

Abstract
Enterotoxigenic Bacteroides fragilis (ETBF) is a commensal bacterium of great importance to human health due to its ability to induce colitis and cause colon tumor formation in mice through the production of B. fragilis toxin (BFT). The formation of tumors is dependent on a pro-inflammatory signaling cascade, which begins with the disruption of epithelial barrier integrity through cleavage of E-cadherin. Here, we show that BFT increases levels of glucosylceramide, a vital intestinal sphingolipid, both in mice and in colon organoids (colonoids) generated from the distal colons of mice. When colonoids are treated with BFT in the presence of an inhibitor of glucosylceramide synthase (GCS), the enzyme responsible for generating glucosylceramide, colonoids become highly permeable, lose structural integrity, and eventually burst, releasing their contents into the extracellular matrix. By increasing glucosylceramide levels in colonoids via an inhibitor of glucocerebrosidase (GBA, the enzyme that degrades glucosylceramide), colonoid permeability was reduced, and bursting was significantly decreased. In the presence of BFT, pharmacological inhibition of GCS caused levels of tight junction protein 1 (TJP1) to decrease. However, when GBA was inhibited, TJP1 levels remained stable, suggesting that BFT-induced production of glucosylceramide helps to stabilize tight junctions. Taken together, our data demonstrate a glucosylceramide-dependent mechanism by which the colon epithelium responds to BFT.
Keywords: Bacteroides fragilis toxin, glucosylceramides, organoids, permeability, tight junctions
1 |. INTRODUCTION
Bacteroides fragilis is a human commensal bacterium with two main molecular subtypes, each with opposing effects on human health. The first, non-toxigenic B. fragilis (NTBF), is a beneficial symbiote that promotes the expansion of anti-inflammatory regulatory T-cells (Treg), while also suppressing pro-inflammatory T-helper 17 (Th17) cells known to aid in colon tumorigenesis.1,2 The second subtype, enterotoxigenic B. fragilis (ETBF), promotes colitis and colon tumorigenesis through a Th17-dependent mechanism.3 The main difference between the NTBF and ETBF subtypes is the production of B. fragilis toxin (BFT) by ETBF strains.4 BFT is a 20 kDa zinc-dependent metalloprotease that binds to an unknown receptor on colon epithelial cells and triggers a cascade of pro-inflammatory signaling events.5 One of the first events is the cleavage of E-cadherin, which leads to an increase in paracellular epithelial permeability.6,7 This increased permeability causes loss of barrier function in the colon and stimulates an immune response that is required for ETBF-induced tumorigenesis.8
Beyond E-cadherin, tight junctions also play a critical role in maintaining epithelial cell barrier function and additionally serve to control the passage of molecules through the paracellular space.9 Tight junctions are comprised of several proteins,9,10 but we focused on three tight junction-associated proteins that are highly expressed in the distal colon: claudin-3, occludin, and tight junction protein 1 (TJP1, also known as zonula occludens-1, or ZO-1.11 Claudin-3 belongs to the claudin family of transmembrane proteins that regulate paracellular permeability and, similar to occludin, has the ability to bind to TJP1.9,10 Occludin is another transmembrane protein that binds to TJP1 and regulates tight junctions.12 TJP1 serves as a scaffolding protein, holding together transmembrane tight junction proteins (such as occludin and claudins), while also anchoring them to the colon epithelial cell (CEC) cytoskeleton.13 Importantly, in response to epithelial cell damage, TJP1 and E-cadherin colocalize and reestablish cell-to-cell junctions.14
Herein, we focused on sphingolipids, a class of lipids containing a sphingoid backbone that has a wide range of roles in healthy cells such as controlling growth and differentiation, regulating apoptosis, and maintaining structural integrity.15,16 Of particular interest in the intestines is glucosylceramide, a sphingolipid involved in inflammation, cell adhesion, and enterocyte function.15,17 Glucosylceramide is generated by glucosylceramide synthase (GCS) and broken down by glucocerebrosidase (GBA). These enzymes attach (GCS) or remove (GBA) a glucose molecule to or from the sphingolipid, ceramide.18 Although global GCS knockout in mice is embryonic lethal, targeted GCS knockout in mouse enterocytes leads to intestinal distress, dysfunction, and soon after, death.17,19 In colon cancer, GCS is frequently overexpressed and can lead to multidrug resistance, evasion of apoptosis, and increased cellular proliferation.15,16,20–24
Considering the epithelial-specific signaling events caused by BFT, and the importance of glucosylceramide in maintaining the structural integrity of the intestinal epithelium, we hypothesized that BFT may alter glucosylceramide synthase levels. To test this hypothesis we utilized colon organoids (or “colonoids”), an ex vivo model for a healthy colon.25 The utilization of colonoids afforded us the ability to interrogate the importance of sphingolipids in healthy CECs exposed to BFT. Using pharmacological inhibitors of GCS and GBA, we have defined a novel and functionally significant role for glucosylceramide in cells undergoing BFT challenge.
2 |. MATERIALS AND METHODS
2.1 |. Animal experiments
Animal experiments were approved by the Johns Hopkins University Institutional Animal Care and Use Committee and performed at Johns Hopkins University School of Medicine. Five-week-old C57BL/6J mice (Jackson Laboratory) were given a 1-week course of antibiotics, 0.1 mg/mL of clindamycin, and 2 mg/mL of streptomycin delivered orally through the drinking water, to enhance bacterial colonization. After a 1-week course of antibiotics, mice were colonized with 1×108 CFU/100 μL ETBF (86-5443-2-2),26 ETBFΔbft,8 or 100 μL of phosphate-buffered saline (PBS, Gibco 10010023) via oral gavage. After 1 week, mice were euthanized, and colons were extracted for lipid analysis.
2.2 |. Lipidomic analysis
Lipids were extracted as previously described,27 and analyzed by ultra-performance liquid chromatography-electrospray tandem mass spectrometry (Waters ACQUITY Xevo TQ-S) based on the method described by Merrill et al.28 Briefly, tissue was homogenized using a BeadBug bead homogenizer (Thomas Scientific D1030-E), sonicated, and resuspended in 0.1X PBS. Colonoids were isolated from 3D-growth matrix, pelleted, and resuspended in 0.1X PBS. Resuspended colonoids were then lysed by sonication. Total protein was determined and quantified using the Bio-Rad DC Protein Assay (Bio-Rad 5000116). For each experiment, all samples submitted for mass spectrometry analysis contained the same amount of protein (typically a minimum of 40 μg/sample). Resuspended tissue/cells were added to 2 mL of lipid extraction mixture (isopropanol:water:ethyl acetate at 30:10:60 v/v/v), vortexed, sonicated, and then incubated for 1 hour with shaking. Samples were centrifuged and the upper organic layer was collected. The remaining sample was re-extracted using the above methods and the resulting organic layers from the two extractions were combined. The combined organic layers were dried using nitrogen gas and then resuspended in the mobile phase buffer. Mobile phase A consisted of 70:30 w/w water:acetonitrile with 0.2% formic acid, 10 mmol/L ammonium formate, and 1 mmol/L methylphosphonic acid. Mobile phase B was comprised of 90:10 w/w isopropanol:methanol with 0.2% formic acid, 10 mmol/L ammonium formate, and 1 mmol/L methylphosphonic acid. Samples were filtered using a 96 well 0.45 μm hydrophilic MultiScreen Solvinert Filter Plate (Millipore Sigma MSRLN0410) and stored at −20°C until they were analyzed. Data from tissue or cell lysates are represented as picomoles of lipid per milligram of protein.
2.3 |. Colonoid isolation and culture
Colonoids were isolated from the distal colons of C57BL/6J mice (Jackson Laboratory). The isolation protocol and media components have both been described previously.29,30 Briefly, the distal colon was extracted and cut into 1-mm pieces, washed 5–6 times in cold chelating solution (CCS),30 and incubated in CCS with 10 mmol/L EDTA to release colon tissue crypts. Fetal bovine serum (FBS) was added to aid in the collection of the crypts. Crypts were pelleted and resuspended in Matrigel (Corning 356 231). Resuspended crypts were plated within 3D domes using 25 μL Matrigel/well on a pre-warmed 24-well plate (Genesee Scientific 25–107) and placed at 37°C for 15 minutes to allow the Matrigel to solidify. Once solidified, 500 μL complete medium with growth factors (CMGF+) was added to each well and refreshed every other day. By Day 3, crypts began forming colonoids. After 1 week, colonoids were passaged and expanded for experimental use.
Complete medium without growth factors (CMGF−) and CMGF + growth media were prepared fresh weekly. CMGF− basal media consisted of Advanced DMEM/F12 (Gibco 12 634 010), GlutaMAX (2 mmol/L; Gibco 35050061), HEPES (10 mmol/L; Gibco 15630080), Penicillin/streptomycin (100 U/mL; Gibco 15140122). CMGF + growth media consisted of CMGF− (17.5% v/v), L-WNT3A-conditioned media (50% v/v; provided by the Zachos Laboratory), R-Spondin-conditioned media (20% v/v; provided by the Zachos Laboratory), Noggin-conditioned media (10% v/v; provided by the Zachos Laboratory), B27 (1X; Invitrogen 17504-044), N-acetylcysteine (1 mmol/L; Sigma A9165), epidermal growth factor (EGF; 50 ng/mL; R&D 236-EG-01M), [Leu15] Gastrin (10 nmol/L; Sigma G9145), A83–01 (500 nmol/L; Tocris 2939), SB202190 (10 μmol/L; Sigma S7067), and Primocin (100 μg/mL; InvivoGen ant-pm-2).
2.4 |. Bacterial culture and preparation of concentrated bacterial culture supernatants
Enterotoxigenic B. fragilis (ETBF, 86-5443-2-2),26 enterotoxigenic B. fragilis Δbft (ETBFΔbft),8 recombinant ETBFΔbft expressing BFT2 (rETBFΔbft bft-2; unpublished), non-toxigenic B. fragilis (NTBF, NCTC 9343),31 recombinant NTBF expressing BFT2 (rNTBF bft-2),31 and recombinant NTBF expressing a mutated and inactive form of BFT (rNTBF bft-2 H352Y)32 were all obtained from the Sears Lab and grown in brain heart infusion broth (BHI; BD Bacto 237200) with hemin (Sigma H9039), vitamin K1 (Sigma V3501), and clindamycin hydrochloride (Sigma C5269) under anaerobic conditions. To generate concentrated bacterial culture supernatants from B fragilis strains, bacteria were grown in 500 mL of BHI media for 24 hours. The bacterial suspension was centrifuged at 4000g for 20 minutes to pellet the cells. The supernatant was collected and the bacterial pellet was discarded. The resulting supernatant was then filtered using a 0.45 μm syringe filter (GE Healthcare 6780-2504) to remove residual cells or debris. After this, the filtered supernatant was added to a 10 kDa centrifuge filter tube (Amicon UFC901024) and centrifuged at 4000g for 30 minutes according to the manufacturer’s protocol. The flowthrough was discarded and the remaining media is referred to as concentrated bacterial culture supernatant. In this study, concentrated bacterial culture supernatant from ETBF is referred to as BFT, while concentrated bacterial culture supernatant from ETBFΔbft is referred to as control. The protein content of concentrated bacterial culture supernatants was determined and quantified using the Bio-Rad DC Protein Assay (Bio-Rad 5000116).
2.5 |. Validation of toxin presence and activity in concentrated bacterial culture supernatants
To confirm that concentrated bacterial culture supernatant from ETBF contained BFT, a western blot was performed using an anti-BFT antibody to detect the 20 kDa protein. BFT was detected only in our ETBF preparation, as expected (Figure S2A). Next, we sought to determine whether the concentrated supernatant contained the active toxin. Based on previous studies showing that BFT triggers cleavage of E-cadherin in HT29/C1 colonic carcinoma cells,6 we treated HT29/C1 cells with our concentrated bacterial culture supernatants and measured cleaved E-cadherin by western blot. We found that our concentrated ETBF culture supernatant effectively cleaves E-cadherin, while the concentrated ETBFΔbft culture supernatant shows only minimal cleavage (Figure S2B). We evaluated BFT activity by measuring E-cadherin cleavage with numerous dilutions of concentrated bacterial culture supernatants and found that 100 μg/mL (total protein/mL of concentrated bacterial culture supernatant) delivered a sufficient amount of biologically active BFT to cells, and all subsequent experiments used this concentration. To further confirm that observed responses were due to the presence of toxin, and not due to other bacterial proteins or factors in the media, we visualized colonoids using confocal microscopy and tracked colonoid morphology changes to assess toxin activity. To enhance the activity of BFT on colonoids, CMGF + medium was replaced with CMGF− basal cell medium containing concentrated bacterial culture supernatants for the first 6 hours. After 6 hours, media was removed and fresh growth media without concentrated bacterial culture supernatant was added to colonoids. Colonoids treated with BFT display dramatic morphology changes, highlighted by the rounding of cells and an overall lack of epithelial cell membrane structure as soon as 1 hour, and persisting for up to 48 hours. Figure S2C shows an example of the morphology changes that occur after the addition of concentrated bacterial culture supernatant from ETBF for 6 hours. In comparison, the addition of concentrated ETBFΔbft bacterial culture supernatant does not alter the appearance of the colonoids (Figure S2D). Using concentrated bacterial supernatant from a recombinant ETBFΔbft strain expressing bft (rETBFΔbft bft-2), morphology changes similar to those in the BFT treatment were observed (Figure S2E). Since ETBF and ETBFΔbft strains are identical except for the expression of bft, we next investigated a non-toxigenic strain that cannot produce toxin (NTBF; NCTC 9343). Concentrated bacterial culture supernatant from the NTBF alone does not cause any morphology changes (Figure S2F). Next, we used concentrated bacterial culture supernatant from a recombinant NTBF strain expressing bft (rNTBF bft-2) and saw morphology changes similar to our BFT treatment (Figure S2G). Finally, we used concentrated bacterial supernatant from a recombinant NTBF strain with a mutated and inactivate form of BFT (rNTBF bft-2 H352Y) and found that it no longer causes morphology changes (Figure S2H). Taken together, concentrated bacterial supernatants derived from wild-type ETBF strain 86-5443-2-2 expressing chromosomal bft, or B fragilis strains genetically engineered to express bft from a plasmid, led to dramatic cell morphology changes. In contrast, concentrated bacterial supernatants from NTBF strains that lack the bft gene or express a mutant bft or ETBF strains with an in-frame chromosomal bft deletion do not yield cellular changes.
2.6 |. Colonoid treatment and glucosylceramide modulation
Colonoids were treated with concentrated bacterial supernatants at a concentration of 100 μg protein/mL or 5 nmol/L purified BFT2 (Obtained from the Sears Laboratory, and purified as previously described).33 For treatment, CMGF + medium was removed and basal CMGF− medium containing concentrated bacterial culture supernatant or purified BFT was added for up to 6 hours, depending on the experiment. Concentrated bacterial supernatants or purified BFT were both added in basal media to enhance the activity of BFT, as early experiments showed that serum, present in CMGF + medium, reduced its activity (data not shown). At 6 hours, media was removed and replaced with CMGF + media for the remainder of the experiment.
In order to inhibit glucosylceramide synthase (GCS) in colonoids, ibiglustat (Venglustat, Sanofi Genzyme; obtained from MedChemExpress HY-16743) was resuspended in DMSO, diluted using CMGF− media, and used at a final concentration of 5 μmol/L. For experiments using ibiglustat, a vehicle control of DMSO was used for all conditions not receiving the drug. Typically, ibiglustat was added 24 hours before the addition of concentrated bacterial culture supernatant so that glucosylceramide lipid levels would be decreased by the start of the assay. When concentrated bacterial supernatant, resuspended in CMGF− media, was added to the colonoids, fresh ibiglustat was added again to maintain the inhibition of GCS.
In order to inhibit glucocerebrosidase (GBA), Conduritol B Epoxide (Cayman Chemical 15 216) was resuspended in water, diluted using CMGF− media, and added at a final concentration of 20 μmol/L. For experiments using CBE, a vehicle control of water, diluted in CMGF− media, was used. A subset of colonoids was cultured with CBE indefinitely, with fresh drug added every other day during passages or media changes.
Glucosylceramide synthase activity was measured by adding C6-ceramide nanoliposomes (C6-CNL, a generous gift from Keystone Nano, State College, PA) to colonoids for 1 hour. Ghost nanoliposomes were used as vehicle control for these experiments. The formulation of the ghost nanoliposomes is identical to that of CNL, but lacks ceramide.
2.7 |. Cell culture of HT29/C1
HT29/C1 cells were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle medium (DMEM, Gibco 11965092) with 10% fetal bovine serum (FBS) and 1% Pen/Strep (Gibco 15140122).
2.8 |. Confocal microscopy
Colonoid images were captured using an Olympus FV3000RS confocal microscope (Johns Hopkins University School of Medicine), an Operetta CLS High-Content Analysis System (University of Virginia Advanced Microscopy Facility, RRID:SCR_018736), or a Zeiss LSM 700 (University of Virginia Advanced Microscopy Facility). Images were illuminated and captured using brightfield at 20x (Olympus FV3000RS) or 10x (Operetta CLS High-Content Analysis System) magnification. For time-lapse experiments, images were captured every 15 (Figure 3A–D) or 20 (Figure 9A–D, Figure 2D, Figure 4C) minutes after the addition of concentrated bacterial supernatant. At 6 hours, between image captures, media was removed and CMGF + media was added. Imaging experiments lasted 48 hours and resulting images were processed with ImageJ34 in order to generate videos. Frames per second (FPS) in all videos was set to 15. In experiments using ibiglustat, cells were pre-treated for 24 hours before observations began.
FIGURE 3.
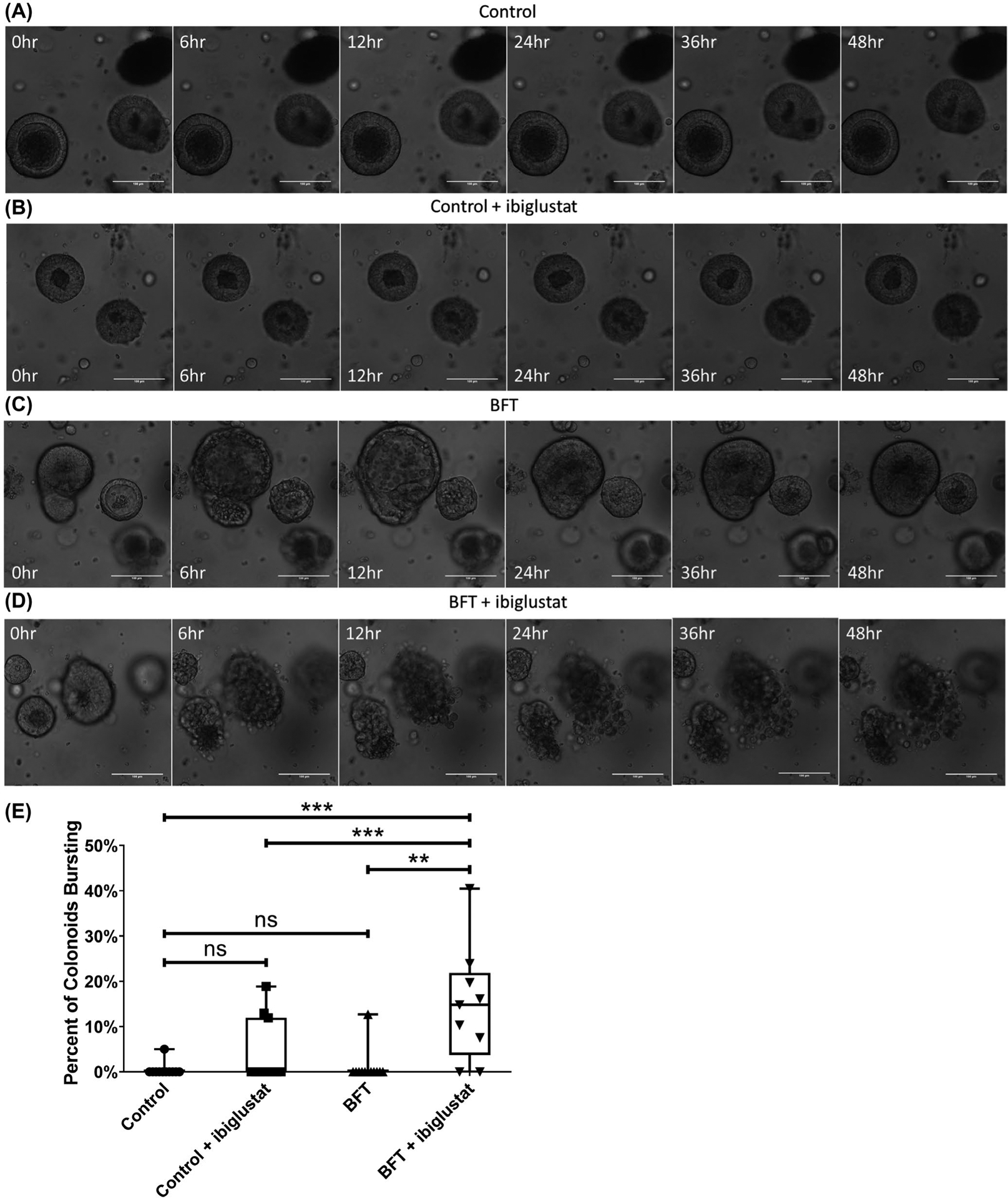
Combination of BFT and ibiglustat causes colonoids to burst. Colonoids treated with concentrated bacterial culture supernatant from ETBFΔbft (control) (A) or control + 5 μmol/L ibiglustat (B) show no obvious morphology changes when visualized by confocal microscopy. Colonoids treated with BFT undergo dramatic swelling and bubbling by 6 h before returning to normal morphology by 48 h (C). Colonoids treated with BFT and 5 μmol/L ibiglustat burst open, spilling contents into the extracellular matrix (D). Colonoids in all videos were tracked and bursting events were counted (E). Colonoids were considered bursting if by 48 h they had lost overall structure and there was no evidence that the outer epithelial membrane was reforming. We counted all of the colonoids in the well and calculated the percentage of colonoids that burst within 48 h (n = 12). Group comparisons were performed using a one-way ANOVA and Tukey’s multiple comparisons test. Statistical significance is indicated by asterisks: **P < .01 or ***P < .001. NS indicates non-significant results. Control represents concentrated bacterial culture supernatant from ETBFΔbft and BFT represents concentrated bacterial culture supernatant from ETBF. Confocal images were captured using 20x magnification. Scale bar indicates a distance of 100 μm. Compiled 48-hour time-lapse videos can be found in Videos S1–S4
FIGURE 9.
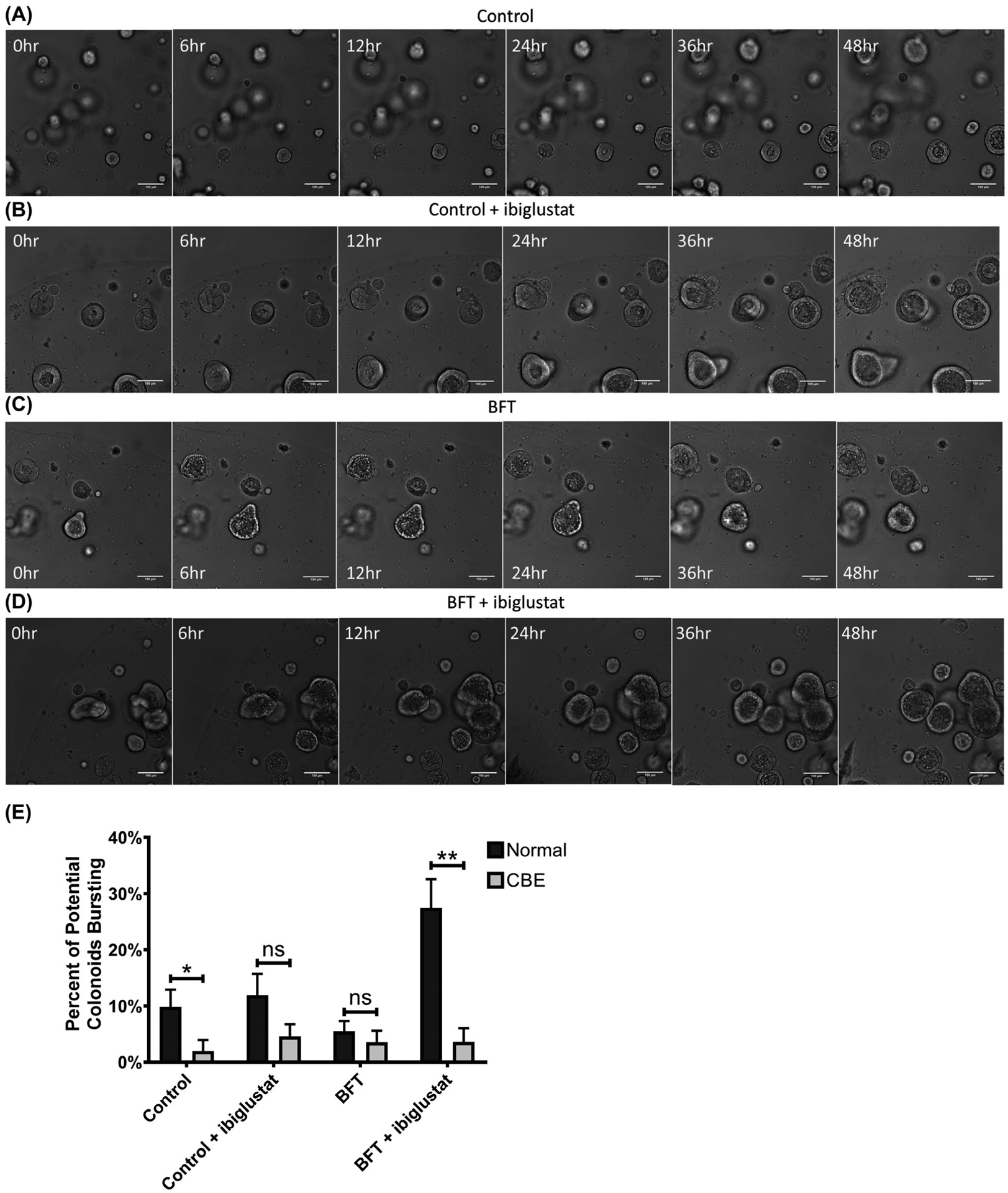
Pharmacological inhibition of glucocerebrosidase protects colonoids from BFT and ibiglustat-induced bursting. Colonoids treated with concentrated bacterial culture supernatant from ETBFΔbft (control) (A) or control + 5 μmol/L ibiglustat (B) showed no obvious morphology changes over 48 h as visualized by confocal microscopy. Colonoids treated with BFT undergo dramatic swelling and bubbling and are still in the process of recovering and returning to normal morphology by 48 h (C). Colonoids treated with BFT and 5 μmol/L ibiglustat also display a normal BFT response and delayed recovery, but no colonoid bursting (D). Colonoids in all videos were tracked and bursting events were counted. Colonoids that appeared to initiate, but not complete, bursting were scored as a potential colonoid explosion. Potential bursting events from normal colonoids and colonoids cultured in CBE were counted and compared across all conditions (E). CBE-treated colonoids overall had less potential bursting events when compared to colonoids without CBE. Further, BFT + ibiglustat-treated colonoids had significantly less potential bursting events in the presence of CBE. Single comparisons were made using an unpaired t-test. Statistical significance for comparison between treatment groups is indicated by asterisks: *P < .05 or **P < .01. Error bars represent the standard deviation of the mean. Normal represents colonoids grown under normal conditions, while CBE represents colonoids grown in CBE. Control represents concentrated bacterial culture supernatant from ETBFΔbft and BFT represents concentrated bacterial culture supernatant from ETBF. Confocal images were captured using 10x magnification. Scale bar indicates a distance of 100 μm. Compiled 48-hour time-lapse videos can be found in Videos S5–S8
FIGURE 2.
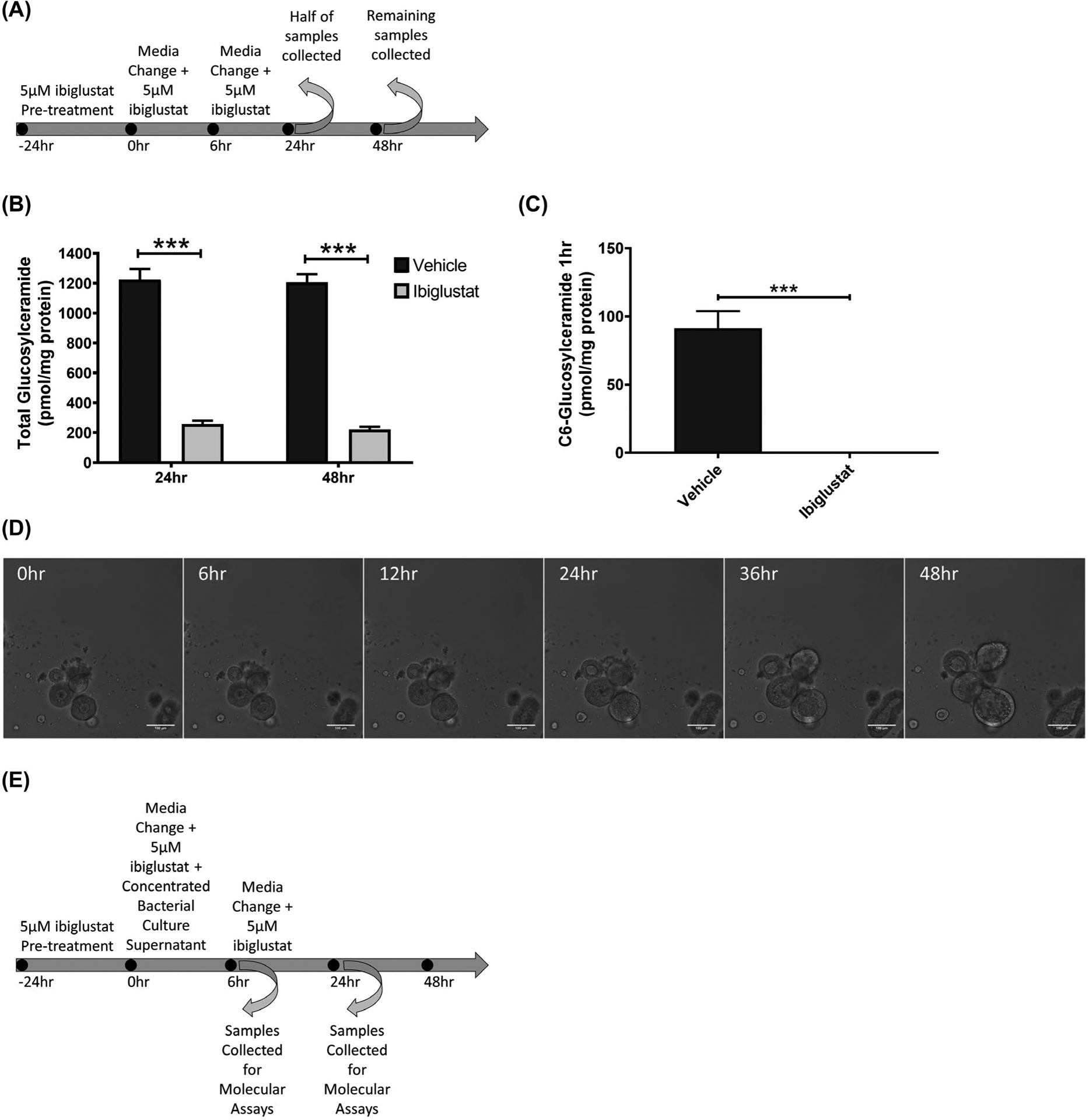
A selective inhibitor of glucosylceramide synthase (GCS) decreases glucosylceramide lipid levels. The treatment regimen for the administration of 5 μmol/L ibiglustat to colonoids is depicted (A). Addition of 5 μmol/L ibiglustat effectively reduced total levels of glucosylceramide (B). Colonoids were pre-treated with 5 μmol/L ibiglustat for 24 h and then treated with C6-ceramide nanoliposomes (CNL) for 1 hour to determine the effectiveness of ibiglustat in blocking GCS activity. Colonoids were collected and lipids were extracted. Presence of ibiglustat completely blocks the ability for colonoids to process C6-ceramide into C6-glucosylceramide, demonstrating the efficacy of ibiglustat at inhibiting GCS activity (C). Pre-treatment of colonoids with 5 μmol/L ibiglustat followed by the addition of concentrated bacterial supernatant from ETBFΔbft does not alter colonoid morphology, visualized by confocal microscopy (D). The treatment regimen for the administration of 5 μmol/L ibiglustat and concentrated bacterial culture supernatant is depicted (E). Single comparisons were made using an unpaired t-test. Statistical significance is indicated by asterisks: ***P < .001. Error bars represent the standard deviation of the mean. Vehicle represents vehicle control (see Materials and Methods). Confocal images were captured using 10x magnification. Scale bar indicates a distance of 100 μm
FIGURE 4.
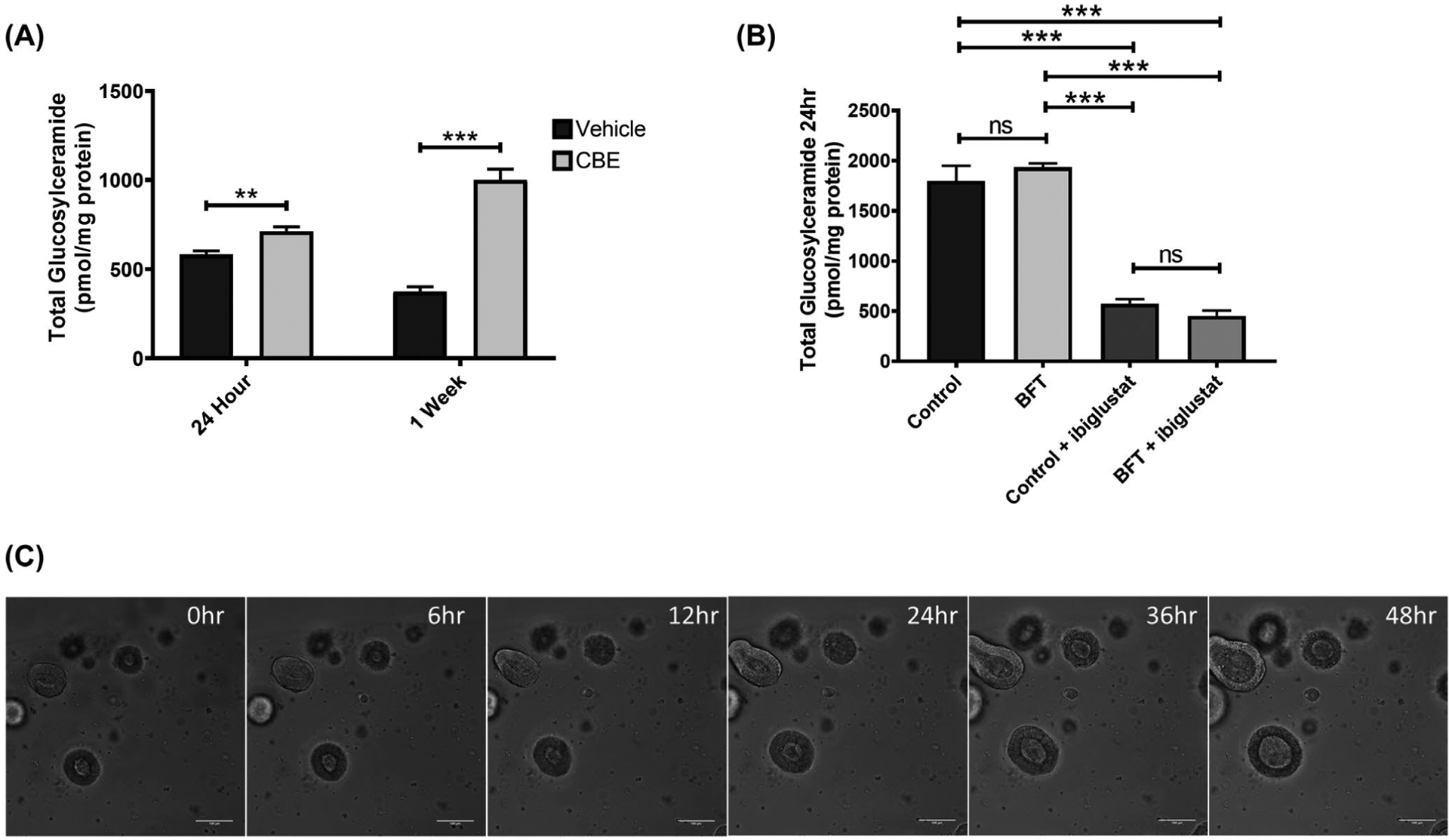
Inhibition of GBA increases glucosylceramide levels but does not alter colonoid morphology. Colonoids treated with 20 μmol/L CBE for 24 h or 1 week have increased levels of glucosylceramide (A). Colonoids cultured in CBE were treated according to the treatment regimen in Figure 2E and collected at 24 h post-concentrated bacterial culture supernatant addition for lipid analysis. Total glucosylceramide levels were similar between control and BFT treatments, but were significantly decreased in the presence of 5 μmol/L ibiglustat (B). Colonoids treated with concentrated bacterial culture supernatant from ETBFΔbft and cultured in CBE did not display any obvious morphological changes when visualized by confocal microscopy (C). Group comparisons were performed using a one-way ANOVA and Tukey’s multiple comparisons test, while single comparisons were made using an unpaired t-test. Statistical significance is indicated by asterisks: **P < .01 or ***P < .001. NS indicates non-significant results. Error bars represent the standard deviation of the mean. Vehicle indicates a vehicle control was used (see Materials and Methods). Control represents concentrated bacterial culture supernatant from ETBFΔbft and BFT represents concentrated bacterial culture supernatant from ETBF. Confocal images were captured using 10x magnification. Scale bar indicates a distance of 100 μm
For the FITC-Dextran permeability assay, images were captured using a Zeiss LSM 700 confocal microscope (University of Virginia Advanced Microscopy Facility) at 10× magnification. In order to prepare samples for imaging, the Matrigel was dissolved using 500 μL of Cultrex Organoid Harvesting Solution (R&D Systems 3700-100-01) for 20 minutes with shaking at 4°C. After 20 minutes, the colonoid suspension was collected and centrifuged at 500g for 5 minutes. After centrifugation, the supernatant was aspirated and the remaining cells were resuspended in 30 μL of FITC-Dextran solution (4 kDa FITC-Dextran resuspended at 2 mg/mL in CMGF + growth medium; FITC-Dextran obtained from Sigma-Aldrich 46944-100MG-F). Colonoids were immediately mounted onto a microscope slide and imaged. Double-sided tape was used to raise the coverslip to prevent the coverslip from crushing the colonoids. For samples treated with EDTA, 2 mmol/L EDTA was prepared from 0.5 mol/L EDTA (Thermo Fisher AM9260G) in ice-cold Hank’s Balanced Salt Solution (HBSS; Gibco 14175095). Quantification of fluorescent signal was performed using ImageJ, and interior colonoid fluorescence was normalized to the exterior signal surrounding the colonoid.
For immunofluorescence imaging, images were captured using a Zeiss LSM 700 confocal microscope (University of Virginia Advanced Microscopy Facility) using the 63x oil-immersion lens. Colonoids were removed from the Matrigel, as in the FITC-Dextran permeability assay above, and centrifuged at 1000g for 5 minutes. Colonoids were resuspended in 2% paraformaldehyde in 100 mmol/L phosphate buffer (pH 7.4) for 30 minutes at room temperature. After fixation, cells were washed in PBS with rocking for 5 minutes at room temperature and then pelleted at 1000G for 5 minutes. The wash step was repeated two more times. After three washes, colonoids were resuspended in 1X PBS/0.3% Triton X-100 for 15 minutes with rocking at room temperature. After this, colonoids were pelleted and resuspended in blocking buffer (1X PBS/5% normal goat serum/0.3% Triton X-100) for 1 hour with rocking at room temperature. After blocking, colonoids were pelleted and resuspended in the primary antibody solution (1X PBS/1% normal goat serum/0.1% Triton X-100) and left overnight, with rocking, at 4°C. The next day, colonoids were washed with 1X PBS/0.1% Triton X-100 with rocking for 5 minutes, repeated two more times for a total of three washes. Colonoids were then resuspended in the secondary antibody solution (1X PBS/1% normal goat serum/0.1% Triton X-100) for one and a half hours, at room temperature with rocking. Colonoids were washed three times with 1X PBS/0.1% Triton X-100 before the addition of Alexa Fluor 633 Phalloidin (resuspended in 1X PBS/1% normal goat serum/0.1% Triton X-100) for 1 hour at room temperature with rocking (Note: phalloidin was only used in conjunction with DAPI, and was excluded from experiments with E-cadherin and TJP1 antibodies due to overlapping fluorescence). Following this, colonoids were washed twice with 1X PBS and then 300 nmol/L DAPI was added for 5 minutes at room temperature. Cells were pelleted, resuspended in 1X PBS, and immediately centrifuged to wash the colonoids. These quick washes were repeated two more times, for a total of three washes. Colonoids were pelleted and resuspended in 15 μL mounting solution (90% glycerol/0.5% N-propyl gallate/20 mmol/L Tris) overnight at room temperature. On the third day, 7 μL of colonoids in mounting solution was added to a slide, surrounded by two pieces of double-sided tape (3M 665-2P12-36), stacked, to prevent the coverslip from crushing the colonoids. The coverslip/sample was sealed using clear nail polish and then imaged. Primary antibodies used for this project were: E-cadherin (Cell Signaling Technology 14 472; 1:100) and TJP1/ZO-1 (Novus Biologicals NBP1-85046; 1:100). Secondary antibodies used were: Goat anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific A-11001; 1 μg/mL) and Goat anti-rabbit Alexa Fluor 594 (Thermo Fisher Scientific A-11012; 2 μg/mL). Dyes/stains used were: Alexa Fluor 633 Phalloidin (Thermo Fisher Scientific A22284) and DAPI (Thermo Fisher Scientific D1306). Phalloidin and DAPI were resuspended according to the manufacturer’s instructions. Other reagents used were: Normal goat serum (Abcam ab7481), N-propyl gallate (Fisher Scientific MP210274780), and Triton X-100 (Sigma-Aldrich TX1568-1).
2.9 |. Quantitative real-time PCR
At the indicated time points, colonoids were harvested and RNA was extracted using the TRIzol reagent following the manufacturer’s instructions (Thermo Fisher Scientific 15596018). cDNA was created using an iScript cDNA Synthesis Kit (Bio-Rad 1708891) according to the manufacturer’s instructions. Quantitative PCR (qPCR) was performed on a Bio-Rad CFX384 following the instructions outlined in the iTaq Universal SYBR Green Supermix Kit (Bio-Rad 1725121). Primers used for this project were purchased from Bio-Rad: GCS (qMmuCID0010046) and reference gene GAPDH (qMmuCED0027497).
2.10 |. Western blot
Colonoids were lysed using RIPA buffer (Alfa Aesar J62524) and protein was quantified using the Bio-Rad DC Protein Assay (Bio-Rad 5000116). Lysed protein samples were loaded onto a NuPAGE 4%−12% Bis-Tris protein gel and run at 100V for two and a half hours. Proteins were transferred to a PVDF membrane using the Trans-Blot Turbo PVDF Transfer Kit (Bio-Rad 1704275) following the manufacturer’s instructions. Membranes were probed using the primary antibodies (listed below) and visualized using Alexa Fluor IgG secondary antibodies (Rabbit Invitrogen A11012, Mouse Invitrogen A11005; 1:10,000) on a Syngene G:BOX. Protein levels were quantified using ImageJ software and normalized to levels of β-actin (Sigma-Aldrich A5441; 1:10 000). Bio-Rad Precision Plus Protein Dual Color Standards ladder was used for all experiments (Bio-Rad 1610374; 5 μL/lane). Primary antibodies used for this project were: E-cadherin (Cell Signaling Technology 14472; 1:1000), TJP1/ZO-1 (Novus Biologicals NBP1-85046; 1:1000), caspase-3 (Cell Signaling Technology 9662; 1:1000), claudin-3 (Abcam ab15102; 1:1000), occludin (Novus Biologicals NBP1-87402; 1:1000), and BFT (Sears Lab; 1:1000).
2.11 |. Flow cytometry
Colonoids were treated with concentrated bacterial culture supernatant ±5 μmol/L ibiglustat, as described previously. At six- or 24-hours post-treatment, colonoid media was aspirated and the Matrigel was dissolved using Cultrex Organoid Harvesting Solution (R&D Systems 3700-100-01) with gentle shaking for 20 minutes at 4°C. After 20 minutes, the suspension was pipetted up and down ~ 30 times in order to break apart the colonoids. Colonoids were moved to 1.7 mL microcentrifuge tubes (Olympus Corporation 24-282) and spun at 1000g for 5 minutes at 4°C. The supernatant was aspirated and the pellet was resuspended in 250 μL TrypLE Express (Gibco 12605010) for 30 minutes at 37°C, with occasional vortexing. Once a single-cell suspension was obtained, cells were pelleted and 500 μL of eBioscience Fixable Viability Dye eFluor 780 (Thermo Fisher Scientific 65-0865-14) was added for 30 minutes at 4°C according to the manufacturer’s instructions. After 30 minutes, cells were spun down, media was aspirated, and cells were resuspended in 100 μL 1X Annexin V Binding Buffer (BD 556454) with 5 μL FITC Annexin V (BD 556419). Samples were incubated for 15 minutes and then 150 μL of 1X Annexin V Binding Buffer was added to each tube. Samples were then immediately run on the Attune Nxt (Life Technologies). Gates were established using forward and side scatters to isolate singlets and eliminate debris.
2.12 |. Statistics
Statistics were performed using GraphPad Prism 8.0.1 for Windows (GraphPad Software). All experiments were performed with a minimum of three replicates unless otherwise stated. Single comparisons were made using an unpaired t-test, while group comparisons were performed using a one-way ANOVA and Tukey’s multiple comparisons test. Statistical significance is indicated by asterisks: *P < .05, **P < .01, or ***P < .001. NS indicates non-significant results. Error bars represent the standard deviation of the mean.
3 |. RESULTS
3.1 |. ETBF, through the production of BFT, increases glucosylceramide levels in mouse distal colon and colonic epithelial cells
We colonized normal C57BL/6J mice with ETBF (strain 86-5443-2-2), a strain with a chromosomal deletion of bft (86-5443-2-2Δbft, herein referred to as ETBFΔbft), or a PBS sham control to determine if BFT alters sphingolipids in the colon. Since previous studies have shown that ETBF-induced tumors occur primarily in the distal colon,3,8,35 we focused on the role of sphingolipids in this location to determine how B fragilis impacts the colon epithelium. After 1 week, ETBF, but not ETBFΔbft or PBS, nearly doubled glucosylceramide levels in the distal colon (Figure 1A). Examination of other major sphingolipid species in the distal colon showed that ceramide and sphingomyelin levels were unchanged across all treatments in the distal colon (Figure S1A,B, respectively). This finding suggests that increased glucosylceramide is a specific event in response to ETBF, dependent on its secreted metalloprotease toxin, BFT.
FIGURE 1.

ETBF, through BFT, increases glucosylceramide levels in mice and in colonoids. Colonization with C57BL/6J mice for 1 week with ETBF increases glucosylceramide levels in the distal colon (A). Treatment of colonoids with concentrated bacterial culture supernatant from ETBF (BFT) significantly increases glucosylceramide levels at 24 h when compared to concentrated bacterial supernatant from ETBFΔbft (Control) (B). Addition of purified Bacteroides fragilis toxin (Purified BFT2, isolated from ETBF strain 86-5443-2-2, see Materials and Methods for details) to colonoids increases glucosylceramide production (C). Treatment of colonoids with BFT increases mRNA expression of glucosylceramide synthase (GCS) at 3 and 6 h, as measured by qPCR (D). Group comparisons were performed using a one-way ANOVA and Tukey’s multiple comparisons test, while single comparisons were made using an unpaired t-test. Statistical significance is indicated by asterisks: *P < .05, **P < .01, or ***P < .001. Error bars represent the standard deviation of the mean. Sham represents PBS control. NT indicates no treatment was added. Control represents concentrated bacterial culture supernatant from ETBFΔbft, BFT represents concentrated bacterial culture supernatant from ETBF, and Purified BFT2 represents purified BFT from ETBF strain 86-5443-2-2 (see Materials and Methods)
We next sought to investigate sphingolipids in CECs after BFT treatment. Due to the inherent dysregulation of sphingolipid metabolism present in cancer cell lines,16,22,36–39 we utilized colonoids from distal colons of C57BL/6J mice as an ex vivo model for normal, healthy CECs.25,40 Colonoids were grown in 3D culture, embedded in Matrigel, following the established protocol by Dr Hans Clevers’ Laboratory.41 In order to replicate our in vivo findings from Figure 1A, we concentrated cell-free bacterial culture supernatants from ETBF as a method to deliver BFT to colonoids (herein referred to as BFT). Furthermore, to focus on BFT specific changes caused by our concentrated bacterial culture supernatants, and to prevent any non-toxic bacterial factors from impacting our results, we utilized concentrated bacterial culture supernatants from ETBFΔbft as the control for all of our colonoid experiments (herein referred to as Control). Concentrated bacterial culture supernatant preparation details, as well as the validation of BFT presence and activity, are detailed in Materials and Methods (Validation of toxin presence and activity in concentrated bacterial culture supernatants). We treated colonoids with our concentrated bacterial culture supernatants, and, similar to our in vivo results (Figure 1A), BFT significantly increased glucosylceramide levels by ~28% in colonoids (Figure 1B). To validate that these results were due to BFT, and not any other bacterial factors present in our concentrated bacterial culture supernatant preparations, colonoids were treated with 5 nmol/L purified BFT2, which was obtained from the Sears lab after an extensive purification process to isolate BFT from ETBF bacterial culture supernatants (see Methods for purification information).33 Importantly, purified BFT alone was sufficient to increase glucosylceramide levels by ~44% in colonoids (Figure 1C).33 We also measured ceramide and sphingomyelin levels in response to concentrated bacterial culture supernatants (Figure S1C,D) or purified BFT (Figure S1E,F), and did not observe any significant changes in either condition. Next, we assessed the mRNA expression of GCS shortly after treatment with BFT (Figure 1D). Consistent with mass spectrometry results showing increased glucosylceramide levels, GCS mRNA expression increased over time in the presence of BFT when compared to the control (Figure 1D). Taken together, these data highlight a BFT-specific increase in glucosylceramide lipids and GCS expression.
3.2 |. Pharmacological reduction of glucosylceramide in the presence of BFT causes colonoids to burst
In order to determine the purpose of increased glucosylceramide in CECs after exposure to BFT, we next utilized a GCS inhibitor to reduce the levels of glucosylceramide and then measured cellular responses. Ibiglustat, an allosteric inhibitor of GCS, currently entering phase III clinical trials for the treatment of autosomal dominant polycystic kidney disease (ClinicalTrials.gov Identifier: NCT03523728), was used. In order to align with our BFT treatment timeline, colonoids received 5 μmol/L ibiglustat for 24 hours in the normal growth medium, then the medium was aspirated and replaced with the basal medium containing ibiglustat for 6 hours, which was subsequently replaced with the growth medium containing ibiglustat again for the remainder of the experiment (18 or 42 hours for 24 or 48 hour time points, respectively). A schematic representation of the treatment regimen can be found in Figure 2A. Using this treatment regimen, we verified through mass spectrometry that levels of glucosylceramide were significantly decreased by nearly 80% at 24 and 48 hours when compared to the vehicle control (DMSO; Figure 2B). To confirm GCS activity was inhibited by ibiglustat, we added 5 μmol/L C6-ceramide, a non-physiological form of ceramide, using C6-ceramide nanoliposomes (C6-CNL) or ghost liposomes (vehicle control that lacks ceramide) to colonoids for 1 hour after a 24-hour pre-treatment with ibiglustat or vehicle control. Conversion of C6-ceramide into C6-glucosylceramide was completely blocked in the presence of ibiglustat, demonstrating that ibiglustat is inhibiting GCS activity (Figure 2C). As expected, ghost liposomes did not return a signal for any C6-sphingolipids (data not shown). Finally, we visualized colonoids treated with 5 μmol/L ibiglustat using confocal microscopy and did not observe any overt morphological changes (Figure 2D).
Once we validated the efficacy of ibiglustat in our model, we added our concentrated bacterial culture supernatants to the treatment regimen mentioned previously (detailed in Materials and Methods; schematic representation is shown in Figure 2E) to determine the effects of BFT, while glucosylceramide levels were reduced. We visualized the colonoids for 48 hours using confocal microscopy, taking images every 20 minutes. Videos were assembled and images from 0, 6, 12, 24, 36, and 48-hour time points are shown in Figure 3A–D (corresponding videos can be found in Videos S1–S4). Figure 3A shows normal colonoid morphology after the control treatment. Similar to Figure 3A, treatment with control and 5 μmol/L ibiglustat did not induce morphological alterations over 48 hours (Figure 3B). However, when BFT was added for 6 hours, colonoids underwent a rapid expansion and cells began to round. This was followed by a recovery and reorganization of the epithelial membrane structure around 36 hours, and a return to normal morphology by 48 hours (Figure 3C). In stark contrast, when BFT is added for 6 hours in the presence of ibiglustat, colonoids lose their structure, and luminal contents escape into the extracellular matrix (Figure 3D). We refer to this phenomenon as “colonoid bursting” to represent the overall loss of the colonoid structural integrity and displacement of cells and other luminal content into the extracellular matrix. When bursting events across all treatments were quantified, BFT and ibiglustat treatment led to a significantly higher level of bursting events (14.77% average) when compared to all other treatments (0.45% average in control, 3.98% average in control + ibiglustat, and 1.06% average in BFT; Figure 3E). Colonoid bursting during BFT and ibiglustat treatment demonstrates that BFT-induced cellular production of glucosylceramide is important for colonoid survival.
3.3 |. Pharmacological inhibition of glucocerebrosidase increases glucosylceramide levels in colonoids but does not alter colonoid morphology
To further examine the importance of glucosylceramide in our model, we utilized conduritol B epoxide (CBE), an inhibitor of glucocerebrosidase (GBA),42 to prevent the breakdown of glucosylceramide and, therefore, increase cellular levels of glucosylceramide. After culturing colonoids for 24 hours with 20 μmol/L CBE, a ~ 22% increase in glucosylceramide levels was observed (Figure 4A). We maintained a subset of colonoids in 20 μmol/L CBE, refreshed every other day. After 1 week, glucosylceramide levels were over twofold higher than colonoids cultured without CBE (Figure 4A). We then measured sphingolipid changes in colonoids cultured in CBE after treatment with control or BFT with or without ibiglustat. We observed that in the presence of CBE, while both the control and the BFT-treated colonoids had elevated levels of glucosylceramide, in contrast to colonoids cultured without CBE (Figure 1B), BFT no longer significantly increased glucosylceramide (Figure 4B). Moreover, ibiglustat remained effective at reducing the levels of glucosylceramide in both treatments, albeit to a lesser degree than that seen in colonoids without CBE (Figure 2B). We visualized colonoids cultured in CBE during treatment with concentrated ETBFΔbft culture supernatant using confocal microscopy and did not observe morphology changes (Figure 4C). Therefore, treatment with CBE is an effective way to increase glucosylceramide levels in colonoids that does not disrupt normal morphology.
3.4 |. Inhibition of GCS enhances BFT-induced early apoptosis
To better understand the bursting events taking place in colonoids treated with ibiglustat and BFT, we next investigated markers of apoptosis and cell viability. We treated colonoids using the same regimen outlined above (Figure 2E), extracted protein from cells collected 6 hours after treatment and measured cleaved caspase-3, a marker of apoptosis,43 by western blot. Because CBE colonoids were cultured with CBE in perpetuity and maintained and passaged separately from the normal subset, we treated them as if they were a different cell line. As such, quantified results from normal and CBE colonoids were normalized to the control treatment within each group. Levels of cleaved caspase-3 were significantly increased in both the BFT and BFT + ibiglustat treatments (Figure 5A,C), indicating an increase in apoptosis. Colonoids cultured in CBE also displayed high levels of cleaved caspase-3 with BFT and BFT + ibiglustat treatment (Figure 5A,C). Total caspase-3 levels were decreased by BFT in normal and CBE colonoids, but this decrease was non-significant (Figure 5A,B).
FIGURE 5.
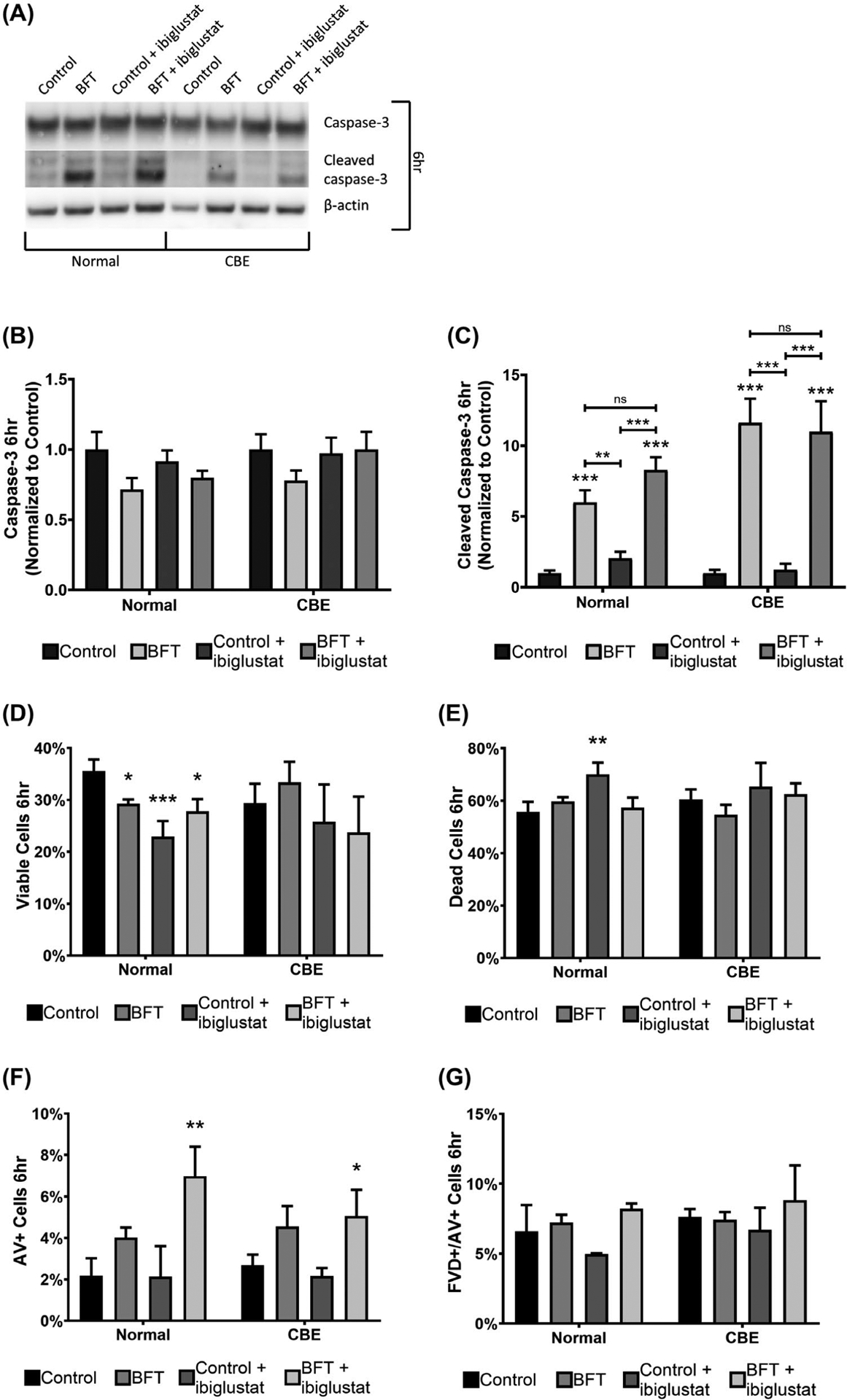
BFT increases caspase-3 cleavage and reduces cell viability in colonoids. As measured by western blots, levels of full-length caspase-3 were unaltered in all conditions in normal and CBE colonoids (A, B). BFT and BFT + 5 μmol/L ibiglustat-treated colonoids have increased levels of cleaved caspase-3 at 6 h in normal and CBE colonoids (A, C). Cell viability and apoptosis were determined by flow cytometry using a fixable viability dye (FVD) and Annexin V (AV) staining, respectively. At 6 h, BFT, control + 5 μmol/L ibiglustat, and BFT + 5 μmol/L ibiglustat caused significant reductions in the number of viable cells in normal colonoids, but did not significantly decrease viability in CBE-treated colonoids (D). The percentage of dead cells was significantly increased in the control + ibiglustat treatment group, but other treatments did not impact the population of dead cells (E). BFT + ibiglustat treatment increased the number of AV + cells, representing cells undergoing early apoptosis, in normal and CBE colonoids (F). FVD+/AV + cells, representing cells in late apoptosis/necrosis, were not significantly altered by any of the treatments in either group (G). Western blot results were compiled among multiple experiments (n = 6 biological replicates), normalized to their respective β-actin to control for loading, and then normalized to the average of their respective controls, where the control value was arbitrarily set to one (normal colonoids were normalized to the average of all control treatments, while CBE colonoids were normalized to the average of all CBE controls). Representative blots are shown for each target. Group comparisons were performed using a one-way ANOVA and Tukey’s multiple comparisons test. Statistical significance of each individual treatment when compared to the respective control is indicated by asterisks: *P < .05, **P < .01, or ***P < .001. Statistical significance between treatment conditions is shown by an asterisk above a line. NS indicates non-significant results. Error bars represent the standard deviation of the mean. Control represents concentrated bacterial culture supernatant from ETBFΔbft and BFT represents concentrated bacterial culture supernatant from ETBF
To determine if caspase-3 cleavage resulted in a decrease in cell viability, and to confirm the increase in apoptosis in response to BFT, cells were stained using a fixable viability dye (FVD; cell viability) and Annexin V (AV; apoptosis) and measured with flow cytometry. At 6 hours, cell viability was significantly reduced in the BFT (~17%) and BFT + ibiglustat (~22%) treatments when compared to control (Figure 5D). Normal and CBE colonoids treated with BFT + ibiglustat had a greater number of cells entering early apoptosis, as indicated by AV + staining (Figure 5F). None of the treatments affected the numbers of cells undergoing late-stage apoptosis/necrosis, indicated by cells positive for AV and FVD, in normal or CBE colonoids (Figure 5G). Interestingly, the control + ibiglustat treatment significantly reduced viability (~35%; Figure 5D), although the number of cells entering early apoptosis or late apoptosis/necrosis was similar to control (Figure 5F,G). Although BFT + ibiglustat increased early apoptosis, this did not significantly reduce viability when compared to BFT alone, suggesting that the induction of early apoptosis is not sufficient for the colonoid bursting events seen in Figure 3D.
3.5 |. Most early cellular responses to BFT are not dependent on glucosylceramide
The morphology changes that occur in colonoids exposed to BFT are highlighted by colonoid swelling (shown previously in human intestinal epithelial cells)44 and cell rounding (Figure 3C). These changes preclude the loss of cell-to-cell adhesion and eventual colonoid bursting when BFT is added in the presence of ibiglustat (Figure 3D). These additional morphological variations, namely colonoid bursting, that occurred when ibiglustat was added prompted us to interrogate whether glucosylceramide in the epithelial membrane of colonoids might contribute to the maintenance of cell adhesion. Membrane polarity of colonoids grown in 3D culture and embedded in Matrigel, according to the established protocol by Dr Hans Clevers’ Laboratory,41 is reversed from the orientation typically found in an intact colon. The apical domain, which is exposed to the digestive tract in the colon, faces the interior of the colonoid and is protected from extracellular factors. Meanwhile, the basolateral membrane, which interacts with the extracellular matrix and is generally protected from environmental factors in the colon, faces the exterior of the colonoid and is exposed to the environment.25 Using confocal microscopy, we stained colonoids with Alexa Fluor 633 phalloidin and confirmed that the apical domain was facing the interior of the colonoid, illustrated by the presence of actin there (Figure 3SA,B).
One of the first molecular events that occur after BFT treatment is the cleavage of E-cadherin,6 and previous studies have shown that BFT displays biological activity on the basolateral side of CEC membranes. Thus, we questioned if glucosylceramide levels would affect E-cadherin cleavage. We examined E-cadherin cleavage via western blot and found that after a 6-hour treatment with our concentrated bacterial culture supernatants, cultured with or without ibiglustat, E-cadherin was only cleaved in the presence of BFT (Figure 6A,B). BFT-induced E-cadherin cleavage was similar in the presence or absence of ibiglustat (Figure 6B). Similarly, when colonoids were cultured with CBE, levels of E-cadherin were decreased in both BFT treatments (Figure 6A,B). To confirm these findings, we used confocal immunofluorescence imaging on colonoids treated for 6 hours (as above) and removed from the Matrigel (see Materials and Methods for a detailed protocol). Consistent with our protein data, E-cadherin was expressed in control and control + ibiglustat treatments (Figure 6F,G middle panel). Further, in the BFT and BFT + ibiglustat treatments, E-cadherin signal was almost completely eliminated (Figure 6H,I middle panel). Collectively, this data suggests that BFT-induced E-cadherin cleavage is not dependent on glucosylceramide, as modulating its levels did not prevent or enhance E-cadherin cleavage.
FIGURE 6.
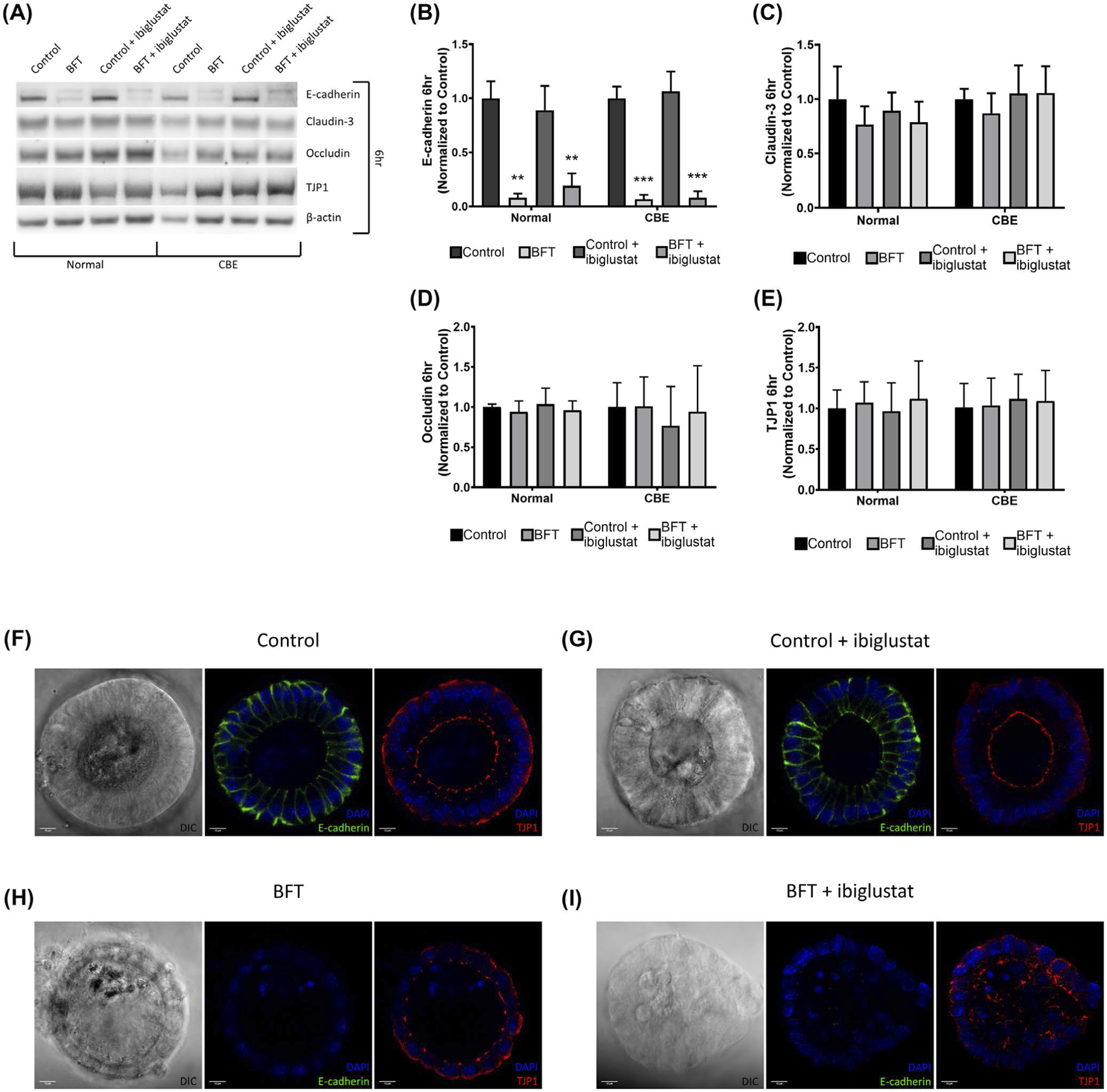
BFT decreases E-cadherin levels in colonoids at 6 h and disrupts TJP1 localization. Colonoids treated with BFT show reduced levels of full-length E-cadherin at 6 h via western blot, but claudin-3, occludin, and TJP1 levels are not significantly impacted by any treatment in normal or CBE colonoids (A). Quantification of E-cadherin western blots shows that BFT significantly decreased levels of full-length E-cadherin in normal and CBE-treated colonoids when compared with their respective controls (B). Claudin-3 (C), occludin (D), and TJP1 (E) levels were all quantified and did not change significantly when compared to the control treatments. Confocal immunofluorescence of colonoids for E-cadherin, TJP1, and DAPI (nuclear stain) confirms western results. Control-treated colonoids displayed normal E-cadherin and TJP1 expression (F). The addition of ibiglustat did not impact E-cadherin or TJP1 expression when compared to the control (G). In colonoids treated with BFT, the E-cadherin signal was almost entirely eliminated. TJP1 localization at the apical membrane was less consistent than in the control treatments (H). BFT + ibiglustat treatment resulted in almost no E-cadherin expression, while TJP1 localization was disorganized and irregular (I). Western blot results were compiled among multiple experiments (n = 6 biological replicates for all targets except occludin, n = 3). Values for each target were normalized to their respective β-actin value to control for loading variability. Finally, adjusted values were then normalized to the average of their respective control, where the control value was arbitrarily set to one (normal colonoids were normalized to the average of all control values, while CBE colonoids were normalized to the average of all CBE control values). Representative blots are shown for each target. Group comparisons were performed using a one-way ANOVA and Tukey’s multiple comparisons test. Statistical significance of each individual treatment when compared to the respective control is indicated by asterisks: **P < .01 or ***P < .001. Error bars represent the standard deviation of the mean. Confocal images were captured using a 63x oil-immersion lens. Scale bar indicates a distance of 10 μm. Normal represents colonoids grown under normal conditions, while CBE represents colonoids grown in CBE. Control represents concentrated bacterial culture supernatant from ETBFΔbft and BFT represents concentrated bacterial culture supernatant from ETBF
Since E-cadherin cleavage was indistinguishable between BFT and BFT + ibiglustat treatments, alteration of E-cadherin alone could not explain the significant difference seen in colonoid bursting events in the presence of ibiglustat (Figure 3E). Thus, we next examined tight junction proteins claudin-3, occludin, and TJP1, as they are located at the apical membrane of cells and function as both a barrier for extracellular factors as well as a CEC structural component to maintain cell polarity.9 Colonoids were cultured with or without CBE, treated as above (Figure 2E), and protein changes in claudin-3, occludin, and TJP1 were measured at 6 hours by western blot. In normal and CBE-treated colonoids, levels of claudin-3 (Figure 6A,C), occludin (Figure 6A,D), and TJP1 (Figure 6A,E) were not changed significantly by any of the treatments.
A previous study by the Sears Lab showed that 1 hour BFT treatment did not alter TJP1 levels in HT29/C1 cells, but did impact the membrane localization of TJP1.6 Therefore, we sought to determine if the membrane localization of TJP1 was different among our treatments using confocal immunofluorescence imaging for TJP1. We observed a strong TJP1 signal at the apical barrier in control (Figure 6F, right panel) and control + ibiglustat (Figure 6G, right panel) treatments. BFT treatment did not seem to modify the levels of TJP1, but we noticed that TJP1 expression at the apical membrane was less consistent in normal colonoids when compared to control treatments (Figure 6H, right panel). In the BFT + ibiglustat treatment, TJP1 expression was normal but appeared to be extremely disorganized, with no obvious apical localization (Figure 6I, right panel). Although TJP1 levels appeared consistent across all conditions, inappropriate localization of the protein during BFT + ibiglustat treatment could increase membrane permeability, making colonoids more susceptible to bursting.
3.6 |. Colonoid viability and tight junction proteins are influenced by glucosylceramide levels 24 hours after BFT treatment
The bursting events seen in the BFT + ibiglustat treatment (Figure 3D) could not be explained by increases in early apoptosis or E-cadherin cleavage, as these events occurred similarly in normal and CBE-treated colonoids. Because of this, we hypothesized that increasing glucosylceramide could be a protective mechanism by colonoids in response to the rapid cellular damage caused by BFT. To test this, we monitored cell viability using flow cytometry 24 hours after BFT treatment. Colonoids were treated using the treatment regimen outlined in Figure 3E, and cell viability was determined using flow cytometry. At 24 hours, the number of viable cells in normal colonoids was significantly decreased by BFT (~32%) and BFT + ibiglustat (~47%) when compared to control (Figure 7A). In colonoids cultured with CBE, the number of viable cells was decreased to a greater degree by BFT (~47%) and BFT + ibiglustat (~56%) when compared to control, but the percentage of viable cells remaining was similar to each respective treatment in normal colonoids (Figure 7A). In general, colonoids treated with CBE had more viable cells (Figure 7A), and less dead cells (Figure 7B), at 24 hours when compared to colonoids cultured normally. Early apoptosis, indicated by AV + staining, was relatively low in normal and CBE colonoids across all treatments (Figure 7C). Late-stage apoptosis/necrosis increased in response to BFT and BFT + ibiglustat (Figure 7D), which was likely the resulting the population of cells undergoing early apoptosis at 6 hours (Figure 5F). Taken together, CBE treatment increases colonoid viability, but this increase does not prevent BFT or BFT + ibiglustat induced death.
FIGURE 7.
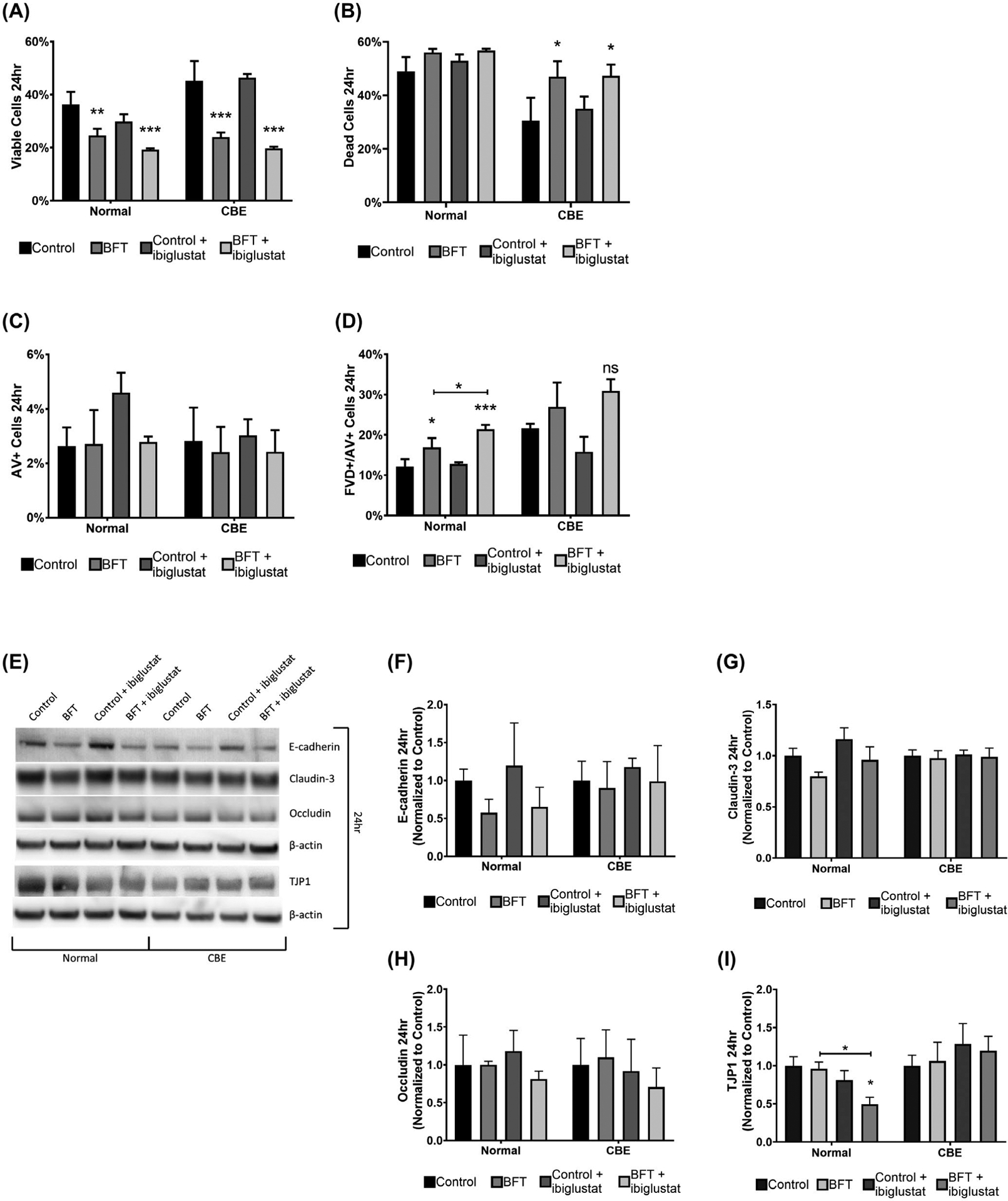
BFT-treated colonoids are less viable and have decreased TJP1 expression when GCS is inhibited. Cell viability and apoptosis were determined by flow cytometry using a fixable viability dye (FVD) and Annexin V (AV) staining, respectively. At 24 h, BFT and BFT + ibiglustat significantly decreased the number of viable cells in both normal, and CBE colonoids (A). BFT and BFT + ibiglustat significantly increased the number of dead cells in CBE colonoids (B). Early apoptotic cells, measured by AV + staining, were not significantly impacted by any treatment in normal or CBE colonoids (C). Colonoids treated with BFT and BFT + ibiglustat had significantly higher percentages of cells in the late apoptosis/necrosis stage in normal colonoids, but non-significantly increased levels in CBE colonoids, as indicated by FVD+/AV + staining (D). Colonoids were collected 24 h after initial BFT treatment for protein and E-cadherin, claudin-3, occludin, and TJP1 were measured (E) and quantified. E-cadherin levels were non-significantly reduced in normal colonoids treated with BFT, but close to baseline in CBE colonoids (F). Claudin-3 (G) and occludin (H) levels were not significantly altered by any treatment. TJP1 expression was significantly decreased in normal colonoids treated with BFT + ibiglustat when compared to control or BFT alone, but not in CBE colonoids (I). Western blot results were compiled among multiple experiments (n = 6 biological replicates for all targets except occludin, n = 3). Values for each target were normalized to their respective β-actin value to control for loading variability. Finally, adjusted values were then normalized to the average of their respective control, where the control value was arbitrarily set to one (normal colonoids were normalized to the average of all control values, while CBE colonoids were normalized to the average of all CBE control values). Representative blots are shown for each target. Group comparisons were performed using a one-way ANOVA and Tukey’s multiple comparisons test. Statistical significance of each individual treatment when compared to the respective control is indicated by asterisks: *P < .05, **P < .01, or ***P < .001. Statistical significance between treatment groups is shown by an asterisk above a line. Error bars represent the standard deviation of the mean. Normal represents colonoids grown under normal conditions, while CBE represents colonoids grown in CBE. Control represents concentrated bacterial culture supernatant from ETBFΔbft and BFT represents concentrated bacterial culture supernatant from ETBF
Next, we evaluated E-cadherin, claudin-3, occludin, and TJP1 levels using a western blot as a way to monitor the adherens and tight junctions within the epithelial membrane 24 hours after initial BFT treatment. E-cadherin levels were still returning to baseline in normal colonoids (Figure 7E,F), and had largely recovered in CBE colonoids (Figure 7E,F) at 24 hours. Claudin-3 (Figure 7E,G) and occludin (Figure 7E,H) levels were mostly unaltered by any of the treatments in normal and CBE colonoids, similar to our results at 6 hours (Figure 6A,C,D). Surprisingly, although TJP1 levels did not change at 6 hours (Figure 6A,E), BFT + ibiglustat treatment in normal colonoids caused a significant drop in TJP1 (Figure 7E,I). This drop was not seen in colonoids cultured with BFT alone, or in any treatment when CBE was present (Figure 7E,I), indicating that glucosylceramide may stabilize TJP1, helping to prevent BFT-induced colonoid burst.
3.7 |. Colonoid permeability increases in colonoids treated with BFT and ibiglustat
To determine if the alterations in TJP1 expression by BFT and ibiglustat were having a functional effect on colonoids, we measured membrane permeability using a modified dextran diffusion assay.45 Because TJP1 is important for maintaining the structural integrity of the membrane, we hypothesized that ibiglustat-induced reduction of TJP1 would increase membrane permeability, an effect that could be prevented by CBE addition. Colonoids were treated as above (Figure 2E) and collected at 24 hours. As colonoids with an intact membrane should exclude fluorescent dye, membrane permeability was determined by measuring the amount of fluorescent dye (FITC-dextran) found within the colonoid. In order to more accurately measure cellular permeability, colonoids that had burst, and would, therefore, be highly permissive to the dye, were excluded. We quantified the internal fluorescence, and, while control-treated colonoids showed very minimal diffusion of FITC-dextran into the lumen (Figure 8A,B), all other treatments caused an increase in permeability, although not always significantly. Ibiglustat addition did not increase permeability in normal colonoids (Figure 8C,K), but did significantly increase permeability in colonoids cultured with CBE (Figure 8D,K). BFT treatment increased permeability in both normal (Figure 8E,K) and CBE (Figure 8F,K) colonoids. However, when BFT was added in combination with ibiglustat, normal colonoids were significantly more permeable (Figure 8G,K), whereas permeability in CBE colonoids was not significantly increased when compared to control (Figure 8H,K). As a positive control for increased permeability, we used EDTA to disrupt tight junctions and, as expected, EDTA treatment significantly increased permeability in both the normal and CBE treatment groups when compared to their respective controls (Figure 8I–K). The increase in colonoid permeability seen with the BFT + ibiglustat treatment further supports our bursting data from Figure 3D,E, and highlights the importance of glucosylceramide in regulating the CEC tight junctional complex.
FIGURE 8.
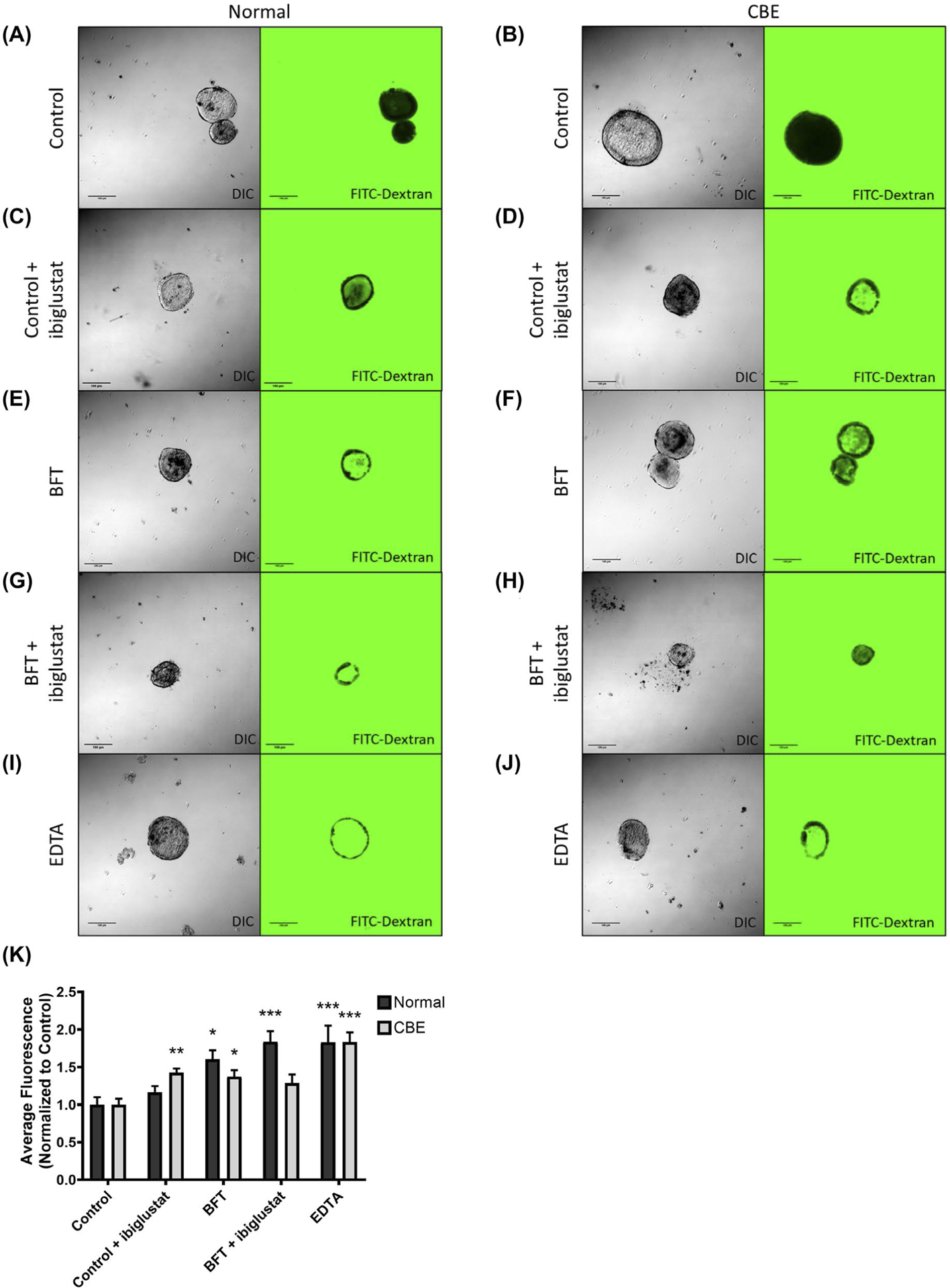
BFT treatment increases colonoid permeability, which is enhanced by GCS inhibition. Epithelial barrier integrity of colonoids grown normally or in 20 μmol/L CBE was assessed using a FITC-Dextran permeability assay. Colonoids were treated as described previously (Figure 2E). Colonoids were removed from the extracellular matrix at 24 h, pelleted, and resuspended in growth media containing 2 mg/mL of 4 kDa FITC-Dextran. Colonoids were mounted onto a slide and immediately imaged using DIC and confocal microscopy (n = 15–29 colonoids/treatment). Control colonoids in normal (A) and CBE (B) groups had very little internal fluorescence. The addition of ibiglustat significantly increased the permeability of CBE colonoids (D), but not normal colonoids (C). BFT treatment caused a significant increase in permeability in normal (E) and CBE (F) colonoids. BFT treatment with ibiglustat caused a significant permeability increase in normal colonoids (G), but not CBE colonoids (H). EDTA-treated colonoids served as a positive control for increased permeability, which was seen in both treatment groups (I, J). The flourescence values from each treatment group were quantified and values were normalized to their respective control (K). Group comparisons were performed using a one-way ANOVA and Tukey’s multiple comparisons test. Statistical significance of each individual treatment when compared to the respective control is indicated by asterisks: *P < .05, **P < .01, or ***P < .001. Error bars represent the standard deviation of the mean. Normal represents colonoids grown under normal conditions, while CBE represents colonoids grown in CBE. Control represents concentrated bacterial culture supernatant from ETBFΔbft and BFT represents concentrated bacterial culture supernatant from ETBF
3.8 |. Increasing levels of glucosylceramide in colonoids prevent BFT-induced bursting
Our data suggest that glucosylceramide synthesis is crucial for the integrity of colonoid structures in the presence of BFT, so we next tested whether increasing glucosylceramide levels protected colonoids from bursting. We visualized colonoids cultured in CBE for 48 hours using confocal microscopy (Figure 9A–D; Videos can be found in Videos S5–S8). In the presence of CBE, colonoids treated with control (Figure 9A) or control + 5 μmol/L ibiglustat (Figure 9B) showed no morphological changes. BFT treatment caused temporary cell rounding and membrane disorganization (Figure 9C), similar to that seen in colonoids without CBE (Figure 3C). The addition of BFT with ibiglustat caused the typical toxin-induced morphology changes, but colonoids did not burst (Figure 9D). This was in stark contrast to those grown without CBE (Figure 3D). Even more striking, across all treatments in the CBE-pre-treated colonoids, we did not observe colonoids bursting. As a more rigorous control, while scoring colonoid bursting events, we also kept track of colonoids that appeared to initiate, but not complete, bursting. These colonoids typically released luminal contents into the extracellular space but would appear to either recover and reconnect their outer epithelial membrane or maintain an ordered outer epithelium that did not fully dissociate by the end of the experiment. For these events, we scored them as “potential colonoid bursts,” indicating that they may or may not recover, but we could not definitively say that they had burst. Within the CBE treatment group, there were no significant changes in potential bursting events (Figure 9E). Further, when compared to colonoids cultured without CBE, it is especially clear that CBE treatment protects colonoids from BFT + ibiglustat-induced burst (Figure 9E). Taken together, cellular increase in glucosylceramide in response to BFT appears to serve as a protective mechanism used to maintain the structural integrity of the colon epithelium. This is highlighted by the fact that colonoids with reduced glucosylceramide have decreased TJP1 expression, increased permeability, and burst under stress in response to BFT, while those pharmacologically treated to maintain functional glucosylceramide levels are able to stabilize TJP1 levels and withstand the assault.
4 |. DISCUSSION
We have demonstrated that ETBF, through BFT, increases glucosylceramide levels in the murine distal colon and in colonoids derived from the murine distal colon. These findings suggest that BFT directly impacts glucosylceramide production in colon epithelial cells. Our results support that the BFT-induced increase in glucosylceramide affords cells a protective mechanism by which they can maintain the structural integrity of the epithelium in response to stressors such as BFT. Pharmacological inhibition of GCS causes BFT-treated colonoids to burst. In contrast to this, those treated with an inhibitor of GBA are protected from BFT-induced burst. These findings support previous studies detailing the importance of glucosylceramide in the intestines.15,17
The utilization of colonoids as a model for healthy colon epithelial cells proved to be incredibly useful in this study, but we would like to comment on some of the shortcomings of colonoid experiments. Because colonoids are grown in 3D culture, any experiments must either be performed by first dissolving the Matrigel and collecting the colonoids, or by performing the assays, while the colonoids remain in the Matrigel. Although colonoids remain intact during the collection process, excess handling can impact downstream assays, especially in experiments using living cells. In the flow cytometry procedure used in this study, colonoids were resuspended in TrypLE to yield a single-cell suspension which could then be stained and analyzed. In an early pilot experiment using only the fixable viability dye, we found that overall viability was higher than in later experiments when we added Annexin V staining (data not shown). The additional handling, pipetting, and time that this staining added likely introduced additional stresses that may have reduced overall viability. Similarly, in immunofluorescence imaging experiments, colonoids imaged within the Matrigel produced low-quality images, requiring the removal of colonoids from the Matrigel to allow for higher resolution imaging. However, colonoids had to be handled very carefully to avoid smashing the colonoids and disrupting their morphology when mounting them on slides and imaging them. Experiments that could be performed on colonoids within the Matrigel (colonoid bursting events and colonoid morphology changes) or in colonoids immediately lysed after removal from the Matrigel (lipid and protein collection) were inherently less prone to alterations caused by handling. Growing colonoids in 2D monolayers may alleviate some of the limitations of 3D culture systems, and protocols detailing the methods to grow colonoids in 2D monolayers have been published recently.46,47
In C57BL/6J mice colonized with ETBF, we showed a significant increase in glucosylceramide levels in the distal colon. In general, sphingolipid levels in the distal colon were higher than that of the proximal colon (data not shown). To our knowledge, this is a novel finding in itself that might warrant further study to determine why sphingolipid levels differ along the colon axis. It has been previously reported that the extracellular sphingolipid sphingosine-1-phosphate (S1P) was upregulated in response to ETBF.48 While we were unable to replicate this finding (data not shown), results from that study were obtained in a cancer model and may not reflect the early changes shown in our non-cancer-ous models.
Our primary focus in this publication was glucosylceramide, but ceramide may also be converted into galactosylceramide if a galactose molecule is added instead of glucose.49 Most of the common methods used for sphingolipid extraction and quantification, including ours, struggle to distinguish galactosylceramide from glucosylceramide.50 Although this means that our “glucosylceramide” changes could have been galactosylceramide or a mix of the two, we felt comfortable reporting our results as glucosylceramide for a few reasons. The first is that our “glucosylceramide” levels dropped after ibiglustat addition (Figure 2B). Ibiglustat is a specific GCS inhibitor, which would not block the activity of galactosylceramide synthase.51 Second, when we measured GCS activity by adding a non-physiological form of ceramide (C6-ceramide) and measured its conversion to glucosylceramide, we found C6-“glucosylceramide,” but when ibiglustat was present, no signal was detected (Figure 2C). Finally, we prevented molecular and phenotypic changes in our model using a GBA inhibitor, which would only impact the GSL pathway at the level of glucosylceramide and any species beyond it (Figures 4–9). Taken together, although we cannot definitively say that galactosylceramide is not included in our results, we feel confident that our results are glucosylceramide dependent.
In order to study the effects of glucosylceramide in CECs exposed to BFT, we utilized inhibitors for the enzymes responsible for creating (ibiglustat; GCS) or breaking down (CBE; GBA) glucosylceramide. In experiments with CBE-treated colonoids, we continued to use ibiglustat in our treatment groups even though these inhibitors should have opposing effects on cells, effectively canceling out their actions. In fact, for the most part, colonoids treated with both CBE and ibiglustat responded similarly to control colonoids without glucosylceramide manipulation. The one exception was where the CBE control + ibiglustat treatment caused a significant increase in permeability when compared to the control alone (Figure 8K). Although this increase in permeability was surprising, it did not impact colonoid bursting events, as shown in Figure 9E. Even though CBE + ibiglustat co-treatment is somewhat counterintuitive, these experiments support and further strengthen the finding that maintaining homeostatic glucosylceramide levels is critically important for colonoids to retain membrane integrity.
Glucosylceramide may be further modified into higher order glycosphingolipids (GSLs) through the attachment of additional carbohydrates (for further explanation of the synthesis of GSLs, see the review by Schnaar and Kinoshita).52 Briefly, GSLs exert many of the same roles as traditional sphingolipids, including modulating proliferation, apoptosis, and cell adhesion.53,54 Perhaps more importantly, however, are the roles that GSLs play in cell-to-cell signaling and pathogen recognition and interaction. GSLs are typical expressed on the outer leaflet of the plasma membrane where they are exposed to the external environment.52 There, they have been shown to interact with other bacterial toxins, such as cholera toxin, tetanus toxin, and botulinum toxin.53 Since there is no known receptor for BFT,5 we hypothesized that GSLs could be serving as a binding partner for the toxin. However, because we still observed toxin activity on cells treated with ibiglustat, which prevents the formation of not only glucosylceramide, but also the higher order GSLs which build upon it, our data suggest the BFT receptor is not a GSL. It is possible that GSLs may help to stabilize the elusive BFT receptor, or simply serve as messengers for cell-to-cell signaling in response to the increased epithelial permeability caused by BFT via cleavage of E-cadherin, but further research will need to be performed to confirm these hypotheses.
While this study focused only on intestinal sphingolipids, B. fragilis itself is able to produce sphingolipids.55–57 These sphingolipids differ from their mammalian counterparts in that their fatty acid chains typically have an odd-chain length (17–19 carbons in length), while mammalian sphingolipids are typically of an even-chain length (18–20 carbons).56,58 One of the most well-studied sphingolipids produced by B fragilis is α-galactosylceramide because of its ability to influence host invariant natural killer T (iNKT) cells.55,57 In addition to modulating host immune responses, bacterial sphingolipids also provide the bacteria with a survival advantage when dealing with the stresses encountered in the gut.56 Since there is already evidence that bacterial sphingolipids can be incorporated into mammalian cells,58 the incorporation of odd-chain length sphingolipids into the membrane has the ability to influence membrane fluidity and structure. Considering the potential disruptions in membrane function caused by bacterial sphingolipids, combined with our findings on the importance of membrane sphingolipids in maintaining gut homeostasis, the inclusion of bacterial sphingolipids into host membranes warrants attention in subsequent studies.
Glucosylceramide modulation in response to BFT-induced epithelial barrier disruption has broad implications for the development of intestinal diseases, such as colitis and colon cancer. ETBF colonization of C57BL/6J mice with ETBF leads to colitis in as little as 1 week.59 Others have shown that the induction of colitis by dextran sodium sulfate (DSS) increases glucosylceramide levels in the colon.60 Our finding that glucosylceramide upregulation protects CECs exposed to BFT is further supported by studies in colitis models where glucosylceramide administration prevented colon epithelial damage and reduced the inflammatory immune response associated with the disease.61,62 In addition, our results showing the stabilization of TJP1 by glucosylceramide allows cells to maintain tight junctions, which are critical for reducing epithelial permeability caused by colitis.63 Persistent colitis caused by ETBF promotes hyperplasia in the colonic crypts of C57BL/6J mice,59 which is a mouse model for colon cancer (Apcmin/+), leads to tumor formation within 4 weeks.3 Additional studies beyond the scope of this project are needed to determine the role of glucosylceramide upregulation in ETBF-induced intestinal disease. However, we can hypothesize that, based on published data, and in conjunction with our results, early glucosylceramide increases are likely serving as a protective mechanism by CECs to prevent colitis. As other studies have shown that increased GCS expression in the colon is pro-tumorigenic,15,16,20–24 persistent and long-term activation of GCS might actually inadvertently support tumor development. Though this study was limited to early BFT-induced molecular alterations in healthy tissue, we suggest that our findings could be applied to a number of inflammatory diseases in the intestines.
In this study, we identify tight junction protein 1 (TJP1) as a putative mediator of glucosylceramide-induced protection and/or stabilization of the colonoids. While we mainly focused on TJP1, claudin-3, and occludin during this study as markers for changes in cellular tight junctions, there are many other proteins involved in the assembly, maintenance, and function of tight junctions. Some other examples include junctional adhesion molecules (JAMs) and other claudin family proteins.9,12,13 In future studies, we plan to revisit these other cell adhesion proteins to determine if they also play a role in epithelial cell response to BFT and pharmacological inhibition of glucosylceramide metabolism.
We propose a model where healthy CECs, exposed to normal microflora, have intact tight junctions and adherens junctions (Figure 10A). However, when ETBF colonizes the gut, B. fragilis toxin is produced and binds to CECs, which triggers E-cadherin cleavage and thus, a reduction of cellular adhesion. Following the loss of E-cadherin, we suggest that glucosylceramide levels are increased and help to stabilize tight junction proteins, mainly TJP1. This stabilization serves as a compensatory response to maintain CEC membrane integrity, cell-to-cell contact, and prevent epithelial breakdown after the loss of E-cadherin (Figure 10B). In contrast, if GCS is inhibited in the presence of BFT, tight junctions are destabilized, paracellular permeability increases and the colon epithelium experiences catastrophic damage, allowing bacteria and bacterial factors to translocate into the lamina propria (Figure 10C). However, if glucosylceramide levels are restored, CECs are protected from BFT-induced damage (Figure 10D). Taken together, this study shows for the first time the protective role of glucosylceramide as an important structural element that protects the colon epithelium from toxic bacterial stress.
FIGURE 10.
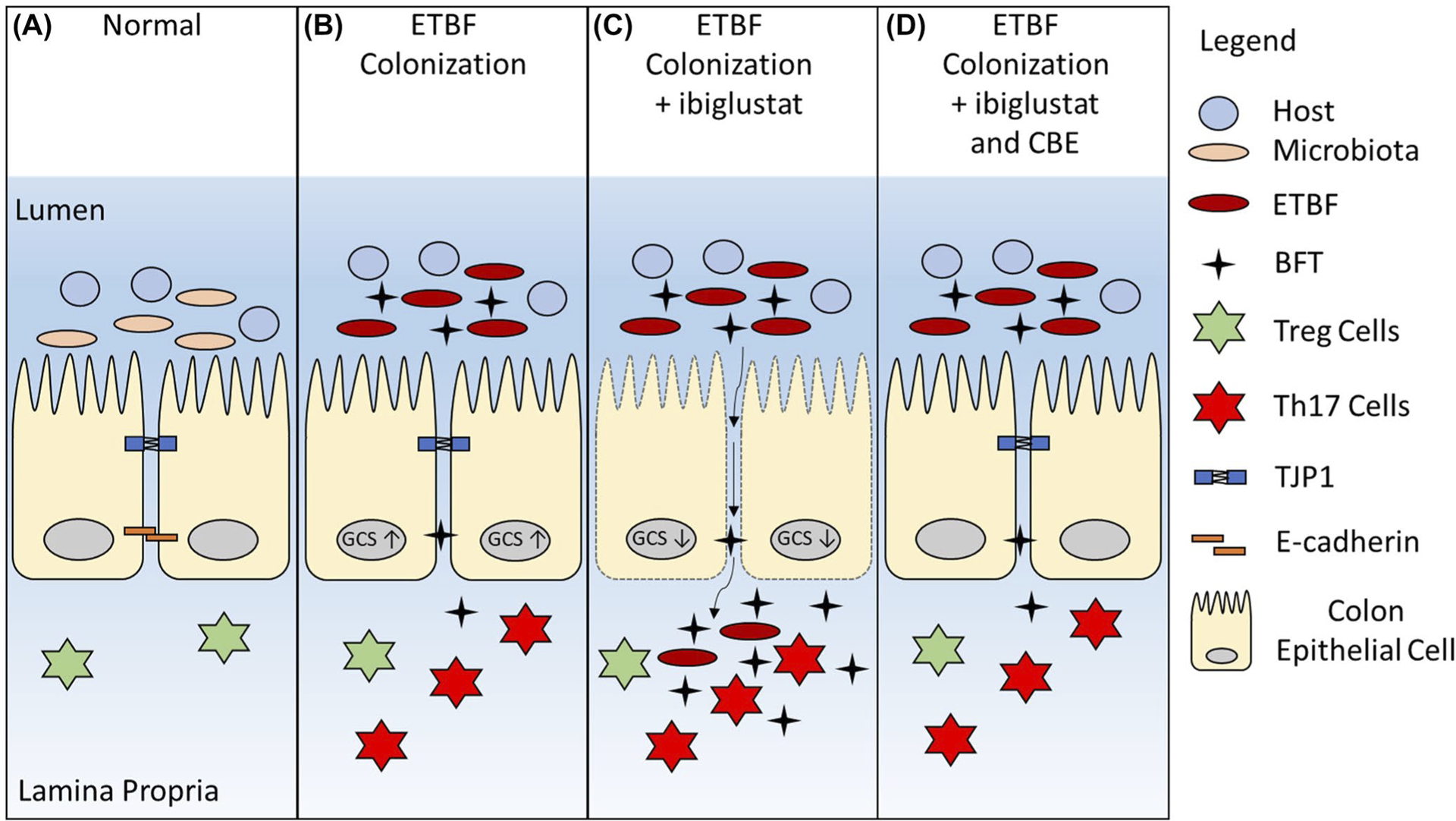
A proposed mechanism for the role of glucosylceramide in response to BFT. In a healthy colon, normal microbiota persists, cells maintain normal levels of glucosylceramide, and adherens junctions and tight junctions ensure cell-to-cell adhesion (A). When the host is colonized with ETBF, ETBF joins the microbiota and begins producing BFT, causing a decrease in CEC E-cadherin levels and an increase in glucosylceramide synthase expression (B). If glucosylceramide synthase is inhibited in a host colonized with ETBF, E-cadherin and tight junction protein 1 levels decrease, which leads to an increase in membrane permeability. Bacterial factors are able to pass through the membrane and interact with host immune cells in the lamina propria, triggering a Th17-driven pro-inflammatory immune response (C). Colonization with ETBF in the presence of CBE protects tight junctions and reduces membrane permeability (D)
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr Adrian Halme and Dr Stacey Criswell with the Advanced Microscopy Facility at UVA (RRID:SCR_018736) for their assistance with confocal imaging. We would also like to thank Michael H. Raymond for his assistance with flow cytometry analysis and Meredith B. Patterson for her help with confocal immunofluorescence imaging and analysis. Confocal imaging with the Operetta was possible thanks to NIH grant S10 OD021723 “Operetta CLS High-content Imaging System,” which funded its purchase.
Funding information
This work was supported by Bloomberg Philanthropies (to CLS). The authors also wish to acknowledge the Integrated Physiology and Imaging Cores of the Hopkins Conte Digestive Disease Basic and Translational Research Core Center (NIH DK-089502) for providing growth factor conditioned media for colonoids and use of Olympus FV3000RS confocal microscope (S10 OD025244).
Abbreviations:
- Apc
adenomatous polyposis coli
- AV
annexin V
- BFT
Bacteroides fragilis toxin
- BHI
brain heart infusion
- C6-CNL
C6-ceramide nanoliposomes
- CBE
conduritol B epoxide
- CCS
cold chelating solution
- CECs
colon epithelial cells
- CMGF−
complete medium without growth factors
- CMGF+
complete medium with growth factors
- DMSO
dimethyl sulfoxide
- DSS
dextran sodium sulfate
- EDTA
ethylenediaminetetraacetic acid
- EGF
epidermal growth factor
- ETBF
enterotoxigenic Bacteroides fragilis
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- FVD
fixable viability dye
- GBA
glucocerebrosidase
- GCS
glucosylceramide synthase
- GSLs
glycosphingolipids
- HBSS
Hank’s Balanced Salt Solution
- iNKT
invariant natural killer T cells
- JAMs
junctional adhesion molecules
- NTBF
non-toxigenic Bacteroides fragilis
- PBS
phosphate-buffered saline
- S1P
sphingosine-1-phosphate
- Th17
T-helper 17 cell
- TJP1
Tight junction protein 1
- Treg
regulatory T cell
Footnotes
CONFLICT OF INTEREST
Penn State Research Foundation has licensed Ceramide nanoliposomes to Keystone Nano Inc, State College PA, MK is CMO and co-founder of KeystoneNano.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCES
- 1.Chan JL, Wu S, Geis AL, et al. Non-toxigenic Bacteroides fragilis (NTBF) administration reduces bacteria-driven chronic colitis and tumor development independent of polysaccharide A. Mucosal Immunol. 2019;12(1):164–177. 10.1038/s41385-018-0085-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Wang K, Mucida D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491(7423):254–258. 10.1038/nature11465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu S, Rhee K, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–1022. 10.1038/nm.2015.A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sears CL. The toxins of Bacteroides fragilis. Toxicon. 2001;39(11):1737–1746. 10.1016/S0041-0101(01)00160-X [DOI] [PubMed] [Google Scholar]
- 5.Wu S, Shin J, Zhang G, Cohen M, Franco A, Sears CL. The Bacteroides fragilis toxin binds to a specific intestinal epithelial cell receptor. Infect Immun. 2006;74(9):5382–5390. 10.1128/IAI.00060-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu S, Lim KC, Huang J, Saidi RF, Sears CL. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc Natl Acad Sci USA. 1998;95(25):14979–14984. 10.1073/pnas.95.25.14979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells C, van de Westerlo E, Jechorek R, Feltis B, Wilkins T, Erlandsen S. Bacteroides fragilis enterotoxin modulates epithelial permeability and bacterial internalization by HT-29 enterocytes. Gastroenterology. 1996;110(5):1429–1437. 10.1053/GAST.1996.V110.PM8613048 [DOI] [PubMed] [Google Scholar]
- 8.Chung L, Thiele Orberg E, Geis AL, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018;23(2):203–214.e5. 10.1016/j.chom.2018.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell HK, Maiers JL, DeMali KA. Interplay between tight junctions & adherens junctions. Exp Cell Res. 2017;358(1):39–44. 10.1016/j.yexcr.2017.03.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyoshi J, Takai Y. Molecular perspective on tight-junction assembly and epithelial polarity. Adv Drug Deliv Rev. 2005;57(6):815–855. 10.1016/j.addr.2005.01.008 [DOI] [PubMed] [Google Scholar]
- 11.Holmes JL, Van Itallie CM, Rasmussen JE, Anderson JM. Claudin profiling in the mouse during postnatal intestinal development and along the gastrointestinal tract reveals complex expression patterns. Gene Expr Patterns. 2006;6(6):581–588. 10.1016/j.modgep.2005.12.001 [DOI] [PubMed] [Google Scholar]
- 12.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778(3):660–669. 10.1016/j.bbamem.2007.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127(11):2525–2532. 10.1038/sj.jid.5700865 [DOI] [PubMed] [Google Scholar]
- 14.Ando-Akatsuka Y, Yonemura S, Itoh M, Furuse M, Tsukita S. Differential behavior of E-cadherin and occludin in their colocalization with ZO-1 during the establishment of epithelial cell polarity. J Cell Physiol. 1999;179(2):115–125. [DOI] [PubMed] [Google Scholar]
- 15.Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19(3):175–191. 10.1038/nrm.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryland LK, Fox TE, Liu X, Loughran TP, Kester M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol Ther. 2011;11(2):138–149. 10.4161/cbt.11.2.14624 [DOI] [PubMed] [Google Scholar]
- 17.Jennemann R, Kaden S, Sandhoff R, et al. Glycosphingolipids are essential for intestinal endocytic function. J Biol Chem. 2012;287(39):32598–32616. 10.1074/jbc.M112.371005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichikawa S, Hirabayashi Y. Glucosylceramide synthase and glycosphingolipid synthesis. Trends Cell Biol. 1998;8(5):198–202. 10.1016/S0962-8924(98)01249-5 [DOI] [PubMed] [Google Scholar]
- 19.Yamashita T, Wada R, Sasaki T, et al. A vital role for glycosphingolipid synthesis during development and differentiation. Proc Natl Acad Sci USA. 1999;96(16):9142–9147. 10.1073/pnas.96.16.9142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Machala M, Procházková J, Hofmanová J, et al. Colon cancer and perturbations of the sphingolipid metabolism. Int J Mol Sci. 2019;20(23):6051. 10.3390/ijms20236051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdel Hadi L, Di Vito C, Riboni L. Fostering inflammatory bowel disease: sphingolipid strategies to join forces. Mediators Inflamm. 2016;2016:3827684. 10.1155/2016/3827684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan RD, Nilsson Å. Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development. Prog Lipid Res. 2009;48(1):62–72. 10.1016/j.plipres.2008.04.003 [DOI] [PubMed] [Google Scholar]
- 23.García-Barros M, Coant N, Truman J-P, Snider AJ, Hannun YA. Sphingolipids in colon cancer. Biochim Biophys Acta. 2014;1841(5):773–782. 10.1016/j.bbalip.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer. 2018;18(1):33–50. 10.1038/nrc.2017.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zachos NC, Kovbasnjuk O, Foulke-Abel J, et al. Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J Biol Chem. 2016;291(8):3759–3766. 10.1074/jbc.R114.635995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers LL, Shoop DS. Association of enterotoxigenic Bacteroides fragilis with diarrheal disease in young pigs. Am J Vet Res. 1987;48(5):774–775. [PubMed] [Google Scholar]
- 27.Pearson JM, Tan S-F, Sharma A, et al. Ceramide analogue SACLAC modulates sphingolipid levels and MCL-1 splicing to induce apoptosis in acute myeloid leukemia. Mol Cancer Res. 2020;18(3):352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merrill AH, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36(2):207–224. 10.1016/J.YMETH.2005.01.009 [DOI] [PubMed] [Google Scholar]
- 29.Allen J, Hao S, Sears CL, Timp W. Epigenetic changes induced by bacteroides fragilis toxin. Infect Immun. 2019;87(6):e00447–e0518. 10.1128/IAI.00447-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.In J, Foulke-Abel J, Zachos NC, et al. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol Gastroenterol Hepatol. 2016;2(1):48–62.e3. 10.1016/j.jcmgh.2015.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franco AA, Cheng RK, Goodman A, Sears CL. Modulation of bft expression by the Bacteroides fragilis pathogenicity island and its flanking region. Mol Microbiol. 2002;45(4):1067–1077. 10.1046/j.1365-2958.2002.03077.x [DOI] [PubMed] [Google Scholar]
- 32.Franco AA, Buckwold SL, Shin JW, Ascon M, Sears CL. Mutation of the zinc-binding metalloprotease motif affects Bacteroides fragilis toxin activity but does not affect propeptide processing. Infect Immun. 2005;73(8):5273–5277. 10.1128/IAI.73.8.5273-5277.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu S, Dreyfus LA, Tzianabos AO, Hayashi C, Sears CL. Diversity of the metalloprotease toxin produced by enterotoxigenic Bacteroides fragilis. Infect Immun. 2002;70(5):2463–2471. 10.1128/IAI.70.5.2463-2471.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Housseau F, Sears CL. Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/−) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle. 2010;9(1):3–5. 10.4161/cc.9.1.10352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gouazé V, Yu JY, Bleicher RJ, et al. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol Cancer Ther. 2004;3(5):633–640. [PubMed] [Google Scholar]
- 37.Kovbasnjuk O, Mourtazina R, Baibakov B, et al. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc Natl Acad Sci USA. 2005;102(52):19087–19092. 10.1073/pnas.0506474102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morjani H, Aouali N, Belhoussine R, Veldman RJ, Levade T, Manfait M. Elevation of glucosylceramide in multidrug-resistant cancer cells and accumulation in cytoplasmic droplets. Int J Cancer. 2001;94(2):157–165. 10.1002/ijc.1449 [DOI] [PubMed] [Google Scholar]
- 39.Beckham TH, Cheng JC, Marrison ST, Norris JS, Liu X. Interdiction of sphingolipid metabolism to improve standard cancer therapies. Adv Cancer Res. 2013;117:1–36. 10.1016/B978-0-12-394274-6.00001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.In JG, Foulke-Abel J, Estes MK, Zachos NC, Kovbasnjuk O, Donowitz M. Human mini-guts: new insights into intestinal physiology and host-pathogen interactions. Nat Rev Gastroenterol Hepatol. 2016;13(11):633–642. 10.1038/nrgastro.2016.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato T, Stange DE, Ferrante M, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and barrett’s epithelium. Gastroenterology. 2011;141(5):1762–1772. 10.1053/j.gastro.2011.07.050 [DOI] [PubMed] [Google Scholar]
- 42.Vardi A, Zigdon H, Meshcheriakova A, et al. Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J Pathol. 2016;239(4):496–509. 10.1002/path.4751 [DOI] [PubMed] [Google Scholar]
- 43.Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6(2):99–104. 10.1038/sj.cdd.4400476 [DOI] [PubMed] [Google Scholar]
- 44.Chambers FG, Koshy SS, Saidi RF, Clark DP, Moore RD, Sears CL. Bacteroides fragilis toxin exhibits polar activity on monolayers of human intestinal epithelial cells (T84 cells) in vitro. Infect immun. 1997;65(9):3561–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Co JY, Margalef-Català M, Li X, et al. Controlling epithelial polarity: a human enteroid model for host-pathogen interactions. Cell Rep. 2019;26(9):2509–2520.e4. 10.1016/j.celrep.2019.01.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.In JG, Foulke-Abel J, Clarke E, Kovbasnjuk O. Human colonoid monolayers to study interactions between pathogens, commensals, and host intestinal epithelium. JOVE-J VIS EXP. 2019; (146). 10.3791/59357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu L, Saitz-Rojas W, Smith R, et al. Mucus layer modeling of human colonoids during infection with enteroaggragative E. coli. Sci Rep 2020.10(1):10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng Z, Mu J, Tseng M, et al. Enterobacteria-secreted particles induce production of exosome-like S1P-containing particles by intestinal epithelium to drive Th17-mediated tumorigenesis. Nat Commun. 2015;6:6956. 10.1038/ncomms7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shayman JA. Glucosylceramide and galactosylceramide synthase. In: Hirabayashi Y, Igarashi Y, Merrill AH, eds. Sphingolipid Biology. Tokyo, Japan: Springer; 2006:83–94. https://link.springer.com/chapter/10.1007%2F4-431-34200-1_6. Accessed August 21, 2020. [Google Scholar]
- 50.von Gerichten J, Schlosser K, Lamprecht D, et al. Diastereomer-specific quantification of bioactive hexosylceramides from bacteria and mammals. J Lipid Res. 2017;58(6):1247–1258. 10.1194/jlr.D076190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Itier J-M, Ret G, Viale S, et al. Effective clearance of GL-3 in a human iPSC-derived cardiomyocyte model of Fabry disease. J Inherit Metab Dis. 2014;37(6):1013–1022. 10.1007/s10545-014-9724-5 [DOI] [PubMed] [Google Scholar]
- 52.Schnaar RL, Kinoshita T, et al. Glycosphingolipids. In: Varki A, Cummings RD, Esko JD, eds. Essentials of Glycobiology, 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2017. 10.1101/glycobiology.3e.011 [DOI] [Google Scholar]
- 53.Daniotti JL, Vilcaes AA, Torres Demichelis V, Ruggiero FM, Rodriguez-Walker M. Glycosylation of glycolipids in cancer: basis for development of novel therapeutic approaches. Front Oncol. 2013;3:306. 10.3389/fonc.2013.00306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hakomori S, Handa K, Iwabuchi K, Yamamura S, Prinetti A. New insights in glycosphingolipid function: “glycosignaling domain”, a cell surface assembly of glycosphingolipids with signal transducer molecules, involved in cell adhesion coupled with signaling. Glycobiology. 1998;8(10):xi–xviii. 10.1093/oxfordjournals.glycob.a018822 [DOI] [PubMed] [Google Scholar]
- 55.An D, Oh S, Olszak T, et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell. 2014;156(1–2):123–133. 10.1016/j.cell.2013.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.An D, Na C, Bielawski J, Hannun YA, Kasper Dl. Membrane sphingolipids as essential molecular signals for Bacteroides survival in the intestine. Proc Natl Acad Sci USA. 2011;108(Suppl):4666–4671. 10.1073/pnas.1001501107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wieland Brown LC, Penaranda C, Kashyap PC, et al. Production of α-Galactosylceramide by a prominent member of the human gut microbiota. PLoS Biol. 2013;11(7):e1001610. 10.1371/journal.pbio.1001610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heaver SL, Johnson EL, Ley RE. Sphingolipids in host–microbial interactions. Curr Opin Microbiol. 2018;43:92–99. 10.1016/J.MIB.2017.12.011 [DOI] [PubMed] [Google Scholar]
- 59.Rhee K-J, Wu S, Wu XinQun, et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun. 2009;77(4):1708–1718. 10.1128/IAI.00814-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zschiebsch K, Fischer C, Pickert G, et al. Tetrahydrobiopterin Attenuates DSS-evoked Colitis in Mice by Rebalancing Redox and Lipid Signalling. J Crohn’s Colitis. 2016;10(8):965–978. 10.1093/ecco-jcc/jjw056 [DOI] [PubMed] [Google Scholar]
- 61.Arai K, Mizobuchi YU, Tokuji Y, et al. Effects of dietary plant-origin glucosylceramide on bowel inflammation in DSS-treated mice. J Oleo Sci. 2015;64(7):737–742. 10.5650/jos.ess15005 [DOI] [PubMed] [Google Scholar]
- 62.Zigmond E, Preston S, Pappo O, et al. Beta-glucosylceramide: a novel method for enhancement of natural killer T lymphoycte plasticity in murine models of immune-mediated disorders. Gut. 2007;56(1):82–89. 10.1136/gut.2006.095497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Landy J, Ronde E, English N, et al. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J Gastroenterol. 2016;22(11):3117–3126. 10.3748/wjg.v22.i11.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.