

Abstract
Lung alveologenesis, formation of the alveolar region, allows sufficient gas exchange surface to be packed inside the chest cavity yet with orderly connection to the trachea. The real-life alveolar region, however, bears little resemblance to idealized cartoons owing to its three-dimensional nature, non-uniform shape, and mostly air-filled void. This morphological complexity is matched by its cellular complexity – comprised of intermixed and often tangled cells of the epithelial, mesenchymal, endothelial, and immune lineages. Modern imaging, genetics, and genomics are shedding light on and updating traditional views of alveologenesis. Accordingly, this review describes a cell-centric 3-phase definition of alveologenesis and discusses its failure in diseases and possible reactivation during regeneration.
Keywords: lung development, AT1 cell, angiogenesis, alveoli, lung morphology, bronchopulmonary dysplasia
Introduction
Situated distally to ramifying airway tubes, the alveolar region of the mammalian lung is packed with ducts and sacs to maximize the gas exchange interface between the air and the blood. These seemingly simple shapes and the vast tissueless airspace belie an engineering feat that allows air and blood to reach millions of termini along orderly and shortest routes. The developmental process that forms this alveolar region – alveologenesis – must be timed with the beginning of extrauterine life, but is interrupted by premature birth and further impeded by mechanical ventilation and oxygen therapy that are often necessary to sustain life in preterm infants, but at times lead to chronic lung diseases.1 The importance of understanding alveologenesis is reinforced by the possibility that the same developmental mechanisms may still be required during homeostasis – but disrupted in emphysematous lungs – and reused during alveolar repair and regeneration.
It is important to recognize that biological structures including the lung alveolar region are composed of and can be distilled to individual cell types and their coordinated behaviors. Such cellular basis of the traditional morphological description of alveologenesis has been unveiling in recent years. Accordingly, this review presents a cell-centric view of alveologenesis, covering distinct cell types in each of the epithelial, mesenchymal, endothelial, and immune lineages.
Section I: Traditional and updated definitions of alveologenesis
The traditional morphological definition of alveologenesis – formation of alveoli
In the traditional 5-stage model of lung development, alveologenesis is preceded by the embryonic, pseudoglandular, canalicular, and saccular stages (Fig. 1A).2 Following its specification from the ventral foregut in the embryonic stage, the lung adopts a pseudoglandular appearance as a result of branching morphogenesis with species-characteristic complexity, ranging from absence of branching in frogs to tens of branch generations in mice and humans.3 The canalicular stage begins when a subset of pseudoglandular tubes elongate, forming canals or alveolar ducts – which are a continuation of the conducting airways but are bordered by epithelial cells of the alveolar fate. Further distally and later in development, the termini of the branching network expand, forming saccules and thus marking the saccular stage. The separations between these initial alveolar sacs are primary septa – different from secondary septa, which form subsequently during the alveolar stage and subdivide the expanding walls of the initial alveolar sacs into individual alveoli. Taken together, alveologenesis is traditionally defined as formation of alveoli, largely on morphological grounds and prior to availability of significant molecular and cellular insights.
Figure 1: Tubes from branching morphogenesis transform into alveolar ducts, sacs, and alveoli in mice.
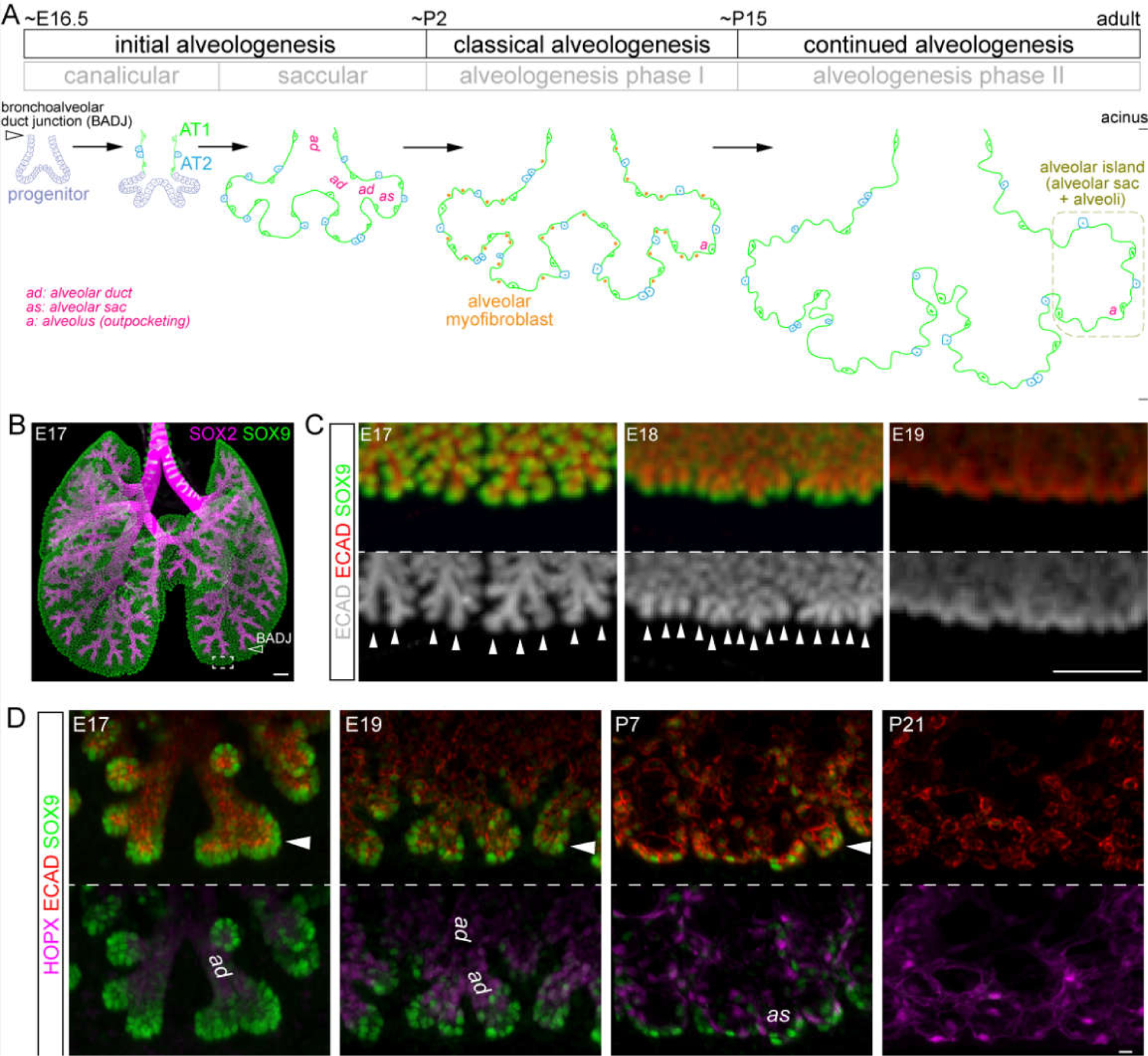
(A) A model depicting the 3 phases of alveologenesis. The initial alveologenesis phase corresponds to the traditional canalicular and saccular stages and is marked by differentiation of AT1 and AT2 cells. Progenitors continue to branch in this phase to form additional alveolar ducts (3 generations shown), but eventually differentiate to form alveolar sacs. The classical alveologenesis phase, corresponding to alveologenesis phase I, is marked by presence of alveolar myofibroblasts. New alveoli or outpocketings continue to form without alveolar myofibroblasts in the continued alveologenesis phase, corresponding to alveologenesis phase II. An alveolar island refers to an alveolar sac and associated alveoli (or outpocketings). The developmental ages of the different phases in mice (E, embryonic day; P, postnatal day) are approximations because the proximal and distal regions develop asynchronously.
(B) Optical project tomography (OPT) images of immunostained mouse lungs at embryonic day (E) 17 when bronchoalveolar duct junctions (BADJ; airway termini marked by SOX2; open arrowhead) form and the non-SOX2 distal region including SOX9 branch tips becomes the future alveolar region (box). Adapted from4.
(C) OPT images of immunostained developing alveolar regions, comparable to the boxed region in (B), showing SOX9 branch tips (arrowheads) continue to branch as alveolar ducts form. See also (D). The epithelium is marked by an epithelial junction protein E-Cadherin (ECAD). Adapted from3.
(D) En face view of confocal images of immunostained alveolar regions, comparable to the boxed region in (B), showing SOX9 progenitors (arrowhead) and morphogenesis of alveolar ducts (ad) and alveolar sacs (as). Differentiating AT1 cells express HOPX. P, postnatal day. Adapted from5.
Scale: B, C 250 um; D 10 um.
This definition, although intuitive conceptually, is challenging to apply practically for the following reasons. (1) An alveolus is not a discrete entity with qualifying dimensions. Although cartooned as uniform semi-spheres, alveoli are tissue outpocketings whose depth and openness are continua even in the mature lung, making it difficult to distinguish an alveolus from a rough sector of the alveolar sac wall. (2) Inferring a process from snapshots – alveologenesis in this case and developmental biology in general – requires comparing “comparables”. On one hand, lung development is spatially asynchronous: proximal regions mature before their distal counterparts. For example, during the canalicular stage when the proximal alveolar ducts are forming, the distal edge is still undergoing branching morphogenesis to form additional generations of future alveolar ducts (Fig. 1B–D).3–5 It is also worth noting that since branches can grow internally, topologically distal regions can be in the center of the lung. On the other hand, the alveolar region is made of non-uniform and non-ideal tubes, sacs, and outpocketings so that two-dimensional (2D) sections of the same object, even if thick, would be different when taken at different angles. Thus, understanding alveologenesis requires comparing three-dimensional (3D) complete views of the alveolar regions at the same position along the proximal-distal axis over developmental time.
An updated cellular definition of alveologenesis – formation of alveolar surface
Recognizing this morphological ambiguity between alveoli and alveolar sacs, and the technical difficulty in obtaining accurate morphological information in 3D, we propose a cellular definition of alveologenesis – formation of alveolar surface. Unlike the 3D alveoli, the alveolar surface is a readily measurable 2D object and has a precise cellular correlate – an epithelium made of alveolar, not airway, cells. More specifically, ultrathin and expansive alveolar type 1 (AT1) cells constitute nearly the entire alveolar surface, notwithstanding a minor contribution from surfactant-secreting alveolar type 2 (AT2) cells. Furthermore, AT1 cells are unique to the lung and allow gas diffusion – whether as part of alveolar ducts, sacs, or alveoli – making the canalicular, saccular, and alveolar stages all part of the process of forming the gas exchange surface. Therefore, we propose that alveologenesis begins with the differentiation of AT1 cells at the canalicular stage and continues as long as the alveolar surface increases; a second cellular feature – the presence of alveolar myofibroblasts – divides alveologenesis into 3 phases, as detailed below (Fig. 1A).
Initial alveologenesis: increase in alveolar surface in the absence of alveolar myofibroblasts (E16.5-P2 in mice)
This phase corresponds to the canalicular and saccular stages that result from alveolar surface expansion, first along the proximal ducts and then at the termini, respectively. This reflects AT1 cell flattening which is characterized by a reduction in lateral aspects and an increase in surface area.6 We consider these changes alveologenesis because, other than their different geometric shapes, alveolar ducts and sacs are made of the same AT1 cells as those of alveoli, and are sufficient to support gas exchange as newborn mice live without alveoli. It is noteworthy that based on counting branch tips and conducting airway termini, all the tubes of the future airway tree have formed by embryonic day (E) 14.5, so that cells subsequently “left behind” by the SRY-box 9 (SOX9)-expressing progenitors will differentiate into alveolar cells; such differentiation, including AT1 cell flattening, begins around E16.5 immediately distal to the conducting airways.4,5 Thus, initial alveologenesis matches the fate switch of epithelial progenitors from airway differentiation to alveolar differentiation.5
Classical alveologenesis: increase in alveolar surface in the presence of alveolar myofibroblasts (P3-P14 in mice)
This is the most studied phase of alveologenesis and is marked by alveolar myofibroblasts – fibroblasts expressing contractile proteins including smooth muscle actin (SMA) in the alveolar region.7 These myofibroblasts form arcs within the aforementioned secondary septa, conceivably restraining the outpocketing of the alveolar surface and thus helping to shape the alveoli, but disappear when the rate of alveolar surface expansion decreases. Similar to the delay between deposition of SOX9 progeny in alveolar ducts and their flattening into AT1 cells, alveolar myofibroblasts may exist as SMA-negative precursors within the secondary septa, or migrate from a more proximal region as proposed.8 It is also important to note that alveolar outpocketing is additionally restrained by capillaries, as detailed in the next section.
Continued alveologenesis: increase in alveolar surface in the absence of alveolar myofibroblasts (P15-adult in mice)
In this phase, the alveolar surface continues to increase, partly due to simple volume expansion concomitant with growth of the lung and the chest cavity, as supported by an average increase in the size of individual outpocketings or alveoli.2,9 However, the number of alveoli continues to increase, suggesting that despite the absence of alveolar myofibroblasts, outpocketing in this phase is still restrained and subdivided into new alveoli, possibly by capillaries. Such opposing actions of volume expansion and subdivision give rise to alveoli of variable depth and openness, causing the aforementioned challenge of defining alveoli by morphological criteria alone. Disappearance of alveolar myofibroblasts provides an operational distinction between classical and continued alveologenesis, which are also called phase I and phase II, respectively, in the literature.10
Section II: Cellular components of alveologenesis
As most mammalian organs, the lung is composed of 4 cell lineages – epithelial, mesenchymal, endothelial, and immune – that are generally considered non-interconvertible but interact via physical and chemical signals. Practically, the epithelial lineage is marked by surface markers Cadherin 1 (CDH1) or Epithelial cell adhesion molecule (EPCAM); the endothelial lineage by Cadherin 5 (CDH5), Platelet and endothelial cell adhesion molecule 1 (PECAM-1, also known as CD31), or Intercellular adhesion molecule 2 (ICAM2); the immune lineage by Protein tyrosine phosphatase receptor type C (PTPRC, also known as CD45); and the mesenchymal lineage is negative for markers of the other 3 lineages. Below, we will describe the structural and signaling contributions of representative cell types in each lineage to alveologenesis.
Epithelial lineage
The alveolar epithelium forms a barrier around the airspace and is made of the aforementioned AT1 and AT2 cells. In our cellular definition, alveologenesis is inherently the differentiation of AT1 and, to a lesser extent, AT2 cells from their SOX9 progenitors.5,11 SOX9 progenitors emerge from the embryonic foregut and always occupy the distal tips of the growing respiratory tree while undergoing branching morphogenesis.5 Early in development, SOX9 progenitors give rise to SOX2+ airway cells including club, ciliated, neuroendocrine, and basal cells, but retain the potential to give rise to alveolar cells, and hence are multipotent; later in development, these SOX9 progenitors give rise to only AT1 and AT2 cells and hence are bipotent.4,5,11,12 As late SOX9 progenitors exit branch tips and become committed AT1 or AT2 precursors/progenitors in the distal tubules of the canalicular stage lung, they upregulate AT1 and AT2 cell markers and hence are efficiently labeled by the corresponding genetic drivers.13,14
Alveolar type 1 cell
Individual AT1 cells can be >10,000 um2 in surface area and collectively constitute ~95% of the alveolar surface, but measure ~0.1 um in thickness to allow passive gas diffusion.6,15 This highly specialized morphology is achieved developmentally via a 2-step process: cell flattening and folding.6 During flattening, columnar progenitors shorten lateral adherence junctions while maintaining apical tight junctions. During folding, flattened AT1 cells expand while being constrained by an irregular mesh of myofibroblasts and capillaries, forming outpocketings, or alveoli, of variable sizes (Fig. 2A–C). In addition to growing the alveoli, AT1 cell expansion provides the epithelial surface that is required for alveolar duct elongation and terminal saccule inflation during the canalicular and saccular stages, respectively. Thus, AT1 cell flattening and folding both fuel and shape most of the alveolar growth with limited contribution from cell proliferation and AT2 cells. Nevertheless, AT1 cell expansion occurs within an intact epithelial sheet, different from the invasive front during collective cell migration, and thus does not necessarily provide an active mechanical force for alveologenesis – which instead could be generated from luminal fluid pressure in embryonic lungs, as supported by a recent study,16 and air pressure in postnatal lungs.
Figure 2: AT1 cells flatten and fold giving rise to the alveolar surface.
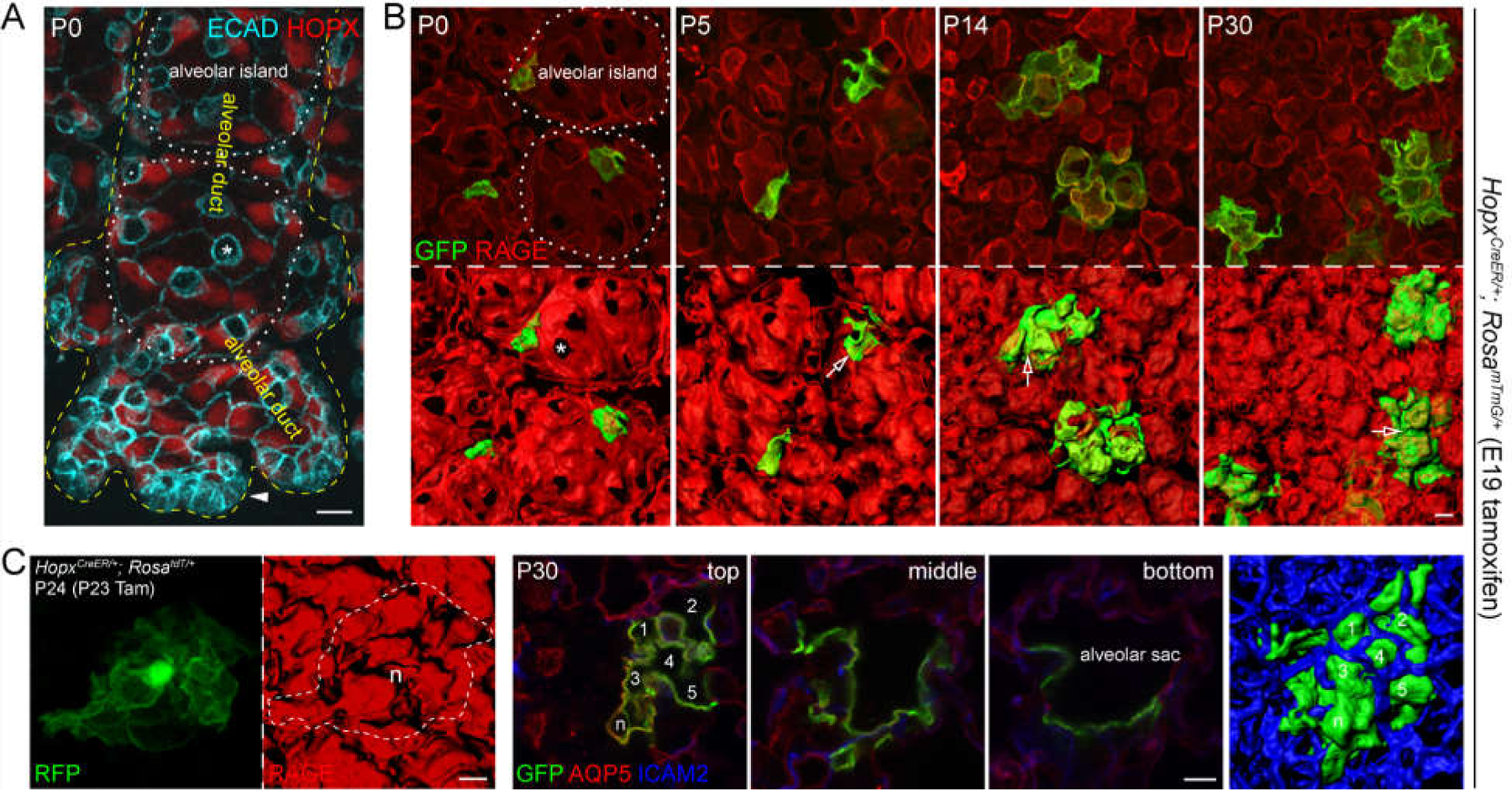
(A) En face view of confocal images of an immunostained alveolar region at the edge of the lung at the saccular stage, showing expanded alveolar ducts in association with flattening of AT1 cells, as outlined by ECAD and marked by HOPX – in contrast to cuboidal AT2 cells (asterisk). Remaining SOX9 progenitors at the distal tips (arrowhead) have not differentiated into AT1 or AT2 cells. Branches growing vertically out of the imaging plane differentiate earlier and have formed alveolar islands, which are made of an alveolar sac and its outpocketings. See also (B).
(B) En face view of confocal images of immunostained alveolar regions with AT1 cells sparsely, genetically labeled with GFP, showing that individual AT1 cells grow in size and fold at secondary septa (open arrow). The alveolar surface is marked by an AT1 membrane protein RAGE. Asterisk, AT2 cell.
(C) Left: A single AT1 cell sparsely labeled with a RosatdT reporter that accumulates in the nucleus (n) and imaged as in (B). Right: A single AT1 cell labeled and imaged as in (B) extends from an alveolar sac and covers multiple alveoli (numbered 1 through 5 and marked by an AT1 membrane protein AQP5) that are demarcated by vessels (ICAM2), as supported by optical sections at different levels (first 3 panels) and the en face view (last panel). (A-C) are adapted from6.
Scale: A-C 10 um.
Blockage of AT1 cell flattening and folding is expected to deprive the building blocks of alveologenesis. An extreme example is seen in the AT1-cell-specific deletion of the lung lineage transcription factor NK2 homeobox 1 (NKX2-1), which is required for 3 defining features of AT1 cells – cell markers, morphology, and quiescence.17 Indeed, classical alveologenesis fails in this mutant, manifesting as dilated airspace with few alveoli. Since NKX2-1 is also required to maintain AT1 cell morphology,17 it will be interesting to delete Nkx2-1 later in development and examine the consequences on continued alveologenesis, as well as maintenance of alveoli formed during classical alveologenesis. On the other end, the contribution of AT1 cells to initial alveologenesis – formation of alveolar ducts and sacs – may be examined by deleting Nkx2-1 in AT1 cell precursors without disrupting its known role in AT2 cells, using genetic drivers that confer AT1-cell-specificity embryonically.13,14 Alternatively, the requirement of Yes-associated protein 1 (YAP) and Transcriptional coactivator with PDZ-binding motif (TAZ) in AT1 cells, but not AT2 cells,18 may be exploited to specifically target AT1 cells; however, their known role in SOX9 progenitors which are still undergoing branching at the distal edge during initial alveologenesis (Fig. 1D, G), might be a confounding factor.
Besides their structural role, AT1 cells are the dominant source of a number of key signaling molecules of alveologenesis, such as Vascular endothelial growth factor A (VEGFA), Platelet derived growth factor A (PDGFA), and Sonic hedgehog (SHH), as supported by publications and public databases.19,20 The implied and demonstrated signaling roles of AT1 cells in alveologenesis will be discussed later.
Alveolar type 2 cell
Like AT1 cells, AT2 cells arise from SOX9-expressing progenitors but distinguish themselves by their lamellar bodies – lysosome-related organelles that process and store surfactants.21 Unlike the columnar morphology adopted by packed progenitors, most AT2 cells contact flattened AT1 cells via apical tight junctions, while their untethered basal-lateral portion becomes cuboidal and is embedded in the interstitium. Such abluminal protrusion has been followed in real time and suggested to shield AT2 cells from the mechanical stretching experienced by AT1 cells.16 In spite of outnumbering AT1 cells and remaining proliferative during alveologenesis, AT2 cells contribute little structurally.6 Nevertheless, their surfactants reduce surface tension and mechanically enable expansion of the alveolar space. Indeed, loss of ATP binding cassette subfamily A member 3 (ABCA3), a surfactant processing protein specifically expressed by AT2 cells, leads to hypercellularity before birth and atelectasis after birth – failure in initial alveologenesis, possibly in association with smaller, incompletely flattened AT1 cells.22,23
Besides producing surfactants, AT2 cells function as facultative stem cells, self-renewing and differentiating into AT1 cells during injury-repair in the adult lung.11,24 Conceptually possible and traditionally believed, AT2 cells could also give rise to AT1 cells during development and in turn contribute to the alveolar surface.25 However, better distinction of SOX9 progenitors and bona fide AT2 cells, neonatal lineage tracing, and AT1 cell number quantification indicate that the majority of AT1 cells do not come from AT2 cells.6,11,13,14,26 Interestingly, loss of Beta-catenin (CTNNB1), the co-transcription factor of canonical Wnt signaling, allows AT2-to-AT1 transition in the neonatal lung, similar to its adult counterpart, although any effect on classical and continued alveologenesis is presumably limited due to the small number of Wnt-responsive AT2 cells – which also do not differ sufficiently from bulk AT2 cells to form a distinct population in scRNA-seq.27–30
Mesenchymal lineage
There is no consensus on a marker of the mesenchymal lineage, which herein refers to fibroblasts that are negative for aforementioned markers of the other 3 lineages. No more consensus has been reached regarding the cell types within the mesenchymal lineage due to paucity of robust markers and possible plasticity in cell fate. This review will focus on the better established players in alveologenesis: alveolar myofibroblasts and pericytes. The remaining major fibroblast population in the alveolar region, often named alveolar lipofibroblasts, is yet to be defined since a commonly used marker Adipose differentiation-related protein (ADRP, also known as PLIN2) is widely and non-specifically expressed based on transcriptomic and genetic analyses.20,31
Alveolar myofibroblast
As indicated by the name, alveolar myofibroblasts are contractile fibroblasts in the alveolar region and herein refer to normally occurring cells during lung development, instead of pathological myofibroblasts in fibrotic lungs.32 They are found within secondary septa and also called secondary crest myofibroblasts, although practically secondary septa incur the same morphological ambiguity as alveoli, where there is no clear distinction between secondary septation and folding of the alveolar epithelium, and secondary septa persist after alveolar myofibroblasts are developmentally cleared, as described below. A more practical definition relies on molecular markers, in particular Platelet derived growth factor receptor alpha (PDGFRA) and SMA, because of their functional roles in cell specification and contractility, respectively.7,33 It is worth pointing out that SMA is also expressed by vascular smooth muscle cells that surround arterioles and venules and extend well into the alveolar region, and that SMA staining do not reliably mark the nucleus to allow cell number quantification. In addition, a slow-turnover Green fluorescent protein (GFP) reporter of PDGFRA marks cells of varying fluorescence intensities, reflecting the net outcome of active transcription and passive dilution from cell division.7,34 These caveats exemplify current challenges in studying alveolar myofibroblasts in particular – and lung mesenchymal cells in general – highlighting the importance of a clear and consistent definition of alveolar myofibroblasts in pinpointing their origin and delineating normal and abnormal alveologenesis.
This PDGFRA/SMA dual positivity marks the classical alveologenesis phase, but intriguingly disappears during continued alveologenesis and in the adult lung. A recent lineage tracing study using Fgf18CreER suggested that the majority of alveolar myofibroblasts are cleared, instead of transdifferentiating into other cell types.35 Nevertheless, ~10% of Fgf18CreER lineage cells persist, possibly due to incomplete developmental clearance or labeling of non-alveolar myofibroblasts, echoing the aforementioned importance and challenge of defining alveolar myofibroblasts, which is also necessary to interpret other lineage tracing studies using Gli1CreER, PdgfrartTA, Fgf10CreER, Plin2CreER, and Tcf21CreER.31,32,36–38 It will be interesting to examine if the PDGFA/PDGFRA-mediated specification of alveolar myofibroblasts is suppressed during their clearance or reactivated during fibrosis.7,8,32,34
The role of alveolar myofibroblasts in alveologenesis is tested using PdgfrartTA-mediated cell ablation and deletion of a key contractility gene Myosin light chain kinase (Mlck). Both genetic alterations lead to failed classical alveologenesis, whereas continued alveologenesis is unaffected at least in the Mlck mutant, as predicted by the developmental clearance of alveolar myofibroblasts.32,39 Intriguingly, Mlck deletion throughout the lung mesenchyme does not affect homeostasis but impairs regrowth post pneumonectomy.39 Although these data are consistent with the idea that alveolar myofibroblasts mechanically constrain epithelial outpocketing during development and regrowth, independent genetic drivers are needed to substantiate the findings given the known low and high expression of PDGFRA34 and likely targeting of non-alveolar myofibroblasts as well as non-lung cells by PdgfrartTA.
Pericyte
Compared to alveolar myofibroblasts, pericytes are easier to define spatially and molecularly. Pericytes wrap around capillaries albeit at a lower density than their proximal counterpart – vascular smooth muscle cells, which surround the macrovasculature. They are marked by Chondroitin sulfate proteoglycan 4 (CSPG4, also known as NG2) and Platelet derived growth factor receptor beta (PDGFRB), the latter of which responds to endothelium-derived Platelet derived growth factor B (PDGFB).40 By virtue of association with capillaries, pericytes are within secondary septa and can physically limit outpocketing.
A more direct role of pericytes in alveologenesis is demonstrated by pericyte-specific loss of YAP/TAZ, which leads to failed classical alveologenesis despite a normal number of pericytes and normal differentiation of alveolar myofibroblasts.41 Instead, these YAP/TAZ deficient pericytes have diminished expression of Hepatocyte growth factor (HGF) and Angiopoietin 1 (ANGPT1) – secreted ligands that can signal to epithelial and endothelial cells via MET and TEK receptors, respectively.41 Despite other local sources of ANGPT1, pericyte-specific deletion of Angpt1 is sufficient to impair classical alveologenesis.41 Future experiments tailoring the timing of gene deletion will test the role of pericytes in initial and continued alveologenesis.
Endothelial lineage
Whereas epithelial tubes are hierarchical and blind-ended to match directional airflow, endothelial tubes in the alveolar region form a mesh to ensure areal coverage by blood flow, and are more numerous yet smaller, making it challenging to visualize the topology of individual endothelial cells relative to the tubes. This gives rise to two enigmas in lung endothelial biology: intussusceptive angiogenesis and capillary maturation. Recent 3D visualization and identification of endothelial cell heterogeneity in the lung microvasculature shed light on both.
Car4 endothelial cell
As other organs, the lung has macrovasculature – arteries, arterioles, veins, and venules – and microvasculature – capillaries.42 Apart from this, the lung vasculature has been considered homogeneous and practically used as a convenient source of primary endothelial cells. Prompted by the observation that a subset of capillaries are more sensitive to loss of AT1-cell-secreted VEGFA, we used single cell RNA-sequencing (scRNA-seq) to profile freshly purified lung endothelial cells and indeed found two populations of capillary endothelial cells – Car4 endothelial cells (~15%) and Plvap endothelial cells (~85%), named after their respective marker gene Carbonic anhydrase 4 and Plasmalemma vesicle associated protein – as well as arterial and venous macrovascular endothelial cells marked by Von Willebrand factor (VWF), and lymphatic endothelial cells marked by Prospero homeobox 1 (PROX1).43
Car4 endothelial cells have a unique location, morphology, ontogeny, and function in alveologenesis.43 They preferentially occupy secondary septa, but not primary septa, and are closer to the epithelium without intervening pericytes (Fig. 3A, B). Individual Car4 endothelial cells have elaborate extensions, contributing to 5–10 vessel segments, although the same tube can also have smaller Plvap endothelial cells (Fig. 3C). Car4 endothelial cells emerge when initial alveologenesis starts and AT1 cells begin to flatten and express VEGFA. Remarkably, without epithelial VEGFA, Car4 endothelial cells, but not Plvap endothelial cells, are completely missing and classical alveologenesis is compromised as evidenced by dilated airspace and less prominent alveolar outpocketing – despite the normal appearance of alveolar myofibroblasts.43 Interestingly, initial alveologenesis in the Vegfa mutant is largely unaffected even though Car4 endothelial cells would be present in this phase, perhaps because the airspace expansion is limited then so that the epithelium has not come into immediate contact with the endothelium. Similarly, the involvement of Car4 endothelial cells in continued alveologenesis and injury-repair is unclear, although they are present in the adult lung.44
Figure 3: Car4 endothelial cells (ECs) cover alveolar islands and a model that links alveolar outpocketing to intussusceptive angiogenesis with implications for single-versus double-layered capillaries.
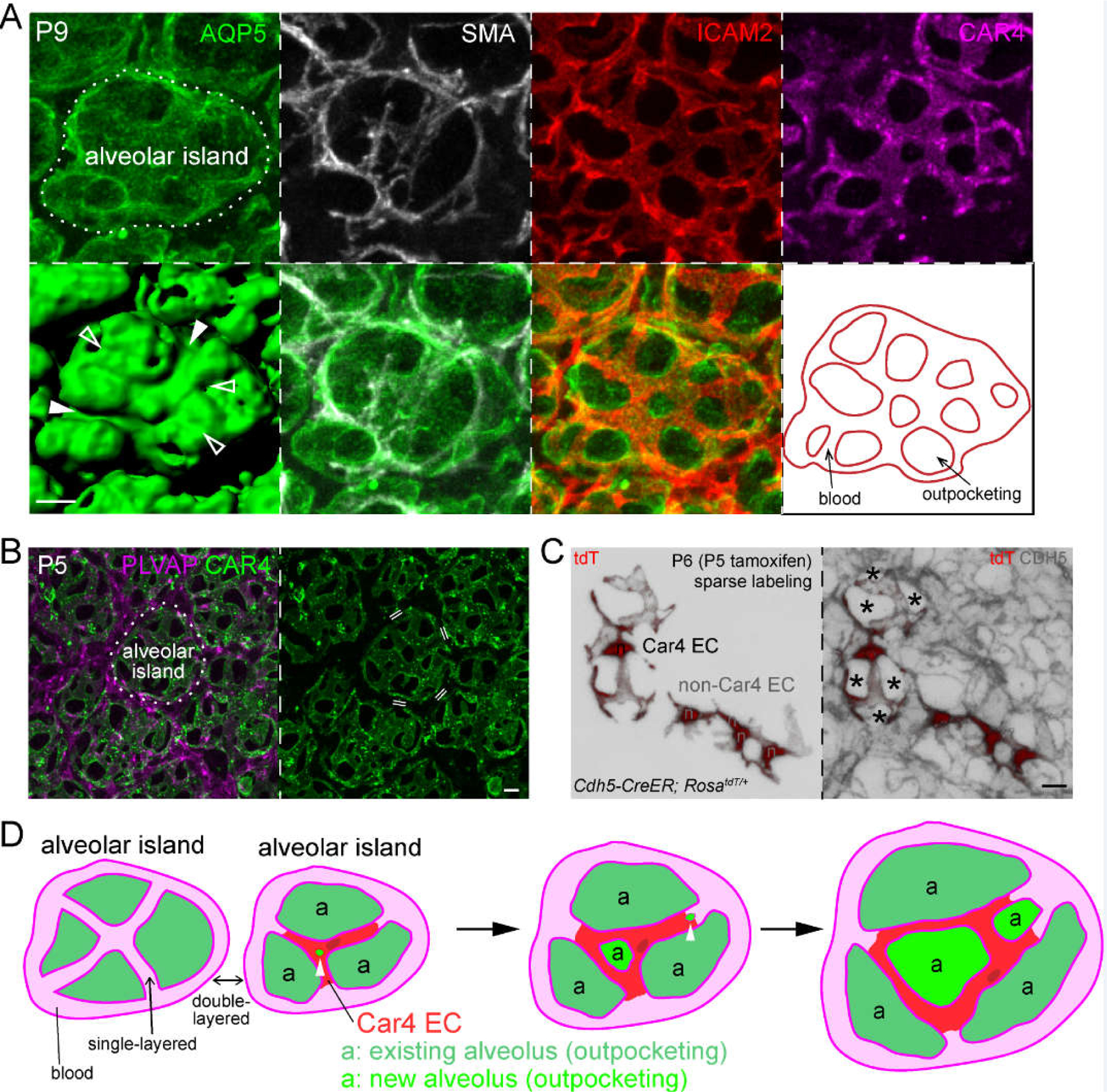
(A) En face view of confocal images of an immunostained alveolar region (AQP5) showing grooves with (filled arrowhead) or without (open arrowhead) alveolar myofibroblasts (marked by SMA), but invariably associated with vessels (ICAM2; also traced for the indicated alveolar island in the last panel). Car4 endothelial cells (CAR4) are specifically located over the alveolar island.
(B) En face view of confocal images of an immunostained alveolar region showing Car4 ECs specifically cover alveolar islands, but do not surround them as Plvap ECs do. Double line: double-layered capillaries are between alveolar islands, whereas vessels on alveolar islands are single-layered.
(C) En face view of confocal images of an immunostained alveolar region with individual endothelial cells (n, nucleus) sparsely, genetically labeled with a RosatdT reporter. The Car4 EC, as identified by costaining in the original publication,43 has cellular extensions contributing to 5–10 vessel segments, which are marked by Cadherin 5 (CDH5). Asterisks: alveolar outpocketings. Non-Car4 ECs have limited projections. (A-C) are adapted from43.
(D) A model depicting 2 alveolar islands with associated vessels and alveoli (outpocketings). The alveolar island on the right is shown to form 2 new alveoli, possibly coinciding with intussusception (arrowhead). Depending on whether intussusception occurs in the middle or at the cell junction of a Car4 EC (darker patch: nucleus), the resulting Car4 EC forms an auto-cellular loop or cellular extensions, respectively. Such a model of alveologenesis does not involve bending capillaries into a hairpin conformation so that single-layered capillaries on alveolar islands remain single-layered but increase in number as a result of intussusception, which dilutes the double-layered capillaries between alveolar islands over time.
Scale: 10 um.
Implication for intussusceptive angiogenesis and capillary maturation
Different from outward spreading led by tip endothelial cells in sprouting angiogenesis, the capillary mesh in the lung needs to amplify itself “internally” to maintain its coverage density over the expanding epithelial surface during alveologenesis. This is proposed to be achieved via intussusceptive angiogenesis, where small trans-vessel holes or “pillars” form and then expand to split existing vessels into two, although little cellular and molecular information is available.45,46 Intriguingly, Car4 endothelial cells reside where intussusceptive angiogenesis is expected to occur, and their extended morphology contributing to multiple vessels might be the result of intussusceptive angiogenesis (Fig. 3D). It is tempting to speculate that Car4 endothelial cells are the tip cell equivalent in the lung because both are VEGFA dependent and share a subset of tip cell genes such as Apelin (Apln).43 Future research is needed to dissect the divergent signaling events downstream of VEGFA that generate organ specific endothelial cell specialization.
Another intriguing idea about the lung vasculature is capillary maturation where double-layered capillary tubes fuse and become single-layered, which has been suggested to mark the end of alveologenesis and thus the maturation of alveoli.45,47 This idea is based on the arguments that (1) conceptually, secondary septation could fold a vessel into a hair-pin, forming a double-layered structure10 and (2) as the lung matures, more single-layered vessels and fewer double-layered vessels are observed. However, available data can be explained without invoking vessel folding and fusion (Fig. 2D). For the first argument, as two epithelial surfaces covered with a vascular mesh are brought into close proximity owing to airspace expansion, double-layered capillaries are expected. However, such juxtaposition occurs within primary septa, but not necessarily within secondary septa whose width fits a single vessel. Additionally, vessels run parallel to the septa, like a river carving a canyon, instead of perpendicularly being bent by the septa. Therefore, double-layered vessels occur within primary septa and single-layered vessels occur within secondary septa. For the second argument, as more secondary septa form during alveologenesis and even without a change in primary septa – specifically vessel fusion, there will appear to be fewer double-layered vessels due to a simple dilution effect. Indeed, intussusceptive angiogenesis, possibly involving Car4 endothelial cells, is expected to amplify the existing vascular mesh – all single-layered, which then becomes part of the secondary septa and constrains outpocketing.
Immune lineage
Resident and circulating immune cells in the lung are seeded overtime as a result of hematopoiesis in the yolk sac, fetal liver, and adult bone marrow. They include lymphoid cells, such as B cells, T cells, and NK cells, and myeloid cells, such as alveolar macrophages, interstitial macrophages, dendritic cells, neutrophils, and monocytes. Immune cells abound in the lung in part due to the mere necessity of oxygenation that requires all the blood carrying the circulating immune cells to passively transit through the lung – without signaling interactions. Meanwhile, exposed to environmental elements including pathogens and allergens, the lung needs to be under constant surveillance by immune cells that extravasate and reside in the interstitial and air spaces. The initial exposure to the environment at the first breath and the associated maturation of specific resident immune cells occur during alveologenesis. This review will focus on alveolar macrophages and platelets, whose roles in alveologenesis are better understood.
Alveolar macrophage
Macrophages are phagocytic and often adopt tissue-specific identities, as exemplified by microglia in the brain, Langerhans cells in the skin, Kupffer cells in the liver, and alveolar macrophages in the lung. Unlike infiltrating macrophages that respond to inflammation, tissue resident macrophages are not maintained by circulating adult monocytes, but arise from fetal monocytes and self-maintain via proliferation.48 Accordingly, alveolar macrophages arrive at the lung interstitium embryonically and relocate to the airspace within days after birth – a maturation process that depends on Granulocyte-macrophage colony-stimulating factor (GM-CSF) and Transforming growth factor beta (TGF-beta).49–54
Although alveolar macrophage deficient lungs develop pulmonary alveolar proteinosis – excessive accumulation of surfactant due to its failed catabolism by alveolar macrophages, no alveologenesis phenotype has been reported.53–55 Nevertheless, clodronate depletion of phagocytic cells – including alveolar macrophages – rescues defects in alveologenesis caused by genetic perturbations of airway or alveolar cell development or high environmental oxygen.56–58 Conceivably, in response to tissue damage, infiltrating and altered resident macrophages could release inflammatory molecules that interfere with normal alveologenesis directly or recruit other immune cells to do so. Careful lineage and single-cell transcriptomic analyses of diverse and dynamic clodronate-sensitive macrophages are necessary to unravel the seemingly non-specific phenotypic rescues.
Platelet
Anucleate fragments shed by circulating megakaryocytes, platelets are best known for their roles in hemostasis and thrombosis, but they also contain signaling molecules.59 Intriguingly, activation of C-type lectin domain family 1 member B (CLEC1B, also known as CLEC-2) on platelets by Podoplanin (PDPN, also known as T1alpha or Gp38) on lymphatic cells is necessary for initial alveologenesis, as the corresponding mutant lungs have limited alveolar duct and sac spaces and fail to inflate upon birth.60 The same CLEC1B-PDPN signaling interaction is required for the separation of lymphatics from veins, whereas loss of lymphatics leads to a similar defect in airspace expansion possibly due to inability to drain the luminal fluid of the embryonic lung near birth.61,62 Besides this mechanical role in lymphatics-mediated fluid-handling, platelets have also been suggested to signal to endothelial cells via C-X-C motif chemokine ligand 12 (CXCL12, also known as SDF-1) during pneumonectomy-induced alveologenesis; however, additional work is needed to address how the otherwise fast-transiting platelets would accumulate CXCL12 locally in a sufficient amount for signaling.63
Extracellular matrix
Besides individual cell types of the 4 lineages, evidence abounds for the roles of extracellular matrix components in alveologenesis, as partly expected from the mechanical forces necessary to support the largely air-filled alveolar region and to sustain the incessant breathing motion.64 Nevertheless, the extracellular matrix still has a cellular basis – secreted by cells and modified by enzymes of cells. Accordingly, cell-type-specific analysis of individual extracellular matrix components is needed to pinpoint their cellular sources and potentially distinct regulation of embedded cells. Compared to the epithelial and endothelial basement membranes and interstitial collagen lattices, the elastin network and its assembly are more specific to the lung and will be discussed here.
Elastin
Similar to its role in accommodating pulsatile blood flow in large vessels, the elastin network supports and constrains alveolar tissue expansion during breathing. Elastin fibers are assembled when secreted soluble tropoelastin monomers are incorporated into a microfibrillar scaffold, and crosslinked to become an insoluble polymer.65 Notwithstanding redundancy, misregulation of any components of this assembly process will impact the alveolar region: loss of tropoelastin (ELN) itself represents the extreme disruption. Notably, the Eln mutant manifests airspace dilation upon birth – presumably due to mechanical failure from the first breath – before classical alveologenesis takes place and alveolar myofibroblasts appear, suggesting that the elastin network can function independent of alveolar myofibroblasts.66 Even during classical alveologenesis, without cell-type-specific deletion, it is unclear which components of the elastogenesis pathway are functionally expressed by alveolar myofibroblasts. Moreover, the normally localized elastin bundles67 have often been described as diffuse, discontinuous, or disorganized in alveologenesis mutants; however, this aberrant appearance could merely reflect elastin bundles coursing through tortuous alveolar tissue that is now of variable thickness and borders dilated airspaces. Therefore, the temporal dissociation between elastin functions and alveolar myofibroblasts, the lack of precise genetic analyses, and the technical challenge in quantifying elastin bundling leave open the possibility that the elastin’s elasticity and alveolar myofibroblast’s contractility represent distinct mechanisms controlling alveologenesis. Intriguingly, the Hox5 triple mutant lungs have reduced elastin without affecting other extracellular matrix components, as well as reduced adhesion of alveolar myofibroblasts to fibronectin in culture, although it is unclear whether these two defects are related and which is primary.68
After assembly, the elastin network in the mature lung is actively guarded against inflammatory destruction, such as degradation by neutrophil elastase. This is evidenced by progressive emphysema in alpha-1 antitrypsin deficient humans and mouse mutants for the 5 corresponding paralogs, although it is unclear – as in studies of proteases and their inhibitors in general – if other proteases and extracellular matrix components are also affected.69,70
Section III: Alveologenesis in diseases and after injury
The myriad cell types involved, their intricate intercellular signaling, and the resulting delicate tissue structure, render alveologenesis sensitive to perturbations – most notably premature birth and the associated necessary interventions. In addition, the alveolar surface is at constant risk of injury by being exposed to several liters of air per minute and, when damaged, needs to be repaired in situ or to form de novo elsewhere to compensate for the loss (Fig. 4).
Figure 4: In situ repair versus de novo growth as distinct modes of lung regeneration.
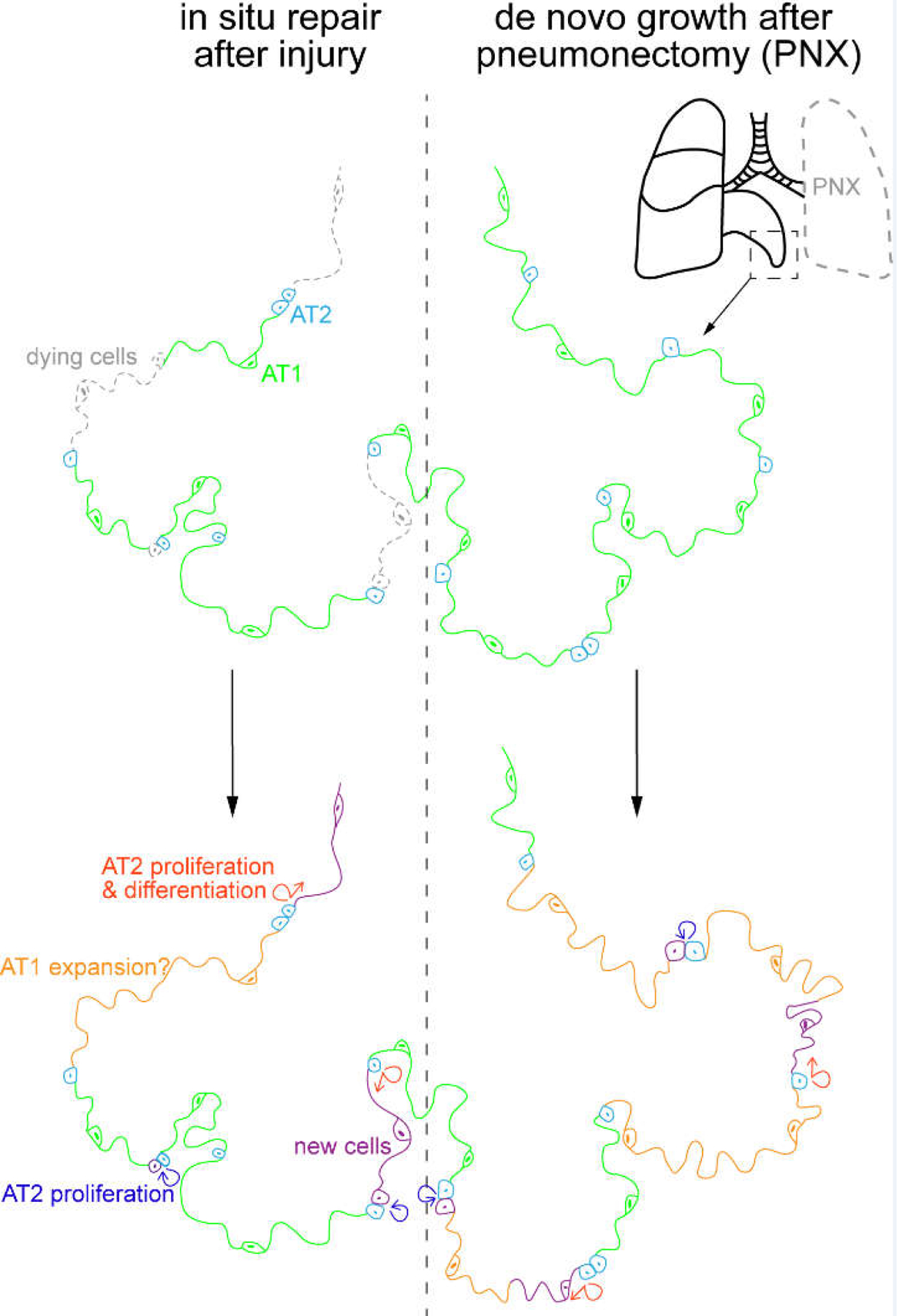
Two extreme modes of lung regeneration. Left: epithelial cell loss without affecting the extracellular matrix can be replaced in situ via AT2 cell proliferation and their differentiation into AT1 cells, as well as possible AT1 cell expansion. Not depicted is the contribution of airway-derived cells, such as p63-expressing atypical basal cells forming dysplastic clusters. Right: In pneumonectomy, where the left lobe is removed, the uninjured lobe grows to compensate for the loss in gas exchange capacity. This growth in tissue volume, surface area, and cell number occurs de novo in the available empty space, possibly due to the reactivation of developmental programs. Cellular changes are color-coded as in the left schematic. Chemical or pathogen-induced lung injury likely activates both modes of lung regeneration.
Bronchopulmonary dysplasia (BPD)
Forced transition to extrauterine life due to premature birth disrupts alveologenesis by exposing otherwise fluid-submerged epithelium to inhaled air or supplemental oxygen and by switching haphazard fetal breathing movements to on-demand, sometimes ventilator-driven, constant respirations. Such life-supporting oxygenation and ventilation used to be so aggressive that the lung often developed marked fibrosis with extensive inflammation – key features of “old” BPD.71 Medical advances including surfactant supplementation and antenatal glucocorticoids have reduced the incidence of old BPD and allowed survival of much younger infants – born around 23 weeks of gestational age – who instead often develop “new” BPD, characterized by alveolar simplification and dysmorphic vasculature and clinically diagnosed by a dependency on supplemental oxygen at corrected 36 weeks of gestational age.72,73 These new BPD individuals are born at the canalicular and saccular stages – during the initial alveologenesis phase when AT1 cells start to flatten, distal epithelial progenitors could still be branching and forming alveolar ducts, and alveolar macrophages are maturing (Fig. 1).
Unlike genetic diseases or infections by specific pathogens, BPD is triggered by aberrant developmental timing and generic environmental factors, thereby affecting multiple cell types. Accordingly and perhaps unsurprisingly, numerous causes and interventions of BPD have been put forward1; however, besides independent validation, it is important to pinpoint the responsible cell types or environmental factors, such as oxidative damage.
Catch-up growth in BPD is incomplete such that in adulthood, affected individuals still have lower pulmonary functions and higher susceptibility to pulmonary diseases and infections74 – consistent with a developmental origin of adult diseases as in the Barker hypothesis.75 It is unclear if the catch-up growth occurs during continued alveologenesis, and potentially beyond this phase, and how it stops. More work is needed to define the persistent changes in the alveolar structure and cell types to discern whether the adulthood pulmonary deficiencies are due to a non-specific, lower functional reserve versus specific, aberrant gas diffusion or responses to perturbations – such as exaggerated inflammation or limited stem cell renewal.75–77
In situ repair versus de novo growth
Despite having limited cell turnover at baseline at least in laboratory settings, the adult lung efficiently recovers from injuries by mobilizing regional progenitor/stem cells – most notably AT2 cells and basal cells for the epithelium.78,79 At one extreme, a decellularized lung that has been artificially stripped of its cells and only retains its extracellular matrix can be repopulated by exogenous epithelial and endothelial cells and briefly support gas exchange.80 At the other extreme, removal of the left lung lobe stimulates expansion and new alveoli formation in the uninjured right lobes.81 More commonly, chemical or pathogen-induced injuries could trigger both recellularization of damaged existing alveolar surface and de novo construction of alveoli, perhaps near expandable termini of the respiratory tree and in response to extra mechanical stretching.82 The associated cellular and molecular mechanisms are potentially distinct: the in situ repair of the alveolar epithelium could involve competition among differentiating AT2 cells, expanding AT1 cells, and atypical basal cells that form dysplastic “Krt5+ pods”78; whereas the compensatory de novo growth could recapitulate the signaling and morphogenetic events of developmental alveologenesis albeit without bona fide developmental progenitors (Fig. 4). Therefore, it is important to recognize repair versus growth as distinct forms of alveolar regeneration. Notably, recent studies have identified intermediate cell states as progenitor/stem cells transition to AT1 cells and failure in such a transition can lead to pulmonary fibrosis.83–87
Summary
This review (1) highlights the difficulty in defining alveoli of variable sizes and shapes; (2) proposes a 3-phase definition of alveologenesis – initial, classical, and continued phases – that includes alveolar ducts and sacs and is based on measurable changes in AT1 cells and alveolar myofibroblasts; (3) challenges the capillary maturation model as prompted by recent 3D imaging and single-cell analyses; and (4) discusses developmental and cell biology concepts in alveologenesis, alveolar regeneration and diseases.
Key Findings.
We propose a cell-centric 3-phase definition of lung alveologenesis to account for the morphological ambiguity of alveoli.
The initial alveologenesis phase begins with alveolar type 1 cell flattening and forms alveolar ducts and alveolar sacs.
The classical alveologenesis phase is marked by alveolar myofibroblasts.
The continued alveologenesis phase increases alveolar surface and outpocketing in the absence of alveolar myofibroblasts.
A distinct Car4 endothelial cell population demarcates and is required for alveolar outpocketing, and may underlie intussusceptive angiogenesis but does not support the model of double- to single-layered capillary conversion.
Grants
This work was supported by the University of Texas MD Anderson Cancer Center Start-up and Retention Fund, an American Lung Association Innovation Award (#626356) and National Institutes of Health R01HL130129 and R01HL153511 (JC).
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Lignelli E, Palumbo F, Myti D, Morty RE. Recent advances in our understanding of the mechanisms of lung alveolarization and bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. December 1 2019;317(6):L832–L887. 10.1152/ajplung.00369.2019. [DOI] [PubMed] [Google Scholar]
- 2.Schittny JC. Development of the lung. Cell Tissue Res. March 2017;367(3):427–444. 10.1007/s00441-016-2545-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang DR, Martinez Alanis D, Miller RK, et al. Lung epithelial branching program antagonizes alveolar differentiation. Proc Natl Acad Sci U S A. November 5 2013;110(45):18042–51. 10.1073/pnas.1311760110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alanis DM, Chang DR, Akiyama H, Krasnow MA, Chen J. Two nested developmental waves demarcate a compartment boundary in the mouse lung. Nature communications. 2014;5:3923. 10.1038/ncomms4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J, Chen J. Developmental programs of lung epithelial progenitors: a balanced progenitor model. Wiley interdisciplinary reviews Developmental biology. Sep-Oct 2014;3(5):331–47. 10.1002/wdev.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang J, Hernandez BJ, Martinez Alanis D, et al. The development and plasticity of alveolar type 1 cells. Development. January 1 2016;143(1):54–65. 10.1242/dev.130005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Branchfield K, Li R, Lungova V, Verheyden JM, McCulley D, Sun X. A three-dimensional study of alveologenesis in mouse lung. Dev Biol. January 15 2016;409(2):429–41. 10.1016/j.ydbio.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bostrom H, Willetts K, Pekny M, et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell. June 14 1996;85(6):863–73. https://doi.orgS0092-8674(00)81270-2 [pii]. [DOI] [PubMed] [Google Scholar]
- 9.Schittny JC. How high resolution 3-dimensional imaging changes our understanding of postnatal lung development. Histochem Cell Biol. December 2018;150(6):677–691. 10.1007/s00418-018-1749-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schittny JC, Mund SI, Stampanoni M. Evidence and structural mechanism for late lung alveolarization. Am J Physiol Lung Cell Mol Physiol. February 2008;294(2):L246–54. 10.1152/ajplung.00296.2007. [DOI] [PubMed] [Google Scholar]
- 11.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. March 13 2014;507(7491):190–4. 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rawlins EL, Clark CP, Xue Y, Hogan BL. The Id2+ distal tip lung epithelium contains individual multipotent embryonic progenitor cells. Development. November 2009;136(22):3741–5. https://doi.org136/22/3741 [pii] 10.1242/dev.037317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frank DB, Penkala IJ, Zepp JA, et al. Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc Natl Acad Sci U S A. March 5 2019;116(10):4362–4371. 10.1073/pnas.1813952116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez R, Leaffer D, Chapin C, Gillespie AM, Eckalbar W, Dobbs L. Cell fate analysis in fetal mouse lung reveals distinct pathways for TI and TII cell development. Am J Physiol Lung Cell Mol Physiol. November 1 2019;317(5):L653–L666. 10.1152/ajplung.00503.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weibel ER. The mystery of “non-nucleated plates” in the alveolar epithelium of the lung explained. Acta Anat (Basel). 1971;78(3):425–43. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Wang Z, Chu Q, Jiang K, Li J, Tang N. The Strength of Mechanical Forces Determines the Differentiation of Alveolar Epithelial Cells. Dev Cell. February 5 2018;44(3):297–312 e5. 10.1016/j.devcel.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 17.Little DR, Gerner-Mauro KN, Flodby P, et al. Transcriptional control of lung alveolar type 1 cell development and maintenance by NK homeobox 2-1. Proc Natl Acad Sci U S A. October 8 2019;116(41):20545–20555. 10.1073/pnas.1906663116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nantie LB, Young RE, Paltzer WG, et al. Lats1/2 inactivation reveals Hippo function in alveolar type I cell differentiation during lung transition to air breathing. Development. November 9 2018;145(21). 10.1242/dev.163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrisey EE, Cardoso WV, Lane RH, et al. Molecular determinants of lung development. Ann Am Thorac Soc. April 2013;10(2):S12–6. 10.1513/AnnalsATS.201207-036OT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ardini-Poleske ME, Clark RF, Ansong C, et al. LungMAP: The Molecular Atlas of Lung Development Program. Am J Physiol Lung Cell Mol Physiol. November 1 2017;313(5):L733–L740. 10.1152/ajplung.00139.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beers MF, Moodley Y. When Is an Alveolar Type 2 Cell an Alveolar Type 2 Cell? A Conundrum for Lung Stem Cell Biology and Regenerative Medicine. Am J Respir Cell Mol Biol. July 2017;57(1):18–27. 10.1165/rcmb.2016-0426PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheong N, Zhang H, Madesh M, et al. ABCA3 is critical for lamellar body biogenesis in vivo. J Biol Chem. August 17 2007;282(33):23811–7. https://doi.orgM703927200 [pii] 10.1074/jbc.M703927200. [DOI] [PubMed] [Google Scholar]
- 23.Besnard V, Matsuzaki Y, Clark J, et al. Conditional deletion of Abca3 in alveolar type II cells alters surfactant homeostasis in newborn and adult mice. Am J Physiol Lung Cell Mol Physiol. May 2010;298(5):L646–59. 10.1152/ajplung.00409.200900409.2009 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. July 1 2013;123(7):3025–36. 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adamson IY, Bowden DH. Derivation of type 1 epithelium from type 2 cells in the developing rat lung. Lab Invest. June 1975;32(6):736–45. [PubMed] [Google Scholar]
- 26.Treutlein B, Brownfield DG, Wu AR, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. May 15 2014;509(7500):371–5. 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. March 9 2018;359(6380):1118–1123. 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frank DB, Peng T, Zepp JA, et al. Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Rep. November 22 2016;17(9):2312–2325. 10.1016/j.celrep.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zacharias WJ, Frank DB, Zepp JA, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. March 8 2018;555(7695):251–255. 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Travaglini KJ, Nabhan AN, Penland L, et al. A molecular cell atlas of the human lung from single cell RNA sequencing. bioRxiv. 2019:742320. 10.1101/742320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ntokou A, Szibor M, Rodriguez-Castillo JA, et al. A novel mouse Cre-driver line targeting Perilipin 2-expressing cells in the neonatal lung. Genesis. December 2017;55(12). 10.1002/dvg.23080. [DOI] [PubMed] [Google Scholar]
- 32.Li R, Bernau K, Sandbo N, Gu J, Preissl S, Sun X. Pdgfra marks a cellular lineage with distinct contributions to myofibroblasts in lung maturation and injury response. eLife. September 4 2018;7. 10.7554/eLife.36865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gouveia L, Betsholtz C, Andrae J. PDGF-A signaling is required for secondary alveolar septation and controls epithelial proliferation in the developing lung. Development. April 10 2018;145(7). 10.1242/dev.161976. [DOI] [PubMed] [Google Scholar]
- 34.Endale M, Ahlfeld S, Bao E, et al. Temporal, spatial, and phenotypical changes of PDGFRalpha expressing fibroblasts during late lung development. Dev Biol. May 15 2017;425(2):161–175. 10.1016/j.ydbio.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hagan AS, Zhang B, Ornitz DM. Identification of a FGF18-expressing alveolar myofibroblast that is developmentally cleared during alveologenesis. Development. January 17 2020;147(2). 10.1242/dev.181032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C, Li M, Li S, et al. Progenitors of secondary crest myofibroblasts are developmentally committed in early lung mesoderm. Stem Cells. March 2015;33(3):999–1012. 10.1002/stem.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El Agha E, Moiseenko A, Kheirollahi V, et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell. February 2 2017;20(2):261–273 e3. 10.1016/j.stem.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park J, Ivey MJ, Deana Y, et al. The Tcf21 lineage constitutes the lung lipofibroblast population. Am J Physiol Lung Cell Mol Physiol. May 1 2019;316(5):L872–L885. 10.1152/ajplung.00254.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li R, Li X, Hagood J, Zhu MS, Sun X. Myofibroblast contraction is essential for generating and regenerating the gas-exchange surface. J Clin Invest. June 1 2020;130(6):2859–2871. 10.1172/JCI132189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. June 1999;126(14):3047–55. [DOI] [PubMed] [Google Scholar]
- 41.Kato K, Dieguez-Hurtado R, Park DY, et al. Pulmonary pericytes regulate lung morphogenesis. Nature communications. Jun 22 2018;9(1):2448. 10.1038/s41467-018-04913-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stevens T, Phan S, Frid MG, Alvarez D, Herzog E, Stenmark KR. Lung vascular cell heterogeneity: endothelium, smooth muscle, and fibroblasts. Proc Am Thorac Soc. September 15 2008;5(7):783–91. 10.1513/pats.200803-027HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vila Ellis L, Cain MP, Hutchison V, et al. Epithelial Vegfa Specifies a Distinct Endothelial Population in the Mouse Lung. Dev Cell. March 9 2020;52(5):617–630 e6. 10.1016/j.devcel.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niethamer TK, Stabler CT, Leach JP, et al. Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. Elife. February 24 2020;9. 10.7554/eLife.53072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caduff JH, Fischer LC, Burri PH. Scanning electron microscope study of the developing microvasculature in the postnatal rat lung. Anat Rec. October 1986;216(2):154–64. 10.1002/ar.1092160207. [DOI] [PubMed] [Google Scholar]
- 46.Djonov V, Schmid M, Tschanz SA, Burri PH. Intussusceptive angiogenesis: its role in embryonic vascular network formation. Circ Res. February 18 2000;86(3):286–92. [DOI] [PubMed] [Google Scholar]
- 47.Burri PH. Postnatal growth and maturation of the lung. Chest. February 1975;67(2 Suppl):2S–3S. 10.1378/chest.67.2_supplement.2s. [DOI] [PubMed] [Google Scholar]
- 48.Hashimoto D, Chow A, Noizat C, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. April 18 2013;38(4):792–804. 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guilliams M, De Kleer I, Henri S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. September 23 2013;210(10):1977–92. 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan SY, Krasnow MA. Developmental origin of lung macrophage diversity. Development. April 15 2016;143(8):1318–27. 10.1242/dev.129122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dranoff G, Crawford AD, Sadelain M, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science. April 29 1994;264(5159):713–6. 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 52.Yu X, Buttgereit A, Lelios I, et al. The Cytokine TGF-beta Promotes the Development and Homeostasis of Alveolar Macrophages. Immunity. November 21 2017;47(5):903–912 e4. 10.1016/j.immuni.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Dranoff G, Mulligan RC. Activities of granulocyte-macrophage colony-stimulating factor revealed by gene transfer and gene knockout studies. Stem Cells. 1994;12 Suppl 1:173–82; discussion 182–4. [PubMed] [Google Scholar]
- 54.Stanley E, Lieschke GJ, Grail D, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A. June 7 1994;91(12):5592–6. 10.1073/pnas.91.12.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uchida K, Nakata K, Trapnell BC, et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood. February 1 2004;103(3):1089–98. 10.1182/blood-2003-05-1565. [DOI] [PubMed] [Google Scholar]
- 56.Branchfield K, Nantie L, Verheyden JM, Sui P, Wienhold MD, Sun X. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science. February 12 2016;351(6274):707–10. 10.1126/science.aad7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plosa EJ, Young LR, Gulleman PM, et al. Epithelial beta1 integrin is required for lung branching morphogenesis and alveolarization. Development. December 2014;141(24):4751–62. 10.1242/dev.117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalymbetova TV, Selvakumar B, Rodriguez-Castillo JA, et al. Resident alveolar macrophages are master regulators of arrested alveolarization in experimental bronchopulmonary dysplasia. J Pathol. June 2018;245(2):153–159. 10.1002/path.5076. [DOI] [PubMed] [Google Scholar]
- 59.Kamykowski J, Carlton P, Sehgal S, Storrie B. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet alpha-granules. Blood. August 4 2011;118(5):1370–3. 10.1182/blood-2011-01-330910. [DOI] [PubMed] [Google Scholar]
- 60.Tsukiji N, Inoue O, Morimoto M, et al. Platelets play an essential role in murine lung development through Clec-2/podoplanin interaction. Blood. September 13 2018;132(11):1167–1179. 10.1182/blood-2017-12-823369. [DOI] [PubMed] [Google Scholar]
- 61.Bertozzi CC, Schmaier AA, Mericko P, et al. Platelets regulate lymphatic vascular development through CLEC-2-SLP-76 signaling. Blood. July 29 2010;116(4):661–70. 10.1182/blood-2010-02-270876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jakus Z, Gleghorn JP, Enis DR, et al. Lymphatic function is required prenatally for lung inflation at birth. J Exp Med. May 5 2014;211(5):815–26. 10.1084/jem.20132308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rafii S, Cao Z, Lis R, et al. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat Cell Biol. February 2015;17(2):123–136. 10.1038/ncb3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mizikova I, Morty RE. The Extracellular Matrix in Bronchopulmonary Dysplasia: Target and Source. Front Med (Lausanne). 2015;2:91. 10.3389/fmed.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yanagisawa H, Davis EC. Unraveling the mechanism of elastic fiber assembly: The roles of short fibulins. Int J Biochem Cell Biol. July 2010;42(7):1084–93. 10.1016/j.biocel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol. September 2000;23(3):320–6. [DOI] [PubMed] [Google Scholar]
- 67.Luo Y, Li N, Chen H, et al. Spatial and temporal changes in extracellular elastin and laminin distribution during lung alveolar development. Sci Rep. May 29 2018;8(1):8334. 10.1038/s41598-018-26673-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hrycaj SM, Marty-Santos L, Cebrian C, et al. Hox5 genes direct elastin network formation during alveologenesis by regulating myofibroblast adhesion. Proc Natl Acad Sci U S A. November 6 2018;115(45):E10605–E10614. 10.1073/pnas.1807067115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Strnad P, McElvaney NG, Lomas DA. Alpha1-Antitrypsin Deficiency. N Engl J Med. April 9 2020;382(15):1443–1455. 10.1056/NEJMra1910234. [DOI] [PubMed] [Google Scholar]
- 70.Borel F, Sun H, Zieger M, et al. Editing out five Serpina1 paralogs to create a mouse model of genetic emphysema. Proc Natl Acad Sci U S A. March 13 2018;115(11):2788–2793. 10.1073/pnas.1713689115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Northway WH Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med. February 16 1967;276(7):357–68. 10.1056/NEJM196702162760701. [DOI] [PubMed] [Google Scholar]
- 72.Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res. December 1999;46(6):641–3. [DOI] [PubMed] [Google Scholar]
- 73.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. June 2001;163(7):1723–9. 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 74.Malleske DT, Chorna O, Maitre NL. Pulmonary sequelae and functional limitations in children and adults with bronchopulmonary dysplasia. Paediatr Respir Rev. March 2018;26:55–59. 10.1016/j.prrv.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 75.Weiss ST. Lung function and airway diseases. Nat Genet. January 2010;42(1):14–6. 10.1038/ng0110-14. [DOI] [PubMed] [Google Scholar]
- 76.Davidson LM, Berkelhamer SK. Bronchopulmonary Dysplasia: Chronic Lung Disease of Infancy and Long-Term Pulmonary Outcomes. J Clin Med. January 6 2017;6(1). 10.3390/jcm6010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Balinotti JE, Chakr VC, Tiller C, et al. Growth of lung parenchyma in infants and toddlers with chronic lung disease of infancy. Am J Respir Crit Care Med. May 15 2010;181(10):1093–7. 10.1164/rccm.200908-1190OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen J Origin and regulation of a lung repair kit. Nat Cell Biol. July 28 2017;19(8):885–886. 10.1038/ncb3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zepp JA, Morrisey EE. Cellular crosstalk in the development and regeneration of the respiratory system. Nat Rev Mol Cell Biol. September 2019;20(9):551–566. 10.1038/s41580-019-0141-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song JJ, Kim SS, Liu Z, et al. Enhanced in vivo function of bioartificial lungs in rats. Ann Thorac Surg. September 2011;92(3):998–1005; discussion 1005–6. 10.1016/j.athoracsur.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 81.Brown LM, Rannels SR, Rannels DE. Implications of post-pneumonectomy compensatory lung growth in pulmonary physiology and disease. Respir Res. 2001;2(6):340–7. 10.1186/rr84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Y, Tang Z, Huang H, et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc Natl Acad Sci U S A. March 6 2018;115(10):2407–2412. 10.1073/pnas.1719474115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strunz M, Simon LM, Ansari M, et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nature communications. July 16 2020;11(1):3559. 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kobayashi Y, Tata A, Konkimalla A, et al. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol. August 2020;22(8):934–946. 10.1038/s41556-020-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiang P, Gil de Rubio R, Hrycaj SM, et al. Ineffectual Type 2-to-Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8(hi) Transitional State. Am J Respir Crit Care Med. June 1 2020;201(11):1443–1447. 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Choi J, Park JE, Tsagkogeorga G, et al. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell. September 3 2020;27(3):366–382 e7. 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu H, Yu Y, Huang H, et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell. January 9 2020;180(1):107–121 e17. 10.1016/j.cell.2019.11.027. [DOI] [PubMed] [Google Scholar]