Algorithms | ||
| Graph Pad Prism 7 | Graphpad Software | https://www.graphpad.com |
| Other | ||
| Regular Chow Diet (RC) | Harlan Teklad | Cat# 2018 |
| High Fat Diet (HFD) | Research Diets | Cat# D12492i |
Context and Significance.
Glucagon-like peptide 1 (GLP-1)-based drugs are effective anti-diabetic therapies. How and where GLP-1 and its related proteins are naturally produced in the body has been the topic of some debate. Investigators at the University of Michigan and University of Toronto re-examined some of their previous conclusions that the pancreas (which also produces insulin) is the main source of GLP-1 in mice. Although small amounts of active GLP-1 can be detected in both the mouse and human pancreas, disruption of the GLP-1-producing gene in the mouse gut had pronounced adverse effects on glucose control. The results reinforce the importance of gut GLP-1 secretion, physiologically and potentially pharmacologically, for control of metabolism.
ACKNOWLEDGMENTS
D.J.D. is supported in part by a Novo Nordisk, Banting and Best Diabetes Center Chair in incretin biology and CIHR Foundation grant 154321. Mt. Sinai Hospital receives support for studies of incretin biology in the Drucker lab from Novo Nordisk. Y.S. is supported by a fellowship program from the BBDC and the Kangbuk Samsung Hospital. The human pancreatic specimens were collected using funding provided by the NIDDK-supported Human Islet Research Network (HIRN, RRID: SCR_014393; https://hirnetwork.org; UC4 DK104211 and DK108120), DK106755, and DK20593. We thank Rita Bottino (Allegheny Health Network, Pittsburgh, PA) for processing the human pancreata.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2019.08.009.
DECLARATION OF INTERESTS
D.J.D. has served as an advisor, consultant, or speaker in the past 12 months to Forkhead Biotherapeutics, Heliome, Inc., Intarcia Therapeutics, Kallyope, Eli Lilly, Merck Research Laboratories, Novo Nordisk, Inc., Pfizer, Inc., and Sanofi, Inc. Neither D.J.D. nor his family members hold stock directly or indirectly in any of these companies.
REFERENCES
- Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, Bottino R, Campbell-Thompson M, Aramandla R, Poffenberger G, et al. (2018). Alpha cell function and gene expression are compromised in type 1 diabetes. Cell Rep 22, 2667–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, and Drucker DJ (2013). Pharmacology physiology and mechanisms of incretin hormone action. Cell Metab 17, 819–837. [DOI] [PubMed] [Google Scholar]
- Capozzi ME, Svendsen B, Encisco SE, Lewandowski SL, Martin MD, Lin H, Jaffe JL, Coch RW, Haldeman JM, MacDonald PE, et al. (2019a). Beta cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi ME, Wait JB, Koech J, Gordon AN, Coch RW, Svendsen B, Finan B, D’Alessio DA, and Campbell JE (2019b). Glucagon lowers glycemia when beta-cells are active. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AP, Sorrell JE, Haller A, Roelofs K, Hutch CR, Kim KS, Gutierrez-Aguilar R, Li B, Drucker DJ, D’Alessio DA, et al. (2017). The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab 25, 927–934.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Taylor AJ, and Verchere CB (2018). Islet prohormone processing in health and disease. Diabetes Obes. Metab 20, 64–76. [DOI] [PubMed] [Google Scholar]
- Drucker DJ (2006). The biology of incretin hormones. Cell Metab 3, 153–165. [DOI] [PubMed] [Google Scholar]
- Drucker DJ (2007). The role of gut hormones in glucose homeostasis. J. Clin. Invest 117, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker DJ (2013). Incretin action in the pancreas: potential promise, possible perils, and pathological pitfalls. Diabetes 62, 3316–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, et al. (2011). Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med 17, 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami A, Seino Y, Ozaki N, Yamamoto M, Sugiyama C, Sakamoto-Miura E, Himeno T, Takagishi Y, Tsunekawa S, Ali S, et al. (2013). Ectopic expression of GIP in pancreatic beta-cells maintains enhanced insulin secretion in mice with complete absence of proglucagon-derived peptides. Diabetes 62, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galsgaard KD, Winther-Sørensen M, Ørskov C, Kissow H, Poulsen SS, Vilstrup H, Prehn C, Adamski J, Jepsen SL, Hartmann B, et al. (2018). Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. Endocrinol. Metab 314, E93–E103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass LL, Calero-Nieto FJ, Jawaid W, Larraufie P, Kay RG, Göttgens B, Reimann F, and Gribble FM (2017). Single-cell RNA-sequencing reveals a distinct population of proglucagon-expressing cells specific to the mouse upper small intestine. Mol. Metab 6, 1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasset E, Puel A, Charpentier J, Collet X, Christensen JE, Tercé F, and Burcelin R (2017). A specific gut microbiota dysbiosis of type 2 diabetic mice induces GLP-1 resistance through an enteric NO-dependent and gut-brain axis mechanism. Cell Metab. 25, 1075–1090.e5. [DOI] [PubMed] [Google Scholar]
- Habener JF, and Stanojevic V (2017). Pancreas and not gut mediates the GLP-1-induced glucoincretin effect. Cell Metab 25, 757–758. [DOI] [PubMed] [Google Scholar]
- Hansen AM, Bödvarsdottir TB, Nordestgaard DN, Heller RS, Gotfredsen CF, Maedler K, Fels JJ, Holst JJ, and Karlsen AE (2011). Upregulation of alpha cell glucagon-like peptide 1 (GLP-1) in Psammomys obesus–an adaptive response to hyperglycaemia? Diabetologia 54, 1379–1387. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Yamamoto M, Mizoguchi H, Watanabe C, Ito R, Yamamoto S, Sun XY, and Murata Y (2009). Mice deficient for glucagon gene-derived peptides display normoglycemia and hyperplasia of islet {alpha}-cells but not of intestinal L-cells. Mol. Endocrinol 23, 1990–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RS, and Aponte GW (1995). Intra-islet regulation of hormone secretion by glucagon-like peptide-1-(7–36)amide. Am. J. Physiol 269, G852–G860. [DOI] [PubMed] [Google Scholar]
- Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, Cho KR, and Fearon ER (2007). Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 67, 9721–9730. [DOI] [PubMed] [Google Scholar]
- Jorsal T, Rhee NA, Pedersen J, Wahlgren CD, Mortensen B, Jepsen SL, Jelsing J, Dalbøge LS, Vilmann P, Hassan H, et al. (2018). Enteroendocrine K and L cells in healthy and type 2 diabetic individuals. Diabetologia 61, 284–294. [DOI] [PubMed] [Google Scholar]
- Kilimnik G, Kim A, Steiner DF, Friedman TC, and Hara M (2010). Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic alpha-cells in mouse models of ss-cell regeneration. Islets 2, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreymann B, Yiangou Y, Kanse S, Williams G, Ghatei MA, and Bloom SR (1988). Isolation and characterisation of GLP-1 7–36 amide from rat intestine. Elevated levels in diabetic rats. FEBS Lett. 242, 167–170. [DOI] [PubMed] [Google Scholar]
- Larraufie P, Roberts GP, McGavigan AK, Kay RG, Li J, Leiter A, Melvin A, Biggs EK, Ravn P, Davy K, et al. (2019). Important role of the GLP-1 axis for glucose homeostasis after bariatric surgery. Cell Rep. 26, 1399–1408.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GE, and Brubaker PL (2006). Glucagon-like peptide 1 secretion by the L-cell: the view from within. Diabetes 55, S70–S77. [Google Scholar]
- Maida A, Lovshin JA, Baggio LL, and Drucker DJ (2008). The glucagon-like peptide-1 receptor agonist oxyntomodulin enhances {beta}-cell function but does not inhibit gastric emptying in mice. Endocrinology 149, 5670–5678. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, Del Guerra S, D’Aleo V, Piro S, Marselli L, et al. (2012). A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 55, 3262–3272. [DOI] [PubMed] [Google Scholar]
- Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, and Habener JF (1986). Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J. Biol. Chem 261, 11880–11889. [PubMed] [Google Scholar]
- Mojsov S, Kopczynski MG, and Habener JF (1990). Both amidated and nonamidated forms of glucagon-like peptide I are synthesized in the rat intestine and the pancreas. J. Biol. Chem 265, 8001–8008. [PubMed] [Google Scholar]
- Nie Y, Nakashima M, Brubaker PL, Li QL, Perfetti R, Jansen E, Zambre Y, Pipeleers D, and Friedman TC (2000). Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J. Clin. Invest 105, 955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orskov C, Holst JJ, Knuhtsen S, Baldissera FGA, Poulsen SS, and Nielsen OV (1986). Glucagon-like peptides GLP-1 and GLP-2, predicted products of the glucagon gene, are secreted separately from pig small intestine but not pancreas. Endocrinology 119, 1467–1475. [DOI] [PubMed] [Google Scholar]
- Orskov C, Holst JJ, Poulsen SS, and Kirkegaard P (1987). Pancreatic and intestinal processing of proglucagon in man. Diabetologia 30, 874–881. [DOI] [PubMed] [Google Scholar]
- Orskov C, Rabenhøj L, Wettergren A, Kofod H, and Holst JJ (1994). Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 43, 535–539. [DOI] [PubMed] [Google Scholar]
- Patterson JT, Ottaway N, Gelfanov VM, Smiley DL, Perez-Tilve D, Pfluger PT, Tschöp MH, and Dimarchi RD (2011). A novel human-based receptor antagonist of sustained action reveals body weight control by endogenous GLP-1. ACS Chem. Biol 6, 135–145. [DOI] [PubMed] [Google Scholar]
- Patzelt C, and Schiltz E (1984). Conversion of proglucagon in pancreatic alpha cells: the major endproducts are glucagon and a single peptide, the major proglucagon fragment, that contains two glucagon-like sequences. Proc. Natl. Acad. Sci. USA 81, 5007–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson RA, Satkunarajah M, McIntosh CH, Scrocchi LA, Flamez D, Schuit F, Drucker DJ, and Wheeler MB (1998). Enhanced glucose-dependent insulinotropic polypeptide secretion and insulinotropic action in glucagon-like peptide 1 receptor −/− mice. Diabetes 47, 1046–1052. [DOI] [PubMed] [Google Scholar]
- Roberts GP, Larraufie P, Richards P, Kay RG, Galvin SG, Miedzybrodzka EL, Leiter A, Li HJ, Glass LL, Ma MKL, et al. (2019). Comparison of human and murine enteroendocrine cells by transcriptomic and peptidomic profiling. Diabetes 68, 1062–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval DA, and D’Alessio DA (2015). Physiology of proglucagon peptides: role of glucagon and GLP-1 in health and disease. Physiol. Rev 95, 513–548. [DOI] [PubMed] [Google Scholar]
- Schirra J, Nicolaus M, Roggel R, Katschinski M, Storr M, Woerle HJ, and Göke B (2006). Endogenous glucagon-like peptide 1 controls endocrine pancreatic secretion and antro-pyloro-duodenal motility in humans. Gut 55, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen B, Pedersen J, Albrechtsen NJ, Hartmann B, Toräng S, Rehfeld JF, Poulsen SS, and Holst JJ (2015). An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology 156, 847–857. [DOI] [PubMed] [Google Scholar]
- Svendsen B, Larsen O, Gabe MBN, Christiansen CB, Rosenkilde MM, Drucker DJ, and Holst JJ (2018). Insulin secretion depends on intra-islet glucagon signaling. Cell Rep 25, 1127–1134.e2. [DOI] [PubMed] [Google Scholar]
- Timper K, Dalmas E, Dror E, Rütti S, Thienel C, Sauter NS, Bouzakri K, Bédat B, Pattou F, Kerr-Conte J, et al. (2016). Glucose-dependent insulinotropic peptide stimulates glucagon-like peptide 1 production by pancreatic islets via interleukin 6, produced by alpha cells. Gastroenterology 151, 165–179. [DOI] [PubMed] [Google Scholar]
- Traub S, Meier DT, Schulze F, Dror E, Nordmann TM, Goetz N, Koch N, Dalmas E, Stawiski M, Makshana V, et al. (2017). Pancreatic alpha cell-derived glucagon-related peptides are required for beta cell adaptation and glucose homeostasis. Cell Rep 18, 3192–3203. [DOI] [PubMed] [Google Scholar]
- Vahl TP, Paty BW, Fuller BD, Prigeon RL, and D’Alessio DA (2003). Effects of GLP-1-(7–36)NH(2), GLP-1-(7–37), and GLP-1-(9–36)NH(2) on intravenous glucose tolerance and glucose-induced insulin secretion in healthy humans. J. Clin. Endocrinol. Metab 88, 1772–1779. [DOI] [PubMed] [Google Scholar]
- van der Wielen N, Ten Klooster JP, Muckenschnabl S, Pieters R, Hendriks HF, Witkamp RF, and Meijerink J (2016). The noncaloric sweetener rebaudioside A stimulates glucagon-like peptide 1 release and increases enteroendocrine cell numbers in 2-dimensional mouse organoids derived from different locations of the intestine. J. Nutr 146, 2429–2435. [DOI] [PubMed] [Google Scholar]
- Vasu S, Moffett RC, Thorens B, and Flatt PR (2014). Role of endogenous GLP-1 and GIP in beta cell compensatory responses to insulin resistance and cellular stress. PLoS One 9, e101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte AB, Grybäck P, Jacobsson H, Näslund E, Hellström PM, Holst JJ, Hilsted L, and Schmidt PT (2011). Involvement of endogenous glucagon-like peptide-1 in regulation of gastric motility and pancreatic endocrine secretion. Scand. J. Gastroenterol 46, 428–435. [DOI] [PubMed] [Google Scholar]
- Zhang X, Young RL, Bound M, Hu S, Jones KL, Horowitz M, Rayner CK, and Wu T (2019). Comparative effects of proximal and distal small intestinal glucose exposure on glycemia, incretin hormone secretion, and the incretin effect in health and Type 2 diabetes. Diabetes Care 42, 520–528. [DOI] [PubMed] [Google Scholar]
- Zhu L, Dattaroy D, Pham J, Wang L, Barella LF, Cui Y, Wilkins KJ, Roth BL, Hochgeschwender U, Matschinsky FM, et al. (2019). Intra-islet glucagon signaling is critical for maintaining glucose homeostasis. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.