

Abstract
SARS-CoV-2 has exploded throughout the human population. To facilitate efforts to gain insights into SARS-CoV-2 biology and to target the virus therapeutically, it is essential to have a roadmap of likely functional regions embedded in its RNA genome. In this report, we used a bioinformatics approach, ScanFold, to deduce the local RNA structural landscape of the SARS-CoV-2 genome with the highest likelihood of being functional. We recapitulate previously-known elements of RNA structure and provide a model for the folding of an essential frameshift signal. Our results find that SARS-CoV-2 is greatly enriched in unusually stable and likely evolutionarily ordered RNA structure, which provides a large reservoir of potential drug targets for RNA-binding small molecules. Results are enhanced via the re-analyses of publicly-available genome-wide biochemical structure probing datasets that are broadly in agreement with our models. Additionally, ScanFold was updated to incorporate experimental data as constraints in the analysis to facilitate comparisons between ScanFold and other RNA modelling approaches. Ultimately, ScanFold was able to identify eight highly structured/conserved motifs in SARS-CoV-2 that agree with experimental data, without explicitly using these data. All results are made available via a public database (the RNAStructuromeDB: https://structurome.bb.iastate.edu/sars-cov-2) and model comparisons are readily viewable at https://structurome.bb.iastate.edu/sars-cov-2-global-model-comparisons.
INTRODUCTION
SARS-CoV-2 is the infectious agent responsible for COVID-19, a globally distributed disease that has upended human civilization. This recent outbreak has massively reiterated the need for research on potential human pathogens and focused attention on the importance of RNA biology to this understanding. SARS-CoV-2 is a roughly 30 kb, positive sense (i.e. translation competent), 5′ capped single-stranded RNA molecule, which utilizes RNA throughout its biology. RNA structural elements have been described in the original SARS-CoV (1,2) and each is broadly conserved within the SARS-CoV-2 genome (see several Rfam (3–10) entries at https://rfam.xfam.org/covid-19), presumably performing essential functions. For example, translation of essential regions of the SARS-CoV genome (e.g. the RNA dependent RNA polymerase) depends on a process of –1 programmed ribosomal frameshifting (–1 PRF) that makes use of a highly-structured frameshift stimulatory element (FSE), which impedes ribosomes allowing for ‘slippage’ to a new reading frame. This FSE was previously studied extensively (11) and even targeted with small molecules to inhibit –1 PRF (12) for potential therapeutic discovery. The homologous region in SARS-CoV-2 is similar in sequence and is capable of forming a near-identical pseudoknot structure (Rfam ID# RF00507) that has become a target of intensive structural analysis (13): indeed, it was recently characterized in 3D using cryo-EM (14).
A sequence region upstream of the pseudoknot in the FSE is presumed to function as an attenuator for –1 PRF and is less well conserved in sequence (13). Our preliminary structural analyses of SARS-CoV-2 provided evidence for this attenuator having highly stable and ordered secondary structure (15), the model of which (Figure 1) was used to design a small molecule (targeting a UU internal loop) that is able to efficiently suppress –1 PRF in vitro (16)—raising the hope that small-molecule regulators of SARS-CoV-2 RNA biology can be discovered that may prove to be effective therapeutics. With such a potent example of a high-value structural element in SARS-CoV-2, multiple groups have undertaken intensive research to characterize the SARS-CoV-2 RNA structurome using high-throughput biochemical probing coupled to experimentally-informed secondary structural modeling (17–20). Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) probing of transcripts from infected human cell lines, which maps regions of RNAs that are structurally flexible (i.e. unpaired regions), and dimethyl sulfate (DMS), which interrogates the Watson–Crick face of adenosine and cytosine (preferentially modifying unpaired or loosely structured bases (21)), have been used to generate robust models of RNA secondary structures found throughout the viral genome/transcriptome.
Figure 1.

In silico ScanFold-Fold predicted secondary structure for the SARS-CoV-2 frameshift stimulatory element (FSE) spanning nts 13422–13547. Average z-scores are overlaid on each nt via a heat map ranging from –2.34 (red) to 0.00 (blue). Top 10% of reactivities are shown for Manfredoina et al. (squares, pentagons and diamonds), Huston et al. (circles), Sun et al. (stars) and Lan et al. (triangles) at their corresponding nt positions (17–20). The attenuator hairpin and UU internal loop, recently targeted with small molecule inhibitors of –1 PRF (16), are depicted in blue shaded boxes and the slippery sequence in a gold shaded box. The interactions of the pseudoknot proposed by other groups (17–20) are shown with solid and dashed gray lines and the specific base pairing pattering are also shown in an inset. The smaller pseudoknot structure as determined by cyro-EM (14,73) is highlighted in lavender (dashed line and inset). The two orange colored pairs at the top of Stem 1 were not detected by Bhatt et al. (73) in their cryo-EM and the two red pairs at the base of stem 3 were not detected by either Bhatt et al. or Zhang et al. (14,73). All significantly covarying bps (R-scape APC corrected G-test; E < 0.05) have been highlighted with a green box.
SARS-CoV-2 appears to have an unusually structured RNA genome with a multitude of exciting target motifs; for example, 106 predicted conserved secondary structures were previously identified via the motif discovery tool RNAz (22–25). Approaches to rank target motifs are essential for driving additional research, particularly in efforts to drug RNA. We previously developed a computational approach known as ScanFold which aids in such efforts by highlighting local RNA motifs with unusually stable base pairs. Unusual stability, as measured by a significantly negative thermodynamic z-score is a hallmark feature of functional RNAs; we partition this value via nucleotides and base pairs to facilitate the model building process. Recently, we showed that this process identifies structures that are more consistently observed in RNA probing experiments (26). Such information then, serves as a valuable complement to other analyses by proposing RNA structures that are not only likely to represent native conformations (27,28) but also those with the greatest potential for being ordered/structured for performing biological functions (29). In this report, we detail the results of a ScanFold analysis of SARS-CoV-2, perform comparisons to available experimental probing analyses, and ultimately use these results to identify eight novel RNA structures with significant evidence of structural conservation (using R-scape (30,31) and CaCoFold (32)). These results provide a roadmap that can be used to drive future studies of SARS-CoV-2 by enumerating local structural motifs with exceptional prediction metrics that are robustly supported by multiple sources of experimental and phylogenetic data.
MATERIALS AND METHODS
ScanFold analyses of SARS-CoV-2
The SARS-CoV-2 reference genome sequence (NC_045512.2) was downloaded from the NCBI nucleotide database. For the standard, purely in silicoScanFold analysis we used the parameters that were most successful at modeling the known functional structures in the HIV and Zika virus genomes (27) as depicted in their experimentally derived global secondary structures (33,34); a range of window sizes (from 120 to 600 nt) and different shuffling routines (mononucleotide or dinucleotide) were tested as well. For the standard run, ScanFold-Scan was used with a 120 nt window moving with a single nucleotide step size and 100 mononucleotide randomizations. Each window was analyzed using the RNAfold algorithm implemented in the ViennaRNA package (35) (Version 2.4.14). For each window the minimum free energy (MFE) ΔG° structure and value was predicted using the Turner energy model (36) at 37°C. To characterize the MFE, a ΔG° z-score is calculated for each window: each MFE predicted for the native sequence (MFEnative) is compared to MFE values calculated for 100 mononucleotide (or dinucleotide when specified) shuffled versions of the sequence with the same nucleotide composition (MFErandom) as shown in Equation 1; using an approach adapted from Clote et al. (37). Here, the standard deviation (σ) is calculated across all MFE values.
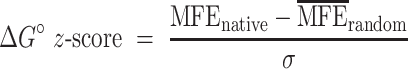 |
(1) |
The P-value corresponds to the number of MFErandom values which were more stable (more negative) than the MFEnative. In addition to these metrics, RNAfold partition function calculations (38) were utilized to characterize the potential structural diversity of the native sequence. These include the ensemble diversity (ED) and the centroid structure. The centroid structure depicts the base pairs which were ‘most common’ (i.e. had the minimal base pair distance) between all the Boltzmann-ensemble conformations predicted for the native sequence. The ED then attempts to quantify the variety of different structures which were present in the ensemble (where higher numbers indicate multiple structures unique from the predicted MFE and low numbers indicate the presence of a dominant MFE structure highly represented in the ensemble (39,40)).
Alignment and conservation analyses
Individual motifs were analyzed for covariation using the cm-builder Perl script, which builds off the RNAFramework toolkit (41) and was recently introduced in (18). This script utilizes Infernal (here using release 1.1.2; (42,43)) to build and search for covariation models of each ScanFold motif's secondary structure. The coronavirus sequence database referenced by Infernal was built using the ViPR database (44,45) (accessed in October 2020) and was composed of 25571 Coronaviridae sequences. For successful covariation models, the resulting structural alignment files (in Stockholm format) were tested for covarying base pairs and analyzed with the CaCoFold algorithm using R-scape (version 1.5.16); statistical significance was evaluated by the APC corrected G-test (30,31) using the default E value of 0.05. All Stockholm alignments and R-scape/CaCoFold results can be found at https://structurome.bb.iastate.edu/sars-cov-2-structure-extracts.
Soft constraint analyses
ScanFold was updated to allow the incorporation of SHAPE reactivities values (from a two or three column reactivity file, where the reactivity values are in the right most column) via RNAfold’s core library functions. By defining slope and intercept parameters for the Deigan (46) or Zarringhalam (47) pseudo-energy model, the corresponding reactivity values are passed into each ScanFold analysis window and incorporated during the native sequence MFE calculation.
We ran ScanFold at varying scanning analysis window sizes (120, 200, 300, 400, 500 and 600 nt) with the in vitro and in vivo SHAPE reactivity data sets generated by Manfredonia et al.. We used the Deigan pseudo-energy model (46) with a slope and intercept of 0.8 and –0.2 (as reported in Manfredonia et al.) respectively. The output data files are formatted the same as standard ScanFold analyses, but the resulting ScanFold-Fold models are now informed by the SHAPE reactivity data sets.
Hard constraint analyses
The SHAPE reactivity datasets for the Incarnato (18), Pyle (17) and Zhang (20) labs are publicly available at http://www.incarnatolab.com/datasets/SARS_Manfredonia_2020.php, https://github.com/pylelab/SARS-CoV-2_SHAPE_MaP_structure, and http://rasp.zhanglab.net/ respectively. The DMS reactivity dataset from the Rouskin Lab (19), was obtained by request. Reactivity and constraint values for each data set were analyzed and characterized using Excel and R. Constraint files were generated which constrained the top 10% of reactivities as being unpaired for select data sets (for Manfredonia et al.’s in vitro SHAPE data, Huston et al.’s in vivo SHAPE data, and Lan et al.’s in vivo DMS data), individually, along with a combined file containing all unique constraints from the top 10% of reactivities. The individual and combined constraint files were then used as hard constraints with ScanFold to analyze the SARS-CoV-2 genome, using the same parameters as described for the standard ScanFold analysis.
ROC analysis
ScanFold-Fold generated SARS-CoV-2 genome secondary structure models (both in silico and soft constrained models at varying analysis windows) along with available global models from Manfredonia et al. and Huston et al. had their corresponding secondary structures (as depicted in connectivity table or ‘CT’ data files) cross referenced to varying SHAPE and DMS reactivity data sets generated from SARS-CoV-2 probing experiments. By sequentially setting reactivity value thresholds from lowest to highest values (at 1% intervals; i.e. 1, 2, 3… 100%) to define a nucleotide as being paired and checking their consistency with base pair coordinates in the reference CT file, we can perform a receiver operator characteristic (ROC) analysis. In this analysis, the true positive rate (TPR) and false positive rate (FPR) are represented by equations 2 and 3 below:
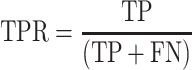 |
(2) |
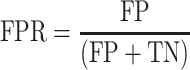 |
(3) |
Here, the true positive (TP) is defined as being paired in the given CT file and paired at the defined reactivity threshold, the false negative (FN) is paired in the CT and unpaired at the reactivity threshold, the false positive (FP) is unpaired in the CT and paired at the reactivity threshold, and the true negative (TN) is unpaired in CT and unpaired at the given reactivity threshold. With these definitions, when the threshold is set to 0%, TPR and FPR will be equal to zero and when the reactivity threshold is set to 100%, TPR and FPR will equal one. If a given RNA secondary structure model is truly random, when compared to increasing reactivity thresholds from a probing data set, then the TPR and FPR should increase proportionately. However, if the RNA secondary structure model agrees with the reactivity data set the TPR should initially rise faster than the FPR, creating a larger area under the curve (AUC). In this way, we can quantitatively assess and compare each model's ability to fit the data via their respective AUCs.
Comparisons of RNA secondary structural models
Comparisons of ScanFold CT files from in silico and constrained runs, along with other global models were done using the script ct_compare.py, which checks every position in the reference CT file (whether paired or unpaired) and reports whether that position is similarly paired or unpaired in the target CT file. The positive predictive value (PPV; Equation 4) and sensitivity (Equation 5) between varying models were generated using the script ct_sensitivity_ppv.py, which is based off of the RNAstructure’s (48–50) scorer script (modified to allow any size CT files) and based on the given comparison will treat one model as the ‘predicted model’ and the other as the ‘known model’.
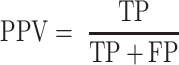 |
(4) |
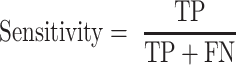 |
(5) |
Z-score binning analyses
To characterize how well negative and positive ΔG° z-score regions predicted by ScanFold agree with SHAPE and DMS informed models, we performed an analysis where each nt position of the SARS-CoV-2 genome was binned based on its in silicoScanFold average nt z-score (Zavg) value (from < –2 to > +2 at intervals of 1) and then cross referenced to positions of structural conflict that exist between in silico (i.e. unconstrained with standard parameters) ScanFold models and SHAPE/DMS informed global models of Manfredonia et al.. The ct_compare.py script (mentioned above) generates and outputs a list of all the conflicting position between two alternative model CT files generated for the same input sequence. ScanFold models, generated at varying window sizes (120, 200, 300, 400, 500, 600), were each compared to the SHAPE in vitro, SHAPE in vivo, and DMS in vitro informed global models proposed by Manfredonia et al. and the SHAPE in vivo global model from Huston et al. The Zavg is a ScanFold calculated metric found in the final partners output file from the ScanFold-Scan analysis (Dataset S1). Using the zscore_conflict_analyzer.py script, the in silicoScanFoldZavg values (at window sizes of 120, 200, 300, 400, 500, 600) were cross referenced to the list of conflicting nts, reported in the ct_compare.py output, for the various model comparisons. The resulting output shows the percent agreement between each Zavg bin and the number of positions present in each bin.
Data availability
All Datasets (S1-S3) associated with this study are available at: https://structurome.bb.iastate.edu/sars-cov-2. Python scripts used in analyses can be found at https://github.com/moss-lab/SARS-CoV-2.
RESULTS AND DISCUSSION
Global assessment of structural propensity in the SARS-CoV-2 genome
Thermodynamics-based RNA folding algorithms (e.g. Mfold (51–54), RNAstructure (50,55–56), and RNAfold (35,57)) utilize experimentally derived parameters to approximate the free energy of formation (ΔG°) for a given RNA secondary structure. Traditionally, these algorithms have been used to find the most stable secondary structure that a sequence can form (i.e. the structure with the most minimum free energy of formation or MFE) in the hopes that this captures the true structure. Due to several limitations (e.g. molecular crowding in vivo, trans-factor interactions, multiple conformations of folding and the inability to natively account for tertiary structures such as pseudoknots), the MFE is not always a reliable method for predicting exactly how an RNA will fold in the cell. The algorithm is, however, able to accurately approximate the folding energy of a sequence, as the ΔG° of the true structure does tend to be fairly close to the predicted MFE ΔG°: for many RNAs there is a < 5% difference between the ΔG° of the true secondary structure and the predicted MFE ΔG° (36). So, even though the predicted structure may be imprecise, the predicted thermodynamics are robust and can be reliable metrics for characterizing the local thermodynamic properties of RNA (36). Informed by these limitations, ScanFold was not explicitly designed to predict global RNA secondary structures, instead, ScanFold utilizes thermodynamic values to detect any local RNA structural elements with unusual stability (with an emphasis on analyzing large sequences such as viral genomes (26–27,58)). In the first step, a sequence is scanned stepwise using a small analysis window and in the second step, any regions of the sequence contributing to unusual thermodynamics are highlighted and modeled. ScanFold-Scan then, is the first step in this process and performs a high volume of overlapping RNA folding calculations in order to (i) generate a local thermodynamic RNA folding profile and (ii) highlight regions of the sequence yielding particularly interesting structural thermodynamics (Figure 2).
Figure 2.
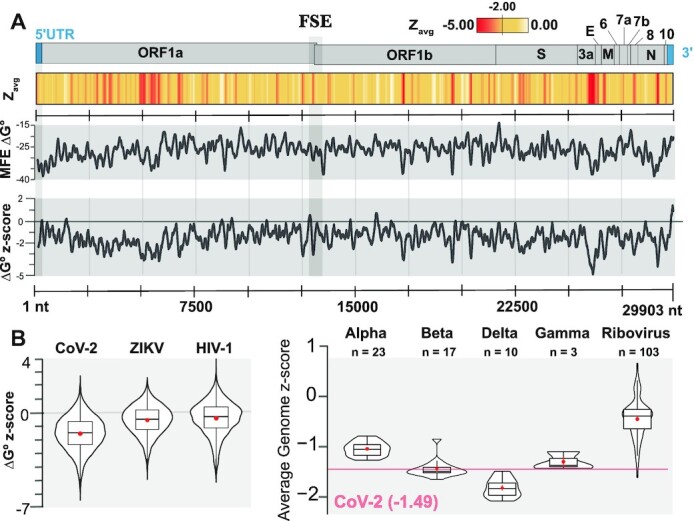
Global results for SARS-CoV-2 and comparisons to other viral genomes. (A) At the top is a cartoon depiction of the SARS-CoV-2 genome with the major regions annotated. Below this is a heatmap of average per nt z-scores (Zavg) with colors ranging from red (–5.00) to white (>=0.00) with yellow set to midrange (–2.00). Next, the MFE ΔG° for each ScanFold analysis window is shown across the genome (black line demarcated every 2500 nts) with values ranging from –47.50 to –8.80 (kcal/mol); here, a moving average of MFE values calculated across 120 nts is shown. Further down is a depiction of the ΔG° z-score for each ScanFold analysis window across the genome and values range from –6.40 to +2.74 with an average of –1.49; here, a moving average of ΔG° z-scores calculated across 120 nts is shown. Finally, there is a positional track from 1 to 29 903 nts with markers spaced 2500 nts apart. Just past the 12500 mark, there is a region shaded with a light gray box that represents the location of the frameshift stimulatory element (FSE). (B) On the left, violin plots depicting the distribution of ΔG° z-scores for ScanFold analysis windows are shown for SARS-CoV-2, ZIKV, and HIV-1. On the right, violin plots depicting the average genome ΔG° z-scores for all NCBI Coronaviridae reference genomes, along with the NCBI reference sequences for all human ribovirus genomes for comparison (genomes accessed from NCBI on 20 March 2020). The number of genomes included in the analysis is shown above each plot. The red line represents the average genome z-score, –1.49, of SARS-CoV-2.
An initial ScanFold-Scan was conducted on the SARS-CoV-2 genome (using the previously optimized parameters of a 120 nt window and single nt step size (26–27,59); details in Materials and Methods) resulting in 29784 almost fully overlapping analysis windows spanning the genome (full results at https://structurome.bb.iastate.edu/sars-cov-2 and in Supplementary Table S1). An overview of the ScanFold-Scan results is given in Figure 2A. The predicted MFE across all windows ranged from –8.8 to –47.5 and averaged –26.1 kcal/mol (somewhat more stable than expected for this window size with a 37% GC-content genome (60)). The key metric calculated by ScanFold-Scan, however, is the thermodynamic z-score, which compares the native sequence's predicted MFE to that of matched randomized samples with the same nt content: here, a negative value indicates the number of standard deviations more stable than random a native sequence is. This unusual stability can be taken to indicate the sequence is ordered (potentially by evolution) to have a functionally significant sequence/structure relationship that is disturbed upon shuffling. The z-scores across the genome ranged from −6.40 to +2.74 and yielded an average of −1.50 with local regions of highly negative z-scores (< –2) and stable MFE values found throughout the entirety of the genome (Figure 2A).
Positive z-score regions can be seen throughout the genome as well, but are less frequent and smaller in size. A previous analysis (26) found that such regions were more likely to be reactive to structure probing molecules (i.e. suggesting they are unstructured or highly dynamic)—potentially to facilitate intermolecular or long-range intra-genomic interactions. For example, of the 118 windows overlapping the start codon of ORF1a, 70 windows (60%) had positive z-scores suggesting a preference for weak structures localizing around the start codon; consistent with previous analyses of RNA folding near start codons (29,61–62). Another notable region is the 3′ UTR, which was found to yield mostly positive z-scores. Given the high GC content for this region (0.45 on average; Supplementary Table S1), MFE values here were less stable than expected, averaging z-scores of +0.98 (or roughly one standard deviation less stable than random).
Globally, however, one of the striking results of our analysis was the extreme overall shift in the global z-score values for the SARS-CoV-2 genome. The mean z-score for all windows was –1.50 with 88.9% of the analysis windows being negative. For comparison, this value is one standard deviation more stable on average than the previously scanned RNA genomes (e.g. in (27,58)) of HIV-1 and ZIKV which had average z-scores of −0.45 and −0.55, respectively. In each case, the z-scores are normally distributed around a negative median value (Figure 2B) however, SARS-CoV-2 is sufficiently shifted into the negative to be classified as having globally ordered RNA structure (63–66). This unusual propensity for ordered RNA structure appears to be a particular feature of coronaviruses: all clades were significantly more negative than 103 other Riboviria RefSeq genomes with the delta and beta clades having the greatest negative shift (Figure 2B).
While low z-score windows and their associated minimum free energy secondary structure predictions suggest functional regions and models, an innovation of ScanFold is its -Fold module, which uses the scanning window data to generate unique z-score weighted consensus secondary structure models. These models are comprised of base pairs that most contribute to unusual thermodynamic stability of low z-score windows and are presumably the key interactions in functional RNA structural motifs. This approach was successfully able to deduce known and novel motifs in other viral genomes (27–28,58) and, additionally, preliminary work on SARS-CoV-2 was able to model and home in on key structural elements within the FSE (Figure 1) that could be targeted with small molecules to disrupt –1 PRF (16). This highlights a key benefit of the ScanFold-Fold analysis, which can not only be useful for generating models of local structure, but can also rapidly deduce sub-domains of larger RNA elements with particular indications of functionality. This is key for the functional annotation of an RNA as large as the SARS-CoV-2 genome, which is predicted to contain multiple large structural domains (67).
Evaluation of available experimental data
The SARS-CoV-2 genome has been under intense study and several high-quality experimental RNA structure probing datasets are available for comparison to ScanFold results (17–20). The agreement of each experimental dataset with respect to the in silicoScanFold-Fold models (z-score weighed consensus folds across all windows) was evaluated using a receiver-operating characteristic (ROC) analysis (Figure 3). Here, the effects of increasing the stringency of reactivity cutoffs, which consider a site to be paired in the model, provides a measure of the consistency of probing data with regard to ScanFold models (see Material and Methods). After calculating the AUC for each set of results, all were found to be above 0.5, indicating global consistency of the data with ScanFold results. AUC values ranged from a minimum value of 0.629 from an in vivo SHAPE dataset (Huston et al.) to a maximum value of 0.783 for a in vivo DMS dataset (Lan et al.). No trends were apparent in comparing AUC’s between DMS and SHAPE results; for example, the second highest AUC (0.756) was for the in vivo SHAPE data of Sun et al. Likewise, no large differences were observed comparing in vitro to in vivo datasets: e.g. the Manfredonia et al. SHAPE data yielded AUCs of 0.689 and 0.675 for in vitro and in vivo results, respectively. These findings indicate that ScanFold is detecting the most robust local elements that do not vary between experimental conditions.
Figure 3.
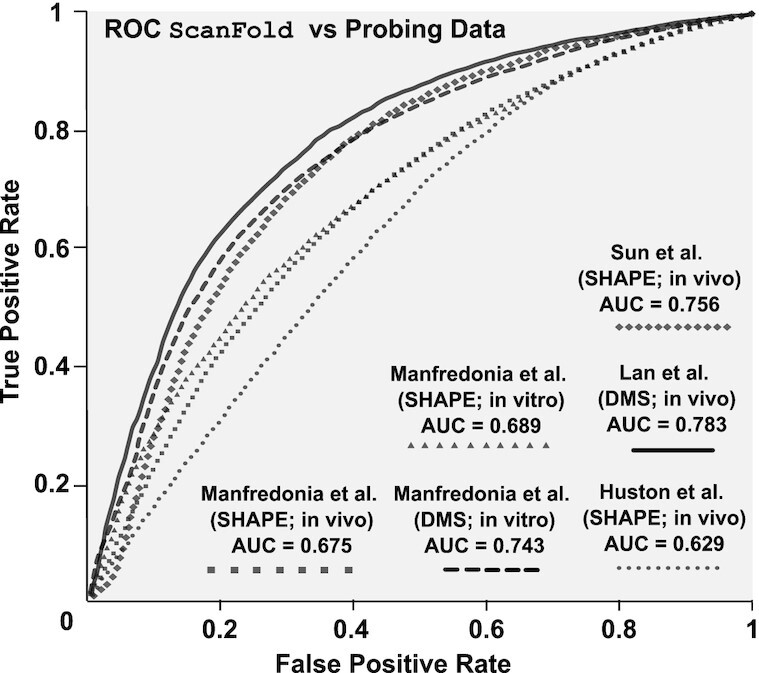
Comparisons of ScanFold vs. experimental data. Receiver-operating characteristic (ROC) analysis of the in silico (at a 120 nt analysis window) ScanFold-Fold predicted base pair structure of SARS-CoV-2 against SHAPE and DMS reactivity data sets generated from SARS-CoV-2 probing experiments. Reactivities are progressively evaluated from the lowest reactivity values to the highest, at intervals of 1% of the total number of reactivity values (see Materials and Methods) and compared to the ScanFold predicted secondary structure yielding a true positive rate (y axis) and a false positive rate (x axis). Progressively increasing reactivity thresholds have their respective TPR and FPR plotted from 0% (coordinate (0,0)) to 100% (coordinate (1,1)) and each respective dataset is indicated by a line with a unique marker (see figure legend). The area under the curve (AUC) is calculated for each curve (listed in the figure legend and Supplementary Table S6) and is an indication of how well the reactivity datasets agree with the in silicoScanFold-Fold predicted structure.
To allow users to integrate experimental results directly into ScanFold calculations, the program has been modified to accept both hard and soft (pseudoenergy) constraints (see Materials and Methods). To compare the behaviors of available experimental datasets with in silicoScanFold, each one was incorporated as soft constraints during ScanFold-Scan steps (for an analysis of the behavior of hard constraints in ScanFold and the effects of larger window sizes, please see the Supplementary Results and Discussion). Predictions were made for all overlapping analysis windows with the predicted folding energy being informed by their chemical reactivity: i.e. highly reactive bases were biased to be unpaired. Inclusion of these data led to varying effects on predicted structure (Table 1 and Supplementary Table S2); however, after ScanFold-Fold model building, a core set of 10702 base pairs remained invariant between all models (Supplementary Table S3). Significantly, the majority (69%) of these common base pairs are predicted by in silicoScanFold-Fold alone, which is consistent with our previous analyses of other viruses (26,27) that showed ScanFold-Fold models of low z-score regions robustly predicted highly structured elements (Supplementary Results and Discussion). Significantly, in SARS-CoV-2, we found that the z-score metric was largely unaffected by probing data and that significantly low z-score motifs are in high agreement with reactivity-informed models (Figure 4A, Dataset S1), we thusly use the in silico only ScanFold results for our subsequent analyses. Furthermore, with our focus on smaller structural elements, which facilitate analyses of druggability, structure/function assays, and biophysical studies, we elected to use results from 120 nt scanning windows (as opposed to larger window sizes) because of their lower false positive rate (Figure 4B; Supplementary Results and Discussion).
Table 1.
Sensitivity and PPV from comparisons of SARS-CoV-2 secondary structure models. PPV values (Equation 4) are shown in the bottom right and sensitivity values (Equation 5) are shown in the top left.
| Sensitivity | |||||||
| D-SF | 0.59 | 0.62 | 0.60 | 0.56 | 0.85 | 0.83 | - |
| SC-vitro-SF | 0.58 | 0.62 | 0.62 | 0.55 | - | 0.79 | |
| SC-vivo-SF | 0.58 | 0.64 | 0.60 | 0.54 | - | 0.80 | |
| M-DMS-vitro | 0.61 | 0.76 | 0.80 | - | 0.71 | 0.71 | 0.69 |
| M-SHAPE-vitro | 0.61 | 0.81 | - | 0.74 | 0.73 | 0.74 | 0.69 |
| M-SHAPE-vivo | 0.63 | - | 0.79 | 0.69 | 0.76 | 0.73 | 0.70 |
| H-SHAPE-vivo | - | 0.67 | 0.64 | 0.59 | 0.74 | 0.74 | 0.71 |
| D; Default (i.e. standard) | H-SHAPE-vivo | M-SHAPE-vivo | M-SHAPE-vitro | M-DMS-vitro | SC-vivo-SF | SC-vitro-SF | D-SF |
| SF; ScanFold | |||||||
| SC; Soft constraints | |||||||
| M; Manfredonia | |||||||
| H; Huston | |||||||
| PPV | |||||||
Figure 4.
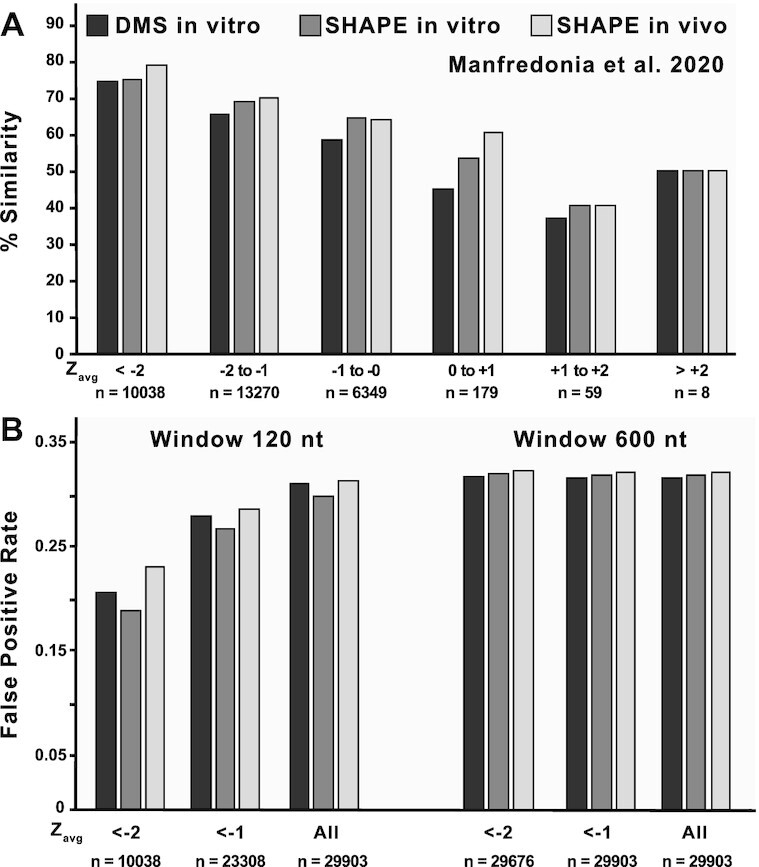
Comparisons of in silicoScanFoldZavg values against three different reactivity-informed secondary structural models of SARS-CoV-2. (A) In silicoScanFold Zavg values were binned based on their magnitude from < -2 to > +2 at intervals of 1 and are labelled across the x axis along with the number of values that are present in each bin. The positions corresponding to each Zavg value were cross referenced between the in silicoScanFold predicted secondary structure of SARS-CoV-2 and the three model conditions proposed by Manfredonia et al. (DMS in vitro, black shading; SHAPE in vitro, dark gray shading; SHAPE in vivo, light gray shading) to calculate a percent similarity which is plotted on the y axis. Across all three model conditions, the lowest Zavg bins consistently have the highest similarity to the reactivity informed global models. (B) The < -1 and < -2 binned Zavg values for in silicoScanFold models of SARS-CoV-2, at both a 120 and 600 nt analysis window, were compared to the three separate models from Manfredonia et al. and a false positive rate (FPR) was calculated. The Zavg bins are labelled across the x axis along with the number of nt positions associated with each bin and the FPR is plotted along the y axis. For the 120nt scanning analysis window, the most negative Zavg bin (i.e. < -2) had the lowest FPR compared to the < -1 Zavg bin and the All Zavg bin (which had the highest FPR). The distribution of Zavg values for the ScanFold model utilizing a 600 nt analysis window were significantly shifted to be more negative, resulting in almost all (>99%) of the Zavg values to be in the < -2 bin, therefore there is little variation in the FPR for these Zavg bins and the FPR in all bins are higher compared to the < -2 bin utilizing the 120 nt analysis window.
Identification of local motifs with high likelihood of functionality
ScanFold-Fold identified 524 uniquely-stable structures (with at least one Zavg < –1 bp); approximately one ordered structural element every 57 nt (Dataset S2). Here, we (i) determine if any of these locally stable structures have evidence of conservation in other Coronaviridae genomes (ii) report which of these structures (if any) are present in known structural regions or (iii) have been recently reported as significant by other groups.
These 524 locally stable structures (as well as the ScanFold model of the FSE; Figure 1) were assessed for evidence of statistically significant structure-preserving sequence covariations (see Material and Methods). Briefly, the ScanFold model was used to build a structural covariation model (cm) with Infernal (42,43), drawing from over 25000 Coronaviridae sequences in the ViPR database (44,45). If a covariation model was successfully constructed, the resulting structural/sequence alignment (in Stockholm format) was tested for significantly covarying base pairs via R-scape’s G-test (31). We found that Infernal was able to create covariation models for 355 of the structures. Of these, we found that 57 of the tested ScanFold structures (and the FSE model) contained at least one pair with significant evidence of conservation (Dataset S3 and https://structurome.bb.iastate.edu/sars-cov-2-structure-extracts). This is in line with Manfredonia et al. results, which found ∼10% of their regions identified as highly structured had evidence of specific base pair conservation (18).
Identification of previously described elements
The full ScanFold-Scan results for the 5′ UTR can be seen in Figure 5A. ScanFold-Fold modeled four of the known stem loops in the 5′ UTR leader region with z-scores < -2. Only one of these (SL4) had evidence of specific base pair conservation. Downstream of this, we find that the start codon has been modeled as unpaired, as opposed to experimental/conservation models which place the start codon within a large multibranch structure (known as SL5) (1,68–69). As reported above, the scanning data around the start codon resulted in positive ΔG° z-scores, which in this case favor the formation of a small hairpin where the 5′ end of the SL5 basal stem would form, keeping the start codon nucleotides unpaired (Figure 5B). Since the basal stem base pairs span >120 nt (the window size used), we would not expect ScanFold to identify it. This stem can, however, be predicted using larger analysis window sizes; https://structurome.bb.iastate.edu/sars-cov-2-global-model-comparisons). The ScanFold model then, leaves 75% of the basal stem nucleotides unpaired, indicating that these predicted local folds may not strongly compete against formation of the larger stem. Further, though the basal stem of SL5 is not present in the ScanFold model, the terminal stem loops are found to be uniquely stable (SL5a-c) and are modeled consistently with recent models of SL5 (68,69), which supports a recent finding that these structures are the most structured portions of SL5 (i.e. are the most sensitive to cleavage by RNAse V1 (70)). We also found that SL5a had three significantly covarying base pairs indicating its particular structural importance.
Figure 5.

Full analysis of the 5′ UTR of SARS-CoV-2. (A) The results of the full ScanFold pipeline are shown. ScanFold metrics and base pairs have been loaded into the IGV desktop browser (91). Metric type and ranges are shown on the left side of the panel (metric descriptions can be found in Material and Methods). Here the start codon has been highlighted with a green bar and structures which correspond to previously named elements have been annotated. (B) ScanFold RNA 2D structures are shown for the 5′ UTR. All base pairs shown are consistent between SARS-CoV and SARS-CoV-2, and nucleotide variations which are present within structures have been highlighted with green circles. Structures have been visualized here using VARNA (92). Top 10% of reactivities are shown for Manfredoina et al. (squares, pentagons and diamonds), Huston et al. (circles), Sun et al. (stars) and Lan et al. (triangles) at their corresponding nt positions (17–20).
The FSE is an RNA structural motif which incorporates nucleotides from the overlapping reading frames of ORF1a and ORF1b (nt 13476–13542). The FSE falls within a low z-score region and the base pairs which correspond to these negative values are shown in the ScanFold-Fold model (Figure 1). The ScanFold-Fold model of the FSE is largely consistent with recent models (68,71); consisting of two stable hairpins—the first of which contains a loop sequence that forms the proposed pseudoknot (13–14,72–73) by pairing with nucleotides upstream of the second hairpin (Figure 1). We also found that this stem was highly conserved, having four base pairs with evidence of covariation. ScanFold cannot predict the pseudoknot directly, however, the generated model does leave the pseudoknot forming nucleotides sufficiently unpaired to allow for the interaction to occur. Comparing the non-pseudoknotted base pairs predicted by ScanFold to two models built using cryo-EM data (one for ribosome-bound RNA (74) and one for free RNA (14), we find that ScanFold predicts only slightly different helixes from either other model (Figure 1). Specifically, the ribosome-bound model did not contain the two closing base pairs of the Stem 1 terminal loop, while both the ribosome-bound and free RNA models did not have the two basal pairs predicted by ScanFold in Stem 3 (both had G13503 base paired to C13476 to extend Stem 1). Interestingly, mutations converting G13486 to an A (consistent with base pairing to U) or C (inconsistent with base pairing to U) both significantly reduced frameshifting in an in vitro assay (73). This supports the ribosome bound model of these bases occurring in a loop where G13486 is proposed to be flipped out to make contacts with the ribosome. It is, however, possible for this base flipping to be stimulated by interactions with the ribosome; in both our model and that of the free RNA, these nts are modeled as forming stable base pairs.
Functional elements upstream of these hairpins are placed into an alternative model by ScanFold. Here, the attenuator hairpin is embedded in a multibranched structure along with the slippery sequence, which is predicted to form a small three base pair stem. Indeed, the only bps which had average z-scores < –2 for the FSE region are found in the basal stem of this previously unreported multibranched structure. In support of this model, both the attenuator hairpin and the basal stem of this upstream structure were found to have evidence of specific base pair conservation (Figure 1). These findings (along with several other reports (17,19,75)) suggest the full frameshift element may incorporate more upstream nucleotides than previously described. Notably, this ScanFold attenuator hairpin model has been used to identify small molecules which specifically bind its UU internal loop (Figure 1) and inhibit –1 PRF (76).
The 3′ UTRs of Sarbecovirus genomes contain three RNA structural elements; a 3′ UTR pseudoknot structure, presumably required for replication (77), a bulged stem loop (BSL), and a mobile genetic element with an undetermined function known as the 3′ stem–loop II-like motif (s2m) embedded in a hypervariable region (HVR) (78). Under the current genome annotation (NC_045512.2) much of the previous 3′ UTR sequence is now found within an upstream open reading frame named ORF10 (however, this has been recently reported as being an untranslated ORF (79)). ScanFold’s model partially recapitulates a recent model of the region (68) (blue pairs; Supplementary Figure S1A) detecting the BSL and the non-pseudoknotted pairs of the pseudoknot stem (though this pseudoknot has recently been called into question (17,20,75)). The overall metrics for the region however, including high ensemble diversity values and positive ΔG° z-scores (Supplementary Table S1) suggest the region is locally unstructured and/or highly dynamic. Indeed, while portions are structurally conserved (18,20) much of the 3′ UTR has been shown to form long range interactions beyond the BSL and pseudoknot structures (75). As such, the ScanFold model predicts the downstream region (composed of the HVR and s2m) to be mostly absent of locally structured elements.
Identification of recently reported structures
Manfredonia et al. predicted 87 high-confidence structured regions based on their in vitro SHAPE-derived modelling (defined as having low Shannon entropy and lower than average SHAPE reactivity (18)). ScanFold-predicted motifs with Zavg < –1 correlated with all but one of these structures (one was correctly modeled by ScanFold, but with Zavg values above –1). Of these 86 correlated motifs, 34 of them were identical between ScanFold and Manfredonia et al. (Supplementary Table S4). Most of the disagreements between models were simply due to ScanFold’s smaller base pair span (eleven of the Manfredonia et al. structures were larger than 120 nt) and only one ScanFold structure was completely different (nt 652–723). However, even larger structures that could not be fully predicted with smaller window sizes were composed of multiple ScanFold structures (e.g. the region spanning nt 7923 to 8127 and the region spanning 23969 to 24097; Supplementary Figure S2A). Other disagreements arose due to ScanFold predicting structures which were simply larger (e.g. the regions spanning nt 8392–8428 and 6260–6320; Supplementary Figure S2B). Despite select disagreements, ScanFold’s ability to model these experimentally derived structured regions (18) averaged a PPV (Equation 4) and sensitivity (Equation 5) of 0.90 and 0.85, respectively (Supplementary Table S4). Huston et al. also reported several well folded regions (defined similarly to Manfredonia et al. as having low Shannon entropy and low SHAPE reactivity) throughout ORF1a/b (17). The well-folded regions here were defined to encompass more nucleotides than Manfredonia et al. averaging 198 nt long (as opposed to 66 nt in Manfredonia et al.Supplementary Table S3). Again, all but one of these regions corresponded to Zavg < –1 ScanFold structures. However, in this case the overall similarity to ScanFold models was somewhat lower with a sensitivity and PPV of 0.71 and 0.82 respectively (Supplementary Table S4).
Structured region conservation
The structured regions defined in this study, and others, were all tested for evidence of specific base pair conservation (by analyzing their respective sequence alignments using R-scape). Evidence of structural conservation suggests that base pairing is being maintained; presumably because it is evolutionarily advantageous (e.g. serves some functional role). Of the 524 ScanFold motifs, 57 had evidence of statistically significant conservation. Several groups have reported conservation of RNA secondary structures in SARS-CoV-2 (20,22) and here we compare our findings to two of them (17,18). The workflow we used to detect conservation was first laid out in Manfredonia et al., where Infernal(42,43) and R-scape (30,31) were used to find that 8 of their 87 structured regions had evidence of conservation (18). A similar approach was used in Huston et al (which looked for conservation within the Betacoronaviridae clade alone) which found 3 of their 40 well-folded regions had evidence of structural conservation (17). Each method found that ∼10% of the independently defined structured regions had evidence of structural conservation and, surprisingly, there is little overlap between these structures. Of the 57 conserved motifs detected by ScanFold, for example, only two were found to overlap any other group: motif-5 (i.e. SL5a) from the 5′ UTR and motif-491 (nt 28066–28118) in ORF8. Between Manfredonia et al. and Huston et al., only one conserved structure was shared (embedded in ORF1a from nt 8144 to 8220).
Evidence of structural conservation via R-scape can be further evaluated by considering the power of the respective alignment (i.e. how many base pairs would be expected to covary given the amount of sequence variation observed in the alignment) (30). Using this, we can determine how many motifs have evidence that they are not under evolutionary pressure because the alignment had enough variation to detect conservation but failed to do so. Of the 298 structures with no evidence of conservation, 56 were expected to detect at least one pair and, significantly, 39 were expected to detect more than one covarying pair but failed to do so (Supplementary Table S5). So, while most motifs lacking evidence of conservation simply lacked alignments powerful enough to detect it, these 39 (or ∼7% of) ScanFold-predicted structured motifs have evidence for not being under evolutionary pressure to maintain their structure (30) throughout Coronaviridae genomes (Supplementary Table S5).
Identification of novel structural motifs in SARS-CoV-2
ScanFold identified 53 uniquely stable and potentially conserved structures (Supplementary Table S5) which have yet to be fully characterized. We further filtered this list to only structures with more than one covarying base; ultimately homing in on nine structures (one was the previously reported: SL5a from the 5′ UTR); the ScanFold-Fold models for these eight remaining motifs are shown in Figure 6. These represent higher priority targets for additional analyses and their identification illustrates the ability of the in silicoScanFold pipeline to rapidly deduce local high-value motifs. Notably, these motifs are invariant with inclusion of experimental constraints (Supplementary Table S3): e.g. models in Figure 6 are annotated with highly reactive sites from six RNA probing datasets (17–19). It is important to note that, even for elements predicted without statistically significant covariation, the ScanFold method calculates metrics that can help prioritize motifs: e.g. by low z-scores and ED values.
Figure 6.

ScanFold predicted motifs annotated with conservation and probing data. Nucleotides are colored based on the Zavg value predicted in the standard ScanFold run. Particularly interesting base pairs which were identified in CaCoFold models are shown in blue. Significantly covarying bps (R-scape APC corrected G-test; E<0.05) have been highlighted with a green box. Nucleotide coordinates are relative to the SARS-CoV-2 reference genome NC_045512.2. Top 10% of reactivities are shown for Manfredoina et al. (squares, pentagons and diamonds), Huston et al., (circles), Sun et al. (stars) and Lan et al. (triangles) at their corresponding nt positions (17–20).
One potential source of modeling error was addressed in the eight high value motifs: the tendency of ScanFold-Fold consensus structures to model ambiguously paired nt as single stranded in the consensus fold (because no pairing arrangements dominated). This issue was addressed by refolding these motifs using the CaCoFold algorithm (32), where the input were the sequence alignments generated for analysis of covariation (see Materials and Methods). CaCoFold generates folding models based on the evolutionary signal of RNA structure conservation (i.e. base pairs are selected/removed based on positive or negative covariation signals). Models built using this orthogonal approach recapitulate the ScanFold-Fold pairs (Figure 6; Dataset S3), while ‘filling in’ potentially artifactually missed pairs (due to the consensus modelling of recurring low z-score base pairs)—indeed, CaCoFold is even able to suggest potential non-Watson–Crick base pairs. For example, the ScanFold model for motif-56 predicts a large (18 nt) terminal loop, which CaCoFold models as ‘zipped up’ into a pentaloop that is stabilized by three consecutive non-Watson–Crick base pairs (two CA pairs followed by a GU ‘wobble’ pair) flanked by canonical AU pairs (Figure 6). A single, highly reactive site was identified by both the Huston et al. (SHAPE in vivo) and Manfredonia et al. (SHAPE in vitro) datasets, which occurs on the U of the GU wobble pair (with two more reactive sites identified in the CaCoFold predicted pentaloop). Indeed, when all available experimental data are assessed vs these model structures, they largely support the ScanFold-Fold, and CaCoFold, predictions (top 10% of reactivities from probing data sets; Figure 6).
These eight motifs can serve as the starting point for biological hypothesis generation or for therapeutic targeting (e.g. for small-molecule degraders of RNA (80–83)). For example Sun et al. recently targeted motif-522 (Figure 6; nt 29504 to 29539) with an antisense oligonucleotide (ASO) which resulted in decreased viral infection in cells (75); two other structures were successfully targeted as well, both of which were identified in our set of 524 structured motifs (motif-134 which had no covarying pairs and 179 which had one covarying pair) showing that even when lacking covariation support ScanFold-predicted motifs may serve as potential targets for ASOs. The remaining 7 motifs are scattered throughout the genome. Several motifs are found in ORF1ab: two are found in relatively close proximity to each other around nt 3000 in (motif-56 and -58), suggesting this area could benefit from extra scrutiny; one is relatively small and found around nt 6300 (Figure 6; motif-132) but is part of a larger structured region identified by Huston et al.; the only other structure (besides the known structures in FSE and 5′ UTR) with three covarying bps is found around nt 9050 and has a large internal loop with lower Zavg values (Figure 6 motif-174); the largest motif is found around nt 12 100 and is riddled with bulges and internal loops, with covariation occurring in the lowest Zavg nts suggesting the stable and conserved basal portion of the structure may have been preserved to support an otherwise unstable structure (Figure 6; motif-219). The remaining three structures are found within the last 3000 nts of the genome: motif-456 is embedded in the E protein's relatively short CDS; motif-479 is found just downstream of ORF7a's start codon; motif-522 has already been successfully targeted with an ASO in cells (20) and is found directly overlapping the N protein's stop codon.
ScanFold-Fold motifs can facilitate the study of larger structural domains in SARS-CoV-2 by helping to define core elements of particular significance for additional functional studies. For example, one of the eight unusually stable, experimentally supported, and conserved motifs (Figure 5B) occurs within the 5′ UTR of SARS-CoV-2 and is part of a larger structural domain (18–19,22,67,84); this motif occurs as a named element (SL5a) within the global 5′ UTR model. Our results highlight this as a likely key structural and functional element within the 5′ UTR. Indeed, only in the FSE pseudoknot-presenting hairpin, did we identify a helix with more phylogenetic support (Figure 1).
It is also worth noting another feature of our results that has only begun to be explored. Although the viral genome shows extreme biases in being ordered to form stable RNA secondary structures (Figure 2), interspersed throughout the genome are regions of unusual instability (indicated by their positive thermodynamic z-scores). These regions are particularly notable for their rarity in SARS-CoV-2 and their apparent ordering to not form stable RNA structures suggests potential functions—perhaps in maintaining accessibility for long-range or intermolecular interactions with host and/or viral biomolecules (75,85). The functions of these sites of unusual instability requires additional study and these regions may also prove useful in efforts to combat COVID-19. The interface of RNA-protein interactions could be targeted using small molecule drugs or antisense oligonucleotides (86–89). Unstructured and accessible sites predicted by ScanFold may also facilitate the design of assays and biosensors to detect infection that rely on the recognition/binding of viral RNA (90).
CONCLUSION
With our bioinformatics program, ScanFold, we sought to define the local thermodynamic landscape of the SARS-CoV-2 genome to enumerate well-structured, potentially functional motifs that could serve as ideal targets for RNA targeting therapeutics. The SARS-CoV-2 genome proved to be exceptionally structured (an apparent feature of CoVs), with many highly-negative thermodynamic z-score regions, an indication of ordered stability and functional propensity. In efforts to enhance our structural modeling, ScanFold was updated to allow the inclusion of experimental reactivities as soft constraints during the scanning process. Interestingly, the inclusion of experimental data did not significantly alter the z-score trends across the SARS-CoV-2 genome or affect the final list of high value motifs. This analysis shows that ScanFold can rapidly highlight regions of highly ordered structures and produce models of sufficient quality to serve as guides for additional studies; indeed, structures we highlight have already been successfully targeted with small molecule inhibitors of viral gene regulation (16) and antisense oligonucleotides (20,89). All ScanFold-Scan and ScanFold-Fold results are available and organized on the RNAStructuromeDB (https://structurome.bb.iastate.edu/sars-cov-2) to maximize their utility in future efforts to understand the roles of RNA folding in this unusually structured RNA virus and, hopefully, to develop novel RNA-targeting therapeutics.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Silvi Rouskin for facilitating the analysis in this paper by sharing the raw DMS reactivity values ahead of their publication. We also thank the Roy J. Carver Charitable Trust for their support.
Contributor Information
Ryan J Andrews, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
Collin A O’Leary, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
Van S Tompkins, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
Jake M Peterson, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
Hafeez S Haniff, Department of Chemistry, The Scripps Research Institute, Jupiter, FL 33458, USA.
Christopher Williams, Department of Chemistry, The Scripps Research Institute, Jupiter, FL 33458, USA.
Matthew D Disney, Department of Chemistry, The Scripps Research Institute, Jupiter, FL 33458, USA.
Walter N Moss, Roy J. Carver Department of Biophysics, Biochemistry and Molecular Biology, Iowa State University, Ames, IA 50011, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
NIH [R00GM112877, R01GM133810] to WNM and NIH [R01CA249180, R35NS116846] to MDD.
Conflict of interest statement. The authors declare the following competing financial interest(s): Matthew D. Disney is a founder of Expansion Therapeutics.
REFERENCES
- 1. Yang D., Leibowitz J.L.. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015; 206:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Madhugiri R., Fricke M., Marz M., Ziebuhr J.. Ziebuhr J. Advances in Virus Research. 2016; 96:Academic Press; 127–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kalvari I., Argasinska J., Quinones-Olvera N., Nawrocki E.P., Rivas E., Eddy S.R., Bateman A., Finn R.D., Petrov A.I.. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018; 46:D335–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nawrocki E.P., Burge S.W., Bateman A., Daub J., Eberhardt R.Y., Eddy S.R., Floden E.W., Gardner P.P., Jones T.A., Tate J.et al.. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015; 43:D130–D137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burge S.W., Daub J., Eberhardt R., Tate J., Barquist L., Nawrocki E.P., Eddy S.R., Gardner P.P., Bateman A.. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013; 41:D226–D232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lessa F.A., Raiol T., Brigido M.M., Neto D.S.B.M., Walter M.E.M.T., Stadler P.F. Clustering Rfam 10.1: clans, families, and classes. Genes-Basel. 2012; 3:378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gardner P.P., Daub J., Tate J.G., Nawrocki E.P., Kolbe D.L., Lindgreen S., Wilkinson A.C., Finn R.D., Griffiths-Jones S., Eddy S.R.et al.. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009; 37:D136–D140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griffiths-Jones S., Moxon S., Marshall M., Khanna A., Eddy S.R., Bateman A.. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005; 33:D121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Griffiths-Jones S. Annotating non-coding RNAs with Rfam. Curr. Protoc. Bioinformatics. 2005; doi:10.1002/0471250953.bi1205s9. [DOI] [PubMed] [Google Scholar]
- 10. Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R.. Rfam: an RNA family database. Nucleic Acids Res. 2003; 31:439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Plant E.P., Perez-Alvarado G.C., Jacobs J.L., Mukhopadhyay B., Hennig M., Dinman J.D.. A three-stemmed mRNA pseudoknot in the SARS coronavirus frameshift signal. PLoS Biol. 2005; 3:e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park S.-J., Kim Y.-G., Park H.-J.. Identification of RNA Pseudoknot-Binding Ligand That Inhibits the −1 Ribosomal Frameshifting of SARS-Coronavirus by Structure-Based Virtual Screening. J. Am. Chem. Soc. 2011; 133:10094–10100. [DOI] [PubMed] [Google Scholar]
- 13. Kelly J.A., Olson A.N., Neupane K., Munshi S., San Emeterio J., Pollack L., Woodside M.T., Dinman J.D.. Structural and functional conservation of the programmed -1 ribosomal frameshift signal of SARS coronavirus 2 (SARS-CoV-2). J Biol Chem. 2020; 295:10741–10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang K., Zheludev I.N., Hagey R.J., Wu M.T., Haslecker R., Hou Y.J., Kretsch R., Pintilie G.D., Rangan R., Kladwang W.et al.. Cryo-electron microscopy and exploratory antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome. 2020; bioRxiv doi:20 July 2020, preprint: not peer reviewed 10.1101/2020.07.18.209270. [DOI] [PMC free article] [PubMed]
- 15. Andrews R.J., Peterson M.P., Haniff H.S., Chen J.L., Williams C., Grefe M., Disney M.D., Moss W.N.. An in silico map of the SARS-CoV-2 RNA structurome. 2020; bioRxiv doi:18 April 2020, preprint: not peer reviewed 10.1101/2020.04.17.045161. [DOI] [PMC free article] [PubMed]
- 16. Haniff H.S., Tong Y., Liu X., Chen J.L., Suresh B.M., Andrews R.J., Peterson J.M., O’Leary C.A., Benhamou R.I., Moss W.N.et al.. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Central Sci. 2020; 6:1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huston N.C., Wan H., Strine M.S., de Cesaris Araujo Tavares R., Wilen C.B., Pyle A.M.. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell. 2021; 81:584–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manfredonia I., Nithin C., Ponce-Salvatierra A., Ghosh P., Wirecki T.K., Marinus T., Ogando N.S., Snijder E.J., van Hemert M.J., Bujnicki J.M.et al.. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically-relevant elements. Nucleic Acids Res. 2020; 48:12436–12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lan T.C.T., Allan M.F., Malsick L.E., Khandwala S., Nyeo S.S.Y., Bathe M., Griffiths A., Rouskin S. Structure of the full SARS-CoV-2 RNA genome in infected cells. 2020; bioRxiv doi:19 February 2021, preprint: not peer reviewed 10.1101/2020.06.29.178343. [DOI]
- 20. Sun L., Li P., Ju X., Rao J., Huang W., Ren L., Zhang S., Xiong T., Xu K., Zhou X.et al.. In vivo structural characterization of the SARS-CoV-2 RNA genome identifies host proteins vulnerable to repurposed drugs. Cell. 2021; 184:1865–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitchell D. 3rd, Assmann S.M., Bevilacqua P.C.. Probing RNA structure in vivo. Curr Opin Struct Biol. 2019; 59:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rangan R., Zheludev I.N., Hagey R.J., Pham E.A., Wayment-Steele H.K., Glenn J.S., Das R.. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses: a first look. RNA. 2020; 26:937–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gruber A.R., Findeiss S., Washietl S., Hofacker I.L., Stadler P.F.. RNAz 2.0: improved noncoding RNA detection. Pac. Symp. Biocomput. 2010; 2010:69–79. [PubMed] [Google Scholar]
- 24. Gruber A.R., Neubock R., Hofacker I.L., Washietl S.. The RNAz web server: prediction of thermodynamically stable and evolutionarily conserved RNA structures. Nucleic Acids Res. 2007; 35:W335–W338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Washietl S., Hofacker I.L.. Identifying structural noncoding RNAs using RNAz. Curr. Protoc. Bioinformatics. 2007; Chapter 12:Unit 12 17. [DOI] [PubMed] [Google Scholar]
- 26. Andrews R.J., Baber L., Moss W.N.. Mapping the RNA structural landscape of viral genomes. Methods. 2020; 183:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Andrews R.J., Roche J., Moss W.N.. ScanFold: an approach for genome-wide discovery of local RNA structural elements—applications to Zika virus and HIV. PeerJ. 2018; 6:e6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andrews R.J., Baber L., Moss W.N.. Mapping the RNA structural landscape of viral genomes. Methods. 2019; 183:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O’Leary C.A., Andrews R.J., Tompkins V.S., Chen J.L., Childs-Disney J.L., Disney M.D., Moss W.N.. RNA structural analysis of the MYC mRNA reveals conserved motifs that affect gene expression. PLoS One. 2019; 14:e0213758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rivas E., Clements J., Eddy S.R.. Estimating the power of sequence covariation for detecting conserved RNA structure. Bioinformatics. 2020; 36:3072–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rivas E., Clements J., Eddy S.R.. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat Methods. 2017; 14:45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rivas E. RNA structure prediction using positive and negative evolutionary information. PLOS Computat. Biol. 2020; 16:e1008387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watts J.M., Dang K.K., Gorelick R.J., Leonard C.W., Bess J.W., Swanstrom R., Burch C.L., Weeks K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009; 460:711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huber R.G., Lim X.N., Ng W.C., Sim A.Y.L., Poh H.X., Shen Y., Lim S.Y., Sundstrom K.B., Sun X., Aw J.G.et al.. Structure mapping of dengue and Zika viruses reveals functional long-range interactions. Nat. Commun. 2019; 10:1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mathews D.H., Sabina J., Zuker M., Turner D.H.. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999; 288:911–940. [DOI] [PubMed] [Google Scholar]
- 37. Clote P., Ferre F., Kranakis E., Krizanc D. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA. 2005; 11:578–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCaskill J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990; 29:1105–1119. [DOI] [PubMed] [Google Scholar]
- 39. Lorenz R., Wolfinger M.T., Tanzer A., Hofacker I.L.. Predicting RNA secondary structures from sequence and probing data. Methods. 2016; 103:86–98. [DOI] [PubMed] [Google Scholar]
- 40. Freyhult E., Gardner P.P., Moulton V.. A comparison of RNA folding measures. BMC Bioinformatics. 2005; 6:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Incarnato D., Morandi E., Simon L.M., Oliviero S.. RNA Framework: an all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Res. 2018; 46:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nawrocki E.P., Eddy S.R.. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nawrocki E.P., Kolbe D.L., Eddy S.R.. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009; 25:1335–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pickett B.E., Greer D.S., Zhang Y., Stewart L., Zhou L., Sun G., Gu Z., Kumar S., Zaremba S., Larsen C.N.et al.. Virus pathogen database and analysis resource (ViPR): a comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses. 2012; 4:3209–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pickett B.E., Sadat E.L., Zhang Y., Noronha J.M., Squires R.B., Hunt V., Liu M., Kumar S., Zaremba S., Gu Z.et al.. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012; 40:D593–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deigan K.E., Li T.W., Mathews D.H., Weeks K.M.. Accurate SHAPE-directed RNA structure determination. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zarringhalam K., Meyer M.M., Dotu I., Chuang J.H., Clote P.. Integrating chemical footprinting data into RNA secondary structure prediction. PLoS One. 2012; 7:e45160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Z.Z., Mathews D.H.. Secondary structure prediction of single sequences using RNAstructure. Methods Mol. Biol. 2016; 1490:15–34. [DOI] [PubMed] [Google Scholar]
- 49. Mathews D.H. RNA secondary structure analysis using RNAstructure. Curr. Protoc. Bioinformatics. 2014; 46:doi:10.1002/0471250953.bi1206s46. [DOI] [PubMed] [Google Scholar]
- 50. Mathews D.H. RNA secondary structure analysis using RNAstructure. Curr. Protoc. Bioinformatics. 2006; Chapter 12:Unit 12 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zuker M., Stiegler P.. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981; 9:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Markham N.R., Zuker M.. UNAFold: software for nucleic acid folding and hybridization. Methods Mol. Biol. 2008; 453:3–31. [DOI] [PubMed] [Google Scholar]
- 54. Walter A.E., Turner D.H., Kim J., Lyttle M.H., Muller P., Mathews D.H., Zuker M.. Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:9218–9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bellaousov S., Reuter J.S., Seetin M.G., Mathews D.H.. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2013; 41:W471–W474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Reuter J.S., Mathews D.H.. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010; 11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P.. Fast folding and comparison of Rna secondary structures. Monatsh Chem. 1994; 125:167–188. [Google Scholar]
- 58. Andrews R.J., O’Leary C.A., Moss W.N.. A survey of RNA secondary structural propensity encoded within human herpesvirus genomes: global comparisons and local motifs. PeerJ. 2020; 8:e9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lange S.J., Maticzka D., Mohl M., Gagnon J.N., Brown C.M., Backofen R.. Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012; 40:5215–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Trotta E. On the normalization of the minimum free energy of RNAs by sequence length. PLoS One. 2014; 9:e113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Peeri M., Tuller T.. High-resolution modeling of the selection on local mRNA folding strength in coding sequences across the tree of life. Genome Biol. 2020; 21:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gu W., Zhou T., Wilke C.O.. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput. Biol. 2010; 6:e1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Simmonds P. Pervasive RNA secondary structure in the genomes of SARS-CoV-2 and other coronaviruses. mBio. 2020; 11:e01661-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Simmonds P., Tuplin A., Evans D.J.. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. RNA. 2004; 10:1337–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Davis M., Sagan S.M., Pezacki J.P., Evans D.J., Simmonds P.. Bioinformatic and physical characterizations of genome-scale ordered RNA structure in mammalian RNA viruses. J Virol. 2008; 82:11824–11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Priore S.F., Moss W.N., Turner D.H.. Influenza B virus has global ordered RNA structure in (+) and (−) strands but relatively less stable predicted RNA folding free energy than allowed by the encoded protein sequence. BMC Res Notes. 2013; 6:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huston N.C., Wan H., Tavares Araujo, R.d.C. Wilen, Pyle A.M. Comprehensive in-vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell. 2021; 81:584–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rangan R., Zheludev I.N., Das R.. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses. RNA. 2020; 26:937–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Michael W. Evolutionarily conserved RNA structures in the upstream regions of Wuhan seafood market pneumonia virus (Wuhan-nCoV) and SARS virus. figshare. 2020; doi:10.6084/m9.figshare.11659575.v1. [Google Scholar]
- 70. Miao Z., Tidu A., Eriani G., Martin F.. Secondary structure of the SARS-CoV-2 5′-UTR. RNA Biology. 2021; 18:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kelly J.A., Dinman J.D.. Structural and functional conservation of the programmed -1 ribosomal frameshift signal of SARS-CoV-2. J. Biol. Chem. 2020; 295:10741–10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Omar S.I., Zhao M., Sekar R.V., Moghadam S.A., Tuszynski J.A., Woodside M.T.. Modeling the structure of the frameshift-stimulatory pseudoknot in SARS-CoV-2 reveals multiple possible conformers. PLoS Comput. Biol. 2021; 17:e1008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bhatt P.R., Scaiola A., Loughran G., Leibundgut M., Kratzel A., McMillan A., O’ Connor K.M., Bode J.W., Thiel V., Atkins J.F.et al.. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. 2020; bioRxiv doi:26 October 2020, preprint: not peer reviewed 10.1101/2020.10.26.355099. [DOI] [PMC free article] [PubMed]
- 74. Omar S.I., Zhao M., Sekar R.V., Moghadam S.A., Tuszynski J.A., Woodside M.T.. Modeling the structure of the frameshift-stimulatory pseudoknot in SARS-CoV-2 reveals multiple possible conformers. PLOS Comput. Biol. 2021; 17:e1008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ziv O., Price J., Shalamova L., Kamenova T., Goodfellow I., Weber F., Miska E.A.. The short- and long-range RNA-RNA interactome of SARS-CoV-2. Mol. Cell. 80:1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Haniff H.S., Tong Y., Liu X., Chen J.L., Suresh B.M., Andrews R.J., Peterson J.M., O’Leary C.A., Benhamou R.I., Moss W.N.et al.. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent. Sci. 2020; 6:1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Williams G.D., Chang R.Y., Brian D.A.. A phylogenetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J Virol. 1999; 73:8349–8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tengs T., Jonassen C.M.. Distribution and evolutionary history of the mobile genetic element s2m in coronaviruses. Diseases. 2016; 4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kim D., Lee J.-Y., Yang J.-S., Kim J.W., Kim V.N., Chang H.. The architecture of SARS-CoV-2 transcriptome. 2020; 181:914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Costales M.G., Suresh B., Vishnu K., Disney M.D.. Targeted degradation of a hypoxia-associated non-coding RNA enhances the selectivity of a small molecule interacting with RNA. Cell Chem. Biol. 2019; 26:1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Costales M.G., Aikawa H., Li Y., Childs-Disney J.L., Abegg D., Hoch D.G., Pradeep Velagapudi S., Nakai Y., Khan T., Wang K.W.et al.. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:2406–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Meyer S.M., Williams C.C., Akahori Y., Tanaka T., Aikawa H., Tong Y., Childs-Disney J.L., Disney M.D.. Small molecule recognition of disease-relevant RNA structures. Chem Soc Rev. 2020; 49:7167–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ursu A., Childs-Disney J.L., Andrews R.J., O’Leary C.A., Meyer S.M., Angelbello A.J., Moss W.N., Disney M.D.. Design of small molecules targeting RNA structure from sequence. Chem. Soc. Rev. 2020; 49:7252–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Miao Z., Tidu A., Eriani G., Martin F.. Secondary structure of the SARS-CoV-2 5′-UTR. RNA Biol. 2020; 18:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Iserman C., Roden C., Boerneke M., Sealfon R., McLaughlin G., Jungreis I., Park C., Boppana A., Fritch E., Hou Y.J.et al.. Specific viral RNA drives the SARS CoV-2 nucleocapsid to phase separate. 2020; bioRxiv doi:12 June 2020, preprint: not peer reviewed 10.1101/2020.06.11.147199. [DOI]
- 86. Baldassarre A., Paolini A., Bruno S.P., Felli C., Tozzi A.E., Masotti A.. Potential use of noncoding RNAs and innovative therapeutic strategies to target the 5′UTR of SARS-CoV-2. Epigenomics. 2020; 12:1349–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rehman Sayeed u., Tabish M.. Alternative splicing of ACE2 possibly generates variants that may limit the entry of SARS-CoV-2: a potential therapeutic approach using SSOs. Clin. Sci. 2020; 134:1143–1150. [DOI] [PubMed] [Google Scholar]
- 88. Singh N.N., Shishimorova M., Cao L.C., Gangwani L., Singh R.N.. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009; 6:341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lulla V., Wandel M.P., Bandyra K.J., Ulferts R., Wu M., Dendooven T., Yang X., Doyle N., Oerum S., Beale R.et al.. The stem loop 2 motif is a site of vulnerability for SARS-CoV-2. 2021; bioRxiv doi:11 March 2021, preprint: not peer reviewed 10.1101/2020.09.18.304139. [DOI] [PMC free article] [PubMed]
- 90. Moitra P., Alafeef M., Dighe K., Frieman M.B., Pan D. Selective naked-eye detection of SARS-CoV-2 mediated by N gene targeted antisense oligonucleotide capped plasmonic nanoparticles. ACS Nano. 2020; 14:7617–7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Darty K., Denise A., Ponty Y.. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All Datasets (S1-S3) associated with this study are available at: https://structurome.bb.iastate.edu/sars-cov-2. Python scripts used in analyses can be found at https://github.com/moss-lab/SARS-CoV-2.