

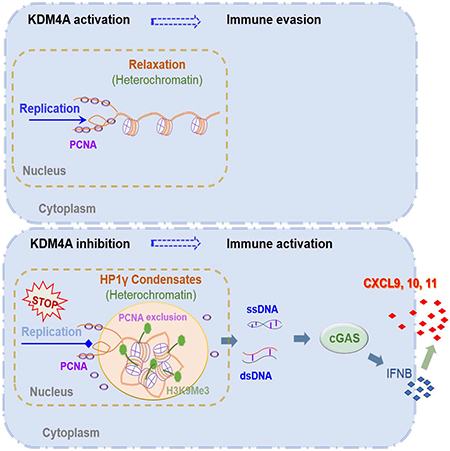
Summary
Developing strategies to activate tumor cell-intrinsic immune response is critical for improving tumor immunotherapy by exploiting tumor vulnerability. KDM4A, as a H3K9me3 demethylase, has been found to play a critical role in squamous cell carcinoma (SCC) growth and metastasis. Here we report that KDM4A inhibition promoted heterochromatin compaction and induced DNA replication stress which elicited antitumor immunity in SCC. Mechanistically, KDM4A Inhibition promoted the formation of liquid-like HP1γ puncta on heterochromatin and stall DNA replication which resultantly activated tumor cell-intrinsic cGAS-STING signaling by replication stress-induced cytosolic DNA accumulation. Moreover, KDM4A inhibition collaborated with PD1 blockade to inhibit SCC growth and metastasis by recruiting and activating CD8+ T cells. In vivo lineage tracing demonstrated that KDM4A inhibition plus PD1 blockade efficiently eliminated cancer stem cells. Taken together, our results demonstrate that targeting KDM4A can activate anti-tumor immunity and enable PD1 blockade immunotherapy by aggravating replication stress in SCC cells.
Graphical Abstract
eTOC BLURB
Zhang et al. show that targeting KDM4A activated tumor cell-intrinsic immunity by inducing heterochromatin compaction and replication stress. The combination of PD1 blockade and KDM4A inhibition potently inhibited SCC growth and lymph node metastasis by recruiting new CD8+ T cells and eliminating cancer stem cells.
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is a highly malignant cancer worldwide with a low 5-year survival rate and poor prognosis (Bray et al., 2018). SCC also frequently occurs in the skin, lung and esophagus. SCC is very invasive and frequently metastasizes to regional lymph nodes (Chinn and Myers, 2015; Sacco and Cohen, 2015). It is well known that tumor cells are potentially immunogenic and the immune system plays a critical role in the surveillance against tumor initiation. Tumor cells must be able to escape from immune surveillance when epithelial dysplasia is transformed to SCC (Economopoulou et al., 2016; Sanmamed and Chen, 2018). Multiple oncogenes and signaling pathways are activated and cell cycle checkpoints are dysregulated during SCC development (Pai and Westra, 2009). Those oncogenes or pathways could be vulnerable targets for immune surveillance and immunotherapy, and ablating them might activate tumor-intrinsic immunity.
Recently, immune checkpoint inhibitors targeting PD1/PD-L1 and CTLA4 have achieved good success in several solid tumors such as melanoma and HNSCC (Ferris et al., 2016; Harrington et al., 2017; Sanmamed and Chen, 2018; Sharma et al., 2017; Zou et al., 2016). Although anti-PD1 therapy has been approved for treating recurrent or metastatic SCC, the objective response rate is less than 20% (Ferris et al., 2016). Growing evidence suggests that the unresponsive tumors are probably deficient of CD8+ T cell infiltration upon PD1 blockade therapy (Tumeh et al., 2014). Thus, reversing the hypo-immunogenicity of SCC cells has become increasingly imperative to ongoing cancer immunotherapy (Sharma et al., 2017). In mouse models of various cancers, it has been found that Th1-type chemokines facilitated the intratumoral infiltration of CD8+ T cells and stimulated anti-tumor immunity, thereby improving the efficacies of checkpoint blockade (Dangaj et al., 2019; Peng et al., 2015). Consistently, the levels of CD8+ T cells-attracting chemokines in tumor cells is positively correlated with the responsiveness of human cancers to PD1 blockade (Dangaj et al., 2019). Therefore, improving tumor cell intrinsic ability to attract new T cells was critical for anti-PD1 therapy by exploiting tumor vulnerability. However, very few vulnerable targets have been identified for improving immunotherapy of SCC.
Histone demethylases KDM4A (also known as JMJD2A), acting as the eraser of H3K9me3, is overexpressed in several human cancers. It has been found that KDM4A stimulated S phase progression and altered replication timing of specific genomic foci by antagonizing HP1γ in a demethylase activity-dependent manner and controlling chromatin compaction as well as accessibility (Black et al., 2010). Moreover, KDM4A overexpression could prompt site-specific copy gain and re-replication of regions amplified in tumors by inducing a DNA replication-favoring chromatin state that can be antagonized by SUV39H1 or HP1γ overexpression (Black et al., 2013). We have also identified that KDM4A as a key epigenetic factor, stimulated the transcription of the invasive genes to promote SCC invasion and metastasis by erasing repressive H3K9me3 marks (Ding et al., 2013). To further determine whether KDM4A is an important target for SCC treatment, we took advantage of the 4-nitroquinoline 1-oxide (4-NQO)-induced mouse model of SCC, which fully simulates SCC initiation, progression and lymph node metastasis in a syngeneic and immunocompetent tumor microenvironment. We found that KDM4A inhibition not only significantly inhibited SCC invasive growth, but also epigenetically activated antitumor immunity. Targeting KDM4A promoted the intratumor infiltration of new CD8+ T cells and collaborated with PD1 blockade to inhibit SCC invasive growth and metastasis.
RESULTS
Suppression of SCC Invasion and Metastasis in vivo by Kdm4a Deletion
To demonstrate that KDM4A is an important therapeutic target for SCC, we examined whether genetic deletion of Kdm4a could inhibit SCC growth and metastasis using 4-NQO-induced immunocompetent and autochthonous mouse model of HNSCC. In this model, mice were given drinking water containing 4-NQO for 16 weeks, and then afterwards returned to normal water (Figures S1A and S1F). Primary mouse tongue SCCs began to develop at 20 weeks, and metastasized to cervical lymph nodes to form metastatic foci at 24 to 26 weeks (Chen et al., 2017; Vitale-Cross et al., 2012). Immunohistochemistry staining revealed that mouse KDM4A protein levels were markedly higher in both epithelial dysplasia and SCC tissues than that in the normal epithelium (Figure S1B), which was consistent with the increased expression of KDM4A in human HNSCC (Figure S1C) (Ding et al., 2013). To inducibly delete Kdm4a in tongue squamous epithelia upon tamoxifen treatment, we crossed Keratin 14 (K14) Cre/ERT2 mice (K14Cre-ERT2) with Kdm4af/f mice which possess loxP sites flanking exon 3 of Kdm4a (Zhang et al., 2011) to generate K14Cre-ERT2;Kdm4af/f and K14Cre-ERT2;Kdm4aw/w mice. Upon tamoxifen treatment, KDM4A proteins were drastically deleted in tongue epithelium from K14Cre-ERT2;Kdm4af/f (4AKO) mice compared with K14Cre-ERT2;Kdm4aw/w (Cre) mice as determined by Western blot analysis (Figures S1D and S1G). We first deleted Kdm4a when we started to feed mice with 4NQO drinking water. 26 weeks after 4NQO treatment, the primary tumor phenotypes including the number of dysplasia and SCC, the SCC area, tumor invasion degree, and lymph node metastasis were analyzed by histology as described before. Histological analysis revealed that epithelial cells-specific deletion of Kdm4a significantly reduced the SCC number (Figure 1A), but not the dysplasia number (Figure S1E), indicating that KDM4A promotes oncogenic transformation from dysplasia to SCC in vivo. Both SCC area and tumor invasion degree were markedly lessened by KDM4A ablation, confirmed by immunohistochemistry staining of pan-cytokeratin (PCK) (Figures 1B–1D). The lymph node metastasis is considered a crucial predictor for mortality in HNSCC patients (Ho et al., 2017). We further analyzed the number of metastatic lymph nodes and the area of lymphatic metastasis. 4AKO mice harbored a much smaller number of metastatic lymph nodes than the control Cre mice (Figure 1E). The area of lymphatic metastasis was significantly decreased over 7-fold by KDM4A ablation, as determined by immunostaining of PCK in lymph nodes (Figures 1F and 1G).
Figure 1. Epithelial KDM4A Deletion Inhibits Mouse SCC Growth and Lymph Node Metastasis.
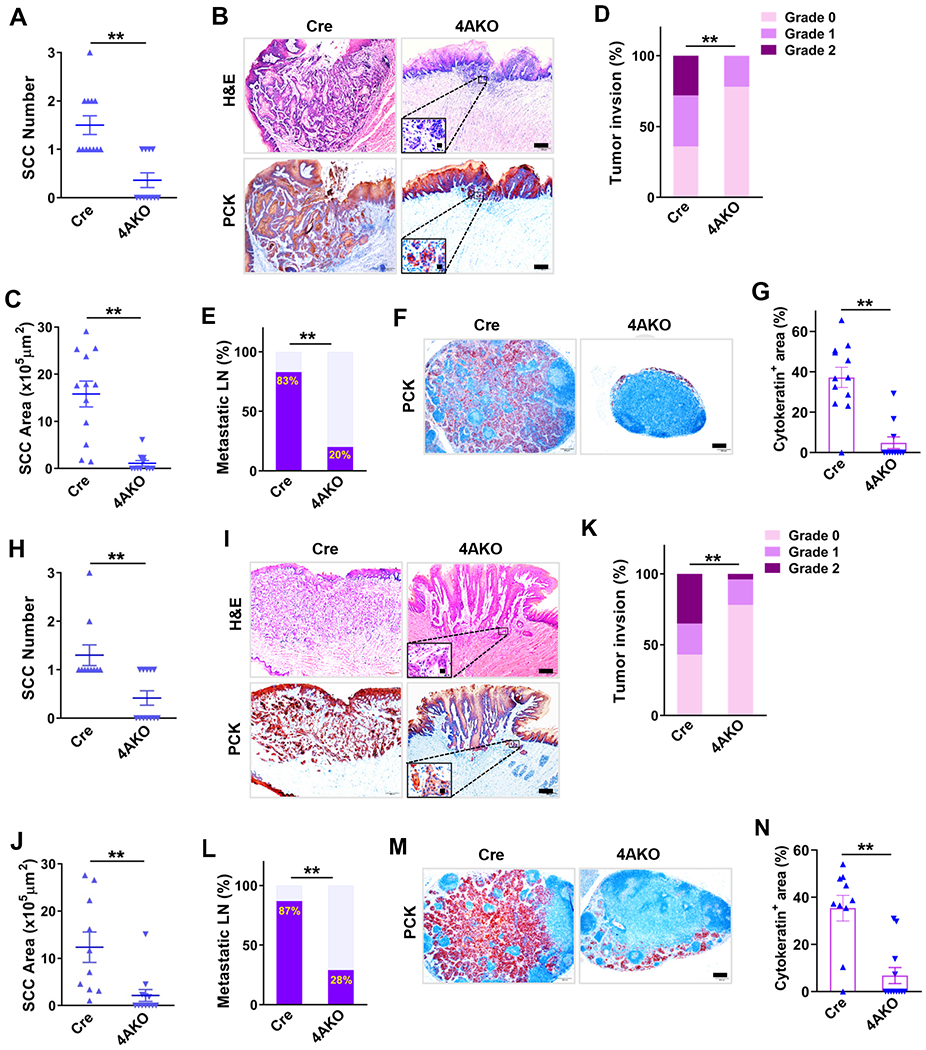
(A) Quantification of tongue SCC number in both control (Cre, n = 12) and 4AKO (n = 11) mice. Values represent mean values ± SD from the pool of two independent experiments. **p < 0.01 by Student’s t test.
(B) Representative images of H&E staining and immunostaining of PCK. Scale bar, 200 μm. Enlarged images are shown in the inserts. Scale bar in inserts, 10 μm.
(C) Quantification of SCC area in both control (n = 12) and 4AKO (n = 11) mice. **p < 0.01 by Student’s t test.
(D) Histogram of primary mouse tumor invasion degree in both control (n = 12) and 4AKO (n = 11) mice. **p < 0.01 by Cochran-Armitage test.
(E) Percentage of metastatic lymph nodes in both control (n = 12) and 4AKO (n = 11) mice. **p < 0.01 by Chi-square test.
(F) Immunostaining of PCK in lymph nodes in both control and 4AKO mice. Scale bar, 200μm.
(G) Quantification of metastatic areas in lymph nodes from both control (n = 12) and 4AKO (n = 11) mice. Values are mean ± SEM. **p < 0.01 by Student’s t test.
(H) Primary tongue SCC numbers in both control (n = 10) and 4AKO (n = 12) mice. Mean values ± SD are shown from the pool of two independent experiments. **p < 0.01 by Student’s t test.
(I) Representative images of H&E staining and PCK staining of SCCs from mice. Scale bars, 200 μm. Inserts show the enlarged images with 10 μm scale bars.
(J) Quantification of primary mouse SCC areas in both control and 4AKO mice. Values are mean ± SD from the pool of two independent experiments. **p < 0.01 by Student’s t test.
(K) Histogram of primary tumor invasion degrees. **p < 0.01 by Cochran-Armitage test.
(L) Quantification of metastatic lymph nodes. **p < 0.01 by Chi-square test.
(M) Representative PCK immunostaining of lymph nodes. Scale bar, 200μm.
(N) Quantification of metastatic areas in lymph nodes. Values are mean ± SEM. **p < 0.01 by Student t test.
Also see Figure S1.
Based on the ineffectiveness of KDM4A ablation on dysplasia number, we were interested in determining whether the deletion of KDM4A could inhibit SCC growth and metastasis when KDM4A expression was already increased in tumor cells (Figure S1B). We treated mice with tamoxifen on the 20th-week of SCC induction when SCCs began to develop (Figures S1F and S1G). 6 weeks after KDM4A deletion, we compared the SCC growth and progression in 4AKO mice with those in Cre mice. While the numbers of epithelial dysplasia were comparable in 4AKO versus Cre mice (Figure S1H), the SCC numbers were significantly reduced in 4AKO mice compared to those in Cre mice (Figure 1H). Both SCC size and invasion degree were also significantly alleviated by KDM4A deletion in tumor cells (Figures 1I–1K). Furthermore, lymphatic metastasis was also significantly inhibited in 4AKO mice (Figures 1L–1N).
Stimulating Anti-tumor Immunity and Inhibiting Cancer Stemness by Tumor Cell Kdm4a Deletion
To explore the underlying mechanisms by which Kdm4a deletion ameliorated the primary mouse SCC, we performed the whole transcriptome analysis of primary SCC after Kdm4a deletion using RNA sequencing (RNA-Seq). Primary mouse SCCs were collected from both control and 4AKO mice after 10 days of the tamoxifen administration (Figure S1F). As shown by both gene ontology (GO) analysis and gene set enrichment analysis (GSEA), KDM4A deletion significantly suppressed the gene expression associated with cell migration and epithelial mesenchymal transition in primary SCC (Figures S2A and S2B). We designed two guide-RNAs (4AKO1 and 4AKO2) to specifically inhibit KDM4A expression. KDM4A expression was efficiently inhibited by CRISPR-Cas9-mediated knockout in SCC23 cells (Figure S2C). As compared with scramble control (Scr) cells, KDM4A-deficient SCC23 cells had higher levels of H3K9me3, suggesting the loss of KDM4A demethylase activity (Figure S2C). Consistent with our previous studies (Ding et al., 2013), KDM4A deletion significantly inhibited the expression of JUN (Figure S2D), a critical component of the transcription factor activator protein 1 (AP1), and suppressed SCC23 cell invasion by Matrigel invasion assays (Figure S2E). Additionally, the mRNA and protein levels of FOSL1 (also known as Fra1), were robustly decreased in 4AKO mice versus control mice (Figures S2F and S2G), entailing the study whether AP1 reduction accounts for the suppression of invasive growth in KDM4A deficient SCC cells. To this end, we stably expressed exogeneous FOSL1 and JUN proteins in both Scr and 4AKO SCC23 cells (Figure S2H). The expression of exogenous AP1 restored KDM4A loss-induced suppression of invasion in SCC23 cells (Figure S2I). Interestingly, we previously showed that AP-1 also regulated the properties of BMI1+ cancer stem cells (CSCs) (Chen et al., 2017). We thus interrogated the effects of KDM4A depletion on CSCs. Two siRNAs (Si1-4A and Si2-4A) were designed to specifically inhibit KDM4A expression in SCC cells (Figure S2J). KDM4A knockdown significantly inhibited cancer stemness in vitro, as determined by tumor sphere formation (Figures 2A, S2K and S2L). Recently, we have identified that BMI1+ CSCs play a critical role in SCC development and therapeutic resistance by in vivo lineage tracing CSCs in BMI1Cre-ERT2;tdTomato mice (Chen et al., 2017; Jia et al., 2020). To explore whether KDM4A ablation inhibited cancer stemness in vivo, we crossmated BMI1Cre-ERT2;tdTomato mice with Kdm4af/f mice to generate BMI1Cre-ERT2;Kdm4af/f;tdTomato and BMI1Cre-ERT2;Kdm4aw/w;tdTomato mice. Upon tamoxifen treatment, BMI1+ CSCs and their progenies were labeled by Tomato expression in both mice (Figure 2B). Immunostaining confirmed that KDM4A expression was markedly reduced in BMI1Cre-ERT2;Kdm4af/f;tdTomato mice compared with that in BMI1Cre-ERT2;Kdm4aw/w;tdTomato mice (Figure S2M). KDM4A ablation not only significantly reduced tdTomato+ tumor cells, but also inhibited the individual clonal numbers of tdTomato+ tumor cells in primary SCCs of BMI1Cre-ERT2;Kdm4af/f;tdTomato mice versus BMI1Cre-ERT2;Kdm4aw/w;tdTomato mice (Figure 2C).
Figure 2. Tumor Cell KDM4A Ablation Suppresses Cancer Stemness and Stimulates Anti-tumor Immunity in Primary Mouse SCC.
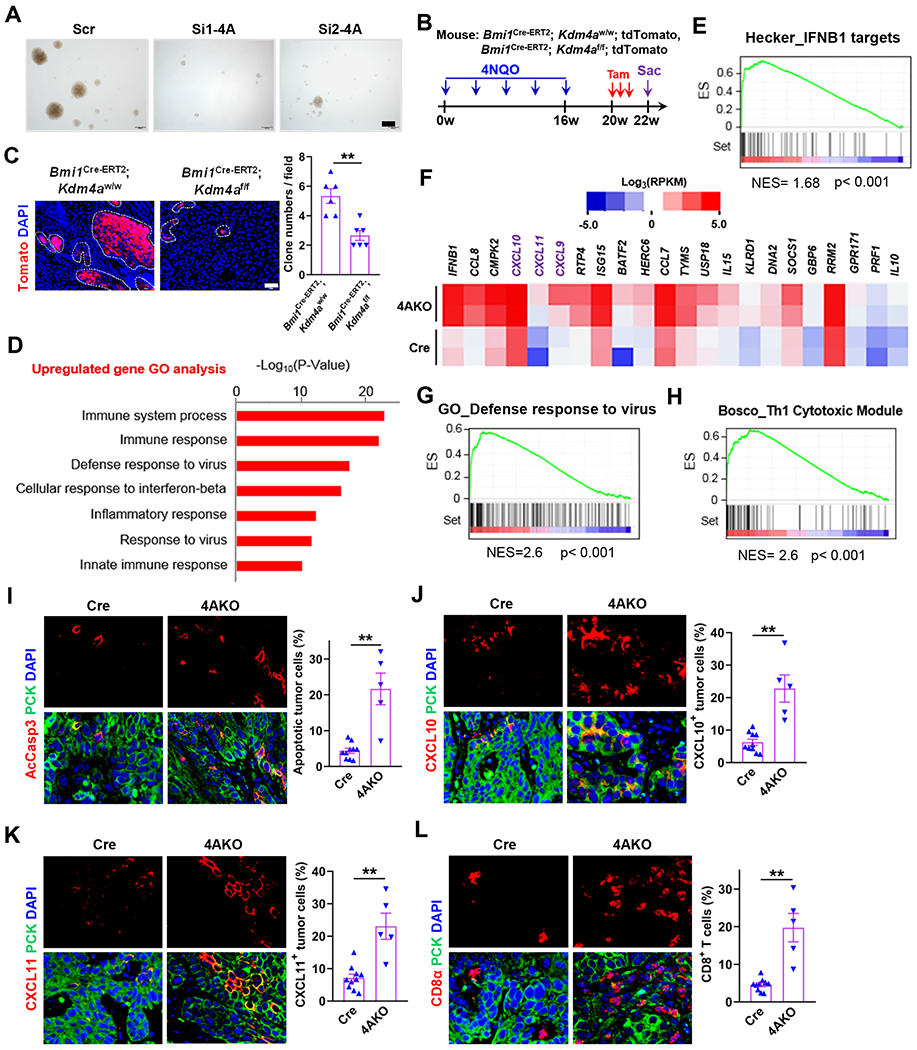
(A) Representative images of tumor spheres derived from Si-Scr, Si1-4A and Si2-4A ALDHhigh SCC23 cells. Scale bar, 200 μm.
(B) Experimental schemes of Kdm4a knockout and simultaneous lineage tracing of CSCs in primary SCC.
(C) Representative fluorescence images and quantification of BMI1+ CSC-derived clones in primary mouse SCCs. Values represent mean ± SEM from the pool of two independent experiments. n = 6:6. Scale bar 200 μm. **p<0.01 by Student t test.
(D) Histogram showing top 7 upregulated biological processes in primary SCC of 4AKO mice. Samples were analyzed in duplicate.
(E) Gene set enrichment analysis (GSEA) plot of IFNB1 targets of primary SCC in 4AKO mice versus control mice. ES, enrichment score. NES, normalized enrichment score. Samples were analyzed in duplicate.
(F) Heat map showing enrichment of at least 3-fold upregulated genes inside the Bosco_Th1 cytotoxic module in 4AKO SCC. RPKM, Reads Per Kilobase of transcript, per Million mapped reads.
(G) Gene set enrichment analysis (GSEA) plot of defense response to virus-related genes of primary SCC in 4AKO mice versus control mice.
(H) Gene set enrichment analysis (GSEA) plot of Th1 cytotoxic genes of primary SCC in 4AKO mice versus control mice.
(I) Immunofluorescence staining and quantification of AcCasp3-positive apoptotic cells in SCCs from both control and 4AKO mice.
(J and K) Immunofluorescence staining and quantification of CXCL10 (J) and CXCL11(K) in primary mouse SCCs
(L) Immunofluorescence staining and quantification of CD8+ T cells in primary mouse SCCs.
(D-H) Tumor tissues were pooled for RNA sequencing (RNA-Seq), n = 2 (control mice); n = 2 (4AKO mice). (I-L) Values are mean ± SEM from two independent experiments. n = 10 (control mice); n = 5 (4AKO mice). **p < 0.01 by Student’s t test. All scale bars, 10 μm. Of note, we totally utilized 10 mice for the control group and 12 mice for the 4AKO group, respectively. However, the samples with SCCs only were stained and analyzed because we could not detect SCCs in some mice.
Also see Figure S2
Unexpectedly, RNA-seq analysis found that a set of genes associated with immune response was robustly upregulated in primary SCC from 4AKO mice versus control mice. Type I interferon (IFN) production and signaling were one of most prominently upregulated processes as shown by both GO (Figure 2D) and GSEA analysis (Figures 2E–2G). Of most interest, Th1-type cytotoxic immunity, one of potently tumoricidal immune response, was also activated by KDM4A ablation (Figures 2F and 2H). Consistently, apoptosis of tumor cells was shown higher levels in 4AKO mice than control mice by co-immunostaining of PCK and active caspase 3 (AcCasp3) (Figure 2I). Since RNA-Seq results demonstrated that KDM4A deletion increased the mRNA levels of Th1-type chemokines including CXCL9, CXCL10 and CXCL11 in primary SCC (Figure 2F), we further examined their protein changes in tumor cells from 4AKO mice versus control mice. Immunostaining confirmed that both CXCL10 (Figure 2J) and CXCL11 (Figure 2K) proteins were substantially increased in 4AKO tumor cells, compared with those in control tumor cells. Of note, CXCL9 protein was expressed by immune cells in lymph nodes (Figure S2N) but was too low to be detected in tumor cells (Figure S2O). We also observed that CD8+ T cells were abundant within and around the tumor islets in 4AKO mice, but scarce in tumors of Cre mice through immunostaining of CD8α (Figure 2L).
Induction of Replication Stress and DNA Damage by Inhibiting KDM4A
By the co-immunoprecipitation assay, we found that KDM4A interacted with both minichromosome maintenance proteins (MCM) 2 and MCM7 in SCC cells (Figure 3A), consistent with previous studies on tight association of KDM4A with replication forks (Black et al., 2010; Black et al., 2013). Therefore, we explored whether KDM4A depletion induced replication stress and consequent DNA damage in SCC cells. To evaluate the effects of KDM4A on DNA replication at single molecule resolution, active replication forks were sequentially labeled by two thymidine analogs, 5-chloro-2’-deoxyuridine (CldU) and 5-iodo-2’-deoxyuridine (IdU), in SCC23 (Figure S3A) and SCC1 cells (Figure S3B). DNA replication rate and stalled fork number were determined by DNA fiber assays. The rate of the replication fork progression declined by 50% (Figure 3B), while the number of stalled forks rose over 3 times more (Figure 3C) in 4AKO SCC23 cells in comparison with those in Scr SCC23 cells. Consistently, KDM4A reduction by siRNA also significantly slowed down the rate of replication fork progression but raised the stalled forks in SCC1 cells (Figures S3C–S3E). These results corroborate that KDM4A loss severely impedes DNA replication and thus gives rise to replication stress in SCC cells. In addition, fluorescence staining of DAPI revealed that chromatin bridges were significantly increased in 4AKO SCC23 cells compared with Scr SCC23 cells (Figure 3D), probably due to the incomplete DNA replication and the ensuing premature S phase exit in the KDM4A-depleted cancer cells. Because chromatin bridges can lead to DNA damage (Fernandez-Casanas and Chan, 2018), we then examined whether DNA damages in the 4AKO cells were increased using Comet assays. We found that KDM4A deletion significantly induced immense DNA strand breakage in both SCC23 (Figure 3E) and SCC1 (Figure S3F) cells, which was further affirmed by examining the phosphorylation of H2AX at Ser 139 (γH2A.X). KDM4A ablation significantly increased the levels of γH2A.X proteins in both SCC23 (Figure 3F) and SCC1 cells (Figure S3G). Furthermore, the majority of γH2A.X foci co-localized with H3K9me3 in 4AKO SCC23 cells, as shown by confocal imaging (Figures 3G and 3H), demonstrating the close association of DNA damage formation with condensed chromatin. KDM4A deletion significantly elevated γH2A.X protein levels in tumor cells in vivo (Figure 3I).
Figure 3. KDM4A Inhibition in SCC cells Induces Replication Stress and DNA Damage.
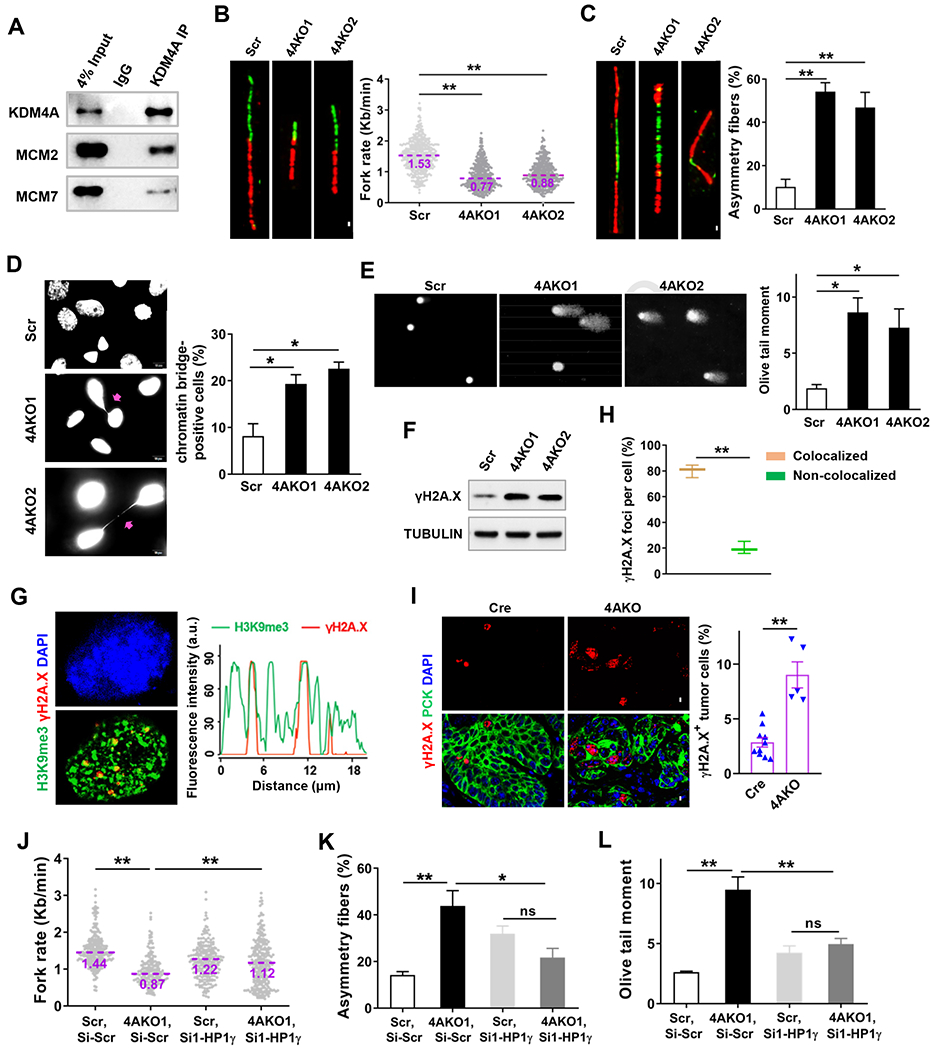
(A) Western blot analysis of the Co-immunoprecipitation of endogenous KDM4A with MCM2 and MCM7 in SCC23 cells.
(B) Representative images of DNA fibers and the quantification of replication fork progress rate in Scr and 4AKO SCC23 cells. The midlines show median values of each group. Scale bar, 5μm. **p<0.01 by Mann-Whitney test.
(C) Representative images of bidirectional DNA fibers and the quantification of asymmetry forks (stalled forks) in Scr and 4AKO SCC23 cells. Fibers with more than 30% of asymmetry were considered. Value represents mean ± SD. Scale bar, 5 μm. **p < 0.01 by one-way ANOVA test.
(D) Immunofluorescence staining of DAPI and quantification of chromatin bridges in Scr and 4AKO SCC23 cells. Arrows indicate the chromatin bridges. Values represent mean ± SEM from at least two independent experiments. *p<0.05 by one-way ANOVA. Scale bar, 10 μm.
(E) Representative images and quantification of DNA Comet assays in Scr and 4AKO SCC23 cells. More than 300 cells were analyzed in every group. Values represent mean ± SEM from three independent experiments. Scale bar, 50 μm. *p<0.05 by one-way ANOVA test.
(F) γH2A.X protein levels in Scr and 4AKO SCC23 cells by Western blot.
(G) Representative confocal images showing DAPI (left upper panel) and the co-localization (left lower panel) of γH2A.X (red) with H3K9me3 (green), and localization analysis for γH2A.X and H3K9me3 along the indicated line scan in 4AKO SCC23 cells. Scale bar, 5 μm.
(H) Quantification of γH2A.X foci in the experiment of (G). Values represent mean ± SEM from three independent experiments. **p < 0.01 by paired Student’s t test.
(I) Immunofluorescence staining and quantification of γH2A.X in primary mouse SCCs. values are mean ± SEM from two independent experiments. n = 10 (control mice); n = 5 (4AKO mice). **p < 0.01 by Student’s t test. All scale bars, 10 μm.
(J) Quantification of replication fork progress rate in both Scr and 4AKO SCC23 cells transfected with Si-scr or Si1-HP1γ. The midlines show median values of each group. **p<0.01 by two-way ANOVA.
(K) Quantification of asymmetry forks in Scr and 4AKO SCC23 cells transfected by Si-scr or Si1-HP1γ. Mean ± SD are shown. ns, not significant, *p < 0.05 and **p < 0.01 by by Mann-Whitney test.
(L) Quantification of DNA Comet assays in Scr and 4AKO SCC23 cells transfected by Si-scr or Si1-HP1γ. Data, shown by mean ± SEM, were collected from three independent experiments. ns, not significant and **p<0.01 by two-way ANOVA.
Also see Figure S3
HP1γ protein is recruited by H3K9me3 and cooperated with it to establish and sustain the compactness of heterochromatin (Black et al., 2010; Machida et al., 2018). We next studied whether HP1γ was engaged in KDM4A deficiency-induced replication stress and DNA damage in SCC23 cells. Two siRNAs (Si1-HP1γ and Si2-HP1γ) were designed for specifically knocking HP1γ down. After the HP1γ targeting-siRNA treatment, SCC23 cells contained strikingly lower levels of HP1γ mRNA in comparison with scramble siRNA (Si-Scr)-transfected cells (Figure S3H). As shown by DNA fiber assay, DNA replication fork progression was slowed down by KDM4A deletion in Si-Scr SCC23 cells, but the fork progression rate significantly rose up when HP1γ expression was blocked in 4AKO SCC23 cells (Figures 3J and S3I). Reasonably, the KDM4A loss-induced upregulation of stalled forks disappeared in Si-HP1γ SCC23 cells (Figures 3K and S3J). In addition, Comet assays showed that KDM4A deletion promoted DNA damage in Si-Scr SCC23 but did not in Si-HP1γ SCC23 (Figures 3L and S3K).
Activating cGAS-STING Signaling and Tumor Cell-intrinsic Immune Response by KDM4A Ablation-induced DNA Replication Stress
Unresolved replication stress and DNA damage in the nucleus could bring about the cytosolic accumulation of DNA fragments (Li and Chen, 2018; Ubhi and Brown, 2019). Accordingly, we examined the change in cytosolic DNA after the loss of KDM4A. Single strand DNA (ssDNA) and double strand DNA (dsDNA) were respectively stained with fluorescent dyes Oligreen and Picogreen in live cells. Mitochondrial DNA was visualized by Mito Tracker staining for excluding its interference during the quantification of cytosolic DNA. KDM4A depletion really increased both cytosolic ssDNA and dsDNA in SCC23 (Figure 4A) and SCC1 (Figure 4B) cells. The accumulation of cytosolic DNA can activate the cyclic GMP-AMP synthase/stimulator of interferon genes (cGAS-STING) signaling axis, shown by the sequential phosphorylation of STING, TBK1 and IRF3, and results in induction of interferon-stimulated genes (ISG) including Th1-type chemokines CXCL9, CXCL10 and CXCL11 (Li and Chen, 2018). As expected, Western blot analysis revealed that the phosphorylated protein levels of IRF3, TBK1 and STING were significantly enhanced by KDM4A knockdown in both SCC23 and SCC1 cells (Figures 4C and 4D). KDM4A knockdown robustly induced the gene expression of IFNB as well as CXCL9, CXCL10 and CXCL11 in both SCC23 (Figure 4E) and SCC1 cells (Figure 4F). Furthermore, the cGAS specific inhibitor, RU.521, significantly lessened KDM4A knockdown-induced increase of IFNβ, CXCL10 and CXCL11 in SCC23 cells (Figure 3G). We next utilized IFNβ neutralizing antibodies to affirm its engagement in the induction of CXCL10 and CXCL11 in 4AKO SCC23 cells. Anti-IFNβ antibodies did not alter its own gene expression, however they robustly decreased mRNA levels of CXCL10 and CXCL11 in 4AKO SCC23 cells (Figure 3H). Moreover, HP1γ knockdown significantly suppressed the expression of IFNB, CXCL10 and CXCL11 in 4AKO SCC23, but not in Scr SCC23 cells (Figures 4I and S3L).
Figure 4. KDM4A Inhibition Promotes Th1-type Chemokine Expression by Activation of cGAS-STING Signaling.
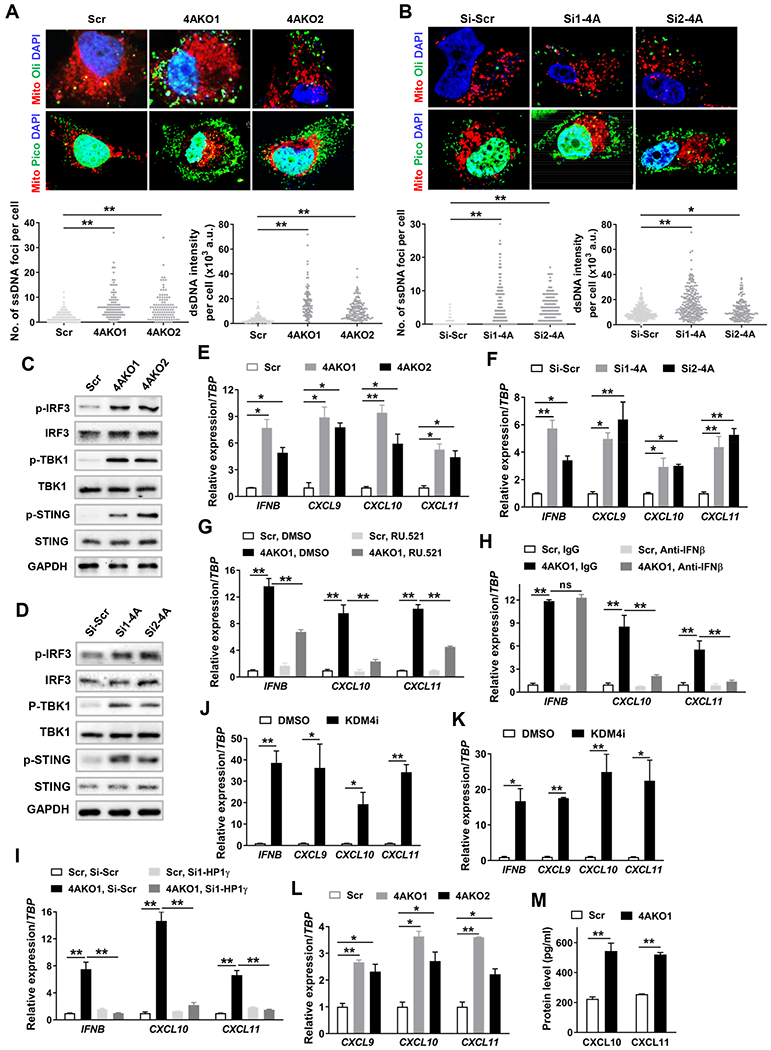
(A and B) Representative confocal images showing cytosolic DNA and mitochondria (red) as well as nuclei (blue), and quantification of cytosolic ssDNA (lower left panel) and dsDNA (lower right panel) in Scr and 4AKO SCC23 cells (A) and in SCC1 cells transfected with Si-Scr, Si1-4A and Si2-4A (B). Scale bar, 10 μm. Values are mean ± SD from the pool of three independent experiments. *p < 0.05, **p < 0.01 by one-way ANOVA.
(C and D) Western blot analysis of phosphorylated and total IRF3, TBK1 and STING in Scr and 4AKO SCC23 cells (C) and in Si-Scr and Si-4A SCC1 cells (D).
(E and F) Gene expression analysis of IFNB and Th1-type chemokines (CXCL9, CXCL10, CXCL11) in Scr and 4AKO SCC23 cells (E) and in Si-Scr and Si-4A SCC1 cells (F). Values are mean ±SD from three independent experiments. *p<0.05 and **p<0.01 by one-way ANOVA.
(G) Gene expression analysis of IFNB, CXCL10 and CXCL11 in both Scr and 4AKO1 SCC23 cells treated with either DMSO or 10 μM RU.521. Values are mean ± SD from three independent experiments. **p<0.01 by two-way ANOVA.
(H) Gene expression analysis of IFNB, CXCL10 and CXCL11 in both Scr and 4AKO1 SCC23 cells treated with either IgG or anti-IFNβ. Values are mean ± SD from three independent experiments. **p<0.01 and ns, not significant by two-way ANOVA.
(I) qRT-PCR analysis of IFNB, CXCL10 and CXCL11 in both Scr and 4AKO1 SCC23 cells transfected with either Si-Scr or Si1-HP1γ. Values are mean ± SD from three independent experiments. **p<0.01 by two-way ANOVA.
(J and K) Gene expression analysis of IFNB and Th1-type chemokines in SCC23 cells (J) and SCC1 cells (K). Values are mean ±SD from three independent experiments. *p<0.05 and **p<0.01 by Student’s t test.
(L) Gene expression analysis of IFNγ-induced Th1-type chemokines in Scr and 4AKO SCC23 cells. Values are mean ± SD from three independent experiments. *p<0.05 and **p<0.01 by one-way ANOVA.
(M) Analysis of CXCL10 and CXCL11 protein levels with ELISA assay in IFNγ-treated Scr and 4AKO1 SCC23 cells. Values are mean ± SD from three independent experiments. **p<0.01 by Student’s t test.
Also see Figure S3
Recently, a chemical inhibitor, termed here as KDM4i, was found to selectively inhibit the activity of KDM4 family proteins. It was also shown that KDM4i represents favorable pharmacokinetics properties and is well tolerated in mice, and significantly inhibit tumor growth via blocking KDM4A-meidated transcription (Metzger et al., 2017). To exploit DNA replication stress responses for cancer immunotherapy, we examined whether the inhibition of KDM4A could induce the expression of Th1-type chemokines. qRT-PCR found that KDM4i treatment also stimulated the expression of IFNB, CXCL9, CXCL10 and CXCL11 in both SCC23 (Figure 4J) and SCC1 cells (Figure 4K). Moreover, the KDM4A deletion also enhanced the gene expression of CXCL9, CXCL10 and CXCL11 induced by IFNγ (Figure 4L), and increased protein levels of CXCL10 and CXCL11, shown by ELISA assay in SCC23 cells (Figure 4M).
Inducing Liquid-like HP1γ Puncta by KDM4A Ablation
Recently, members of HP1 family were proven as organized by liquid-liquid phase separation (LLPS) using heterochromatin DNA as a scaffold (Strickfaden et al., 2020). These liquid-like puncta of HP1 molecules endow heterochromatin with repressive properties by excluding crucial molecules, such as transcription inhibition by excluding TFIIB (Larson et al., 2017; Wang et al., 2019). We found that the majority of γH2A.X foci localized in the H3K9me3-enriched heterochromatic area in 4AKO SCC23 cells (Figures 3G and 3H), indicating the important impact of KDM4A loss on heterochromatin.
We measured the distribution of HP1γ using the immunofluorescence staining in both control and KDM4A deficient cells. HP1γ localized across the nucleus and mostly distributed in a dispersive manner in SCC cells (Figures 5A and S4A). Interestingly, HP1γ was also present in the discrete and prominently bright puncta with relatively spherical shape, indicating that liquid-like condensates of HP1γ could form in SCC cells (Figures 5A and S4A). Moreover, KDM4A deletion significantly increased the numbers of HP1γ puncta in both SCC23 and SCC1 cells (Figures 5A and S4A). Consistently, KDM4i treatment also significantly induced more numbers of HP1γ puncta compared with vehicle-treated control cells. (Figures S4B and S4C). As reflected by immunofluorescence staining of HP1γ and H3K9me3, H3K9me3 was not only positive in HP1γ puncta, but also its levels were markedly higher than that outside HP1γ puncta (Figures 5B and S4D), suggesting the accumulation and compaction of heterochromatin in HP1γ puncta. In addition, we found that 1,6-hexanediol, usually used to disrupt weak hydrophobic interactions, completely abolished the HP1γ puncta in both SCC23 and SCC1 cells (Figures S4E and S4F), consistent with that HP1-mediated liquid-like condensates were maintained by hydrophobic interactions. Liquid-like condensates are dynamic, mirrored by exchange with their surrounding area and fusion of each other. To observe the dynamic change of HP1γ puncta in real time, we directly transfected GFP-tagged HP1γ (GFP-HP1γ) expression plasmids in both control and 4AKO SCC23 cells. KDM4A loss significantly increased the numbers of GFP-HP1γ puncta (Figure S4G), consistent with the upregulated puncta of endogenous HP1γ in 4AKO cells. According to the levels of GFP-HP1γ, cells were dichotomous as high and low expression groups. The numbers of GFP-HP1γ puncta were substantially more in high GFP-HP1γ expression groups versus in low expression groups (Figure S4G), suggesting that HP1γ dose-dependently formed the condensates in line with concentration-dependent property of LLPS. As liquid-like condensates are dynamic, GFP fluorescence in HP1γ puncta efficiently recovered after bleaching by high intensity of laser beams in both SCC23 and SCC1 cells on the timescale of seconds (Figures 5C, S4H and S4I). GFP-HP1γ puncta were also capable of fusing together, despite that it occurred relatively slowly (Figures 5D and S4J).
Figure 5. KDM4A Loss Increases Liquid-like HP1γ puncta in SCC cells.
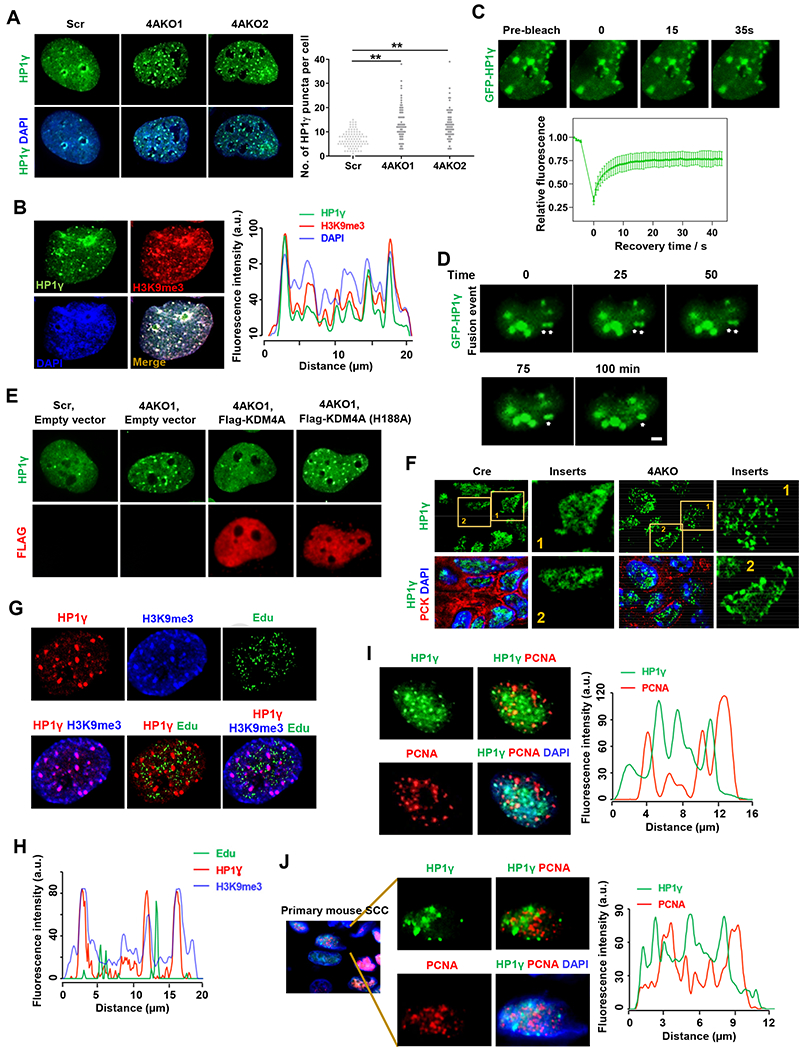
(A) Immunofluorescence staining of HP1γ and DAPI, and quantification of HP1γ puncta in Scr and 4AKO SCC23 cells. Values showed by mean ± SD were pooled from three independent experiments. Scale bar, 5 μm. **p<0.01 by one-way ANOVA.
(B) Representative immunofluorescence images of HP1γ (green), H3K9me3 (red), DAPI (blue) and their merge, and localization analysis for HP1γ puncta, H3K9me3 and DAPI along the indicated line scan (white arrow) in 4AKO SCC23 cells. Scale bar, 2 μm.
(C) Representative fluorescence images and quantitative analysis of fluorescence recovery after photobleaching (FRAP) on GFP-HP1γ puncta in 4AKO SCC23 cells. Arrowheads indicated the puncta that were analyzed by FRAP assay. Scale bar, 2 μm. Values were shown by mean ± SD (n=12, GFP-HP1γ puncta) from one representative of three independent experiments.
(D) Live-cell imaging of 4AKO SCC23 cells expressing GFP-HP1γ. The arrows indicate two HP1γ puncta that are fusing over time. Three independent experiments show similar results. Scale bar, 5 γm.
(E) Representative immunofluorescence staining of HP1γ and FLAG in Scr and 4AKO SCC23 cells transfected with empty or Flag-KDM4A expression vectors. Scale bar, 5 μm.
(F) Representative confocal fluorescence images showing HP1γ (green), H3K9me3 (red) and DAPI (blue) in primary SCCs from both control (Cre) and 4AKO mice. Enlarged images were shown in the inserts. Scale bar, 2 μm. Scale bar in the insert, 1 μm.
(G) Representative confocal fluorescence images of HP1γ (red), H3K9me3 (blue), Edu (green) and their merges in 4AKO SCC23 cells. Scale bar, 2 μm.
(H) Localization analysis for Edu, HP1γ and H3K9me3 along the indicated line scan (white arrow) in (G).
(I) Representative immunofluorescence images of HP1γ (green), PCNA (red), DAPI (blue) and their merge, and localization analysis for HP1γ puncta and PCNA along the indicated line scan (white arrow) in 4AKO SCC23 cells. Scale bars, 2 μm.
(J) Immunofluorescence staining of HP1γ (green), PCNA (red) and DAPI (blue), and localization analysis for HP1γ condensates and PCNA along the indicated line scan (white arrow) in primary SCCs from 4AKO mice. Enlarged images of single channels and merge were shown in inserts. Scale bars for main images, 5 μm. Scale bars for inserts, 2 μm.
To further explore mechanisms by which KDM4A regulated the phase separation of HP1γ, we restored KDM4A expression by transfecting plasmids encoding for wild type and demethylase-inactivated mutant (H188A) Flag-tagged KDM4A in 4AKO SCC23 cells. As shown by the immunofluorescence staining of HP1γ and FLAG, HP1γ puncta and Flag-KDM4A were readily visualized (Figure 5E). The increase in HP1γ condensates in 4AKO cells were significantly reduced by restoring KDM4A, but not demethylase-inactivated KDM4A (Figures 5E and S4K), suggesting that KDM4A demethylase activity was required for counteracting the formation of HP1γ puncta. Consistent with our in vitro results, immunostaining revealed that HP1γ puncta were also enriched with H3K9me3-marked heterochromatin in primary SCCs of 4AKO mice (Figure S4L). KDM4A ablation also significantly upregulated the numbers of HP1γ puncta in primary mouse SCC cells in vivo (Figures 5F and S4M).
To examine whether liquid-like HP1γ puncta impeded DNA replication, SCC cells were incubated with 5-Ethynyl-2-′-deoxyuridine (Edu) for a short time to track DNA replication, followed by immunofluorescence staining against HP1γ and H3K9me3. As shown by the immunofluorescence results and co-localization analysis, while HP1γ puncta always co-localized with H3K9me3-enriched heterochromatin, most of them (>90%) did not incorporate Edu in both control and KDM4A-depleted SCC cells (Figures 5G, 5H and S5A–S5D), suggesting that the liquid-like HP1γ puncta on heterochromatin slowed DNA replication. Of note, DNA could be efficiently replicated on the chromatin area outside HP1γ puncta, where HP1γ molecules were dispersedly present (Figures 5G, 5H and S5A–S5D), highlighting that the liquid-like puncta structure of HP1γ, instead of HP1γ molecules themselves, slowed DNA replication. Proliferating cell nuclear antigen (PCNA) acts as a crucial DNA replication factor, dictating both the assembly and progress of replication forks by recruiting and activating DNA polymerases as well as orchestrating replication events (Choe and Moldovan, 2017). We thus performed the co-immunofluorescence staining of HP1γ puncta with PCNA in 4AKO SCC23 cells. The co-localization analysis revealed that HP1γ and PCNA puncta almost completely excluded each other, more than 95% of HP1γ puncta were proven PCNA-negative in 4AKO SCC23 cells (Figures 5I and S5E). Like in human cancer cells, HP1γ puncta were also found deficient of PCNA recruitment in primary mouse SCC cells of 4AKO mice in vivo (Figures 5J and S5F). However, the area with dispersed HP1γ distribution were enriched with PCNA (Figures 5I and 5J).
To study whether other members of KDM4 family, except for KDM4A, also play the important roles on HP1γ puncta formation in SCC cells, SCC23 cells were transfected with siRNAs targeting KDM4B, KDM4C and KDM4D genes. The expression of KDM4B KDM4C, and KDM4D were efficiently inhibited by siRNA transfection (Figure S5G). however, the numbers of HP1γ puncta were not significantly affected (Figure S5H), suggestive of the unique effects of KDM4A on HP1γ puncta formation in SCC cells. In addition, we found that the numbers of HP1γ puncta were significantly upregulated by KDM4D knockdown, but not by KDM4A reduction in HeLa cells (Figures S5I and S5J), showing that different types of cancer cells could use the different members of KDM4 family to regulate HP1γ puncta formation.
CXCR3-mediated CD8+ T Cell Infiltration Required for Inhibiting SCC by KDM4A Deletion
CXCR3, activated by its chemokine ligands CXCL9, CXCL10 and CXCL11, mediates the recruitment of CD8+ T cells into tumor bulk (Mikucki et al., 2015). Next, we investigated whether CXCR3-mediated CD8+ T infiltration was responsible for SCC regression in vivo. At the 20th week of SCC induction, when tamoxifen was administered to delete KDM4A in tumor cells, mice were also given anti-CXCR3 neutralizing antibodies (Figure 6A). The immunofluorescence staining of CD8α confirmed that anti-CXCR3 treatment markedly reduced the numbers of intratumoral CD8+ T cells in 4AKO mice (Figure 6B). Anti-CXCR3 treatment significantly increased the numbers of SCCs in 4AKO mice compared with isotype IgG treatment (Figure 6C). Moreover, anti-CXCR3 treatment also significantly increased SCC growth and invasion in 4AKO mice compared with the isotype IgG treatment. Yet the tumor growth and invasion were still partially inhibited in anti-CXCR3-treated 4AKO mice, as compared with that in anti-CXCR3-treated control mice (Figures 6D–6F), due to the fact that KDM4A deletion also impaired SCC invasive growth in a cancer cell-autonomous manner. To account for the restoration of SCC growth in anti-CXCR3-treated 4AKO mice compared with those in IgG-treated 4AKO mice, we measured the tumor cell apoptosis in primary SCCs. Anti-CXCR3 significantly alleviated apoptosis in 4AKO mice compared with IgG treatment (Figure 6G), which substantiated that CD8+ T cell infiltration was indispensable for tumor cell apoptosis induced by KDM4A loss. In addition, tumor cell KDM4A deficiency-induced amelioration of lymphatic metastasis was significantly impaired by anti-CXCR3 treatment, as mirrored by more metastatic lymph nodes and larger metastatic areas in anti-CXCR3-treated 4AKO mice than those in IgG-treated 4AKO mice (Figures 6H–6J). We also noticed that both the numbers of metastatic lymph nodes and the area of lymphatic metastasis remained smaller in 4AKO mice than those in control mice following anti-CXCR3 treatment.
Figure 6. Th1-type Chemokines-induced CD8+ T Cell Infiltration Accounts for the Inhibitory Impacts of KDM4A Deletion on Primary Mouse SCC Growth and Lymphatic Metastasis.
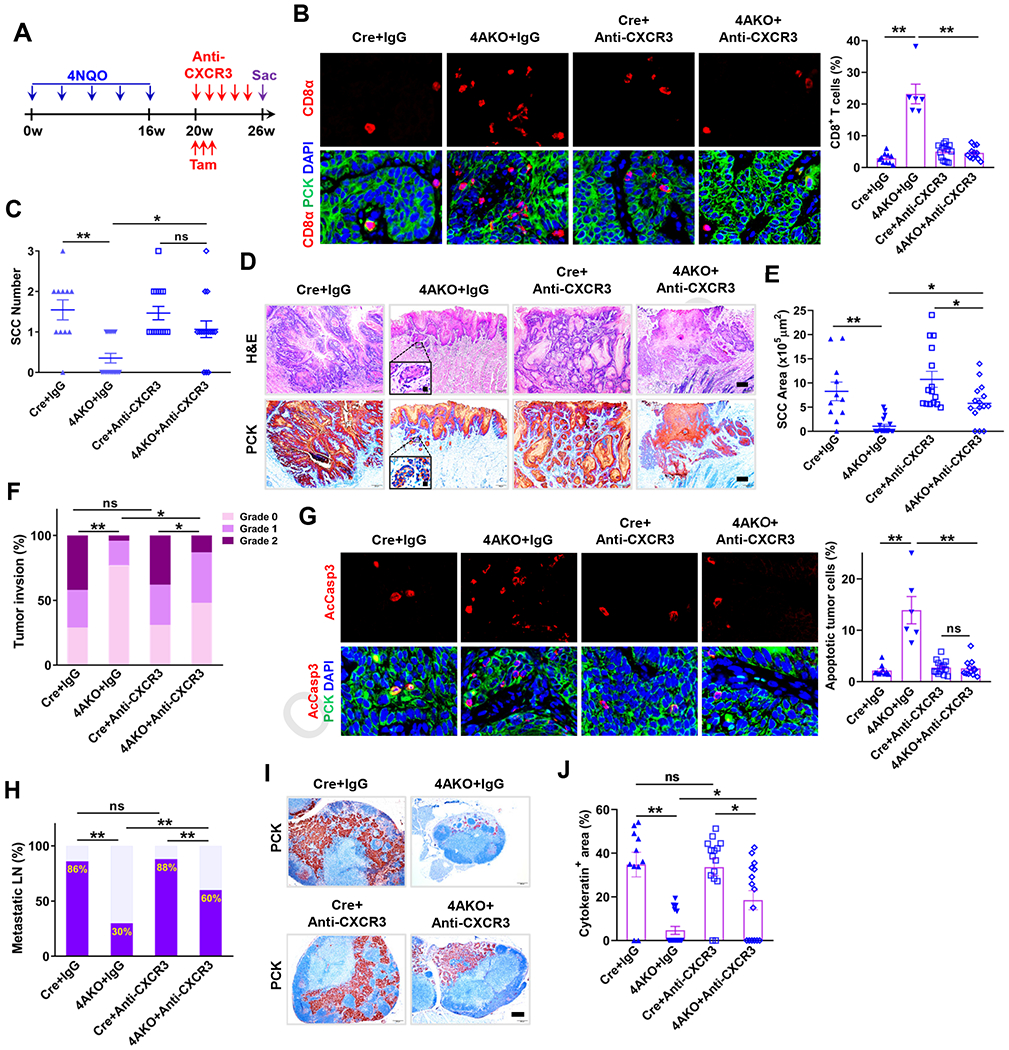
(A) Schematic diagram for anti-CXCR3 treatment in Cre and 4AKO mice with 4-NQO-induced SCCs.
(B) Immunofluorescence staining and quantification of CD8+ T cell infiltration within primary mouse SCC in both Cre and 4AKO mice treated by IgG or anti-CXCR3. Scale bars, 10 μm. Values represent mean ± SEM from the pool of two independent experiments. n = 10:6:15:12. **p < 0.01 by two-way ANOVA.
(C) Quantification of primary mouse SCC number in both Cre and 4AKO mice treated by either IgG or anti-CXCR3. Values are mean ±SD from the pool of two-independent experiments. n = 11:17:15:15. ns, not significant, *p<0.05 and **p<0.01 by two-way ANOVA.
(D) Representative images showing H&E staining (upper) and PCK immunostaining (lower) of SCCs in both Cre and 4AKO mice treated by either IgG or anti-CXCR3. Scale bars, 200 μm. Inserts are enlarged images with 10 μm scale bars.
(E) Quantification of primary SCC areas. *p<0.05 and **p<0.01 by two-way ANOVA.
(F) Histogram analysis of tumor invasion degree. *p < 0.05, **p < 0.01 and ns, not significant by Cochran-Armitage test.
(G) Immunofluorescence staining and quantification of AcCasp3–positive tumor cells. Scale bars, 10μm. Values represent mean ± SEM from the pool of two independent experiments. n = 10:6:15:12. **p < 0.01 and ns, not significant by two-way ANOVA.
(H) Quantification of metastatic lymph nodes. ns, not significant and **p < 0.01 by Chisquare test.
(I) Immunostaining of PCK in lymph nodes. Scale bar, 200 μm.
(J) Quantification of metastatic areas in lymph nodes. Values represent mean ± SEM. n=11:17:15:15. ns, not significant, *p<0.05 and **p<0.01 by two-way ANOVA.
Overcoming PD1 Blockade Therapy Resistance and Eliminating CSCs by Targeting KDM4A
Increasing evidence from both clinical data and mouse experiments supports that intratumoral infiltration of new CD8+ T cells dictates the response to PD1 blockade therapy and its efficacy on various cancers (Tang et al., 2016; Tumeh et al., 2014; Yost et al., 2019). Since KDM4A deficiency induced tumor cell-intrinsic immune response to recruit CD8+ T cells into the primary mouse SCC, we hypothesized that pharmacological inhibition of KDM4A potentiated the efficacy of PD1 blockade in vivo. To affirm the hypothesis, we carried out the combination treatment of KDM4i and PD1 neutralizing antibodies (Anti-PD1) (Figure S6A). Of note, after the endpoint of 26 weeks, we observed that KDM4A ablation alone had significantly inhibited SCC growth. In order to compare the efficacy of combination treatment with the single therapy of KDM4i or PD1 blockade very well, we extended our experiment for additional 4 weeks. Consistent with KDM4A deletion, the treatment with KDM4i alone significantly reduced SCC number, size and its invasive degree compared with vehicle control (Figures 7A–7C). While PD1 blockade did not affect SCC growth, its combination with KDM4i significantly reduced SCC number, size, and invasive degrees. Moreover, the combination treatment also had more significant effect on the inhibition of SCC growth than KDM4i alone (Figures 7A–7C). Considering long duration of the combination treatment, we also monitored mouse tolerance to treatment. Mouse weight was not significantly changed by KDM4i or its combination with Anti-PD1 (Figure S6B), suggesting that mice tolerated KDM4i and anti-PD1 treatment.
Figure 7. Targeting KDM4A Overcomes SCC Resistance to PD1 Blockade and Eliminates CSCs by Recruiting CD8+ T cells into Tumors.
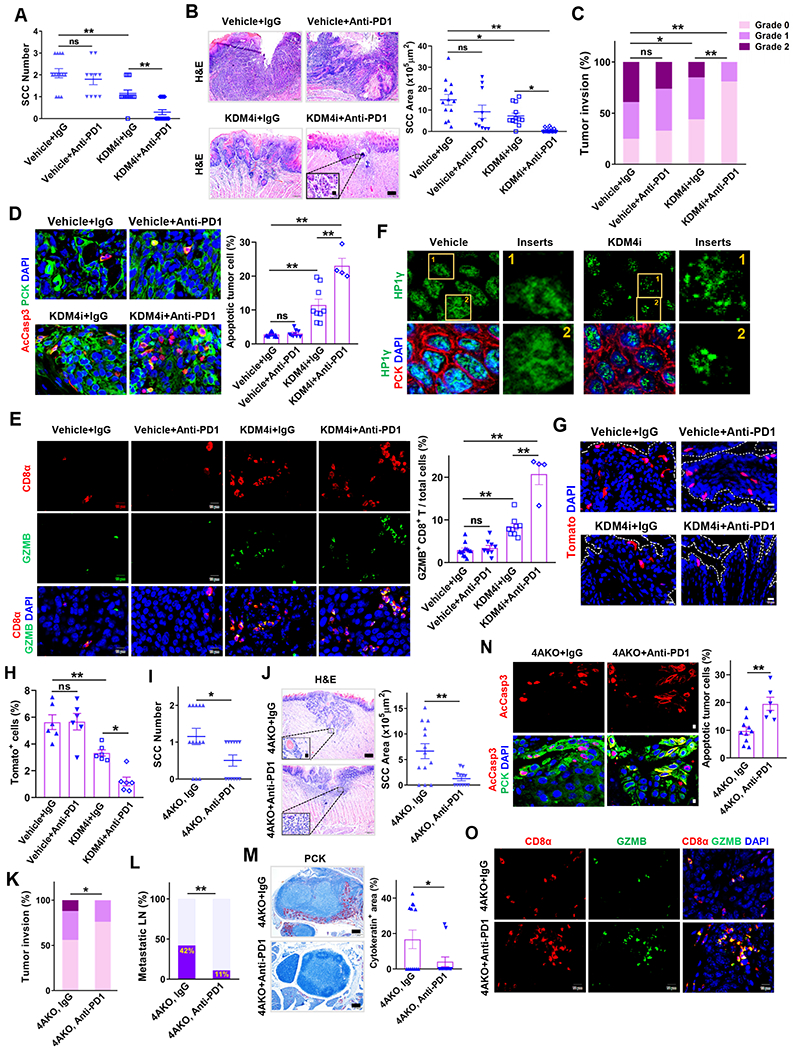
(A) Quantification of primary SCC number in mice treated with anti-PD1, KDM4i or their combination. Values represent mean ± SD from the pool of two independent experiments. n=13:10:13:17. ns, not significant, and **p < 0.01 by one-way ANOVA.
(B) H&E staining and area quantification of primary SCCs. Scale bar, 200 μm. Enlarged images were shown in inserts with 10 μm scale bar. n = 13:10:13:17. ns, not significant, *p < 0.05 and **p<0.01 by one-way ANOVA.
(C) Histogram analysis of primary tumor invasion grades. ns, not significant, *p<0.05 and **p<0.01 by Cochran-Armitage test.
(D) Immunofluorescence staining and quantification of apoptotic tumor cells. Values are mean ± SEM from the pool of two-independent experiments. n = 12:9:9:4. ns, and **p < 0.01 by one-way ANOVA.
(E) Immunofluorescence staining and quantification of GZMB+CD8+ T cells within and around tumor islets. Values represent mean ± SEM from the pool of two independent experiments. Scale bars, 10 μm. n = 12:9:9:4. ns, not significant, and **p<0.01 by one-way ANOVA.
(F) Representative immunofluorescence staining of HP1γ (green), PCK (red) and DAPI (blue) in primary SCCs of mice. Scale bar, 2 μm. Enlarged images were shown in inserts with 1 μm scale bar.
(G and H) Representative fluorescence images (G) and quantification (H) of tdTomato+ cells (BMI1+ CSCs) in primary SCCs of mice. n = 6:6:6:6. Scale bar 10 μm. ns, not significant, *p<0.05 and **p<0.01 by one-way ANOVA.
(I) Quantification of primary tongue SCC number in 4AKO mice treated with anti-PD1 or IgG. Values are mean ± SD from the pool of two independent experiments, n = 13:12. *p < 0.05 by Student’s t test.
(J) H&E staining and area quantification of primary SCCs. Scale bar for the main image, 200 μm. Inserts are enlarged images with 10 μm scale bar. Values are mean ± SD from the pool of two independent experiments, n = 13:12. **p < 0.01 by Student’s t test.
(K) Quantification of primary tumor invasion degree in 4AKO mice treated with anti-PD1 or IgG. n = 13:12. *p < 0.05 by Cochran-Armitage test.
(L) Percentage of metastatic lymph nodes in 4AKO mice treated with anti-PD1 or IgG. Data are collected from two independent experiments. n = 13:12. **p < 0.01 by Chisquare test.
(M) PCK immunostaining and quantification of metastatic areas in lymph nodes from 4AKO mice. n = 13:12. Values represent mean values ± SEM from two independent experiments. *p < 0.05 by Student’s t test. Scale bars, 200 μm.
(N) Immunofluorescence staining and quantification of apoptotic tumor cells in primary mouse SCCs in 4AKO mice treated by either anti-PD1 (n = 6) or IgG (n = 10). Values are mean ± SEM from the pool of two independent experiments. Scale bars, 10μm. **p<0.01 by Student’s t test.
(O) Immunofluorescence staining of GZMB+CD8+ T cells within and around tumor islets. Scale bars, 10 μm.
Also see Figures S6 and S7.
We examined apoptosis and CD8+ T cell activation in SCCs by immunostaining AcCasp3, CD8α and granzyme B (GZMB), respectively. Unlike PD1 blockade alone, KDM4i treatment significantly induced apoptosis in SCCs. However, the combination treatment of KDM4i and anti-PD1 collaboratively induced more apoptosis in SCCs compared with KDM4i treatment alone (Figure 7D). Activated CD8+ T cells express GZMB to induce apoptosis in tumor cells. PD1 blockade did not alter the frequency of GZMB+CD8+ T cells in primary SCCs (Figure 7E). No surprisingly, KDM4i treatment alone increased CD8+ T cell infiltration as well as activating GZMB+CD8+ T cells. However, the combination treatment of KDM4i and PD1 blockade significantly further increased CD8+ cells and GZMB+CD8+ T cells compared with just KDM4Ai alone (Figures 7E, S6C and S6D). These results indicated that KDM4A inhibition overcame SCC cell-intrinsic resistance to PD1 blockade by recruiting and activating CD8+ T cells. As expected, KDM4i treatment increased the numbers of HP1γ puncta (Figures 7F and S6E), induced DNA damage (Figure S6F) and increased the protein levels of CXCL10 (Figure S6G) and CXCL11 (Figure S6H) in tumor cells in vivo. Furthermore, the combination treatment of KDM4i and PD1 blockade also collaboratively eliminated lymph node metastasis of SCCs as determined by anti-PCK immunostaining (Figures S6I and S6J).
To further demonstrated that KDM4i overcame the SCC resistance to PD1 blockade by promoting CXCR3-mediated CD8+ T cell infiltration, we examined whether KDM4i could potentiated anti-PD1-mediated inhibition of SCC growth and metastasis in the presence anti-CXCR3 neutralizing antibodies (Figure S7A). As compared to the treatments of KDM4i alone and together with Anti-PD1 (Figures 7E, S6C and S6D), anti-CXCR3 treatment significantly abolished the CD8+ T cell recruitment induced by KDM4i plus anti-PD1 treatment (Figures S7B and S7C). KDM4i was unable to collaborate with anti-PD1 to inhibit SCC growth or lymph node metastasis in the presence of anti-CXCR3 (Figures S7D–S7J). To further examine whether KDM4i plus anti-PD1 treatment could eliminate CSCs, we induced SCC in Bmi1Cre-ERT2;tdTomato mice and treated with KDM4i and anti-PD1 for 8 weeks. One day before sacrificing mice, mice were given tamoxifen to label BMI1+ CSCs as described before (Chen et al., 2017; Jia et al., 2020). Histological analysis found that anti-PD1 did not affect BMI1+ CSCs and KDM4i treatment reduced BMI1+ CSCs in primary SCCs. The combination of KDM4i and anti-PD1 more drastically reduced BMI1+ CSCs in primary SCCs compared with anti-PD1 or KDM4i alone (Figures 7G and 7H).
To rule out the non-specific effect of KDM4i, we also determined whether the specific deletion of KDM4A in tumor cells rendered SCC sensitive to PD1 blockade treatment. 4AKO mice were treated with PD1 blockade or isotype IgG control for 8 weeks (Figure S7K). While PD1 blockade did not ameliorate primary SCC growth and lymph node metastasis in the control Cre mice (Figures 7A–7C, S6I and S6J), PD1 blockade significantly reduced the SCC number, area and its invasion degree (Figures 7I–7K) in 4AKO mice. Similarly, PD1 blockade also potently suppressed the lymphatic metastasis, as mirrored by the fewer metastatic lymph nodes and the smaller size of lymphatic metastasis in anti-PD1-treated 4AKO mice, compared with IgG control (Figures 7L and 7M). Immunostaining revealed that PD1 blockade significantly induced more apoptosis in SCCs of 4AKO mice, compared with IgG treatment (Figure 7N). Consistently, PD1 blockade significantly increased CD8+ T cell infiltration as well as GZMB+CD8+ T cells in 4AKO mice, compared with IgG treatment (Figures 7O and S7L).
To examine whether KDM4A levels in tumor cells were negatively associated with CD8+ T cell infiltration in human HNSCC samples, TCGA database was first explored. We evaluated the association of KDM4A gene amplification with CD8+ T cell infiltration. TCGA data analysis revealed that there was a negative association between KDM4A gene amplification and CD8+ T cells in HPV− HNSCC sample (Figure S7M). However, the negative correlation did not exist in HPV+ HNSCC samples (Figure S7M), possibly due to the HPV E7 protein-induced loss of cGAS-STING signaling pathway (Shaikh et al., 2019). To affirm the negative correlation of KDM4A expression and CD8+ T cell infiltration, we collected 100 HPV− HNSCC samples from the UCLA translational pathology core of and carried out the immunohistochemical staining to measure protein levels of KDM4A, CXCL10 and CD8α. Human HNSCCs abundant with KDM4A proteins lacked CXCL10 expression and CD8+ T cell infiltration (Figures S7N, S7P and S7Q). On the contrary, human HNSCCs with low levels of KDM4A proteins had increased CXCL10 expression and CD8+ T cell infiltration (Figures S7O–S7Q). The expression of CXCL10 in human HNSCCs was positively associated with CD8+ T cell infiltration (Figure S7R).
DISCUSSION
Immunotherapy has emerged as a new type of clinical treatment for cancer. However, a significant proportion of patients do not respond to immunotherapy. There are several strategies to improve anti-tumor immunity. Targeting the DNA damage response (DDR) proteins PARP and checkpoint kinase 1 could improve anti-tumor effect and augment cytotoxic T-cell infiltration (Chabanon et al., 2019; Pantelidou et al., 2019; Sen et al., 2019). DNA demethylating agents induced double-stranded RNA expression, activation of the cytosolic pattern recognition receptor MDA5, and downstream activation of MAVS and IRF7 (Chiappinelli et al., 2015). Recently, it has been found that the histone H3K4 demethylase LSD1 suppressed endogenous expression of endogenous retroviral element transcripts and IFN responses in tumor cells. The inhibition of LSD1 induced double-stranded RNA stress and activated anti-tumor T cell immunity (Sheng et al., 2018). Unlike LSD1, KDM4A is a JMJD domain family histone demethylase which erases H3K9me3 associated with heterochromatin formation. Our results demonstrate that the inhibition of KDM4A utilized a completely different mechanism from LSD1 to induce anti-tumor immunity.
DNA replication stress frequently occurred in highly proliferating cancer cells including SCC cells. Heterochromatin area are fragile sites where replication stress is readily induced. Liquid-like HP1γ puncta exclude PCNA and stalled DNA replication, suggesting that LLPS of heterochromatin components could account for the vulnerability of heterochromatin to DNA replication stress. It has been shown that LLPS renders heterochromatin to repress gene transcription by excluding TFIIB (Larson et al., 2017; Wang et al., 2019). Very recently, the common therapeutic agent cisplatin was found to exhibit its cytotoxicity by aggregating in the mediator of RNA polymerase II transcription subunit 1 (Med1) transcriptional condensates while diffusing freely through HP1a condensates, suggesting the specificity of the therapeutic effect of cisplatin to a distinct type of condensates (Klein et al., 2020). Our study here not only highlighted that targeting DNA replication helped the cancer immunotherapy, but also demonstrated that spatial organization of heterochromatic components can be harnessed to activate antitumor immunity and improve cancer immunotherapy. Given that epigenetic programs are extensively dysregulated in various cancers (Valencia and Kadoch, 2019), such as KDM4A overexpression in HNSCC, it is compelling to investigate whether induction of HP1γ puncta on heterochromatin can also unleash antitumor immune response in other types of cancers.
Very recently, our preclinical model showed that BMI1+ CSCs were resistant to PD1 blockade (Jia et al., 2020). KDM4A inhibition could inhibit tumorsphere formation in vitro and reduce CSC numbers of SCC in vivo, suggesting that KDM4A could directly inhibit cancer stemness. Interestingly, KDM4A inhibition cooperated with anti-PD1 to eliminate CSCs of SCC in vivo that might explain the synergetic effect of KDM4i and anti-PD1 on the inhibition of SCC growth and metastasis. Currently, most of studies have focused on developing strategies for blocking inhibitory receptors. Given the low response rate of PD1 blockade immunotherapy, our studies suggest that the inhibition of KDM4A might help to improve the efficacy of PD1 blockade for the larger patient population with refractory HNSCC. In summary, our findings have important implications for harnessing nuclear condensates for cancer immunotherapy by aggravating replication stress to induce tumor cell-intrinsic immune responses via exploiting tumor vulnerability.
Limitations of Study
There are some limitations in our mouse model. Tumor immune profiles between human papillomavirus negative (HPV−) and HPV+ HNSCC may be different. However, our study focused on HPV− HNSCC by using 4-NQO mouse model. The effects of KDM4A on HPV+ HNSCC need further studies. While we showed that KDM4A inhibition unleashed tumor cell-intrinsic immunity by inducing replication stress in HNSCC, it is unknown whether our findings can extend to other solid cancers. Finally, we need to develop a more specific inhibitor for targeting KDM4A to translate our findings into clinical setting.
STAR ★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Cun-Yu Wang (cunywang@ucla.edu).
Materials Availability
Cell lines and plasmids used in this study are described in the Key Resource Table and available upon request. Bmi1Cre-ERT2;R26tdTomato and K14Cre-ERT2;Kdm4af/f mice are available upon request with the approved animal protocol. All requests need to execute a suitable Materials Transfer Agreement.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-pan cytokeratin | Abcam | Cat#ab86734; RRID: AB_10674321 |
| Mouse monoclonal anti-pan cytokeratin | Santa Cruz | Cat#sc-8018; RRID: AB_627396 |
| Rabbit polyclonal anti-wide spectrum Cytokeratin | Abcam | Cat#ab9377; RRID: AB_307222 |
| Rabbit polyclonal anti-Active Caspase-3 | Abcam | Cat#ab13847; RRID: AB_443014 |
| Rabbit polyclonal anti-CD8 | Abcam | Cat#ab203035; RRID: N/A |
| Rabbit monoclonal anti-CD8α (clone D8A8Y) | Cell Signaling Technology | Cat#85336s; RRID: AB_2800052 |
| Mouse monoclonal anti-α-tubulin | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Goat polyclonal anti-CXCL9 | R&D Systems | Cat#AF-492-NA; RRID: AB_2086734 |
| Rabbit polyclonal anti-CXCL10 | Bioss Antibodies | Cat#BS-1502R; RRID: AB_10859700 |
| Mouse monoclonal anti-IP10 | Santa Cruz | Cat#sc-101500; RRID: AB_1564138 |
| Rabbit polyclonal anti-CXCL11 | MyBioSource | Cat#MBS7049567; RRID: N/A |
| Mouse monoclonal anti-phospho-Histone H2A.X (Ser139) Antibody (clone JBW301) | Upstate | Cat#05-636; RRID: AB_309864 |
| Rabbit polyclonal anti-gamma-H2AX | Bethyl Laboratories | Cat#IHC-00059; RRID: AB_533402 |
| Rabbit monoclonal anti-KDM4A | Abcam | Cat#ab191433; RRID: N/A |
| Rabbit polyclonal anti-JMJD2A | Bethyl Laboratories | Cat#IHC-00188; RRID: AB_873052 |
| Rabbit polyclonal anti-Histone H3 (tri methyl K9) | Abcam | Cat#ab8898; RRID: AB_306848 |
| Rabbit polyclonal anti-Histone H3 (tri methyl K36) | Abcam | Cat#ab9050; RRID: AB_306966 |
| Rat monoclonal anti-BrdU | Abcam | Cat#ab6326; RRID: AB_305426 |
| Mouse monoclonal anti-BrdU (clone B44) | BD Biosciences | Cat#347580; RRID: AB_400326 |
| Mouse monoclonal anti-IFNβ (clone 76703) | R&D Systems | Cat# MAB814; RRID: AB_2122895 |
| Rat monoclonal anti-PD1 (clone 29F.1A12) | BioXCell | Cat#BE0273; RRID: AB_2687796 |
| Armenian Hamster monoclonal anti-CXCR3 (clone CXCR3-173) | BioXCell | Cat# BE0249; RRID: AB_2687730 |
| Goat polyclonal anti-Granzyme B | NOVUS Biologicals | Cat#AF1865; RRID: AB_2294988 |
| Rabbit polyclonal anti-FOSL1 | Cell Signaling Technology | Cat#5281S; RRID: AB_10557418 |
| Rabbit polyclonal anti-JUN | Abcam | Cat#ab31419; RRID: AB_731605 |
| Rabbit polyclonal anti-p-IRF3 | Cell Signaling Technology | Cat#4947S; RRID: AB_823547 |
| Rabbit polyclonal anti-IRF3 | Cell Signaling Technology | Cat#4302S; RRID: AB_1904036 |
| Rabbit polyclonal anti-p-TBK1 | Cell Signaling Technology | Cat#5483S; RRID: AB_10693472 |
| Rabbit polyclonal anti-TBK1 | Cell Signaling Technology | Cat#3504S; RRID: AB_2255663 |
| Rabbit polyclonal anti-p-STING | Cell Signaling Technology | Cat#19781S; RRID: AB_2737062 |
| Rabbit polyclonal anti-STING | Cell Signaling Technology | Cat#13647S; RRID: AB_2732796 |
| Mouse monoclonal anti-KDM4A | Abcam | Cat#ab105953; RRID: AB_10864568 |
| Rabbit polyclonal anti-MCM2 | Cell Signaling Technology | Cat#3619S; RRID: AB_2142137 |
| Rabbit polyclonal anti-MCM7 | Cell Signaling Technology | Cat#3735S; RRID: AB_2142705 |
| Mouse monoclonal anti-HP1γ | Santa Cruz | Cat#sc-398562; RRID: N/A |
| Rabbit polyclonal anti-HP1γ | Abcam | Cat#ab10480; RRID: AB_297219 |
| Rabbit polyclonal anti-PCNA | Abcam | Cat#ab92552; RRID: AB_10561973 |
| Goat polyclonal anti-RFP | Rockland | Cat#200-101-379; RRID: AB_2744552 |
| Goat polyclonal anti-Rabbit IgG HRP conjugate | Promega | Cat#W401B; RRID: N/A |
| Goat polyclonal anti-Mouse IgG HRP conjugate | Promega | Cat#W402B; RRID: N/A |
| Bacterial and Virus Strains | ||
| TOP10 Chemically Competent E. coli | Thermo Fisher Scientific | Cat# C404003 |
| Biological Samples | ||
| Human HNSCC embedded tumor samples | UCLA translational pathology core laboratory | http://pathology.ucla.edu/tpcl |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Monoclonal rat IgG2a (clone 2A3) | BioXCell | Cat# BE0089; RRID: AB_1107769 |
| Normal Rabbit IgG | Millipore | Cat#12-370; RRID: AB_145841 |
| Mouse IgG1 Isotype Control | Cell Signaling Technology | Cat# 5415S; RRID: AB_10829607 |
| Recombinant Human IFN-γ | PeproTech | Cat# 300-02 |
| QC6352 | MedChemExpress | Cat#HY-104048 |
| Ru.521 | AOBIOUS | Cat# AOB37877 |
| MitoTracker™ Red CMXRos | Thermo Fisher Scientific | Cat# M7512 |
| 1,6-Hexanediol | Sigma-Aldrich | Cat# 240117 |
| G418 | Sigma-Aldrich | Cat# 04727878001 |
| DMEM | Thermo Fisher Scientific | Cat#11995065 |
| M-MuLV Reverse Transcriptase | New England Biolabs | Cat#M0253L |
| Random Primers | Thermo Fisher Scientific | Cat#48190011 |
| dNTP | Thermo Fisher Scientific | Cat#18427013 |
| Dimethyl 3,3'-Dithiobispropionimidate.2HCl | Thermo Fisher Scientific | Cat#20665 |
| Dynabeads™ Protein A | Thermo Fisher Scientific | Cat#10002D |
| Hot Start DNA Polymerase | New England Biolabs | Cat#M0481L |
| RNase Inhibitor | New England Biolabs | Cat#0307L |
| BsmBI | New England Biolabs | Cat#0580S |
| T4 Polynucleotide Kinase | New England Biolabs | Cat#0201S |
| T4 DNA Ligase | New England Biolabs | Cat#0202S |
| 4-Nitroquinoline N-oxide (4NQO) | Santa Cruz | Cat#sc-256815 |
| Fisherbrand Superfrost Plus Microscope Slides | Thermo Fisher Scientific | Cat#12-550-15 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Cat#13778150 |
| ProLong™ Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat#P36962 |
| RIPA Buffer | Sigma-Aldrich | Cat#R0278 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596026 |
| PhosSTOP™ | Sigma-Aldrich | Cat#4906845001 |
| B-27 supplement | Thermo Fisher Scientific | Cat#17504044 |
| N-2 supplement | Thermo Fisher Scientific | Cat#17502048 |
| Insulin | Sigma-Aldrich | Cat#I0516 |
| Halt™ Protease Inhibitor Cocktail | Thermo Fisher Scientific | Cat#78430 |
| 5-Iodo-2’-deoxyuridine | Sigma-Aldrich | Cat#I7125 |
| 5-Chloro-2’-deoxyuridine | Sigma-Aldrich | Cat#C6891 |
| Tamoxifen | Sigma-Aldrich | Cat#5648 |
| Recombinant Human HGF Protein | R&D Systems | Cat#294-HG-005 |
| BioCoat™ Matrigel® Invasion Chambers | Corning | Cat#354480 |
| SYBR™ Green I Nucleic Acid Gel Stain | Thermo Fisher Scientific | Cat#S7563 |
| Critical Commercial Assays | ||
| Human CXCL10/IP-10 Quantikine ELISA Kit | R&D Systems | Cat# DIP100 |
| Human CXCL11/I-TAC Quantikine ELISA Kit | R&D Systems | Cat# DCX110 |
| Comet SCGE Assay Kit | Enzo Life Sciences | Cat#ADI-900-166 |
| Quant-iT™ OliGreen™ ssDNA Assay Kit | Thermo Fisher Scientific | Cat# O11492 |
| Quant-iT™ PicoGreen™ dsDNA Assay Kit | Thermo Fisher Scientific | Cat# P11496 |
| ChIP DNA Clean & Concentrator | Zymo Research | Cat#D5205 |
| Quant-iT™ dsDNA Assay Kit | Thermo Fisher Scientific | Cat#Q33130 |
| Pierce™ BCA protein Assay Kit | Thermo Fisher Scientific | Cat#23227 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69504 |
| Click-iT™ EdU Alexa Fluor™ 488 Imaging Kit | Thermo Fisher Scientific | Cat#C10337 |
| Deposited data | ||
| RNA-Seq for this study | This paper | GSE137953 |
| Whole exome sequencing of SCC1 cell | This paper | PRJNA701731 |
| Whole exome sequencing of SCC23 cell | Sequence Read Archive | PRJNA525437 (Mann et al., 2019) |
| Experimental Models: Cell Lines | ||
| Human: UM-SCC23 (SCC23) | Laboratory of Thomas Carey | N/A |
| Human: UM-SCC1 (SCC1) | Laboratory of Thomas Carey | N/A |
| Human: 293T cells | ATCC | Cat#CRL-3216; RRID: CVCL_0063 |
| Phoenix-Ampho 293T cells | ATCC | Cat#CRL-3213 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tg (KRT14-cre/ERT)20Efu/J | The Jackson Laboratory | JAX: 005107 |
| Mouse: Bmi1tm1(cre/ERT)Mrc/J | The Jackson Laboratory | JAX: 010531 |
| Mouse: Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX: 007914 |
| Mouse: KDM4Afl/fl: KDM4A tm1Zpl | Laboratory of Liu, ZP (Zhang et al., 2011) | JAX: 029424 |
| Oligonucleotides | ||
| Primer for CXCL9, forward: GTGGTGTTCTTTTCCTCTTGGG | This paper | N/A |
| Primer for CXCL9, Reverse: ACAGCGACCCTTTCTCACTAC | This paper | N/A |
| Primer for CXCL10, forward: GCAAGCCAATTTTGTCCACG | This paper | N/A |
| Primer for CXCL10, Reverse: ACATTTCCTTGCTAACTGCTTTCAG | This paper | N/A |
| Primer for CXCL11, forward: CAGAATTCCACTGCCCAAAGG | This paper | N/A |
| Primer for CXCL11, Reverse: GTAAACTCCGATGGTAACCAGCC | This paper | N/A |
| Primer for IFNB, forward: GCTTGGATTCCTACAAAGAAGCA | This paper | N/A |
| Primer for IFNB, Reverse: ATAGATGGTCAATGCGGCGTC | This paper | N/A |
| Primer for KDM4A, forward: CCTCACTGCGCTGTCTGTAT | This paper | N/A |
| Primer for KDM4A, Reverse: CCAGTCGAAGTGAAGCACAT | This paper | N/A |
| Primer for KDM4B, forward: ACTTCAACAAATACGTGGCCTAC | This paper | N/A |
| Primer for KDM4B, Reverse: CGATGTCATCATACGTCTGCC | This paper | N/A |
| Primer for KDM4C, forward: CGAGGTGGAAAGTCCTCTGAA | This paper | N/A |
| Primer for KDM4C, Reverse: GGGCTCCTTTAGACTCCATGTAT | This paper | N/A |
| Primer for KDM4D, forward: TAGAAGGCGTCAATACACCCT | This paper | N/A |
| Primer for KDM4D, Reverse: GGGGCACCACATACCAAGTT | This paper | N/A |
| Primer for HP1γ, forward: TAGATCGACGTGTAGTGAATGGG | This paper | N/A |
| Primer for HP1γ, Reverse: TGTCTGTGGCACCAATTATTCTT | This paper | N/A |
| Primer for TBP, forward: GCACAGGAGCCAAGAGTGA | This paper | N/A |
| Primer for TBP, Reverse: GTTGGTGGGTGAGCACAAG | This paper | N/A |
| siRNA targeting sequence, Si1-KDM4A: GUUCGAGAGUUCCGCAAGA | This paper | N/A |
| siRNA targeting sequence, Si2-KDM4A: AACACAGUUAUUGACCAUACU | This paper | N/A |
| siRNA targeting sequence, Si1-KDM4B: CCUCUUCACGCAGUACAAU | This paper | N/A |
| siRNA targeting sequence, Si2-KDM4B: CCUGCUACGAAGUGAACUU | This paper | N/A |
| siRNA targeting sequence, Si1-KDM4C: CCAGGCUCUCCAGUACAUU | This paper | N/A |
| siRNA targeting sequence, Si2-KDM4C: GCCAGGCUCUCCAGUACAU | This paper | N/A |
| siRNA targeting sequence, Si1-KDM4D: GCAACCACGAUCUUUACAA | This paper | N/A |
| siRNA targeting sequence, Si1-KDM4D: CCAGAGAGACCUAUGAUAA | This paper | N/A |
| siRNA targeting sequence, Si1-HP1γ: GGAAUCCCUGUUGCUACAU | This paper | N/A |
| siRNA targeting sequence, Si2-HP1γ: CCGGAAACACCCGAAUCAA | This paper | N/A |
| sgRNA targeting sequence, KDM4A #1: ACTCCGCACAGTTAAAACCA | This paper | N/A |
| sgRNA targeting sequence, KDM4A #2: TTTCGGAACTCTTCCATAGT | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: lentiCRISPR v2- sgRNA-hKDM4A | This paper | N/A |
| Plasmid: psPAX2 | AddGene | Cat#12260 |
| Plasmid: pCMV-VSV-G | AddGene | Cat#8454 |
| Plasmid: GFP-HP1γ | AddGene | Cat#17650 |
| Plasmid: pcDNA-flag-hKDM4A-polyA | AddGene | Cat#101051 |
| Plasmid: pcDNA-flag-hKDM4A(H188A)-polyA | AddGene | Cat#101052 |
| Plasmid: MSCV-IP N-HAonly FOSL1 | AddGene | Cat#34897 |
| Plasmid: pCLXSN-c-JUN | AddGene | Cat#102758 |
| Software and Algorithms | ||
| CellSens | Olympus | http://www.olympus-lifescience.com/en/software/cellsens/ |
| LAS X | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| CASP | Laboratory of A. Wojcik | http://casplab.com/ |
| GraphPad Prism 8 | GraphPad Prism | https://www.graphpad.com/ |
| CRISPR Design | Broad Institute | http://crispr.mit.edu |
| BLOCK-iT™ RNAi Designer | Thermo Fisher Scientific | https://rnaidesigner.thermofisher.com/rnaiexpress/ |
| TIMER | Laboratory of S. Liu | https://cistrome.shinyapps.io/timer/ |
Data and Code Availability
The authors declare that all relevant data are available within the article and its supplementary information files or from the corresponding author upon reasonable request. The accession number for the RNA-seq datasets reported in this paper is GEO: GSE137953. Exome sequencing data was deposited in the sequence read archive (SRA) BioProject ID: PRJNA701731.
EXPERIMNTAL MODEL AND SUBJECT DETAILS
Mice
Kdm4af/f mice were described as before (Zhang et al. 2011), and K14Cre-ERT2 mice (Cat#005107) were purchased from The Jackson Laboratory. Their eight-week-old male and female littermates that were 129SvEv/CD-1 mixed backgrounds were randomly assigned to the groups. Both Bmi1Cre-ERT2 mice (Cat#010531, Jackson Laboratory) and RosatdTomato mice (Cat#007914, Jackson Laboratory) were described as before, and were backcrossed to C57BL/6J backgrounds (Chen et al., 2017), and their eight-week-old male and female littermates were used. All mice were housed under specific-pathogen-free (SPF) condition in the UCLA animal facility. For the induction of autochthonous mouse HNSCC, mice were consecutively treated by 40 μg/ml 4-NQO (Cat#sc-256815, Santa Cruz)-containing drinking water for 16 weeks and then by normal drinking water for another 10-14 weeks. All experiments were carried out in accordance with the protocol approved by UCLA Animal Research Committee.
Cell lines
Human HNSCC cell lines SCC1 and SCC23 were obtained from University of Michigan. These two cell lines were routinely used in our laboratory because they carried the majority of frequent and pathogenic single nucleotide variants in HNSCC as determined by whole exome sequencing (Table S1). The whole exome sequencing of SCC1 cells was performed at UCLA, and the whole exome sequencing data of SCC23 cells was downloaded from Sequence Read Archive (Mann et al., 2019). 293T cells (Cat#CRL-3216) and Phoenix-AMPHO cells (Cat#CRL-3213) were purchased from ATCC and were cultured in normal DMEM supplemented with 10% FBS at 37 °C in 5% CO2 atmosphere.
Human HNSCC samples
The use of human HNSCC pathological samples was approved by the UCLA Institutional Review Board. The paraffin-embedded blocks of human HNSCC tumor samples were retrieved from the tissue bank of the Department of Pathology and sectioned by the UCLA Translational Pathology Core Laboratory. A total of 100 sample comprised of HNSCC without lymph node metastasis (n=32), HNSCC with lymph node metastasis (n=34), and lymph node metastasis (n=34) were used in this study without any exclusion of subjects. All of the HNSCC samples were HPV negative.
METHOD DETAILS
4-NQO mouse model of HNSCC, mouse treatment, tumor assessment and lineage tracing of BMI1+ cancer stem cells
For in vivo genetic deletion of tumor cell KDM4A, mice were intraperitoneally administered three consecutive injections of 25 mg/mL tamoxifen (Cat#5648, Sigma). For in vivo pharmacological inhibition of KDM4A activity, mice were intraperitoneally injected with KDM4i (8 mg/kg body weight; QC6352, Cat#HY-104048, MedChem Express) every other day, meanwhile, 100 μl of KDM4i solution (1 mg/mL) was applied over the whole mouse tongue once a day. To block the intratumoral CD8+ T cell infiltration, anti-CXCR3 antibodies (300 μg; Cat#BE0249, Bio X Cell) were intraperitoneally administered to each mouse three times per week. For PD1 blockade immunotherapy, mice were intraperitoneally injected with anti-PD1 neutralizing antibodies (300 μg; Cat#BE0273, Bio X Cell) once every 3-day.
After mice were sacrificed, both tongue and cervical lymph nodes were immediately dissected, then fixed in 10% buffered formalin solution for 48 h and embedded in paraffin. Longitudinal tongue section was carried out throughout the tumor area. H&E staining and PCK immunohistochemistry staining (IHC) were performed on both sections of tongue and lymph node. 3 representative sections of one mouse were used for each staining. Based on the results of H&E staining and PCK immunostaining of these sections, the numbers of dysplasia, SCC and metastatic lymph nodes were counted, SCC area and lymphatic metastasis size were calculated using cellSens software. If multiple SCCs were present in one mouse, their total SCC area were shown and used for statistical analysis. Tumor invasion degree was evaluated on dysplasia and SCC area according to the published criterions (Chen et al., 2017). In brief, epithelial cell dysplasia but clearness of basement membrane (grade 0), tumor cells spread out of basement membrane but just reach at the superficial sites of tongue muscle (grade 1), tumor cells invade into the deep area of tongue muscle (grade 2). If there are multiple SCCs in one mouse, the tumor with most severe invasiveness was recorded.
For lineage tracing of BMI1+ cancer stem cells in primary mouse SCCs, 25 mg/mL tamoxifen (Cat#5648, Sigma) was administered to induce tdTomato expression in Bmi1Cre-ERT2;Kdm4af/f;tdTomato and BMI1Cre-ERT2;Kdm4aw/w; tdTomato mice. After mice were sacrificed, their tongues were dissected and fixed in 4% paraformaldehyde in PBS for 48 h at 4 °C. Sample were then soaked in 30% sucrose in PBS for another 48 h at 4 °C. OCT embedding and cryosection were carried out by the UCLA Translational Pathology Core Laboratory. 8 μm frozen sections were generated and stored at −20 °C. For visualizing tdTomato and cell nuclei, frozen sections were first washed in PBS at room temperature, and then mounted with DAPI-containing anti-fade mounting medium (Cat#P36962, Thermo Fisher Scientific). Images were taken under the conventional fluorescence microscope. At least 3 images were taken for each SCC. The numbers of BMI1+ CSCs-derived clones were manually counted.
Immunohistochemistry and immunofluorescence staining
For immunostaining of tissue sections, 5 μm paraffin-embedded sections were used in this study and sequentially deparaffinized by xylene, rehydrated through a series of graded ethanol until PBS. Tissue antigens were retrieved by heating in the pressure cooker in pH 6.1 citrate buffer (Cat#S1699, Agilent Dako). Both endogenous peroxidase and alkaline phosphorylase were inhibited by incubation with blocking agents (Cat#S200380-2, Agilent Dako) for 10 min. tissue sections were incubated with the primary antibodies overnight at 4°C. Horseradish peroxidase (HRP)- and fluorochrome-conjugated 2nd antibodies were then respectively applied in the immunohistochemistry and immunofluorescence staining. Sections with the HRP-conjugated immunocomplex was visualized by 3,3′-Diaminobenzidine (DAB) chromogen (Cat#K3464, Agilent Dako), then counterstained with hematoxylin and lastly mounted with aqueous permanent mounting medium (Cat#S196430-2, Agilent Dako). Sections with the fluorochrome-conjugated immunocomplex was then mounted with DAPI-containing anti-fade medium (Cat#P36962, Thermo Fisher Scientific). Except for anti-HP1γ (Cat#sc-398562, 1:1000 and Cat#ab10480, 1;2000) and anti-PCNA (Cat#ab92552, 1:1000), all antibodies were diluted as a ratio of 1:100. The antibody dilution buffer was prepared by dissolving 0.1% Triton X-100 and 1% bovine serum albumin (BSA) in PBS. The detailed information for the 1st and 2nd antibodies was provided in the Key Resource Table. For most samples, at least 3 images were taken on each primary SCC area. For a few samples with limited SCC area, 2 images were taken. The percentages of AcCasp3+, CXCL10+, CXCL11+, CD8α+, γH2A.X+, HP1γ puncta-positive, GZMB+ and FOSL1+ tumor cells were manually analyzed.
IHC staining in human HNSCC samples was scored as described before (Pirker et al., 2012). In brief, the staining intensity of KDM4A and CXCL10 was categorized at four levels, no staining (0), weak staining (1+), intermediate staining (2+) and strong staining (3+). The percentage of cells with different staining intensity were visually measured. The IHC scores were calculated with the formula, 1 × (the percentage of cells with weak staining) + 2 × (the percentage of cells with intermediate staining) + 3 × (the percentage of cells with strong staining).
For the immunofluorescence staining on cells, cells were cultured on the glass coverslips (Cat#354087, Corning). After treatment, cells were washed with PBS, and then fixed with 4% paraformaldehyde (PFA) for 10min, and permeabilized by 0.1% triton X-100. Non-specific binding was blocked through incubation with 1% donkey serum for 30 min. Cells were stained with 1st antibodies overnight at 4°C, and then incubated with fluorochrome-conjugated 2nd antibodies for 1 h at room temperature. Except for anti-HP1γ (Cat#sc-398562, 1:1000 and Cat#ab10480, 1;2000) and anti-PCNA (Cat#ab92552, 1:2000), all antibodies were diluted as a ratio of 1:100. Nucleus was visualized by mounting with DAPI-containing anti-fade medium. Pictures were taken by conventional and confocal fluorescence microscopes. At least 3 images were taken on each sample. The numbers of γH2A.X foci and HP1γ puncta were manually counted, the localization analysis was carried out using Image J software.
Tumorsphere formation assay
ALDHhigh SCC23 cells were fluorescently labeled using ALDEFLUOR™ kit (Cat#01700, STEMCELL Technologies) and then isolated by fluorescence-activated cell sorting. The sorted cells were cultured in low attachment 6-well plate overnight for recovery before the transfection of scramble and KDM4A targeting siRNAs. Tumorsphere formation medium was prepared in serum-free DMEM/F12 medium (Cat#11330-032, Thermo Fisher Scientific) supplemented with 1% N2 supplement (Cat#17502048, Thermo Fisher Scientific), 1% B27 supplement (Cat#17504044, Thermo Fisher Scientific), 10 ng/mL human recombinant basic fibroblast growth factor (Cat#233-FB-025/CF, R&D Systems), 20 ng/mL human recombinant epidermal growth factor (Cat#236-EG-01M, R&D Systems) and 4 μg/mL insulin (Cat#I0516, Sigma). Fresh medium was regularly added until the spheres were formed. At least 3 images were taken for each group. We only counted the spheres with no less than 50 μm diameters.
DNA fiber assays
DNA fiber assays were performed according to the published protocol (Quinet et al., 2017). In brief, cells were sequentially labeled by two consecutive thymidine analogs, 25 μM CldU (Cat#C6891, Sigma) and 250 μM IdU (Cat#I7125, Sigma). Labeled cells were mixed with unlabeled cells in PBS as a ratio of 1:2. The mixed cells were then lysed on slides in the buffer comprising of 50 mM EDTA, 0.5% SDS and 200 mM Tris-HCl (pH 7.4). DNA fiber tracks were obtained on the slides through DNA spreading and fixed in the mixed solution of methanol and acetic acid (3:1) for 10 min. Then, DNA fibers were denatured by 2.5 M HCl and blocked for 2 h in 5% BSA-containing PBS buffer at room temperature, and sequentially stained with primary antibodies against CldU (1:100, Cat#ab6326, Abcam) and IdU (1:100, Cat#347580, BD Biosciences) for 2 h as well as with secondary antibodies of Cy2-conjugated anti-rat (1:100, 712-225-150, Jackson ImmunoResearch Laboratories) and Cy3-conjugated anti-mouse (1:100, 715-167-003, Jackson ImmunoResearch Laboratories) for 2 h at room temperature. Lastly, slides were mounted with anti-fade solution (Cat#P36961, Thermo Fisher Scientific) and images were taken using Olympus fluorescence microscope. The length of DNA fibers was measured using Olympus cellSens software.
Cytosolic DNA imaging and measurement
Live cells were incubated with 3 μL/mL fluorescent dye OliGreen (Cat#O11492, Thermo Fisher Scientific) and PicoGreen (Cat#P11496, Thermo Fisher Scientific) for 1 h in complete culture medium at 37 °C and in 5% CO2 atmosphere to visualize cytosolic ssDNA and dsDNA, respectively. 100 nM Mito-tracker dye (Cat#M7512, Thermo Fisher Scientific) was used to stain mitochondria. After the live cell staining, cells were fixed in 4% PFA for 10 min and mounted with DAPI-containing anti-fade medium (Cat#P36962, Thermo Fisher Scientific). At least 3 Images were taken for each sample by confocal fluorescence microscope. The number of ssDNA foci was manually counted, and the fluorescence intensity of dsDNA was quantified using LAS X software.
Comet SCGE assays
Commercial comet assay kits (Cat#ADI-900–166, Enzo Life Sciences) were utilized in this study, and the assay was performed per the manufacturer’s instruction. Briefly, cells were harvested through scraping dishes and suspended in PBS, then combined with molten LMAgarose at 37 °C. The molten LMAgarose was gelled on slides after chilling for 30 min at 4 °C. The slides were sequentially immersed in lysis buffer at 4 °C for 1 h and in alkaline solution (pH > 13) at room temperature for another 1 h. After cell lysis and DNA denature, electrophoresis was then carried out in the alkaline buffer (pH > 13) for 30 min at 20 V voltage. Lastly, DNA was stained with CYGREEN dye for 30 min as a dilution ratio of 1:100,000. Slides were completely air-dried before taking images. At least 5 images were taken by the fluorescence microscope and analyzed using CASP software.
Gene deletion by CRISPR-Cas9 and gene knockdown by siRNAs
To delete KDM4A gene, 2 KDM4A-specific guide RNA (gRNA) oligonucleotides were designed using online resource (Broad institute GPP). Plasmids were constructed as described before (Sanjana et al., 2014). Briefly, the lentiviral transfer plasmid (lentiCRISPR V2-Puromycin+; Cat#52961, AddGene) were digested by BsmBI enzyme (Cat#R0580S, New England BioLabs) for 30 min at 37 °C. Pairs of gRNA oligonucleotides were phosphorylated and annealed in T4 ligation buffer. The BsmBI-digested lentiCRISPR V2 plasmids were then ligated with gRNA oligonucleotides at room temperature for 10 min. After plasmid construction, the lentiCRISPR V2 plasmids (10 μg) were transfected together with packaging (5 μg psPAX2, Cat#12260, AddGene) and envelop plasmids (5 μg pVSVg, Cat#8454, AddGene) into 293T cells for lentivirus generation. Lentivirus were harvested twice at 48h and 72h after transfection, and immediately used for infection on SCC23 cells. SCC23 cells were then selected with 2 μg/mL puromycin. Infected survival cells were seeded at a super-low density to allow single cell colony formation. KDM4A deletion in these cells was validated by Western blot analysis. To knock down KDM4A, KDM4B, KDM4C, KDM4D and HP1γ genes in SCC cell lines, 2 specific siRNAs (Sigma) for each target gene were designed and synthesized. 60 pmol siRNAs were transfected into cells on each well of 6-well plate using 6 μL Lipofectamine RNAiMAX Reagent (Cat#13778150, Thermo Fisher Scientific).
Stable and transient expression of exogenous genes
To stably express FOSL1 and JUN in human HNSCC cells, amphotropic retroviral expression system was utilized. Human FOSL1 gene was cloned in the plasmid of pRetroX-IRES-ZsGreen1(Cat#632520, Takara Bio). The human JUN-expressing vector was purchased from AddGene (Cat#102758). Vectors were amplified in TOP10 chemically competent cells (Cat#C404003, Thermo Fisher Scientific) and purified using Wizard Plus SV minipreps DNA purification kits (Cat#A1460, Promega). 5 μg of FOSL1 and JUN-expressing plasmids were transfected with Lipofectamine 2000 (Cat#11668–019, Invitrogen) into phoenix-AMPHO cells (Cat#CRL3213, ATCC), and retroviruses were harvested twice at 48h and 72h after transfection. After 12 hours of retrovirus infection, human HNSCC cells were washed with PBS and proliferated for 2 days in complete culture media. Lastly, 500 μg/mL G418 (Cat#0472787800, Sigma) were used for selection of human JUN-overexpressing cells. Human FOSLI-overexpressing cells were enriched by flow cytometry and cell sorting using ZsGreen as the reporter.
GFP-tagged human HP1γ (GFP-HP1γ) expression plasmids (Cat#17650, AddGene) were transfected in both control and KDM4A-deficient HNSCC cells. 1.0 μg of GFP-HP1γ plasmids were used for transfection in 2.0× 105 cells. To recover KDM4A expression in CRISPR-Cas9-mediated KDM4A knockout SCC23 cells, Cas9 inhibitor (Cat#S8844, Selleck Chemicals) were first added for inhibition of Cas9 activity, and Flag-tagged human KDM4A (Cat#101051, AddGene) and Flag-tagged enzyme-inactive KDM4A (Cat#101052, Addgene) expression plasmids were then transfected. 6 μL lipofectamine 2000 reagent (Cat#11668–019, Invitrogen) was used for 1 μg plasmid transfection in the Opti-MEM reduced serum media (Cat#31985–062, Gibco). After 12 hours of transfection, cells were washed with PBS and cultured for another 36 hours in the complete culture media.
Fluorescence imaging of live cells and Fluorescence recovery after photobleaching (FRAP) assay
HNSCC cells were cultured on 35 mm glass bottom dishes (Cat#P35GC-1.5–14-C, MatTek), and transfected by 0.4 μg GFP-HP1γ expression vectors in Opti-MEM reduced serum media for 12 hours, and then washed with PBS and continually cultured in complete cell culture media. 36 hours later, fluorescence imaging and FRAP assay were performed on live cells at 37 °C in 5% CO2 atmosphere. Fluorescence images were regularly taken at 25 min interval for observing the fusion process of HP1γ puncta. FRAP assay was carried out with the confocal microscope (SP8-STED, Leica) equipped with 100× oil immersion objective. 488 nm laser was chosen for GFP excitation. 80% and 30% laser intensity were respectively used for bleaching GFP fluorescence and taking images on HP1γ puncta. GFP fluorescence on HP1γ puncta was shortly monitored and then bleached by 3 repeats of 488 nm laser pulse. GFP fluorescence recovery from bleaching was recorded for the indicated time.
EdU incorporation and imaging
HNSCC cells were incubated with 10 μM 5-ethynyl-2′-deoxyuridine (EdU) for 5 min, and then washed twice with PBS. EdU imaging were carried out strictly following the manufacturer’s instructions (Cat#C10337, Thermo Fisher Scientific). Briefly, cells were fixed by 4% PFA for 15 min and permeabilized by 0.5% Triton X-100 for 20 min, lastly incubated with fluorochromes-containing reaction buffer for 30 min at room temperature to fluorescently visualize EdU. Afterwards, immunofluorescence staining against HP1γ and H3K9me3 was performed on cells as described above. Fluorescence images were taken using the confocal microscope. Localization analysis was carried out with Image J software.
qRT-PCR, RNAseq and whole exome sequencing
Cells were directly lysed by TRIzol reagents (Cat#15596026, Thermo Fisher Scientific) after PBS washing, and total RNA was extracted according to the manufacturer’s instruction. mRNA concentration was measured via 3rd generation of NanoDrop machine. 1-2 μg of mRNA were used for the reverse transcription reaction with random primer (Cat#48190011, Thermo Fisher Scientific), dNTP mix (Cat#18427013, Thermo Fisher Scientific), and M-MuLV Reverse Transcriptase (Cat#M0253L, New England BioLabs). SYBR green supermix (Cat#S7563, Thermo Fisher Scientific) was used in qRT-PCR with TBP gene as an internal control. Gene specific primers for qRT-PCR were listed in the Key Resources Table. qRT-PCR was performed using the Bio-Rad CFX96 machines. Data were analyzed using 2-ΔΔCt method.
For RNAseq, primary SCC tumors were isolated from 2 control (Cre) mice and 2 mice of tumor cell KDM4A knockout (4AKO) (n=2:2). In each mouse, two tumors were isolated and pooled together for RNA extraction. RNA quality for sequencing was examined using an Agilent 2100 Bioanalyzer. Library was constructed using the KAPA RNA-Seq Library Preparation Kits (Cat#07960140001, KAPA Biosystems) at UCLA sequencing core facilities, and RNAs were single-end sequenced on Illumina HiSeq 3000 machines. Analysis of RNA-seq data was done using the cummeRbund package in R as described before (Chen et al., 2017). Reads per kilobase of transcript, per million mapped reads (RPKM) were used for identification of differentially expressed genes. The fold changes of gene expression were calculated from the RPKM means in 4AKO mice divided by the RPKM means in Cre mice. Gene ontology (GO) term and gene set enrichment analysis (GSEA) were utilized to find enriched functional annotations for differentially expressed genes. GO term analysis were carried out only for gene transcripts both upregulated and downregulated by at least 2-fold in primary SCCs of 4AKO mice. GO analysis was carried out by database for annotation, visualization and integrated Discovery (DAVID) version 6.8. GSEA was performed by using the annotated gene sets in molecular signatures database version 7.1.
For whole exome sequencing (WES), the genomic DNA of SCC1 cells were extracted using DNeasy Blood & Tissue kit (Cat#69504, Qiagen). The sequencing library was prepared using NimbleGen Seqcap kit at the UCLA sequencing core. Whole exome sequencing was performed using pair end sequencing with read length of 2 × 150 bp based on the Illumina HiSeq3000 platform. In addition to SCC1 WES data, we also analyzed the whole exome sequencing data of SCC23 cells from Sequence Read Archive under the accession number SRA: SRS4436944 (BioProject ID: PRJNA525437) (Mann et al., 2019). Briefly, sequencing reads were aligned to hg19 reference genome using BWA software package. Variant calling and annotation were respectively performed using the VarDict and ANNOVAR software tools.
Western blot analysis, Co-immunoprecipitation (Co-IP) and ELISA assays
Tissues were homogenized and lysed in RIPA lysis buffer (150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 50Mm Tris, pH8.0) supplemented with a cocktail of protease inhibitors (Cat#78430, Thermo Fisher Scientific) and phosphatase inhibitors (Cat#4906845001, Sigma-Aldrich). The supernatant of tissue lysate was collected, and the protein concentration was measured using Pierce™ BCA protein assay kit (Cat#23225, Thermo Fisher Scientific). For the protein extraction of SCC cells, cells were lysed in RIPA lysis buffer. Protein extracts were mixed with protein sample loading buffer, boiled for 10 min at 95 °C and separated by SDS-PAGE and then transferred to a PVDF membrane using the semi-dry transfer apparatus. After blocking with 5% milk for 1 h, the membrane was probed overnight at 4 °C with 1st antibodies including anti-KDM4A (1:1000, Cat#ab191433, Abcam), anti-H3K9me3 (1:10000, Cat#ab8898, Abcam), anti-FOSL1 (1:1000, Cat#5281S, Cell Signaling Technology), anti-JUN (1:1000, Cat#ab31419, Abcam), anti-p-IRF3 (1:1000, Cat#4947S, Cell Signaling Technology), anti-IRF3 (1:1000, Cat#4302S, Cell Signaling Technology), anti-p-TBK1 (1:1000, Cat#5483S, Cell Signaling Technology), anti-TBK1 (1:1000, Cat#3504S, Cell Signaling Technology), anti-p-STING (1:1000, Cat#19781S, Cell Signaling Technology) and anti-STING (1:1000, Cat#13647S, Cell Signaling Technology), and followed by incubation with HRP-conjugated 2nd antibodies for 2 h at room temperature. Lastly, the immunocomplexes were detected via the chemiluminescence method.
For Co-IP assay, human HNSCC cells were twice washed with cold PBS and lysed in IP lysis buffer (Cat#87787, Thermo Fisher Scientific) supplemented by a cocktail of protease inhibitors and phosphatase inhibitors. Cell lysate was incubated with anti-KDM4A antibody (1:100, Cat#ab105953, Abcam) and species-matched IgG overnight at 4 °C. Protein G magnetic beads (Cat#10004D, Thermo Fisher Scientific) were then added for pulling out the immunoprecipitated complex. After washing with IP lysis buffer, proteins were eluted out from magnetic beads with 1× SDS loading buffer and denatured by heating at 95 °C for 10 min. Lastly, protein samples were separated by the SDS-PAGE and measured by Western blotting using antibodies against KDM4A (1:1000, Cat#ab191433, Abcam), MCM2 (1:1000, Cat#3619S, Cell Signaling Technology) and MCM7 (1:1000, Cat#3735S, Cell Signaling Technology).
For ELISA assays, SCC cell conditioned media were collected after treatment, and the protein levels of both CXCL10 and CXCL11 protein levels were measured using ELISA kits (Cat#DIP100, Cat#DCX110, R&D Systems) per the manufacturer’s instruction, respectively. Absorbance was measured at 450 nm and 550 nm using the microplate reader.
Tumor invasion assays
After 20 ng/ml of HGF (Cat#294-HG-005, R&D Systems) treatment, SCC cells were trypsinized and seeded in the upper compartment of BioCoat Matrigel invasion chamber with 8 μm pores (Cat#354480, Corning). DMEM supplemented with 10% FBS was added into the lower compartment of the chamber as the attractant. 12 hours later, Cells in the lower side of insert membrane were fixed in 70% ethanol and stained with 1% crystal violet, followed by counting under the bright-field microscope.
Analysis of TCGA databases for HNSCC
The online resource of TIMER version 1 was utilized for analyzing the correlations of KDM4A copy number amplification with the numbers of intra-tumoral CD8+ T cells. Two-sided Wilcoxon rank-sum test was used for evaluating the significance.
QUANTIFICATION AND STATISTICS ANALYSES
All data were shown as mean ± SD or mean ± SEM. The details about particular statistic parameters were specified in the figure legends. All statistical analysis was performed using GraphPad Prism 8 software. All in vitro experiments were performed at three times independently, and in vivo results were the pool of two independent experiments. Two-tailed Student’s t-test was performed between two groups and a difference was considered statistically significant with p < 0.05. For significance analysis of differences from anti-CXCR3 treatment in Cre and 4AKO mice, two-way ANOVA was used. For comparison of differences from KDM4i and anti-PD1 treatment, one-way ANOVA was used, followed by the Tukey’s HSD post-hoc tests to minimize type I errors. For both one-way and two-way ANOVA analyses, normal distribution of data was validated using Shapiro-Wilk test. The Cochran-Armitage test was utilized to compare SCC invasion grades between different groups. Chi-square test was utilized for comparison of numbers of metastatic lymph nodes. The correlation between different proteins in human HNSCC samples was determined using the Pearson correlation coefficient of liner regression. GSEA and the statistical analyses were performed with GSEA software (http://www.broad.mit.edu/GSEA) and a two-tailed t-test, respectively. p value of less than 0.05 was considered significant.
Supplementary Material
Highlights.
KDM4A controls invasive growth and immune evasion during SCC development
KDM4A loss induces liquid-like HP1γ puncta and DNA replication stress in SCC
KDM4A loss activates tumor cell-intrinsic immunity by replication stress
Targeting KDM4A enhances anti-PD-1 therapy and eliminates cancer stem cells.
ACKNOWLEDGEMENTS
We thank Dr. Zhi-Ping Liu from UT Southwestern Medical Center for providing Kdm4af/f mice. This work was supported by NIH/NIDCR grants R01DE15964 and R01DE043110 and NIH/NCI grant R01CA236878. Confocal laser scanning microscopy was performed at the Advanced Light Microscopy/Spectroscopy Laboratory and the Leica Microsystems Center of Excellence at the California NanoSystems Institute at UCLA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Black JC, Allen A, Van Rechem C, Forbes E, Longworth M, Tschop K, Rinehart C, Quiton J, Walsh R, Smallwood A, et al. (2010). Conserved antagonism between JMJD2A/KDM4A and HP1gamma during cell cycle progression. Mol. Cell 40, 736–748. [DOI] [PubMed] [Google Scholar]
- Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, Pineda CM, Murphy N, Daniels DL, Montagna C, et al. (2013). KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 154, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, Lamb A, Henon C, Dorvault N, Rouanne M, et al. (2019). PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Invest. 129, 1211–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Wu M, Li Y, Chang I, Yuan Q, Ekimyan-Salvo M, Deng P, Yu B, Yu Y, Dong J, et al. (2017). Targeting BMI1(+) Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell 20, 621–634 e626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinn SB, and Myers JN (2015). Oral Cavity Carcinoma: Current Management, Controversies, and Future Directions. J. Clin. Oncol. 33, 3269–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe KN, and Moldovan GL (2017). Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 65, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, Lanitis E, Duraiswamy J, Tanyi JL, Benencia F, et al. (2019). Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 35, 885–900 e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Pan H, Li J, Zhong Q, Chen X, Dry SM, and Wang CY (2013). Epigenetic activation of AP1 promotes squamous cell carcinoma metastasis. Sci. Signal. 6, ra28 21-13, S20–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economopoulou P, Perisanidis C, Giotakis EI, and Psyrri A (2016). The emerging role of immunotherapy in head and neck squamous cell carcinoma (HNSCC): anti-tumor immunity and clinical applications. Ann. Transl. Med 4, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Casanas M, and Chan KL (2018). The Unresolved Problem of DNA Bridging. Genes (Basel) 9, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. (2016). Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 375, 1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington KJ, Ferris RL, Blumenschein G Jr., Colevas AD, Fayette J, Licitra L, Kasper S, Even C, Vokes EE, Worden F, et al. (2017). Nivolumab versus standard, single-agent therapy of investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): health-related quality-of-life results from a randomised, phase 3 trial. Lancet Oncol. 18, 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AS, Kim S, Tighiouart M, Gudino C, Mita A, Scher KS, Laury A, Prasad R, Shiao SL, Van Eyk JE, and Zumsteg ZS (2017). Metastatic Lymph Node Burden and Survival in Oral Cavity Cancer. J. Clin. Oncol. 35, 3601–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Zhang W, and Wang CY (2020). BMI1 Inhibition Eliminates Residual Cancer Stem Cells after PD1 Blockade and Activates Antitumor Immunity to Prevent Metastasis and Relapse. Cell Stem Cell 27, 238–253 e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Boija A, Afeyan LK, Hawken SW, Fan M, Dall’Agnese A, Oksuz O, Henninger JE, Shrinivas K, Sabari BR, et al. (2020). Partitioning of cancer therapeutics in nuclear condensates. Science 368, 1386–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida S, Takizawa Y, Ishimaru M, Sugita Y, Sekine S, Nakayama JI, Wolf M, and Kurumizaka H (2018). Structural Basis of Heterochromatin Formation by Human HP1. Mol. Cell 69, 385–397 e388. [DOI] [PubMed] [Google Scholar]
- Mann JE, Kulkarni A, Birkeland AC, Kafelghazal J, Eisenberg J, Jewell BM, Ludwig ML, Spector ME, Jiang H, Carey TE, and Brenner JC (2019). The molecular landscape of the University of Michigan laryngeal squamous cell carcinoma cell line panel. Head Neck 41, 3114–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Stepputtis SS, Strietz J, Preca BT, Urban S, Willmann D, Allen A, Zenk F, Iovino N, Bronsert P, et al. (2017). KDM4 Inhibition Targets Breast Cancer Stem-like Cells. Cancer Res. 77, 5900–5912. [DOI] [PubMed] [Google Scholar]
- Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. (2015). Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 6, 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai SI, and Westra WH (2009). Molecular pathology of head and neck cancer: implications for diagnosis, prognosis, and treatment. Annu. Rev. Pathol 4, 49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. (2019). PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 9, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017). DNA Fiber Analysis: Mind the Gap! Methods Enzymol 591, 55–82. [DOI] [PubMed] [Google Scholar]
- Sacco AG, and Cohen EE (2015). Current Treatment Options for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 33, 3305–3313. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmamed MF, and Chen L (2018). A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 175, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. (2019). Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 9, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh MH, Bortnik V, McMillan NA, and Idris A (2019). cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb. Pathog. 132, 162–165. [DOI] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu PH, et al. (2018). LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–563 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickfaden H, Tolsma TO, Sharma A, Underhill DA, Hansen JC, and Hendzel MJ (2020). Condensed Chromatin Behaves like a Solid on the Mesoscale In Vitro and in Living Cells. Cell 183, 1772–1784 e1713. [DOI] [PubMed] [Google Scholar]
- Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, Wang J, Wang X, and Fu YX (2016). Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell 29, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubhi T, and Brown GW (2019). Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res. 79, 1730–1739. [DOI] [PubMed] [Google Scholar]
- Valencia AM, and Kadoch C (2019). Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat. Cell Biol. 21, 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale-Cross L, Molinolo AA, Martin D, Younis RH, Maruyama T, Patel V, Chen W, Schneider A, and Gutkind JS (2012). Metformin prevents the development of oral squamous cell carcinomas from carcinogen-induced premalignant lesions. Cancer Prev. Res. (Phila) 5, 562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gao Y, Zheng X, Liu C, Dong S, Li R, Zhang G, Wei Y, Qu H, Li Y, et al. (2019). Histone Modifications Regulate Chromatin Compartmentalization by Contributing to a Phase Separation Mechanism. Mol. Cell 76, 646–659 e646. [DOI] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QJ, Chen HZ, Wang L, Liu DP, Hill JA, and Liu ZP (2011). The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J. Clin. Invest. 121, 2447–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, and Chen L (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med 8, 328rv324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all relevant data are available within the article and its supplementary information files or from the corresponding author upon reasonable request. The accession number for the RNA-seq datasets reported in this paper is GEO: GSE137953. Exome sequencing data was deposited in the sequence read archive (SRA) BioProject ID: PRJNA701731.