

Abstract
PURPOSE
Epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors (TKIs) show efficacy in treating patients with lung adenocarcinoma with EGFR-activating mutations. However, a significant subset of targeted patients fail to respond. Unlike acquired resistance (AR), intrinsic resistance (IR) remains poorly understood. We investigated whether epigenomic factors contribute to patient-to-patient heterogeneity in the EGFR-TKI response and aimed to characterize the IR subpopulation that obtains no benefit from EGFR-TKIs.
PATIENTS AND METHODS
We conducted genome-wide DNA methylation profiling of 79 tumors sampled from patients with advanced lung adenocarcinoma before they received EGFR-TKI treatment and analyzed the patient responses. Pyrosequencing was performed in a validation cohort of 163 patients with EGFR-activating mutations.
RESULTS
A DNA methylation landscape of 216 CpG sites with differential methylation was established to elucidate the association of DNA methylation with the characteristics and EGFR-TKI response status of the patients. Functional analysis of 37 transcription-repressive sites identified the enrichment of transcription factors, notably homeobox (HOX) genes. DNA methylation of HOXB9 (cg13643585) in the enhancer region yielded 88% sensitivity for predicting drug response (odds ratio [OR], 6.64; 95% CI, 1.98 to 25.23; P = .0009). Pyrosequencing validated that HOXB9 gained methylation in patients with a poor EGFR-TKI response (OR, 3.06; 95% CI, 1.13 to 8.19; P = .019).
CONCLUSION
Our data suggest that homeobox DNA methylation could be a novel tumor cellular state that can aid the precise categorization of tumor heterogeneity in the study of IR to EGFR-TKIs. We identified, for the first time, an epigenomic factor that can potentially complement DNA mutation status in discriminating patients with lung adenocarcinoma who are less likely to benefit from EGFR-TKI treatment, thereby leading to improved patient management in precision medicine.
INTRODUCTION
Adenocarcinoma is the most common subtype of non–small-cell lung cancer (NSCLC), which has the highest cancer mortality worldwide.1 High intratumor heterogeneity of lung adenocarcinoma has been documented, and several targetable oncogenic mutations2-9 have been characterized. Inhibition of epidermal growth factor receptor (EGFR) kinase activity by EGFR-tyrosine kinase inhibitors (TKIs), such as erlotinib, gefitinib, and afatinib, was effective in patients with NSCLC with EGFR-activating mutations. However, despite the remarkable clinical success, the treatment efficacy was still limited to 50%-80%.10,11 The sizable percentage of nonresponders (20%-30%) suggested intrinsic TKI resistance and substantial heterogeneity among tumors, even among EGFR-mutant tumors, highlighting the need for reliable predictive biomarkers.
CONTEXT
Key Objective
A sizable portion (20%-30%) of patients with epidermal growth factor receptor (EGFR)–mutant non–small-cell lung cancer have no good initial clinical response to EGFR-tyrosine kinase inhibitors (TKIs), and how to predict intrinsic drug resistance accurately is challenging. Global DNA methylation landscape of tumors from patients with lung adenocarcinoma before TKI treatment was analyzed to investigate the association with EGFR-TKI responses.
Knowledge Generated
A total of 216 TKI response–associated methylated sites were identified, and functional analysis revealed the enrichment of homeobox genes. In particular, increased methylation of HOXB9 correlated with higher rate of intrinsic resistance (IR) to EGFR-TKI.
Relevance
DNA methylation provides a different dimension to complement the DNA mutation–based markers for understanding the mechanism of IR. Evaluation of the methylation level on HOXB9 may be incorporated in the management of lung adenocarcinoma to aid the prediction of EGFR-TKI response.
The comprehensive molecular profiling of pretreatment lung adenocarcinoma to identify inherently TKI-resistant cases can aid the development of potential strategies to manage such cases. Recently, genomic profiling of advanced NSCLC with EGFR mutations at baseline has identified multiple genetic, phenotypic, and functional mechanisms that may contribute to intrinsic resistance (IR).12 Whole-exome sequencing on untreated EGFR-mutant NSCLC tumors13 and the detection of co-occurring genetic alternations, such as MET, PIK3KA, CDK4, CDK6, and NF1, in the cfDNA of advanced-stage patients before treatment with EGFR-TKI14 suggest many DNA-based biomarkers for IR prediction. On the other hand, tumor suppressor genes involved in the alternative mechanisms of IR may be inactivated by epigenetic mechanisms that result in phenotypic or functional changes.15 However, although studies have reported that epigenetic changes in tumor participate in the evolution of acquired drug resistance through regulating gene expression patterns,16 epigenomic data associated with IR to TKI are lacking. Modification of methylation on DNA is stable and abnormal methylation represents an early event for cancer diagnosis, making methylation aberrations equally suitable candidates for recurrence detection and prediction of patient survival.17-21 Therefore, we undertook a genome-wide approach to investigate DNA methylation patterns associated with IR to TKI.
DNA methylation that occurs at cytosines of CpG dinucleotides, especially within CpG islands in the promoter region, can lock genes in off status, resulting in a transcriptionally silent state.22,23 Although DNA methylation is an important mechanism for maintaining normal development and cellular homeostasis, aberrant DNA methylation–mediated silencing of tumor suppressor genes has been reported to be associated with cell survival and progression in cancer.24 DNA methylation profiling of tumors, including those of glioma, acute myeloid leukemia, and colorectal and lung cancers, has aided the identification of cancer subtypes correlated with clinical outcomes.25-29
In this study, we aimed to identify epigenetic markers for predicting drug efficacy in patients with lung adenocarcinoma. We conducted genome-wide DNA methylation profiling of tumors from patients before their EGFR-TKI therapy and established a DNA methylation landscape to elucidate the association of DNA methylation with the EGFR-TKI response status of patients via a pipeline of statistical analysis, gene ontology (GO), and bioinformatics analysis. Our study identified a DNA methylation marker for predicting drug response in lung adenocarcinoma and provides insight into the epigenetic regulation of IR to EGFR-TKIs.
PATIENTS AND METHODS
Tumor samples were obtained from two cohorts of patients with lung adenocarcinoma before EGFR-TKI therapy. Detailed accounts of sample collection and protocols for DNA extraction, bisulfite conversion, DNA methylation analysis, pyrosequencing along with statistical analysis, bioinformatics analysis, and data availability are presented in the Data Supplement.
RESULTS
Clinicopathologic Features of the Patients
Table 1 lists the clinicopathologic features of the two cohorts. Patients in the discovery cohort were at the advanced stage (IIIB or IV), and most EGFR mutations were L858R point mutations and exon 19 deletions. No significant differences were found in the distribution of tumor stage, sex, or smoking behavior between EGFR mutant and EGFR wild type. The validation cohort consisted of 163 EGFR-mutant patients and the majority (85.28%) were at stage IV. The TKI response assessment—progressive disease (PD), stable disease (SD), partial response (PR), or complete response (CR)—was determined according to the RECIST guidelines.30 The PD group, defined at the first scan done at 8 weeks following the start of EGFR-TKI, is considered as patients with IR. We determined the disease control rate (DCR) by comparing the number of patients with SD, PR, or CR with those with PD to study intrinsic drug resistance.
TABLE 1.
Clinicopathologic Characteristics of the Patients with Lung Adenocarcinoma

Differential DNA Methylation Sites Associated With EGFR-TKI Response Heterogeneity Were Enriched in Transcription Factors
Following the flowchart in Figure 1, the DNA methylation profiles of the 79 tumors in the discovery cohort were assessed. Only probes showing a high variation across tumors were retained. Using the top 5% coefficient of variation as the cutoff, 24,121 probes with the greatest variability were analyzed, and 391 probes were found correlated with EGFR-TKI response. To identify the subpopulation of intrinsic drug-resistant patients obtaining no benefit from TKIs, we further analyzed DCR by univariate and multivariate logistic regression. Of 391 probes, 216 were found significant (Data Supplement). Interestingly, 30 of the 216 probes (13.88%) were annotated to transcription factors (TFs). Comparison with the percentage of probes in the Illumina Infinium HumanMethylation450 BeadChip annotated to TFs (34,129/482,421; 7.07%) revealed that the enrichment was highly significant (binomial P = .0001).
FIG 1.
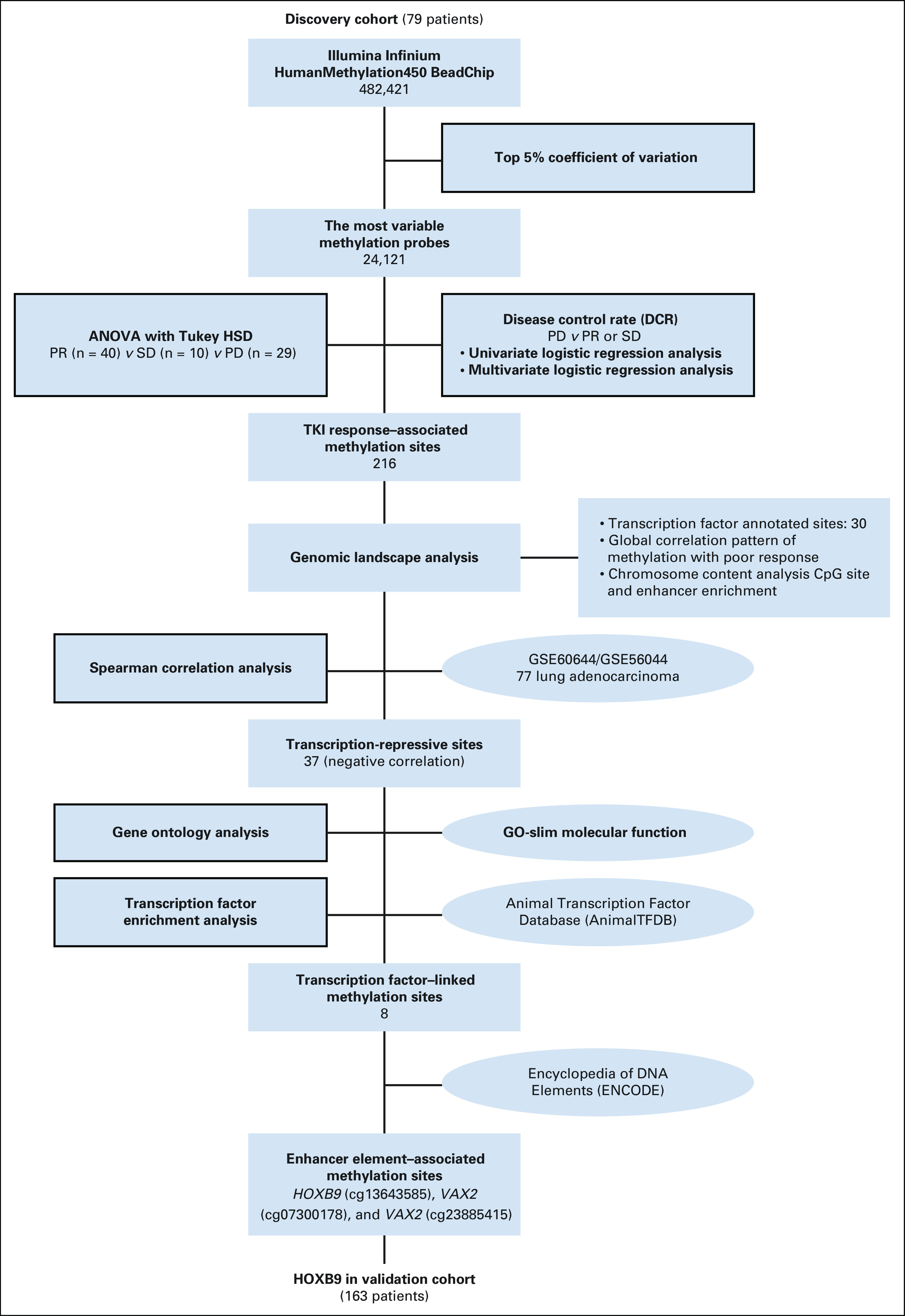
Flowchart of the study design and discovery cohort analysis. Whole-genome DNA methylation analysis was conducted in 79 patients to investigate the association of DNA methylation with EGFR-TKI response. Starting from a total of 482,421 CpG probes, a series of statistical and bioinformatic procedures were conducted to filter out the less-relevant probes. The numbers show the number of CpG probes remaining after passing each selection procedure. Statistical tests are shown in the blocks with black borders. Publicly available databases are shown in the ovals in the diagram. ANOVA, analysis of variance; DCR, disease control rate; EGFR, epidermal growth factor receptor; HSD, honestly significant difference; PD, progressive disease; PR, partial response; SD, stable disease; TKI, tyrosine kinase inhibitor.
DNA Methylation Landscape of 216 Probes Associated With Differential EGFR-TKI Responses
The patient DNA methylation profiles with the 216 TKI response–associated methylated probes were established (Fig 2A). The majority of the probes (203) had higher DNA methylation in patients with a poor response than in those with a favorable response; hypermethylation correlated with poor response. Only a group of 13 probes (bottom of the plot) showed the opposite trend; hypomethylation correlated with poor response. The probes were grouped by their locations relative to the CpG island content and by the chromosome content relative to the transcription start site. We also rearranged this plot according to patient characteristics (Data Supplement) and conducted a correlation analysis by linear regression to find the significantly correlated probes (P < .05). We found 11 probes correlated with EGFR status, 54 probes with sex, and 32 probes with smoking behavior. A global view of the DNA methylation distribution contrasting the patients with PD against those with PR showed that all but 13 probes were located above the diagonal line, elucidating a clear pattern of methylation gain in patients with PD (Fig 2B-D). The tumors of patients who were more likely to be resistant to EGFR-TKIs tended to have higher pretreatment methylation levels.
FIG 2.

DNA methylation landscape of 79 patients with lung adenocarcinoma with different EGFR-TKI responses. (A) DNA methylation heatmap of 216 EGFR-TKI response–associated sites for DCR prediction. The color-coded beta value at each selected probe ranged between 0 (blue) and 1 (red). The patients’ characteristics, smoking behavior (P1), sex (P2), EGFR status (P3), and EGFR-TKI response (P4), are shown by the bars at the top of the heatmap. Probes were grouped by CpG island content (left bar) and by chromosome content (right bar). (B-D) DNA methylation density for PD versus PR. For each CpG probe, the average beta value across patients with PD was plotted against that across patients with PR and is shown as a smooth kernel scatter plot. The set of probes used is indicated at the top of each plot, along with the number of probes. In the gradient scale, red represents the densest region, whereas purple represents the sparsest region; PR (red), SD (yellow), and PD (blue). (E) Genomic context distributions of CpG methylation sites. The probe distributions in the CpG context and the gene context are shown on the left and right, respectively, for all probes in the array (top), for the most variably methylated probes (upper middle), for the TKI response–associated methylation sites (lower middle), and for the transcription-repressive sites (bottom). (F) Functional enrichment analysis of genes linked to the 37 transcription-repressive sites. All 186 molecular function categories in GO-slim were evaluated, and the top 5 most enriched categories are shown, along with fold changes and P values. The GO terms involving transcription factors were GO:0003676, GO:0003677, and GO:0003700. (G) F value of ANOVA for the eight TF-linked sites. *P < .05; **P < .01; $enhancer (ENCODE). ANOVA, analysis of variance; DCR, disease control rate; EGFR, epidermal growth factor receptor; ENCODE, Encyclopedia of DNA Elements; GO, gene ontology; PD, progressive disease; PR, partial response; SD, stable disease; TF, transcription factor; TKI, tyrosine kinase inhibitor.
Chromosomal Context Analysis of Candidate CpG Sites Showed Enrichment in CpG Islands and Gene Promoter Regions
The CpG sites were assigned to the annotated categories according to their chromosome positions relative to the nearby transcription start sites and the closest CpG islands (Data Supplement). We examined the changes in the proportion of each category during our probe selection and found an increasing trend in CpG islands and the gene promoter region TSS1500 (between 1,500 bp and 200 bp upstream of the transcription start site). The 216 EGFR-TKI response prediction sites were highly enriched in CpG islands (81.94%) compared with only 31.15% sites initially in CpG islands. Similarly, the percentage of sites in the TSS1500 region increased from 11.65% to 18.06%. In addition, the proportion of probes in the open sea region decreased sharply from 35.89% to 5.56%. For the 37 transcription-repressive sites, the enrichment pattern in CpG islands was retained and that in TSS1500 was increased to 27.03% (Fig 2E).
Identification of Transcription-Repressive Methylation Sites
We investigated the potential of the 216 methylation sites in cis-regulation of gene expression by correlating publicly accessible mRNA gene expression data (GSE60644) with DNA methylation data (GSE56044) in lung adenocarcinoma (Data Supplement). We computed the Spearman rank correlation between DNA methylation and gene expression to select the methylation sites that showed evidence of repressing downstream transcript expression. A total of 37 sites were identified as transcription-repressive sites (Data Supplement).
Functional Enrichment Analysis of Transcript-Linked Methylation Showed Eight Probes Linked to TFs
To evaluate the molecular function of the genes mapped by the 37 transcription-repressive sites, GO enrichment analysis was conducted. Three top significant molecular function categories of GO slim terms, nucleic acid binding (GO:0003676), DNA binding (GO:0003677), and sequence-specific DNA binding TF activity (GO:0003700), were all related to TFs (Fig 2F and Data Supplement). Comparisons of the percentage of TFs in the 37 transcription-repressive sites (21.62%; 8/37) with that in all probes (7.07%; 34,129/482,421) and in the most variable probes (11.59%; 2,796/24,121) revealed significant enrichment (P values = .0025 and .036, respectively; Data Supplement). Furthermore, using Animal Transcription Factor Database (AnimalTFDB), we found that eight of the 37 sites were annotated to five TFs: IKZF1, HOXB9, SP8, LASS4, and VAX2 (Table 2 and Data Supplement). Figure 2G shows the F statistic and the corresponding P value from analysis of variance for each of the eight sites, along with the location and chromosome context information. We found that seven of the eight TF-linked sites are located in the context of CpG islands and five in the transcription start site (TSS) region (Table 2). Sites located in the TSS region were what we focused on next.
TABLE 2.
Transcription Factor–Linked Methylation Sites

Identification of the DNA Methylation Site cg13643585 (HOXB9) With Predictive Ability Regarding EGFR-TKI Response
Among the five sites in the TSS region, three sites, cg13643585 (HOXB9), cg07300178 (VAX2), and cg23885415 (VAX2), are located in enhancer regions annotated by the Encyclopedia of DNA Elements (ENCODE) Consortium. The DNA methylation levels of cg13643585, cg07300178, and cg23885415 were the highest in the group of patients with PD (Fig 3A and Data Supplement). HOXB9 methylation (cutoff beta value = .15) predicted disease control by EGFR-TKIs with 88% sensitivity (area under the receiver operating characteristic curve [AUROC], 0.6917; odds ratio [OR], 6.64; 95% CI, 1.98 to 25.23; P = .0009) (Figs 3B and 3C). The two VAX2 methylation sites also showed a moderate ability to predict the EGFR-TKI response (Data Supplement).
FIG 3.
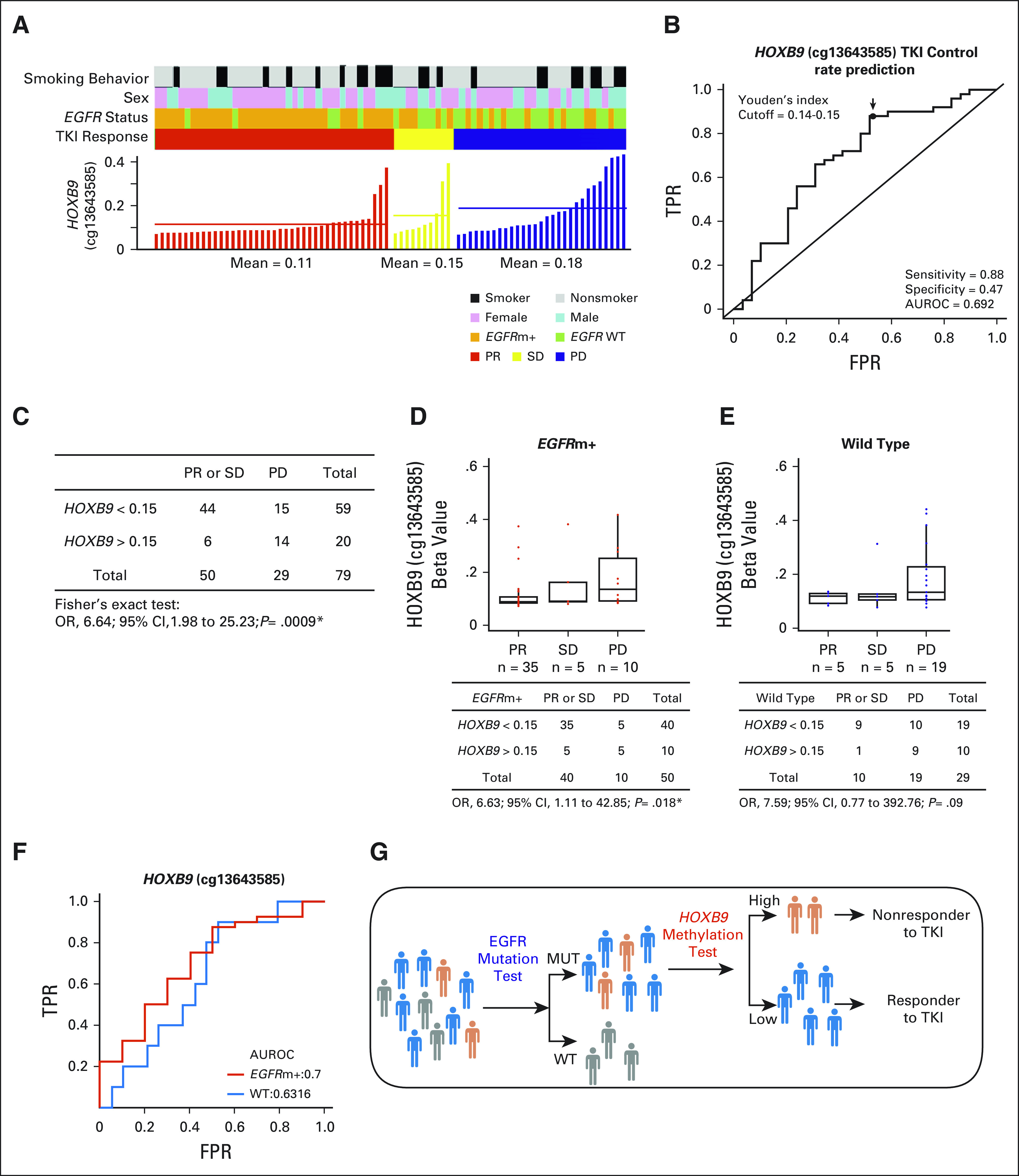
DNA methylation of HOXB9 (cg13643585) was correlated with EGFR-TKI response. (A) The HOXB9 beta values of 79 patients with NSCLC with PR (red), SD (yellow), and PD (blue). (B) AUROC and (C) Fisher’s test of HOXB9 methylation for predicting the EGFR-TKI response. . The optimal cutoff points were determined by the Youden's index, which maximizes the sum of the specificity and sensitivity. (D-F) Stratified analysis. Patients were classified as EGFRm+ (red) or EGFR wild type (blue). Comparison of HOXB9 beta value among PR, SD, and PD. (G) Strategy using HOXB9 methylation complementing EGFR to classify the subpopulation of patients likely to be nonresponders to EGFR-TKI. AUROC, area under the receiver operating characteristic curve; EGFR, epidermal growth factor receptor; FPR, false positive rate; NSCLC, non–small-cell lung cancer; OR, odds ratio; PD, progressive disease; PR, partial response; SD, stable disease; TKI, tyrosine kinase inhibitor; TPR, true positive rate; WT, wild type.
We further performed stratification analysis by classifying patients as those with EGFR-activating mutations and those without (Figs 3D-F). Increased methylation of HOXB9 (cg13643585) was observed in PD patients. The OR (6.63) between disease control and progression in the EGFR mutation group was significant (P = .018, Fig 3D). For the EGFR wild-type group, the OR (7.59) was comparable, but the P value was .09, likely because of the small sample size (Fig 3E). On the other hand, DNA methylation of VAX2 (Data Supplement) exhibited no discriminatory power in the EGFR mutation group (OR, 2.29 and 2.27; P = .31 and .17 for cg07300178 and cg23885415, respectively; Data Supplement) but performed better in the wild-type group (OR, 11.07 and 6.06; P = .011 and .046, respectively; Data Supplement).
Validation of HOXB9 as a DNA Methylation Marker
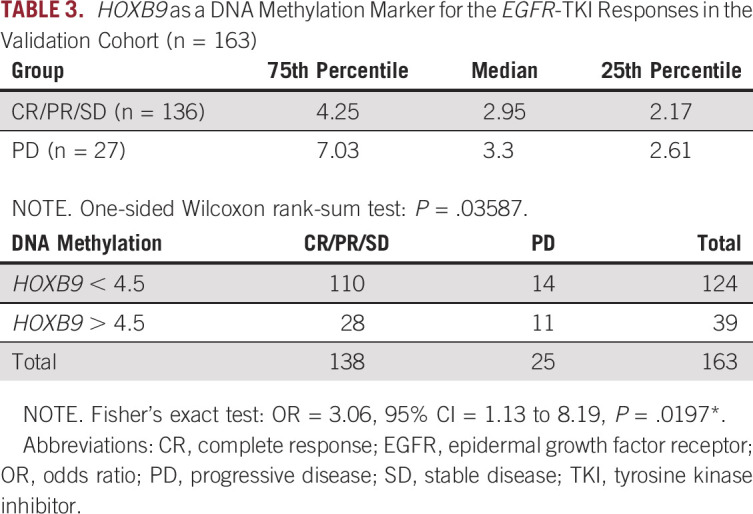
We conducted pyrosequencing to quantify the DNA methylation level of HOXB9 (cg13643585) in an independent cohort of 163 patients with EGFR-activating mutations who were receiving EGFR-TKI therapy. Tumor DNA was obtained before treatment. The data in Table 3 confirmed the pattern of increased HOXB9 methylation in the EGFR-TKI–resistant (PD) group compared with the disease control group (CR, PR, or SD). The PD group was ranked first in each quartile, and the one-sided rank-sum test for between-group differences indicated a significant difference (P = .036) (Data Supplement). In addition, analysis of the AUROC showed that HOXB9 methylation statistically significantly increased the predictive precision of EGFR-TKI resistance for the DCR (Table 3). Overall, the ratio of disease control to PD was approximately 5.5:1. For patients with lower HOXB9 methylation levels (< 4.5), the ratio of disease control to PD increased to approximately 8:1. In contrast, for patients with higher HOXB9 methylation levels (> 4.5), the ratio was greatly reduced—only approximately 2.5:1 (OR, 3.06; 95% CI, 1.13 to 8.19; P = .02). This result confirmed the benefit of using epigenomic markers complementing DNA markers to identify subpopulations of patients with higher-than-average susceptibility to intrinsic EGFR-TKI resistance (Fig 3G).
TABLE 3.
HOXB9 as a DNA Methylation Marker for the EGFR-TKI Responses in the Validation Cohort (n = 163)
DISCUSSION
The paradigm shift in treating lung adenocarcinoma with EGFR-targeted therapy is a major success in precision medicine. However, drug resistance remains a pertinent issue that hinders further improvement in the management of EGFR-targeted therapy. Although significant advances have been made in understanding acquired resistance (AR), the causes of IR remain unclear.12 In addition to being classified as AR versus IR, resistance mechanisms can be classified in terms of on-target versus off-target,31 suggesting the activation of collateral signaling. T790M mutation in exon 20 of EGFR is the most common mechanism for resistance to first- and second-generation EGFR-TKI. Several third-generation EGFR-mutant selective TKIs such as AZD9291 (osimertinib) have been approved for EGFR T790M–positive NSCLC treatment.12 As Oxnard32 summarized, the well-known EGFR T790M erlotinib-resistance mutation occurs both as the result of subclonal presence of T790M followed by outgrowth and as a de novo consequence of mutation in persister cells. Existence of any subclonal variation of methylation for either AR or IR posts an interesting issue to explore further. Moreover, where genomic resistance has been found, epigenomic modulation has been proposed as the potential mechanism. Changes in resistant phenotypes, including epithelial-mesenchymal transition (EMT) and cancer stemness shift, have been found to be driven by epigenetic remodeling. TKI-induced DNA methylation changes have been reported in advanced EGFR-mutated lung cancer.33 Decitabine, the DNA methyl transferase inhibitor, could reverse the sensitivity of EGFR-TKI–resistant NSCLC cell line PC9/GR through demethylation of RASSF1A and GADD45β.34 The combination of TKIs with epigenetic drugs has shown promise as a treatment in preclinical and clinical studies.33-35
In this study, we conducted clinical oncological investigation on the potential role of DNA methylation in mediating IR to EGFR-TKI treatment in patients with advanced lung adenocarcinoma. Aberrant DNA methylation is one of the most classical events that occurs during lung cancer development.36 Many studies have shown altered methylation patterns in lung cancer, indicating roles of epigenetic biomarkers and therapeutic targets.37-39 Earlier study by Zhu et al40 focused on the methylation patterns of Wnt antagonists, showing the association of methylated SFRP5 with shortened progression-free survival under EGFR-TKI treatment, but not with IR to TKI. Epigenome-wide analysis has demonstrated that homeobox genes can act as potential DNA methylation markers for the early diagnosis of lung cancer.41 Moreover, Sandoval et al42 identified a hypermethylated five-gene signature associated with shorter relapse-free survival times of patients with stage I NSCLC without adjuvant chemotherapy. Interestingly, two of those five genes, AXL1 and HOXA9, are homeobox TFs. Our pursuit of the primary EGFR-TKI–resistant methylation markers also identified enrichment of homeobox genes. Among the 30 TKI-associated methylation probes annotated to TFs, 11 accounted for nine homeobox genes (Data Supplement).
We identified and confirmed the correlation of HOXB9 DNA methylation with an increased rate of IR to EGFR-TKIs. HOXB9 is involved in cell development and proliferation43 and is suggested to function as a TF that can induce the expression of EMT genes and several angiogenic factors, such as VEGF, IL-8, and TGFβ, resulting in the activation of EGFR and ERBB2.44-46 EGFR signaling is connected to the NF-κB pathway, giving the role in IR or AR to EGFR inhibitors.47 However, the molecular mechanisms by which HOXB9 contributes to carcinogenesis are debated.48,49 The overexpression of HOXB9 can suppress the AKT/NF-κB/Snail pathway and inhibit the proliferation of gastric carcinoma cells.50 We analyzed the correlation between EGFR signaling and NF-κB–dependent pathways (GSE60644) and found that expression of HOXB9 negatively associated with that of KIAA1199 (Cell migration–inducing hyaluronidase 1; Data Supplement). Through protein-protein interaction (PPI) analysis, we found that HOXB9 might cross talk with both IR and AR to EGFR-TKI through EZH2, SIRT1, and EGR2 (Data Supplement). Therefore, the regulation of HOXB9 is crucial in the cooperated oncogenic loops.12 Our data suggested that HOXB9 hypermethylation may be a novel tumor cellular state that is useful for precise categorization of tumor heterogeneity in the study of intrinsic EGFR-TKI resistance via off-target effects such as redundant or compensating signaling. In addition, the pattern was consistent between patients with EGFR-activating mutations and patients with wild-type EGFR, implying that the regulatory effect of DNA methylation of HOXB9 may be independent of EGFR activity.
DNA methylation changes can be accurately detected in tumors and liquid biopsies. Such detection is promising for the development of biomarkers for cancer screening.51 DNA methylation in distal regulatory sites, such as enhancer regions, plays important roles in gene regulation through the binding of cell type–specific TFs and interaction with promoters.52-54
We validated a DNA methylation site in the enhancer region of HOXB9 that can help the prediction of nonresponse to EGFR-TKI. In cancer, aberrant DNA methylation at enhancers couples with recruitment of coactivators or corepressors, forming networks of cancer-associated TFs and their targeted genes.55-57 Stone et al58 defined hypermethylation enhancers that correlate with sensitivity to endocrine therapy, suggesting the impact of enhancer status on the drug treatment response. Therefore, DNA methylation at enhancers could regulate downstream gene expression, although the underlying mechanisms require further study.
The combination of genetic aberrations, gene expression, and DNA methylation highlights the potential of the identified candidates in the development of biomarkers for tumor diagnosis or prognosis. Additionally, clinical applications of biomarkers in public health, including the effect size, therapeutic drugs, or measurable signals, need to be considered. In this study, with a focus on medical actionability, we discovered that HOXB9 methylation could be a biomarker useful for discriminating patients with TKI resistance from those with TKI sensitivity, especially patients whose tumors harbor EGFR-activating mutations. Although improving the sensitivity and specificity of HOXB9 methylation is recommended, our work provided a preliminary proof of concept on the usefulness of HOXB9 methylation for opening up more clinical options to manage lung adenocarcinoma. For example, in accordance with the current clinical standard of treating EGFR-mutant patients with EGFR-TKI, for patients with HOXB9 hypermethylation, combination treatment such as EGFR-TKI plus antiangiogenic therapy59 or EGFR-TKI plus chemotherapy60 may be another option to overcome the resistance and improve the response rate. A larger cohort study with the inclusion of HOXB9 methylation in addition to other genomic aberrations may be designed to investigate how to select patients for combination treatment.
ACKNOWLEDGMENT
The authors thank all the patients and research staff who participated in this work.
SUPPORT
Supported by grants from Academia Sinica AS-104-TP-A07; the Ministry of Science and Technology MOST 105-2911-I-002-302, MOST 106-2319-B-002-002, and MOST 107-2314-B-002-226; and National Taiwan University, 105R7557.
S.-F.S., C.-H.L., and C.-L.C. contributed equally to this work as first authors. S.-L.Y., G.-C.C., and K.-C.L. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Sheng-Fang Su, Chia-Hsin Liu, Chiou-Ling Cheng, Tsung-Ying Yang, Kun-Chieh Chen, Gee-Chen Chang, Sung-Liang Yu, Ker-Chau Li
Financial support: Sheng-Fang Su, Sung-Liang Yu, Ker-Chau Li
Administrative support: Ker-Chau Li
Provision of study materials or patients: Chao-Chi Ho, Tsung-Ying Yang, Kun-Chieh Chen, Kuo-Hsuan Hsu, Sung-Liang Yu, Ker-Chau Li
Collection and assembly of data: Chiou-Ling Cheng, Tsung-Ying Yang, Kun-Chieh Chen, Kuo-Hsuan Hsu, Jeng-Sen Tseng, Gee-Chen Chang, Ker-Chau Li
Data analysis and interpretation: Sheng-Fang Su, Chia-Hsin Liu, Chao-Chi Ho, Tsung-Ying Yang, Kun-Chieh Chen, Huei-Wen Chen, Gee-Chen Chang, Sung-Liang Yu, Ker-Chau Li
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Chao-Chi Ho
Consulting or Advisory Role: Boehringer Ingelheim, Eli Lilly, Roche/Genentech/Chugai, BMS, Ono
Speakers' Bureau: Boehringer Ingelheim, Eli Lilly, Roche/Genentech/Chugai, MSD, Pfizer, Novartis, BMS, Ono
Research Funding: AstraZeneca
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A.Cancer statistics, 2019 CA Cancer J Clin 697–342019 [DOI] [PubMed] [Google Scholar]
- 2.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR N Engl J Med 3622380–23882010 [DOI] [PubMed] [Google Scholar]
- 3.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial Lancet Oncol 11121–1282010 [DOI] [PubMed] [Google Scholar]
- 4.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study Lancet Oncol 12735–7422011 [DOI] [PubMed] [Google Scholar]
- 5.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial Lancet Oncol 13239–2462012 [DOI] [PubMed] [Google Scholar]
- 6.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma N Engl J Med 361947–9572009 [DOI] [PubMed] [Google Scholar]
- 7.Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study Ann Oncol 261883–18892015 [DOI] [PubMed] [Google Scholar]
- 8.Yang JCH, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6 Lancet Oncol 16830–8382015 [DOI] [PubMed] [Google Scholar]
- 9.Yang JCH, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials Lancet Oncol 16141–1512015 [DOI] [PubMed] [Google Scholar]
- 10.Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: A meta-analysis J Clin Oncol 331958–19652015 [DOI] [PubMed] [Google Scholar]
- 11.Lee CK, Davies L, Wu YL, et al. Gefitinib or erlotinib vs chemotherapy for EGFR mutation-positive lung cancer: Individual patient data meta-analysis of overall survival J Natl Cancer Inst 109djw2792017 [DOI] [PubMed] [Google Scholar]
- 12.Santoni-Rugiu E, Melchior LC, Urbanska EM, et al. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: Differences and similarities with acquired resistance. Cancers. 2019;11:923. doi: 10.3390/cancers11070923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma Nature 511543–5502014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blakely CM, Watkins TBK, Wu W, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers Nat Genet 491693–17042017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glasspool R, Teodoridis J, Brown R.Epigenetics as a mechanism driving polygenic clinical drug resistance Br J Cancer 941087–10922006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown R, Curry E, Magnani L, et al. Poised epigenetic states and acquired drug resistance in cancer Nat Rev Cancer 14747–7532014 [DOI] [PubMed] [Google Scholar]
- 17.Baylin SB, Jones PA.A decade of exploring the cancer epigenome-biological and translational implications Nat Rev Cancer 11726–7342011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heyn H, Esteller M.DNA methylation profiling in the clinic: Applications and challenges Nat Rev Genet 13679–6922012 [DOI] [PubMed] [Google Scholar]
- 19.Brock MV, Hooker CM, Ota-Machida E, et al. DNA methylation markers and early recurrence in stage I lung cancer N Engl J Med 3581118–11282008 [DOI] [PubMed] [Google Scholar]
- 20.Laird PW.Principles and challenges of genome-wide DNA methylation analysis Nat Rev Genet 11191–2032010 [DOI] [PubMed] [Google Scholar]
- 21.Balgkouranidou I, Liloglou T, Lianidou ES.Lung cancer epigenetics: Emerging biomarkers Biomark Med 749–582013 [DOI] [PubMed] [Google Scholar]
- 22.Lay FD, Liu Y, Kelly TK, et al. The role of DNA methylation in directing the functional organization of the cancer epigenome Genome Res 25467–4772015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones PA, Issa JPJ, Baylin S.Targeting the cancer epigenome for therapy Nat Rev Genet 17630–6412016 [DOI] [PubMed] [Google Scholar]
- 24.De Carvalho D, Sharma S, You JS, et al. DNA methylation screening identifies driver epigenetic events of cancer cell survival Cancer Cell 21655–6672012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma Cancer Cell 22425–4372012 [DOI] [PubMed] [Google Scholar]
- 26.Hinoue T, Weisenberger DJ, Lange CPE, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer Genome Res 22271–2822012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen H, Fridley BL, Song H, et al. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat Commun. 2013;4:1628. doi: 10.1038/ncomms2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stefansson OA, Moran S, Gomez A, et al. A DNA methylation-based definition of biologically distinct breast cancer subtypes Mol Oncol 9555–5682015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heller G, Babinsky VN, Ziegler B, et al. Genome-wide CpG island methylation analyses in non-small cell lung cancer patients Carcinogenesis 34513–5212013 [DOI] [PubMed] [Google Scholar]
- 30.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer 45228–2472009 [DOI] [PubMed] [Google Scholar]
- 31.Rotow J, Bivona TG.Understanding and targeting resistance mechanisms in NSCLC Nat Rev Cancer 17637–6582017 [DOI] [PubMed] [Google Scholar]
- 32.Oxnard GR.The cellular origins of drug resistance in cancer Nat Med 22232–2342016 [DOI] [PubMed] [Google Scholar]
- 33.Angus SP, Zawistowski JS, Johnson GL.Epigenetic mechanisms regulating adaptive responses to targeted kinase inhibitors in cancer Annu Rev Pharmacol Toxicol 58209–2292018 [DOI] [PubMed] [Google Scholar]
- 34.Hou T, Ma J, Hu C, et al. Decitabine reverses gefitinib resistance in PC9 lung adenocarcinoma cells by demethylation of RASSF1A and GADD45β promoter Int J Clin Exp Pathol 124002–40102019 [PMC free article] [PubMed] [Google Scholar]
- 35.Niu X, Liu F, Zhou Y, et al. Genome-wide DNA methylation analysis reveals GABBR2 as a novel epigenetic target for EGFR 19 deletion lung adenocarcinoma with induction erlotinib treatment Clin Cancer Res 235003–50142017 [DOI] [PubMed] [Google Scholar]
- 36.Nelson HH, Marsit CJ, Christensen BC, et al. Key epigenetic changes associated with lung cancer development: Results from dense methylation array profiling Epigenetics 7559–5662012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liloglou T, Bediaga NG, Brown BRB, et al. Epigenetic biomarkers in lung cancer Cancer Lett 342200–2122014 [DOI] [PubMed] [Google Scholar]
- 38.Mehta A, Dobersch S, Romero-Olmedo AJ, et al. Epigenetics in lung cancer diagnosis and therapy Cancer Metastasis Rev 34229–2412015 [DOI] [PubMed] [Google Scholar]
- 39.Ansari J, Shackelford RE, El-Osta H.Epigenetics in non-small cell lung cancer: From basics to therapeutics Transl Lung Cancer Res 5155–1712016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu J, Wang Y, Duan J, et al. DNA methylation status of Wnt antagonist SFRP5 can predict the response to the EGFR-tyrosine kinase inhibitor therapy in non-small cell lung cancer. J Exp Clin Cancer Res. 2012;31:80. doi: 10.1186/1756-9966-31-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rauch T, Wang Z, Zhang X, et al. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay Proc Natl Acad Sci U S A 1045527–55322007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandoval J, Mendez-Gonzalez J, Nadal E, et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer J Clin Oncol 314140–41472013 [DOI] [PubMed] [Google Scholar]
- 43.Hayashida T, Takahashi F, Chiba N, et al. HOXB9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis Proc Natl Acad Sci U S A 1071100–11052009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhan J, Wang P, Niu M, et al. High expression of transcriptional factor HoxB9 predicts poor prognosis in patients with lung adenocarcinoma Histopathology 66955–9652015 [DOI] [PubMed] [Google Scholar]
- 45.Nguyen DX, Chiang AC, Zhang XHF, et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis Cell 13851–622009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiba N, Comaills V, Shiotani B, et al. Homeobox B9 induces epithelial-to-mesenchymal transition-associated radioresistance by accelerating DNA damage responses Proc Natl Acad Sci U S A 1092760–27652012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shostak K, Chariot A.EGFR and NF-κB: Partners in cancer Trends Mol Med 21385–3932015 [DOI] [PubMed] [Google Scholar]
- 48.Sha S, Gu Y, Xu B, et al. Decreased expression of HOXB9 is related to poor overall survival in patients with gastric carcinoma Dig Liver Dis 45422–4292013 [DOI] [PubMed] [Google Scholar]
- 49.Song J, Wang T, Xu W, et al. HOXB9 acetylation at K27 is responsible for its suppression of colon cancer progression Cancer Lett 42663–722018 [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, Wu Q, He C, et al. HOXB9 inhibits proliferation in gastric carcinoma cells via suppression of phosphorylated-Akt and NF-κB-dependent Snail expression Dig Liver Dis 51157–1652019 [DOI] [PubMed] [Google Scholar]
- 51.Locke WJ, Guanzon D, Ma C, et al. DNA methylation cancer biomarkers: Translation to the clinic. Front Genet. 2019;10:1150. doi: 10.3389/fgene.2019.01150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones P.Functions of DNA methylation: Islands, start sites, gene bodies and beyond Nat Rev Genet 13484–4922012 [DOI] [PubMed] [Google Scholar]
- 53.Blattler A, Farnham PJ.Cross-talk between site-specific transcription factors and DNA methylation states J Biol Chem 28834287–342942013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiench M, John S, Baek S, et al. DNA methylation status predicts cell type-specific enhancer activity EMBO J 303028–30392011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013;14:R21. doi: 10.1186/gb-2013-14-3-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taberlay PC, Statham AL, Kelly TK, et al. Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA methylation of enhancers and insulators in cancer Genome Res 241421–14322014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yao L, Shen H, Laird PW, et al. Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol. 2015;16:105. doi: 10.1186/s13059-015-0668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stone A, Zotenko E, Locke WJ, et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat Commun. 2015;6:7758. doi: 10.1038/ncomms8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saito H, Fukuhara T, Furuya N, et al. Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR-positive advanced non-squamous non-small-cell lung cancer (NEJ026): Interim analysis of an open-label, randomised, multicentre, phase 3 trial Lancet Oncol 20625–6352019 [DOI] [PubMed] [Google Scholar]
- 60.Hosomi Y, Morita S, Sugawara S, et al. Gefitinib alone versus gefitinib plus chemotherapy for non-small-cell lung cancer with mutated epidermal growth factor receptor: NEJ009 study J Clin Oncol 38115–1232020 [DOI] [PubMed] [Google Scholar]