

Abstract
Alternative splicing of messenger RNA can generate an array of mature transcripts, but it is not clear how many go on to produce functionally relevant protein isoforms. There is only limited evidence for alternative proteins in proteomics analyses and data from population genetic variation studies indicate that most alternative exons are evolving neutrally. Determining which transcripts produce biologically important isoforms is key to understanding isoform function and to interpreting the real impact of somatic mutations and germline variations. Here we have developed a method, TRIFID, to classify the functional importance of splice isoforms. TRIFID was trained on isoforms detected in large-scale proteomics analyses and distinguishes these biologically important splice isoforms with high confidence. Isoforms predicted as functionally important by the algorithm had measurable cross species conservation and significantly fewer broken functional domains. Additionally, exons that code for these functionally important protein isoforms are under purifying selection, while exons from low scoring transcripts largely appear to be evolving neutrally. TRIFID has been developed for the human genome, but it could in principle be applied to other well-annotated species. We believe that this method will generate valuable insights into the cellular importance of alternative splicing.
INTRODUCTION
Alternative splicing (AS) is estimated to occur in almost all multi-exon human genes (1) and it has been suggested that alternative splicing of messenger RNA is a major source of cellular protein diversity (2,3). The human reference databases (4–6) contain >100 000 alternative transcripts and even greater numbers of alternative transcripts have been reported in large-scale experiments (7,8). As a result, the number of annotated alternative coding transcripts in the human genome is likely to grow considerably in the coming years.
It is theoretically possible that all predicted coding transcripts are translated into functional proteins. Alternative splicing can generate many different transcripts, and many of the alternative splice events would produce substantially different proteins. These large differences may allow alternative isoforms to have a diverse range of cellular effects. Alternative isoforms have been invoked to explain differences between tissues (9) and even between species (10,11).
There are examples of differences of function between protein splice isoforms both in vivo and in vitro (12,13) and alternative proteins have been shown to bind different targets in vitro (14). While there is little doubt that alternative isoforms would behave differently if they were present in cells, it is still not clear how many alternative transcripts are actually translated into stable functioning proteins.
It ought to be possible to validate most alternative isoforms through mass spectrometry-based proteomics experiments, as long as alternative transcripts are translated into protein products in sufficient quantities. However, there is considerably less evidence for alternatively spliced protein products in proteomics experiments than would be expected (15,16), and in fact, there is much less evidence for alternative isoforms at the protein level than there is for alternative transcripts in transcriptomics studies (17). Instead, for a large majority of coding genes, proteomics data supports a single ‘principal’ isoform regardless of cell type (15,18).
The relative importance of alternative protein isoforms has become a controversial issue (17,19–21). While a number of alternative isoforms do appear to have tissue specific functional roles (22), a growing number of papers suggest that alternative splicing at the transcript level may be noisy. Transcripts sampled from simulated complementary DNA libraries (23) and variation patterns in alternative splice sites (24) strongly suggest that the majority of annotated splice variants are the result of noise from the splicing machinery, while accumulated transcriptomics evidence suggests that many of the transcripts produced by alternative transcription initiation (25) and alternative polyadenylation (26) are the result of molecular errors. Analysis of cross-species conservation (16,22) and human genetic variation (19,27) support the possibility that most alternative exons are unlikely to be under purifying selective pressure.
There are fewer alternative isoforms detected in proteomics experiments than would be expected from transcription levels (16), even when technical issues are taken into account (28). It is not clear why so few alternative isoforms are detected. Missing alternative isoforms might, for example, be expressed in low abundance, or in limited tissues or may have short half-lives post-translation (29–30). Some transcripts may not be translated and some may also play roles at the transcript level rather than at the protein level (31).
The relative importance of individual alternative variants is an issue that is becoming ever more pertinent as the number of annotated alternative transcripts grows. The most recent GENCODE reference proteome (release v37, Ensembl 103) (5,32) officially recognises 19 951 protein-coding genes and 86 054 coding transcripts. How many of these transcripts code for functional proteins is still an open question.
There are vast numbers of both annotated and unannotated alternative transcripts in eukaryotic species, yet almost nothing is known about the cellular function of their protein isoforms. Although individual research groups have recorded functional differences between splice isoforms, and many of these are listed in collations of isoform function (12,13), this is only scratching at the surface, and leaves plenty of room for computational prediction methods.
Attempts to predict functional roles for protein isoforms can be split into three categories. The first category of predictor simply attempts to predict the functional role of all splice isoforms. Alternative isoform function predictors have gained in popularity in recent years, and a number of tools for estimating function roles have been developed (33–39). These methods assume a priori that all alternative isoforms have functional roles. They have to negotiate one important hurdle: there is no real training data since very few alternative isoforms have known functions.
The second category of predictor attempts to predict a main functional isoform. The general consensus here is that many protein-coding genes have a single main protein isoform (15,40,41), though different approaches have been taken to predict this main isoform. Approaches based principally on transcript level information (40,41) turn out to not correlate well at the protein level (15,19). APPRIS (18) predicts principal isoforms based on the preservation of protein features and cross-species conservation. When we compared principal isoforms selected by APPRIS with the isoforms with most peptide evidence and with unique CCDS (42) variants (consensus CDS sequences based on cDNA evidence whose coding sequences are agreed on by distinct groups of manual curators), the agreement was overwhelming, over 99.5% (15). Such unanimity between three orthogonal methods demonstrated two clear facts: firstly, a large majority of coding genes have a single main protein isoform and secondly that APPRIS is the best predictor of this isoform.
The final class of function prediction methods predicts the relative functional importance of alternative splice isoforms. Even though evidence from human population variation studies suggests that most alternative exons are not under selective pressure (19,27), proteomics experiments and conservation data show that a substantial number of genes are likely to have more than one functionally important protein isoform. The prediction of biological relevance faces similar obstacles to the prediction of isoform function. The biggest difficulty in the prediction of whether a protein isoform is functionally important is that there is no negative training set; it is almost impossible to demonstrate that alternative isoforms do not have some function.
To date, just one method has been developed to predict the functional relevance of alternative isoforms, PULSE (43). PULSE got around the lack of a negative training set by using a semi-supervised training algorithm and training only on positive examples. Unfortunately, PULSE had a number of flaws. The main problem was that the 145 positive instances used to train the algorithm (44) were based loosely on a set of ‘named’ alternative splice isoforms from UniProtKB (4). That isoforms are named in UniProtKB does not imply that they have cellular functions. Indeed, some were clearly not positive cases; PULSE included translations from 18 nonsense mediated decay transcripts in its positive set. PULSE positive instances were supposed to differ from the display isoforms by a single exon skipping event, though several are actually generated by alternative poly-adenylation events and are mistakenly tagged as frame shifts.
Here, we have developed a random forest (RF)-based predictor of splice isoform functional importance based on unbiased data from large-scale mass spectrometry proteomics experiments. TRIFID uses protein isoforms detected in proteomics experiments as a proxy for functional importance at the protein level and is able to distinguish protein isoforms that are under selective pressure from those that are not.
MATERIALS AND METHODS
Defining the training sets
Machine learning methods require positive and negative sets in order to train a model. One of the most difficult steps in designing a method to predict the functional importance of protein isoforms is defining the training sets (43). Positive training cases are hard to find because there are relatively few alternative isoforms with known cellular function (45). Negative training sets are even harder to define because there are no known cases of non-functional isoforms and it is almost impossible to demonstrate that an alternative isoform has no cellular function at all.
Although it would have been possible to generate a positive training set of likely functional alternative isoforms by manual curation of scientific papers, the equivalent negative training set does not exist. The solution to the problem was to use proteomics evidence to seed both the positive and negative sets.
Using peptide evidence as a proxy for functional importance is based on the reasoning that gene products detected in proteomics experiments are highly likely to be functionally important Almost all exons with peptide evidence are from principal isoforms (15) and principal isoforms as a whole are under purifying selection (19), while exons from likely non-coding genes that are not detected in proteomics experiments appear to be evolving neutrally (46). Few alternative exons are detected in proteomics experiments and most are not under purifying selection (19). There are exceptions to the rule though: we have shown that SINE Alu exons are detected in proteomics experiments, but there is no evidence to suggest that these SINE Alu insertions have any functional role in the cell (47).
A negative training set can also be proposed, but only for those genes that have good peptide coverage. The logic is that any isoform from these genes that is not detected, is either harder to detect for technical reasons, or is not expressed in sufficient quantity to be detected at by mass spectrometry. There are two provisos that make this work. Firstly, the proteomics data must cover as many tissues as possible, because alternative protein isoforms are often tissue specific (22), and preferably should have replicate experiments to avoid missing peptides. Secondly, isoforms that are harder to detect for technical reasons, should not be included in the negative set.
The use of the word ‘negative’ in this paper should not be taken to imply that these non-detected alternative isoforms have no function. They may be translated in isolated tissues or under certain conditions or developmental stages. They might be translated, but have a short half-life. They may be translated in undetectable quantities. Or they may not be translated at all. Some of them, particularly some of the nonsense mediated decay (NMD) targets (31), may have functional roles as transcripts, but not as proteins. Although the presence of an isoform in the negative set does not necessarily mean that it is non-functional, the fact that we do not detect it, despite close to complete peptide coverage for at least one other isoform from the same gene, means that it is measurably different from other isoforms in the same gene.
Training the model based on genes with peptide evidence means that both the positive and negative classes are populated entirely with isoforms from genes that are well expressed. It is reasonable to assume that isoforms in less well-expressed genes will behave in a similar way. The model was not trained with proteomics data and although transcript expression data was one of the features, it was always normalized by gene. This means that TRIFID has no way of knowing the expression level of the gene.
We generated the positive and negative training sets from genes detected in a large-scale tissue-based proteomics analysis (Figure 1). We included isoforms from genes that either had peptide evidence for two or more protein isoforms or that had one isoform with at least 80% peptide coverage. The negative and positive training sets were produced from a total of 421 genes. We eliminated 396 duplicated isoforms from these genes. Where there was an identical sequence to another isoform in the same gene, we prioritized the isoform with the higher transcript support level (5).
Figure 1.

Schema and model selection, training and feature importance in the final RF model. (A) A simplified schema of the design of the TRIFID algorithm. (B) Isoforms in the training set were annotated with features. (C) Nested cross validation (CV) strategy using an external test set to evaluate the performance of the model (to overcome the risk of test set bias). (D) Precision-recall curves from stratified 10-fold cross validation for the best model selected in the inner loop (75% of the training set, 2062 isoforms) once the hyperparameter tuning step has been performed. (E) Graphical representation of the RF training process. The RF had 400 de-correlated decision trees, and the best split of each tree was based on the Gini impurity function. At each leaf node, the minimum number of samples was set to 7, which also helps to avoid overfitting. (F) The predicted functionality score of an input isoform is the average predicted class probabilities of the trees in the forest.
The positive classification dataset was made up of those protein isoforms that were detected in the large-scale proteomics experiments. The only filter step that affected the positive training set was the removal of duplications.
The negative training set was formed of isoforms from the same genes that were not detected. The negative training set had an extra filter; we excluded some isoforms that went undetected in the proteomics analyses from the negative set because if they were present in the cell, it would have been difficult to discriminate these isoforms in standard proteomics experiments.
The proteomics experiments used trypsin to cleave peptides and trypsin cleaves after lysines and arginines in the sequence. When lysines and arginines coincide with splice junctions (28), it is harder to detect discriminating peptides because the only peptides with missed cleavages can distinguish the splice events. Since it is difficult to tell whether they ought to be in the positive or negative set, we removed splice events with lysines or arginines in their splice junctions from the negative set. We also removed isoforms where lysines and arginines were either too far apart (regions where the only possible discriminating tryptic peptide would be longer than 40 residues), or too close together (where discriminating peptides would be shorter than 7 residues).
We removed a total of 171 alternative isoforms from the negative training set for these reasons. The final classification data set contained 2,790 isoforms. The positive instances for training the classifier totalled 712 isoforms (25.5%), while there were 2,078 (74.5%) in the negative training set.
Proteomics analysis
We analysed spectra from 79 experiments from one of the largest tissue-based proteomics analyses to date (48). These tissues are histologically normal and comprise 49 experiments carried out on adult tissues, 12 experiments with hematopoietic cells and 18 experiments with foetal tissue. There are 30 distinct tissues and cell types in total and each is represented by at least two replicates. We downloaded the data from ProteomeXchange (49) with the identifier PXD000561.
We worked with v27 of the GENCODE manual annotation of the human reference gene set in this analysis. GENCODE v27 (5) is equivalent to Ensembl 90. We used the official GENCODE v27 translations in the proteomics analysis and took training features from the official GENCODE v27 gtf for these translations. The GENCODE v27 translated gene set contained 20 250 coding genes, which include read-through genes, polymorphic pseudogenes, and immunoglobulin and t-cell receptor fragments. These coding genes were annotated with 95 584 translated transcripts, of which 14,200 were predicted to be NMD transcripts.
We used Comet (50) to map spectra from the experiments to the GENCODE human reference gene set, version v27. Comet was run with the default parameters, allowing oxidation of methionines. After the search, we generated posterior error probability (PEP) values using Percolator (51). We limited peptide spectrum matches (PSM) to those that had a PEP value of 0.001, while peptides had to be fully tryptic with a maximum of one missed cleavage, no shorter than seven residues and no longer than 40. There were minor differences between experiments, but we found that a PEP value of 0.001 corresponded to a PSM q-value of ∼0.0001.
As a further step to reduce false positive identifications, we required peptides to have at least one valid PSM in two of the 79 experiments. All filtering steps were applied with one idea in mind - to reduce false positives. Combining many experiments will inflate the peptide false discovery rate. Most valid peptides will appear in multiple experiments, while the vast majority of false positive matches will be different in each experiment. This means that the proportion of detected false positive matches will increase with each added experiment (52). The conservative PEP value and the requirement for a minimum of two supporting PSMs at least partially compensates for this. Since each tissue has at least two replicates, this rule does not eliminate the possibility of detecting tissue specific splicing (22). Though we could have included tissue specificity as a feature, the isoforms we detected were considered to be functional whether they were tissue specific or not.
Peptides that mapped to more than one gene were discarded. The remaining 130 212 validated peptides were used to identify protein isoforms for each gene. We required that valid peptides mapped to both sides of a splice event to identify an alternative splice event (15).
Training features from the GENCODE reference
Training features taken from the GENCODE v27 gtf included transcript support level (TSL, 5), the GENCODE Basic category (5) labels and the CCDS (42) labels. CCDS labels are generated by the Consensus CDS Project and a CCDS tag indicates agreement between Ensembl/GENCODE and RefSeq manual annotators over the entire coding sequence of the transcript. Agreed on CCDS transcripts are generally supported by full length cDNA evidence, but the RefSeq and Ensembl gene models may have different 5′ and 3′ UTR (untranslated regions).
Features taken from the GENCODE gtf are detailed in the Supplementary Material.
Features from the APPRIS database
APPRIS (18) annotates splice isoforms with protein structural and functional information and assigns a score representing cross-species conservation. APPRIS also selects a single protein sequence unique isoform as the ‘principal’ isoform for all coding genes (53). Translations that are sequence identical have the same APPRIS tag (unless they are annotated as NMD), so genes can have more than one (sequence identical) Principal isoform. APPRIS defined 26 653 GENCODE v27 isoforms as ‘Principal’.
APPRIS features included SPADE (a measure of the effect of splicing on functional domain composition), Matador3D (a measure of the effect of splicing on protein structure) and CORSAIR. CORSAIR is a measure of cross-species conservation that counts the number of homologues that align without gaps. All features taken from APPRIS are detailed in the Supplementary Material.
Transcript expression
To capture differences between isoforms at the transcript level, we created a score from RNA-seq data from a large-scale study by the Human Protein Atlas (54) in which RNA-seq was performed on 36 different tissues samples from 122 human individuals.
We downloaded data from the Human Protein Atlas RNA-seq experiments. To align this data to GENCODE v27 we used STAR 2.6 (55). To avoid unwanted alignments to repetitive regions, we forced end to end read alignments and set the maximum number of multiple alignments allowed to 50. The remaining parameters were set by default.
We used the collapsed CDS splice junction outputs to calculate a score per transcript. We used CDS junctions only because these junctions are translated to protein. We recorded the maximum number of reads over all the tissues for each splice junction. Once we had a score for each splice junction, we calculated the mean for each gene over all the splice junctions. Each transcript was represented by its least supported splice junction (the weakest evidence for its expression). However, since we trained the method on a subset of highly translated genes, we had to adjust these scores against the relative expression of each gene. So, the final score for each transcript was the number of reads that supported the lowest scoring junction divided by the average read count of all the CDS in the gene. To add further information, we also calculated a normalized splice junction score for each transcript normalized against the highest scoring transcript in each gene.
Details on the implementation and interpretation of this method are available in Supplementary Material.
Other predictive features
We have previously noted that alternative isoforms detected in proteomics experiments are highly enriched in certain features such as cross-species conservation and the non-disruption of Pfam (56) functional domain composition (16). We added further measures of domain composition and conservation to the features in the predictor.
We generated a series of features such as how much the Pfam domain composition changed after a splice event (Pfam domain impact), the number of residues lost from Pfam domains (Pfam residues lost), the type of splicing event (e.g. indel, substitution) and the length differences between isoforms (length delta score). These features are detailed in Supplementary Material.
We also added two measures of conservation as features, PhyloCSF (57) and Alt-CORSAIR. PhyloCSF (57) is a comparative genomics method that can help distinguish protein coding and non-coding regions. Alt-CORSAIR is a method based on the CORSAIR module in APPRIS. Alt-CORSAIR carries out BLAST searches against RefSeq protein sequences and looks for orthologues that align completely without gaps. The Alt-CORSAIR score is the age of the last common ancestor of the most distant orthologue that fulfils the search criteria. Details on all these methods can be found in the Supplementary Material.
Generating predictive features for the model
For those features that were numeric, we also generated normalized scores by rescaling against the highest scoring isoform in each gene. For example, for normalization of the length feature, we took the longest isoform as the reference isoform. Normalized scores were rescaled between 0 (no score) and 1 (the reference value). Features were normalized in order to better capture differences between alternative isoforms of the same gene, and both normalized and raw feature scores were used in the predictor. This rescaling allowed us to quantify the local effect of splicing on the predictive features and added insights that would have been hard to detect with the non-normalized scores.
In total, we collected 45 predictive features for both canonical (APPRIS principal) and alternative splice isoforms. Features were divided into five main categories: annotation, evolution, expression, structure, and splicing (Figure 1A). Structural characteristics were taken from the APPRIS database and capture how much an isoform deviates from the highest scoring isoform in terms of mapping to functional domains, known protein structures, functionally important residues and trans-membrane helices. Annotation features are those that came from the GENCODE/Ensembl annotation of the human reference set and include attributes such as transcript support level, length, transcript type, or CCDS support. Splicing features were those that characterized the direct effects of the splicing events, such as the number of lost residues with respect to the longest isoform, length difference, and the effect of the splicing event on Pfam functional domains. Expression features came principally from RNAseq analyses. Last, but not least, the evolutionary features were made up of sequence conservation scores from Alt-CORSAIR, APPRIS and PhyloCSF.
More detailed information on the predictive features used and the normalization processes can be found in Supplementary Material. The relationship between the 45 features in TRIFID can be found in Supplementary Figure S1.
Predictor selection
We benchmarked a range of machine learning approaches for their capacity to combine the 45 predictive features. To overcome the risk of test set bias, we used an inner-outer (nested) cross validation strategy using an external test set to evaluate model performance (Figure 1C). For the inner loop hyperparameter tuning step, we split our data into 10 stratified folds, preserving the percentage of samples for each class. We performed cross-validation by iteratively training with nine folds and using the remaining fold to test. The outer loop was used to estimate the predictor performance. Here the training set was created from four fifths of the samples. Given the slightly imbalanced ratio (1:3) for the majority class (negative instances) in the training set, we evaluated binary classification performance with both Matthews Correlation Coefficient (MCC; 58) and an Area Under the Precision–Recall Curve (AUC-PR; 59). Model selection, parameter tuning and detailed evaluation can be found in the Supplementary Material.
We selected the RF-based algorithm as the final model. It was one of the best performing methods, though there was little difference between the best methods. RF algorithms are widely used in binary classification tasks due to their robust performance across a wide range of data sets. Moreover, RF algorithms have the ability to handle categorical, Boolean and continuous features and do not require aggressive feature selection to reach adequate performance. They can handle correlated features, and do not need a high number of hyperparameters to avoid overfitting. RF models provide ways to perform reliable predictions even with instances of missing data. In theory, RF models can handle sets that do not have complete coverage of all predictive features, so they could be exported to other species and annotation sets.
Non-linear predictive models like RF used to have the disadvantage of being less interpretable, but recent advances in interpretability of tree-based models have improved the ability to explain both global influences of model features, and the influence of features on individual predictions (60).
Feature importance calculation
We applied the SHAP feature importance calculation (60) to measure overall feature importance in TRIFID. The SHAP feature importance calculation is a recent advance in the interpretability of tree-based models. It is a game theoretic approach that explains models globally by combining local contributions of individual features and is supposed to perform better than any other global approximation. The algorithm returns measures of global feature importance and can also provide clues as to the influence of each feature within individual predictions.
Generating a non-redundant set of isoforms from GENCODE v27 coding transcripts
More than a third of GENCODE v27 coding transcripts differ only in their 5′ and 3′ untranslated regions (UTR) or would produce translations that are fragments of other proteins, so the set of GENCODE v27 translations is redundant. For the purpose of analysis of our model we generated a non-redundant set of protein isoforms. We filtered out translations from the same gene with identical protein sequences and also those that were fragments of longer protein sequences and that were tagged as being incomplete by GENCODE. Incomplete sequences are tagged as ‘cds_end_NF’ or ‘cds_start_NF’ in the gtf annotation file and total 27 727 transcripts.
We also filtered translations that came from genes not recognised as protein coding, such as immunoglobulin or T-cell receptor fragments, and those translations tagged with labels such as ‘nonsense mediated decay’, ‘non-stop decay’ or ‘readthrough’ that are highly unlikely to produce functional proteins. The filtering left us with 57 367 non-redundant translations from just 19 327 coding genes (46). Non-redundant alternative isoforms totalled 38,040.
Genetic variation
As part of the analysis of TRIFID predictions, we calculated non-synonymous to synonymous rates across different sets of exons. To calculate genetic variation rates, we used the human variation data from the 2504 individuals in phase 3 of the 1000 Genomes Project (61) remapped from GRCh37 to GRCh38 using dbSNP v149 (62). More than 99% of the variants successfully mapped from GRCh37 to GRCh38 (46).
For the analysis, GENCODE v27 exons were separated into principal or alternative according to their annotation in APPRIS. Principal exons were those that generated the principal isoform. These made up the vast majority of the exons (∼90%). The remaining exons were tagged as alternative exons. The effect of the variants on the GENCODE v27 transcripts was predicted using VEP (63). We calculated the ratio of non-synonymous to synonymous variants for both rare and common allele frequencies. We defined common alleles as those with allele frequencies >0.005, while rare alleles were those with allele frequencies <0.005.
RESULTS
The final TRIFID model configuration achieved a Matthew's correlation coefficient (MCC) of 0.9 ± 0.027 over the inner 10-fold cross validation, and of 0.904 ± 0.021 over the outer 5-fold cross validation. The average precision (AUC-PR) for the final model was 0.981 ± 0.001 (Figure 1D) over the inner 10-fold cross validation and 0.984 ± 0.001 over the outer 5-fold cross validation. The fact that the RF algorithm yields an AUC-PR of 0.984 for the training set indicates that the features used to train the classifier are able to distinguish positive from negative cases.
It might be possible to argue that the restrictions put in place to limit false positives and to validate the translation of alternative isoforms might have been too strict and that TRIFID might have benefited from a larger training set. The number of protein isoforms in the training set was relatively small, 2790 isoforms between the positive and negative training sets, fewer than 5% of the non-redundant isoforms annotated in the human reference set.
To test the impact of using a limited training set, we trained the model using sub-samples of the final training set in increments of 10%. We found that the training set MCC reached a plateau with <50% of the training data (Figure 2A) and that the model could even have made reliable predictions with few data points. The model had an MCC of over 0.88 and an AUC-PR of almost 0.98 with just 20% of the training set (Figure 2B). Hence, adding further isoforms to the training data is unlikely to lead to much improvement in the model. The training set was more than sufficient to train a stable model.
Figure 2.

TRIFID learning curve and feature importance. The Matthews correlation coefficient (A) and the average precision (B) for the training score and cross-validation score using subsets of the data set to train the model. Results clearly show that the model is stable even with smaller subsets. (C) The SHAP feature importance calculation (60) is a game theoretic approach that explains models globally by combining local contributions of individual features and is supposed to perform better than any other global approximation. The top 18 features are divided into five sub-types (evolutionary, annotation, structure/functional, expression and splicing effects) as described in the methods section. A lower case ‘n’ indicates that the feature was normalized.
Model feature importance
With the SHAP scores, we were able to quantify the importance of the different features, and the features that best distinguished positive from negative isoforms in the training sets were conservation-based (Figure 2C). The greater the evidence of cross-species conservation, the higher the TRIFID score.
CORSAIR and Alt-CORSAIR both capture information from cross-species alignments; CORSAIR is part of APPRIS (18) and Alt-CORSAIR was developed recently (see method section) and is based on CORSAIR. For both CORSAIR and Alt-CORSAIR, scores that were normalized against the highest scoring isoform in each gene discriminated better than the raw scores.
Other important features include the length difference between the tested isoform and the longest isoform and whether or not the transcript has a CCDS (42). A CCDS tag indicates agreement on the coding sequence between Ensembl/GENCODE and RefSeq manual annotators. Length difference was the most important feature in PULSE (43), the only previous method developed for the prediction of functional isoform relevance; here it is only the 4th most important feature. The conservation of protein structure and functional domains, and transcript support level and expression also contributed to the decision making.
TRIFID produces raw and normalized scores
With the development of TRIFID we have carried out in depth analyses into the relationship between TRIFID scores and the functionality of individual genes and isoforms. Although we know that all coding genes ought to have at least one functional translation, we found that some genes had nothing but low scoring isoforms; in 399 genes all isoforms scored less than 0.25 while 1955 genes had isoforms with TRIFID scores of less than 0.5. One reason for these low scores might be that these genes are not coding. Indeed, we predicted that 171 of the genes with TRIFID scores lower than 0.2 (74.7%) might not code for proteins (46). Fifty of these genes (21.8%) have already been reclassified as not coding by GENCODE. Other genes might have nothing but low scoring TRIFID isoforms because their gene model is incomplete; for example, MYCBP had the lowest TRIFID scores in the whole gene set because in v27 its main transcript was mistakenly tagged as readthrough.
However, there are also genes where all isoforms are low scoring that are clearly coding and that have no clear error in their gene models. One such case is TTN. No titin isoform has a raw TRIFID score of over 0.3, yet TTN is clearly coding and must have (at least) one functionally important isoform. The isoforms in TTN have low scores in CORSAIR and Alt-CORSAIR principally because the length and the number of exons makes it difficult to accurately determine gene models. In general, the longer an isoform is, the more likely that it has large indels relative to orthologues in other species.
We found that a number of genes (often, but not always, genes with larger isoforms) had low CORSAIR and Alt-CORSAIR conservation scores for all isoforms even if the gene could trace its ancestry back to distant species. These low conservation scores were reflected in low TRIFID scores for all the isoforms in those genes. The longer the protein, the less chance of generating CORSAIR and Alt-CORSAIR scores for whole sequences, and the lower the CORSAIR and Alt-CORSAIR scores, the worse the TRIFID scores. This bias meant that relative biological importance predicted by TRIFID was often gene dependent. This was not just true for more recently evolved genes, but was also true for some ancient genes as can be seen Figure 3.
Figure 3.
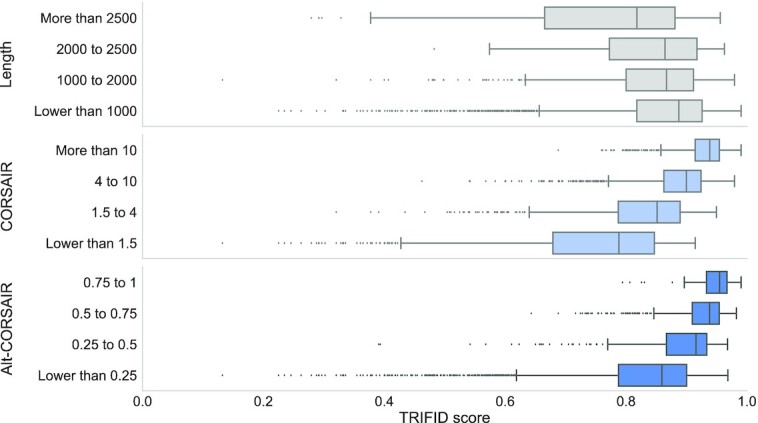
Length and conservation scores versus TRIFID scores. Boxplots of the highest TRIFID score per gene against the length of the longest isoform in each gene and against the highest scoring CORSAIR and Alt-CORSAIR values in each gene. Results are only shown for singleton genes with a last common ancestor before the split with Bilateria. Bilateria gene family age was calculated from Ensembl Compara in a previous study (32,46). Box plots show the interquartile range, median, 95% confidence interval and outliers as black dots. We binned genes by the length of their longest isoform and by the highest CORSAIR and Alt-CORSAIR scores, and calculated average TRIFID scores for each of these bins. Since these genes are conserved back to Bilateria, we would expect them all to have the highest possible CORSAIR and Alt-CORSAIR scores. However, many CORSAIR and Alt-CORSAIR scores are lower than expected. The longer the protein and the lower the conservation scores, the lower the TRIFID scores. Genes with lower conservation scores had substantially lower TRIFID values.
In order to take account of low scoring coding genes like TTN, we generated a second set of TRIFID scores. For each gene we normalized isoform scores against the highest scoring isoform for that gene on the assumption that each bona fide coding gene will code for at least one functional isoform. For genes where the highest scoring isoform is below 0.5, we normalized using 0.5 as the highest score. We do this to avoid inflating scores for those genes that are either composed entirely of NMD transcripts (220 genes are composed entirely of NMD transcripts in GENCODE v27), that have incomplete gene models (where the correct principal isoform is not yet annotated), or that have features to suggest they might not be coding. If we normalise against the highest scoring isoform in these genes, we may end up predicting that most isoforms in these genes are functionally important. TRIFID, CORSAIR and Alt-CORSAIR distributions for known coding genes and potential non-coding genes are shown in Supplementary Figure S2.
We calculated raw and normalized TRIFID scores for the GENCODE v27 translations. Raw TRIFID scores had a bimodal distribution with a huge peak of isoforms predicted as not functionally important below 0.05 and a smaller peak at ∼0.9 (Supplementary Figure S3). The bi-modal distribution of TRIFID scores comes from the clear separation of principal isoforms and alternative isoforms; most low scoring isoforms are alternative isoforms, while most high scoring isoforms are APPRIS principal isoforms (Supplementary Figure S4). Reassuringly, isoforms from transcripts that are unlikely to be translated, such as nonsense mediated decay and non-stop decay transcripts, scored <0.05 (Supplementary Figure S5).
Normalized TRIFID scores are always higher than raw TRIFID scores (Supplementary Figure S6), but the distribution of normalized TRIFID scores for principal and alternative isoforms has the same pattern as those of the raw scores (Figure 4 and Supplementary Figure S7). Principal and alternative isoforms have clearly distinct distributions. Most principal isoforms have high scores: 17 791 (92.1%) have normalized scores greater than 0.8 and 16 839 (87.1%) have scores above 0.95. Just 557 APPRIS principal isoforms (2.9%) have normalized scores <0.5.
Figure 4.
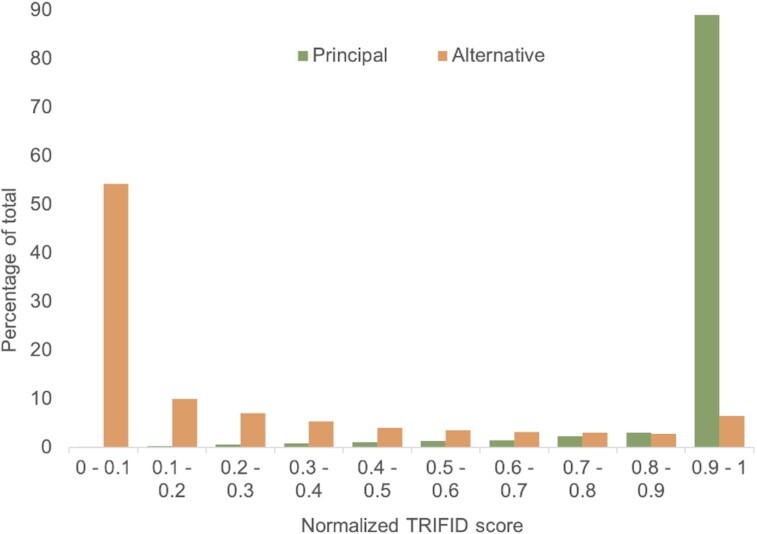
Normalized TRIFID scores and for alternative and principal isoforms. Non-redundant isoforms were divided into principal or alternative according to their annotation in APPRIS. Normalized TRIFID scores for the alternative and principal isoforms were binned in increments of 0.1 and the percentage of all isoforms in each bin plotted. Most alternative isoforms have TRIFID scores <0.1. Almost all principal isoforms have predictor scores above 0.9.
By way of contrast, most alternative isoforms have low normalized TRIFID scores (Figure 4). A total of 17 847 alternative isoforms (46.9%) have scores below 0.05, and 30,758 (80.9%) have normalized scores <0.5. At the same time, 5,944 alternative isoforms (15.6%) have normalized TRIFID scores above 0.6, and 3,567 (9.4%) scored higher than 0.8. The pattern of the raw scores is similar (Supplementary Figure S7).
Normalized TRIFID scores are stable across different annotations. The vast majority of scores for identical isoforms in the v27 and v35 versions of the human reference set are almost unchanged (Supplementary Figure S8) and the two sets of normalised TRIFID scores have a Pearson correlation coefficient of 0.99. Most large changes in TRIFID scores are due to changes in the principal isoform. For example, CCNP/CNTD2 (the gene changed its name between v27 and v35) has the same number of isoforms in both annotations, but one isoform was extended to complete a Pfam domain (Supplementary Figure S9). Before it was extended, it had a normalised TRIFID score of 0, in v35 the score was 0.742. As a result, two of the three unchanged isoforms had much lower normalised TRIFID scores in v35, one even dropping from 1.0 to 0.214.
After testing with both normalized scores and the raw TRIFID score, we believe that relative functionality is best represented by the normalized TRIFID score. Raw TRIFID scores will be most useful if researchers are interested in a set of reliable gene models and do not require a complete gene set.
Below, we discuss the cases of genes ERRC6 and FGFR1 in order to illustrate the utility of TRIFID, the use of normalized and raw TRIFID scores, and the challenges of predicting the functional importance of protein isoforms.
DNA excision repair protein ERCC-6 and TRIFID score normalization
DNA excision repair protein ERCC-6 (gene ERCC6) is a chromatin remodelling protein implicated in transcription elongation and DNA damage repair. It has important roles in a range of cellular processes (64–67). ERCC6 is annotated with six translations in GENCODE v27. One is an NMD target, a second is a 3′ CDS incomplete sequence fragment and a third has a downstream ATG (and is no longer annotated as part of this gene). Two of the remaining isoforms are sequence identical, so there are only two distinct full-length proteins annotated for ERCC6.
The two isoforms both appear to be functionally important, but both have raw TRIFID scores below 0.5. The principal isoform, ENSP00000348089 (1,493 amino acids), scores just 0.48, while the alternative isoform, ENSP00000387966 (1,061 amino acids), scores 0.444. These relatively low raw scores occur because neither ERCC6 isoform scores well in conservation measures CORSAIR and Alt-CORSAIR, even though orthologues for the main isoform can be traced back to yeast (Rad26) (68). While other TRIFID features such as CCDS and protein structure and function support the functional importance of the two isoforms, the low conservation scores depress the final raw TRIFID scores (Supplementary Figure S10).
Normalising the TRIFID scores for the two ERCC6 isoforms raises their scores to 0.96 and 0.89 and this fits with the functional information available for the two isoforms. Both isoforms have an N-terminal chromatin remodelling domain and the principal isoform has approximately a thousand residues in a C-terminal that includes an SNF2 family N-terminal domain and a helicase domain (Figure 5). The principal isoform is clearly highly important. Most of the sequence is conserved in Fungi and mutations in this isoform are known to cause Cockayne syndrome, a severe neurological disorder.
Figure 5.
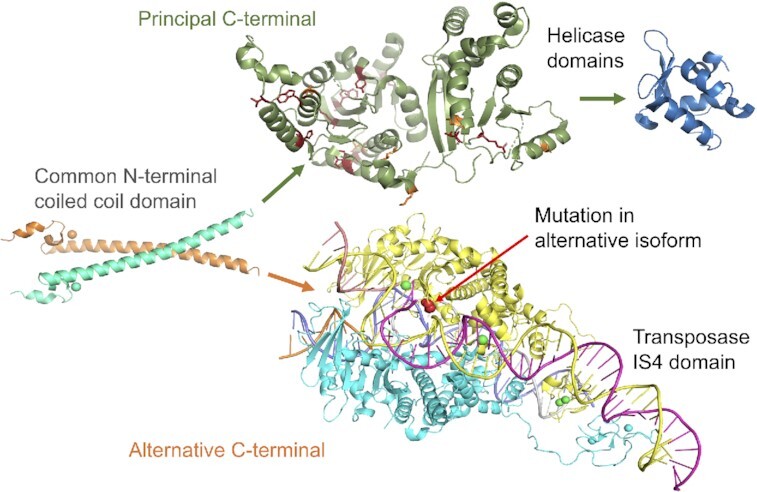
A schematic illustration of the two functionally important ERCC6 isoforms. Both isoforms have a common N-terminal, represented by the resolved coiled coil structure of residues 84 to 160 from PDB (73) structure 4CVO, left. The principal isoform (above right, red arrows) is represented by structures of the SNF2 family N-terminal domain (PDB: 5HZR) and C-terminal helicase domain (PDB: 6A6I). Pathogenic mutations from ClinVar (74) that map to the N-terminal domain are shown in red (stop gained) and yellow (missense). The alternative isoform (below right, blue arrows) is represented by the structure of the transposase IS4 domain (PDB: 6×67). The pathogenic mutation that affects ovary function (72) is mapped to this domain and shown in red. Mapping to the PDB structures where necessary was carried out using HHPRED (75) and all images were generated using PyMol.
The C-terminal of the alternative isoform is ∼600 residues long and includes a piggybac-derived transposase domain (PGBD3). The ancestor of PGBD3 was incorporated before the split between cnidaria and bilateria >650 million years ago. A fusion between ERCC6 and the transposon-derived PGBD3 seems to have taken place in the ancestor of primates (69). Domain fusion has the potential to create new functions, though here the addition of a domain seems to have generated new functionality via alternative splicing, just as with genes TMPO and ZNF451 (70). The functional role of this protein isoform (CSB-PGBD3 fusion protein) is still not wholly clear, but it has been suggested that it may work in tandem with the principal isoform (71). Mutations specific to this isoform cause ovarian failure (72).
TRIFID scores for fibroblast growth factor receptor 1
Fibroblast growth factor receptor 1 (FGFR1) is a membrane-bound tyrosine-protein kinase. Fibroblast growth factor receptors play a crucial role during development and they are associated with the formation of solid tumours (76,77). Fibroblast growth factor receptors have three extracellular immunoglobulin-like receptor domains linked to a cytosolic tyrosine kinase domain by a single trans-membrane helix (Figure 6A).
Figure 6.

Model predictions for four fibroblast growth factor receptor 1 (FGFR1) isoforms. (A) A representation of the architecture of fibroblast growth factor receptors with domains shown in red. The protein forms dimers through its kinase domain. The extracellular region is shaded. (B) A comparison of principal transcript (ENST00000447712) and alternative transcript ENST00000356207. The top half of the panel shows the extracellular region coding exon composition. ENST00000356207 loses an exon with respect to ENST00000447712 (shown with a gold box), the effect of which would be to remove the first immunoglobulin domain, coloured in gold on the model of the extracellular portion of fibroblast growth factor receptor 1. (C) A comparison of principal transcript and alternative transcript ENST00000397103. The top of the panel shows the coding exon composition. ENST00000397103 loses the same exon as ENST00000356207, but would also swap exon 8 (blue box) for exon 9 (coloured in blue) and lose six bases as a result of NAGNAG splicing (shown by an arrow). The effect on the isoform would be to remove domain 1 (gold), two residues in the region between domains 1 and 2 (shown by arrow), and to generate a distinct but homologous version of domain 3 (residues that would differ in the domain are shown in blue). (D) A comparison of the principal transcript and alternative transcript ENST00000619564. The top of the panel shows the coding exon composition. ENST00000619564 loses exon 8 and all downstream exons and replaces them with a shorter non-homologous exon (shown in green). The effect on fibroblast growth factor receptor 1 would be to damage domain 3 (residues lost from domain 3 in green) and eliminate the entire downstream sequence of the protein, including the trans-membrane helix and the tyrosine kinase domain.
FGFR1 is annotated with 23 coding transcripts that would produce 10 distinct isoforms. Five isoforms have normalized TRIFID scores higher than 0.5. TRIFID predicts that the principal transcript, ENST00000447712, produces a functionally important protein with a score of 0.973. Alternative transcript ENST00000356207 (Figure 6B) skips the second coding exon and as a result would generate an isoform without the first immunoglobulin domain. TRIFID gives the isoform a normalized score of 0.692. This isoform has been shown to be functional with a higher affinity for fibroblast growth factors (FGF) than the principal isoform (78).
A third transcript, ENST00000619564, would give rise to an isoform without the first immunoglobulin domain and with a C-terminal truncation in the middle of the final extracellular domain (Figure 6C). A priori, this isoform seems less likely to be functional: if this isoform were translated, it would be an entirely extra-cellular protein and would be unable to bind FGF. There is no conservation evidence at all for this isoform (Supplementary Figure S11). It has a normalized TRIFID score of 0.006.
Transcript ENST00000397103 (Figure 6D) is an interesting case. As with ENST00000356207 it also skips the second coding exon. In addition, it substitutes exon 8 for homologous exon 9. These two exons generate distinct versions of domain 3, a domain that plays a role in determining FGF binding specificity (79). Exon 9 is conserved back to a last common ancestor with vertebrates and homologues FGFR2 and FGFR3 express the equivalent of exon 9 in a tissue-specific manner, so this exon ought to be functionally important in FGFR1 too. However, TRIFID gives this isoform a normalized score of 0.062, in part because the particular exon combination in this transcript is not annotated in many other species (Supplementary Figure S11), and in part because there is no CCDS (RefSeq does not annotate this transcript).
Despite the conservation evidence for exon 9, it has no experimental support and nor could we find published evidence for the function of the isoform. At the transcript level, the exon is practically not detected in either Human Protein Atlas (54) or GTEx RNA seq experiments (80), and there are no peptides for this region in PeptideAtlas (81). This isoform, like the equivalent isoform in FGFR2, is supposed to be exclusively expressed in the mesenchyme (82), so this may be why there is little experimental evidence. Nevertheless, the evidence for the functionality of this isoform is conflicting; GNOMAD (83) records five potential loss of function variants for exon 9, three more than any other FGFR1 exon.
Comparison with PULSE
We compared TRIFID with PULSE, the only previous attempt to predict the functional relevance of splice isoforms, over GENCODE v27 alternative isoforms that had a prediction in PULSE. There are just 2692 non-NMD sequences in common between the two analyses because PULSE made predictions for transcripts detected in the Human Body Map (84) analysis and many of these are not annotated in GENCODE. The majority (51.3%) of alternative isoforms in this reduced set score >0.6 in PULSE (this was the cut-off above which an isoform was considered functional by the authors of PULSE), while just a quarter (24.9%) have a normalized TRIFID score >0.6.
Despite this, the comparison (Figure 7A) suggests that there is some relationship between PULSE and normalized TRIFID scores for alternative isoforms: 21% of isoforms score >0.6 in both methods while 44.9% of isoforms score <0.6 in both. However, >3 in 10 isoforms score more than 0.6 in PULSE and <0.6 in TRIFID. The Pearson correlation between normalized TRIFID score and PULSE score was 0.51.
Figure 7.

A comparison between TRIFID and PULSE. (A) A scatter plot of PULSE and TRIFID scores over alternative isoforms that coincide between the two analyses. The comparison was carried out over 2692 sequences present in both data sets. The distribution of scores for the predictors is shown above or to the right of the graphic. Spearman's rank correlation between the two sets was 0.504. (B) The 346-residue splice variant of IL1RAP mapped onto PDB structure 5VI4. This isoform is generated from an exon skip that changes the frame of the protein. The exon skip occurs in the middle of the third immunoglobulin domain (in purple) and as a result of the frame shift, the variant loses half of the domain (lost region shown in light grey) and the downstream trans-membrane helix and downstream TIR domain. The interaction with interleukin-33 (yellow) and interleukin 1 receptor like 1 (teal) will also be affected. The isoform is annotated only in the human genome. PULSE predicts that this isoform is functional (0.831), while TRIFID does not (0.002). (C) The 475-residue splice variant of ATE1 mapped onto PDB structure 2ATR using HHPRED. This isoform is generated from an exon skip that removes 41 residues including the first part of the Arginine-tRNA-protein transferase domain (lost region shown in light grey). This splice event skips a pair of mutually exclusively spliced exons that appear to be important in substrate selection (85) and that are conserved even in Orb weaver spiders. It seems unlikely that such important exons can be skipped without consequence for the function of the protein. PULSE predicts that this isoform is functional (0.707) and TRIFID does not (0.037). (D) Two splice variants of MACROH2A1 mapped onto PDB structure 6fy5 using HHPRED. The first isoform is generated from an exon skip that changes the frame at the start of the macro domain. The section of the structure that would be maintained is shown in teal, the remainder (in yellow and light grey) would be replaced by 27 residues as a result of the frame shift. PULSE predicts that this isoform is functional (0.676), while TRIFID does not (0.184). A second exon skip produces another frame shift that affects the same domain. Here the conserved region is shown in purple and yellow, and the region of the domain replaced by frame-shifted residues in light grey. Neither method predicts that this isoform is functional, but the PULSE score for this improbable protein is much higher, 0.484 against 0.008. All images were generated using PyMol.
The most important feature in PULSE (by a large margin) was similarity in length. Length delta was also an important feature in TRIFID, but other important features such as CCDS annotation and conservation were also important. Indeed, four of the six most important features in TRIFID according to the SHAP scores were conservation-based. As a comparison, the correlation between PULSE score and three conservation features used by PULSE was 0.204, 0.106 and 0.082. PULSE also reported that 36% of functional splice variants had events that fell inside Pfam domain boundaries. This is very close to the proportion of events you would expect to find if the events were chosen at random: 37% of splice events in the human reference set fall inside Pfam domains (16). PULSE used six domain features to train their model and correlation with the PULSE score ranged between −0.272 and +0.078, which suggests that domain features had little bearing on the prediction of functional importance in PULSE. In TRIFID, alternative isoforms predicted as functionally important have significantly fewer altered functional domains (11.3%) than alternative isoforms predicted as non-functional (40.6%, Fisher's exact test P < 0.00001). It is not surprising then that the isoforms that PULSE predicts as functional and that TRIFID predicts as non-functional, tend to have broken functional domains and little cross-species conservation (Figure 7). Further examples are shown in Supplementary Figure S12.
Validating the results against an external source of information
Although the model evaluation shows that TRIFID is able to distinguish efficiently between positive and negative instances in the training set, we required orthogonal evidence strands to validate our predictions for the whole genome. We used germline variation data to calculate rates of genetic variation for alternative and principal isoforms. All exons and exon fragments that overlapped principal transcripts were classified as principal exons, while those exons and exon fragments that were exclusive to alternative transcripts were classified as alternative. That meant that ∼90% of exons were classified as principal. We evaluated principal and alternative exons separately because otherwise the results would be dominated by the variants in the principal exons.
Exons from principal and alternative transcripts were each binned in five subsets by TRIFID score. We calculated non-synonymous to synonymous substitution rates for common and rare alleles from the human variation data from the 1000 Genomes Project (61) for each of the subsets. Exons under selective pressure should have non-synonymous to synonymous ratios that are significantly lower for common allele frequencies than for rare allele frequencies.
The results are shown in Figure 8. Non-synonymous to synonymous ratios for exons derived from principal transcripts decrease notably as normalized TRIFID score increases. In each set of principal exons with scores >0.2, the non-synonymous to synonymous ratio is significantly lower for common alleles than it is for rare alleles, as would be expected if these exons were under selective pressure (see Supplementary Material for more details). The only exceptions are those with normalized scores <0.2. Almost three quarters of principal isoforms with normalized scores <0.2 are from coding genes tagged as potential non-coding in a previous study (46), suggesting that many of these lowest scoring principal isoforms are in fact from genes that were mis-classified as coding.
Figure 8.
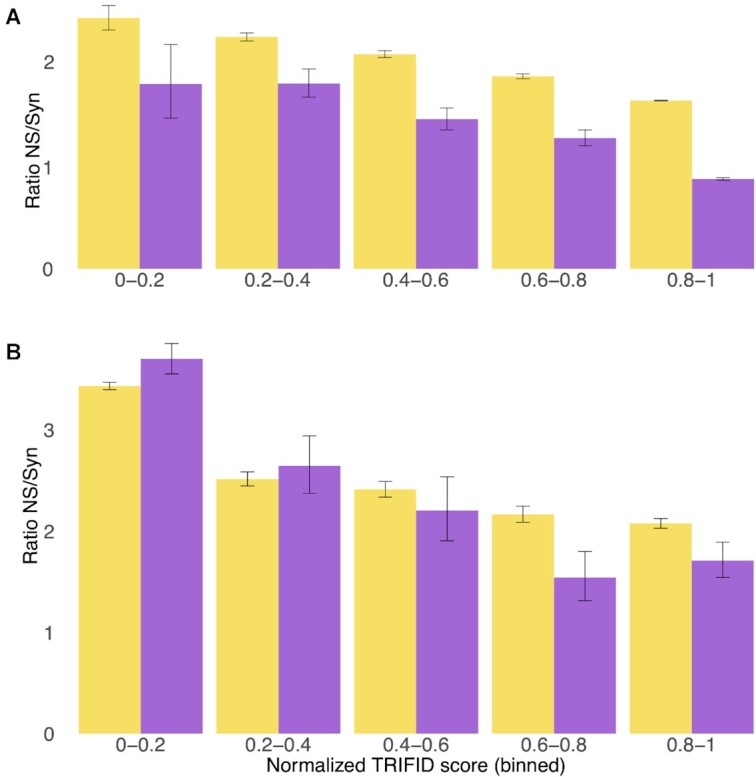
TRIFID scores and genomic variation for principal and alternative exons. (A) Non-synonymous to synonymous ratios for rare (yellow) and common allele frequencies (purple) for exons from principal transcripts binned by the TRIFID score of the transcript. (B) Non-synonymous to synonymous ratios for rare (yellow) and common allele frequencies (purple) for exons that do not overlap principal transcripts binned by the TRIFID score of their transcript. Error bars show the confidence intervals for each subset of exons.
Non-synonymous to synonymous ratios for exons derived from alternative transcripts also decrease with increasing TRIFID score. Since there are comparatively fewer alternative exons, there are relatively few common variants in each bin (as reflected by the larger confidence intervals). Despite this, non-synonymous to synonymous ratios are significantly lower for common alleles than for rare alleles at normalized TRIFID scores of 0.6–0.8 and >0.8. This suggests that at least a considerable fraction of alternative transcripts with normalized TRIFID scores >0.6 are under selective pressure.
Non-synonymous to synonymous ratios are not significantly lower for common alleles than for rare alleles in those exons from transcripts with normalized TRIFID scores of <0.6, even if the statistical power is larger given the number of instances in this category. This result suggests that most of these exons are not under selective pressure. Isoforms translated from these lowest scoring exons make up almost 85% of all alternative isoforms.
Exporting the TRIFID model to the genomes of other species
We trained TRIFID with splice variants from the human genome. The human genome is more curated and has more supporting evidence than any other genome. Several of the features that we used to train TRIFID (for example CCDS number, transcript support and RNA support) are either not available or are less complete for non-human reference sets. However, eight of the ten most important features are available for all eukaryotic species that can be annotated in APPRIS.
We retrained TRIFID with the 25 features that would be available for all species in order to analyse the effectiveness of a generic predictor of functional isoforms. We compared precision recall curves from the generic predictor with those of the human-specific predictor. In this case, we used the whole training set (outer loop) to validate the general performance over the whole set of training isoforms. In this configuration, the overall AUC-PR of the human-specific TRIFID is 0.985, while the AUC-PR of the generic TRIFID is lower, but only drops to 0.974 (Supplementary Figure S13). This suggests that there ought to be enough discriminatory power in the remaining features to allow us to export TRIFID to other species.
DISCUSSION
In order to understand the true complexity of the proteome and to detect function-altering mutations and variants in clinical practice, it is important to determine which protein isoforms are biologically relevant and which are not. For that reason, we have developed a machine learning algorithm to classify all isoforms for a given gene and to predict which isoforms are most likely to be functionally important.
The predictor, TRIFID, uses proteomics evidence as a proxy for functionality and was trained and validated on peptide data. The model had an overall AUC-PR of 0.985. We found that TRIFID scores were gene dependent because the CORSAIR and Alt-CORSAIR modules in TRIFID do not always detect cross-species conservation. This can be overcome somewhat by using the normalized scores, at the risk of over-predicting functionally important variants.
Analysis of non-synonymous to synonymous ratios of germline variants shows that alternative isoforms with normalized TRIFID scores of more than 0.6 are under selective pressure and that most alternative isoforms with lower TRIFID scores appear to be evolving neutrally. These results demonstrate that TRIFID can distinguish functionally important isoforms. Normalized TRIFID score can be used to select those alternative isoforms that are more likely to have biological roles; 15.6% of alternative isoforms in the GENCODE human gene set have a normalized TRIFID score greater than 0.6.
Cross-species conservation of isoforms was the most important feature for distinguishing biological relevance; the greater the evidence of cross-species conservation, the more likely the isoform was predicted to be functional. Isoforms that were predicted as functional impacted Pfam domains significantly less often than isoforms predicted as non-functional.
Although TRIFID predicts that fewer than 1 in 6 alternative proteins are functionally important, there are several caveats. Firstly, many alternative isoforms are predicted as functionally important based on their similarity to the main isoform. It is not clear how many of those isoforms that differ by micro-indels of four or fewer amino acids really are functional. At the same time, the conservation-based metrics in TRIFID do not always detect cross species conservation, and even though this is partly corrected through normalization of the TRIFID score, it almost certainly means that TRIFID will miss some functional isoforms.
Finally, it should be pointed out that transcripts may have functional roles that do not involve a protein product (33), TRIFID does not predict functional importance at the transcript level. Also, although TRIFID was trained on tissue-based proteomics data, it does not predict tissue specificity.
We have shown that TRIFID could also be exported to other species even though some features are not available for all species. This could make TRIFID a useful tool for genome annotation, though it is important to note that the predictor will work better on well annotated species. The method arrives just in time to deal with the likely explosion of new transcript models from short and long-read sequencing studies (86,87).
The results of our research provide important insights into understanding the importance of alternative splicing at the protein level. From a clinical standpoint, a method that can predict the relative functional importance of protein isoforms will be a particularly valuable tool to help understand the pathogenic effects of mutations on splice variants. Potential pathogenic mutations are of clinical interest, but it is important to know if these mutations affect exons from biologically relevant splice variants, as in the example of ERCC6.
We believe our dataset of likely functional isoforms would be of great value to better our understanding of alternative splicing, for example focusing on datasets with less noise and enriched in real functional events.
DATA AVAILABILITY
The datasets supporting the conclusions of this article are available in the gitlab repository at https://gitlab.com/bu_cnio/trifid.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Irwin Jungreis for making the PhyloCSF data available.
Contributor Information
Fernando Pozo, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), Madrid, Spain.
Laura Martinez-Gomez, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), Madrid, Spain.
Thomas A Walsh, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), Madrid, Spain.
José Manuel Rodriguez, Cardiovascular Proteomics Laboratory, Centro Nacional de Investigaciones Cardiovasculares (CNIC), Madrid, Spain.
Tomas Di Domenico, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), Madrid, Spain.
Federico Abascal, Somatic Evolution Group, Wellcome Sanger Institute, Hinxton CB10 1SA, UK.
Jesús Vazquez, Cardiovascular Proteomics Laboratory, Centro Nacional de Investigaciones Cardiovasculares (CNIC), Madrid, Spain.
Michael L Tress, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), Madrid, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Human Genome Research Institute of the National Institutes of Health [2 U41 HG007234]; Spanish Ministry of Science, Innovation and Universities [PGC2018-097019-B-I00]; Carlos III Institute of Health-Fondo de Investigación Sanitaria PRB3 [IPT17/0019 - ISCIII-SGEFI / ERDF, ProteoRed]; ‘la Caixa’ Banking Foundation [HR17-00247].
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B.. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Black D.L. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000; 103:367–370. [DOI] [PubMed] [Google Scholar]
- 3. Graveley B.R. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001; 17:100–107. [DOI] [PubMed] [Google Scholar]
- 4. The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright .J, Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sayers E.W., Beck J., Brister J.R., Bolton E.E., Canese K., Comeau D.C., Funk K., Ketter A., Kim S., Kimchi A.et al.. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2020; 48:D9–D16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hu Z., Scott H.S., Qin G., Zheng G., Chu X., Xie L., Adelson D.L., Oftedal B.E., Venugopal P., Babic M.et al.. Revealing missing human protein isoforms based on ab initio prediction RNA-seq and proteomics. Sci. Rep. 2015; 5:10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pertea M., Shumate A., Pertea G., Varabyou A., Breitwieser F.P., Chang Y.C., Madugundu A.K., Pandey A., Salzberg S.L.. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018; 19:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buljan M., Chalancon G., Eustermann S., Wagner G.P., Fuxreiter M., Bateman A., Babu M.M.. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell. 2012; 46:871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calarco J.A., Xing Y., Cáceres M., Calarco J.P., Xiao X., Pan Q., Lee C., Preuss T.M., Blencowe B.J.. Global analysis of alternative splicing differences between humans and chimpanzees. Genes Dev. 2007; 21:2963–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Merkin J., Russell C., Chen P., Burge C.B.. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science. 2012; 338:1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhuiyan S.A., Ly S., Phan M., Huntington B., Hogan E., Liu C.C., Liu J., Pavlidis P.. Systematic evaluation of isoform function in literature reports of alternative splicing. BMC Genomics. 2018; 19:637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kelemen O., Convertini P., Zhang Z., Wen Y., Shen M., Falaleeva M., Stamm S.. Function of alternative splicing. Gene. 2013; 514:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang X., Coulombe-Huntington J., Kang S., Sheynkman G.M., Hao T., Richardson A., Sun S., Yang F., Shen Y.A., Murray R.R.et al.. Widespread expansion of protein interaction capabilities by alternative splicing. Cell. 2016; 164:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ezkurdia I., Rodriguez J.M., Carrillo-de Santa Pau E., Vázquez J., Valencia A., Tress M.L.. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res. 2015; 14:1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abascal F., Ezkurdia I., Rodriguez-Rivas J., Rodriguez J.M., del Pozo A., Vázquez J., Valencia A., Tress M.L.. Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comp. Biol. 2015; 11:e1004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tress M.L., Abascal F., Valencia A.. Most alternative isoforms are not functionally important. Trends Biochem. Sci. 2017; 42:408–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodriguez J.M., Rodriguez-Rivas J, Di Domenico T., Vázquez J., Valencia A., Tress M.L.. APPRIS 2017: principal isoforms for multiple gene sets. Nucleic Acids Res. 2018; 46:D213–D217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tress M.L., Abascal F., Valencia A.. Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci. 2017; 42:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blencowe B.J. The Relationship between alternative splicing and proteomic complexity. Trends Biochem. Sci. 2017; 42:407–408. [DOI] [PubMed] [Google Scholar]
- 21. Wan Y., Larson D.R.. Splicing heterogeneity: separating signal from noise. Genome Biol. 2018; 19:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodriguez J.M., Pozo F., di Domenico T., Vazquez J., Tress M.L.. An analysis of tissue-specific alternative splicing at the protein level. PLoS Comp. Biol. 2020; 16:e1008287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Melamud E., Moult J.. Stochastic noise in splicing machinery. Nucleic Acids Res. 2009; 37:4873–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saudemont B., Popa A., Parmley J.L., Rocher V., Blugeon C., Necsulea A., Meyer E., Duret L.. The fitness cost of mis-splicing is the main determinant of alternative splicing patterns. Genome Biol. 2017; 18:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu C., Park J.K., Zhang J.. Evidence that alternative transcriptional initiation is largely nonadaptive. PLoS Biol. 2019; 17:e3000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu C., Zhang J.. Alternative polyadenylation of mammalian transcripts is generally deleterious, not adaptive. Cell Syst. 2018; 6:734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu T., Lin K.. The distribution pattern of genetic variation in the transcript isoforms of the alternatively spliced protein-coding genes in the human genome. Mol. Biosyst. 2015; 11:1378–1388. [DOI] [PubMed] [Google Scholar]
- 28. Wang X., Codreanu S.G., Wen B., Li K., Chambers M.C., Liebler D.C., Zhang B.. Detection of proteome diversity resulted from alternative splicing is limited by trypsin cleavage specificity. Mol. Cell. Proteomics. 2018; 17:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang S.H., Hsiao C.J., Khan Z., Pritchard J.K.. Post-translational buffering leads to convergent protein expression levels between primates. Genome Biol. 2018; 19:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Inada T. The ribosome as a platform for mRNA and Nascent polypeptide quality control. Trends Biochem. Sci. 2017; 42:5–15. [DOI] [PubMed] [Google Scholar]
- 31. Lareau L.F., Brenner S.E.. Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Mol. Biol. Evol. 2015; 32:1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R.et al.. Ensembl 2020. Nucleic Acids Res. 2020; 48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eksi R., Li H.D., Menon R., Wen Y., Omenn G.S., Kretzler M., Guan Y.. Systematically differentiating functions for alternatively spliced isoforms through integrating RNA-seq data. PLoS Comp. Biol. 2013; 9:e1003314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li W., Kang S., Liu C.C., Zhang S., Shi Y., Liu Y., Zhou X.J.. High-resolution functional annotation of human transcriptome: predicting isoform functions by a novel multiple instance-based label propagation method. Nucleic Acids Res. 2014; 42:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Panwar B., Menon R., Eksi R., Li H-D., Omenn G.S., Guan Y.. Genome-wide functional annotation of human protein-coding splice variants using multiple instance learning. J. Proteome Res. 2016; 15:1747–1753. [DOI] [PubMed] [Google Scholar]
- 36. Chen H., Shaw D., Zeng J., Bu D., Jiang T.. DIFFUSE: predicting isoform functions from sequences and expression profiles via deep learning. Bioinformatics. 2019; 35:i284–i294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu G., Wang K., Domeniconi C., Guo M., Wang J.. Isoform function prediction based on bi-random walks on a heterogeneous network. Bioinformatics. 2020; 36:303–310. [DOI] [PubMed] [Google Scholar]
- 38. Shaw D., Chen H., Jiang T.. DeepIsoFun: a deep domain adaptation approach to predict isoform functions. Bioinformatics. 2019; 35:2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang K., Wang J., Domeniconi C., Zhang X., Yu G.. Differentiating isoform functions with collaborative matrix factorization. Bioinformatics. 2020; 36:1864–1871. [DOI] [PubMed] [Google Scholar]
- 40. Gonzàlez-Porta M., Frankish A., Rung J., Harrow J., Brazma A.. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol. 2013; 14:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li H.D., Menon R., Govindarajoo B., Panwar B., Zhang Y., Omenn G.S., Guan Y.. Functional networks of highest-connected splice isoforms: from the chromosome 17 human proteome project. J. Proteome Res. 2015; 14:3484–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harte R.A., Farrell C.M., Loveland J.E., Suner M.M., Wilming L., Aken B., Barrell D., Frankish A., Wallin C., Searle Set al.. Tracking and coordinating an international curation effort for the CCDS Project. Database. 2012; 2012:bas008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hao Y., Colak R., Teyra J., Corbi-Verge C., Ignatchenko A., Hahne H., Wilhelm M., Kuster B., Braun P., Kaida D.et al.. Semi- supervised learning predicts approximately one third of the alternative splicing isoforms as functional proteins. Cell Rep. 2015; 12:183–189. [DOI] [PubMed] [Google Scholar]
- 44. Hegyi H., Kalmar L., Horvath T., Tompa P.. Verification of alternative splicing variants based on domain integrity, truncation length and intrinsic protein disorder. Nucleic Acids Res. 2011; 39:1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ezkurdia I., del Pozo A., Frankish A., Rodriguez J.M., Harrow J., Ashman K., Valencia A., Tress M.L.. Comparative proteomics reveals a significant bias toward alternative protein isoforms with conserved structure and function. Mol. Biol. Evol. 2012; 29:2265–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abascal F., Juan D., Jungreis I., Martinez L., Rigau M., Rodriguez J.M., Vazquez J., Tress M.L.. Loose ends: almost one in five human genes still have unresolved coding status. Nucleic Acids Res. 2018; 46:7070–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martinez-Gomez L., Abascal F., Jungreis I., Pozo F, Kellis M., Mudge J.M., Tress M.L.. Few SINEs of life: Alu elements have little evidence for biological relevance despite elevated translation. NAR Genom. Bioinform. 2020; 2:lqz023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim M.S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S.et al.. A draft map of the human proteome. Nature. 2014; 509:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deutsch E.W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D.S., Bernal-Llinares M., Okuda S., Kawano S.et al.. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017; 45:D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eng J.K., Jahan T.A., Hoopmann M.R.. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013; 13:22–24. [DOI] [PubMed] [Google Scholar]
- 51. The M., MacCoss M.J., Noble W.S., Käll L.. Fast and accurate protein false discovery rates on large-scale proteomics data sets with Percolator 3.0. J. Am. Soc. Mass. Spectrom. 2016; 27:1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ezkurdia I., Calvo E., Del Pozo A., Vázquez J., Valencia A., Tress M.L.. The potential clinical impact of the release of two drafts of the human proteome. Expert Rev. Proteomics. 2015; 12:579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rodriguez J.M., Maietta P., Ezkurdia I., Pietrelli A., Wesselink J.J., Lopez G., Valencia A., Tress M.L.. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 2013; 41:110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 55. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A.et al.. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lin M.F., Jungreis I., Kellis M.. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011; 27:275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chicco D., Jurman G.. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics. 2020; 21:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chicco D. Ten quick tips for machine learning in computational biology. BioData Min. 2017; 10:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lundberg S.M., Erion G., Chen H., DeGrave A., Prutkin J.M., Nair B., Katz R., Himmelfarb J., Bansal N., Lee S.-I.. From local explanations to global understanding with explainable AI for trees. Nat. Mach. Intell. 2020; 2:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F.. The Eensembl variant effect predictor. Genome Biol. 2016; 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu J., Wang W., Xu L., Chen J.Y., Chong J., Oh J., Leschziner A.E., Fu X.D., Wang D.. Cockayne syndrome B protein acts as an ATP-dependent processivity factor that helps RNA polymerase II overcome nucleosome barriers. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:25486–25493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Feng E., Batenburg N.L., Walker J.R., Ho A., Mitchell T.R.H., Qin J., Zhu X.D.. CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J. Cell Sci. 2020; 133:jcs234914. [DOI] [PubMed] [Google Scholar]
- 66. Okur M.N., Lee J.H., Osmani W., Kimura R., Demarest T.G., Croteau D.L., Bohr V.A.. Cockayne syndrome group A and B proteins function in rRNA transcription through nucleolin regulation. Nucleic Acids Res. 2020; 48:2473–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhu Q., Ding N., Wei S., Li P., Wani G., He J., Wani A.A.. USP7-mediated deubiquitination differentially regulates CSB but not UVSSA upon UV radiation-induced DNA damage. Cell Cycle. 2020; 19:124–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Duan M., Selvam K., Wyrick J.J., Mao P.. Genome-wide role of Rad26 in promoting transcription-coupled nucleotide excision repair in yeast chromatin. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:18608–18616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Newman J.C., Bailey A.D., Fan H.Y., Pavelitz T., Weiner A.M.. An abundant evolutionarily conserved CSB-PiggyBac fusion protein expressed in Cockayne syndrome. PLos Genet. 2008; 4:e1000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Abascal F., Tress M.L., Valencia A.. Alternative splicing and co-option of transposable elements: the case of TMPO/LAP2α and ZNF451 in mammals. Bioinformatics. 2015; 31:2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bailey A.D., Gray L.T, Pavelitz T., Newman J.C., Horibata K., Tanaka K., Weiner A.M.. The conserved Cockayne syndrome B-piggyBac fusion protein (CSB-PGBD3) affects DNA repair and induces both interferon-like and innate antiviral responses in CSB-null cells. DNA Repair (Amst.). 2012; 11:488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Qin Y., Guo T., Li G., Tang T.S., Zhao S., Jiao X., Gong J., Gao F., Guo C, Simpson J.L., Chen Z.J.. CSB-PGBD3 mutations cause premature ovarian failure. PLoS Genet. 2015; 11:e1005419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Burley S.K., Berman H.M., Kleywegt G.J., Markley J.L., Nakamura H., Velankar S.. Protein Data Bank (PDB): the single global macromolecular structure archive. Methods Mol. Biol. 2017; 1607:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W.et al.. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gabler F., Nam S.Z., Till S., Mirdita M., Steinegger M., Söding J., Lupas A.N., Alva V.. Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinformatics. 2020; 72:e108. [DOI] [PubMed] [Google Scholar]
- 76. Turner N., Grose R.. Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer. 2010; 10:116–129. [DOI] [PubMed] [Google Scholar]
- 77. Wang S., Ding Z.. Fibroblast growth factor receptors in breast cancer. Tumour Biol. 2017; 39:1010428317698370. [DOI] [PubMed] [Google Scholar]
- 78. Olsen S.K., Ibrahimi O.A., Raucci A., Zhang F., Eliseenkova A.V., Yayon A., Basilico C., Linhardt R.J., Schlessinger J., Mohammadi M.. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:935–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Olsen S.K., Li J.Y., Bromleigh C., Eliseenkova A.V., Ibrahimi O.A., Lao Z., Zhang F., Linhardt R.J., Joyner A.L., Mohammadi M.. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 2006; 20:185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Deutsch E.W. The PeptideAtlas Project. Methods Mol. Biol. 2010; 604:285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zinkle A., Mohammadi M.. Structural biology of the FGF7 subfamily. Front. Genet. 2019; 10:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cummings B.B., Karczewski K.J., Kosmicki J.A., Seaby E.G., Watts N.A., Singer-Berk M., Mudge J.M., Karjalainen J., Satterstrom F.K., O’Donnell-Luria A.H.et al.. Transcript expression-aware annotation improves rare variant interpretation. Nature. 2020; 581:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L.. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011; 25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang J., Pejaver V.R., Dann G.P., Wol M.Y., Kellis M., Huang Y., Garcia B.A., Radivojac P., Kashina A.. Target site specificity and in vivo complexity of the mammalian arginylome. Sci. Rep. 2018; 8:16177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tardaguila M., de la Fuente L., Marti C., Pereira C., Pardo-Palacios F.J., Del Risco H., Ferrell M., Mellado M., Macchietto M., Verheggen K.et al.. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 2018; 28:396–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang X., You X., Langer J.D., Hou J., Rupprecht F., Vlatkovic I., Quedenau C., Tushev G., Epstein I., Schaefke B.et al.. Full-length transcriptome reconstruction reveals a large diversity of RNA and protein isoforms in rat hippocampus. Nat. Commun. 2019; 10:5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article are available in the gitlab repository at https://gitlab.com/bu_cnio/trifid.