

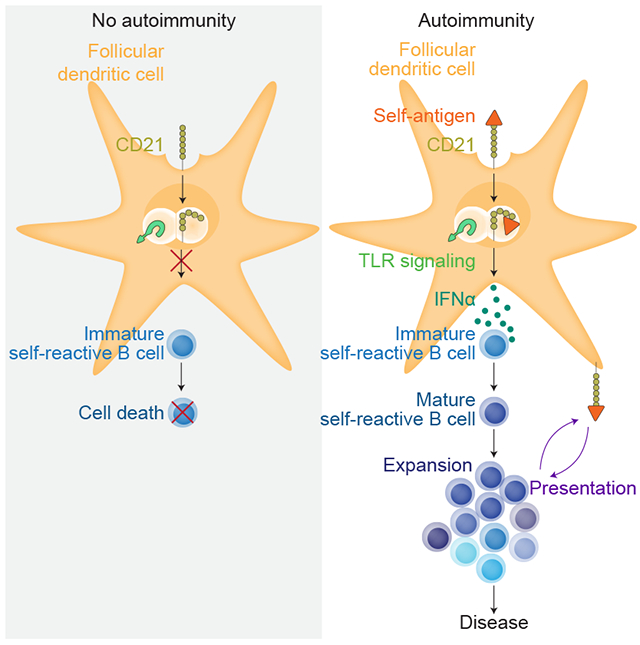
SUMMARY:
A hallmark of autoimmunity in murine models of lupus is the formation of germinal centers (GC) in lymphoid tissues where self-reactive B cells expand and differentiate. In the host response to foreign antigens, follicular dendritic cells (FDCs) are essential for the maintenance of GCs through their uptake and cycling of complement-opsonized immune complexes. Yet, whether FDCs retain self-antigens and are required for self-reactive GC maintenance and autoantibody secretion is not well understood. Here, we determined if FDCs were required for the survival of mature self-reactive B cells and their differentiation in the GC using lupus-prone mice. We found that FDCs took up and retained self-immune complexes composed of ribonucleotide proteins (RNP), autoantibody, and complement (referred to as RNP-IC). This uptake, which was mediated through CD21, triggered endosomal TLR7, leading to the secretion of IFNα via an IRF5-dependent pathway. Blocking of FDC secretion of IFNα in lupus mice restored B cell tolerance, with a significant reduction in the amount of GCs and pathogenic autoantibody. Thus, FDCs are a critical functional source of the IFNα driving autoimmunity in this lupus model. This pathway is conserved in humans, suggesting that it may be a viable therapeutic target in human lupus.
Graphical Abstract
INTRODUCTION:
Systemic lupus erythematosus (SLE) is a multigenic, incurable autoimmune disease of unknown etiology (Cotran et al., 1994; Hahn, 1998). It is characterized by the loss of B-cell tolerance and the secretion of isotype-switched antibodies against self- or neo-antigen from the nuclei of cells from diverse organs such as the kidney, skin, lungs, and joints. In mice, SLE is dependent on B cells (Shlomchik et al., 1994), and a major event leading to disease is when the self-reactive B cells differentiate within the germinal center (GC) into memory cells and IgG-secreting plasma cells (Banchereau et al., 2016; Casellas et al., 2001; Li et al., 2001; Melamed et al., 1998; Rifkin et al., 2005).
GCs are where activated B cells undergo cellular division, class-switch recombination, and somatic hypermutation. GCs form in the B-cell follicles of secondary lymphoid tissues during infection and immunization with foreign antigen (Allen et al., 2007a; Allen et al., 2007b; Hauser et al., 2007; Schwickert et al., 2007), but they are also common in murine models of lupus (Chatterjee et al., 2013; Cohen et al., 2002; Hanayama et al., 2004; Nanda et al., 2011). Although their specificity is less well defined, GC occur frequently in patients with chronic infection and autoimmune diseases such as arthritis, Sjogren’s syndrome, and SLE where their presence correlates with the production of pathogenic isotype-switched autoantibodies (Humby et al., 2009; Pitzalis et al., 2014; Salomonsson et al., 2003; Stott et al., 1998; Vinuesa et al., 2009).
Follicular dendritic cells (FDCs) are essential for the maintenance of GCs, as they retain antigen for extended periods and secrete cytokines, such as interleukin (IL)-6 and B-cell activating factor (BAFF), that promote B cell differentiation and survival (Garin et al., 2010; Wang et al., 2011). Ablating FDCs results in a rapid loss of GCs and changes follicular architecture(Cremasco et al., 2014; Wang et al., 2011). Recent studies have found that immune complexes (ICs) coated with complement C3 (C3) bind to CD21 receptors on FDC and are internalized into a cycling endosomal compartment (Heesters et al., 2013). This periodic cycling of foreign antigen complexes to the cell surface could explain how antigen is retained for long periods yet is accessible on the cell surface for acquisition by cognate B cells (Barrington et al., 2002; Gray, 2002; Steiner and Eisen, 1967). It is unknown whether this process impacts responses to self-antigen in autoimmune settings.
A current model of autoimmunity asserts that chromatin and nucleolar material have “danger-associated molecular patterns” (DAMPs) that bind toll-like receptors (TLR), much like pathogen antigens (Green et al., 2012; Leadbetter et al., 2002). DAMPs released by dying or apoptotic cells may act on multiple cell types including dendritic cells and B cells. It has been proposed that inappropriate uptake of apoptotic material by dendritic cells induces type I interferon (IFN) release, which could drive further inflammation, activation of hematopoietic cells, and differentiation of self-reactive B cells (Krieg, 2007). Furthermore, self-reactive B cells could be rescued from anergy by the activation of TLR7 following the B-cell receptor (BCR) internalization of nuclear material (Lau et al., 2005; Leadbetter et al., 2002). In the autoimmune mouse strain 564 Igi, in which the B cells express a BCR specific for nuclear antigen, isotype-switched IgG autoantibody titers are dependent on TLR7 and TLR8 signaling, though the signaling cell type is unknown (Berland et al., 2006; Umiker et al., 2014).
Type I interferon acts synergistically with DAMPs to increase the sensitivity of TLR7 signaling in B cells, leading to their escape of anergy and the secretion of antibody (Green et al., 2012). Exogenous administration of IFNα to lupus-prone mice can accelerate development of autoantibody production (Liu et al., 2011), and in the pristane model of lupus, IFNα promotes the differentiation of self-reactive B cells (Lee et al., 2008; Thibault et al., 2009). Inducing anti-IFNα antibodies in NZB/NZW F1 mice partly rescues the autoimmune phenotype (Zagury et al., 2009), and in the autoimmune BXSB strain, which bears a duplicated Tlr7 locus, anti-type-I-interferon-receptor (anti-IFNAR) antibody reduces autoantibody levels (Baccala et al., 2012). One proposed source of IFNα are plasmacytoid dendritic cells (pDCs) that have become activated by apoptotic debris (Krieg, 2007; Rowland et al., 2014; Sisirak et al., 2014), but other sources, such as lymphoid stromal cells, have not been investigated.
Here, we determined if FDCs were required for the survival of mature self-reactive B cells and their differentiation in the GC using the lupus-prone strain 564 Igi (Berland et al., 2006; Chatterjee et al., 2013). We found that FDCs took up and retained self-immune complexes composed of ribonucleotide proteins (RNP), autoantibody, and complement (referred to as RNP-IC). This uptake was mediated through CD21- triggered endosomal TLR7, leading to the secretion of IFNα via an IRF5-dependent pathway. Blocking of FDC secretion of IFNα in 564 Igi mice restored B cell tolerance, with a significant reduction in the amount of GCs and pathogenic autoantibody. Thus, FDCs are a critical functional source of the IFNα driving autoimmunity in this lupus model. This pathway is conserved in humans, suggesting that it may be a viable therapeutic target in human lupus.
RESULTS
B cell tolerance escape is dependent on IFNAR in lupus mice
Elevated levels of type I IFNs are common among a significant subpopulation of lupus patients, a characteristic called the “interferon signature” (Banchereau et al., 2016; Crow, 2009; Obermoser and Pascual, 2010). Elevated levels of IFNα are also present in murine models of lupus, such as BXSB (Baccala et al., 2012), NZB/NZW F1, and 564 Igi mice (Han et al., 2014; Umiker et al., 2014). 564 Igi mice have increased IFNα expression by neutrophils and macrophages in their bone marrow (BM), blood, and spleen, suggesting its importance in activating B cells, neutrophils, and monocytes (Han et al., 2014). However, the mechanism of this overexpression is unknown.
To identify the source of IFNα expression in lupus-prone mice, we first confirmed that Ifna is overexpressed in the spleens of NZB/NZW F1 mice (8–12 weeks) and 564 Igi mice bred onto the C57BL6 (B6) background using real-time quantitative PCR (RT-PCR). We found negligible expression of Ifna in WT mice, but 3- to 4-fold overexpression in the autoimmune strains (Figure 1A). Moreover, Mx1, a known IFNα response gene, was upregulated 5- to 10-fold compared to WT. We next used flow cytometry to analyze a single-cell suspension of spleen cells permeabilized and stained with an antibody specific for multiple isoforms of IFNα.
Figure 1: IFNα is required for the maintenance of self-reactive B cells.
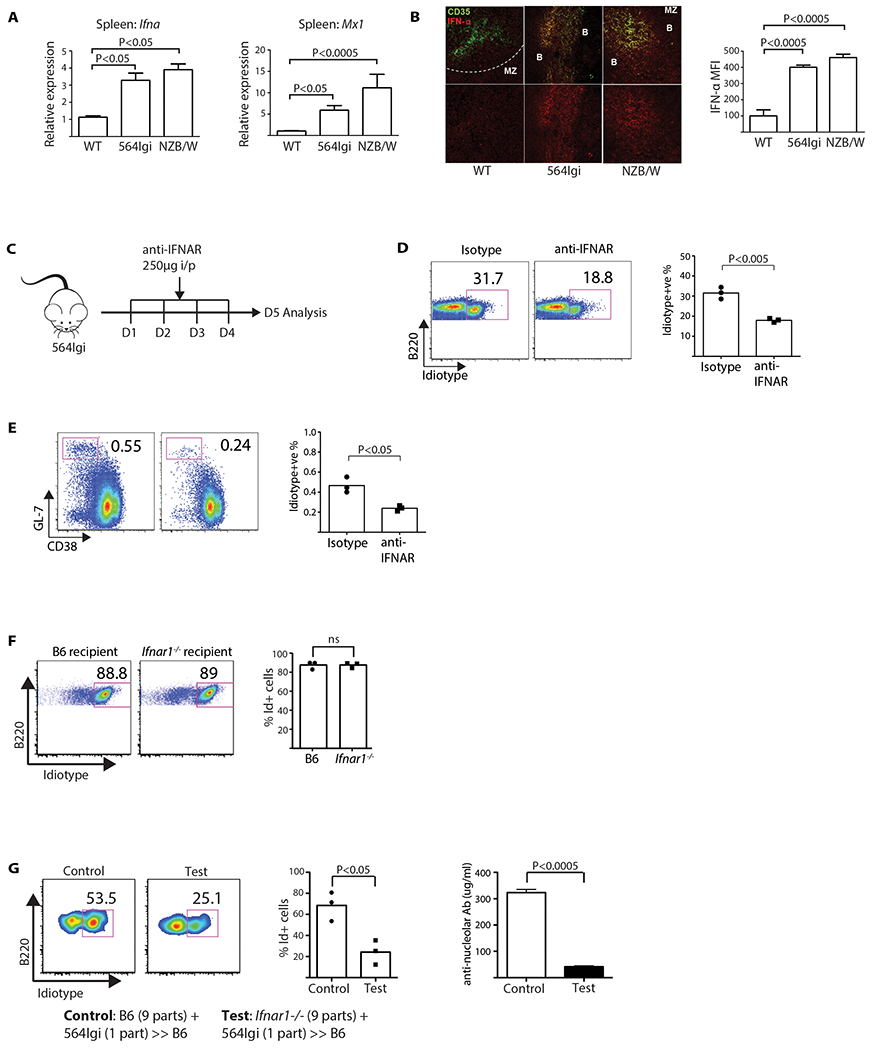
(A) Ifna and Mx1 expression measured by RT-PCR using splenic RNA from respective mice strains. Results representative of two experiments (one-way ANOVA, n = 6). (B) Spleen sections stained with anti-CD35 (green) and anti-IFNα (red), imaged by LSCM. Bar graph shows the MFI of IFNα (one-way ANOVA, n = 3). (C) Timeline of anti-IFNAR treatment. (D) FACS plots of mature Id+ B cell % in 564 Igi mice treated with anti-IFNAR (Student’s T test, n = 3, P < 0.005). Plots show two groups of mice gated on B220+ AA4.1 low cells. Each data point is one mouse. (E) Flow cytometry of spleen GC B cell % in anti-IFNAR treated 564 Igi mice (Student’s t test, n = 3, P < 0.05). Plots shown are gated on B220+ cells. Each data point is one mouse. (F) Plots are spleen cells gated on B220+ AA4.1 low cells. Data representative of two experiments. P values were calculated by Student’s t test; ns = not significant. (G) 564 Igi BM cells were mixed with either B6 (Ifnar1+/+) or Ifnar1−/− BM cells in a 1:9 ratio and transferred into irradiated B6 recipients. Left, representative flow-cytometry plots of mature Id+ B cells. Middle, mature Id+ cells in B6 controls vs. Ifnar1−/− BM donors (Student’s t test, n = 3, P < 0.05). Right, anti-nucleolar antibody titers between B6 controls (Ifnar1+/+) and Ifnar1−/− BM donors (Student’s t test, n = 3, P < 0.0005). Data representative of two experiments.
Plasmacytoid dendritic cells (PDCA-1+; pDCs) expressed IFNα, as expected (Blasius et al., 2004; Eloranta et al., 2010; Jego et al., 2003; Lee et al., 2008) (Figure S1A), with 564 Igi mice having 80-fold more expression than the WT B6 mice, according to RT-PCR on sorted spleens (Figure S1B). In parallel, splenic cryo-sections from the three strains were analyzed using laser scanning confocal microscopy (LSCM), revealing that IFNα co-stained with CD35+ FDCs (Figure 1B). Staining with control rabbit sera or the secondary antibody alone had negligible effects, whereas absorption with recombinant IFNα blocked binding confirming the specificity of the primary anti-IFNα pan antibody (Figure S2A).
IFNα co-staining with FDCs was unexpected, as pDCs were thought to be the major source of type I interferon in autoimmune mice and SLE patients (Crow, 2009; Davison and Jorgensen, 2015). To examine whether IFNα expression by FDCs is common to all GCs, WT B6 mice were immunized with two doses (10 μg each) of NP-KLH three weeks apart to induce GC formation, which was confirmed by LSCM on spleen sections. In contrast to 564 Igi mice, FDC secretion of IFNα in B6 mice was negligible (Figure S2B), as previously reported (Chatterjee et al., 2013). Thus, FDCs do not secrete IFNα in GCs arising from protein antigen in the absence of adjuvant. Yet, they do secrete IFNα in GCs arising from bacterial, and possibly food, antigens as indicated by staining the Peyer’s patches, mesenteric LNs, and inguinal LNs of 564 Igi and B6 mice (Figure S2C).
To elucidate how IFNα may contribute to lupus phenotypes in mice, we needed to determine if IFNα has a role in the escape of tolerance by self-reactive B cells at the immature stage. We decided to focus on 564 Igi mice, because the self-reactive B cells of NZB/NZW F1 mice have not been well described. The 564 Igi mice are an immunoglobulin heavy and light chain insertion model (Igi), in which the BCR recognizes a conserved protein-RNA complex with ribonuclear proteins like the SSB/La complex (Chatterjee et al., 2013). The self-reactive B cells can be identified using an anti-idiotype (Id+) antibody. The younger mice do not show overt signs of peripheral inflammation or pathology that might confound B cell differentiation. However, with age, they develop isotype-switched (i.e., IgG 2a and 2b) autoantibodies that activate complement and lead to renal dysfunction (Berland et al., 2006).
Blocking IFNAR ameliorates disease in the lupus BXSB strain (Baccala et al., 2012). Thus, we treated 564 Igi mice with a IFNAR-blocking antibody (Sheehan et al., 2006) for 4 days or 2 weeks (Figure 1C). Using flow cytometry on the single-cell suspensions of the spleen cells, we found that while the total number of B cells remained similar, both anti-IFNAR treatments reduced the frequency of mature (AA4.1 low) Id+ B cells (Figure 1D and S3), meaning IFNα impacts immature B cells. . Moreover, the 4-day treatment reduced the frequency of Id+ GC cells (B220+GL7+CD38neg) (Figure 1E),
To test whether the effects of blocking IFNAR manifested in BM-derived cells or stromal cells, Ifnar1−/− or B6 mice were lethally irradiated and reconstituted with 564 Igi BM. Both sets of chimeras developed an autoimmune phenotype, implicating the BM cells (Figure 1F). By contrast, mixed chimeras in which B6 mice were reconstituted with a mixture of 9 parts Ifnar1−/− BM plus 1 part 564 Igi BM had a significant reduction in mature Id+ B cells, compared to those reconstituted with 9 parts B6 BM (Figure 1G). Consistent with a reduction in self-reactive B cells, the total IgG anti-nucleolar titers were reduced ~7–8 fold (Figure 1G). These results support a model in which the maintenance of circulating, mature self-reactive B cells is dependent on IFNα and signaling through IFNAR by BM-derived cells.
IFNα expression by pDCs does not affect B cell tolerance
pDCs respond to viral infections through TLR7 and 9, and express robust levels of type I interferon (Colonna et al., 2004; Iwasaki and Medzhitov, 2004). They can be identified by the unique cell-surface marker Siglec-H (Blasius et al., 2004; Blasius et al., 2006), which signals through the adaptor DAP-12 to dampen IFNα expression. A specific antibody (440c) binding to Siglec-H shuts off type I interferon expression (Blasius et al., 2006).
To determine if pDCs are an important functional source of IFNα, we first treated 564 Igi mice with a neutralizing dose (500 μg/day) of anti-Siglec-H (440c) for 4 days as described (Ochando et al., 2006) (Figure 2A). At day five, treated and non-treated controls were sacrificed and single-cell suspensions of spleen cells were prepared. Flow analysis of the CD11c+PDCA-1+ pDCs isolated from the controls confirmed that this population was positive for IFNα (Figures 2B) while the cells from anti-Siglec-H-treated 564 Igi mice were negative (Figure 2B). These results were confirmed by LSCM analysis of splenic tissue (Figure S4), and this treatment was confirmed to not affect expression of IFNα by FDCs (Figure S4).
Figure 2: Absence of IFNα secretion by pDCs does not affect mature self-reactive B cells.
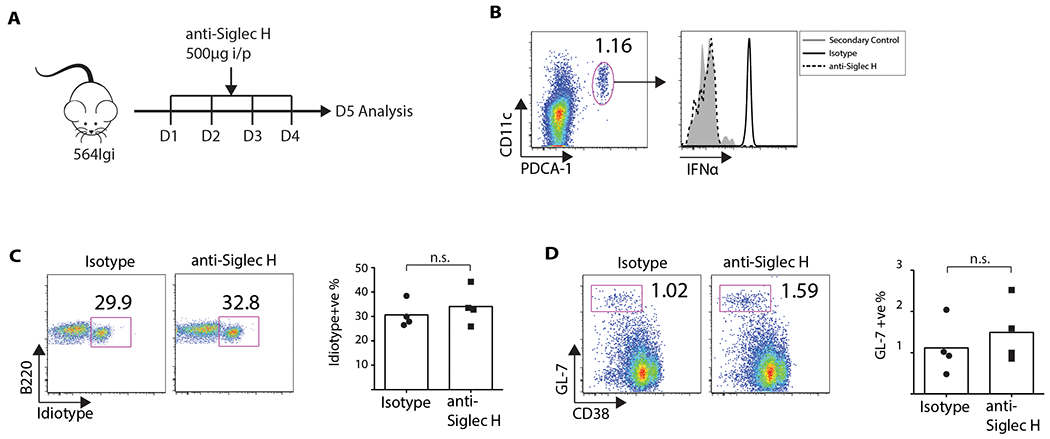
(A) 564 Igi mice were injected with 500 μg of anti-Siglec H (clone 440c). (B) Day 5, cells were stained for B220, CD11 c, and PDCA-1, followed by fixation-permeablization and staining for IFNα. Left, pDC gating. Right, IFNα MFI comparison. There were four mice per group. (C) FACS plots showing % mature Id+ B cells (Student’s t test, n = 4, P > 0.05). Gated on mature B220+ AA4.1 low cells. Bar graph has four mice per group. (D) FACS plots showing % GC B cells (Student’s t test, n = 4, P > 0.05). Gated on B220+ cells. Bar graph has four mice per group.
Next, spleen cells from the two groups were analyzed for the frequency of Id+ cells. Blocking IFNα expression by pDCs had a negligible effect on the frequency of Id+ B cells in the 564 Igi mice (Figure 2C), and staining of the spleen cells (B220+GL7+CD38neg) showed that treatment with anti-Siglec-H antibody did not affect the frequency of Id+ GC B cells (Figure 2D). Altogether, the results demonstrate that although IFNα expression is elevated in pDCs, they are not critical for mediating the escape of self-reactive B cells from negative selection or for forming GCs in 564 Igi mice.
FDCs are a functional source of IFNα
Immunostaining of splenic cryosections prepared from 564 Igi and NZB/NZW F1 mice suggests FDCs may be a functional source of IFNα (Figure 1B).To examine this source of IFNα in more detail, we isolated FDCs from the spleens of 564 Igi and B6 controls and sorted them using flow cytometry (Figures 3A and S1D). We extracted the RNA and analyzed the transcriptome using a TaqMan Fluidigm gene array, identifying increased expression of at least five isoforms of Ifna (Figure 3B) and a two-fold increase in IFNα–stimulated genes such as Mx1, Cxcl10, and Ifit1 (Figure 3C).
Figure 3: FDCs express Ifna in response to self-antigen (RNP)–IC and TLR7 activation, and at comparable levels to pDCs in the 564 Igi mouse.
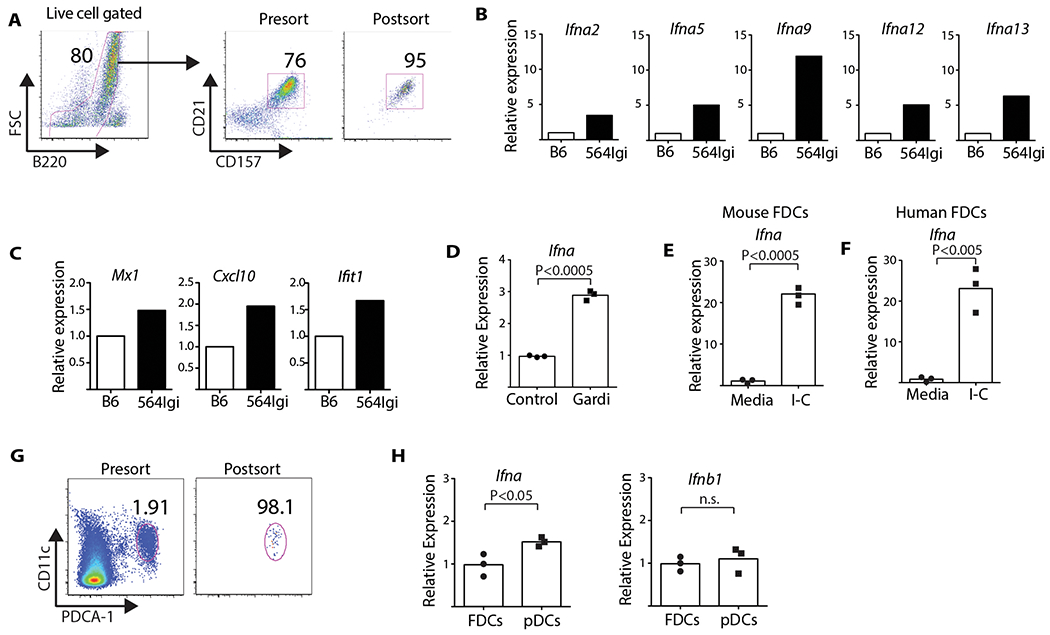
(A) FDCs were purified as described in Methods. (B) Fluidigm analysis of mRNA expression of different Ifna isoforms. (C) IFNα-inducible genes expressed by FDCs: Mx1, Cxcl10, and Ifit1. (D) B6 FDCs from spleens treated with gardiquimod (1 μg/ml) for 6 hours. Bar graph shows relative level of Ifna (Student’s t test, n = 3, P < 0.0005). Data from three experiments. (E) Murine FDCs treated with RNP-IC. RT-PCR of Ifna levels (Student’s t test, n = 3, P < 0.0005). Data representative of three experiments. (F) FDCs isolated from human spleen incubated with RNP-IC. Primers designed to detect all isoforms of Ifna (Student’s t test, n = 3, P < 0.005). (G) Splenic pDCs (B220+CD11intPDCA-1+) isolated from 564 Igi mice were purified by sorting. FDCs were also isolated from the same mice. (H) Expression levels of Ifna and Ifnb1 measured by RT-PCR. Bar graphs show expression of Ifna and Ifnb1 in FDCs and pDCs (Student’s t test, n = 3, P < 0.05). Data representative of two experiments.
To validate the gene array results and to test for functional activation of TLR7, splenic FDCs from B6 mice were stimulated for 6 hours with a TLR7-specific agonist (gardiquimod). We analyzed the FDC lysates using RT-PCR, identifying a significant upregulation of Ifna RNA, as compared to the no-ligand controls (Figure 3D). To determine if self-antigen can stimulate IFNα production, we exposed B6 FDCs to complement-opsonized immune complexes composed of RNP and antibody (RNP-IC). After overnight culture, cells were washed and RNA extracted for RT-PCR analysis. We identified an approximate 20-fold increase in the expression of Ifna in cultures treated with RNP-IC relative to the no-IC controls (Figure 3E). Thus, FDCs express Ifna in vivo in 564Igi mice and ex vivo when stimulated with complement-opsonized RNP complexes.
This suggests a novel pathway for “sensing” DAMPs and secreting proinflammatory cytokines. To test if a similar pathway was conserved in human FDCs, CD35+ stromal cells were harvested from splenic tissue, seeded on cover slips, and cultured overnight with nucleolar IC. We extracted and analyzed the RNA, finding an approximate 20-fold increase in Ifna expression relative to the no-IC controls (Figure 3F). Thus, as observed in mice, human FDCs can “sense” DAMPs taken up by CD21, and they respond through activation of endosomal TLRs.
To estimate the relative amounts of Ifna and Ifnb1 expressed, we harvested FDCs and pDCs from 564 Igi spleens and analyzed the RNA using RT-PCR (Figure 3G and H), finding a ~1.5× increase in Ifna expression by pDCs relative to FDCs, but similar levels of Ifnb1. Ifna expression by splenic FDCs and pDCs was enriched about 80- and 120-fold, respectively, when compared to total spleen; while Ifnb1 expression was enriched about 50-fold (Figure S1C). Although both pDCs and FDCs expressed Ifnb1 the levels were substantially less than those of Ifna based on CT values (Figure S1C). Overall, FDCs isolated from 564 Igi mice spontaneously expressed elevated levels of Ifna relative to B6 mice and that level is comparable to that of pDCs. Moreover, stimulation of murine and human FDCs in ex vivo cultures with complement-opsonized RNP-IC induces a robust Ifna response, supporting the in vivo findings.
FDCs internalize and cycle self-antigen immune complexes
FDCs take up foreign-antigen immune complexes via the CD21 receptor and retain the intact complexes for extended periods within a cycling non-degradative compartment (Heesters et al., 2013; Heesters et al., 2014). To test whether self-antigen IC is opsonized with complement and internalized in a similar manner, splenic FDCs were isolated from 564 Igi mice, stained with a panel of antibodies specific for CD21, autoantibody (idiotype), and RNP, and analyzed by LSCM (results not shown). RNP antigen and the CD21 receptor were co-localized (results not shown), suggesting that self-antigen IC was taken-up by CD21. To confirm this, 564 Igi FDCs were cultured overnight with fluorescently labeled transferrin ligand (Tf) – a molecule that binds to transferrin receptor and is constitutively internalized and cycled in a non-degradative early endosomal compartment (Boulant et al., 2011; Heesters et al., 2013). The FDCs were then stained and analyzed by LSCM for idiotype, LAMP1 (lysosome marker), and the nucleus. There was significant co-localization between Id+ RNP-IC and Tf (Figure 4A) and limited co-localization between Id+ RNP-IC and the degradative LAMP1 compartment. Thus, self-antigen ICs are bound by CD21 and internalized for the most part into non-degradative cycling compartments, much like foreign antigen.
Figure 4: FDCs cycle self-antigen after internalization through CD21 and activate TLR7.
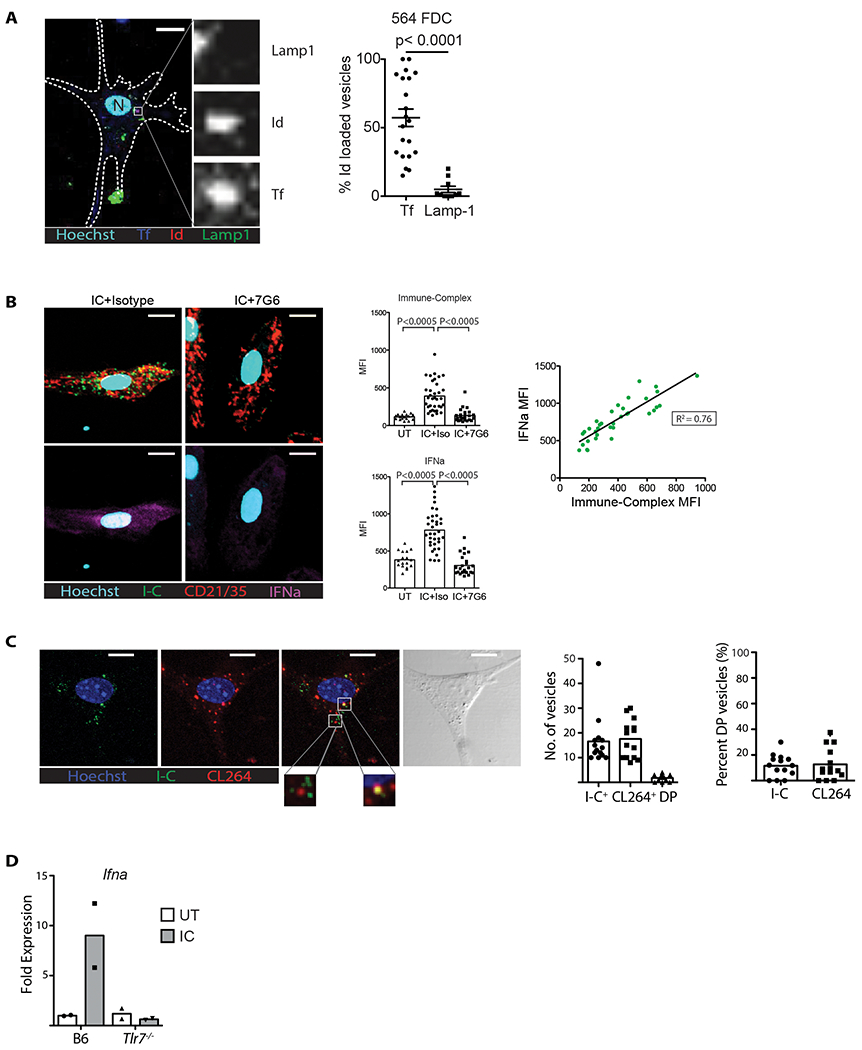
(A) Purified 564 Igi FDCs were treated with fluorescent-labeled transferrin ligand, stained for RNP-IC and Lamp1, and imaged by LSCM. Left, representative confocal image with Id+, transferrin ligand (Tf), or lysosome (Lamp1) stain. Tf (blue); Id (red); Lamp1 (green); Nucleus (cyan). Scale: 10 μm. Insets identify one Id+ Tf+ vesicle at higher magnification. Right, quantification of Id+ vesicles. Each data point is one cell (Student’s T test, P < 0.05). (B) B6 splenic FDCs treated with RNP-IC followed by anti-CD21 (7G6 clone). Representative LSCM panels; nucleus (cyan); Id+ vesicles (green); CD21/35 (red); IFNα (magenta). Scale: 10 μm. Top bar graph, MFI of Id+ vesicles in each cell quantified from three groups: untreated (UT), RNP-IC, and RNP-IC followed by purge with anti-CD21 (7G6) or isotype control (Iso) (Student’s t test, n = 35, P < 0.0005). Bottom bar graph, MFI of IFNα signal in each cell quantified from the same 3 groups (Student’s t test, n = 35, P < 0.0005). The dot plot shows the correlation between the RNP-IC and IFNα MFIs of each cell quantified. (C) Cultured B6 splenic FDCs treated with RNP-IC before labeling with CL264-rhodamine. Left, representative LSCM; nucleus (blue); Id+ (IC+) vesicles (green); CL264+ vesicles (red); combined red and green, and bright-field image. Scale: 10 μm. Insets show magnified examples of CL264+ vesicles with or without RNP-IC. Right, bar graphs compare the number of vesicles per cell stained for Id+ (IC+) alone, CL264+ alone, or both. Bar graph on far right compares % of double-positive vesicles of total Id+ (IC+) or CL264+. Representative of two experiments. (D) FDCs isolated from B6 or Tlr7−/− mice treated with RNP-IC overnight, then RT-PCR of Ifna levels. Data representative of two experiments.
Although self-antigen ICs co-localized primarily with CD21, it is possible that some were internalized via other pathways. To test this, splenic FDCs isolated from B6 mice were loaded with C3-opsonized RNP-IC for 60 minutes, then treated with a CD21-blocking antibody (clone 7G6) that disrupts C3d-bound ICs or treated with an Ig isotype control (Heyman et al., 1990). After washing, fixing, and imaging, we found a negligible amount of self-antigen ICs in cultures treated with anti-CD21, while the isotype controls still had it (Figure 4B). Staining with anti-IFNα and analysis with LSCM identified a punctate pattern that correlated with CD21 expression, uptake of RNP-IC, and total level of IC present (R2=0.76) (Figure 4B). Treatment of the loaded FDCs with anti-CD21 reduced expression of both RNP-IC and IFNα, as expected (Figure 4B). Thus, anti-CD21 treatment of the live cell cultures purged RNP-ICs from the FDCs and “shut-off” expression of the IFNα protein. The results not only confirm that self-antigen ICs are primarily retained by CD21 and are periodically accessible on the FDC surface, but they suggest a novel pathway by which DAMPs internalized via CD21 trigger endosomal TLR and induce expression of IFNα.
Activation of endosomal TLR7 leads to activation of type I interferons and the NF-kB pathway (Barton and Medzhitov, 2003). Our results suggest that CD21+ vesicles containing RNP-IC complexes intersect with the TLR endosomal compartment and activate TLR7. To test this, we cultured B6 FDCs for 30 minutes with labeled RNP-IC, cultured them for 30 minutes with rhodamine-labeled CL264 (a ligand specific to TLR7), fixed the cultures, counterstained for nuclei with Hoechst, and imaged the cells. We identified distinct vesicles containing RNP-IC or CL264 (Figure 4C), with ~10% of these vesicles being co-localized. These results suggest that a fraction of the CD21-RNP-IC vesicles intersect with the TLR7 compartment, meaning DAMPs are potentially recognized by TLR7, which could induce IFNα expression (Figure 4C).
If the major pathway for FDC expression of IFNα is via TLR7 then TLR7-deficient FDCs will not respond to the uptake of RNP-IC via CD21. Using a similar approach as described above, B6 and B6 Tlr7−/− FDCs were cultured with complement-opsonized RNP-IC overnight, and Ifna RNA expression was analyzed. As expected, there was a significant increase in Ifna expression by B6 FDCs treated with RNP-IC, but Tlr7−/− FDCs failed to respond to RNP-IC and expressed negligible levels of Ifna (Figure 4D).
Thus, as observed for foreign antigen, FDCs took up complement-opsonized self-antigen complexes via CD21 into a similar compartment as transferrin ligand. We observed that a fraction of the RNP-IC-positive vesicles intersected with the TLR7 endosomal compartment and triggered IFNα expression. Furthermore, FDCs deficient in TLR7 failed to express Ifna following culture with complement-opsonized DAMPs.
Autoantibody production in 564 Igi mice is dependent on the FDC MYD88 pathway
MYD88 is an essential adaptor for signaling through endosomal TLR 3, 5, 7, 8, and 9. To test the importance of endosomal TLR signaling by FDCs in the lupus model, mice bearing a floxed MYD88 locus (Myd88fl/fl (Hou et al., 2008)) were bred with a Cd21cre strain (Kraus et al., 2004). In this strain, Cd21cre expression is limited to FDCs, B cells, and to an undefined cell type in the CNS (Moriyama et al., 2011; Victoratos et al., 2006). To further limit MYD88 blocking to FDCs, a BM chimera was prepared in which Cd21creMyd88fl/fl mice were lethally irradiated and reconstituted with 564 Igi BM (Figure 5A). Thus, all BM-derived cells, including pDCs and self-reactive B cells, had sufficient MYD88 while FDCs were deficient. MYD88 also participates in the TLR4 pathway; however, TIRAP is required for activation (Barton and Medzhitov, 2003). To distinguish between the endosomal TLR and TLR4 pathways, additional control BM chimeras were constructed by reconstituting Tirap−/− mice with 564 Igi BM (Figure 5A).
Figure 5: FDCs maintain self-reactive B cells via the MyD88 pathway.
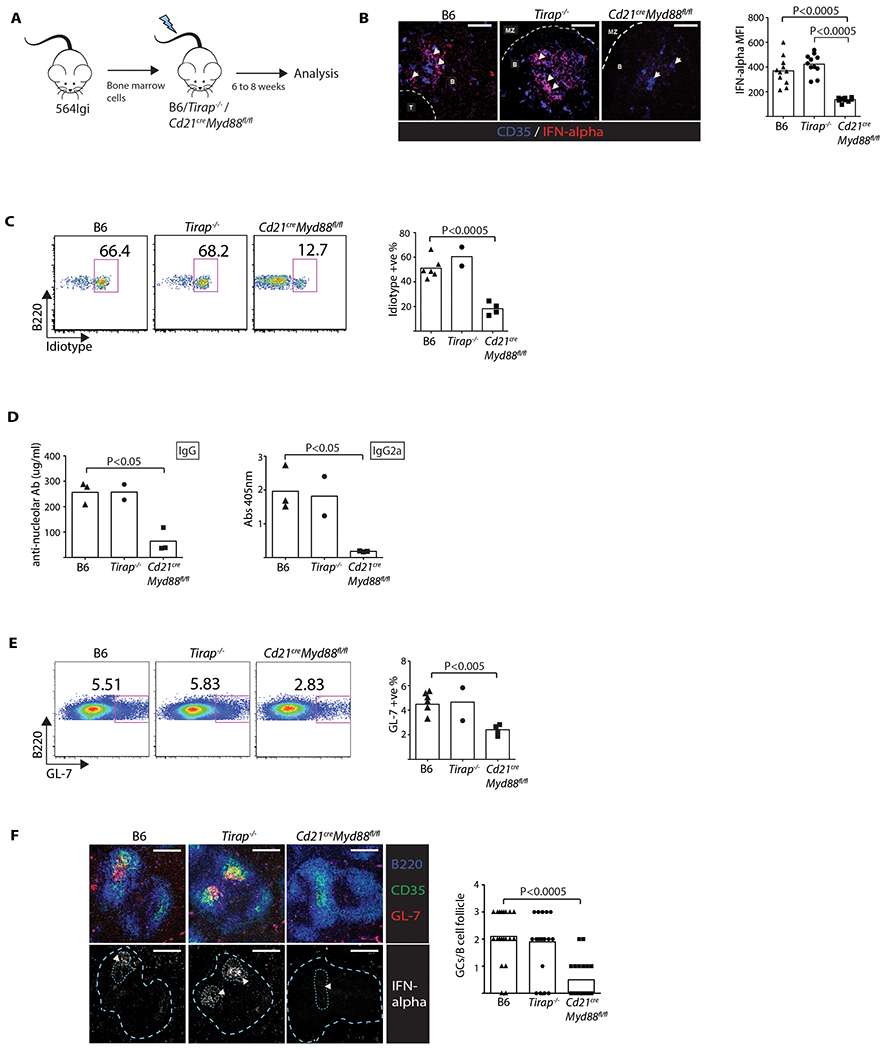
(A) Diagram of BM chimeras. (B) LSCM of spleen sections (60×) from BM recipients (one-way ANOVA, n = 11, P < 0.0005). Scale: 50 μm. Arrowheads indicate FDC networks. Data representative of two experiments. Bar graph of MFI of IFNα. Each data point is one continuous FDC area. (C) Flow cytometry plots showing mature % Id+ B cells in BM chimeras gated on mature B220 positive AA4.1 low cells (one-way ANOVA). Data from two experiments. (D) ELISA serum anti-nucleolar IgG (left panel) and IgG2a (right panel) levels in the BM chimeras (one-way ANOVA, n = 3, P < 0.05). (E) Flow cytometry plots showing GL7+ B cell frequencies (one-way ANOVA). Representative plots are gated on B220+GL7+ splenic B cells. Data from two experiments. (F) LSCM of spleen cryosections (20×) from the BM chimeras (one-way ANOVA, n = 20, P < 0.0005). Scale: 200 μm. Arrowheads indicate FDC/GC area. GC areas and follicle boundaries are indicated by dotted lines in second row of images. Data representative of two experiments. Bar graph indicates number of GCs per B-cell follicle. Each data point is one follicle.
The spleens were harvested from the three groups of BM chimeras, and cryosections stained with anti-IFNα. In B6 and B6 Tirap−/− recipient spleens, follicular IFNα co-localized with CD35+ reticular cells. By contrast, negligible IFNα staining of FDCs was observed in the Cd21cre Myd88fl/fl recipient spleens (Figure 5B). Thus, as observed in the in vitro experiment using TLR7-deficient FDCs, blocking MYD88 expression in FDCs led to impaired IFNα expression in the spleens of the 564 Igi BM chimeras.
To determine whether blocking FDC endosomal TLRs affected the escape from B cell tolerance, a single-cell suspension was prepared from the spleens of the three sets of 564 Igi BM chimeras and analyzed by flow cytometry. There was a spontaneous return of Id+ self-reactive B cells in the B6 control chimeras, as expected (Figure 5C), and a similar frequency of Id+ cells in the Tirap−/− chimeras, confirming that TLR4 activation via MYD88 was not required for escape, though TLR4 can also be activated independently of MYD88 via the TRIF adaptor. Strikingly, the 564 Igi BM chimeras prepared with Cd21creMyd88fl/fl recipients had a reduced frequency of mature Id+ B cells relative to the control and Tirap−/− chimeric mice (Figure 5C).
To look for autoantibody secretion in the 564 Igi BM chimeras, serum prepared from the 3 groups was assayed by ELISA for binding to nucleoli-coated plates. As predicted, both B6 and Tirap−/− chimeras expressed similar levels of IgG autoantibody as 564 Igi mice, which ranged between 200 to 300 μg/ml (Figure 5D). By contrast, mouse chimeras bearing the FDC-specific impairment of MYD88 signaling (Cd21creMyd88fl/fl) expressed ~4-fold lower levels of total IgG autoantibody. Analysis of the pathogenic IgG2a isotype revealed a 5–6 fold reduction in the anti-nucleolar titer (OD405 nm) (Figure 5D). This isotype is particularly relevant since it activates the inflammatory complement system, and activating Fc-receptors can be pathogenic in autoimmune disease such as lupus (Banchereau et al., 2016).
We also determined the frequency of splenic GC B cells by flow cytometry and LSCM. Consistent with a reduction in the anti-nucleolar autoantibody of the switched IgG2a isotype, the frequency of GL7+ GC B cells was significantly reduced in the Cd21creMyd88fl/fl recipients relative to the B6 and Tirap−/− recipients in both spleen (Figure 5E) and skin-draining LNs (data not shown), and there was a 4-fold reduction in the GCs per B-cell follicle count in splenic tissue (Figure 5F). IFNα expression by FDCs within the GC follicular/GC areas was prominent in sections prepared from the B6 and Tirap−/− recipients but negligible in the Cd21creMyd88fl/fl recipients (Figure 5F), as expected.
Although FDCs are best characterized in their role of retaining antigen in secondary lymphoid tissues, such as the spleen, LNs, and Peyer’s patches, they may have a role in the negative selection of 564 Igi B cells in the BM during the immature stage (Allman et al., 2001). We found a similar frequency of Id+ immature B cells (AA4.1hi B220+) among the 3 chimeras (data not shown), approximately 80%, which is similar to the range identified earlier for immature B cells isolated from the spleens of 564 Igi mice (Chatterjee et al., 2013). Thus, the MYD88 signaling by FDC is unlikely to play a role in the elimination of immature self-reactive B cells in the BM and more likely to be affecting their maturation and differentiation instead.
Together, these results support a model in which expression of IFN I by FDCs is critical for the escape of tolerance by self-reactive B cells and their maturation and differentiation in GCs, as well as their eventual secretion of pathogenic IgG autoantibody.
TLR7 signaling by FDCs is dependent on IRF5
Activation of TLR7 by agonists, such as gardiquimod, triggers the IFNα pathway in pDCs, which is dependent on the transcription factor IRF7 (Honda et al., 2005) and is regulated by IRF5, whose deficiency in lupus-prone mice is protective (Watkins et al., 2015; Yasuda et al., 2013; Yasuda et al., 2007; Yasuda et al., 2014). To determine if IRF5 or IRF7 is required in the FDC response to RNP-IC, FDCs were isolated from B6 controls or mice bearing a knockout of either transcription factor. We treated ex vivo FDC cultures with RNP-ICs overnight and analyzed their RNA by RT-PCR for Ifna and Il6 expression. Ifna expression in Irf5−/− FDCs was ~10-fold less than B6 FDCs, whereas the response of Irf7−/− FDCs was less robust (Figure S5A). Similarly, Il6 expression was reduced in Irf5−/− and Irf7−/− FDCs relative to B6 controls (Figure S5A). Thus, activation of FDCs and the induction of Ifna and Il6 by the TLR7 agonist are dependent on both IRF5 and IRF7.
To test whether IRF5 is required in stromal cells for IFNα production in vivo, Irf5−/− and control B6 mice were lethally irradiated and reconstituted with 564 Igi BM (Figure S5B). IFNα expression was reduced in Irf5−/− FDCs (Figure S5C), as determined by staining, and there were fewer mature Id+ B cells in the Irf5−/− recipients, as determined by flow cytometry (Figure S5D). Concordantly, there was a ~5-fold decrease in serum IgG autoantibody levels in the Irf5−/− recipients, a decrease in the anti-nucleolar autoantibody titer (OD405 nm) of the IgG2a isotype (Figure S5E), a decrease in the frequency of GL7+ GC B cells (Figures S5F), and a decrease in the number of GCs per follicle versus the B6 recipients (Figure S5G). IFNα expression by FDCs co-localized with the splenic GC/follicle areas in the B6 recipients as expected. By contrast, expression was absent in those areas in the Irf5−/− recipients (Figure S5G). Overall, the results suggest that IRF5, and potentially IRF7, are required for TLR7 activation and its downstream affects on the lupus phenotype.
TLR7 expression by stromal cells is essential for the maintenance of self-reactivity
The finding that blocking MYD88 signaling in FDCs shuts off IFNα secretion and leads to a reduction in spontaneous autoimmunity supports a critical role for endosomal TLR signaling in autoimmunity. To provide direct support for TLR7’s effect in lymphoid stromal cells, Tlr7−/− mice were lethally irradiated and reconstituted with 564 Igi BM (Figure 6A), meaning that the hematopoietic cells, including pDCs and B cells, were derived from the 564 Igi donor strain (and were Tlr7+/+), while the radiation-resistant stromal cells, including FDCs, were TLR7-deficient. If endosomal TLR7 signaling by FDCs is the major pathway for IFNα secretion, we would expect that splenic follicles in the Tlr7−/− BM chimeras would have negligible amounts of IFNα protein. Indeed, splenic sections prepared from the Tlr7−/− recipients had significantly less FDC IFNα expression (Figure 6B) than the 564 Igi BM chimeras where the recipients were TLR7 sufficient.
Figure 6: Autoreactive B cells and autoantibody production is dependent on TLR7 expression by stromal cells.
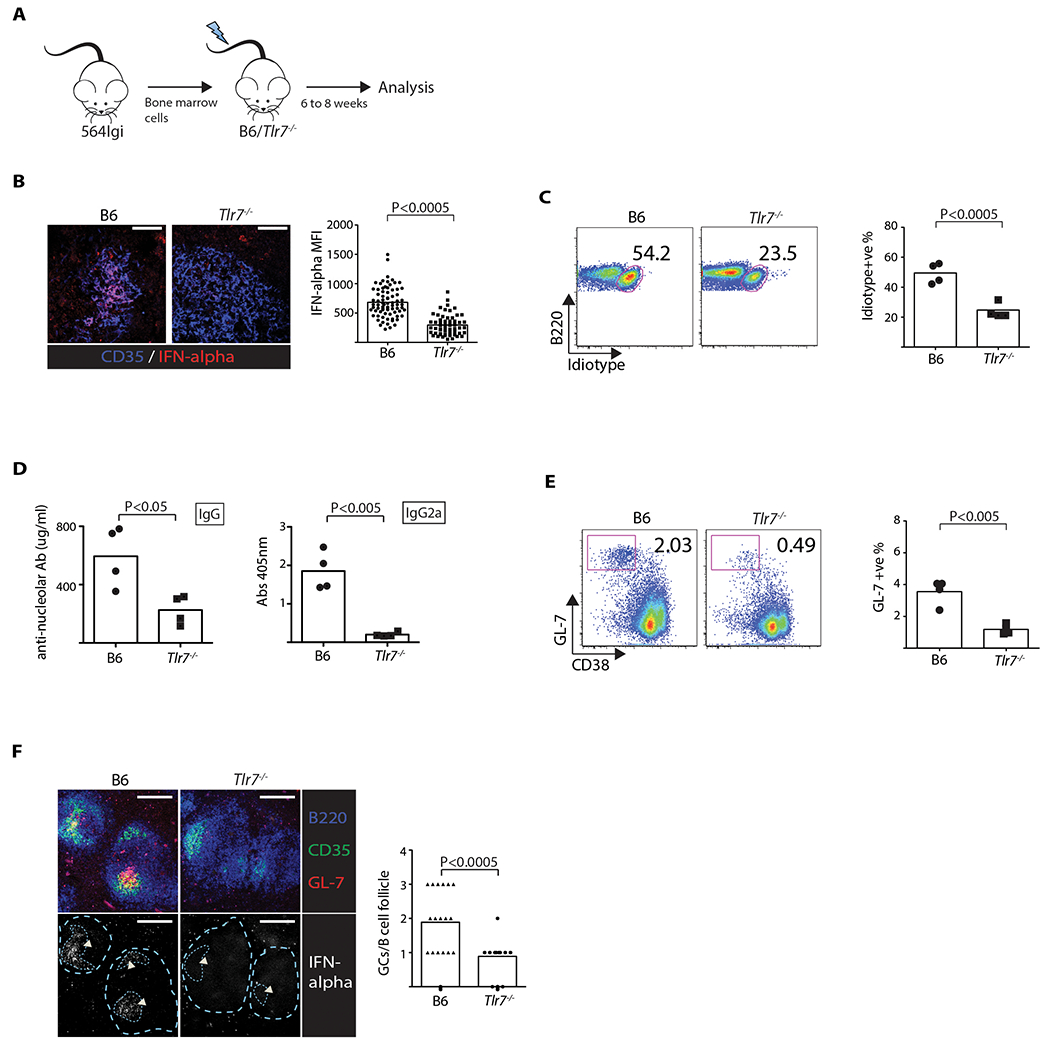
(A) Diagram of BM chimeras. (B) IFNα expression within FDC network based on LSCM of spleen sections (60×) (Student’s t test, n = 75, P < 0.0005). Scale: 50 μm. Data representative of two experiments. Bar graph of MFI of IFNα. Each data point is one continuous FDC area. (C) FACS plots showing mature Id+ B cell frequencies (Student’s t test, n = 4, P < 0.0005). Plots shown are gated on B220+ AA4.1 low cells. Each data point is one mouse. Data representative of two experiments. Bar graphs represent pooled results. (D) ELISA data showing serum anti-nucleolar IgG (left panel) and IgG2a (right panel) (Student’s t test, n = 4, P < 0.05). (E) FACS of splenic GL7+ CD38− GC B cell frequencies (Student’s t test, n = 4, P < 0.005). Plots shown are gated on B220+ cells. Data representative of three experiments. Each data point is one mouse. (F) LSCM of spleen sections (20×) (one-way ANOVA, n = 18, P < 0.0005). Scale: 200 μm. Arrowheads indicate FDC/GC area. GC areas and follicle boundaries are indicated by dotted lines in second row of images. Data representative of two experiments. Bar graph indicates number of GCs per B-cell follicle. Each data point is one B-cell follicle.
Flow cytometry of splenic cells found that the Tlr7−/− recipients had a reduced frequency of mature self-reactive (Id+) B cells compared to B6 mice reconstituted with 564 Igi BM (Figure 6C) and reduced IgG and IgG2a titers of anti-nucleolar autoantibody (Figure 6D). As expected from the IgG autoantibody titers, the frequency of GL7+ CD38 lo GC B cells was reduced in the Tlr7−/− recipients relative to B6 recipients (Figures 6E), and LSCM analysis confirmed a similar reduction in the frequency of GCs (Figure 6F). Notably, IFNα staining colocalized with the FDC region in the B6 recipients, but was absent in follicles of Tlr7−/− recipients (Figure 6F). To test if the Tlr7−/− stromal cells in the BM chimeras also led to reduced deposition of Id+ immune complexes, kidneys were harvested from the two chimeras and cryosections stained with anti-Id antibody. LSCM analysis identified deposits of Id+ antibody in glomeruli of all four B6 564 Igi BM chimeras, but only one of the four kidneys of Tlr7−/− 564 Igi BM chimeras (Figure S6), which is consistent with the reduction in IgG2a anti-nucleolar antibody titers.
The combined results demonstrate that the Tlr7−/− recipients had a significant reduction in autoimmunity relative to the B6 controls, confirming the importance of TLR7 activation in stromal cells during the autoantibody response.
DISCUSSION:
We examined the importance of follicular dendritic cells (FDCs) in the 564 Igi murine model of lupus. As observed in the well-characterized lupus strain NZB/NZW F1, we found that autoimmunity was dependent on type I interferon (IFN I). Blocking IFNα receptor (IFNAR) signaling, either with a blocking antibody or genetic deficiency in bone marrow–derived cells, reduced the loss of B cell tolerance, reduced spontaneous GC formation, and reduced by 4–5 fold the secretion of pathogenic autoantibody. Remarkably, plasmacytoid dendritic cells (pDCs), which are a major source of IFN I in lupus mouse models and lupus patients, were not an essential source here. Instead, FDC expression was crucial to spontaneous autoimmunity. We propose that the underlying mechanism is due to FDCs internalizing ribonucleolar immune complexes (RNP-IC) via CD21, which triggers the endosomal TLR7 pathway and induces expression of IFNα and IFNβ.
We presented several lines of evidence to support our model. First, FDCs isolated from 564 Igi mice, but not WT mice, spontaneously expressed Ifna and Ifnb1. Second, stimulation of WT FDCs with gardiquimod, a specific ligand for TLR7, upregulated Ifna mRNA ~3-fold over a 6-hour period. Third, B6 FDCs (and human FDCs) that have taken up RNP-IC opsonized with complement expressed Ifna mRNA 20-fold more than non-activated controls. Moreover, in vitro treatment of FDCs loaded with RNP-IC with a blocking anti-CD21 antibody eliminated self-antigen complexes and reduced IFNα expression. Fourth, spontaneous autoimmunity was dramatically reduced in BM chimeric mice in which the FDCs were derived from Tlr7−/− or Cd21cre Myd88fl/fl mice. Finally, IRF5 deficiency in stromal cells reduced IFNα expression by FDCs and, correspondingly, reduced autoimmunity in BM chimeric mice.
Previous studies proposed a two-signal model in which self-reactive B cells are rescued from anergy by the activation of TLR7 following the BCR internalization of nuclear material (Lau et al., 2005; Leadbetter et al., 2002). It has been proposed that the uptake of IC containing apoptotic material by pDCs induces IFNα secretion that enhances B cell sensitivity to TLR7 activation and triggers their differentiation into Ig-producing cells (Krieg, 2007). In the BXSB.DTR model, ablating pDCs prior to disease onset reduces renal injury and, to some extent, autoantibody titers, though the frequency of autoreactive B cells and the GC response have not been examined (Rowland et al., 2014). Likewise, impairing pDCs in lupus strains that overexpress Tlr7 or express the Sle1.Sle3 susceptibility genes reduces autoimmunity (Sisirak et al., 2014). These results are not inconsistent with our observations, as our Siglec-H treatment blocks type I interferon expression and is not thought to impair pDC function.
A possible role for FDCs in autoimmunity was first proposed by Victoratos et al., after they found that an intact FDC network was required for autoimmunity in the K/BxN T cell transgenic arthritis model (Victoratos and Kollias, 2009). They proposed that FDCs were required for the recruitment of arthritogenic T follicular helper cells. Whether self-antigen complexes in the K/BxN model are acquired by FDCs and trigger TLRs was not reported, although elevated expression of type I interferon could help explain autoimmunity in their model.
Using a novel BM chimeric model in which FDCs in the recipient mice express a membrane form of duck egg lysozyme (DEL), Yau et al. constructed chimeric mice in which the B cell compartment developed from donor marrow prepared from SwHEL BCR transgenic strain (Yau et al., 2013). In this mouse, FDC expression of the DEL self-antigen leads to deletion of the self-reactive B cells at the T2 transitional stage, although not all self-reactive B cells are eliminated and a small fraction differentiate to maturity. In the 564 Igi model, where self-antigen immune complexes are retained by FDCs, we also observed a reduced number of self-reactive B cells at the transitional stage but some did escape tolerance. An important difference between the two models is that, in the 564 Igi model, the self-antigen, which includes DAMPs, is internalized and triggers FDC endosomal TLR7, leading to expression of cytokines, such as IFNα, that enhance the escape of tolerance. Thus, the nature of the antigen retained by FDCs can influence their role in the negative selection of self-reactive B cells.
Based on our results, we propose that internalizing DAMPs opsonized with complement via CD21 by FDCs activates endosomal TLRs and triggers the secretion of IFNα. The fact that C3-opsonized DAMPs can be retained and cycled periodically over extensive periods explains the chronic cytokine secretion that “drives” differentiation of self-reactive B cells, spontaneous GC formation, and possibly epitope spreading in mice heterozygous for the Ig BCR. While the staining of FDCs with anti-IFNα was primarily with GCs in the follicles, it seems likely that the response to DAMPs and the activation of TLR7 is not limited to GC FDCs but can occur in primary FDCs following internalization of nucleolar material, as observed in the in vitro experiments.
Although the 564 Igi mice develop a mild form of disease, the secretion of anti-nucleolar IgG leads to kidney deposits of IgG immune complexes and complement C3 by nine months of age (Chatterjee et al., 2013). Eventually, the mice develop renal pathology and dysfunction by 12–15 months of age (Berland et al., 2006). Blocking TLR7 signaling in FDCs dramatically reduces IgG2a autoantibody production and deposition of Id+ immune complexes in the glomeruli of 564 Igi BM chimeras, thus it would be expected to protect against renal injury. This observation combined with our finding that human FDCs also respond to DAMPs and express IFNα in ex vivo cultures suggests this pathway may be an important target for therapy.
In summary, we identified an unexpected role for FDCs as sensors of self-antigen DAMPs, which leads to the secretion of IFNα and the enhancement of autoreactive GCs and autoantibody titers. It is important to learn if additional sensors of innate immunity are triggered via internalization of DAMPs via the CD21 cycling receptor.
EXPERIMENTAL PROCEDURES:
Mice.
We used 564 Ig heavy and light chain knock-in (564 Igi) mice (Berland et al., 2006). Mice were heterozygous for the transgenic (IgMa) and endogenous (IgMb) IgH alleles and were aged 6 to 8 weeks (both males and females). C57BL/6, Ifnar1−/−, Cd21cre, and Myd88fl/fl mice were purchased from Jackson Laboratories. All mice were maintained in specific-pathogen-free facilities at Harvard Medical School. The Institutional Animal Care and Use Committee (IACUC) at PCMM and Harvard Medical School approved all animal experimental procedures. Irf5−/− (Takaoka et al., 2005) and Irf7−/− mice (Honda et al., 2005) were maintained at Boston University animal facility. The Irf5−/− mice used were backcrossed 15 generations with B6 and expressed the normal WT allele for Dock2 (dedicator of cytokinesis)(Yasuda et al., 2013).
Flow cytometry.
Single-cell suspensions were stained with anti-564 (anti-idiotype; IgG1) monoclonal antibody (clone 9D11) or antibodies to CD3, CD19 (1D3), CD21/35 (7G6), CD35 (8C12), CD23, IgD, GL-7, CD38, CD93 (AA4.1), or B220 (RA3-6B2). Antibodies were purchased from BioLegend. Stained cells were acquired using a FACS Calibur or a FACS Canto (BD Biosciences), and data were analyzed using FlowJo (Tree Star, Inc.) software.
Generation of BM chimeric mice.
Recipient mice were prepared as described (Chatterjee et al., 2013). In short, recipient mice were lethally irradiated, and the next day, 1×107 donor-derived BM cells were injected. Recipient mice were fed with sulfamethoxazole/trimethoprim-containing water for two weeks following reconstitution and analyzed 6–8 weeks after reconstitution.
FDC isolation and ex vivo culture.
FDCs were purified by in vivo CD35/CD21 labeling. In short, mice were injected with 10 μg of anti-CD35/CD21 (8C12) labeled with biotin. Spleens were harvested and digested with Collagenase P, Dispase, and DNase I. FDCs were obtained by labeling with streptavidin-coated magnetic beads (Miltenyi) and passing the cell suspension through EasySep magnet. 50000 cells were plated on collagen(Roche)-coated coverslips (Warner Instruments). Cells were cultured on coverslips for 5 to 10 days to allow recovery and to regain their dendritic morphology.
Antibody treatment of 564Igi mice.
To block type I interferon signaling in 564Igi strain, mice were injected with 250μg of anti-IFNAR(Clone MAR1-5A3, a kind gift from Medimmune, LLC, originally developed in the lab of Dr. Robert D. Schreiber) or isotype control (Clone 1A7, a kind gift from Medimmune, LLC, originally developed in the lab of Dr. Michael S. Kinch), intra-peritoneally for 4 consecutive days (Sheehan et al., 2006). On day 5 mice were euthanized and cells were isolated for analysis of frequency of idiotype positive B cells and germinal center B cells. For long-term treatment, 564Igi mice were treated for two weeks with anti-IFNAR. The mice were injected with 250μg of the anti-IFNAR or isotype control antibody intra-peritoneally on days 0, 4, 8 and 12. The spleens were harvested and analyzed on day14.
Anti-Siglec H treatment is known to block IFNα production by pDCs. 564Igi mice were injected with 500μg of anti-Siglec H(Bio X Cell, Clone 440c, originally developed in the lab of Dr. Marco Collona) or isotype control (Bio X Cell, Clone LTF-2), intra-peritoneally for 4 consecutive days (Ochando et al., 2006) (Blasius et al., 2004; Blasius et al., 2006). On day 5 mice were euthanized and cells were isolated for analysis. For intracellular staining of IFNα, 10 X 106 cells were resuspended in 1ml of DMEM supplemented with 10%FBS containing 10μg/ml of Brefeldin A. The cells were incubated for 3 hours at 37°C. After the incubation, cells were stained for surface markers and fixed with fixation buffer (Biolegend). Cells were then permeabilized using permeabilization buffer (Biolegend) following manufacturer’s protocol and then stained for IFNα.
Anti-nucleolar ELISA.
For measuring levels of auto-antibodies against nucleoli, an indirect ELISA approach was used. ELISA plates were coated with nucleoli isolated from Raji cells (as described in supplemental experimental methods). After blocking, serum samples and standard antibody (564 anti-RNP; C11 clone) were added to their designated wells. For detection, either alkaline phosphatase (AP) labeled anti-IgG or anti-IgG2a was used. In case of IgG level analysis, 564-anti-nucleolar antibody (clone C11) was used as standard for calculation of serum levels.
Immunohistology and confocal imaging.
Cryosections of spleen and LN tissues were prepared and stained as described previously (Chatterjee et al 2013). Images were acquired with a FluoView FV1000 confocal microscope (Olympus) and processed with FluoView software (Olympus). Data were analyzed with Volocity software (Perkin-Elmer) or ImageJ.
Gene expression analysis
Gene expression analysis was performed using TaqMan Gene Expression Assays in the BioMark 48.48 Dynamic Array chips (Fluidigm Corp.) Delta-delta Cts (ΔΔCt) were calculated using the mean of two reference genes (18S and GAPDH) and converted to fold change by the 2−ΔΔCt formula relative to the B6 control. qPCR (RT-PCR) analyses of Ifna, Il6, and Ifnb1 were performed using the SYBR-green system. The primers used were Ifna: 5’-cttccacaggatcactgtgtacct-3’ and 5’-ttctgctctgaccacctccc-3’, Il6: 5’-tagtccttcctaccccaatttcc-3’ and 5’-ttggtccttagccactccttc-3’, and Ifnb1: 5’–actagaggaaaagcaagaggaaag-3’ and 5’-ccaccatccaggcgtagc-3’.
Statistical Analysis.
Results were expressed as the mean ± the standard error of the mean (SEM). Differences between groups were analyzed using the Student’s t test. One-way ANOVA was used for multi-group comparisons.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank all members of the M.C.C. lab for suggestions and help with experiments and reading of the manuscript. We acknowledge the valuable assistance of Elisabeth M. Carroll and the editorial assistance from Dr. Christina Usher. The authors thank Drs. T. Taniguchi and T. Mak for access to Irf5−/− and Irf7−/− mice. This work was supported by grants from NIH (AI039246) to M.C.C. and from MedImmune LLC.
Footnotes
COMPETING FINANCIAL INTERESTS
R.H., N.M., and G.C. are employees of MedImmune, LLC
REFERENCES:
- Allen CD, Okada T, and Cyster JG (2007a). Germinal-center organization and cellular dynamics. Immunity 27, 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CD, Okada T, Tang HL, and Cyster JG (2007b). Imaging of germinal center selection events during affinity maturation. Science 315, 528–531. [DOI] [PubMed] [Google Scholar]
- Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, and Hardy RR (2001). Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. Journal of immunology 167, 6834–6840. [DOI] [PubMed] [Google Scholar]
- Baccala R, Gonzalez-Quintial R, Schreiber RD, Lawson BR, Kono DH, and Theofilopoulos AN (2012). Anti-IFN-alpha/beta receptor antibody treatment ameliorates disease in lupus-predisposed mice. Journal of immunology 189, 5976–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, Cepika AM, Acs P, Turner J, Anguiano E, et al. (2016). Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 165, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrington RA, Pozdnyakova O, Zafari MR, Benjamin CD, and Carroll MC (2002). B lymphocyte memory: role of stromal cell complement and FcgammaRIIB receptors. J Exp Med 196, 1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, and Medzhitov R (2003). Toll-like receptor signaling pathways. Science 300, 1524–1525. [DOI] [PubMed] [Google Scholar]
- Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, and Imanishi-Kari T (2006). Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 25, 429–440. [DOI] [PubMed] [Google Scholar]
- Blasius A, Vermi W, Krug A, Facchetti F, Cella M, and Colonna M (2004). A cell-surface molecule selectively expressed on murine natural interferon-producing cells that blocks secretion of interferon-alpha. Blood 103, 4201–4206. [DOI] [PubMed] [Google Scholar]
- Blasius AL, Cella M, Maldonado J, Takai T, and Colonna M (2006). Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood 107, 2474–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Kural C, Zeeh JC, Ubelmann F, and Kirchhausen T (2011). Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat Cell Biol 13, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R, Shih TA, Kleinewietfeld M, Rakonjac J, Nemazee D, Rajewsky K, and Nussenzweig MC (2001). Contribution of receptor editing to the antibody repertoire. Science 291, 1541–1544. [DOI] [PubMed] [Google Scholar]
- Chatterjee P, Agyemang AF, Alimzhanov MB, Degn S, Tsiftsoglou SA, Alicot E, Jones SA, Ma M, and Carroll MC (2013). Complement C4 maintains peripheral B-cell tolerance in a myeloid cell dependent manner. European journal of immunology 43, 2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, and Reap EA (2002). Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. The Journal of experimental medicine 196, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Trinchieri G, and Liu YJ (2004). Plasmacytoid dendritic cells in immunity. Nat Immunol 5, 1219–1226. [DOI] [PubMed] [Google Scholar]
- Cotran RS, Kumar V, and Robbins SL (1994). Pathologic basis of disease, 5th edn (Philadelphia: W.B. Saunders; ). [Google Scholar]
- Cremasco V, Woodruff MC, Onder L, Cupovic J, Nieves-Bonilla JM, Schildberg FA, Chang J, Cremasco F, Harvey CJ, Wucherpfennig K, et al. (2014). B cell homeostasis and follicle confines are governed by fibroblastic reticular cells. Nature Immunology 15, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow MK (2009). Interferon-alpha: a therapeutic target in systemic lupus erythematosus. Rheum Dis Clin North Am 36, 173–186, x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison LM, and Jorgensen TN (2015). Sialic acid-binding immunoglobulin-type lectin H-positive plasmacytoid dendritic cells drive spontaneous lupus-like disease development in B6.Nba2 mice. Arthritis Rheumatol 67, 1012–1022. [DOI] [PubMed] [Google Scholar]
- Eloranta ML, Franck-Larsson K, Lovgren T, Kalamajski S, Ronnblom A, Rubin K, Alm GV, and Ronnblom L (2010). Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis 69, 1396–1402. [DOI] [PubMed] [Google Scholar]
- Garin A, Meyer-Hermann M, Contie M, Figge MT, Buatois V, Gunzer M, Toellner KM, Elson G, and Kosco-Vilbois MH (2010). Toll-like receptor 4 signaling by follicular dendritic cells is pivotal for germinal center onset and affinity maturation. Immunity 33, 84–95. [DOI] [PubMed] [Google Scholar]
- Gray D (2002). A role for antigen in the maintenance of immunological memory. Nat Rev Immunol 2, 60–65. [DOI] [PubMed] [Google Scholar]
- Green NM, Moody KS, Debatis M, and Marshak-Rothstein A (2012). Activation of autoreactive B cells by endogenous TLR7 and TLR3 RNA ligands. The Journal of biological chemistry 287, 39789–39799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn BH (1998). Antibodies to DNA. N Engl J Med 338, 1359–1368. [DOI] [PubMed] [Google Scholar]
- Han JH, Umiker BR, Kazimirova AA, Fray M, Korgaonkar P, Selsing E, and Imanishi-Kari T (2014). Expression of an anti-RNA autoantibody in a mouse model of SLE increases neutrophil and monocyte numbers as well as IFN-I expression. European journal of immunology 44, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, and Nagata S (2004). Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304, 1147–1150. [DOI] [PubMed] [Google Scholar]
- Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, von Andrian UH, Shlomchik MJ, and Haberman AM (2007). Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity 26, 655–667. [DOI] [PubMed] [Google Scholar]
- Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, and Carroll MC (2013). Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity 38, 1164–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesters BA, Das A, Chatterjee P, and Carroll MC (2014). Do follicular dendritic cells regulate lupus-specific B cells? Mol Immunol,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman B, Wiersma EJ, and Kinoshita T (1990). In vivo inhibition of the antibody response by a complement receptor-specific monoclonal antibody. J. Exp. Med. 172, 665–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, and Taniguchi T (2005). IRF7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. [DOI] [PubMed] [Google Scholar]
- Hou B, Reizis B, and DeFranco AL (2008). Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity 29, 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, Spencer J, and Pitzalis C (2009). Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med 6, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, and Medzhitov R (2004). Toll-like receptor control of the adaptive immune responses. Nat Immunol 5, 987–995. [DOI] [PubMed] [Google Scholar]
- Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, and Banchereau J (2003). Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19, 225–234. [DOI] [PubMed] [Google Scholar]
- Kraus M, Alimzhanov MB, Rajewsky N, and Rajewsky K (2004). Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell 117, 787–800. [DOI] [PubMed] [Google Scholar]
- Krieg AM (2007). The toll of too much TLR7. Immunity 27, 695–697. [DOI] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, and Marshak-Rothstein A (2005). RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med 202, 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, and Marshak-Rothstein A (2002). Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607. [DOI] [PubMed] [Google Scholar]
- Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, Kellner ES, Nacionales D, Barker T, Kelly-Scumpia K, et al. (2008). TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. The Journal of experimental medicine 205, 2995–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Jiang Y, Prak EL, Radic M, and Weigert M (2001). Editors and editing of anti-DNA receptors. Immunity 15, 947–957. [DOI] [PubMed] [Google Scholar]
- Liu Z, Bethunaickan R, Huang W, Lodhi U, Solano I, Madaio MP, and Davidson A (2011). Interferon-alpha accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis and rheumatism 63, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed D, Benschop RJ, Cambier JC, and Nemazee D (1998). Developmental regulation of B lymphocyte immune tolerance compartmentalizes clonal selection from receptor selection. Cell 92, 173–182. [DOI] [PubMed] [Google Scholar]
- Moriyama M, Fukuhara T, Britschgi M, He Y, Narasimhan R, Villeda S, Molina H, Huber BT, Holers M, and Wyss-Coray T (2011). Complement Receptor 2 Is Expressed in Neural Progenitor Cells and Regulates Adult Hippocampal Neurogenesis. J Neurosci 31, 3981–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, Arthur JS, and Cohen P (2011). Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. The Journal of experimental medicine 208, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermoser G, and Pascual V (2010). The interferon-alpha signature of systemic lupus erythematosus. Lupus 19, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, Angeli V, Li Y, Boros P, Ding Y, et al. (2006). Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol 7, 652–662. [DOI] [PubMed] [Google Scholar]
- Pitzalis C, Jones GW, Bombardieri M, and Jones SA (2014). Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol 14, 447–462. [DOI] [PubMed] [Google Scholar]
- Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, and Marshak-Rothstein A (2005). Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunological reviews 204, 27–42. [DOI] [PubMed] [Google Scholar]
- Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, and Colonna M (2014). Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med 211, 1977–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomonsson S, Jonsson MV, Skarstein K, Brokstad KA, Hjelmstrom P, Wahren-Herlenius M, and Jonsson R (2003). Cellular basis of ectopic germinal center formation and autoantibody production in the target organ of patients with Sjogren’s syndrome. Arthritis Rheum 48, 3187–3201. [DOI] [PubMed] [Google Scholar]
- Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, and Nussenzweig MC (2007). In vivo imaging of germinal centres reveals a dynamic open structure. Nature 446, 83–87. [DOI] [PubMed] [Google Scholar]
- Sheehan KC, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, and Schreiber RD (2006). Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res 26, 804–819. [DOI] [PubMed] [Google Scholar]
- Shlomchik MJ, Madaio MP, Ni D, Trounstein M, and Huszar D (1994). The role of B cells in lpr/lpr-induced autoimmunity. J. Exp. Med. 180, 1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, D’Agati V, Elkon KB, and Reizis B (2014). Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med 211, 1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner LA, and Eisen HN (1967). Sequential changes in the relative affinity of antibodies synthesized during the immune response. J Exp Med 126, 1161–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott DI, Hiepe F, Hummel M, Steinhauser G, and Berek C (1998). Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease. The salivary glands of patients with Sjogren’s syndrome. J Clin Invest 102, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, and Taniguchi T (2005). Integral role of IRF5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249. [DOI] [PubMed] [Google Scholar]
- Thibault DL, Graham KL, Lee LY, Balboni I, Hertzog PJ, and Utz PJ (2009). Type I interferon receptor controls B-cell expression of nucleic acid-sensing Toll-like receptors and autoantibody production in a murine model of lupus. Arthritis Res Ther 11, R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umiker BR, Andersson S, Fernandez L, Korgaokar P, Larbi A, Pilichowska M, Weinkauf CC, Wortis HH, Kearney JF, and Imanishi-Kari T (2014). Dosage of X-linked Toll-like receptor 8 determines gender differences in the development of systemic lupus erythematosus. European journal of immunology 44, 1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoratos P, and Kollias G (2009). Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity 30, 130–142. [DOI] [PubMed] [Google Scholar]
- Victoratos P, Lagnel J, Tzima S, Alimzhanov MB, Rajewsky K, Pasparakis M, and Kollias G (2006). FDC-specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity 24, 65–77. [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, Sanz I, and Cook MC (2009). Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol 9, 845–857. [DOI] [PubMed] [Google Scholar]
- Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, and Cyster JG (2011). Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. The Journal of experimental medicine 208, 2497–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins AA, Yasuda K, Wilson GE, Aprahamian T, Xie Y, Maganto-Garcia E, Shukla P, Oberlander L, Laskow B, Menn-Josephy H, et al. (2015). IRF5 deficiency ameliorates lupus but promotes atherosclerosis and metabolic dysfunction in a mouse model of lupus-associated atherosclerosis. J Immunol 194, 1467–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Nundel K, Watkins AA, Dhawan T, Bonegio RG, Ubellacker JM, Marshak-Rothstein A, and Rifkin IR (2013). Phenotype and function of B cells and dendritic cells from interferon regulatory factor 5-deficient mice with and without a mutation in DOCK2. Int Immunol 25, 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Richez C, Maciaszek JW, Agrawal N, Akira S, Marshak-Rothstein A, and Rifkin IR (2007). Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. Journal of immunology 178, 6876–6885. [DOI] [PubMed] [Google Scholar]
- Yasuda K, Watkins AA, Kochar GS, Wilson GE, Laskow B, Richez C, Bonegio RG, and Rifkin IR (2014). Interferon regulatory factor-5 deficiency ameliorates disease severity in the MRL/lpr mouse model of lupus in the absence of a mutation in DOCK2. PLoS One 9, e103478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau IW, Cato MH, Jellusova J, Hurtado de Mendoza T, Brink R, and Rickert RC (2013). Censoring of self-reactive B cells by follicular dendritic cell-displayed self-antigen. Journal of immunology 191, 1082–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagury D, Le Buanec H, Mathian A, Larcier P, Burnett R, Amoura Z, Emilie D, Peltre G, Bensussan A, Bizzini B, et al. (2009). IFNalpha kinoid vaccine-induced neutralizing antibodies prevent clinical manifestations in a lupus flare murine model. Proceedings of the National Academy of Sciences of the United States of America 106, 5294–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.