

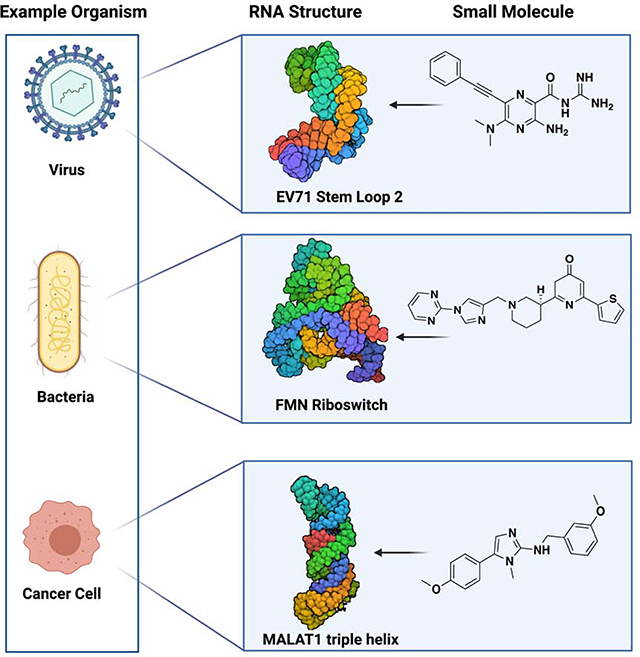
Summary
Initial successes in developing small molecule ligands for non-coding RNAs have underscored their potential as therapeutic targets. More recently, these successes have been aided by advances in biophysical and structural techniques for identification and characterization of more complex RNA structures; these higher-level folds present protein-like binding pockets that offer opportunities to design small molecules that could achieve a degree of selectivity often hard to obtain at the primary and secondary structure level. More specifically, identification of RNA tertiary and quaternary structures and targeting these structures with small molecule ligands have allowed researchers to probe several human diseases and have resulted in promising clinical candidates. In this review we highlight a selection of diverse and exciting successes and the experimental approaches that led to their discovery. These studies include examples of recent developments in RNA-centric assays and ligands that provide insight into the features responsible for the affinity and biological outcome of RNA-targeted chemical probes. This report highlights the potential and emerging opportunities to selectively target RNA tertiary and quaternary structures as a route to better understand and, ultimately, treat many diseases.
eTOC:
Zafferani et al. review diverse approaches that resulted in the successful small molecule targeting of RNA tertiary and quaternary structures. This review highlights the opportunities and challenges in developing chemical probes that target the exponentially growing number of functionally characterized RNA tertiary and quaternary structures across all domains of life.
Graphical Abstract
Introduction
“There is no question, then, that RNA presents a filing system perfectly capable of handling any load of learning and memory which the human being is likely to put upon it—and a billion times more than that quantity, too.” Isaac Asimov, 1966. While insightful, such a statement covered only a minute part of the soon-to-be discovered roles and complexities of RNA. Indeed, at the time RNA was still perceived as a passive messenger of DNA genetic information, with proteins as the “ultimate product”. Recent reports have changed the perception of RNA from a simple carrier of information to a much more active player, whose intricacies we are only beginning to discover (Cech and Steitz, 2014). The finding that while most of our genome is transcribed, less than ~3% is translated opened a new door of exploration, including the discovery of the pervasiveness long non-coding RNA (lncRNA), which are key players in many cellular pathways and epigenetic regulation (Hu et al., 2018). In recent years, RNA has been increasingly recognized as an attractive target to modulate disease-related pathways, including those dependent on undruggable proteins.
The fast-growing interest in RNA targeting has met with several challenges as compared to protein targeting, with the lack of diversity in building blocks and increased structural flexibility of RNA at the forefront. Initial efforts in small molecule development focused on designing motif-specific probes for RNA secondary structures such as bulges, stems, apical loops, and internal loops. However, the repetition of many secondary motifs within a given cell led to concerns around selectivity and off-target effects. In an effort to increase specificity, sequence-based inhibitors called antisense oligonucleotides (ASO) were developed to target RNA sequences. ASOs have played a key role in elucidating RNA function and have yielded some promising clinical candidates (Kole et al., 2012; Shen and Corey, 2017). Nevertheless, their generally problematic clinical delivery has led to renewed interest in small molecules as complementary probes to ASOs that target tertiary structures and/or the dynamic landscape of RNA, moving beyond sequence or simple secondary motifs. The development of new biophysical and structural techniques aimed at obtaining a more complete picture of the folding of long transcripts has revealed short- and long-range interactions that create unique, higher order folding structures as well as the presence of biologically relevant conformational landscapes (Mustoe et al., 2019). These insights have provided scientists with a new opportunity for selective RNA targeting with small molecules, which can be tuned toward bioavailability and cell permeability.
In this review, we will discuss successes in targeting RNA tertiary and quaternary structures. In particular, we will highlight diverse and inspiring approaches proven to be effective for complex RNA structures. Specifically, we consider tertiary structures elements such as RNA triple helices, g-quadruplexes, pseudoknots, co-axial stacking, and kissing loops (Butcher and Pyle, 2011). Quaternary structures are defined as RNA-RNA, DNA-RNA, or RNA-protein assemblies, and we will discuss ligands aimed at stabilizing or destabilizing the complexation of the two binding partners in biologically and disease relevant settings (Jones and Ferré-D’Amaré, 2015). Some of the first examples of RNA targeting in an RNA:protein complex came from antibacterial small molecules targeting ribosomal RNA (rRNA). While these examples helped highlight the potential of RNA as a drug target, rRNA targeting has been recently reviewed and, therefore, will not be discussed among quaternary structures (Hong et al., 2014). For the purpose of this review small molecules are defined as compounds with a molecular weight less than ~800 amu. Compounds developed to target repeating secondary structures will not be discussed herein, as they have been extensively reviewed recently (Costales et al., 2020).
Tertiary Structures
RNA tertiary structure is defined as the arrangement and interaction of secondary structure building blocks in three-dimensional space (Butcher and Pyle, 2011). As the RNA field optimizes and gains new tools to characterize structural features, new and more complex structural arrangements are revealed, increasing the targets available for selective probing. Recent advances in designing and tuning small molecules for tertiary motifs highlighted the potential of targeting higher-level folds to achieve a degree of selectivity that proved to be challenging for secondary structures (Fedorova et al., 2018). Herein, we highlight examples of biologically relevant tertiary structures that have been successfully targeted and the strategies that yielded these promising chemical probes.
Triple Helices
Base triples occur frequently in RNA and usually result from the insertion of a third single RNA strand into a duplex in the minor groove, via A-minor interactions, or in the major groove through Hoogsteen H-bonds (Conrad, 2014). Base triples can be found as part of other tertiary and quaternary structures to enhance their stability, such as within pseudoknots or ribosomal subunits (Butcher and Pyle, 2011). Stand-alone triple helices have recently been mapped within several non-coding RNAs (ncRNA) and are linked to an increase in the half-life of the transcript by protecting the transcript from degradation (Brown, 2020; Conrad, 2014). Despite their clear biological relevance, biophysical and structural methods for their characterization in vitro and in vivo are still limited. Triple helices have mainly been structurally investigated through NMR, FRET experiments, UV-Vis and X-ray diffraction, which have limitations in the throughput and/or reflection of the conformer population. Despite the limited set of tools for characterization of RNA triple helices, recent successes in small molecule targeting of this structure, vide infra, have brought the attention of the field to the feasibility and biological potential of targeting triple helix motifs within longer transcripts.
One example of a biologically relevant triple helix is the one present at the 3’-end of the metastasis-associated lung adenocarcinoma transcript 1 (MALAT1). This lncRNA gained attention as a therapeutic target after it was found to be overexpressed in several cancer types and has now been implicated in other disease-related pathways (Abdulle et al., 2019; Wilusz et al., 2012). Specifically, the 8.7 kb long immature MALAT1 transcript undergoes a multiple step processing, including the cleavage of a 3’-end 61-nucleotide (nt) tRNA like structure. The process yields the blunt formation of a 3’-end triple helix that has been associated with nuclear accumulation of the transcript and as an essential element for protection against degradation (Conrad, 2014). A reported crystal structure obtained from a truncated version of the wild-type (WT) triple helix revealed 9 U•A-U base triples resulting from the major groove insertion of a genomically encoded A-rich 3’-tail, interrupted by an essential +C•G-C triple and a C-G doublet (Brown et al., 2014). Recently, two notable examples of small molecule targeting have been reported for the MALAT1 triple helix utilizing a focused library and high throughput approach, respectively.
Donlic et al. approached targeting the triple helix from a focused library standpoint (Donlic et al., 2018). In the study, a 33-member library was synthesized by decorating a known RNA binding scaffold with different functionalities and regioisomer architectures. The scaffold furamidine was previously reported as a DNA and RNA duplex binder, including a reported T-A-T DNA triplex binder (Chaires et al., 2004), making it an ideal starting point for selective tuning for U-A-U triple helices. UV-melts and fluorescence titrations identified a nanomolar binder (1, EC50 = 36 nM) selective for the MALAT1 triple helix construct over other sequences, including a MALAT1 stem loop proxy created by deleting the A-rich tail (Figure 1A). Interestingly, after analyzing the focused library for small molecule 3D shape utilizing principal moments of inertia, a trend between affinity and rod-like shape of the ligand was observed, potentially suggesting a shape bias or complementarity requirement for selective in vitro targeting. This study was the first report of a small molecule targeting a ncRNA triple helix (Donlic et al., 2018). In their follow-up work, Donlic et al. performed a structure-activity relationship (SAR) study of the previously reported lead (1) to gain a better understanding of the moieties responsible for its affinity and selectivity (Donlic et al., 2020). While none of the newly synthesized analogues exhibited better affinity or selectivity parameters, melting experiments revealed an interesting discrepancy between affinity and stabilization, with a mediocre binder resulting in the greatest increase in melting temperature (ΔTm), a measure of thermal stability. A previously reported in vitro degradation assay that utilizes exonuclease RNase R was performed to reconcile this discrepancy. Indeed, the small molecules that stabilized the triple helix to the greatest extent resulted in greatest protection from enzymatic degradation of the construct over time. The authors screened the newly synthesized analogues against a similar triple helix motif found in the lncRNA NEAT1. While the best MALAT1 binder (1) also displayed affinity for the NEAT1 triple helix via fluorescence titration, it retained higher affinity for MALAT1, thereby suggesting that it is possible to tune small molecules to preferentially bind one of two closely related triple helices. Overall, stability and functional in vitro assays provided insight into the fitness of the analogues, while docking studies supported ligand pre-organization as a potential factor in small molecule binding and effects on construct stabilization.
Figure 1.
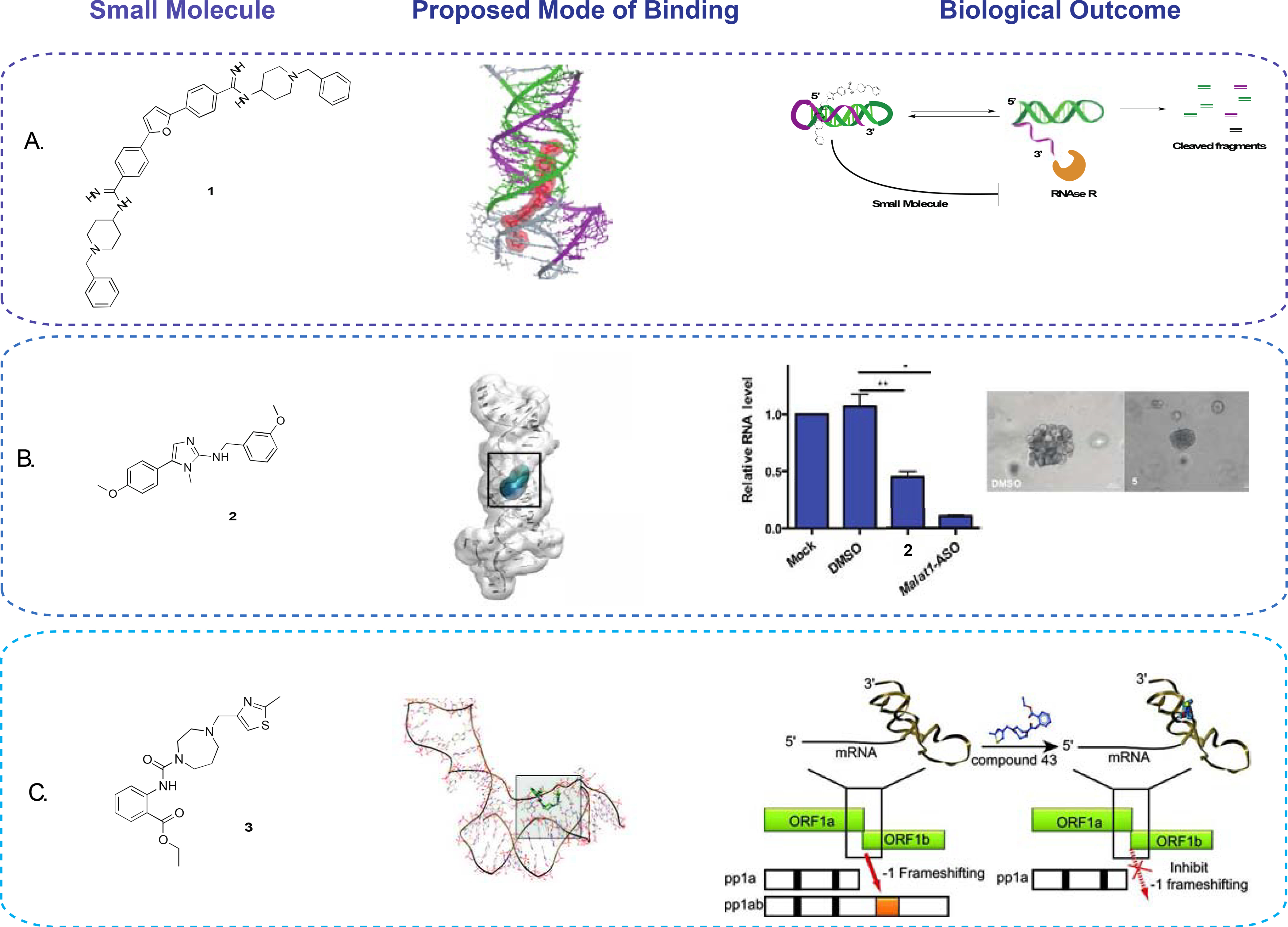
(A) MALAT1 triplex binder with highest affinity and selectivity (left), proposed mode of binding obtained through docking (center), and representative assay set up to test the effects of the small molecule on RNAse R-induced MALAT1 degradation (right) (Donlic et al., 2020). (B) Small molecule lead found through HTS (left), proposed mode of binding obtained through docking (middle), and effect of the small molecule on MALAT1 RNA levels and branching in organoid ex vivo models (right) (Abulwerdi et al., 2019). (C) Small molecule reported binder (left) of SARS-CoV pseudoknot element discovered through a high-throughput docking effort (middle), which resulted in a decrease in frameshifting upon binding (right) (Park et al., 2011).
Concurrently, Abulwerdi et al. reported a different MALAT1 ligand. A high throughput screen employing a small molecule microarray was used to evaluate a 26,000 compound library against a fluorescently labeled MALAT1 triple helix construct and yielded a 0.7% hit rate (Abulwerdi et al., 2019). Hit small molecules were selected based on their selectivity against DNA and RNA targets, including other biologically occurring RNA triple helices. Two small molecules hits were further validated through a two-body FRET labeled construct of the MALAT1 triple helix and isothermal titration calorimetry (ITC). Markedly, FRET experiments showed significant conformational changes of the MALAT1 triple helix upon small molecule treatment. Organoid branching studies performed from a mammary cancer murine model, with ASO as a positive control, showed a significant decrease in branching as well as a decrease in MALAT1 transcript abundance as shown by RT-qPCR upon treatment with 2 (Figure 1B). Conversely, RT-qPCR revealed little to no change in the levels of NEAT1 RNA, supporting the selectivity of the small molecule in ex vivo models (Abulwerdi et al., 2019). RNAse R in vitro degradation assays reported in the follow-up study of Donlic et al. revealed a significant increase in RNA degradation upon treatment with 2 (Donlic et al., 2020).
These successes not only open the door for the development of selective MALAT1 chemical probes but highlight the targetability of triple helices. Importantly, MALAT1 is only one of a few triple helices identified to date (Conrad, 2014). We foresee that the impact of the discovery and optimization of small molecules to target triple helices in ncRNA will bring about a surge of interest in this class of tertiary structures and, with it, a push for development of more computational and experimental methods to map and target triple helices across the transcriptome.
Pseudoknot
Pseudoknots are RNA tertiary structures that arise from energetically favorable long-range interactions between loops and hairpins that are stabilized by coaxial stacking of the newly formed RNA helices (Butcher and Pyle, 2011; Pleij et al., 1985). Such structures have been found and mapped across many species and RNAs, from human telomerase RNA to viral genomes, with biological functions that are still being elucidated (Mihalusova et al., 2011). Notably, pseudoknots have been found to be essential for viral replication in a number of viruses, including SARS-CoV, where their main function is in −1 programmed ribosomal frameshifting. Frameshifting is a desirable target in some viruses as essential proteins encoded by the secondary open reading frame (ORF1b) region are normally out of frame, thus requiring a re-arrangement of the ribosome to proceed with elongation and translation (Thulson et al., 2020). In coronaviruses, re-arrangement takes place through the presence of a slippery site, a linker region and the downstream regulatory pseudoknot (Dinman et al., 1998). While normal ribosome positioning starts at the zero position of the slippery site, the encounter between the ribosome and the pseudoknot tertiary structure causes the ribosome to pause and re-arrange at the −1 position, which puts ORF1b in frame and allows for ribosomal elongation upon pseudoknot unwinding. Mutational studies as well as antisense peptide nucleic acid (PNA) targeting of the pseudoknot revealed its potential as an antiviral target to dramatically decrease replication and virion production (Ahn et al., 2011; Plant et al., 2010).
Park et al. performed a high throughput virtual ligand screening on an 80,000-member library of commercially available compounds against a computationally predicted model of the SARS-CoV pseudoknot (Figure 1C) (Park et al., 2011). The top scoring small molecules were validated using a dual reporter frameshifting assay. Small molecules that reduced frameshifting from the reporter assay were further validated by an SDS-PAGE in vitro assay to ensure inhibition of translation. The same in vitro assay was used to ensure specificity against the SARS-CoV pseudoknot over other naturally occurring pseudoknots to yield a single lead probe (3). Finally, dual luciferase transfected HEK cells showed a concentration-dependent decrease in frameshifting upon small molecule treatment with an IC50 of 0.45 μM (Park et al., 2011). As the pseudoknot is traditionally known as a frameshifting stimulatory element, a follow-up study aimed at understanding the mode of action of the ligand through single-molecule force spectroscopy (Ritchie et al., 2014). Interestingly, Ritchie et al. found a concentration-dependent decrease in conformational plasticity of the pseudoknot induced by small molecule binding. Because the ribosome has to be able to unwind the pseudoknot to proceed with elongation, conformational plasticity has been proposed as an important feature in frameshifting efficiency (Ritchie et al., 2014).
Park et al. not only identified the pseudoknot as a potential antiviral target, but also underscored the success of in silico screening of small molecules against higher order RNA structures. The successful targeting of coronavirus pseudoknot elements offers inspiration and opportunities to target other members of the coronavirus family, including SARS-CoV-2, the viral agent behind the world-wide COVID19 pandemic. Given the efforts and focus of the field on biophysical characterization and targeting of the RNA elements of SARS-CoV2 genome, we foresee an exponential increase in successes in targeting CoV viral motifs with a plethora of strategies. Finally, while their conformational plasticity has been a hurdle in the 3D characterization of pseudoknots, we can expect that the rapid advancement of chemical probing and electron microscopy techniques will provide crucial insights into 3D shape of pseudoknots and allow for the creation of even more accurate models.
G-quadruplexes
Initially discovered in DNA decades ago, G-quadruplexes (G4s) are folding structures formed within a G-rich strand or between two G-rich strands. Generally, a tetrad of guanines arranges co-planarly via Hoogsteen H-bond interactions further stabilized by interactions with a cation. Critical work by Siddiqui-Jain et al. confirmed the presence of G4s in cells and their pivotal role in c-MYC transcription, which could be modulated by small molecule binding, thereby making G4s a desirable therapeutic candidate (Siddiqui-Jain et al., 2002). Importantly, computational and experimental methods have helped map G4 structures in the promoter region of many eukaryotic and prokaryotic genes, with the most notable one being telomeric DNA. G4 overall structure depends on strand polarity, orientation of connecting loops, and cation stabilization, thereby making it a diverse topological family (Varshney et al., 2020). Recent studies have supported the existence of G4s in several ncRNA, though these differ from DNA G4s as they can be transient and highly dynamic. Remarkably, RNA G4s have showed higher stability than their DNA counterparts due to the added H-bonds of the 2′-OH, as well as stronger stacking of the G-quartets. These key differences ultimately distinguish RNA G4s unique topological features, allowing for small molecule discrimination between RNA and DNA (Arora and Maiti, 2009). Recent technical advances in detecting and monitoring RNA G4s in vitro and in cellulo include 19F-labeling of RNA, which capitalizes on the lack of natural presence of fluorine in cells to yield a high-quality signal with no interference (Bao and Xu, 2018). Other strategies involve the use of an RNA G4-selective red fluorescent probe for real-time tracking of G4 folding and unfolding in cellulo (Chen et al., 2018). Finally, chemical crosslinking followed by affinity capture and sequencing increases the scale of G4 identification to the entire transcriptomic landscape (Yang et al., 2018). The application of these techniques allowed for the discovery of the increased presence of G4 structures in cancer cells over non-neoplastic tissues and pinpointed the role of many G4s in ncRNA to be tied to regulation of transcription of several oncogenes and tumor suppressors (Biffi et al., 2014). To this end, small molecules have been developed to either stabilize or destabilize the target structure based on the desired biological outcome. In the following sections we will highlight notable examples of both classes of small molecules and the successful strategies that led to their discovery.
G-quadruplex Stabilizers
Stabilizing a G4 structure is an effective approach when targeting common tumorigenic pathways to inhibit the translation of pro-oncogenes. One of the most well-studied and characterized RNA G4s is transcribed from sub-telomeric DNA regions, yielding telomeric repeat sequences RNA (TERRA). G4s within the ncRNA TERRA have been identified as essential players in heterochromatin formation and, as such, they constitute an attractive therapeutic candidate for small molecule modulation. Garavís et al. used a fragment-based drug discovery strategy (FBDD) through an in vitro high-throughput screen (HTS) utilizing a 355-compound library (Garavís et al., 2014). As every molecule contained one fluorine atom, binding could be monitored by 19F NMR and, subsequently, validated by 1H NMR. This study provided diverse small molecule hits that showed selectivity against other DNA and RNA constructs but not DNA G4s thereby highlighting the need for more selective probes (Garavís et al., 2014).
Selectivity of RNA over its parent DNA G4 was achieved by Katsuda et al. who employed a novel reverse transcriptase (RTase) in vitro assay for HTS. The study aimed at targeting the RNA G4 at the 5’ terminal region of the proto-oncogene NRAS mRNA (Katsuda et al., 2016). The authors used the known TERRA RNA G4 as a model which, analogously to the NRAS G4, is a stable RNA tertiary structure that hinders elongation of the complementary cDNA by RTases; therefore, stabilizing small molecules result in lower levels of full-length cDNA amplifiable by qPCR. Out of an initial library of 8,000 compounds, one ligand (4) was identified as selective binder with a Kd of 5.9 μM for the RNA TERRA over a plethora of parallel, antiparallel, and hybrid DNA G4 (Figure 2A). Analogously, compound 4 was able to decrease the expression of NRAS mRNA in a dose dependent manner. Notably, Tm studies on the NRAS mRNA, aimed at confirming its binding target, revealed that the small molecule did not act through binding to the known G-tetrad. Instead, the ligand bound to a previously uncharacterized second RNA G4 in the 5’-UTR of NRAS. This study revealed the potential of small molecules to discover novel G4s not predicted by computational methods and to uncover the biological relevance of G4s when more than one is present on the same transcript (Katsuda et al., 2016).
Figure 2.

(A) NRAS mRNA G4-binder (left) reported to decrease the expression of NRAS (right) (Katsuda et al., 2016). (B) TERRA mRNA G4 stabilizer (left), which decreases increases the affinity of the structure for TRF2, thereby hijacking it from binding telomeric DNA (right) (Zhang et al., 2017). (C ) Small molecule (left) reported to destabilize ADAM10 and increase its expression in AD models (right) (Dai et al., 2015). (D) VEGF G4 binder (left) that results in the destabilization of the G4 structure thereby reducing expression of the angiogenic protein (right) (Wang et al., 2017b).
Recent efforts have focused on mapping the pathway of the TERRA RNA G4 and its role in heterochromatin formation. Specifically, the folding of the TERRA sequence in a G4 has been proven necessary for its binding to telomeric repeat factor 2 (TRF2). In the absence of the G4, TRF2 binds telomeric DNA duplex and inhibits the activation of the DNA damage response (DDR), thereby halting apoptosis. The TERRA G4 has been implicated in allosterically diminishing TRF2 binding affinity for telomeric DNA. Accordingly, a small molecule stabilizer would increase the TERRA-TRF2 complexation and decrease protection of telomeric DNA from DDR pathways (Biffi et al., 2012). Zhang et al. performed an HTS of natural products and their derivatives utilizing a stability FRET assay, which allowed the authors to rank ligands based on their degree of melting stabilization (Tm) (Zhang et al., 2017). A surface plasmon resonance (SPR) assay was performed by tethering targets of interest on a chip; specifically, the authors used it to assess the selectivity of hits against TERRA over other DNA and RNA constructs. Notably, Zhang et al. observed 10 times stronger KD of their ligand (5) to the TERRA G4 when screened using microscale thermophoresis (MST, Kd = 200 nM) compared to SPR (Kd = 2.56 μM). The authors highlight that, especially with tertiary structures, immobilization may affect both binding and RNA folding, making MST more sensitive and reliable as it does not require immobilization. MST confirmed 5 as a selective TERRA G4 binder over its parent DNA G4. While circular dichroism revealed very little selectivity of the small molecule for the TERRA G4 over other known RNA G4s, chromatin immunoprecipitation (ChIP) experiments in U2OS cells showed a dose-dependent disassociation of TRF2-bound telomeric DNA (Figure 2B). Finally, western blots and MTT assays displayed an increase in triggering of apoptosis and a dose-dependent decrease inhibition of cell proliferation, respectively. In addition, flow cytometry revealed an increase in the percentage of cells in growth arrest phases (Zhang et al., 2017). Overall Zhang et al. provided an excellent example of a bioactive RNA G4-binder with improved selectivity over DNA G4.
G-quadruplex Destabilizers
The increase in discovery and classification of G4s among the human transcriptome also called for probes that could destabilize G4 structures to increase the translation of disease-relevant proteins in an attempt to rescue the non-diseased phenotype. Specifically, Alzheimer’s disease (AD) is a neurodegenerative disorder proposed to be a direct cause of the overproduction of the neurotoxic amyloid peptide (Aβ). ADAM10, a member of disintegrin and metalloproteinases, has been shown to increase activity of a protease that yields the non-neurotoxic isoform of Aβ-peptide (Lammich et al., 2011). Dai et al. sought to find a destabilizing ligand of the ADAM10 mRNA G4 through a focused library approach to ultimately increase translational efficiency and promote cleavage of the amyloid precursor protein (sAPPα) by α-secretase (Dai et al., 2015). A 52-compound synthetic focused library was screened against the wild-type biotinylated ADAM10 G4 and a non-functioning mutant via SPR to identify selective ligands. A structure-activity relationship study on a lead compound afforded a ligand (6), with a 10-fold higher affinity for the wild-type (Kd = 6.1 μM) over the mutant. Dual-luciferase reporter assays confirmed a significant increase in translation efficiency for ADAM10 mRNA while little to no effect was observed when the same assay was used with other known RNA G4s. While RT-qPCR showed no significant changes in ADAM10 mRNA abundance, western blots performed in HEK-APP cells reported a dose-dependent increase in ADAM10 protein, confirming the mode of action through the mRNA G4 at the translational level (Figure 2C). Finally, to confirm the biological relevance of their findings, Dai et al. treated HEK-APP cells with increasing concentrations of 6 and analyzed the levels of Aβ40 peptide, the neurotoxic isoform, via ELISA. At 1.5 μM of 6, well below its MTT-tested cytotoxicity, the small molecule reduced the Aβ40 peptide concentration by approximately 41%, eliciting the desired biological effect (Dai et al., 2015).
The most commonly assigned mode of action of G4s is through translational repression. A notable exception is a G4 present at the 5’-UTR of the vascular endothelial growth factor (VEGF), which promotes translation (Bhattacharyya et al., 2015). As an aberrantly expressed factor involved in angiogenesis, reduction of VEGF levels is an appealing avenue to pursue for anticancer drug design. Wang et al. aimed at finding a G4 selective small molecule that could put into focus the biological relevance of the structure as well as possibly find compounds with antitumor activity (Wang et al., 2017b). A 144-member library of natural products and derivatives was screened against the biotinylated WT, a non-functional mutant of VEGF G4 and a DNA hairpin using SPR, ultimately identifying hit compounds, including one nanomolar lead (7, Kd= 928 nM). MST was employed to confirm the hit compounds and identified 7 to be a selective lead, which was revealed to destabilize the G4 by a ΔTm decrease of 18 °C (7, Figure 2D). Upon small molecule treatment, the wild type construct showed inhibition of translation of VEGF in dual luciferase assays, but not in constructs without the G4 motif, confirming the mode of action through destabilization of the G4. In contrast, when the same dual-luciferase reporter assay was performed with a known G4 stabilizer, an enhancement in VEGF-A protein expression was reported. These results not only confirm the discovery of a G4 destabilizing small molecule probe, but also corroborate the importance of the G4 structure in the translation process. Notably, MCF-7 cell-based assays and nude mice assays were performed to confirm the in vivo biological activity of the compound as well as compare its effects to an siRNA designed to disrupt the VEGF G4 structure. The small molecule led to a dose-dependent decrease in cell viability and wound closure to a similar extent as siRNA, which had a previously reported impressive potency, yet problematic delivery (Bugaut and Balasubramanian, 2012).
Overall, these studies highlighted the applicability and success both of scaffold-based focused library design and curation and of high-throughput screening of larger libraries. Both approaches yielded chemically distinct selective small molecules with biological relevance and selectivity comparable to that of antisense oligonucleotides. Furthermore, notable differences between in vitro affinity-based techniques like SPR and MST suggests that immobilization-based techniques may significantly affect apparent Kd and should be validated with a secondary method. The previously discussed advances in biophysical tools to characterize G4s in vitro and in vivo hold the promise to increase the potential to evaluate small molecule engagement of G4s in cells and, therefore, immensely increase the insight needed to continue the development of selective bioactive G4 binders.
Riboswitches
Riboswitches are regulatory ncRNAs found in the untranslated regions of mRNAs usually formed by multi-helical junctions and pseudoknots and are found in prokaryotes, fungi, and plants. This class of ncRNA is distinguished by its ability to regulate transcription and translation merely through reversible changes in conformation, without the intervention and mediation of proteins (Breaker, 2012). Riboswitches often act through a feedback loop by binding to a cognate ligand in the aptamer domain. This binding event leads to a conformational change that regulates the expression platform, resulting in abolition of transcription or translation (Figure 3A, B). Remarkably, riboswitches that recognize the same cognate ligand or control the same gene can adopt vastly different architectures based on the respective species, making it a diverse topological class (Serganov and Nudler, 2013). To date, the ability of riboswitches to reversibly adopt vastly different conformations has been used to develop and optimize structural and probing techniques. X-ray crystallography has been the most commonly used technique to obtain high resolution structures of the aptamer-ligand complexes. Recent advances in probing protocols have provided a better understanding of the kinetics that regulate ligand binding as well as the link between co-transcriptional folding and gene expression (Watters et al., 2016). Ultimately, the discovery of riboswitches proved that there are tertiary structures of RNA, like proteins, that have evolved to possess highly specific binding pockets for a small molecule or co-factor and that conformational trapping could be a promising avenue to control ncRNA biological function (Warner et al., 2018). Accordingly, small molecule targeting has the potential to lead to selective regulation of the genes associated with riboswitches and yield novel antibacterial and antifungal agents (Blount and Breaker, 2006; Machtel et al., 2016; Panchal and Brenk, 2021). Two main strategies have been adopted in reaching this goal: mimicry of natural ligand and high throughput screening (Furukawa et al., 2012; Rizvi et al., 2018; Wang et al., 2017a). In the following sections we will highlight noteworthy examples of each strategy
Figure 3.

(A) Representative effect of a small molecule that stabilizes the inactive state of a riboswitch resulting in a decrease in transcription/translation adapted from Mulhbacher et al. (B) Schematic of a small molecule that favors the active conformation of a riboswitch increasing transcription/translation. (C) Small molecule lead (right) and its binding pocket (light blue) on PreQ1 riboswitch of T. tencongensis PDB: 3Q51 (Connelly et al., 2019). (D) Small molecule fragment (magenta) bound to E.coli thiM TPP riboswitch (PDB: 4NYB) (Warner et al., 2014). While not being the best fragment from Cressina et al. it was chosen for representation due to its detailed follow-up by Warner et.al. (E) Ribocil-C (magenta) binding-site on FMN riboswitch of F. nucleatum (magenta) PDB 5C45 (Vicens et al., 2018). (F) Modeled small molecule (magenta) bound to FMN riboswitch with improved cellular accumulation and activity against gramnegative bacteria PBD: 6BFB (Motika et al., 2020; Rizvi et al., 2018).
High-throughput Screening
The conformational trapping of riboswitches by their cognate ligand makes them particularly suitable for X-ray crystallography, which is especially needed when trying to design mimics of native ligands. At the same time, X-ray diffraction structures do not encompass all the possible folding states of the riboswitch, and they are not always attainable. High throughput screens can allow for fast identification of riboswitch regulators in the absence of an X-ray diffraction structure, generally without consideration of chemical and binding mode similarity to the natural ligand. In a notable example, Connelly et al. used a small molecule microarray to identify binders for the PreQ1 riboswitch (Connelly et al., 2019). This RNA structure regulates the expression of genes implicated in the biosynthesis of queuosine (Q), essential in translational fidelity and reading of degenerate codons. Given a fluorescently labeled RNA of interest, small molecules with a chemical handle (usually an amine) can be tethered to a microarray slide and incubated with the fluorescently labeled construct. Upon washing the slide to eliminate nonspecific binders, the fluorescence output and location can be used to pinpoint the lead small molecule. This strategy was employed to screen a 25,000 small molecule library against the PreQ1 riboswitch of Bacillus subtilis and yielded a 0.93% hit rate. Hits were refined by counter-screening with two other well characterized riboswitches, SAM and TPP. While orthogonal fluorescence titration experiments identified a lead with nanomolar affinity (Kd= 534 nM), termination experiments did not show any effect on PreQ1 mediated transcription. In-line probing assays revealed a decrease in cleavage upon treatment of the RNA constructs with small molecule (8); however, the cleavage pattern was different when compared to the cognate PreQ1 ligand, possibly explaining the lack of activity in the first two strains tested—B. subtilis and T. tengcongensis. Subsequent termination assays of the lead molecule (8) against PreQ1 riboswitches from different strains reported a significant decrease in full length transcript. SAR studies revealed that the analogue with the highest affinity measured by fluorescence titration resulted in the lowest effect on transcription efficiency (Figure 3C). X-ray crystal structures revealed that while both the native and synthetic ligand (8) occupied the same binding site, they engaged in different interactions within the binding pocket (Connelly et al., 2019). While successful, the binding-based HTS underscored that binding to the intended riboswitch does not directly translate to the desired activity.
The recurring theme of discrepancies between affinity and activity is exemplified by the work of Cressina et al. (Cressina et al., 2011). In their study, the authors used a plethora of analytical and in vitro techniques to validate hits identified through an HTS using a fragment library against the only riboswitch found in both prokaryotes and eukaryotes: the thiamine pyrophosphate riboswitch (TPP). A fragment-based library was screened in cocktails of five compounds using competitive dialysis. Quantification of the levels of radiolabeled native ligand inside and outside the membrane allowed for identification of fragments that displaced the native probe. Cressina et al. identified several selective leads for the TPP riboswitch over other riboswitches. TPP binders were also confirmed through the NMR method Water LOGSY and ITC as leads with micromolar affinity (9, Kd= 100μM) (Figure 3D). Strikingly, no effect was observed when assayed in an in vitro transcription-translation experiment. A follow-up X-ray crystallographic study revealed the fragments bind, indeed, in the same binding site as the cognate thiamine; however, SHAPE-based probing experiments indicated that the fragments induced a conformation distinct from the native ligand (Warner et al., 2014). This report underscores the diverse folding landscape accessible to riboswitches, even though only a few conformations affect transcriptional or translational outcomes.
The HTS methods presented so far, namely small molecule microarray and fragment-based competitive dialysis, enriched the field with novel riboswitch binders and provided important insights into kinetic and thermodynamic factors that influence the activity of new binders. Notably, both HTS methods described are in vitro, affinity-based techniques. A recent effort by Howe et al. used phenotypic HTS method to screen a 57,000-member internal library of antibacterial compounds (Howe et al., 2015). The study ultimately identified a bioactive small molecule that regulates the flavin mononucleotide (FMN) riboswitch and presents a distinct chemical architecture relative to the cognate riboflavin ligand. FMN riboswitches play an essential role in regulating the expression of genes needed for flavin biosynthesis. Cultures of hyper-permeable and efflux deficient E. coli allowed for the identification of competitive inhibitors that displayed antibacterial activity in absence of exogenous riboflavin but no activity upon riboflavin addition. One small molecule, Ribocil, displayed antibacterial activity in minimal media but not in rich media—implying that the effects could be reversed with the addition of exogenous riboflavin. To further confirm the mode of action, Ribocil was tested against an E. coli mutant that lacked the FMN riboswitch and no antibacterial effects were observed. Co-crystal structure of one enantiomer, Ribocil B, showed this small molecule in the same binding pocket but with a different H-bond pattern than that of the cognate ligand. No co-crystal structure was observed for the other enantiomer, Ribocil A. The two enantiomers were subsequently separated, and antibacterial and affinity-based assays confirmed Ribocil-B as the enantiomer with higher affinity and antibacterial activity (Kd= 6.6 nM, IC50= 0.13 μM) (Howe et al., 2015). In a follow-up study, the same chiral bias was observed with the higher affinity and activity analogue Ribocil-C (10, IC50= 50 nM) (Wang et al., 2017a) (Figure 3E).
It is noteworthy that the lead small molecule Ribocil-C (10) performed well in gram-positive (GP) bacteria containing FMN riboswitches and in the E.coli gram-negative (GN) mutant with compromised efflux pumps. The efflux pumps have been shown to prevent membrane penetration and reduce intracellular accumulation of the compound in the wild-type gram negative strain. Motika et al. attempted to design a cell-permeable GN-active analogue of 10 based on their previous findings of common physicochemical properties (eNTRy) among GN antibacterial agents, including rotatable bonds, globularity and ionizable nitrogen groups. A lead small molecule was designed to fit these parameters while still retaining most of the core of 10 (Motika et al., 2020). The co-crystal structure previously published was utilized to identify moieties that could tolerate the introduction of an ionizable nitrogen group. Promisingly, the lead compound 11 showed approximately a 10-fold decrease in minimum inhibitory concentration (MIC) against the wild-type E. coli strain when compared to 10 (Figure 3F). The probe (11) retained the same chiral specificity as its precursor Ribocil-C (10) and showed activity against a panel of known multi-drug resistant GN bacteria. The authors generated spontaneous E. coli resistant mutants against 10. Sequencing of the 20 isolated resistant strains revealed mutations in the FMN riboswitch in all the strains, thereby highlighting that antibacterial resistance must be considered in RNA targeting as well as protein targeting. Overall, application of the eNTRy rules to riboswitch binders successfully guided synthetic tuning and broadened the therapeutic applicability of a previously reported FMN ligand, thereby offering an avenue to re-evaluate GP antibacterial agents and expand their activity to GN strains by increasing their intracellular accumulation (Motika et al., 2020).
Mimicry of Natural Ligand
Multi-drug resistance (MDR) in bacteria can be pinpointed to the aberrant use of antibiotics in agriculture and animal production. Mulhbacher et al. proposed riboswitches as an alternative antibacterial target, along with the hypothesis that maximizing retention of natural ligand-riboswitch interactions would minimize off-target binding (Mulhbacher et al., 2010). Taking advantage of the X-ray structure of the bound guanine riboswitch, in silico ligand design led to the synthesis and testing of two pyrimidine analogues. Both small molecules retained the same H-bond pattern at the three-way junction as the cognate ligand while having minor modifications. In-line probing experiments indicated a comparable level of stabilization and conformational change in the presence of the synthetic ligands versus guanine. Strain selectivity tests performed utilizing a lacZ reporter assay demonstrated selectivity of the lead analogue (12) for C. difficile and S. aureus over other guanine riboswitch containing bacteria (Figure 4A). The two strains that exhibited antibacterial activity have a guanine riboswitch-regulated guaA gene, previously shown to be essential for virulence in porcine and murine models. The compounds exhibited activity only in minimal medium, with bacterial growth being restored in rich guanine-containing medium. Altogether, this data suggests that the compounds act by inhibiting de novo guanine synthesis via riboswitch binding but do not affect normal processing when exogenous guanine is introduced in the media. This study provided evidence that riboswitch targeting with cognate ligand analogs can yield narrow spectrum antibacterial probes, thereby reducing resistance pressure for non-targeted bacteria (Mulhbacher et al., 2010).
Figure 4.
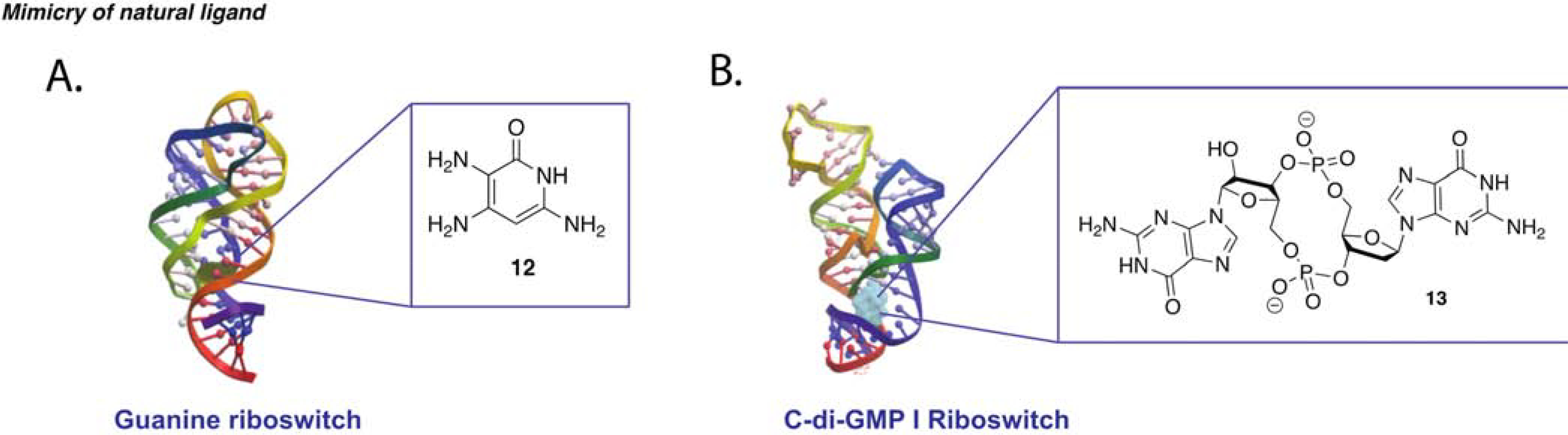
(A) Representative effect of a small molecule that stabilizes the inactive state of a riboswitch resulting in a decrease in transcription/translation adapted from Mulhbacher et al. (B) Schematic of a small molecule that favors the active conformation of a riboswitch increasing transcription/translation. (C) Reported small molecule analogue binder of guanine riboswitch PDB: 1U8D (Mulhbacher et al., 2010). (D) Cyclic C -di-GMP analogue bound to V. choloeae riboswitch PDB: 3IRW (Furukawa et al., 2012).
Another example is riboswitches for circular RNA dinucleotides, which can act as second messengers to mediate several pathways involved in virulence and biofilm formation. Specifically, the concentration of c-di-GMP, formed by two GTP molecules, is known to bring about several physiological changes. Though it has been reported to be implicated in various enzyme-mediated pathways, c-di-GMP is also known to bind two classes of riboswitches. Furukawa et al. aimed at synthesizing riboswitch binding analogs of c-di-GMP to observe the phenotypes associated with their regulation (Furukawa et al., 2012). Given the synthetic impracticality of circular nucleic acids, a few analogues were synthesized with modifications at either the sugar, backbone, or base. In this report, two c-di-GMP-binding riboswitches of V. choleae and C. difficile were chosen for analog testing in the hope that, given their vastly different riboswitch conformational architecture, selectivity for one class over the other could be achieved. The riboswitches were tested with increasing concentration of small molecules in an in-line probing assay with 32P-labeled RNA, which allowed for comparison of small molecule affinity and effect on riboswitch folding. Upon identification of essential and modifiable moieties, a SAR study was conducted on a linear form of the analogues due to their synthetic feasibility, decreased susceptibility to cleavage, and increased cell penetration. After screening of 17 analogues, a few compounds were identified with low micromolar affinity and, most importantly, with 3 to 10-fold selectivity for one of the two riboswitches (13, Figure 4B). Although transcription termination assays confirmed the tightest circular binders as having the greatest effect on transcription regulation, the same cannot be said for the linear analogues, which exhibited close to no inhibition of transcription despite their having Kd in the same micromolar range as the circular analogues. This might suggest that the linear compounds cannot bind at sufficient speed to significantly affect the transcription process. Given that both in-line probing and termination assays put more emphasis on thermodynamic equilibrium, Furukawa et al. devised a fast, 15-minute assay that involves the fusion of the riboswitch of interest with a hammerhead ribozyme. According to this design, small molecules that stabilize the riboswitch allosterically inhibit the ribozyme and suppress cleavage. Termination and ribozyme assays showed a similar yet not perfect trend between the ranking of the best stabilizers and molecules with lowest Kd. The authors highlighted that inconsistencies between assays can be explained by the fact that the function of riboswitches may be heavily dictated by kinetic factors such as rates of RNA folding, RNA transcription and aptamer-ligand association (Furukawa et al., 2012).
A recent joint effort by the Ferré-D’Amaré and Schneekloth labs allowed for a direct comparison between synthetic and high-throughput approaches (Tran et al., 2020). In the study, the authors aimed at targeting a purine-related riboswitch, the ZTP riboswitch. Interestingly, both a SMM microarray high-throughput and a mimicry of natural ligand approach were adopted with the same goal: to find novel binders of the riboswitch that would have in vitro and in vivo activity. Both methods yielded medium and high affinity compounds, but the only ones that resulted in in vivo activity were synthetic analogues of the cognate ligand ZMP. Notably, the analogue found to be 3-fold more potent than the cognate ligand in vivo reported a 2-fold lower affinity than ZMP to the riboswitch in vitro, thereby highlighting once again the importance of not relying on affinity to rank compounds fitness when targeting riboswitches (Tran et al., 2020).
Despite their impressive cross-species sequence/function conservation, recent successes have proven that the differences in 3D conformations of riboswitches can be harnessed to develop ligands with high specificity for a single species-specific topology. Furthermore, comparison of affinity-based and phenotypic-based assays have highlighted the importance of taking into consideration that effective ligands might not be the ones with higher affinity under thermodynamic equilibrium conditions, but the ones that associate the fastest, highlighting the kinetic perspective of targeting RNA constructs.
Other Multi-junctional Structures
While not falling within a traditional tertiary structure class, group II introns—a large class of self-splicing ribozymes—have been found to participate in intricate and unique tertiary interactions. These autocatalytic RNA constructs have been found across several species. In pathogenic yeast, for example, they regulate splicing of genes essential for respiration, thereby making it an attractive topological class for the development of antifungal probes (Pyle, 2010). The targetability potential of yeast group II introns was successfully harnessed by Fedorova et al. in their attempt to target unique RNA structures in pathogenic yeast splice sites (Fedorova et al., 2018). Group II introns are ribozymes found in the mitochondrial genome of plants and fungi that regulate genes essential for survival. This ribozyme class is known for its complex tertiary structure folding, and X-ray crystallography revealed several binding pockets for co-factor or inhibitor binding, suggesting a potential target for antifungal agents. HTS of a 10,000-member library was performed using the previously characterized ai5 ribozyme. The flanking exons were removed, and a FRET pair was incorporated to report cleavage. Upon filtering of lead compounds, a SAR study was performed by changing moieties at the five distinct architectural features of the small molecule. As group II introns regulate the COX1 gene essential for respiration and survival in S. cerevisiae, this organism was chosen to test potency of lead compounds. Selectivity of the analogues was tested by creating an intronless mutant. The most selective analogue (14) inhibited growth in the wild type but not the intronless variant (Figure 5). Compounds that showed antifungal activity also caused an accumulation of unspliced COX1 transcript as measured via RT-qPCR. The most promising probe (14) contained a catechol moiety found essential for intron binding. Incidentally, polyphenolic compounds are often eliminated from screening libraries and classified as PAINS, promiscuous probes that bind multiple classes of biological targets. Due to this, 14 was tested in human HEK293 T-cell lines and no toxicity was observed, highlighting its specificity for yeast and warning against excluding possible PAINS compounds solely on the presence of common moieties (Fedorova et al., 2018).
Figure 5.

Group II intron binder (left) recently reported to prevent splicing in COX1-containing C. parapsilosis (Fedorova et al., 2018)
RNA Quaternary Structures
RNA quaternary structures are defined as the association of distinct RNAs or RNA-protein complexes. Targeting higher order complexes by stabilizing or destabilizing their formation may hold the key to therapeutic design for a plethora of diseases (Jones and Ferré-D’Amaré, 2015). Specifically, RNA-protein interactions, such as those in splicing complexes, offer several opportunities for small molecule targeting through the RNA-protein interface, conformational trapping to enhance or decrease binding, or destabilization of the RNA structural motif responsible for protein recognition. While stabilizing RNA-protein complexes offers opportunities for binders that engage both parties, destabilizing or preventing the complex formation usually involves the design or discovery of a small molecule that binds one of the two partners. Consequently, the sections below will discuss a few very recent and notable examples that highlight the feasibility and applicability of small molecule targeting of biologically relevant RNA-protein complexes, such as splicing complexes, through RNA structure modulation. While the biological relevance of RNA-RNA quaternary structures has been recently catalogued, small molecule targeting of this class has not been well explored and, therefore, will not be discussed in the section below.
Splice Sites
RNA splicing is a eukaryotic process utilized to increase the coding potential of the genome and the diversity of the transcriptome and proteome (Kim et al., 2007). Differential incorporation of exons in the mature transcript leads to alternative splicing. Intron exclusion can either occur by self-splicing, as in the case of ribozymes, or through complexation of the spliceosome at the introns/exon junction. Recognition of splicing enhancers by proteins occurs through sequence recognition or often through RNA structure detection (Tazi et al., 2009). Lack or increase in recognition of splice sites by the splicing complex has been linked to a variety of human diseases, increasing the interest in splice sites as therapeutic targets. Impactful advances in structure characterization of select disease-relevant transcripts and mapping of dynamic conformational ensembles has enabled the identification of specific folded states that bring about the splicing event of interest (Tomezsko et al., 2020). Therefore, depending on whether we want to promote or prevent a splicing event, small molecules can be devised to stabilize or destabilize the desired RNA tertiary structure conformation and modulate protein binding. While at its early stages, small molecule targeting of RNA splice sites has provided exciting and successful approaches and leads, including FDA approved drugs, emphasizing the potential of chemical probes in regulating RNA-driven biological processes.
SMA is a degenerative disease caused by a deletion mutation in the survival motor neuron 1 (SMN1) gene, responsible for encoding the motor protein. While humans have two paralogous SMN genes, namely SMN1 and SMN2, the former leads to the expression of full-length protein necessary for muscle strength. The latter differs by few nucleotides, including a C840T mutation near the 5’-end of exon 7 splice site (5’-ss) that leads to the expression of a shortened less stable SMN protein due to the exclusion of exon 7 (Darras et al., 2015). Several approaches have been explored in recent years to treat this condition including antisense oligonucleotides (Spinraza), gene editing strategies, and small molecule modulation of splice variants. Despite their great success and clinical applications, ASO and gene-editing systems often come with problematic delivery, especially across the blood-brain barrier, and the high cost reduces their population-wide application (Wurster and Ludolph, 2018). In a study published in 2014, a team of PTC-Roche researchers identified small molecules that modulate the SMN2 mRNA to promote exon 7 inclusion in SMA patients (Naryshkin et al., 2014). The team designed a luciferase-based assay in HEK293H embryonic kidney cells transfected with a SMN2 minigene. An HTS was conducted with an in-house 200,000-member library and identified three related compounds that significantly increased exon 7 inclusion with nanomolar EC50 values (Figure 6A). The clinical relevance of these compounds was confirmed by testing the effect of the ligands on the SMN pathways in several patient-derived cell lines and tissues, highlighting their bioavailability along with brain and spinal tissue permeability (Wurster and Ludolph, 2018). Remarkably, all three analogues increased full-length mature mRNA and SMN protein expression. The compounds altered the expression of only six other genes, which could each be linked to the SMN pathway, implying selectivity of the small molecule (15). Treatment of mice models of various SMA types increased body weight, life expectancy, and motility, making small molecule 15 a promising clinical candidate (Naryshkin et al., 2014).
Figure 6.

(A) First small molecule reported binder (left) to promote full length expression of SMN1 protein in SMA patients (right) (Naryshkin et al., 2014) and its reported mode of binding (center) (Campagne et al., 2019). (B) SMN 5’-splice site binder(left) and its reported mode of binding (center) (Palacino et al., 2015).
Concurrently to the work at Naryshkin et al., a Novartis research team published the discovery of a distinct small molecule that regulates exon 7 incorporation in SMN2 splicing (Palacino et al., 2015). In their study, Palacino et al. also utilized a complementary luciferase reporter to conduct an HTS against NSC34 motor neuron cells containing the SMN2 minigene. Two nanomolar analogs were reported, and the compounds displayed high bioavailability and promoted full-length SMN protein expression (16, EC50 = 5nM) (Figure 6B). An SMN2 mouse model showed an increase in body weight, life expectancy, and SMN protein expression. Global gene expression analysis via RNA-Seq revealed alteration in expression of 23 genes.
Structural information on the binding mode and action of the SMN ligands 15, 16, and related analogues were elucidated in the work of Palacino et al. as well as follow-up studies of Campagne et al. (Campagne et al., 2019; Sivaramakrishnan et al., 2017) Given the importance of the C840T mutation in exon 7 inclusion, it is plausible that the small molecules bind near or at the intron-exon junction and induce a conformational change to restore the strength of the splice site. A study of small molecule activity with different SMN mutants, completed by Palacino et al., confirmed the binding site of 16 to be at the interface of the 5’ end of intron 7 and 3’ end of exon 7 (Figure 6B). SPR experiments with the RNA construct and U1 small nuclear ribonucleoprotein (snRNP), which can bind the 5’-ss to promote inclusion of the exon, revealed an increased binding affinity of the protein for the RNA in the presence of 16. On the other hand, no binding to the RNA or protein alone was observed for 16. Collectively, this data suggested a cooperative stabilization of the 5’-ss and U1 snRNP, increasing the strength of the splice site (Palacino et al., 2015). Despite having distinct chemical architecture, the ligand reported by Roche (15) was found to also positively cooperate in enhancing splicing by promoting recognition of the weak 5’-ss by U1 snRNP (Sivaramakrishnan et al., 2017). In NMR studies, the main core of the compound 15 bound the major groove at the intron-exon junction of the 5’-ss and the positively charged piperazine moiety was found to be essential in recruiting the U1-C zinc finger to the RNA by making contacts with the ribose phosphate backbone of U1 snRNP (Campagne et al., 2019). The most potent ligands of Roche and Novartis had a mapped binding site at the extremity of the intron-exon junction, while the other less potent analogues are proposed to bind a few nucleotides upstream of the junction. These results suggest a correlation between potency and proximity of binding to the RNA-protein interface (Campagne et al., 2019). Ultimately, the PTC-Roche studies resulted in the FDA approval of Risdiplam, an analogue of the small molecules presented and currently sold as Evrysdi, a modulator of splicing events in SMA (Mullard, 2020). Evrysdi constitutes the first small molecule treatment for this neurodegenerative and, in some forms, fatal disease. The studies presented herein highlight the new and exciting possibility of modulating splicing events by harnessing the unique RNA folding at splice sites, thus bypassing targeting protein members of the spliceosome that control splicing of multiple genes and would lead to off-target effects.
Other RNA-protein Complexes
The recently reported small molecule modulators of splicing complexes at the RNA-protein interface provided the scientific community with the basis to pursue other diseases linked to aberrant splicing, as well as other RNA-protein complexes across organisms.
For example, research based on discovery of new biomarkers for specific tumors led to the identification of several dysregulated lncRNAs, many of which bind chromatin modifying-proteins (Hu et al., 2018). This discovery implicated lncRNAs in the epigenetic overhaul that often leads to over-expression of oncogenes and under-expression of tumor suppressors (Schmitt and Chang, 2013). HOTAIR is among the best characterized lncRNA and is mainly known for its association with polycomb repressive complex 2 (PRC2) and its role in chromatin remodeling of many genes. Upon binding EZH2, the catalytic subunit of PRC2, HOTAIR mediates silencing of nemo-like kinase (NLK), a tumor suppressor gene. Thus, inhibiting the complexation of HOTAIR and EZH2 may lead to enhanced chromatin remodeling and initiation of transcription. To test this hypothesis, Ren et al. performed a virtual ligand screening on the previously reported HOTAIR fragment identified as the minimal unit needed for binding to EZH2 (Ren et al., 2019). 3D models of the construct were created by taking advantage of MCfold and MC-Sym programs, and the entire PubChem library was docked against the fragment. An NLK luciferase assay was designed for four human cell lines and true hits were identified as the compounds that exhibited high fluorescence in all four cell lines and that increased HOTAIR target gene NLK mRNA levels but not HOTAIR itself. RNA immunoprecipitation (RIP) and chromatin immunoprecipitation (ChIRP) revealed decreased occupancy of EZH2 on HOTAIR upon treatment with hit ligand 17. Noticeably, EZH2 is known to bind at least five other lncRNAs including Xist, MALAT1, and HOXA11 and while EZH2 occupancy of HOTAIR was drastically reduced, no effects were observed in any of the other lncRNAs, indicating selectivity of ligand 17 (Figure 7A). While no biophysical experiments were performed to characterize the small molecule:RNA complex, the authors performed mutational studies to further confirm the proposed binding site of 17. Specifically, mutants of the nucleotides hypothesized to be involved in RNA-small molecule interactions were made in silico and the compound was re-docked. The predicted binding energy increased for every mutation thereby corroborating the hypothesized binding site to the EZH2 binding RNA fragment. The in silico observations were experimentally confirmed via immunoprecipitation. The WT and mutants tested in silico were transfected in MB-231 cells. Notably, EZH2 associated with both the mutant and the WT construct, but 17 reduced the binding efficiency of EZH2 to the WT and not the mutant. A SAR study was performed on the lead compound revealing the importance of the nitro-substitution for activity. Specifically, while its deletion resulted in decreased activity, its replacement with methyl or chloro-moieties completely abrogated the activity. Cell based assays reported a significant decrease in invasion, metastasis, and wound healing upon small molecule treatment (17). Finally, RNA in situ hybridization and immunofluorescence showed that while the DMSO control showed significant nuclear co-localization of EZH2 and HOTAIR, treatment with 17 redistributed EZH2 to the cytosol, further corroborating their dissociation. The compound was effective in reducing tumor volume in lung cancer murine model, and while the dosage needed to rescue the non-metastatic phenotype did not make it a viable candidate for clinical application, it represented the first small molecule binder for the heavily researched lncRNA HOTAIR (Ren et al., 2019).
Figure 7.

(A) small molecule binder (left) reported to decrease the affinity of HOTAIR essential binding element (center) for EZH2 and promote NLK expression (right) (Ren et al., 2019). (B) Small molecule (left) found to induce a conformational change (center) that exposes AUF1 binding site but not the hnRNP A1 site, thereby halting viral proliferation (right) (Davila-Calderon et al., 2020).
Targeting of key RNA-protein complexes has also been investigated as an avenue to design novel antiviral agents. Specifically, a recent study by Davila-Calderon et al. aimed at targeting the association of host RNA-binding proteins to human enterovirus 71 (EV71) RNA. The 5’-UTR of EV71 is predicted to fold into six stem loop structures, where stem loop I promotes genome replication and stem loops II-VI are involved in cap-independent translation via a type I internal ribosome entry site (IRES) (Davila-Calderon et al., 2020). Protein association with stem loop II (SLII) was shown to stabilize conformations that either promote translation (hnRNP A1) or repress translation (AUF1) (Hung et al., 2016). Davila-Calderon et al. first screened a small molecule focused library for binding to SLII. The focused library was built by decorating the amiloride scaffold, a previously reported RNA-binding scaffold that showed potential for tunability and selectivity (Patwardhan et al., 2017). Small molecule hits were identified through an in vitro FRET-based displacement assay. Identified leads were then validated via NMR experiments and ITC (Figure 7B), and the biological activity of the lead compounds was examined through a cell-based dual luciferase reporter assay. Specifically, Firefly luciferase (FLuc) translation was rendered EV71 IRES-dependent while Renilla luciferase (Rluc) translation was driven by the normal host cap-dependent process. Out of the top hits identified, only 18 significantly reduced IRES dependent but not cap-dependent translation, revealing its therapeutic potential and corroborating its mode of action through specific IRES binding. The 3D models obtained via NMR showed that 18 induced a conformational change in the RNA that is consistent with an exposed AUF1 binding site. Furthermore, while 18 showed moderate affinity for SLII via ITC (Kd=520 nM), it caused the most significant chemical shift perturbation to the construct when compared to the other initial hit analogues. Calorimetric titrations of the AUF1 RNA-binding domain into the SLII-18 complex showed an increase in relative binding affinity of the protein for the RNA as a function of 18 concentration. Finally, to confirm the suspected mode of action, in vitro pull-down and in cellulo immunoprecipitation assays were performed to test the stability of the SLII-AUF1 complex as a function of 18. Both in vitro and in cellulo experiments confirmed an increase in stability of the SLII-AUF1 complex in the presence of small molecule. The tuning of a known RNA binding scaffold through synthesis of a focused library yielded an RNA-protein allosteric binder that decreased viral replication in cells with an IC50 of 7.54 μM, thereby providing a promising lead in better understanding IRES-mediated translation, as well as a potential antiviral small molecule for EV71, a virus with no approved antivirals or vaccine to date.
Significance
The continuous development of biophysical and structural techniques is enriching the field with a growing body of complex RNA structures resulting from intricate networks of short- and long-range base pairing, offering opportunities to design small molecule modulators of function as has been done for proteins for decades. Recent studies highlighted the feasibility of targeting higher level folded RNA structures at both the tertiary and quaternary structure level.
Bioactive small molecules targeting RNA tertiary structures, as exemplified by RNA G-quadruplexes, have been discovered via high throughput and focused library approaches, underscoring the potential of both avenues to yield a biological probe. In addition, in silico screening was successful at providing the first SARS-CoV pseudoknot binder, inspiring attempts at targeting similar elements of other viruses, including the current pandemic-causing SARS-CoV2. Selectivity for SARS-CoV2 functional elements could be achieved via synthetic tuning of known SARS-CoV ligands, and indeed, this strategy was successfully employed in the design and discovery of probes selective for MALAT1 over other closely related triple helix motifs.
For RNA quaternary structures, in silico, focused library, and HTS screening have yielded bioactive small molecules targeting RNA-protein complexes, as reported for HOTAIR, EV71, and SMN ligands, respectively. In the case of SMN ligands, which stabilized a splicing complex for SMN2 RNA, the studies not only provided one of the first RNA-targeted small molecule splicing modulators but also the first FDA approved small molecule for the treatment of SMA. The efforts of PTC-Roche and Novartis provided the scientific community with direct evidence that small molecules can modulate the strengths of RNA splice-sites and ultimately opened the door to new pursuits of RNA-targeting drugs.
While several RNA-centric approaches have been developed in the last decade, many traditional drug discovery approaches, such as those used for SMN, have produced promising bioactive ligands. One observation that resulted from the SMN ligands developed by the two industry teams was that, while possessing a different chemical architecture, both Novartis and PTC-Roche probes resulted in exon 7 incorporation in SMA models. The finding that chemically distinct small molecules can elicit similar effects in a biological setting was also seen in synthetic versus cognate riboswitch binders. What is more, these studies, as in the case of FMN riboswitch ligands, raised the possibility that similarity between native and synthetic RNA binders may not be the best predictor of affinity or activity.
Traditional approaches were also exemplified by the work of Fedorova et al. aimed at targeting the group II fungal intron, which combined HTS followed by medicinal chemistry optimization to yield a novel bioactive ligand. The authors posit that while there are important structural differences between RNA and proteins, such features should be used as an opportunity for further optimization of small molecules without the need for re-inventing the wheel of drug discovery approaches. Recently, notable efforts have been made to find differences between known protein binders and RNA binders. The study by Fedorova et al., among many others, highlights that the question of what makes a probe an RNA binder over a protein binder is far from being answered. Furthermore, the authors warn against excluding compounds from screening based on previously reported protein activity, as exemplified by their PAINS-like initial hit. This observation also sparks the question: What factors play a role in small molecule selectivity in a biological setting? While preliminary studies evaluated selectivity based on the affinity of a probe for one construct over another, part of the answer might lie in the abundance of the intended target relative to other possible binding partners within the cell.
Finally, while the successes highlighted herein prove that RNA can be targeted by molecules traditionally classified as ‘drug-like’, we should not limit ourselves to such traditional classifications and parameters. As the field progresses in the understanding of kinetic processes and thermodynamic modulation of the conformational landscape of RNA, more opportunities will arise to better design small molecule ligands as well as exponentially increase our ability to refine the definition of what makes a small molecule a ‘good’ RNA binder.
The accomplishments presented herein underscore the potential of higher-level folding RNA structures as promising avenues for the development of selective biologically relevant probes in a manner more comparable to the canonical protein-targeting approach over simple RNA secondary motif targeting. Importantly, the notable advances and new additions to the set of biophysical and structural tools available to characterize tertiary and quaternary motifs have recently yielded novel promising structures, exponentially increasing the opportunities for small molecule targeting of RNA for many years to come.
Acknowledgments
We are grateful to the members of the Hargrove lab for their support and feedback and especially to Sarah Wicks and Aline Umuhire Juru, whose insight was crucial in defining the initial framework of this review. The graphical abstract was made using BioRender. This work was funded by Duke University and the U.S. National Institutes of Health (R35 GM124785).
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdulle LE, Hao JL, Pant OP, Liu XF, Zhou DD, Gao Y, Suwal A, and Lu CW (2019). MALAT1 as a Diagnostic and Therapeutic Target in Diabetes-Related Complications: A Promising Long-Noncoding RNA. Int J Med Sci 16, 548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abulwerdi FA, Xu W, Ageeli AA, Yonkunas MJ, Arun G, Nam H, Schneekloth JS, Dayie TK, Spector D, Baird N, et al. (2019). Selective Small-Molecule Targeting of a Triple Helix Encoded by the Long Noncoding RNA, MALAT1. ACS Chemical Biology 14, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn D-G, Lee W, Choi J-K, Kim S-J, Plant EP, Almazán F, Taylor DR, Enjuanes L, and Oh J-W (2011). Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res 91, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora A, and Maiti S (2009). Differential Biophysical Behavior of Human Telomeric RNA and DNA Quadruplex. The Journal of Physical Chemistry B 113, 10515–10520. [DOI] [PubMed] [Google Scholar]
- Bao H-L, and Xu Y (2018). Investigation of higher-order RNA G-quadruplex structures in vitro and in living cells by 19F NMR spectroscopy. Nature Protocols 13, 652–665. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya D, Diamond P, and Basu S (2015). An Independently folding RNA G-quadruplex domain directly recruits the 40S ribosomal subunit. Biochemistry 54, 1879–1885. [DOI] [PubMed] [Google Scholar]
- Biffi G, Tannahill D, and Balasubramanian S (2012). An Intramolecular G-Quadruplex Structure Is Required for Binding of Telomeric Repeat-Containing RNA to the Telomeric Protein TRF2. Journal of the American Chemical Society 134, 11974–11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, Tannahill D, Miller J, Howat WJ, and Balasubramanian S (2014). Elevated Levels of G-Quadruplex Formation in Human Stomach and Liver Cancer Tissues. PLOS ONE 9, e102711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount KF, and Breaker RR (2006). Riboswitches as antibacterial drug targets. Nat Biotechnol 24, 1558–1564. [DOI] [PubMed] [Google Scholar]
- Breaker RR (2012). Riboswitches and the RNA world. Cold Spring Harb Perspect Biol 4, a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA (2020). Unraveling the structure and biological functions of RNA triple helices. WIREs RNA 11, e1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Bulkley D, Wang J, Valenstein ML, Yario TA, Steitz TA, and Steitz JA (2014). Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nature structural & molecular biology 21, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugaut A, and Balasubramanian S (2012). 5’-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res 40, 4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SE, and Pyle AM (2011). The Molecular Interactions That Stabilize RNA Tertiary Structure: RNA Motifs, Patterns, and Networks. Accounts of Chemical Research 44, 1302–1311. [DOI] [PubMed] [Google Scholar]
- Campagne S, Boigner S, Rüdisser S, Moursy A, Gillioz L, Knörlein A, Hall J, Ratni H, Cléry A, and Allain FHT (2019). Structural basis of a small molecule targeting RNA for a specific splicing correction. Nature Chemical Biology 15, 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech Thomas R., and Steitz Joan A. (2014). The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell 157, 77–94. [DOI] [PubMed] [Google Scholar]
- Chaires JB, Ren J, Hamelberg D, Kumar A, Pandya V, Boykin DW, and Wilson WD (2004). Structural Selectivity of Aromatic Diamidines. Journal of Medicinal Chemistry 47, 5729–5742. [DOI] [PubMed] [Google Scholar]
- Chen X-C, Chen S-B, Dai J, Yuan J-H, Ou T-M, Huang Z-S, and Tan J-H (2018). Tracking the Dynamic Folding and Unfolding of RNA G-Quadruplexes in Live Cells. Angewandte Chemie International Edition 57, 4702–4706. [DOI] [PubMed] [Google Scholar]
- Connelly CM, Numata T, Boer RE, Moon MH, Sinniah RS, Barchi JJ, Ferré-D’Amaré AR, and Schneekloth JS (2019). Synthetic ligands for PreQ1 riboswitches provide structural and mechanistic insights into targeting RNA tertiary structure. Nature Communications 10, 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad NK (2014). The emerging role of triple helices in RNA biology. WIREs RNA 5, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costales MG, Childs-Disney JL, Haniff HS, and Disney MD (2020). How We Think about Targeting RNA with Small Molecules. Journal of Medicinal Chemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressina E, Chen L, Abell C, Leeper FJ, and Smith AG (2011). Fragment screening against the thiamine pyrophosphate riboswitchthiM. Chemical Science 2, 157–165. [Google Scholar]
- Dai J, Liu ZQ, Wang XQ, Lin J, Yao PF, Huang SL, Ou TM, Tan JH, Li D, Gu LQ, et al. (2015). Discovery of Small Molecules for Up-Regulating the Translation of Antiamyloidogenic Secretase, a Disintegrin and Metalloproteinase 10 (ADAM10), by Binding to the G-Quadruplex-Forming Sequence in the 5’ Untranslated Region (UTR) of Its mRNA. J Med Chem 58, 3875–3891. [DOI] [PubMed] [Google Scholar]
- Darras BT, Markowitz JA, Monani UR, and De Vivo DC (2015). Chapter 8 - Spinal Muscular Atrophies. In Neuromuscular Disorders of Infancy, Childhood, and Adolescence (Second Edition), Darras BT, Jones HR, Ryan MM, and De Vivo DC, eds. (San Diego: Academic Press; ), pp. 117–145. [Google Scholar]
- Davila-Calderon J, Patwardhan NN, Chiu L-Y, Sugarman A, Cai Z, Penutmutchu SR, Li M-L, Brewer G, Hargrove AE, and Tolbert BS (2020). IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nature Communications 11, 4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman JD, Ruiz-Echevarria MJ, and Peltz SW (1998). Translating old drugs into new treatments: ribosomal frameshifting as a target for antiviral agents. Trends Biotechnol 16, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlic A, Morgan BS, Xu JL, Liu A, Roble C Jr., and Hargrove AE (2018). Discovery of Small Molecule Ligands for MALAT1 by Tuning an RNA-Binding Scaffold. Angew Chem Int Ed Engl 57, 13242–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlic A, Zafferani M, Padroni G, Puri M, and Hargrove Amanda E. (2020). Regulation of MALAT1 triple helix stability and in vitro degradation by diphenylfurans. Nucleic Acids Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova O, Jagdmann GE, Adams RL, Yuan L, Van Zandt MC, and Pyle AM (2018). Small molecules that target group II introns are potent antifungal agents. Nature Chemical Biology 14, 1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Gu H, Sudarsan N, Hayakawa Y, Hyodo M, and Breaker RR (2012). Identification of ligand analogues that control c-di-GMP riboswitches. ACS chemical biology 7, 1436–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavís M, López-Méndez B, Somoza A, Oyarzabal J, Dalvit C, Villasante A, Campos-Olivas R, and González C (2014). Discovery of Selective Ligands for Telomeric RNA G-quadruplexes (TERRA) through 19F-NMR Based Fragment Screening. ACS Chemical Biology 9, 1559–1566. [DOI] [PubMed] [Google Scholar]
- Hong W, Zeng J, and Xie J (2014). Antibiotic drugs targeting bacterial RNAs. Acta Pharm Sin B 4, 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JA, Wang H, Fischmann TO, Balibar CJ, Xiao L, Galgoci AM, Malinverni JC, Mayhood T, Villafania A, Nahvi A, et al. (2015). Selective small-molecule inhibition of an RNA structural element. Nature 526, 672–677. [DOI] [PubMed] [Google Scholar]
- Hu G, Niu F, Humburg BA, Liao K, Bendi VS, Callen S, Fox HS, and Buch S (2018). Molecular mechanisms of long noncoding RNAs and their role in disease pathogenesis. Oncotarget 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung C-T, Kung Y-A, Li M-L, Brewer G, Lee K-M, Liu S-T, and Shih S-R (2016). Additive Promotion of Viral Internal Ribosome Entry Site-Mediated Translation by Far Upstream Element-Binding Protein 1 and an Enterovirus 71-Induced Cleavage Product. PLoS Pathog 12, e1005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CP, and Ferré-D’Amaré AR (2015). RNA quaternary structure and global symmetry. Trends Biochem Sci 40, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuda Y, Sato S. i., Asano L, Morimura Y, Furuta T, Sugiyama H, Hagihara M, and Uesugi M (2016). A Small Molecule That Represses Translation of G-Quadruplex-Containing mRNA. Journal of the American Chemical Society 138, 9037–9040. [DOI] [PubMed] [Google Scholar]
- Kim E, Magen A, and Ast G (2007). Different levels of alternative splicing among eukaryotes. Nucleic Acids Res 35, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole R, Krainer AR, and Altman S (2012). RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 11, 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammich S, Kamp F, Wagner J, Nuscher B, Zilow S, Ludwig A-K, Willem M, and Haass C (2011). Translational repression of the disintegrin and metalloprotease ADAM10 by a stable G-quadruplex secondary structure in its 5’-untranslated region. J Biol Chem 286, 45063–45072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machtel P, Bąkowska-Żywicka K, and Żywicki M (2016). Emerging applications of riboswitches – from antibacterial targets to molecular tools. Journal of Applied Genetics 57, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalusova M, Wu JY, and Zhuang X (2011). Functional importance of telomerase pseudoknot revealed by single-molecule analysis. Proceedings of the National Academy of Sciences 108, 20339–20344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motika SE, Ulrich RJ, Geddes EJ, Lee HY, Lau GW, and Hergenrother PJ (2020). A Gram-Negative Antibiotic Active Through Inhibition of an Essential Riboswitch. Journal of the American Chemical Society. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulhbacher J, Brouillette E, Allard M, Fortier L-C, Malouin F, and Lafontaine DA (2010). Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog 6, e1000865–e1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A (2020). FDA approves RNA-targeting small molecule. In Nature Reviews Drug Discovery (Nature). [DOI] [PubMed] [Google Scholar]
- Mustoe AM, Lama NN, Irving PS, Olson SW, and Weeks KM (2019). RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proceedings of the National Academy of Sciences 116, 24574–24582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KKY, Karp GM, Qi H, Woll MG, et al. (2014). SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693. [DOI] [PubMed] [Google Scholar]
- Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Van Hoosear M, Shin Y, Chin DN, Keller CG, et al. (2015). SMN2 splice modulators enhance U1–pre-mRNA association and rescue SMA mice. Nature Chemical Biology 11, 511–517. [DOI] [PubMed] [Google Scholar]
- Panchal V, and Brenk R (2021). Riboswitches as Drug Targets for Antibiotics. Antibiotics 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-J, Kim Y-G, and Park H-J (2011). Identification of RNA Pseudoknot-Binding Ligand That Inhibits the −1 Ribosomal Frameshifting of SARS-Coronavirus by Structure-Based Virtual Screening. Journal of the American Chemical Society 133, 10094–10100. [DOI] [PubMed] [Google Scholar]
- Patwardhan NN, Ganser LR, Kapral GJ, Eubanks CS, Lee J, Sathyamoorthy B, Al-Hashimi HM, and Hargrove AE (2017). Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. Medchemcomm 8, 1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant EP, Rakauskaitė R, Taylor DR, and Dinman JD (2010). Achieving a Golden Mean: Mechanisms by Which Coronaviruses Ensure Synthesis of the Correct Stoichiometric Ratios of Viral Proteins. Journal of Virology 84, 4330–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleij CW, Rietveld K, and Bosch L (1985). A new principle of RNA folding based on pseudoknotting. Nucleic Acids Res 13, 1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle AM (2010). The tertiary structure of group II introns: implications for biological function and evolution. Crit Rev Biochem Mol Biol 45, 215–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Wang Y. f., Zhang J, Wang Q. x., Han L, Mei M, and Kang C. s. (2019). Targeted design and identification of AC1NOD4Q to block activity of HOTAIR by abrogating the scaffold interaction with EZH2. Clinical Epigenetics 11, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie DB, Soong J, Sikkema WKA, and Woodside MT (2014). Anti-frameshifting Ligand Reduces the Conformational Plasticity of the SARS Virus Pseudoknot. Journal of the American Chemical Society 136, 2196–2199. [DOI] [PubMed] [Google Scholar]
- Rizvi NF, Howe JA, Nahvi A, Klein DJ, Fischmann TO, Kim HY, McCoy MA, Walker SS, Hruza A, Richards MP, et al. (2018). Discovery of Selective RNA-Binding Small Molecules by Affinity-Selection Mass Spectrometry. ACS Chem Biol 13, 820–831. [DOI] [PubMed] [Google Scholar]
- Schmitt AM, and Chang HY (2013). Gene regulation: Long RNAs wire up cancer growth. Nature 500, 536–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A, and Nudler E (2013). A Decade of Riboswitches. Cell 152, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, and Corey DR (2017). Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res 46, 1584–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui-Jain A, Grand CL, Bearss DJ, and Hurley LH (2002). Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proceedings of the National Academy of Sciences 99, 11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaramakrishnan M, McCarthy KD, Campagne S, Huber S, Meier S, Augustin A, Heckel T, Meistermann H, Hug MN, Birrer P, et al. (2017). Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nature Communications 8, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazi J, Bakkour N, and Stamm S (2009). Alternative splicing and disease. Biochim Biophys Acta 1792, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulson E, Hartwick EW, Cooper-Sansone A, Williams MAC, Soliman ME, Robinson LK, Kieft JS, and Mouzakis KD (2020). An RNA pseudoknot stimulates HTLV-1 pro-pol programmed −1 ribosomal frameshifting. RNA 26, 512–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomezsko PJ, Corbin VDA, Gupta P, Swaminathan H, Glasgow M, Persad S, Edwards MD, McIntosh L, Papenfuss AT, Emery A, et al. (2020). Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran B, Pichling P, Tenney L, Connelly CM, Moon MH, Ferré-D’Amaré AR, Schneekloth JS, and Jones CP (2020). Parallel Discovery Strategies Provide a Basis for Riboswitch Ligand Design. Cell Chemical Biology 27, 1241–1249.e1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney D, Spiegel J, Zyner K, Tannahill D, and Balasubramanian S (2020). The regulation and functions of DNA and RNA G-quadruplexes. Nature Reviews Molecular Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicens Q, Mondragón E, Reyes FE, Coish P, Aristoff P, Berman J, Kaur H, Kells KW, Wickens P, Wilson J, et al. (2018). Structure-Activity Relationship of Flavin Analogues That Target the Flavin Mononucleotide Riboswitch. ACS chemical biology 13, 2908–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Mann PA, Xiao L, Gill C, Galgoci AM, Howe JA, Villafania A, Barbieri CM, Malinverni JC, Sher X, et al. (2017a). Dual-Targeting Small-Molecule Inhibitors of the Staphylococcus aureus FMN Riboswitch Disrupt Riboflavin Homeostasis in an Infectious Setting. Cell Chemical Biology 24, 576–588.e576. [DOI] [PubMed] [Google Scholar]
- Wang S-K, Wu Y, Wang X-Q, Kuang G-T, Zhang Q, Lin S-L, Liu H-Y, Tan J-H, Huang Z-S, and Ou T-M (2017b). Discovery of Small Molecules for Repressing Cap-Independent Translation of Human Vascular Endothelial Growth Factor (hVEGF) as Novel Antitumor Agents. Journal of Medicinal Chemistry 60, 5306–5319. [DOI] [PubMed] [Google Scholar]
- Warner KD, Hajdin CE, and Weeks KM (2018). Principles for targeting RNA with drug-like small molecules. Nat Rev Drug Discov 17, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner KD, Homan P, Weeks KM, Smith AG, Abell C, and Ferré-D’Amaré AR (2014). Validating fragment-based drug discovery for biological RNAs: lead fragments bind and remodel the TPP riboswitch specifically. Chem Biol 21, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters KE, Strobel EJ, Yu AM, Lis JT, and Lucks JB (2016). Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat Struct Mol Biol 23, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, JnBaptiste CK, Lu LY, Kuhn CD, Joshua-Tor L, and Sharp PA (2012). A triple helix stabilizes the 3’ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26, 2392–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurster CD, and Ludolph AC (2018). Antisense oligonucleotides in neurological disorders. Therapeutic Advances in Neurological Disorders 11, 1756286418776932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, Lejault P, Chevrier S, Boidot R, Robertson AG, Wong JMY, and Monchaud D (2018). Transcriptome-wide identification of transient RNA G-quadruplexes in human cells. Nature Communications 9, 4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zeng D, Cao J, Wang M, Shu B, Kuang G, Ou T-M, Tan J-H, Gu L-Q, Huang Z-S, et al. (2017). Interaction of Quindoline derivative with telomeric repeat–containing RNA induces telomeric DNA-damage response in cancer cells through inhibition of telomeric repeat factor 2. Biochimica et Biophysica Acta (BBA) - General Subjects 1861, 3246–3256. [DOI] [PubMed] [Google Scholar]