

SUMMARY
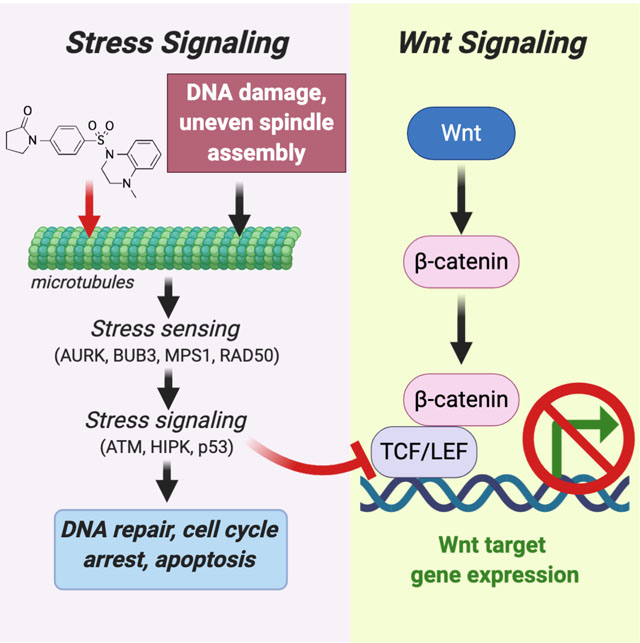
Wnt signaling plays a central role in tissue maintenance and cancer. Wnt activates downstream genes through β-catenin, which interacts with TCF/LEF transcription factors. A major question is how this signaling is coordinated relative to tissue organization and renewal. We used a recently described class of small molecules that binds tubulin to reveal a molecular cascade linking stress signaling through ATM, HIPK2 and p53 to the regulation of TCF/LEF transcriptional activity. These data suggest a mechanism by which mitotic and genotoxic stress can indirectly modulate Wnt responsiveness to exert coherent control over cell shape and renewal. These findings have implications for understanding tissue morphogenesis and small molecule anti-cancer therapeutics.
Keywords: Wnt, beta-catenin, p53, ataxia telangiectasia mutated/ATM, homeodomain interacting protein kinase/HIPK2, aurora kinases, TCF
Graphical Abstract
eTOC Blurb
Cheng et al used high throughput screening to identify a small molecule with dual p53 agonist and Wnt inhibitory activity. Named PAWI, the compounds were used to trace a kinase cascade linking stress signaling to the regulation of TCF/LEF function and the control of Wnt-dependent gene expression.
INTRODUCTION
Small molecules have been important for chemical genetics exploration of intracellular pathways involved in normal and pathological processes. For Wnt signaling, characterized small molecule modulators affect multiple components of the canonical pathway (Voronkov and Krauss, 2013; Zhang and Hao, 2015). These include proteins involved in the secretion of the soluble Wnt protein, as well as the intracellular signaling pathway leading from the Frizzled receptors to the stabilization of β-catenin, which is the effector of Wnt signaling responsible for the transcriptional activation of target genes. Small molecule inhibitors of the core pathway have helped define Wnt/β-catenin signaling as central to maintaining the balance between stem cell renewal and differentiation (Farin et al., 2016; Lien et al., 2014; Sato et al., 2004; Willems et al., 2011) as well as in the uncontrolled cell growth of cancer (Lau et al., 2013; Lei et al., 2015; Liu et al., 2013).
β-Catenin does not recognize sites in DNA directly but interacts with specific genetic loci through physical association with transcription factors, most notably T-cell factor/lymphoid enhancer factor TCF/LEF proteins (TCF1, TCF3, TCF4 and LEF1). Genetic manipulation of TCFs closely resembles those of Wnt ligands and receptors, showing that these proteins are critical mediators of Wnt signaling (Arce et al., 2006; Doumpas et al., 2019).
Understanding TCF regulation is important because diverse inputs, including mitotic stress and cell cycle status, regulate TCF transactivation and could, therefore, provide context-dependent repression or activation of Wnt/β-catenin-responsive loci (Cadigan and Waterman, 2012; Sokol, 2011). For instance, disease states, such as cancer, affect the interaction of TCF proteins with β-catenin and alter Wnt responsiveness (Arce et al., 2006; Nusse and Clevers, 2017). TCF1, 4 and LEF1 are most commonly associated with β-catenin-mediated gene activation, whereas TCF3 and less prevalently TCF4 are linked to gene repression during fetal development (Cadigan and Waterman, 2012). The availability of TCFs for partnering with β-catenin is regulated by phosphorylation by HIPK2 (Hikasa and Sokol, 2011; Lee et al., 2009), however there is only a rudimentary understanding of the cues that dictate TCF availability and contextual competence to respond to Wnt (Nusse and Clevers, 2017).
We have recently reported the anti-cancer activity of a small molecule that binds tubulin and acts as both a p53 activator and Wnt signaling inhibitor (Cheng et al., 2018). Here, we describe studies that used this compound to delineate a kinase cascade that responds to DNA damage and mitotic spindle assembly stress to modulate Wnt signaling. Wnts have been shown to regulate microtubule dynamics (Ciani et al., 2004; Schlesinger et al., 1999; Stanganello et al., 2019); however, to our knowledge, this is the first report to define a molecular cascade from microtubules that regulates Wnt responsiveness. This finding has implications for understanding the influence of stress signaling on cell renewal and tissue morphogenesis.
RESULTS
Small molecules that inhibit Wnt/β-catenin transactivation of target genes
We identified PAWI-1 in a high throughput screen of ~76,000 drug-like compounds designed to discover molecular probes of orthogonal signaling that regulates the ability of Wnt/β-catenin to induce target genes (screen schematic is shown in Fig. 1A and Supplementary Fig. S1). Fig. 1B shows the structures of the screen hit PAWI-1 and a more potent analogue, PAWI-2, developed based on a structure-activity relationship study (Cashman et al., 2016; Okolotowicz et al., 2018). The two analogues functioned identically in all assays tested but with different potencies (IC50 values for inhibition of Wnt3a in HEK293T cells are ~25 and ~11 nM for PAWI-1 and PAWI-2, respectively). The maximal inhibition of both compounds were similar to that observed with an IWR analogue (IC50 = ~24 nM), that blocks Wnt signaling by stabilizing Axin (Chen et al., 2009; Lanier et al., 2012). Consistent with the effects on TCF-luciferase reporters, PAWI-1 also inhibited the expression of genes regulated by endogenous β-catenin signaling in SW480 colorectal adenocarcinoma cells, including c-MYC, CCND and AXIN2 (Fig. 1C). In contrast, PAWI-1 had no effect on the transcriptional responses of NF-kB, Notch, and TGFβ pathways (Fig.1D), suggesting that PAWI-1 did not cause a general inhibition of transcriptional activity.
Figure 1. Chemical structures and effect of Compounds 1 and 2 on Wnt.
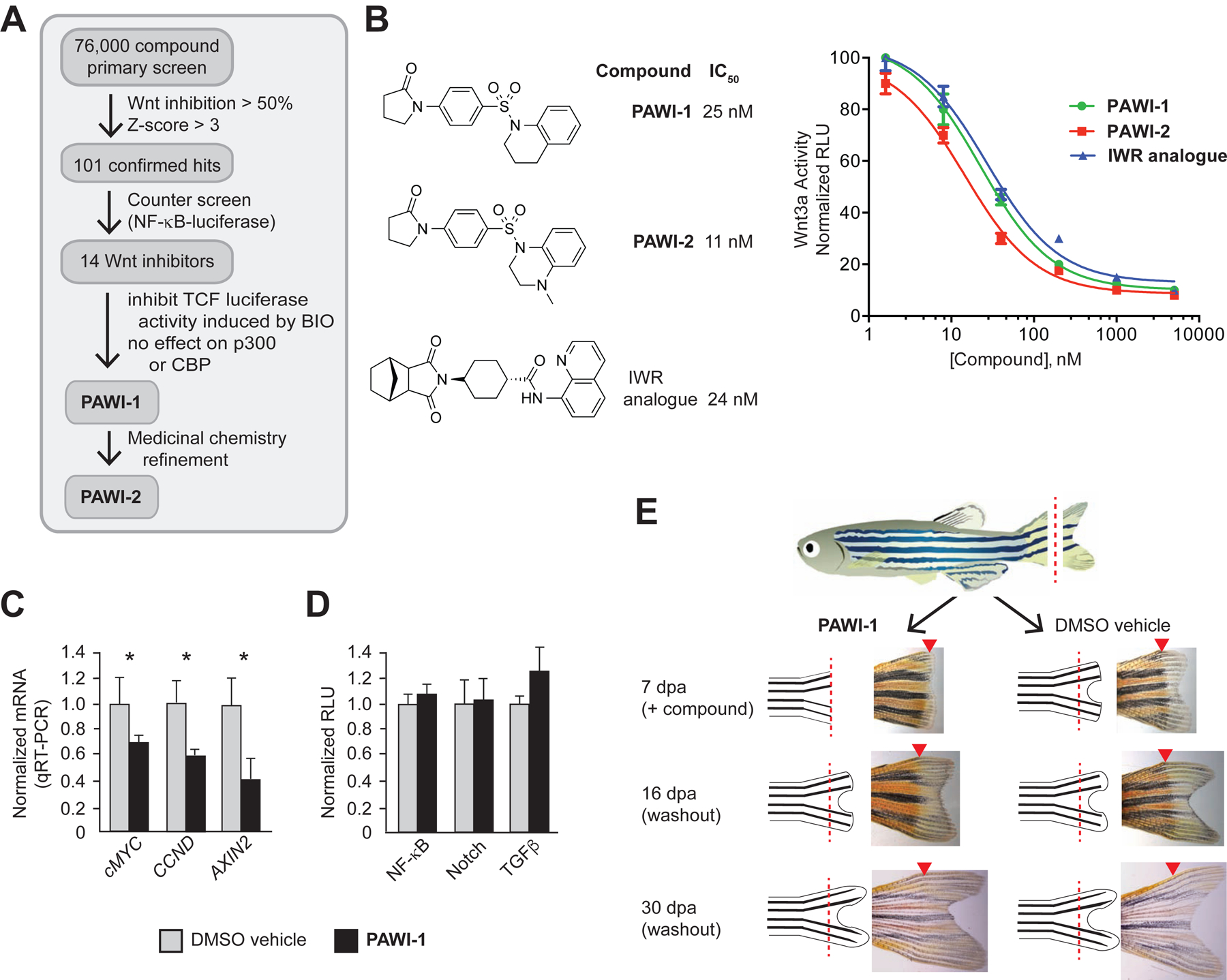
A) Scheme for high throughput screen. The primary assay quantified inhibition of Wnt3a in an RKO cell line engineered with a TCF-responsive firefly luciferase reporter and internal control Renilla luciferase. 76,000 small molecules were screened (see Methods). To rule out non-specific inhibitors, a NF-κB responsive luciferase reporter assay was used as a counter screen (see Methods). Inhibitors likely to act at or downstream of β-catenin stabilization were selected based upon inhibition of the GSK3 inhibitor BIO in the RKO TCF-luciferase primary screen assay.
B) Structures of PAWI-1, PAWI-2 and an IWR-1 analogue (Lanier et al., 2012; Willems et al., 2011). IC50 values for inhibition of Wnt3a (100 ng/ml)-stimulated Super TOPflash TCF-luciferase reporter activity were determined in HEK293T cells; n=6; error bars, s.e.m.
C) PAWI-1 (500 nM) effect on induction of Wnt/β-catenin target genes c-MYC, CCND and AXIN2, as compared to DMSO vehicle alone control, in SW480 cells that have endogenous Wnt signaling. n=4; error bars, s.e.m. * indicates p-value <0.05 (T-test).
D) Effect of PAWI-1 (500 nM) on NF-κB, Notch and TGFβ signaling pathways (black bars), as determined using specific luciferase reporters in HEK293T cells (see Methods) and compared to DMSO vehicle controls (light bars). n=4; error bars, s.e.m. * indicates p-value <0.05 (T-test).
E) Effect of PAWI-1 (500 nM) on adult zebrafish tail fin regeneration. Compound was present for first 7 days post amputation (dpa) and removed for remaining time to 30 dpa (washout). PAWI-1 inhibited regeneration at 7dpa, but regeneration resumed after washout (16 and 30 dpa) indicating reversibility. Arrowheads (red) indicate the point of resection.
We recently reported (Cheng et al., 2018) that these compounds block Wnt signaling and simultaneously activate p53 indirectly by binding to the colchicine site on tubulin (Cheng et al., 2018). Hence, these compounds are termed PAWI-1 and PAWI-2 (p53 activator and Wnt inhibitor-1). To our knowledge, microtubule function or dynamics has not been shown to impact Wnt signaling. Microtubules mediate diverse processes from DNA replication to cell motility and cytoarchitecture that play key roles in development, tissue homeostasis and disease. Hence, revealing links to Wnt and p53 might be fundamentally important.
Reversible inhibition of tissue regeneration in vivo
Zebrafish tailfin regeneration is a common assay of Wnt-responsiveness that can be used to test in vivo efficacy and reversibility. Complete regeneration of the adult zebrafish tailfin occurs within 3–4 weeks after surgical amputation, and depends on Wnt and other signals to sustain a high rate of progenitor cell proliferation in the wound blastema (Stoick-Cooper et al., 2007). PAWI-1 (500 nM) added to aquarium water showed nearly complete block of regeneration that occurred normally in DMSO vehicle-treated control fish (Fig. 1E). To test reversibility, the aquarium water was exchanged for water without PAWI-1 at 7 days post amputation (dpa). Following washout, the amputated tails regenerated substantially by 16 dpa and became indistinguishable from those of DMSO vehicle-treated animals by 30 dpa (Fig. 1E). These data suggest reversibility in vivo.
TCF activity
Based on the potency to inhibit BIO-stimulated TCF activity in HEK293T cells (Supplementary Fig. 1B), we inferred that PAWI-1 and PAWI-2 might function downstream of β-catenin stabilization (diagrammed in Fig. 2A). To confirm this hypothesis, we tested whether PAWI-1 would inhibit a constitutively active form of β-catenin generated by eliminating the phosphorylation sites necessary for targeted degradation (Taelman et al., 2010). As expected for a core pathway inhibitor, the IWR-1 analogue did not inhibit constitutively active β-catenin expressed in HEK293T cells (Fig. 2B). In contrast, PAWI-1 showed dose-dependent inhibition of the constitutively active β-catenin protein (Fig. 2B) with a potency (IC50 = ~45 nM) comparable to that in the Wnt3a luciferase assay (Fig. 1B). PAWI-1 did not alter either the abundance or nuclear localization of β-catenin (Supplementary Fig. S2), suggesting that β-catenin itself was unaffected by PAWI-1.
Figure 2. Inhibition of TCF transcriptional activity.
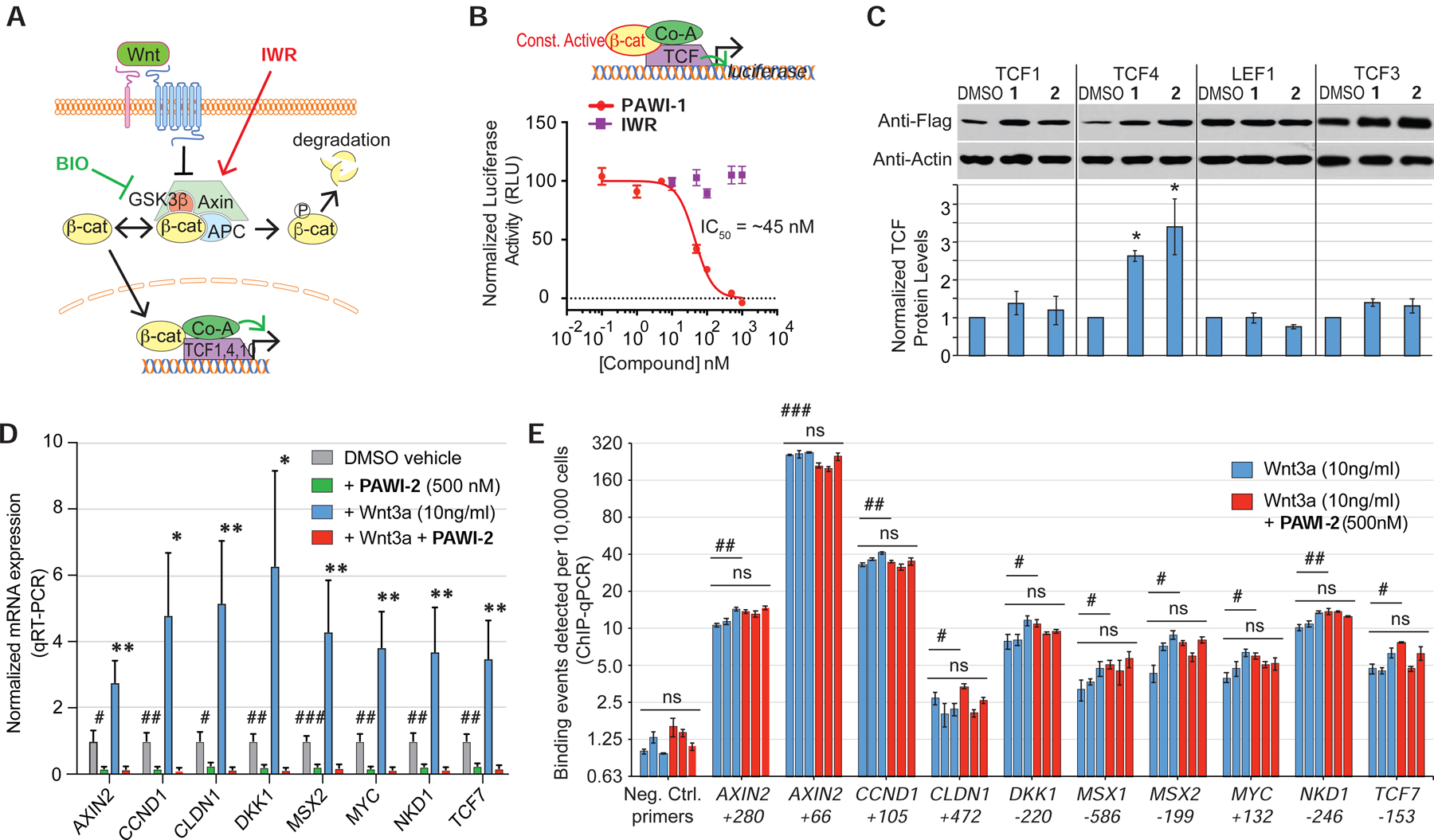
A) Schematic depicting the core canonical Wnt signaling cascade.
B) Scheme depicts β-catenin luciferase assay. Effect of PAWI-1 and an IWR-1 analogue on a constitutively active mutant form of β-catenin protein expressed in HEK29T cells together with a TCF-luciferase reporter gene. PAWI-1 IC50 = 45.5 nM. Error bars represent s.e.m (n=3).
C) Effect of PAWI-1 and PAWI-2 on TCF proteins. HEK293T cells were transfected to express flag-tagged TCF proteins and treated with either DMSO vehicle or compounds (500 nM) for 24 hours. Western blotting shows the levels of the TCF proteins. Error bars represent s.e.m (n=3, TCF1, TCF4, LEF1; n=4, TCF3).
D) RT-qPCR showing gene expression of canonical Wnt-inducible genes in HEK293T cells at following treatment with DMSO (vehicle for PAWI-2), PAWI-2 (500 nM), recombinant Wnt3a (10 ng/ml), and Wnt3a plus PAWI-2. Data were normalized to GAPDH expression and DMSO vehicle. Error bars represent s.e.m. (n=5).
E) ChIP PCR of promoter regions of Wnt/β-catenin target genes in HEK293T cells treated as in panel D. The centers of the amplified regions are indicated. 3 biological replicates (blue and red bars). Error bars represent s.e.m. (n=3 technical replicates).
ns indicates not statistically significant; *,#; **,## and ***,### indicate p-values of <0.05, <0.01 and <0.001, respectively (T-test of biological replicates). In panel D, symbols indicate comparisons of PAWI-2-treated to respective DMSO vehicle (#) or to Wnt3a-induced (*) levels. In panel E, symbols indicate comparisons of specific TCF4 binding sites versus negative control sites (#) or to Wnt3a versus respective Wnt3a + PAWI-2 treated (ns).
We then focused on TCF proteins as distal targets of PAWI-1 and PAWI-2 because they partner with β-catenin on the promoters of Wnt responsive genes. Neither compound decreased the abundance of flag-tagged human TCF proteins in HEK293T cells (Fig. 2C), despite suppression of target gene expression induced by recombinant human Wnt3a (Fig. 2D). Given the lack of an effect on TCF/LEF protein abundance, we tested if PAWI-2 might cause TCF/LEF proteins to disengage from the promoter regions of Wnt responsive genes. We measured occupancy of TCF4 (for which there are ChIP qualified antibodies) by chromatin immunoprecipitation followed by qPCR (ChIP qPCR) at specific TCF4-binding sites of Wnt-responsive genes conserved in HEK293T and other cell types (UCSC genome browser, hg38 assembly). As expected, treatment of HEK293T cells with Wnt3a (10ng/ml) upregulated TCF4 binding (blue bars) at each promoter examined (AXIN2, CCND1, CLND1, DKK1, MSX1, MSX2, MYC, NKD1, TCF7) when compared to a ‘gene desert’ region in the genome (negative control primers, Fig. 2E). The addition of PAWI-2 (500nM) did not alter the promoter occupancy at these sites by TCF4 (red bars). Our findings were consistent with previous data suggesting that Wnt target gene expression is regulated not only by TCF occupancy, but also by the association between TCFs and other proteins located on target promoters (van Tienen et al., 2017).
Regulation of HIPK2
Phosphorylation of TCF proteins affects the responsiveness of genes to Wnt/β-catenin signaling (Hikasa et al., 2010; Hikasa and Sokol, 2011; Ishitani et al., 1999). Among the kinases implicated, homeodomain-interacting protein kinase 2 (HIPK2) is of interest because it integrates cell proliferation, differentiation, ER stress and genotoxic stimuli and, like Wnt signaling, regulates stem cell renewal (Sombroek and Hofmann, 2009). HIPK2 is predominantly nuclear-localized and has been proposed to phosphorylate TCF3, TCF4 and LEF1 proteins regulating their ability to activate genes (Hikasa and Sokol, 2011) (Fig. 3A) as well as enhance Wnt signaling by stabilizing β-catenin and disheveled (Shimizu et al., 2014; Verheyen et al., 2012). PAWI-2 treatment caused a dose-dependent phosphorylation of HIPK2 on Y361 that activates the kinase (van der Laden et al., 2015) (Fig. 3B,C). To test if HIPK2 activation would lead to TCF phosphorylation, we used the only antibody currently reported to recognize a HIPK2 phospho-site in a TCF protein [S209 in TCF3 (Hikasa and Sokol, 2011)]. PAWI-2 indeed caused a dose-dependent accumulation of phospho-S209-TCF3 (Fig. 3 B,C). The coordinated accumulation of phospho-Y362-HIPK2 and phospho-S209-TCF3 implicated HIPK2 as a mediator of the PAWI compounds. Corroborating this conclusion, PAWI-2 increased phosphorylation of the p53 protein on S46, which is also a target of activated HIPK2. (Fig. 3D,E).
Figure 3. Chemical activation of HIPK2.
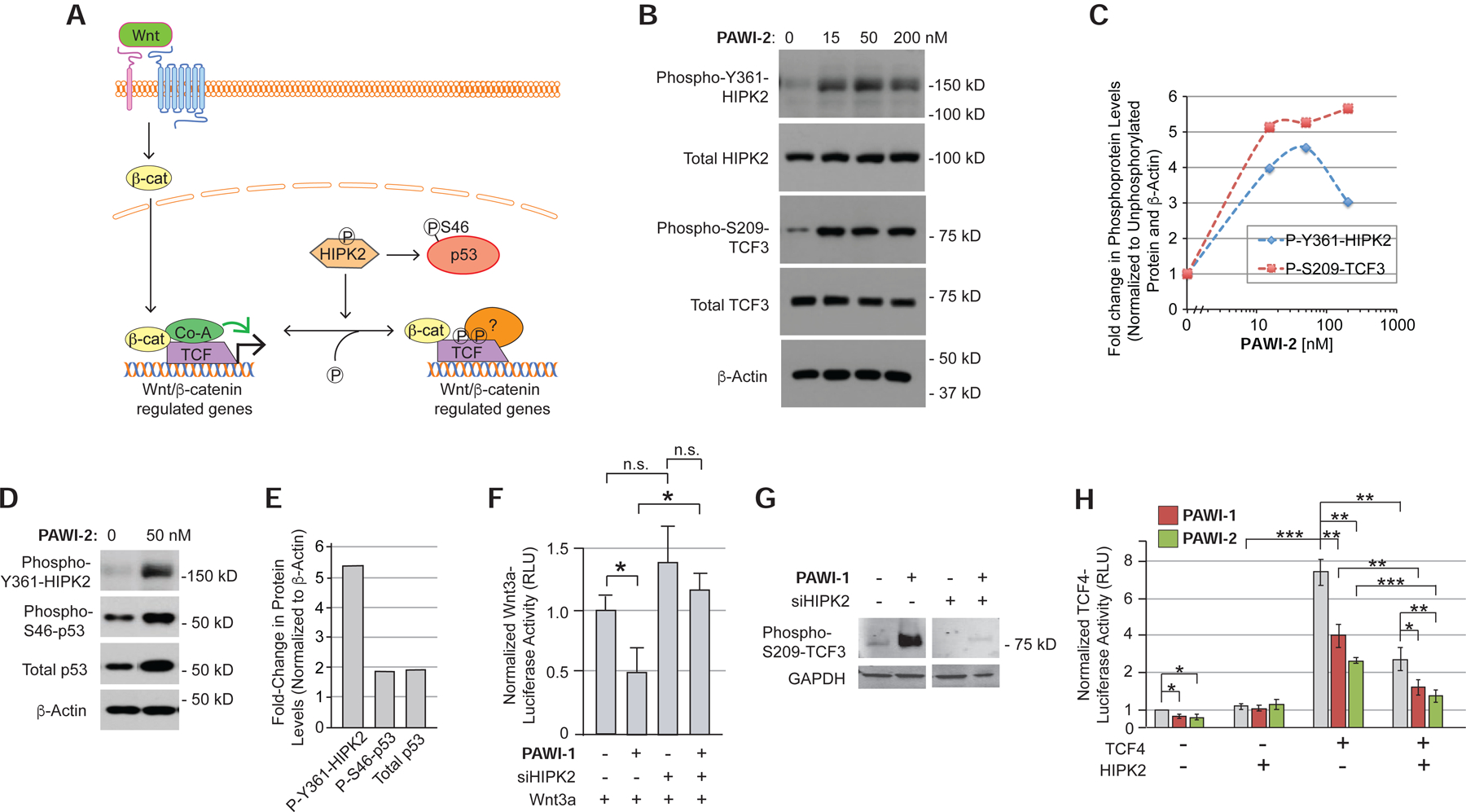
A) Model of the role of HIPK2 in regulating the stabilization of p53 and the localization of TCF proteins on chromatin.
B,C) Effect of PAWI-2 (0–200 nM, for 4 hours) on Y361 phosphorylation of endogenous HIPK2 in non-transfected HEK293T cells and S209 phosphorylation of TCF3 in HEK293T cells transfected to express flag-tagged TCF3 (B) by Western blotting. Densitometry quantification is shown in (C).
D,E) Effect of PAWI-2 (50 nM) on S46 phosphorylation and total protein levels of p53, assayed as in (B).
F) Effect of siRNA knockdown of HIPK2 on the ability of PAWI-1 (200 nM) to inhibit Wnt3a/β-catenin signaling, as measured in HEK293T cells transfected with a TCF-luciferase reporter gene and treated with recombinant Wnt3a (10% conditioned media of HEK293T cells overexpressing Wnt3a). Error bars, s.e.m. (n=6).
G) Effect of siRNA knockdown of HIPK2 on the S209 phosphorylation of TCF3 in response to PAWI-1 (200 nM).
H) Effect of HIPK2 overexpression on PAWI-1 and PAWI-2 (50 nM) inhibition of TCF4 activity, as measured using a TCF-luciferase reporter in HEK293T cells. HIPK2, PAWI-1 and PAWI-2 inhibit TCF4-dependent signaling. Error bars, s.e.m. (n=3).
*, ** and *** indicate p-values <0.05, <0.01, and <0.001 respectively (T-test).
To determine if HIPK2 were required for PAWI-1 and PAWI-2 to inhibit Wnt responsiveness, we transfected HEK293T cells with siRNA to HIPK2 (siHIPK2) together with the TCF-luciferase reporter two days prior to treatment with recombinant Wnt3a (10% conditioned media from HEK293T cells overexpressing Wnt3a). siHIPK2 completely abrogated the inhibitory effect of PAWI-1 on Wnt3a-dependent TCF-luciferase activity (Fig. 3F) whereas siRNAs to the structurally related HIPK1, 3 and 4 did not inhibit this activity (Supplementary Fig. S3A). siHIPK2 also prevented phosphorylation of TCF3 at S209 (the HIPK2 target site) in response to PAWI-1 (Fig. 3G), further supporting the role of HIPK2 as an essential mediator of PAWI-1. Overexpression of HIPK2 exerted the opposite effect, mimicking the ability of PAWI-1 and -2 to suppress TCF4 activity in HEK293T cells (Fig. 3H). Moreover, HIPK2 overexpression in combination with PAWI-1 and -2 potentiated the small molecules’ effects (Fig. 3H). A similar effect was observed using TCF3 (Supplementary Fig. S4A,B). Finally, treating blastula-stage Xenopus embryos with PAWI-1 caused anterior deficits resembling those seen by HIPK2 overexpression (Supplementary Fig. S4C–E). Although PAWI-1 activated HIPK2 in vivo, it did not increase the activity of purified recombinant HIPK2 protein in vitro (Supplementary Fig. S3B). Taken together, we concluded that PAWI-1 and PAWI-2 activate HIPK2 in vivo, but they are unlikely to target HIPK2 directly.
Activation of HIPK2 and ATM kinases
Y361 phosphorylation and activation of HIPK2 occur in response to ATM kinase, which plays a central role in signaling DNA damage and mitotic checkpoint activation (Marechal and Zou, 2013; Shiloh and Ziv, 2013). Therefore, we hypothesized that PAWI-1 and PAWI-2 might target proteins involved in activating, sensing, or mediating the mitotic checkpoint and/or DNA damage response (DDR). To test this hypothesis and identify candidate mediators within this complex target space, we conducted an siRNA screen of 98 proteins involved in sensing and responding to DNA damage and mitotic checkpoint control for their ability to block the inhibitory effect of PAWI-2 on Wnt3a/β-catenin-dependent luciferase activity (Fig. 4A–C and Supplementary Table S2). We reasoned that block of compound activity by an individual siRNA would implicate the corresponding protein as a possible mediator. siRNAs were tested by transfection using the same system as used for the primary assay as diagrammed in Fig. 4A. Two days following transfection, Wnt3a plus either PAWI-2 or DMSO vehicle was added to the cells and the firefly luciferase activity was determined 6 hours later. The effect of individual siRNAs was expressed as a Z-score relative to the effect of a control siRNA (inert sequence) (0% inhibition, Z-score = 0, blue data points) (Fig. 4B). 21 siRNAs (red data points) inhibited PAWI-2 with a Z-score ≥ 2 (Fig. 4B,C). The expanded plot in Fig. 4C shows the % inhibition observed for these siRNAs, clustered according to the major role of their protein targets in sensing or transducing mitotic stress signaling (although many of these proteins play multiple roles).
Figure 4. Activation of the ATM kinase cascade.
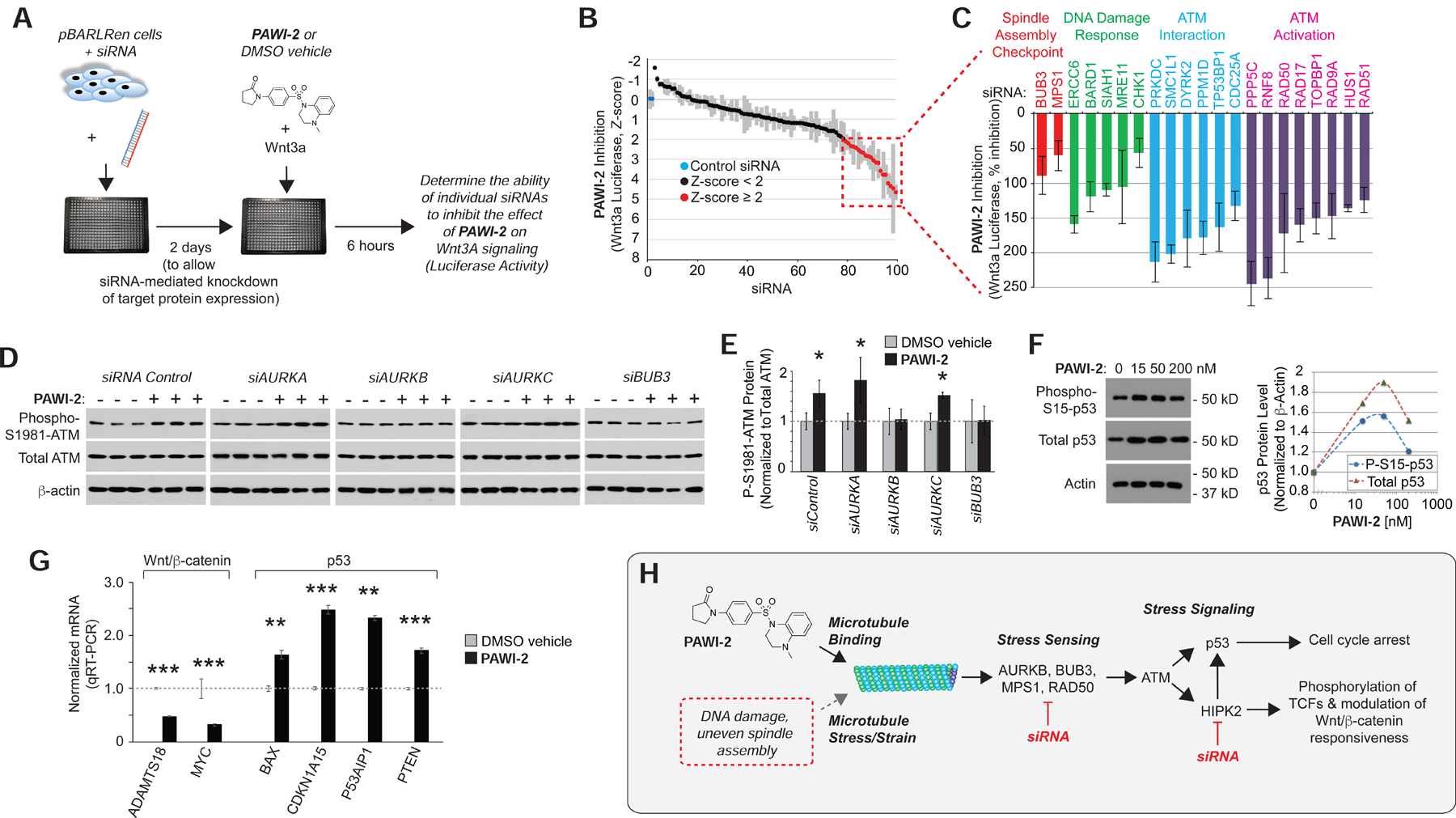
A) Design of the siRNA screen to elucidate target space. siRNAs against 98 proteins involved in ATM regulation were screened for ability to prevent inhibition of Wnt3a/β-catenin firefly luciferase activity by PAWI-2 (100 nM), measured in the TCF-responsive pBARLRen reporter cells and normalized to Renilla luciferase as in the primary screen.
B) Screen results expressed as a Z-score relative to control (inert sequence) siRNA (blue).
C) Plot of % inhibition of Wnt3a-dependent luciferase activity for siRNAs with Z-score ≥ 2 (red). n=4; error bars = s.e.m.
D,E) Western blots showing the effect of siRNA knockdown of Aurora kinases and BUB3 on the ability of PAWI-2 (200 nM, 4 hours) to induce S1981 phosphorylation of ATM in SW480 cells (D). Quantification of phospho-S1981-ATM normalized to total ATM protein levels (E). Note siRNA directed against AURKB and BUB3 prevented phosphorylation of ATM in response to PAWI-2. n=4; * indicates p-value <0.05 (T-test).
F) Western blot showing effect of PAWI-2 (0–200 nM, 4 hours) on S15 phosphorylation and total p53 in HEK293 cells, and quantification of phospho-S15 and total p53 normalized to β-actin levels.
G) PAWI-2 (100 nM, 24 hours) induction of p53 target genes and simultaneous suppression of Wnt/β-catenin target genes in HEK293T cells treated with recombinant Wnt3a (100 ng/ml) by TaqMan qRT-PCR. Error bars = s.e.m. (n=4). *,**, *** indicates p-value <0.05, <0.01, <0.005 (T-test).
H) Model that PAWI-1, 2 mimics stress signaling to activate p53 and modulate the ability of a cell to respond to Wnt. PAWI compounds bind tubulin and activate ATM and HIPK2 at lower concentrations than needed to destabilize microtubules. ATM and HIPK2 phosphorylate and activate p53. HIPK2 also phosphorylates TCF/LEF proteins modifying their availability for Wnt/β-catenin transcriptional activity. Points of siRNA block that were used to substantiate the model in panels B,C,D and Fig. 3F,G are indicated.
siRNAs to MRE11 and RAD50 potently blocked the action of PAWI-2 on the Wnt3a luciferase response (Fig. 4C). These proteins, together with NBS1 (siRNA that did not block PAWI-2, Z-score < 2, Supplementary Table S2), comprise the MRN complex that senses DNA damage and recruits ATM to the site in chromatin (Marechal and Zou, 2013; Shiloh and Ziv, 2013). Other inhibiting siRNAs also targeted proteins involved in the DDR. For instance, RNF8, RAD9A and HUS1 sense or amplify the response, and with RAD51 lead to ATM activation and DNA repair (Marechal and Zou, 2013; Shiloh and Ziv, 2013). siRNAs to PPP5C, PRKDC and SMC1L1 were among the most potent inhibitors (Fig. 4C). PPP5C is a phosphatase that interacts with ATM and regulates the DNA damage checkpoint, such that siRNA or genetic attenuation of PPP5C enhances phosphorylation and activates several stress responsive proteins including p53 and apoptosis signal-regulating kinase 1 (ASK1) that activates HIPK2 (Ali et al., 2004; Kutuzov et al., 2005; van der Laden et al., 2015). PRKDC encodes the catalytic subunit of the DNA-dependent protein kinase DNA-PK, and loss of PRKDC decreases ATM levels and sensitizes cells to DNA damage (Peng et al., 2005). SMC1L1 is a kinetochore-associated protein that maintains sister chromatid cohesion and is phosphorylated by ATM in response to DNA damage and, like DNA-PK, associates with BRCA1 and the E3 ubiquitin ligase BARD1 at sites of DNA damage (Kitagawa et al., 2004) (Yazdi et al., 2002). siRNA to BARD1 also blocked the effect of PAWI-2 on Wnt/β-catenin luciferase (Fig. 4C). BARD1 interacts with BRCA1 downstream of ATM to regulate Aurora A and B kinases to stabilize p53 and induce cell cycle arrest (Bosse et al., 2012; Fabbro et al., 2004; Feki et al., 2005; Ryser et al., 2009) and is also involved in complexes with several proteins (Greenberg et al., 2006) whose corresponding siRNAs also blocked the action of PAWI-2 on Wnt/β-catenin, including TOPBP1, MRE11, RAD50, RAD51, and RNF8 (Fig. 4C). None of the siRNAs had a significant effect on Wnt signaling in the absence of PAWI-2. Taken together, these data suggested that PAWI-2 activates DDR signaling.
Upstream of stress signaling are proteins that sense DNA damage and mitotic spindle assembly (London and Biggins, 2014; Sulli et al., 2012). The proteins BUB3 and MPS1 assemble at the kinetochore upon entry into mitosis and participate in the spindle assembly checkpoint mechanism that checks for even microtubule tension on sister chromatids (London and Biggins, 2014; Musacchio and Salmon, 2007). siRNAs to BUB3 and MPS1 inhibited the effect of PAWI-2 on the Wnt3a-dependent TCF-luciferase response (Fig. 4C). The sensing of uneven tension by these proteins activates CHK1, the Aurora kinases and ATM to pause mitosis until tension is normalized and chromosomes are properly aligned on the mitotic spindle (Musacchio and Salmon, 2007). In the absence of CHK1, cells cannot sustain mitotic arrest in response to the microtubule poison taxol (Zachos et al., 2007). siRNA to CHK1 was also among those that blocked the effect of PAWI-2 (Fig. 4C), consistent with CHK1 acting downstream of its target. Taken together, the siRNA screen suggested that PAWI-2 might act above the sensing mechanisms for proper spindle assembly (e.g., upstream of BUB3, MPS1) and DNA damage (e.g., upstream of CHK1, RAD50 and MRE11).
To confirm that PAWI-2 activates stress signaling, we examined the status of ATM, which integrates the response to DNA damage, mitotic checkpoints and oxidative stress and transduces these stimuli to a range of cellular effectors (Marechal and Zou, 2013; Shiloh and Ziv, 2013). PAWI-2 caused the phosphorylation of ATM on S1981, which is a sign of activation and autophosphorylation (Bakkenist and Kastan, 2003) (Fig. 4D,E, Supplementary Fig. S5A). S1981 phosphorylation of ATM was blocked by siRNAs to BUB3 and Aurora kinase B (AURKB) (Fig. 4D,E), both of which convey spindle defects to ATM (Musacchio and Salmon, 2007), suggesting that PAWI-2 acted upstream of these proteins. Given the evidence for ATM activation, we examined p53 and found that PAWI-2 caused a dose-dependent increase in phosphorylation at S15, which is the target site for ATM (Banin et al., 1998; Canman et al., 1998) (Fig. 4F). PAWI-2 also increased p53 target gene expression (Fig. 4G).
The observation that siRNA-mediated knockdown of proteins involved in sensing mitotic stress, including BUB3, MPS1, CHK1, RAD50 and BARD1, blocked the ability of PAWI-2 to inhibit Wnt3a/β-catenin signaling is consistent with microtubules as a target. In a recent study (Cheng et al., 2018), we showed that PAWI-2 binds tubulin and inhibits tubulin acetylation. However, the EC50 for inhibition of tubulin acetylation or activation of tubulin tyrosination (indicating that PAWI-2 can destabilize microtubules) was 144 and 166 nM, respectively, which was ~14–87 fold greater than IC50 concentrations for Wnt/β-catenin inhibition (11 nM) and p53 stabilization (1.9 nM) [Figs. 1B, 4F and (Cheng et al., 2018)]. Therefore, it appears that low levels of microtubule binding activate the ATM/HIPK2 cascade (leading to inhibition of Wnt/β-catenin signaling and stabilization of p53) but that higher occupancy is necessary for destabilization. To corroborate a link between stress signaling and Wnt/β-catenin inhibition, we tested a bonafide microtubule destabilizer taxol and the DNA damaging agent cisplatin and found that both inhibited Wnt/β-catenin signaling in the pBARLRen cell luciferase assay (IC50 = ~100nM and ~1μM, respectively, Supplementary Fig. S5B). Taken together, these data support model that PAWI-2 binding to microtubules mimics DNA damage and improper mitotic spindle assembly, and that PAWI-2 binding is sensed by the stress signaling machinery and transduces via ATM and HIPK2 to coordinately activate p53 and repress Wnt/β-catenin-dependent transcription (Fig. 4H).
DISCUSSION
Although PAWI-1 and PAWI-2 inhibit Wnt signaling, they are distinguished from core Wnt pathway inhibitors by binding to tubulin and indirectly regulate Wnt signaling. Using these molecules, we traced a signaling cascade that senses mitotic stress and acts through ATM and HIPK2 to phosphorylate TCF/LEF proteins. Consistent with prior studies of HIPK2 (Hikasa and Sokol, 2011; Lee et al., 2009), we postulate that the phosphorylation of TCFs alters their availability to activate Wnt/β-catenin target genes. To our knowledge, this is the first reported chemical biology exploration of the signaling that links stress signaling downstream of microtubule dynamics to TCF/LEF proteins and Wnt/β-catenin signaling.
A number of small molecule Wnt pathway inhibitors target proteins in the core pathway, from the production of Wnt itself to the function of the transcriptional complex and activation of downstream genes. For example, IWP (Chen et al., 2009) inhibits the acyltransferase activity of Porcupine that is needed in the production of secreted Wnt proteins. Small molecules target the PDZ domain of Disheveled and functionally disrupt its interaction with Frizzled receptors (Choi et al., 2016; Grandy et al., 2009; Ma et al., 2018). Further downstream, IWR (Chen et al., 2009) and XAV939 (Huang et al., 2009) target the stability and function of Axin, a protein that interacts with GSK3β to phosphorylate and destabilize β-catenin. Other compounds target the transcriptional co-activators CBP and p300 (e.g., ICG-001 and YH250) (Emami et al., 2004). More recently, small molecules have been developed that or disrupt the physical interaction between β-catenin and TCF (Dietrich et al., 2017) and the interaction between these proteins and the Pol II transcription machinery (Dale et al., 2015). Our data reveal a new class of compounds that inhibit downstream of β-catenin by activating stress signaling indirectly modulating the availability of TCFs.
Mitotic stress has not been mechanistically linked to Wnt and cell fate decisions, but the concept is relevant to the control of fetal development and tissue renewal. Several kinases involved in stress signaling have been implicated in stem and progenitor cell differentiation. For instance, loss of ATM or inhibition of HIPK2 increase proliferation and decrease differentiation of uncommitted adipogenic progenitors and might contribute to insulin resistance and type 2 diabetes (de la Vega et al., 2013; Takagi et al., 2015). Also, the inhibition of HIPK2 similarly affects myogenic progenitors (Sjolund et al., 2014). In addition, the structurally related DYRK2 promotes proliferation and opposes differentiation of human pluripotent stem cells in a Wnt-dependent manner (Hasegawa et al., 2012). Like HIPK2, DYRK2 phosphorylates p53 on S46 (Taira et al., 2007), showing that the two kinases might have similar substrate recognition. Accordingly, knockdown data (Supplementary Fig. S3A) suggested that DYRK2 might also mediate the Wnt inhibitory effect of PAWI-2. Our data showed that stress signaling is linked through these proteins (i.e., ATM, HIPK2, DYRK2) to p53 and Wnt-responsiveness. Consistent with this model, the DNA damaging agent Cisplatin and the microtubule destabilizer Taxol also inhibited Wnt signaling (EC50 values of ~1 μM and ~100nM, respectively, Supplementary Fig. S5B). Since both Wnt and p53 are key players in cell renewal and differentiation (Farin et al., 2016; Lien et al., 2014; Sato et al., 2004; Willems et al., 2011), we propose that mitotic, oxidative and other stresses might regulate the balance of renewal and differentiation in stem/progenitor cell pools by regulating Wnt responsiveness and p53 control of cell cycle.
The complexities of phosphorylation of the four TCF/LEF proteins that partner with β-catenin make control of stress signaling an intriguing means to modulate Wnt/β-catenin activity, both in nature and as a therapeutic approach for cancer. As discussed above, existing small molecule Wnt inhibitors target either Wnt production, core pathway components, or the ability of the β-catenin:TCF complex to activate transcription. As such, these molecules are expected to block Wnt/β-catenin signaling across many contexts. Unlike these inhibitors, activating stress signaling might provide more context-selective inhibition in part because phosphorylation of TCF3, TCF4, and LEF1 whereas TCF1 is reported to lack HIPK2 phosphorylation sites and be unresponsive to HIPK2 (Hikasa et al., 2010; Hikasa and Sokol, 2011; Ishitani et al., 1999). The expression of TCF1 in the intestinal epithelium might explain the absence of significant intestinal toxicity of PAWI-2 (Cheng et al., 2018). Moreover, such compounds would be expected to inhibit the effects of common tumorigenic mutations in the Wnt/β-catenin pathway that occur downstream of the targets of core pathway inhibitors (Zhan et al., 2017).
Interestingly, the PAWI compounds inhibited expression of the Wnt responsive genes in HEK293T cells basally and in response to exogenous recombinant Wnt3a (Fig. 2D). Addition of Wnt3a increased binding of TCF4 to conserved binding sites near the transcriptional start sites of Wnt-responsive genes but binding was unaffected by PAWI-2 (Fig. 2E). Together with the phosphorylation data, we conclude that PAWI compounds affect TCF/LEF function on chromatin and thereby inhibit Wnt responsiveness. This might be mediated by a change in interactions with partnering proteins in the promoter regions (van Tienen et al., 2017). More broadly, the compounds might modulate the activities of TCF/LEF proteins beyond Wnt signaling.
Activators of spindle and mitotic checkpoint signaling have been considered as cancer therapeutics. Specifically, compounds that alter microtubule polymerization and stability induce mitotic arrest of proliferating cells (Dumontet and Jordan, 2010). The observation that siRNA-mediated knockdown of proteins involved in sensing mitotic stress, including BUB3, MPS1, CHK1, RAD50 and BARD1, blocked the ability of PAWI-2 to inhibit Wnt3a/β-catenin signaling is consistent with microtubules as a target. We showed previously that PAWI-1,2 bind to the colchicine site on tubulin (Cheng et al., 2018). However, the IC50 values for inhibition of tubulin acetylation is ~166 nM (Cheng et al., 2018), which is 1–2 orders of magnitude greater than that for Wnt inhibition or p53 stabilization (i.e., 11 nM for Wnt inhibition and 1.9 nM for p53 stabilization, respectively, Figs. 1B and 4F). To reconcile these findings, we propose that low levels of microtubule binding are sufficient to activate stress signaling and inhibit Wnt while considerably higher occupancy is needed for microtubule destabilization. Concomitant activation of p53 and inhibition of aberrant Wnt/β-catenin signaling by PAWI-2 could be therapeutically advantageous in tumors with Wnt/β-catenin dependency, including pancreatic ductal adenocarcinoma, for which Wnt signaling has been associated with resistance to the first-line therapeutic gemcitabine (Singh et al., 2010), and colorectal cancers, which frequently involve mutation and/or overexpression of Wnt/β-catenin pathway components (Schatoff et al., 2017). In a companion paper (Cheng et al., 2018), we showed that PAWI-2 inhibited the migration, invasion and epithelial-mesenchymal transition of colorectal cancer cell lines (HCT-116, DLD-1, SW480, and 10.1) in vitro, and induced cell death and inhibited cell growth in mouse xenografts without damaging DNA or causing significant toxicity.
In summary, we have used PAWI-1 and PAWI-2 as chemical probes to uncover a kinase cascade that associates DNA damage and mitotic stress to the regulation of p53 and the ability of a cell to respond to Wnt. From a biological perspective, the molecular association of genome integrity and microtubule dynamics to that activity of Wnt and p53 is important because it enables cells to exert coherent control over structural and signaling aspects of cell motility, cytoarchitecture and renewal. In terms of translational potential, the results might address the concern that canonical Wnt pathway inhibitors as cancer therapeutics might lack sufficient selectivity and/or have too narrow a therapeutic window (Blagodatski et al., 2014; Kahn, 2014; Lu et al., 2016). Our results might broaden the scope of candidates to include compounds that target signaling that acts orthogonally to the core pathway and coordinates activation of p53 with the inhibition of Wnt signaling.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mark Mercola (mmercola@stanford.edu)
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study. This study did not generate code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and Plasmids
The pBARLRen cell line was engineered from the RKO cell line (male) by Drs. Travis Biechele and Randy Moon at University of Washington, Seattle (Biechele et al., 2009) and kindly provided to us. The NF-κB reporter (293-NF-kB-Luc) expressing NOD1 (HEK293-NF-kB-Luc-NOD1) was provided by Dr. John Reed at Sanford-Burnham-Prebys Medical Discovery Institute. HEK293T (female) and SW480 (male) cell lines were from the ATCC.
Plasmids encoding the MYC-tagged TCF4, the wildtype and constitutively active mutant forms of β-catenin (with mutations of S33A, S37A, T41A, and S45A), CBP (pcDNA3β-FLAG-CBP-HA), p300 (pCMVβ-p300-MYC), HIPK2 (myc-tagged HIPK2) and Wnt3a (pcDNA-Wnt3a) were from Addgene. Plasmids encoding FLAG-tagged TCF1, TCF3, TCF4 and LEF1 were kind gifts from Dr. Sergei Sokol (Mount Sinai School of Medicine). Cells were tested monthly for mycoplasma. Cells (and any data) were discarded if positive.
Cell culture
pBARLRen cells, HEK293T cells, and SW480 cells were cultured at 37 °C in a 5% CO2 incubator in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS for maintenance culture and 5% for the luciferase assays in the screens. HEK293T or SW480 cells were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. For transient transfection luciferase assays, cells were transfected with 20 ng of reporter plasmids and the same amounts of other plasmids as indicated in 24-well plate format. Compound was added immediately following transfection, if not indicated otherwise, and Western blot or luciferase assays were performed 24 hours after transfection. Luciferase assays were performed using either SteadyLite HTS (PerkinElmer) for Firefly luciferase or Dual-Glo Luciferase kit (Promega) for both Firefly and Renilla luciferase.
METHOD DETAILS
High throughput chemical primary screen
The primary screen process is diagrammed in Supplementary Fig. 1 and details are in Supplementary Table 1. ~600 RKO pBARLRen cells were seeded per well in 1536-well clear bottom plates (Corning) in 3 μL of culture medium supplemented with 2% FBS. 24 hours after plating, 12.5 nl of 2 mM compound and 2 μl of 25% Wnt3a conditioned media were added to the individual wells to achieve final concentrations of 5 μM compound and 10% Wnt3a conditioned media, respectively. 24 hours later, a luciferase assay was conducted by addition of 10 μl of SteadyLite HTS solution (PerkinElmer) and luminescence measured on an EnVision plate reader (PerkinElmer). Hits were selected based on inhibition > 50% and Z-score >3 (see Statistics section below). Activities of primary hit compounds were confirmed by re-testing using Dual-Glo (Promega) to measure both Firefly and Renilla luciferase activities.
NF-κB, Notch and TGFβ counter and secondary screening
A counter screen was conducted using HEK293T cells stably transfected with NF-κB responsive luciferase reporter system and NOD1 (to activate the NF-κB reporter, Pubchem AID 1290). Briefly, HEK293-NF-kB-Luc-NOD1 stable cells were seeded at a density of ~600 cells/ well in 1536-well clear bottom plates (Corning) and confirmed hit compounds were added to give a final concentration of 5 μM. A luciferase assay was conducted 24 hours after addition of compounds as described above.
TGFβ reporter assays were conducted in HEK293T cells in DMEM-high glucose supplemented with 1% FBS as in (Willems et al., 2012). Cells were transfected with SBE4-Luciferase (Addgene #16495) and CMV-Renilla-Luciferase (Promega) plasmids with Lipofectamine 2000 (Invitrogen). After 12 hours, cells were induced with TGF-β2 (15 ng/ml) and compounds were added simultaneously. Luminescence was measured 24 hours later using the Dual-Glo luciferase kit (Promega). Notch signaling reporter assays were conducted similarly in HEK293T cells in DMEM-high glucose supplemented with 1% FBS (Diaz-Trelles et al., 2016). Cells were transfected with Hes1-Luciferase (Addgene, #43806) and CMV-Renilla-Luciferase (Promega) plasmids with Lipofectamine 2000 (Invitrogen). Luminescence was measured 24 hours later [endogenous Notch/RBPJ signaling activates the reporter (Diaz-Trelles et al., 2016)] using the Dual-Glo luciferase kit (Promega).
Xenopus and zebrafish studies
Xenopus embryos were fertilized in vitro, dejellied in 2% cysteine–HCl, pH7.8, and maintained in 0.1x MMR and staged according to Nieuwkoop and Faber, as in (Colas et al., 2012). In situ hybridization for Otx2 and Vent2 were done using cRNA probes as in (Colas et al., 2012).
Adult Zebrafish were treated by the addition of PAWI-1 diluted into the aquarium water from 10 mM stock (in DMSO) to achieve a final concentration of 100 or 500 nM. Compounds were removed overnight (16 hours exposure/day) by exchanging water. Exposure continued for the indicated periods. Fish were fed a standard diet.
All animal handling and care followed the NIH Guide for Care and Use of Laboratory Animals. The experimental protocols were approved by Institutional Animal Care and Use Committees of the Sanford-Burnham-Prebys Medical Discovery Institute.
Flow cytometry
Cells were dissociated using 0.25% Trypsin EDTA for flow cytometry in PBS containing 0.1% FBS using either LSRFortessa or FACSAria Flow cytometers (BD Biosciences).
Quantitative real-time PCR
Total RNA was extracted with a miRVana isolation kit (Thermo Fisher) and reverse-transcribed to cDNA with a QuantiTect reverse transcription kit (Qiagen) according to the manufacturer’s instructions. cDNA samples synthesized from 1 μg of total RNA were subjected to RT-qPCR with the Applied Biosystems Quant Studio 7 Flex instrument using the iTaq SYBR Green Supermix (Bio-Rad). Relative gene expression was calculated using the ΔΔCT method normalizing the expression to that of a stable reference gene (GAPDH or as stated). We assumed a primer efficiency of 100% (±10%), according to manufacturer’s guidelines. The primers are listed in Table S3.
TaqMan gene expression assays (Fig. 4) were conducted using probes from ThermoFisher listed in Table S4. They were used following manufacturer’s protocols and run on an Applied Biosystems Quant Studio 7 Flex instrument. Data were analyzed as described above.
Co-immunoprecipitation
2×105 of HEK293T cells transfected with expression plasmids as indicated. Cells were lysed in immunoprecipitation buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.05% NP-40, 5 mM EDTA, proteinase inhibitor and phosphatase inhibitor) and supernatants were collected by centrifugation at 16000× g for 15 minutes at 40°C. The resulting supernatants were incubated with primary antibodies as indicated in addition to Sepharose-conjugated protein G (Sigma) overnight at 4 °C. The beads were rinsed five times in immunoprecipitation buffer, and following the final centrifugation, resuspended in 10 μl loading (Laemli) buffer and analyzed on SDS-PAGE.
ChIP-qPCR assay
HEK293 cells were fixed with 1% formaldehyde for 15 min and quenched with 0.125 M glycine, and sent to Active Motif Services (Carlsbad, CA) to be processed for ChIP-qPCR. In brief, chromatin was isolated by the addition of lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by ethanol precipitation. Pellets were resuspended and the resulting DNA was quantified on a NanoDrop spectrophotometer. Extrapolation to the original chromatin volume allowed quantitation of the total chromatin yield.
An aliquot of chromatin (45 μg) was precleared with protein G agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using 25 μl of antibody against TCF-4 (Santa Cruz Biotechnologies, sc-8631). Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65°C, and ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation.
Quantitative PCR (qPCR) reactions were carried out in triplicate on specific genomic regions using SYBR Green Supermix (Bio-Rad, 170–8882) on a CFX Connect™ Real Time PCR system. The qPCR assay was performed using ten test regions as well as a negative control primer pair that amplifies a region in a gene desert on chromosome 12 (Ctrl primers). The resulting signals were normalized for primer efficiency by carrying out qPCR for each primer pair using Input DNA, yielding binding events per cell count using ChIP-IT qPCR Analysis Kit (Active Motif, 53029).
HIPK2 in vitro kinase assay
The HIPK2 in vitro kinase assay was conducted by Reaction Biology (Malvern, PA). Compounds were assayed in triplicate for a 10-dose IC50 determination with 3-fold serial dilution starting at 20 μM (Staurosporine) or 10 μM (PAWI-1). GST tagged-catalytic domain of HIPK2 fragment (amino acids 165–564) was purified from insect cells. GST-HIPK2 was added to a mixture of 20 μM Myelin Basic Protein (MBP), and then compounds were added using acoustic transfer. The compound, substrate and kinase were incubated for 15 minutes, and then 1 μM γ33-ATP was added to start the reaction. The reaction was carried out for 120 minutes at room temperature and γ33-MBP was measured at termination.
siRNA screen
The stably transfected RKO (pBARLRen) cells were used as described for the chemical compound library screen. Cells were plated at ~2400 per well of 384-well black-walled plates (Greiner) pre-spotted with 10 nanomoles of either individual siRNAs (ON-TARGETplus, Dharmacon/GE Healthcare) or siRNA inert sequence control (Dharmacon) listed in Supplemental Table 1 in Lipofectamine RNAiMAX (ThermoFisher). Cells were cultured for 2 days to allow siRNA-mediated knockdown of cognate protein expression. Recombinant Wnt3a (10% final concentration of Wnt3a conditioned medium, as above) together with either PAWI-2 (100 nM) or DMSO vehicle was added to each well and luciferase activity was determined 6 hours later using SteadyLite HTS (PerkinElmer). PAWI-2 and DMSO vehicle conditions for each siRNA were tested in quadruplicate wells. PAWI-2 activity was normalized relative to the corresponding DMSO control. The effect of each siRNA on PAWI-2 activity was then calculated as % inhibition relative to that observed in the control siRNA samples (0% inhibition). These activities were then expressed as a Z-score to account for variation between plates. Most siRNAs were ± 1 standard deviation from the control siRNA, and hence considered to have negligible activity.
siRNA transfection
5 × 104 SW480 cells were plated in 24-well plates with 400 μl of DMEM medium (with 10% FBS and without antibiotics) per well and incubated at 37°C, 5% CO2 for 24 hours before the transfection. Fresh DMEM medium with 2% FBS was used for tranfection experiment. The transfection complex was made by mixing RNAi in Opti-MEM (ThermoFisher) with Lipofectamine RNAiMAX (ThermoFisher) in Opti-MEM solution at 1:1 ratio and incubating at room temperature for 15 min. 100 μl of the complexes was added dropwise into each well containing cells and medium by gently mixing. The final RNAi concentrations were 20 nM. The cells were then incubated at 37°C, 5% CO2 for 24 hours then treated with either DMSO control (0.5%) or PAWI-2 at a final concentration of 500 nM for 4 hours. Effectiveness of knockdown was confirmed (Supplementary Fig. S5A).
Western blotting
Whole cell extracts were harvested by lysis with RIPA buffer supplemented with protease and phosphatase inhibitors (Sigma). Extracts were resolved by SDS-PAGE (10%) followed by Western blotting using antibodies listed in Materials above. Antibodies were typically diluted 1:1000 in TBST (tris-buffered saline (TBS) with 0.01 % Tween-20, pH 7.4) or with 5 % BSA (in cases of antibodies to phospho-Ser15-p53, phospho-Ser1981-ATM, total ATM). Detection was conducted using ECL Plus detection kit (Abcam) and quantified by densitometry (Image J) or with an Odyssey system (LICOR).
QUANTIFICATION AND STATISTICAL ANALYSIS
Chemical and siRNA screens
For the chemical and siRNA screens, Z-scores were calculated using the luminescence readout and normalized to values for either the DMSO treated wells or siRNA controls, respectively. Z-score is calculated as the number of standard deviations from the mean value of the control wells.
Data standardization and statistical analysis for qRT-PCR was using the ΔΔCT method as described above and in (Willems et al., 2008).
Dose curves and IC50 values were obtained with the Prism statistical analysis package (GraphPad Software Inc.) using the (log) agonist vs. normalized response (variable slope) equation. Data used for dose curves consisted of quadruplicate reads and were normalized to the compound concentration with the highest induction level (designated as 100%).
The number of replicates (n) for each point in each experiment is indicated. Error bars are plotted as standard error of the mean (s.e.m.) unless otherwise indicated. The statistical tests were calculated using Prism statistical analysis package (GraphPad Software Inc.) and the statistical test to determine significance is described in the figure legends. Two-sided T-tests were used for comparisons. The high throughput screen (Supplementary Fig. S1) was conducted once. All other experiments were conducted at least twice and representative examples shown in the figures.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-ATM | Cell Signaling | Cat#2873 |
| anti-β-Actin | Cell Signaling | Cat#3700 |
| anti-β-catenin | Upstate | Cat#06–734 |
| anti-cMYC | Santa Cruz Biotech | Cat#sc-40 |
| anti-FLAG | Sigma-Aldrich | Cat#F3165 |
| anti-GAPDH | Abcam | Cat#ab9485 |
| anti-HDAC2 | Santa Cruz Biotech | Cat#sc-7899 |
| anti-HIPK2 | Abcam | Cat#ab75937 |
| Anti-LEF1 | Bethyl Laboratories | Cat#A303–486A |
| anti-phospho-Y361-HIPK2 | Thermo Scientific | Cat#PA5–13045 |
| anti-phospho-S209-TCF3 | Sergei Sokol (Hikasa and Sokol, 2011) | N/A |
| anti-phospho-S46-p53 | Abcam | Cat#ab76242 |
| anti-phospho-S15-p53 | Cell Signaling | Cat#9284 |
| anti-TCF4 | Abcam | Cat#ab217668 |
| anti-TCF4 (for ChIP qPCR) | Santa Cruz Biotechnologies | Cat# sc-8631 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| miRVana isolation kit | Thermo Fisher | Cat# AM1560 |
| SYBR Green Supermix | BioRad | Cat#1708882 |
| iTaq SYBR Green Supermix | Bio-Rad | Cat#1725120 |
| QuantiTect reverse transcription kit | Qiagen | Cat# 205311 |
| Recombinant human Wnt3a | R&D Systems | Cat#5036-WN |
| Primary screen library | Sanford-Burnham-Prebys Medical Discovery Institute Chemical Genomics core facility | https://www.sbpdiscovery.org/medical-discovery/drug-discovery/prebys-center-for-drug-discovery/overview) |
| Deposited Data | ||
| Not applicable | ||
| Experimental Models: Cell Lines | ||
| pBARLRen cell line (derived from RKO cells) | Moon lab, University of Washington (Biechele et al., 2009) | N/A |
| NF-kB reporter (HEK293-NF-kB-Luc) stably expressing NOD1 | John C. Reed, | https://pubchem.ncbi.nlm.nih.gov/bioassay/1290 |
| HEK293T | ATCC | Cat#CRL-3216 |
| SW480 | ATCC | Cat#CCL-228 |
| Oligonucleotides | ||
| Primers for ChIP qPCR, see Table S3 | This paper | N/A |
| Primers for qRT-PCR, see Table S4 | This paper | N/A |
| Taqman primers for Taqman qRT-PCR, see Table 2 | Thermo Fisher | See Table 2 |
| Recombinant DNA | ||
| pcDNA-Wnt3a | (Najdi et al., 2012) | Addgene Cat#35908 |
| MYC-tagged TCF4 | (Korinek et al., 1997) | Addgene Cat#16512 |
| Constitutively active mutant b-catenin | (Taelman et al., 2010) | Addgene Cat#29684 |
| wildtype b-catenin | (Lee et al., 2001) | Addgene Cat#13434 |
| pcDNA3β-FLAG-CBP-HA | (Zhao et al., 2005) | Addgene Cat#32908 |
| pCMVβ-p300-MYC | (Addgene depositor Tso-Pang Yao) | Addgene Cat#30489 |
| pCTX-myc-HIPK2 WT | (Hikasa et al., 2010) | Addgene Cat#32996 |
| TCF1-FLAG | Sergei Sokol (Hikasa and Sokol, 2011) | N/A |
| TCF3-FLAG | Sergei Sokol (Hikasa and Sokol, 2011) | N/A |
| TCF4-FLAG | Sergei Sokol (Hikasa and Sokol, 2011) | N/A |
| LEF1-FLAG | Sergei Sokol (Hikasa and Sokol, 2011) | N/A |
| Software and Algorithms | ||
| ChIP-IT qPCR Analysis Kit | Active Motif | https://www.activemotif.com/catalog/872/chip-it-qpcr-analysis-kit |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ChemDraw Ultra version 11 | PerkinElmer | https://www.perkinelmer.com/ |
| Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Biorender | Biorender | https://biorender.com/ |
Highlights.
PAWI-1,2 are small molecules with dual p53 agonist and Wnt inhibitor activities
PAWIs bind tubulin and activate stress signaling
Stress signaling through ATM, AURK and HIPK inhibits TCF/LEF transcriptional activity
PAWIs inhibit the ability of a cell to respond to Wnt
SIGNIFICANCE.
Wnt signaling plays a central role in maintaining tissues in the body. When dysregulated, it also can cause cancer, including colorectal and pancreatic tumors. Small molecule inhibitors have been important for revealing the role of the Wnt pathway and are under clinical development. All characterized small molecule inhibitors target proteins in the core Wnt signaling pathway that leads to β-catenin-induced alterations in gene expression. β-Catenin acts on downstream genes through interactions with transcription factors, notably members of the TCF/LEF family, that are themselves regulated by a parallel but poorly defined signaling cascade. We focused a small molecule screen on discovering inhibitors outside of the core Wnt pathway that revealed a new class of potent (IC50 = 11–25 nM) Wnt inhibitors that bind tubulin and stimulate the ataxia-telangiectasia mutated (ATM) protein. Here we describe the use of these new compounds to reveal a kinase cascade that connects microtubules and stress signaling to Wnt signaling. The same cascade regulates p53; thus, the compounds activate p53 with the same nanomolar potencies. The new compounds were named PAWI-1 and PAWI-2 based on the their dual p53 activating and Wnt inhibiting effects. Few Wnt/β-catenin inhibitors have progressed to clinical trials for cancer, and it has been suggested that canonical pathway inhibitors lack sufficient selectivity and/or have too narrow a therapeutic window to be effective anti-cancer agents. The discovery of nanomolar potency compounds that act indirectly to modulate Wnt and p53 should broaden the scope of candidates that could be exploited for chemical biology and therapeutic applications.
ACKNOWLEDGEMENTS
We thank Sergei Sokol (Mount Sinai School of Medicine), John Reed and Shu-Ichi Matsuzawa (Sanford-Burnham-Prebys Medical Discovery Institute) and Karl Willert (University of California, San Diego) for reagents. We also thank Pilar Ruiz-Lozano (Regencor) for critical discussions and editing of the manuscript. For flow cytometry, we are grateful for the expertise of Yoav Altman (Sanford-Burnham-Prebys Medical Discovery Institute) and support of P30CA030199.
This research was supported by the NIH (R37HL059502, R01HL113601, 1R01HL130840, R01HL132225 and P01HL141084 to MM, 1R01HL13967901 to IK, R43CA203566 to KO and R41CA176931 to JC, R01HL126527 and R01HL146690 to JCW, DP2DK098092 and U01DK105541 to PDSD), the California Institute for Regenerative Medicine (CIRM RC1-000132 to MM and CIRM RS1-00169 to JC) the W. M. Keck Foundation (2017-01 to PDSD), and the Diabetes Research Connection (Project #08) to JJL. MM gratefully acknowledges the Joan and Sanford I. Weill Scholars Endowment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
JRC and MM have equity in ChemRegen, Inc. and declare no other competing interests. The other authors declare no competing interests. A patent has been issued for the PAWI compounds.
REFERENCES
- Ali A, Zhang J, Bao S, Liu I, Otterness D, Dean NM, Abraham RT, and Wang XF (2004). Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev 18, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arce L, Yokoyama NN, and Waterman ML (2006). Diversity of LEF/TCF action in development and disease. Oncogene 25, 7492–7504. [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, and Kastan MB (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- Biechele TL, Adams AM, and Moon RT (2009). Transcription-based reporters of Wnt/beta-catenin signaling. Cold Spring Harb Protoc 2009, pdb prot5223. [DOI] [PubMed] [Google Scholar]
- Blagodatski A, Poteryaev D, and Katanaev VL (2014). Targeting the Wnt pathways for therapies. Mol Cell Ther 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse KR, Diskin SJ, Cole KA, Wood AC, Schnepp RW, Norris G, Nguyen le B, Jagannathan J, Laquaglia M, Winter C, et al. (2012). Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res 72, 2068–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, and Waterman ML (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, and Siliciano JD (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Cashman J, Mercola M, Schade D, and Tsuda M (2016). Compounds for inhibition of cancer cell proliferation, U.S.P. Office, ed. (US: Chemregen,Inc.). [Google Scholar]
- Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, et al. (2009). Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 5, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Dwyer M, Okolotowicz KJ, Mercola M, and Cashman JR (2018). A Novel Inhibitor Targets Both Wnt Signaling and ATM/p53 in Colorectal Cancer. Cancer Res 78, 5072–5083. [DOI] [PubMed] [Google Scholar]
- Choi J, Ma S, Kim HY, Yun JH, Heo JN, Lee W, Choi KY, and No KT (2016). Identification of small-molecule compounds targeting the dishevelled PDZ domain by virtual screening and binding studies. Bioorg Med Chem 24, 3259–3266. [DOI] [PubMed] [Google Scholar]
- Ciani L, Krylova O, Smalley MJ, Dale TC, and Salinas PC (2004). A divergent canonical WNT-signaling pathway regulates microtubule dynamics: dishevelled signals locally to stabilize microtubules. J Cell Biol 164, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colas AR, McKeithan WL, Cunningham TJ, Bushway PJ, Garmire LX, Duester G, Subramaniam S, and Mercola M (2012). Whole-genome microRNA screening identifies let-7 and mir-18 as regulators of germ layer formation during early embryogenesis. Genes Dev 26, 2567–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale T, Clarke PA, Esdar C, Waalboer D, Adeniji-Popoola O, Ortiz-Ruiz MJ, Mallinger A, Samant RS, Czodrowski P, Musil D, et al. (2015). A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat Chem Biol 11, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Vega L, Hornung J, Kremmer E, Milanovic M, and Schmitz ML (2013). Homeodomain-interacting protein kinase 2-dependent repression of myogenic differentiation is relieved by its caspase-mediated cleavage. Nucleic Acids Res 41, 5731–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Trelles R, Scimia MC, Bushway P, Tran D, Monosov A, Monosov E, Peterson K, Rentschler S, Cabrales P, Ruiz-Lozano P, et al. (2016). Notch-independent RBPJ controls angiogenesis in the adult heart. Nat Commun 7, 12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich L, Rathmer B, Ewan K, Bange T, Heinrichs S, Dale TC, Schade D, and Grossmann TN (2017). Cell Permeable Stapled Peptide Inhibitor of Wnt Signaling that Targets beta-Catenin Protein-Protein Interactions. Cell Chem Biol 24, 958–968 e955. [DOI] [PubMed] [Google Scholar]
- Doumpas N, Lampart F, Robinson MD, Lentini A, Nestor CE, Cantu C, and Basler K (2019). TCF/LEF dependent and independent transcriptional regulation of Wnt/beta-catenin target genes. EMBO J 38, e98873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumontet C, and Jordan MA (2010). Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Kim HY, Moon SH, et al. (2004). A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A 101, 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro M, Savage K, Hobson K, Deans AJ, Powell SN, McArthur GA, and Khanna KK (2004). BRCA1-BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J Biol Chem 279, 31251–31258. [DOI] [PubMed] [Google Scholar]
- Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DV, de Punder K, Angers S, Peters PJ, Maurice MM, et al. (2016). Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 530, 340–343. [DOI] [PubMed] [Google Scholar]
- Feki A, Jefford CE, Berardi P, Wu JY, Cartier L, Krause KH, and Irminger-Finger I (2005). BARD1 induces apoptosis by catalysing phosphorylation of p53 by DNA-damage response kinase. Oncogene 24, 3726–3736. [DOI] [PubMed] [Google Scholar]
- Grandy D, Shan J, Zhang X, Rao S, Akunuru S, Li H, Zhang Y, Alpatov I, Zhang XA, Lang RA, et al. (2009). Discovery and characterization of a small molecule inhibitor of the PDZ domain of dishevelled. J Biol Chem 284, 16256–16263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, and Livingston DM (2006). Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev 20, 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Yasuda SY, Teo JL, Nguyen C, McMillan M, Hsieh CL, Suemori H, Nakatsuji N, Yamamoto M, Miyabayashi T, et al. (2012). Wnt signaling orchestration with a small molecule DYRK inhibitor provides long-term xeno-free human pluripotent cell expansion. Stem Cells Transl Med 1, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikasa H, Ezan J, Itoh K, Li X, Klymkowsky MW, and Sokol SY (2010). Regulation of TCF3 by Wnt-dependent phosphorylation during vertebrate axis specification. Dev Cell 19, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikasa H, and Sokol SY (2011). Phosphorylation of TCF proteins by homeodomain-interacting protein kinase 2. J Biol Chem 286, 12093–12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. (2009). Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620. [DOI] [PubMed] [Google Scholar]
- Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N, Waterman M, Bowerman B, Clevers H, Shibuya H, et al. (1999). The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta-catenin and transcription factor TCF. Nature 399, 798–802. [DOI] [PubMed] [Google Scholar]
- Kahn M (2014). Can we safely target the WNT pathway? Nat Rev Drug Discov 13, 513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa R, Bakkenist CJ, McKinnon PJ, and Kastan MB (2004). Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev 18, 1423–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutuzov MA, Andreeva AV, and Voyno-Yasenetskaya TA (2005). Regulation of apoptosis signal-regulating kinase 1 (ASK1) by polyamine levels via protein phosphatase 5. J Biol Chem 280, 25388–25395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier M, Schade D, Willems E, Tsuda M, Spiering S, Kalisiak J, Mercola M, and Cashman JR (2012). Wnt inhibition correlates with human embryonic stem cell cardiomyogenesis: a structure-activity relationship study based on inhibitors for the Wnt response. J Med Chem 55, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau T, Chan E, Callow M, Waaler J, Boggs J, Blake RA, Magnuson S, Sambrone A, Schutten M, Firestein R, et al. (2013). A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res 73, 3132–3144. [DOI] [PubMed] [Google Scholar]
- Lee W, Swarup S, Chen J, Ishitani T, and Verheyen EM (2009). Homeodomain-interacting protein kinases (Hipks) promote Wnt/Wg signaling through stabilization of beta-catenin/Arm and stimulation of target gene expression. Development 136, 241–251. [DOI] [PubMed] [Google Scholar]
- Lei B, Chai W, Wang Z, and Liu R (2015). Highly expressed UNC119 promotes hepatocellular carcinoma cell proliferation through Wnt/beta-catenin signaling and predicts a poor prognosis. Am J Cancer Res 5, 3123–3134. [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Polak L, Lin M, Lay K, Zheng D, and Fuchs E (2014). In vivo transcriptional governance of hair follicle stem cells by canonical Wnt regulators. Nat Cell Biol 16, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D, et al. (2013). Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A 110, 20224–20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, and Biggins S (2014). Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol 15, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Green BA, Farr JM, Lopes FC, and Van Raay TJ (2016). Wnt Drug Discovery: Weaving Through the Screens, Patents and Clinical Trials. Cancers (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Choi J, Jin X, Kim HY, Yun JH, Lee W, Choi KY, and No KT (2018). Discovery of a small-molecule inhibitor of Dvl-CXXC5 interaction by computational approaches. J Comput Aided Mol Des 32, 643–655. [DOI] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, and Salmon ED (2007). The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8, 379–393. [DOI] [PubMed] [Google Scholar]
- Nusse R, and Clevers H (2017). Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999. [DOI] [PubMed] [Google Scholar]
- Okolotowicz KJ, Dwyer M, Ryan D, Cheng J, Cashman EA, Moore S, Mercola M, and Cashman JR (2018). Novel tertiary sulfonamides as potent anti-cancer agents. Bioorg Med Chem. [DOI] [PubMed] [Google Scholar]
- Peng Y, Woods RG, Beamish H, Ye R, Lees-Miller SP, Lavin MF, and Bedford JS (2005). Deficiency in the catalytic subunit of DNA-dependent protein kinase causes down-regulation of ATM. Cancer Res 65, 1670–1677. [DOI] [PubMed] [Google Scholar]
- Ryser S, Dizin E, Jefford CE, Delaval B, Gagos S, Christodoulidou A, Krause KH, Birnbaum D, and Irminger-Finger I (2009). Distinct roles of BARD1 isoforms in mitosis: full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds Aurora B and BRCA2. Cancer Res 69, 1125–1134. [DOI] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, and Brivanlou AH (2004). Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10, 55–63. [DOI] [PubMed] [Google Scholar]
- Schatoff EM, Leach BI, and Dow LE (2017). Wnt Signaling and Colorectal Cancer. Curr Colorectal Cancer Rep 13, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger A, Shelton CA, Maloof JN, Meneghini M, and Bowerman B (1999). Wnt pathway components orient a mitotic spindle in the early Caenorhabditis elegans embryo without requiring gene transcription in the responding cell. Genes Dev 13, 2028–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, and Ziv Y (2013). The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14, 197–210. [PubMed] [Google Scholar]
- Shimizu N, Ishitani S, Sato A, Shibuya H, and Ishitani T (2014). Hipk2 and PP1c cooperate to maintain Dvl protein levels required for Wnt signal transduction. Cell Rep 8, 1391–1404. [DOI] [PubMed] [Google Scholar]
- Singh S, Srivastava SK, Bhardwaj A, Owen LB, and Singh AP (2010). CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer 103, 1671–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjolund J, Pelorosso FG, Quigley DA, DelRosario R, and Balmain A (2014). Identification of Hipk2 as an essential regulator of white fat development. Proc Natl Acad Sci U S A 111, 7373–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol SY (2011). Wnt signaling through T-cell factor phosphorylation. Cell Res 21, 1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sombroek D, and Hofmann TG (2009). How cells switch HIPK2 on and off. Cell Death Differ 16, 187–194. [DOI] [PubMed] [Google Scholar]
- Stanganello E, Zahavi EE, Burute M, Smits J, Jordens I, Maurice MM, Kapitein LC, and Hoogenraad CC (2019). Wnt Signaling Directs Neuronal Polarity and Axonal Growth. iScience 13, 318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoick-Cooper CL, Weidinger G, Riehle KJ, Hubbert C, Major MB, Fausto N, and Moon RT (2007). Distinct Wnt signaling pathways have opposing roles in appendage regeneration. Development 134, 479–489. [DOI] [PubMed] [Google Scholar]
- Sulli G, Di Micco R, and d’Adda di Fagagna F (2012). Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer 12, 709–720. [DOI] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, and De Robertis EM (2010). Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira N, Nihira K, Yamaguchi T, Miki Y, and Yoshida K (2007). DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell 25, 725–738. [DOI] [PubMed] [Google Scholar]
- Takagi M, Uno H, Nishi R, Sugimoto M, Hasegawa S, Piao J, Ihara N, Kanai S, Kakei S, Tamura Y, et al. (2015). ATM Regulates Adipocyte Differentiation and Contributes to Glucose Homeostasis. Cell Rep. [DOI] [PubMed] [Google Scholar]
- van der Laden J, Soppa U, and Becker W (2015). Effect of tyrosine autophosphorylation on catalytic activity and subcellular localisation of homeodomain-interacting protein kinases (HIPK). Cell Commun Signal 13, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tienen LM, Mieszczanek J, Fiedler M, Rutherford TJ, and Bienz M (2017). Constitutive scaffolding of multiple Wnt enhanceosome components by Legless/BCL9. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheyen EM, Swarup S, and Lee W (2012). Hipk proteins dually regulate Wnt/Wingless signal transduction. Fly (Austin) 6, 126–131. [DOI] [PubMed] [Google Scholar]
- Voronkov A, and Krauss S (2013). Wnt/beta-catenin signaling and small molecule inhibitors. Curr Pharm Des 19, 634–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems E, Cabral-Teixeira J, Schade D, Cai W, Reeves P, Bushway PJ, Lanier M, Walsh C, Kirchhausen T, Izpisua Belmonte JC, et al. (2012). Small molecule-mediated TGF-beta type II receptor degradation promotes cardiomyogenesis in embryonic stem cells. Cell Stem Cell 11, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems E, Leyns L, and Vandesompele J (2008). Standardization of real-time PCR gene expression data from independent biological replicates. Anal Biochem 379, 127–129. [DOI] [PubMed] [Google Scholar]
- Willems E, Spiering S, Davidovics H, Lanier M, Xia Z, Dawson M, Cashman J, and Mercola M (2011). Small-molecule inhibitors of the Wnt pathway potently promote cardiomyocytes from human embryonic stem cell-derived mesoderm. Circ Res 109, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, and Qin J (2002). SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev 16, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos G, Black EJ, Walker M, Scott MT, Vagnarelli P, Earnshaw WC, and Gillespie DA (2007). Chk1 is required for spindle checkpoint function. Dev Cell 12, 247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N, and Boutros M (2017). Wnt signaling in cancer. Oncogene 36, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Hao J (2015). Development of anticancer agents targeting the Wnt/beta-catenin signaling. Am J Cancer Res 5, 2344–2360. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. This study did not generate code.