

INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) carries one of the highest mortality risks of all cancers. Most patients present with either metastatic PDAC (50%-60%) or locally advanced tumors (30%-40%), for which the median survival is 5-9 months after diagnosis.1,2 Outcomes are still suboptimal for the small subset (10%-20%) of patients who present with resectable tumors confined to the pancreas; < 50% of these patients survive 5 years after surgery despite modern adjuvant chemotherapy.3 Moreover, at autopsy, the disease is metastatic for 88% of recurrences, and > 80% have more than 10 distinct metastatic lesions4 that are genetically related to the primary tumor5 and other metastases.6 These observations indicate that the majority of patients with PDAC harbor nonradiographically evident, micrometastatic disease after resection and that remnant tumor cells evade—or acquire resistance to—adjuvant chemotherapy. Although tropism to specific organs during metastatic spread is still poorly understood, patients with recurrence in the liver or peritoneum7 survive significantly shorter than patients with recurrent lung metastases.8-10
PDAC tumors are commonly classified into two major subtypes: one that shares some features with adenosquamous tumors (squamoid and/or basal) and the other that retains a differentiated, glandular and/or ductal morphology (ductal and/or classical). Squamoid PDAC tumors are more glycolytic,11,12 more hypoxic,13 more likely to metastasize,14,15 recruit inflammatory fibroblasts,16 and yield a poorer prognosis than ductal tumors.17-21 Classical subtype tumors are associated with more tumor-infiltrating leukocytes, denser collagen, and better outcomes,15,22 although significant subtype heterogeneity within tumors13 complicates the relationship between subtype and outcome.
More cytolytic T cells infiltrate PDAC tumors than most other tumors,23 but they are ultimately insufficient to control the cancer. Density of tumor-infiltrating T cells is highly variable among PDAC tumors,24 and abundant CD8+ T cells (along with a high number of neoantigens) can support exceptionally long-term survival.25 However, T cell–mediated tumor immunity may be restricted by several mechanisms: tumor cell downregulation of HLA, immunosuppressive leukocytes, T cell proximity to tumor cells, and T cell exhaustion.26-30 Effective T cell–mediated tumor immunity is enhanced by a high diversity of functional T cell clones, tumor-specific recruitment of effector T cells, and abundant neoantigens.23,25,31 Quantifying the phenotypes, functions, and locations of leukocytes is critical to identify specific tumor immunity required for exceptional positive outcomes.
Rare, exceptional control of PDAC progression may be mediated by equally rare, idiosyncratic patient genetics, tumor-specific somatic alterations and gene expression, and/or stochastically generated tumor immunity. In this report, we present the cases of Pt1, who survived more than 46 months with occult, chemotherapy-resistant metastatic PDAC, and her niece (Pt2) who succumbed to progressing metastatic disease despite aggressive treatment. We compare the clinical outcomes and primary and metastatic tumors from these two patients, identifying major differences in subtype, somatic alterations, and leukocyte lineages that—in some combination or alone—might have led to divergent disease courses.
RESULTS
Disparate Outcomes for Two Related Patients Both Diagnosed With Stage II PDAC
Pt1 is a chronic smoker diagnosed at 64 years of age with stage II PDAC (pT3 N1 M0) and treated 26 days later by surgical resection of a moderately to poorly differentiated, invasive ductal adenocarcinoma, followed by two cycles of gemcitabine and capecitabine. A pulmonary lesion measuring 20 mm × 16 mm was evident by CT scan 27 days prior to resection of the primary tumor, but upon biopsy was negative for malignancy. However, during adjuvant chemotherapy, the lung lesion increased to 23 mm × 18 mm, and a CT and/or PET was suspicious for malignancy with a standardized uptake value (SUV) of 4.5. Based on the imaging, the lung lesion was resected and found to be metastatic PDAC. Pt1 chose to receive no further treatment and was radiographically disease-free for over 2 years (Fig 1) until an isolated, recurrent lung metastasis was identified by CT and FDG-PET (SUV of 9) and then treated by 50 Gy in five fractions using Stereotactic Body Radiotherapy. Four months later, restaging CT revealed that the size of the previously visualized left lower lobe nodule had decreased; there were no additional suspicious lesions, and the patient is asymptomatic 46 months after diagnosis.
FIG 1.
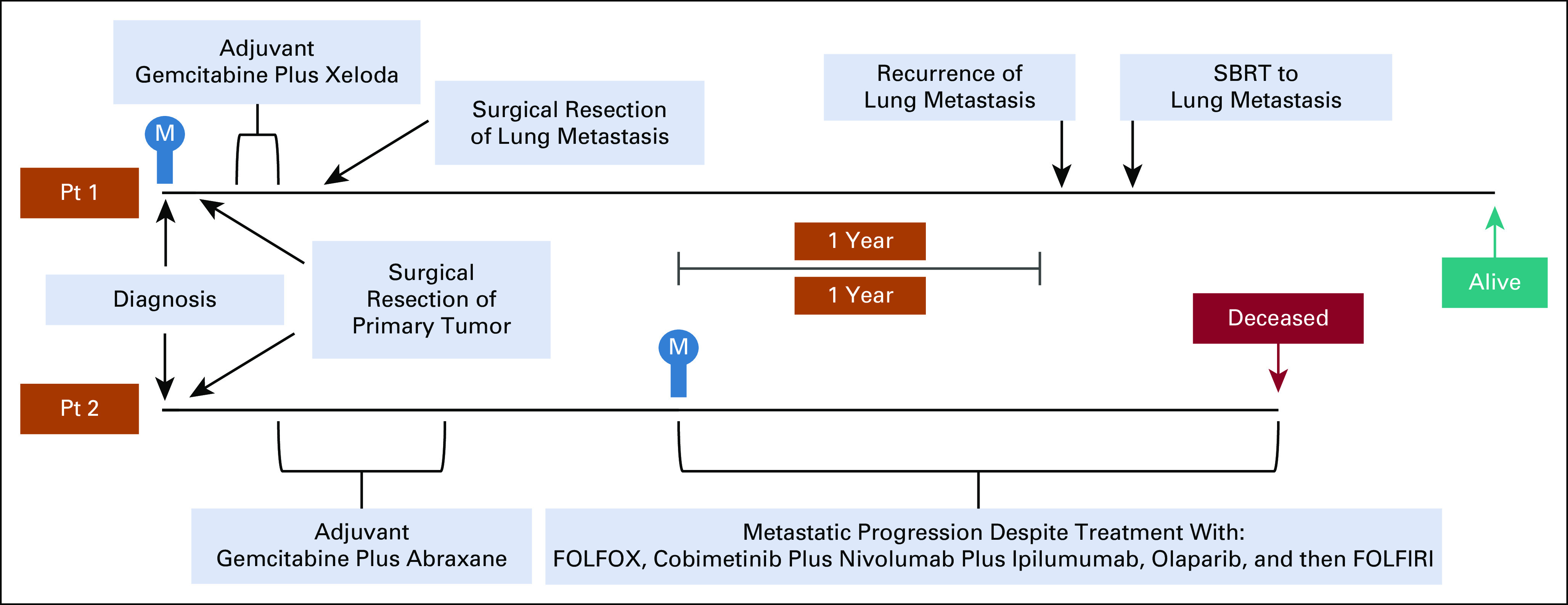
Disease courses for patients 1 (top) and 2 (bottom) aligned by date of diagnosis. The date when metastatic disease was first radiographically evident is indicated with an M in blue. CA19-9 test results are not shown because—for both patients—they were reproducibly approximately 90% lower than the 37 U/mL threshold for normal, even when the primary tumor was present. This result suggests a false negative result because of a Lewisa-b- antigen phenotype, as is the case for 5%-10% of Caucasian patients with PDAC.65
Pt2—a nonsmoker—was diagnosed at 44 years of age with stage II PDAC (pT3 N1 M0) and treated by surgical resection of a moderately differentiated adenocarcinoma with squamoid features as described by Hayashi et al32—this tumor was not qualified as an adenosquamous subtype of ductal adenocarcinoma. Pt2 was treated with six cycles of gemcitabine and nab-paclitaxel (on a clinical trial). A biopsy-proven, right ureter metastasis in the context of widespread peritoneal metastases was identified 1 year after the last dose of adjuvant chemotherapy, and Pt2 was treated with four cycles of fluorouracil and oxaliplatin. Subsequently, a right retroperitoneal metastasis and multiple liver metastases were identified and continued to progress despite further treatment (Fig 1). Pt2 died 19 months after metastatic disease recurrence.
A Shared Germline PRSS1 Mutation and Somatic Alterations Common to PDAC
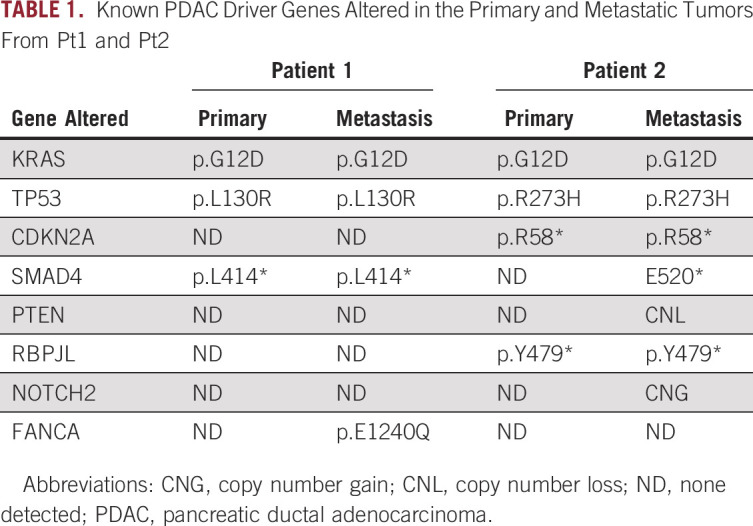
We identified germline PRSS1 p.A16V in both patients and somatic alterations in the two most common PDAC driver genes—KRAS and TP53—in both tumors from each patient (Table 1). The primary tumor from Pt2 also contained a CDKN2A alteration. Metastatic tumors from both patients had all alterations found in their respective primary tumors. Additionally, the Pt1 metastasis contained an FANCA mutation, whereas the Pt2 metastasis contained a PTEN deletion. SMAD4 was altered in both the primary and metastatic tumors from Pt1, but only in the metastasis from Pt2.
TABLE 1.
Known PDAC Driver Genes Altered in the Primary and Metastatic Tumors From Pt1 and Pt2
Evidence of Squamoid Differentiation in Tumors From Patient 2
The histology of Pt1's primary tumor was homogeneously ductal and did not express KRT5, whereas Pt2's primary tumor contained heterogeneous squamoid features and KRT5 expression in approximately 25% of tumor cells (Fig 2A).32 The lung metastasis from Pt1 was ductal, whereas the ureter metastasis from Pt2 was mostly squamoid (Fig 2B).
FIG 2.
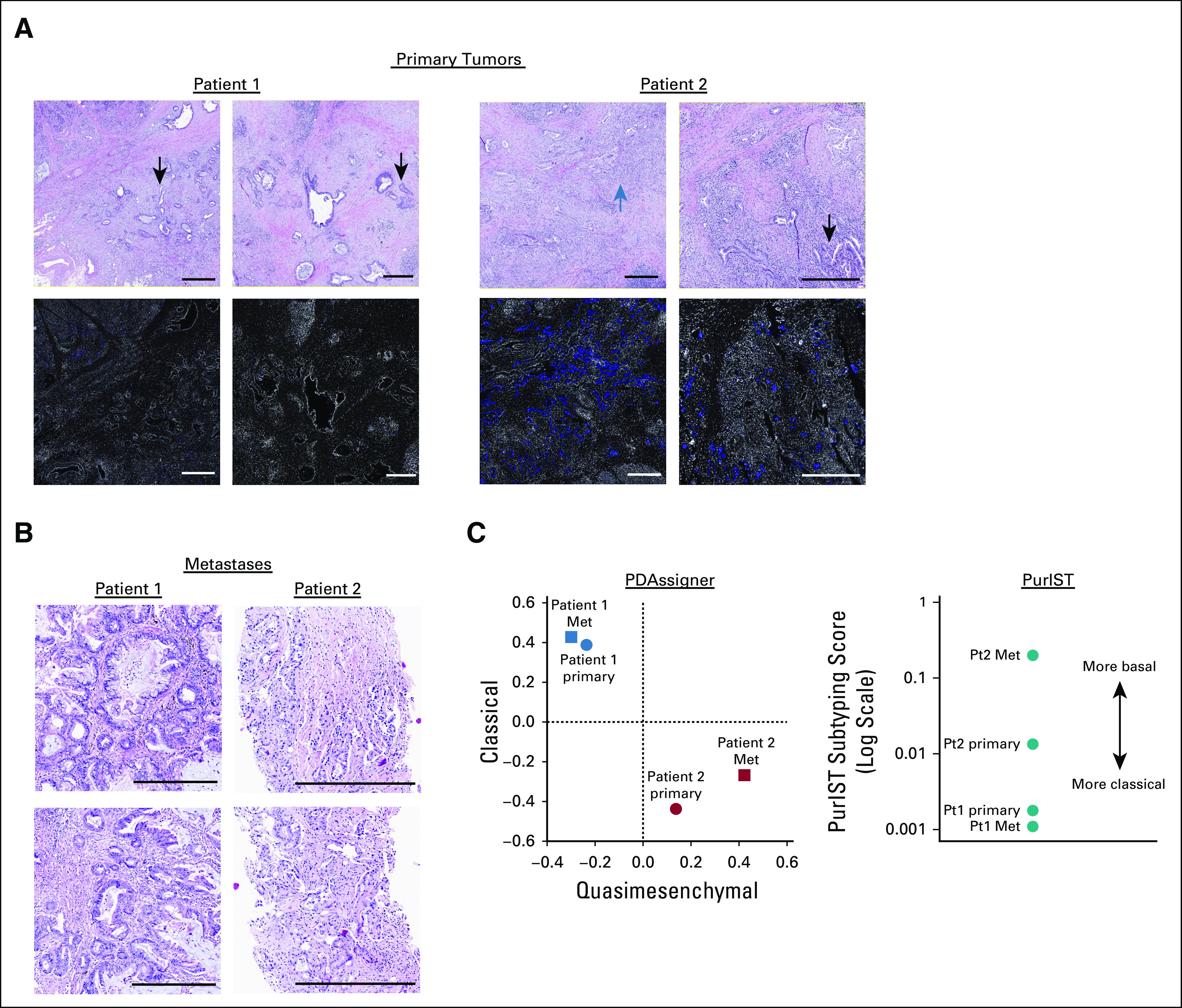
PDAC subtype assignment based on histology and gene expression. (A) H&E stained sections and immunohistofluorescence for KRT5 (blue) and DAPI (white) from primary tumors. Regions of ductal or squamoid histology are indicated by black or blue arrows, respectively. Each scale bar is 0.5 mm. (B) H&E stained sections of metastases from Pt1 (lung) and Pt2 (ureter). Each scale bar is 0.5 mm. (C) PDAC subtyping of each patient's primary and metastatic tumors. Left panel: Spearman correlation coefficients for tumors from Pt1 and Pt2 compared with the published rank order of PDAssigner genes for classical (y-axis) and quasimesenchymal (x-axis) tumors. Right panel: scores from PurIST subtyping for each tumor. PDAC, pancreatic ductal adenocarcinoma.
To further distinguish ductal PDAC from squamoid PDAC, we classified the subtype (classical or quasimesenchymal/basal of each tumor using RNASeq and a previously published gene panel [PDAssigner18]) and the PurIST technique33 that normalizes differences in tumor cellularity (Fig 2C). Both tumors from Pt1 aligned well with the classical subtype, whereas the tumors from Pt2 were more quasimesenchymal and/or basal with the metastasis scoring more basal than the primary tumor.
Leukocyte Types Associated with Immunity in Tumors From Patient 1
We used a multiplex immunohistochemistry (mIHC) workflow34 to quantify the densities of tumor cells, fibroblasts, and 10 leukocyte subsets (Appendix Table A1). We compared the primary tumors from both patients with the metastasis from Pt1, but limited material prevented us from analyzing the metastasis from Pt2. We selected regions of interest (ROIs) within each section that contained both KRT+ epithelial or tumor cells and CD45+ immune cells (Appendix Fig A1), followed by quantitative assessment of each ROI. The metastasis from Pt1 had more epithelial or tumor cells than her primary tumor and fewer fibroblasts than the primary tumor from Pt2 (Fig 3A). A detailed analysis of leukocyte subsets revealed that CD8+ T cells and CD4+ T cells were significantly denser in the metastasis from Pt1 compared with the primary tumor from Pt2 (Fig 3B). Additionally, the primary tumor from Pt1 had a lower density of granulocytes and a higher density of Th1-like macrophages compared with the primary tumor from Pt2 (Fig 3B). Among subsets of CD4+ T cells, we found more pro-inflammatory Th1 T cells in the metastasis from Pt1 than either primary tumor (Fig 3C). Neither Th2 nor Treg cells were significantly different among the tumors. Within CD4+ T cells, we found a greater density of both cytolytic granzyme B+ (GRZB) cells and proliferative Ki67+ cells in the metastasis from Pt1 than the primary tumor from Pt2 and a higher density of proliferative Ki67+ cells in Pt1's primary tumor compared with Pt2's primary tumor (Fig 3D). Consistent with the abundant Th1-like macrophages in Pt1's primary tumor, we found that the ratio of Th1-like to Th2-like macrophages was significantly greater in Pt1's primary tumor relative to the other 2 tumors (Fig 3E). A high granulocyte/neutrophil to lymphocyte ratio is associated with poor outcomes.35,36 Consistent with this, the primary tumor from Pt2 had a significantly higher granulocyte to CD8+ T cell ratio than either tumor from Pt1 (Fig 3E). In addition, across ROIs, there was no correlation between the densities of CD8+ T cells and granulocytes in the tumors from Pt1, but there was a significant positive correlation in Pt2's primary tumor (P < .03, Fig 4A). Furthermore, the abundant granulocytes in Pt2's tumor were proximal to tumor cells and appeared within lumens (Fig 4B) where they may contribute to pathogenesis by occluding ducts.37
FIG 3.
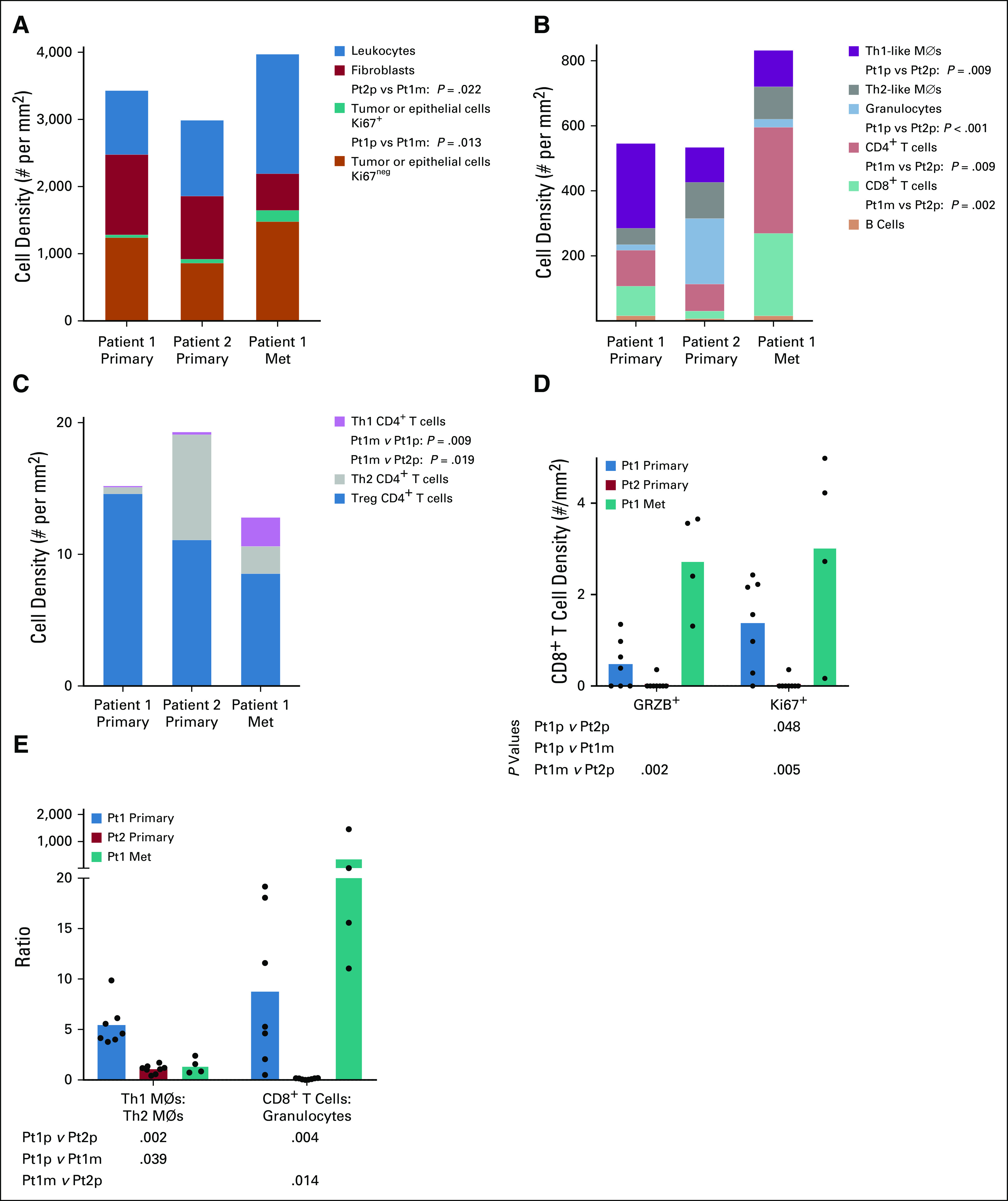
(A) Mean cell density per ROI for mutually exclusive populations of Ki67+ tumor or epithelial cells, Ki67neg tumor or epithelial cells, fibroblasts, and leukocytes. (B) Mean cell density per ROI of selected leukocyte types. (C) Mean cell density per ROI of mutually exclusive CD4+ T cell subsets. (D) Densities of CD8+GRZB+ and CD8+Ki67+ T cells. (E) Relative densities of Th1-like macrophages to Th2-like macrophages and CD8+ T cells to granulocytes. For D and E, each dot represents one ROI and column heights represent the mean of ROIs. All statistical comparisons between each of the three tumors for all cell types were tested by Kruskal-Wallis one-way ANOVA. Only P values < 0.05 are given. ANOVA, analysis of variance; ROI, region of interest.
FIG 4.

(A) Correlation between the densities of CD8+ T cells and granulocytes. Each dot represents one ROI. R2 and P values are based on a simple linear regression. (B) Granulocytes (CD66b+), tumor cells (PanKRT+), and fibroblasts (ACTA2+) in primary tumors from each patient. Scale bars represent 250 μm. ROI, region of interest.
We also observed tertiary lymph structures (TLSs) at the border of both primary tumors, but these were more prevalent in the primary tumor from Pt1 (Appendix Fig A1). TLSs from Pt1 also contained significantly more B cells, CD4+ T cells, and CD8+ T cells (Appendix Fig A2) as previously reported for patients with relatively positive outcomes.24,38
Tissue Acquisition and Patient Consent
Human tissues were obtained with informed consent in accordance with the Declaration of Helsinki and were acquired through the Oregon Pancreas Tissue Registry under Oregon Health & Science University IRB protocol #3609.
DISCUSSION
We investigated multiple modalities and tumor characteristics of PDAC between two related patients. We found several similarities between both patients' cancers, including genes commonly altered in aggressive PDAC (KRAS, TP53, and SMAD439,40) and a shared PRSS1 A16V germline mutation that may predispose to pancreatitis41-44 but is variably penetrant,45 raising the possibility that this specific alteration leads to subclinical pancreatitis that accelerates tumorigenesis, consistent with Pt2's early onset of disease.41,46
The disease course for Pt1 was exceptional in the context of occult, chemotherapy-resistant metastatic disease for nearly 4 years. We identified several differences between Pt1 and Pt2 that may alone—or in combination—account for the divergent outcomes. An FANCA mutation in the metastasis from Pt1 might have sensitized that tumor to adjuvant gemcitabine and Xeloda and later to SBRT; however, Pt1's lung metastasis progressed during adjuvant chemotherapy, and no adjuvant treatment was given after surgical resection of the lung metastasis, suggesting that the indolent disease might have also been controlled by other factors. Both tumors from Pt2 contained an alteration in CDKN2A, and the metastasis from Pt2 had a copy number loss of PTEN; alterations in these genes are associated with poor outcomes and drug resistance.47-49
Pt1's classical subtype tumors and metastases limited to the lungs are both favorable prognostic factors. Patients with classical subtype tumors survive longer (25-30 months) than patients with basal-type tumors (10-15 months)17-19 and are less likely to have recurrent, metastatic disease.14,15 Patients with metastases restricted to the lungs survive on average 23 months from diagnosis8; Pt1 has survived > 46 months with asymptomatic metastatic disease.
Our data link the indolent disease in Pt1 with reported indicators of tumor immunity. Specifically, more prevalent TLSs at the periphery of the primary tumor (Appendix Fig A1) and denser CD8+ T cells (Fig 3B) that did not correlate with the density of granulocytes24,50 (Fig 4A), opening the possibility that tumor cells and/or CD4+ T cells in Pt1's tumors recruit fundamentally different leukocyte cell types from Pt2's tumors.51 Additionally, granulocytes typically promote immunosuppression in late-stage tumors,52 consistent with the relatively low number of T cells in Pt2's tumor.
Classical subtype PDAC with lung metastases and unconventional tumor immunity may lead to exceptional outcomes. Genetic analyses and tumor leukocyte phenotyping of larger cohorts of patients with PDAC lung metastases may reveal whether metastatic organotropism is modulated by tumor immunity and/or PDAC subtype.
METHODS
See Appendix 1, Supplemental Material.
APPENDIX 1. Supplemental Methods
Tumor Somatic Alterations
Somatic alterations were identified by the Knight Diagnostics Lab at Oregon Health & Science University as previously described53,54 using the GeneTrails® Comprehensive Solid Tumor Panel.
Germline variants identified by whole exome sequencing
Genomic DNA was isolated from 10 µm tumor sections macrodissected to enrich for tumor cells. Exome sequencing libraries were prepared with a KAPA Hyper Prep Kit, enriched using the Agilent SureSelectXT Clinical Research Exome, and paired-end sequenced with an Illumina HiSeq 2500 to a high depth of 300 ×. Tumor and autologous normal blood cell whole exome sequencing (WES) data were compared to identify tumor-specific somatic alterations. WES data were processed by aligning reads to the human genome using the Burrows-Wheeler Aligner (bwa)55 and removing duplicate reads with Picard (http://broadinstitute.github.io/picard). Base quality recalibration and small insertion and deletion (indel) realignment were performed using the Genome Analysis Toolkit (GATK).56,57 Single-nucleotide variants (SNVs) were identified by MuTect58, and indels were identified using Pindel.59 All somatic mutation cells were manually reviewed using the Integrative Genomics Viewer.60 Somatic copy number alterations were identified by comparing exome sequencing coverage levels in each sample with those in a pool of normal exomes using the CNVkit algorithm.61
RNA Sequencing and Analysis
RNA was isolated from 10 µm tumor sections macrodissected to enrich for at least 40% tumor cells. RNA sequencing was performed as previously reported.62 Briefly, RNA was extracted using an RNeasy Kit (Qiagen) and evaluated by the OHSU Massively Parallel Sequencing Shared Resource (MPSSR) core facility for library construction with the TruSeq RNA Access Library Prep Kit (Illumina). All RNA samples had a %DV200 > 30% as measured using an Agilent 2100 Bioanalyzer. RNA was sequenced on an Illumina HiSeq 2500/4000 to generate 100 bp paired-end reads for a minimum depth of 40 million reads per sample. RNAseq data were preprocessed using Illumina Sequencing Control Software and CASAVA v1.8 to produce demultiplexed fastq files (http://broadinstitute.github.io/picard). The Bowtie alignment algorithm was used for read realignment to the hg19 human genome.63 UCSC Refseq gene annotations were used to calculate FPKM values.
For gene expression–based subtype assignment, we use the PDAssigner gene list and the average rank order of expression for these genes in published human PDAC samples previously designated as classical or quasimesenchymal.18 Gene expression of tumor samples from Pt1 and Pt2 was normalized to each other by calculating z-scores. We determined the Spearman rank-order correlation coefficient by comparing the average rank of the PDAssigner genes in the previously published classical and quasimesenchymal tumors with the rank of these genes in each of the four tumor samples from Pt1 and Pt2. We followed the published protocol for PurIST subtyping33 and used the same data set as for PDAssigner subtyping.
Tissue processing and immunostaining
Tumor tissue samples were fixed in 10% neutral buffered formalin for 24 hours and embedded in paraffin (FFPE). Five micrometer FFPE tissue sections were affixed to glass slides and incubated at 55°C for 12 hours and then deparaffinized with xylene. Antigen retrieval was performed with both pH 6.0 and pH 9.0 buffers under high heat and pressure. FFPE sections were stained with hematoxylin and eosin, and KRT5 was detected with a rabbit monoclonal antibody (EP1601Y, Abcam) and polyclonal goat anti-rabbit antibodies (Invitrogen). Images were acquired with a Zeiss Axio Scan Slide Scanner. Exposure times and signal thresholds were normalized across all images.
Multiplex Immunohistochemistry, Image Acquisition, and Processing
Multiplex IHC was performed on 5 μm FFPE sections using an adapted protocol based on methodology we previously described.34 Briefly, slides were deparaffinized and stained with hematoxylin (S3301, Dako, Santa Clara, CA), followed by whole slide scanning at 20 × magnification on an Aperio AT2 (Leica Biosystems, Wetzlar, Germany). Tissues were then subjected to 15 minutes of heat-mediated antigen retrieval in pH 6.0 Citra solution (BioGenex, Fremont, CA), 10 minutes of blocking in Dako Dual Endogenous Enzyme Block (S2003, Dako, Santa Clara, CA), and then 10 minutes of protein blocking with 5% normal goat serum and 2.5% BSA in TBST. Primary antibody details, dilution, and incubation times are listed in Appendix Tables A1 and A2. After washing off primary antibody in TBST, anti-rat, anti-mouse, or anti-rabbit Histofine Simple Stain MAX PO horseradish peroxidase (HRP)–conjugated polymer (Nichirei Biosciences, Tokyo, Japan) was applied for 30 minutes at room temperature, followed by AEC chromogen (Vector Laboratories, Burlingame, CA). Slides were digitally scanned following each chromogen development.
Scanned images were registered in MATLAB version R2018b using the SURF algorithm in the Computer Vision Toolbox (The MathWorks, Inc, Natick, MA). Image processing and cell quantification were performed using FIJI (FIJI Is Just ImageJ) CellProfiler Version 3.5.164 and FCS Express 6 Image Cytometry RUO (De Novo Software, Glendale, CA). AEC signal was extracted for quantification and visualization in FIJI using a custom macro for color deconvolution. Briefly, the FIJI plugin Color_Deconvolution [H AEC] was used to separate hematoxylin, followed by postprocessing steps for signal cleaning and background elimination. AEC signal was extracted in FIJI using the NIH plugin RGB_to_CMYK. Color deconvoluted images were processed in CellProfiler to quantify single-cell mean intensity signal measurements for every stained marker. Cells were classified based on hierarchical gating (image cytometry) and defined as described in Appendix Table A2. For visualization of CD45 and PanKRT staining, signal-extracted images were overlaid in pseudocolor in FIJI. QiTissue (Quantitative Imaging Systems) was used to generate pseudocolored images for Figure 4.
Statistics
Leukocyte types were analyzed if each tumor sample had an average of > 10 events per region of interest and if the average cell density for any tumor was > 1% of all leukocytes. Kruskal-Wallis one-way analysis of variance tests were performed using GraphPad Prism.
FIG A1.

(A) KRT (magenta) and CD45 (teal) expression and selected ROIs (yellow rectangles) used for mIHC analyses of tumors (scale bars represent 5 mm). Examples of tertiary lymph structures (TLSs) are indicated with white arrows. ROIs, regions of interest.
FIG A2.

(A) Representative images of lymphocytes within TLSs. Scale bars represent 250 μm. (B) Mean number of B cells, CD4+ T cells, and CD8+ T cells within ROIs containing TLSs. P values are derived from a 2-tailed t test. ROIs, regions of interest; TLS, tertiary lymph structure.
TABLE A1.
Antibodies and Targets Used in mIHC to Identify Phenotypes and Functions of Leukocytes

TABLE A2.
Target Combinations Used to Define the Leukocyte Types Reported

SUPPORT
Supported by the Brenden-Colson Foundation and NCI grant U01 CA224012 to R.C. Sears and L.M. Coussens; and a Brenden-Colson Center for Pancreatic Care pilot project grant to J.M. Link.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/cci/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Carl R. Pelz
Consulting or Advisory Role, Immediate Family Member: Novartis
Charles D. Lopez
Consulting or Advisory Role: Boston Scientific, Celgene, Boston Biomedical, Pfizer, Exelixis, Astellas Pharma
Research Funding: Taiho Pharmaceutical
Travel, Accommodations, Expenses: RenovoRx
Lisa M. Coussens
Employment: Oregon Health & Science University (OHSU)
Honoraria: Prospect Creek Foundation, Lustgarten Foundation for Pancreatic Cancer Research, Syndax Pharmaceuticals, Inc: External Advisory Board, Carisma Therapeutics Inc: Scientific Advisory Board, Verseau Therapeutics, Inc, Scientific Advisory Board, Zymeworks, Inc, Scientific Advisory Board, CytomX Therapeutics, Inc, Kineta Inc, (P30) Koch Institute for Integrated Cancer Research, Massachusetts Inst. of Tech, (P30) Salk Institute Cancer Center, Bloomberg-Kimmel Institute for Cancer Immunotherapy, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Dana Farber Cancer Center Breast SPORE, (P30) Dana Farber/Harvard Cancer Center, (P30) University of California, San Diego Moores Cancer Center, Starr Cancer Consortium, Lustgarten Foundation for Pancreatic Cancer Research, Therapeutics Working Group, NIH/NCI-Frederick National Laboratory Advisory Committee (FNLAC), Susan G Komen Foundation, Komen Scholar
Consulting or Advisory Role: Cell Signaling Technologies, Pharmacyclics, CytomX Therapeutics, Syndax, Carisma Therapeutics, Verseau Therapeutics, Zymeworks, Kineta, Inc, Abbvie, Shasqi Inc
Research Funding: Syndax Pharmaceuticals Inc, Pharmacyclics, Cell Signaling Technologies, Innate Pharma, Deciphera
Travel, Accommodations, Expenses: Cell Signaling Technologies, AstraZeneca, Pharmacyclics, Verseau, Carisma Therapeutics, CytomX Therapeutics, Zymeworks
Other Relationship: Prospect Creek Foundation, Lustgarten Foundation for Pancreatic Cancer Research, (P30) Koch Institute for Integrated Cancer Research, Massachusetts Inst. of Tech. (2012-present; honorarium), (P30) Salk Institute Cancer Center: (2016-2020; honorarium), Bloomberg-Kimmel Institute for Cancer Immunotherapy, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins: (2016-present; honorarium), Dana Farber Cancer Center Breast SPORE: (2017-present; honorarium), (P30) Dana Farber/Harvard Cancer Center: (2019-present; honorarium), (P30) University of California, San Diego Moores Cancer Center (2019-present; honorarium), Cancer Research Institute (CRI): (2013-present; unpaid), The V Foundation for Cancer Research): (2013-present; unpaid), Starr Cancer Consortium: (2011-present, honorarium), Lustgarten Foundation for Pancreatic Cancer Research, Therapeutics Working Group: (2019-present; paid), NIH/NCI-Frederick National Laboratory Advisory Committee (FNLAC): (2016-present; daily honorarium), Susan G Komen Foundation, Komen Scholar (2020 – 2023; honorarium)
Rosalie C. Sears
Consulting or Advisory Role: Novartis
No other potential conflicts of interest were reported
AUTHOR CONTRIBUTIONS
Conception and design: Jason M. Link, Courtney B. Betts, Charles D. Lopez, Lisa M. Coussens, Rosalie C. Sears
Financial support: Lisa M. Coussens, Rosalie C. Sears
Administrative support: Dove Keith
Provision of study materials or patients: Lisa M. Coussens
Collection and assembly of data: Jason M. Link, Shannon M. Liudahl, Kenna R. Leis, Mary McDonnell, Brett Johnson, Kelly J. Hamman, Dove Keith, Terry K. Morgan, Charles D. Lopez, Lisa M. Coussens, Rosalie C. Sears
Data analysis and interpretation: Jason M. Link, Shannon M. Liudahl, Courtney B. Betts, Shamilene Sivagnanam, Carl R. Pelz, Brett Johnson, Kelly J. Hamman, Jone E. Sampson, Charles D. Lopez, Lisa M. Coussens, Rosalie C. Sears
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
REFERENCES
- 1.Hidalgo M, Cascinu S, Kleeff J, et al. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes Pancreatology 158–182015 [DOI] [PubMed] [Google Scholar]
- 2.Cancer Facts & Figures 2018. American Cancer Society; 2018. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2018.html [Google Scholar]
- 3.Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer N Engl J Med 3792395–24062018 [DOI] [PubMed] [Google Scholar]
- 4.Iacobuzio-Donahue CA, Fu B, Yachida S, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer J Clin Oncol 271806–18132009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer Nature 4671114–11172010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makohon-Moore AP, Zhang M, Reiter JG, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer Nat Genet 49358–3662017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katz MHG, Wang H, Fleming JB, et al. Long-term survival after multidisciplinary management of resected pancreatic adenocarcinoma Ann Surg Oncol 16836–8472009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Downs-Canner S, Zenati M, Boone BA, et al. The indolent nature of pulmonary metastases from ductal adenocarcinoma of the pancreas J Surg Oncol 11280–852015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deeb A, Haque SU, Olowokure O.Pulmonary metastases in pancreatic cancer, is there a survival influence? J Gastrointest Oncol 6E48–E512015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasukawa M, Kawaguchi T, Kawai N, et al. Surgical treatment for pulmonary metastasis of pancreatic ductal adenocarcinoma: Study of 12 cases Anticancer Res 375573–55762017 [DOI] [PubMed] [Google Scholar]
- 11.Follia L, Ferrero G, Mandili G, et al. Integrative analysis of novel metabolic subtypes in pancreatic cancer fosters new prognostic biomarkers. Front Oncol. 2019;9:115. doi: 10.3389/fonc.2019.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daemen A, Peterson D, Sahu N, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors Proc Natl Acad Sci U S A 112E4410–E44172015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan-Seng-Yue M, Kim JC, Wilson GW, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution Nat Genet 52231–2402020 [DOI] [PubMed] [Google Scholar]
- 14.Connor AA, Denroche RE, Jang GH, et al. Integration of genomic and transcriptional features in pancreatic cancer reveals increased cell cycle progression in metastases Cancer Cell 35267–282.e72019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puleo F, Nicolle R, Blum Y, et al. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features Gastroenterology 1551999–2013.e32018 [DOI] [PubMed] [Google Scholar]
- 16.Somerville TD, Biffi G, Daßler-Plenker J, et al. Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. eLife. 2020;9:e53381. doi: 10.7554/eLife.53381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer Nature 53147–522016 [DOI] [PubMed] [Google Scholar]
- 18.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy Nat Med 17500–5032011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raphael BJ, Hruban RH, Aguirre AJ, et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma Cancer Cell 32185–203.e132017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Santiago I, Yau C, Heij L, et al. Immunophenotypes of pancreatic ductal adenocarcinoma: Meta-analysis of transcriptional subtypes Int J Cancer 1451125–11372019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma Nat Genet 471168–11782015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knudsen ES, Vail P, Balaji U, et al. Stratification of pancreatic ductal adenocarcinoma: Combinatorial genetic, stromal, and immunologic markers Clin Cancer Res 234429–44402017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balli D, Rech AJ, Stanger BZ, et al. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer Clin Cancer Res 233129–31382017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stromnes IM, Hulbert A, Pierce RH, et al. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma Cancer Immunol Res 5978–9912017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balachandran VP, Łuksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer Nature 551512–5162017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunderson AJ, Kaneda MM, Tsujikawa T, et al. Bruton's tyrosine kinase (BTK)-dependent immune cell crosstalk drives pancreas cancer Cancer Discov 6270–2852016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joyce JA, Fearon DT.T cell exclusion, immune privilege, and the tumor microenvironment Science 34874–802015 [DOI] [PubMed] [Google Scholar]
- 28.Bauer C, Kühnemuth B, Duewell P, et al. Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer Cancer Lett 381259–2682016 [DOI] [PubMed] [Google Scholar]
- 29.Gunderson AJ, Coussens LM.B cells and their mediators as targets for therapy in solid tumors Exp Cell Res 3191644–16492013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of a MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity Gut 631769–17812014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Byrne KT, Yan F, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy Immunity 49178–193.e72018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi A, Fan J, Chen R, et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma Nat Cancer 159–742020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rashid NU, Peng XL, Jin C, et al. Purity independent subtyping of tumors (PurIST), A clinically robust, single-sample classifier for tumor subtyping in pancreatic cancer Clin Cancer Res 2682–922020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsujikawa T, Kumar S, Borkar RN, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis Cell Rep 19203–2172017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y, Wei Q, Fan J, et al. Prognostic role of the neutrophil-to-lymphocyte ratio in pancreatic cancer: A meta-analysis containing 8252 patients Clin Chim Acta 479181–1892018 [DOI] [PubMed] [Google Scholar]
- 36.Giakoustidis A, Neofytou K, Costa Neves M, et al. Identifying the role of neutrophil-to-lymphocyte ratio and platelets-to-lymphocyte ratio as prognostic markers in patients undergoing resection of pancreatic ductal adenocarcinoma Ann Hepatobiliary Pancreat Surg 22197–2072018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leppkes M, Maueröder C, Hirth S, et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis Nat Commun 710973–110132016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation Cancer Immunol Res 2616–6312014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blackford A, Serrano OK, Wolfgang CL, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer Clin Cancer Res 154674–46792009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herman JM, Jabbour SK, Lin SH, et al. Smad4 loss correlates with higher rates of local and distant failure in pancreatic adenocarcinoma patients receiving adjuvant chemoradiation Pancreas 47208–2122018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rebours V, Boutron-Ruault MC, Schnee M, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: A national exhaustive series Am J Gastroenterol 103111–1192008 [DOI] [PubMed] [Google Scholar]
- 42.Weiss FU. Pancreatic cancer risk in hereditary pancreatitis. Front Physiol. 2014;5:70. doi: 10.3389/fphys.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schubert S, Traub F, Brakensiek K, et al. CFTR, SPINK1, PRSS1, and CTRC mutations are not associated with pancreatic cancer in German patients. Pancreas. 2014;43:1078. doi: 10.1097/MPA.0000000000000166. [DOI] [PubMed] [Google Scholar]
- 44.Teich N, Rosendahl J, Tóth M, et al. Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis Hum Mutat 27721–7302006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grocock CJ, Rebours V, Delhaye MN, et al. The variable phenotype of the p.A16V mutation of cationic trypsinogen (PRSS1) in pancreatitis families Gut 59357–3632010 [DOI] [PubMed] [Google Scholar]
- 46.McWilliams RR, Maisonneuve P, Bamlet WR, et al. Risk factors for early-onset and very-early-onset pancreatic adenocarcinoma: A pancreatic cancer case-control consortium (PanC4) analysis Pancreas 45311–3162016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim M, Shah A, Zhang J, et al. Gemcitabine resistance in cultured pancreatic tumor cells is associated with down regulation of PTEN and a mesenchymal phenotype. Cancer Res. 2008;68:2460. [Google Scholar]
- 48.Ferrer M, de Winter JP, Mastenbroek DC, et al. Chemosensitizing tumor cells by targeting the Fanconi anemia pathway with an adenovirus overexpressing dominant-negative FANCA Cancer Gene Ther 11539–5462004 [DOI] [PubMed] [Google Scholar]
- 49.Doyle A, Kubler MM, Harris AC, et al. The impact of CDKN2A mutations on overall survival in pancreatic adenocarcinoma. J Clin Oncol. 2019;37:278. [Google Scholar]
- 50.Chao T, Furth EE, Vonderheide RH.CXCR2-Dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma Cancer Immunol Res 4968–9822016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.SenGupta S, Subramanian BC, Parent CA.Getting TANned: How the tumor microenvironment drives neutrophil recruitment J Leukoc Biol 105449–4622019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lecot P, Sarabi M, Pereira Abrantes M, et al. Neutrophil heterogeneity in cancer: From biology to therapies. Front Immnuol. 2019;10:2155. doi: 10.3389/fimmu.2019.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corless CL.Next-generation sequencing in cancer diagnostics J Mol Diagn 18813–8162016 [DOI] [PubMed] [Google Scholar]
- 54.Mitri ZI, Parmar S, Johnson B, et al. Implementing a comprehensive translational oncology platform: From molecular testing to actionability. J Transl Med. 2018;16:358. doi: 10.1186/s12967-018-1733-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Durbin R.Fast and accurate short read alignment with Burrows–Wheeler transform Bioinformatics 251754–17602009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data Genome Res 201297–13032010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data Nat Genet 43491–4982011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cibulskis K, Lawrence M, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples Nat Biotechnol 31213–2192013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ye K, Schulz MH, Long Q, et al. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads Bioinformatics 252865–28712009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer Nat Biotechnol 2924–262011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Talevich E, Shain AH, Botton T, et al. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daemen A, Griffith OL, Heiser LM, et al. Modeling precision treatment of breast cancer. Genome Biol. 2013;14:R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan A, Prassas I, Dimitromanolakis A, et al. Validation of biomarkers that complement CA19.9 in detecting early pancreatic cancer Clin Cancer Res 205787–57952014 [DOI] [PMC free article] [PubMed] [Google Scholar]