

SUMMARY
Ample evidence indicates that individuals with intellectual disability (ID) are at increased risk of developing stress-related behavioral problems and mood disorders. Yet, a mechanistic explanation for such a link remains largely elusive. Here, we focused on characterizing the syndromic ID gene oligophrenin-1 (OPHN1). We find that Ophn1 deficiency in mice markedly enhances helpless/depressive-like behavior in the face of repeated/uncontrollable stress. Strikingly, Ophn1 deletion exclusively in parvalbumin (PV) interneurons in the prelimbic medial prefrontal cortex (PL-mPFC) is sufficient to induce helplessness. This behavioral phenotype is mediated by a diminished excitatory drive onto Ophn1-deficient PL-mPFC PV interneurons, leading to hyperactivity in this region. Importantly, suppressing neuronal activity or RhoA/Rho-kinase signaling in the PL-mPFC reverses helpless behavior. Our results identify OPHN1 as a critical regulator of adaptive behavioral responses to stress and shed light onto the mechanistic link between OPHN1 genetic deficits, mPFC circuit dysfunction, and abnormalities in stress-related behaviors.
eTOC
Wang et al. show that deficiency of the intellectual disability gene Ophn1 enhances stress-induced helpless/depressive-like behavior. This phenotype is mediated by a diminished excitatory drive onto Ophn1-deficient parvalbumin interneurons in the prelimbic medial prefrontal cortex, leading to hyperactivity in this region. Suppressing neuronal activity or RhoA/Rho-kinase signaling reverses helpless behavior.
Graphical Abstract
INTRODUCTION
Aversive or stressful events are a prominent risk factor for behavioral problems and mood disorders (Kendler et al., 1999; McEwen and Morrison, 2013; Russo et al., 2012). However, not all people who encounter aversive/stressful events develop untoward outcomes, and more individuals show resilience and develop adaptive responses to control aversive situations. Individuals susceptible to stress adapt poorly and express inappropriate responses that can precipitate behavioral problems and/or mood disorders, including depression (Atkinson et al., 2009; Feder et al., 2009; Franklin et al., 2012; Krishnan et al., 2007; Maier, 2015; Southwick and Charney, 2012; Southwick et al., 2005). Great efforts are being devoted to understanding why some individuals are resilient to stress, while others are not. Though recent studies have begun to shed light on the environmental, epigenetic, and neurophysiological factors involved (Bagot et al., 2014; Cathomas et al., 2019; Han and Nestler, 2017; Herman, 2013; Klengel and Binder, 2015; Lammel et al., 2014; Macri, 2012; Maier, 2015; Price and Drevets, 2012; Riga et al., 2014; Wu et al., 2013), the genetic factors that underlie or promote either resilience or susceptibility to stress-induced maladaptive behavioral phenotypes remain largely elusive.
It is now well-known that individuals with intellectual disability (ID) have increased risk of developing stress-related behavioral problems and mood disorders (Baudewijns et al., 2018; Borthwick-Duffy, 1994; Embregts et al., 2010; Gacek et al., 2017; Hartley and Maclean, 2008, 2009; Hurley, 2006; Janssen et al., 2002; Scott and Havercamp, 2014). Such individuals are more often exposed to stressful/uncontrollable events in everyday life, like bullying and demanding problem-solving situations, than those without ID. Moreover, they may have less control over both minor and major daily life decisions (e.g., what to eat or where to live) (Bannerman et al., 1990; Dulin et al., 2013). In addition, people with ID typically have more difficulty effectively coping with stressful negative situations, often using maladaptive coping strategies (Hartley and Maclean, 2008, 2005; Janssen et al., 2002; Scott and Havercamp, 2014; Wayment and Zetlin, 1989). Prevalence studies estimate 10–50% of individuals with ID exhibit some sort of behavioral problems and/or mood disorders (Baudewijns et al., 2018; Bertelli et al., 2012; Borthwick-Duffy, 1994; Cooper et al., 2009; Cooper et al., 2007; Deb et al., 2001; Esbensen and Benson, 2006; Hartley and Maclean, 2009; Scott and Havercamp, 2014; Smiley et al., 2007). Frequently reported problems/symptoms include aggression, self-injury, emotional imbalance/instability, and/or helplessness/hopelessness, all of which have been linked to stress and/or lack of control (Baudewijns et al., 2018; Esbensen and Benson, 2006; Gacek et al., 2017; Hartley and Maclean, 2008, 2009; Janssen et al., 2002; Scott and Havercamp, 2014). A number of genes underlying ID with co-occurrent stress-related behavioral problems and mood disorders have been identified (Chiurazzi and Pirozzi, 2016; Hagerman et al., 2018; Mehregan et al., 2016; Morgan et al., 2008; Plummer et al., 2016). To date, however, little is known as to how perturbations in such genes contribute to or facilitate the development of stress-related behavioral problems/disorders. To gain insight, we focused on the X-linked ID gene oligophrenin-1 (OPHN1 in humans, Ophn1 in mice).
OPHN1, which encodes a Rho GTPase-activating protein (GAP), was the first identified Rho-linked ID gene (Bienvenu et al., 1997; Billuart et al., 1998). Mutations in OPHN1, located at Xp12, cause a syndromic form of X-linked ID, with affected individuals exhibiting mild to moderate/severe ID, vermis and/or hemispheric cerebellar hypoplasia, and behavioral problems (Bergmann et al., 2003; Chabrol et al., 2005; des Portes et al., 2004; Moortgat et al., 2018; Philip et al., 2003; Pirozzi et al., 2011; Santos-Reboucas et al., 2014; Schwartz et al., 2019; Tentler et al., 1999; Zanni et al., 2005). Most OPHN1 mutations identified to date have been shown, or predicted, to result in OPHN1 loss of function (Santos-Reboucas et al., 2014; Schwartz et al., 2019). Behavioral problems include hyperactivity, emotional imbalance, and intolerance to frustration, which can trigger helpless/depressive reactions and are often precipitated or exacerbated by stress (Busa et al., 2017; Moortgat et al., 2018; Philip et al., 2003; Santos-Reboucas et al., 2014; Schwartz et al., 2019; Tentler et al., 1999; Waschbusch et al., 2003; Zanni et al., 2005). The severity of the behavioral phenotypes is typically milder in females than males (Al-Owain et al., 2011; Bienvenu et al., 1997; Moortgat et al., 2018; Schwartz et al., 2019). OPHN1 is highly expressed in the brain, where it is found in neurons of all major brain regions involved in cognitive/adaptive processes, including the hippocampus and cortex (Billuart et al., 1998; Fauchereau et al., 2003; Govek et al., 2004). To date, OPHN1 function has mostly been studied in the hippocampus, with multiple studies unveiling key roles for OPHN1 in regulating hippocampal synaptic structure/function and plasticity, as well as learning and memory (Govek et al., 2004; Khelfaoui et al., 2007; Khelfaoui et al., 2014; Khelfaoui et al., 2009; Nadif Kasri et al., 2009; Nadif Kasri et al., 2011; Nakano-Kobayashi et al., 2009; Nakano-Kobayashi et al., 2014; Powell et al., 2012). Despite the stress-related behavioral problems observed in OPHN1 patients, its role in modulating maladaptive behavioral responses and resilience to stress remains unexplored.
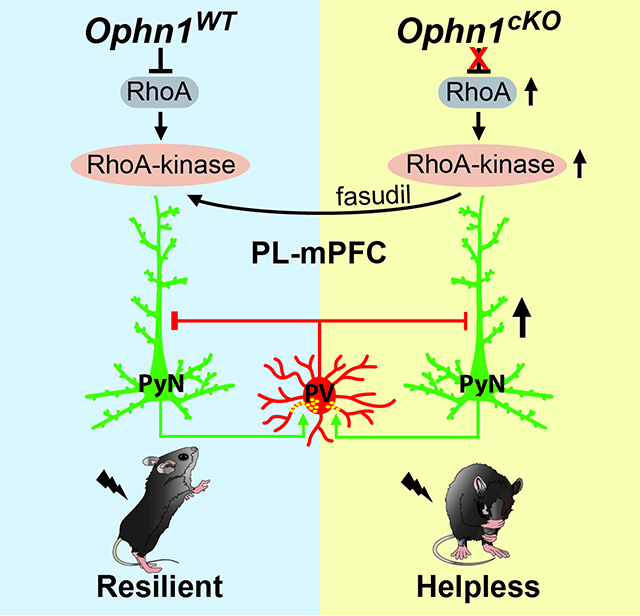
In this study, we genetically ablated Ophn1 either globally or locally in distinct brain regions/neuronal cell types in mice and assessed the effects on performance in the learned helplessness (LH) procedure (Chourbaji et al., 2005; Kim et al., 2016; Li et al., 2011; Maier and Watkins, 2005; Perova et al., 2015; Vollmayr and Henn, 2001; Wang et al., 2014) to determine OPHN1’s involvement in establishing adaptive vs. maladaptive behavioral responses to inescapable/uncontrollable stress. We found that male mice lacking Ophn1 globally or selectively in the prelimbic (PL) region of the medial prefrontal cortex (mPFC) display a marked increase in LH, a “depression-like” phenotype whereby animals show reduced escape from escapable foot shocks. In line with this, Ophn1-deficient mice also exhibited maladaptive behavioral responses in a repeated social defeat stress model. Strikingly, Ophn1 deletion exclusively in PL-mPFC parvalbumin (PV) interneurons (INs), but not somatostatin (SOM) INs or Emx1-expressing pyramidal neurons (PyNs), was sufficient to induce helpless behavior. At a cellular level, we found that excitatory synaptic transmission onto PL-mPFC PV INs lacking OPHN1 is weakened, leading to decreased spike output of inhibitory PV INs and consequently increased activity in the PL-mPFC. Importantly, suppressing PL-mPFC neuronal activity with an inhibitory DREADD (designer receptors exclusively activated by designer drugs) or by increasing the activity of PL-mPFC PV INs with an excitatory DREADD reversed the helpless behavioral phenotype of Ophn1-deficient mice. Furthermore, we uncovered that OPHN1’s effect on neuronal activity and stress-related helpless behavior depends on its inhibition of the RhoA/Rho-kinase pathway. Suppressing this pathway normalized the elevated PL-mPFC neuronal activity and alleviated the helpless behavior of Ophn1-deficient mice. Thus, our results uncover a previously unknown role of OPHN1 in regulating PL-mPFC PV IN activity required for shaping adaptive behavioral responses in the face of stress and provide mechanistic insights into how mutations in OPHN1 may contribute to the behavioral issues in OPHN1 patients.
RESULTS
Global Deletion of Ophn1 Induces Maladaptive Behavioral Responses to Stress
To investigate the role of OPHN1 in regulating stress-related behaviors, we generated a mouse line carrying a floxed allele of Ophn1 (Fig. S1A). Given that the mouse Ophn1 gene, like its human counterpart, is ubiquitously expressed with high levels in the brain (Billuart et al., 1998; Fauchereau et al., 2003; Govek et al., 2004), we crossed Ophn1-flox (Opnn1flox/+) mice with β-actin-Cre (ACTBCre) mice to achieve global Ophn1 deletion. Loss of Ophn1 expression was confirmed by Western blot of OPHN1 using total lysates from whole brain tissue or cerebral cortices of 50-day-old mice (Fig. S1B). Because the mouse Ophn1 gene is located on the X chromosome only male Ophn1 knockout (KO) mice (Ophn1flox//Y;ACTBCre/+) and their wild-type (WT) littermates (Ophn1+/Y;ACTBCre/+) were used unless otherwise specified; for simplicity, we will refer to these as ACTB-Ophn1cKO and ACTB-Ophn1WT, respectively. The ACTB-Ophn1cKO mice were viable, fertile, and without developmental disadvantages and displayed a largely intact brain architecture (Fig. S1C). While some lateral ventricle enlargement was observed (Fig. S1C), the structure and lamination of all major brain regions, including the cerebral cortex and hippocampus, appeared normal overall (Fig. S1D, E). Similar findings were previously reported for conventional Ophn1 KO mice (Khelfaoui et al., 2007). Thus, globally ablating Ophn1 does not majorly affect gross brain development.
To assess if ACTB-Ophn1cKO mice present aberrant behavioral responses to uncontrollable stress, we used the well-established LH procedure. Seven-week-old ACTB-Ophn1cKO mice and their WT littermates were subjected to two successive induction sessions of unpredictable and inescapable foot shocks; then, their coping capabilities were evaluated in a testing session of escapable foot shocks (Fig. S1F). Escape latency and number of escape failures—the most commonly reported indices of LH (Chourbaji et al., 2005; Li et al., 2011; Perova et al., 2015; Wang et al., 2014)—were measured. The mice were classified as learned helpless or resilient by k-means clustering analysis of the number of escape failures and latency to escape, with helpless mice having significantly more failures and longer escape latencies than resilient mice (see Methods; Fig. S1G). Consistent with previous studies (Chourbaji et al., 2005; Perova et al., 2015; Wang et al., 2014), we found that ~24% of ACTB-Ophn1WT mice displayed LH (Fig. 1A). Strikingly, 74% of ACTB-Ophn1cKO mice exhibited LH (Fig. 1A), implying that Ophn1 deficiency promotes helpless behavior in the face of stress.
Figure 1. Ophn1 Deficiency Induces Maladaptive Behavioral Responses to Stress.

(A) Learned helplessness (LH) test of ACTB-Ophn1WT (WT) and ACTB-Ophn1cKO (cKO) male mice. Escape latencies and number of failures are presented. n = 21–23 mice per group; Mann-Whitney test.
(B) Testing session only (see Methods) of the LH test of ACTB-Ophn1WT and ACTB-Ophn1cKO male mice. n = 13 mice per group; Mann-Whitney test.
(C) LH test of Ophn1+/+;ACTBCre/+ (ACTB-Ophn1-WT), Ophn1flox/+;ACTBCre/+ (ACTB-Ophn1-HET), and Ophn1flox/ flox;ACTBCre/+ (ACTB-Ophn1-HOM) female mice. n = 17–22 mice per group; Kruskal-Wallis with Dunn’s post-hoc test.
(D) Social interaction test following 5-d social defeat stress of ACTB-Ophn1WT and ACTB-Ophn1cKO male mice. Left: Schematic of social interaction arena. Right: Representative movement tracks of ACTB-Ophn1WT and ACTB-Ophn1cKO male mice in either the absence or presence of a social defeat target (i.e., CD1 mouse ✿).
(E) Quantification of time spent in interaction zone by non-defeated ACTB-Ophn1WT and ACTB-Ophn1cKO mice. n = 12–13 mice per group; Two-way RM ANOVA (genotype F(1, 23) = 0.1435, p = 0.7) with Bonferroni post-hoc test.
(F, G) Quantification of time spent in interaction zone (F) and corner zone (G) by defeated ACTB-Ophn1WT and ACTB-Ophn1cKO mice. n = 12–16 mice per group; Two-way RM ANOVA (in F, genotype x target interaction F(1, 26) = 7.85, p = 0.0095; in G, genotype x target interaction F(1, 26) = 4.401, p = 0.046) with Bonferroni post-hoc test.
(H) Quantification of total distance traveled by ACTB-Ophn1WT and ACTB-Ophn1cKO mice following social defeat stress. n = 12–16 mice per group; Two-way RM ANOVA (genotype F(1, 26) = 0.2712, p = 0.6) with Bonferroni post-hoc test.
For A-C, data are presented with the median and interquartile ranges shown in red. Closed and open circles represent LH and resilient mice, respectively. Percentage of LH+ mice is indicated at the bottom of each genotype. For E-H, data are mean ± SEM. n.s., p ≥ 0.05, **p < 0.01, ***p < 0.001, #p < 0.05, ##p < 0.01.
See also Figure S1.
To verify that the observed behavioral phenotype was not an indirect consequence of a lack of pain sensitivity in ACTB-Ophn1cKO mice, we subjected the animals to the conventional hot plate test (Bannon, 2001; Shin et al., 2015). We saw no difference in hotplate response latency between ACTB-Ophn1cKO mice and their WT littermates (Fig. S1H), indicating that ACTB-Ophn1cKO mice do not have impaired pain sensitivity. We also validated that ACTB-Ophn1cKO mice do not exhibit defective/decreased locomotor activity in an open field test (Wang et al., 2014) (Fig. S1I). In fact, we found that the mutant mice traveled over a longer distance and at a higher speed than their WT littermates (Fig. S1I). Moreover, while a previous study reported a high occurrence of perseverance behaviors in conventional Ophn1 KO mice using a Y-maze spatial working memory test (Zhang et al., 2017), we observed that ACTB-Ophn1cKO mice displaying helpless behavior typically remain in a corner of the cage and move very little, similar to their WT littermates exhibiting helpless behavior (Videos S1–3). Thus, the behavioral phenotype observed in ACTB-Ophn1cKO mice was not secondary to uncoordinated/indirect movements or perseverative behaviors. We detected no substantial differences in the frequency of visits, time spent, or distance traveled in the center area (the anxiety zone) of the open field between the two genotypes (Fig. S1I), suggesting that naïve ACTB-Ophn1cKO mice do not exhibit increased anxiety-like behavior. To corroborate this, we performed elevated plus-maze and light/dark exploration tests (File et al., 2005; Walsh et al., 2017). In both tests, the performance of ACTB-Ophn1cKO mice was comparable to that of ACTB-Ophn1WT mice (Fig. S1J, K), indicating that under basal conditions, ACTB-Ophn1cKO mice were not more anxious than their control littermates. Finally, we verified that the observed behavioral phenotype was not due to learning deficits in the avoidance task. We subjected the animals to only the testing session (i.e., without preceding unpredictable/inescapable foot shocks). We found that ACTB-Ophn1cKO mice were indistinguishable from their WT littermates with respect to both escape latencies and failures (Fig. 1B), indicating that ACTB-Ophn1cKO mice are principally capable of learning and performing the avoidance task. Thus, the observed increase in LH likely reflects a stress-coping deficit in male ACTB-Ophn1cKO mice.
As aforementioned, mutations in the human OPHN1 gene cause a syndromic form of X-linked ID, with behavioral alterations being milder in females. Hence, we also investigated if Ophn1 deletion in female mice impacts their behavioral responses to stress. We subjected Ophn1 heterozygous (Ophn1flox/+;ACTBCre/+) and homozygous (Ophn1flox/flox;ACTBCre/+) female KO mice and their WT (Ophn1+/+;ACTBCre/+) littermates to the LH procedure. We found that the behavioral responses of Ophn1 heterozygous and homozygous female KO mice were not significantly different from those of their control littermates, with respect to both escape latencies and failures. Only 17% and 22.7% of Ophn1flox/+;ACTBCre/+ and Ophn1flox/flox;ACTBCre/+ mice, respectively, displayed LH, which was not significantly different from the 23.5% seen in the control group (Fig. 1C). Thus, in line with observations in humans, maladaptive behavioral responses are blunted/absent in female Ophn1-deficient mice.
To further reinforce our findings that Ophn1 deficiency in male mice leads to increased stress susceptibility, we implemented an independent animal stress model: the repeated social defeat stress model (Golden et al., 2011; Shin et al., 2015) (Fig. 1D). Male ACTB-Ophn1cKO mice and their control littermates were exposed to a larger, more aggressive male CD1 mouse for 5 days as a social defeat stressor. The mice were then subjected to a social interaction behavior test in an open area with an interaction zone and a corner zone 24 hours after the fifth defeat episode (Fig. 1D). Under non-defeated conditions, baseline social interactions of ACTB-Ophn1cKO mice were similar to those of their WT littermates (Fig. 1E). However, after 5 days of defeat stressor exposure, the ACTB-Ophn1cKO mice spent significantly more time in the corner zone than their WT littermates, maximizing distance from the unfamiliar, aggressive CD1 mouse (Fig. 1F, G). No difference in total distance traveled was observed between the two genotypes (Fig. 1H). These data reveal a strong social avoidance behavior in Ophn1-deficient mice. Of note, this stress model can only be applied using male C57BL/6 mice, mainly because innate aggression of male toward female mice is limited (Beery and Zucker, 2011).
Together, our results from two different stress models indicate that Ophn1-deficient male mice are more susceptible to stressful events and unveil a critical role for OPHN1 in regulating stress-related behaviors.
Ophn1 Deletion in the PL-mPFC and in PV INs Promotes Stress-Induced Helpless Behavior
The Ophn1 gene is broadly expressed in the central nervous system, and the protein is present in both excitatory and inhibitory neurons of all major brain regions (Billuart et al., 1998; Fauchereau et al., 2003; Govek et al., 2004; Powell et al., 2012; Redolfi et al., 2016). Thus, we next sought to determine which brain region(s) and which neuronal cell type(s) contribute to the emergence of maladaptive behavioral responses to stress in ACTB-Ophn1cKO mice.
We initially focused on the mPFC and hippocampal CA1, as both brain regions have been implicated in stress-related behaviors (Amat et al., 2005; Arnsten, 2009; Dias-Ferreira et al., 2009; Franklin et al., 2012; Ghosal et al., 2017; Goldwater et al., 2009; McGuire and Botvinick, 2010; Perova et al., 2015; Schmidt et al., 2010; Seese et al., 2013; Shrestha et al., 2015; Wang et al., 2014; Warden et al., 2012; Yuen et al., 2011) and the OPHN1 protein is abundantly expressed in both these regions (Govek et al., 2004). In particular, we examined if deleting Ophn1 specifically in the PL-mPFC and/or hippocampal CA1 is sufficient to promote helpless behavior. We began with the PL area of the mPFC given that this subregion reportedly plays a role in mediating behavioral responses to stress in the LH procedure (Perova et al., 2015; Wang et al., 2014). An adeno-associated virus (AAV) expressing Cre-GFP or control GFP was bilaterally injected in the PL-mPFC or hippocampal CA1 of Ophn1flox/Y mice at postnatal day 21 (P21) (Fig. 2A, D). About 4 weeks later, the mice were subjected to the LH procedure. OPHN1 depletion in AAV-Cre-GFP-infected cells in the respective areas was confirmed by immunohistochemistry (Fig. 2B, E). Intriguingly, Ophn1 ablation in the PL-mPFC resulted in a significant increase in escape latency and number of escape failures (Fig. 2C). Indeed, 50% of Ophn1flox/Y mice injected with AAV-Cre-GFP displayed LH, while only 24% of Ophn1flox/Y mice injected with control GFP-expressing virus exhibited LH. The behavioral defect significantly correlated with the number of Cre-GFP-expressing neurons in the PL-mPFC (Fig. S2A, B), demonstrating the specificity and potency of our manipulation. In contrast, Ophn1 ablation in hippocampal CA1 did not affect the animals’ escape latency or failures (Fig. 2F). Ablating Ophn1 in either the PL-mPFC or hippocampal CA1 did not affect locomotion or anxiety-like behavior (Fig. S2C–H). Thus, deleting Ophn1 in the PL-mPFC, but not hippocampal CA1, promotes LH.
Figure 2. Ophn1 Deletion in PL-mPFC but not Hippocampal CA1 or IL-mPFC Promotes LH.

(A, D, G) Representative images showing expression of GFP or Cre-GFP in the prelimbic area of the medial prefrontal cortex (PL-mPFC) (A), hippocampal CA1 (D), and infralimbic area of the mPFC (IL-mPFC) (G) of Ophn1flox/Y mice injected with AAV-GFP and AAV-Cre-GFP, respectively. Scale bars, 100 μm.
(B, E, H) Representative images of neurons in the PL-mPFC (B), hippocampal CA1 (E), and IL-mPFC (H) of Ophn1flox/Y mice injected with AAV-GFP or AAV-Cre-GFP with immunostaining for OPHN1. Dashed-line circles depict randomly selected representative cells. Scale bars, 10 μm. (C, F, I) LH test of Ophn1flox//Y mice in which the PL-mPFC (C), hippocampal CA1 (F), or IL-mPFC (H) was injected with AAV-GFP or AAV-Cre-GFP. n = 13–24 mice per group; Mann-Whitney test. Data are presented with the median and interquartile ranges shown in red. Closed and open circles represent LH and resilient mice, respectively. Percentage of LH+ mice is indicated at the bottom of each group. n.s., p ≥ 0.05, **p < 0.01.
See also Figure S2.
It is worth noting that separate, often opposite roles have been reported for the PL-mPFC and the adjacent infralimbic area of the mPFC (IL-mPFC) in affective behaviors, particularly fear expression/extinction (Lammel et al., 2014; Mukherjee and Caroni, 2018; Sierra-Mercado et al., 2011; Vidal-Gonzalez et al., 2006) and more recently, mood disorders (Capuzzo and Floresco, 2020; Hamani et al., 2010a; McKlveen et al., 2015). Hence, we also examined the effect of Ophn1 deletion in the IL-mPFC on animals’ behavioral responses to stress, using a similar approach as described above. We found that Ophn1 ablation in the IL-mPFC (Fig. 2G, H) did not significantly alter the animals’ escape latency and failures (Fig. 2I). Thus, in contrast to the PL-mPFC, Ophn1 deletion in the IL-mPFC does not promote LH.
Regarding the neuronal cell type(s) involved, we first examined the impact of selective Ophn1 deletion in two major interneuron (IN) subtypes—PV and SOM INs—and in Emx1-expressing PyNs, on animals’ behavioral responses to stress. OPHN1 is expressed in all three cell types (Fig. 3A, C, E). To selectively ablate Ophn1 in PV-, SOM-, or Emx1-expressing neurons, we crossed Ophn1flox/+ mice with PV-Cre, SOM-Cre, or Emx1-Cre mice, respectively. Ophn1flox/Y;PValbCre/+ (PV-Ophn1cKO), Ophn1flox/Y;SstCre/+ (SOM-Ophn1cKO), and Ophn1flox/Y;Emx1Cre/+ (Emx1-Ophn1cKO) mutant offspring were all viable and displayed no developmental disadvantages. Also, their brain architecture was overall intact and appeared similar to that of their corresponding WT littermates (PV-Ophn1WT, SOM-Ophn1WT, and Emx1-Ophn1WT) (data not shown). Immunofluorescence staining confirmed that OPHN1 expression was depleted in PV+, SOM+, and Emx1+ neurons (Fig. 3A, C, E).
Figure 3. Ophn1 Deletion in PV INs, but not SOM INs or Emx1-expressing PyNs, Promotes LH.

(A, C, E) Representative images of neurons in the PL-mPFC of P50 PV-Ophn1WT and PV-Ophn1cKO (A), P50 SOM-Ophn1WT and SOM-Ophn1cKO (C), and P50 Emx1-Ophn1WT and Emx1-Ophn1cKO (E) mice with immunostaining for OPHN1 and parvalbumin (PV) (A), somatostatin (SOM) (C), or neurogranin (NG) (E). Scale bars, 10 μm.
(B, D, F) LH test of PV-Ophn1WT and PV-Ophn1cKO (B), SOM-Ophn1WT and SOM-Ophn1cKO (D), and Emx1-Ophn1WT and Emx1-Ophn1cKO (F) mice. n = 17–43 mice per group; Mann-Whitney test.
All data are presented with the median and interquartile ranges shown in red. Closed and open circles represent LH and resilient mice, respectively. Percentage of LH+ mice is indicated at the bottom of each genotype. n.s., p ≥ 0.05, ***p < 0.001.
See also Figure S3.
Seven-week-old PV-Ophn1cKO, SOM-Ophn1cKO, and Emx1-Ophn1cKO mutant mice and their WT littermates were then subjected to the LH procedure. Interestingly, similar to the ACTB-Ophn1cKO mice, PV-Ophn1cKO mice exhibited longer escape latencies and more escape failures than their control littermates (Fig. 3B). At least 69% of the PV-Ophn1cKO mice displayed LH, while only 28% of the PV-Ophn1WT mice reached criteria for LH. In contrast, the behavioral responses of SOM-Ophn1cKO and Emx1-Ophn1cKO mice were indistinguishable from those of their control littermates, with respect to both escape latencies and failures (Fig. 3D, F). None of the mutant mice exhibited altered locomotion or anxiety-like behavior compared to their WT littermates (Fig. S3A–I). These data show that Ophn1 deletion in PV INs, but not SOM INs or Emx1-expressing PyNs, promotes helpless behavior in the face of stress.
Ophn1 Deletion Exclusively in PL-mPFC PV INs is Sufficient to Induce LH
Our above findings demonstrate a key role for OPHN1 in the PL-mPFC and PV INs in mediating adaptive behavioral responses to stress. Yet in these experiments, Ophn1 was deleted either in multiple cell types in the PL-mPFC or in all PV INs throughout the brain. To further investigate if Ophn1 ablation specifically in PL-mPFC PV INs is sufficient to promote LH, we developed a strategy that enables selective deletion of Ophn1 in PV INs in the PL-mPFC. In this strategy, we exploited both the Cre-loxP and Flp-FRT recombination systems by generating Ophn1flox/Y;PV-Flp mice, in which the flippase (Flp) recombinase is selectively expressed in PV INs in an Ophn1flox/Y genetic background. We then bilaterally injected the PL-mPFC of these mice (at P21) with an AAV we engineered to express Cre-GFP in a Flp-dependent manner (AAV-fDIO-Cre-GFP) (Fig. 4A, B). Injecting this virus, but not control virus (AAV-fDIO-mCherry), into the PL area of Ophn1flox/Y;PV-Flp mice led to OPHN1 depletion specifically in PV INs in the PL-mPFC (Fig. 4C, D).
Figure 4. Ophn1 Deletion Selectively in PV INs in PL-mPFC is Sufficient to Induce LH.

(A) AAV vector expressing flippase-dependent Cre-GFP (AAV-fDIO-Cre-GFP) for gene deletion with spatial and cell-type specificity.
(B) Schematic for depleting Ophn1 in PV INs in the PL-mPFC of Ophn1flox/Y;PV-Flp mice (PL, prelimbic; IL, infralimbic; DP, dorsal peduncular; ACd, dorsal anterior cingulate).
(C) Representative images of neurons infected with control AAV-fDIO-mCherry or AAV-fDIO-Cre-GFP in the PL-mPFC. Scale bar, 200 μm.
(D) Representative images showing selective depletion of OPHN1 in PL-mPFC PV INs of Ophn1flox/Y;PV-Flp mice injected with AAV-fDIO-Cre-GFP but not AAV-fDIO-mCherry. Scale bar, 10 μm.
(E) LH test of Ophn1flox/Y;PV-Flp mice in which the PL-mPFC was injected with AAV-fDIO-mCherry or AAV-fDIO-Cre-GFP. n = 19–25 mice per group; Mann-Whitney test. Data are presented with the median and interquartile ranges shown in red. Closed and open circles represent LH and resilient mice, respectively. Percentage of LH+ mice is indicated at the bottom of each group. **p < 0.01.
See also Figure S4.
About 4 weeks post-AAV injection, the mice were subjected to the LH procedure. Strikingly, we found that deleting Ophn1 selectively in PL-mPFC PV INs resulted in a significant increase in escape latency and number of escape failures: 56% of Ophn1flox/Y;PV-Flp mice injected with AAV-fDIO-Cre-GFP virus displayed LH as opposed to just 21% of the control AAV-fDIO-mCherry-injected mice (Fig. 4E). The behavioral defect significantly correlated with the number of GFP-expressing PV INs in the PL-mPFC (Fig. S4A, B). Ablating Ophn1 from PL-mPFC PV INs did not affect locomotion or anxiety-like behaviors (Fig. S4C–E). Thus, deleting Ophn1 exclusively in PV INs in the PL-mPFC is sufficient to promote LH. These data also imply that Ophn1 inactivation during adolescence into adulthood is sufficient to induce helpless behavior and suggests that a circuitry defect is a major contributing factor to the stress-induced helpless behavioral phenotype.
Excitatory Drive onto PL-mPFC PV INs Lacking OPHN1 is Reduced
We next investigated how loss of OPHN1 affects mPFC PV IN function, leading to maladaptive behavioral responses to stress. We first examined if excitatory and/or inhibitory synaptic transmission onto Ophn1-deleted PL-mPFC PV INs was altered. Before doing so, we confirmed by Sholl analysis that Ophn1 deletion does not affect the gross morphology of PL-mPFC PV INs (Fig. S5A–C). To identify PV-expressing cells in electrophysiological measurements, we generated PV-Ophn1cKO;Ai14 and PV-Ophn1WT;Ai14 mice, in which the red fluorescent protein tdTomato is expressed in PV INs following Cre-mediated recombination in the respective genetic backgrounds (Madisen et al., 2010).
Acute brain slices were prepared from both genotypes and whole-cell patch-clamp recordings of layer II/III PV INs in the PL-mPFC region were performed. Miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs, respectively) were recorded. We found that both the frequency and amplitude of mEPSCs onto PL-mPFC PV INs were reduced in PV-Ophn1cKO;Ai14 mice compared to PV-Ophn1WT;Ai14 mice (Fig. 5A, B), while no significant difference in mIPSCs onto these neurons was observed between the two genotypes (Fig. 5C, D). These data indicate that the strength of excitatory synapses onto PV INs in the PL-mPFC is selectively weakened in PV-Ophn1cKO mice. Of note, the resting membrane potential and input resistance of PL-mPFC PV INs were not different between the two genotypes (Fig. S5D, E). Similar results were obtained when recordings were performed on PV INs in the PL-mPFC of Ophn1flox/Y;PV-Flp mice injected with AAV-fDIO-Cre-GFP. Both the frequency and amplitude of mEPSCs onto PL-mPFC PV INs were reduced in AAV-fDIO-Cre-GFP-injected mice compared to AAV-fDIO-mCherry-injected mice (Fig. 5E, F). Contrary to PL-mPFC PV INs, Ophn1 deletion in PL-mPFC SOM INs did not affect the frequency or amplitude of mEPSCs onto such INs (Fig. 5G, H).
Figure 5. Reduced Excitatory Drive onto PL-mPFC PV INs Lacking OPHN1.

(A, C) Representative traces of mEPSCs (A) and mIPSCs (C) recorded from layer (L) II/III PV INs in the PL-mPFC of PV-Ophn1WT;Ai14 (WT) and PV-Ophn1cKO;Ai14 (cKO) mice.
(B, D) Quantification of mEPSC (B) and mIPSC (D) amplitude and frequency. n = 23–41 cells from 3–5 mice for each genotype; Student’s t-test.
(E) Representative traces of mEPSCs recorded from LII/III PV INs in the PL-mPFC region of Ophn1flox/Y;PV-Flp mice injected with AAV-fDIO-mCherry or AAV-fDIO-Cre-GFP.
(F) Quantification of mEPSC amplitude and frequency. n = 22–23 cells from 3 mice for each condition; Student’s t-test.
(G) Representative traces of mEPSCs recorded from LII/III SOM INs in the PL-mPFC region of SOM-Ophn1WT;Ai14 (WT) or SOM-Ophn1cKO;Ai14 (cKO) mice.
(H) Quantification of mEPSC amplitude and frequency. n = 26–27 cells from 3 mice for each genotype; Student’s t-test.
(I) Schematic of the recording configuration for J and K.
(J) Representative traces showing averaged uEPSCs (30 trials) recorded in a PV IN after presynaptic spike from PyN in LII/III of the PL-mPFC of PV-Ophn1WT;Ai14 and PV-Ophn1cKO;Ai14 mice.
(K) Quantification of uEPSC amplitude. n = 8–12 pairs from 5–7 mice for each genotype; Student’s t-test.
(L) Schematic of the recording configuration for M and N.
(M) Representative traces showing averaged uIPSCs (30 trials) recorded in a PyN after presynaptic spike from PV IN in LII/III of the PL-mPFC of PV-Ophn1WT;Ai14 and PV-Ophn1cKO;Ai14 mice.
(N) Quantification of uIPSC amplitude. n = 10–14 pairs from 6–8 mice for each genotype; Student’s t-test.
Data are mean ± SEM. n.s., p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
See also Figures S5 and S6.
To corroborate and extend these findings, we examined the responses of Ophn1-deficient and WT PV INs to excitatory synaptic inputs from neighboring PyNs (Fig. 5I). We simultaneously recorded pairs of adjacent PyNs and PV INs in layers II/III of the PL-mPFC of PV-Ophn1cKO;Ai14 and PV-Ophn1WT;Ai14 mice, in which PyNs were stimulated to generate a single action potential (AP). While no difference in connection probability from PyNs onto PV INs was observed between the two genotypes (WT, 8 of 43 pairs connected; cKO, 15 of 68 connected; Fisher’s exact test, p > 0.05), we found that the average amplitude of unitary EPSCs (uEPSCs) in PV INs of PV-Ophn1cKO;Ai14 mice was markedly smaller than that of PV-Ophn1WT;Ai14 mice (Fig. 5J, K). These data show that excitatory synaptic transmission from PyNs onto PV INs in the PL-mPFC is impaired in PV-Ophn1cKO mice and further substantiate our above findings.
We also examined the responses of PyNs to inhibitory synaptic inputs from neighboring Ophn1-deficient or WT PV INs by performing paired recordings in layers II/III of the PL-mPFC of the two respective mouse genotypes, in which PV INs were stimulated to generate a single AP (Fig. 5L). No differences were observed between the PV-Ophn1cKO;Ai14 and PV-Ophn1WT;Ai14 groups with respect to both connection probability from PV INs onto PyNs (WT, 10 of 39 pairs connected; cKO, 16 of 68 connected; Fisher’s exact test, p > 0.05) and the average amplitude of uIPSCs in PyNs (Fig. 5M, N). These results indicate that GABAergic synaptic transmission from PV INs onto PyNs is intact in PV-Ophn1cKO mice at least when PV INs are stimulated strongly enough to elicit postsynaptic responses. They further imply that OPHN1 depletion in PL-mPFC PV INs does not directly impact their presynaptic function.
Our above findings reveal that Ophn1 deficiency in PL-mPFC PV INs leads to a decrease in excitatory synaptic transmission onto such INs. To gain further insight into the underlying mechanism(s), we examined if the observed decrease was the result of pre- and/or post-synaptic alterations. We first measured the paired-pulse ratio (PPR), an indicator of presynaptic release probability, of evoked EPSCs onto PV INs in the PL-mPFC of PV-Ophn1cKO;Ai14 and PV-Ophn1WT;Ai14 mice. No difference in PPR of evoked EPSCs recorded from PL-mPFC PV INs was seen between the two genotypes (Fig. S5F, G). We then assessed whether the number of excitatory synapses on Ophn1-deficient PV INs was altered. Brain sections from PV-Ophn1cKO;Ai14 and PV-Ophn1WT;Ai14 mice were coimmunostained with antibodies to PV and post- and pre-synaptic markers for excitatory synapses, PSD-95 and VGLUT1, respectively, and the density of PSD-95 puncta colocalizing with VGLUT1 on PV IN cell bodies and dendritic processes in the PL-mPFC was quantified. We found that the density of PSD-95 puncta colocalizing with VGLUT1 was significantly reduced on both the cell body and dendritic processes in the PV-Ophn1cKO;Ai14 group compared to that of the control group (Fig. S5H–J). Thus, the observed decrease in excitatory drive onto Ophn1-deficient PL-mPFC PV INs is likely the result of a reduced number of excitatory synapses on such neurons. Consistent with our electrophysiological data (Fig. 5G, H), the density of PSD-95 puncta colocalizing with VGLUT1 on the cell body and dendritic processes of SOM INs was not significantly different between the SOM-Ophn1cKO;Ai14 and SOM-Ophn1WT;Ai14 groups (Fig. S5K–M).
Ophn1 Deficiency in PV INs Leads to Enhanced Neuronal Activity in the PL-mPFC
Next, we further examined the consequences of reduced excitatory drive onto Ophn1-deficient PL-mPFC PV INs. While Ophn1 deficiency does not appear to directly affect the presynaptic function of PL-mPFC PV INs, we reasoned that the decrease in excitatory drive onto these neurons could affect the activity/output of these cells and consequently alter the activity of neighboring PyNs. To test this hypothesis, we measured the spontaneous firing rate of PV INs in the PL-mPFC of PV-Ophn1cKO;Ai14 and PV-Ophn1WT;Ai14 mice and found that it was significantly reduced in PV-Ophn1cKO;Ai14 mice compared to PV-Ophn1WT;Ai14 mice (Fig. 6A, B), indicating that Ophn1-deficient PV INs are less active.
Figure 6. Ophn1 Deficiency Leads to Less Active PV INs and Increased Activity in Neighboring PyNs in the mPFC.

(A) Representative traces of spontaneous spiking activity in PV INs in the PL-mPFC of PV-Ophn1WT;Ai14 (WT) and PV-Ophn1cKO;Ai14 (cKO) mice.
(B) Quantification of spontaneous spiking frequency. n = 14–17 neurons from 4 mice for each genotype; Student’s t-test.
(C) Representative traces of sEPSCs recorded from LII/III PyNs in the PL-mPFC of PV-Ophn1WT;Ai14 and PV-Ophn1cKO;Ai14 mice.
(D) Quantification of sEPSC amplitude and frequency. n = 24–28 cells from 3 mice for each genotype; Student’s t-test.
(E) Representative traces of sEPSCs recorded from LII/III PyNs in the PL-mPFC of Ophn1flox/Y;PV-Flp mice injected with AAV-fDIO-mCherry or AAV-fDIO-Cre-GFP.
(F) Quantification of sEPSC amplitude and frequency. n = 21–26 cells from 3 mice for each condition; Student’s t-test.
(G) Representative traces of sEPSCs recorded from LII/III PyNs in the PL-mPFC of ACTB-Ophn1WT (WT) and ACTB-Ophn1cKO (cKO) mice.
(H) Quantification of sEPSC amplitude and frequency. n = 21–26 cells from 3 mice for each genotype; Student’s t-test.
Data are mean ± SEM. n.s., p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
See also Figure S6.
We then asked if the decrease in PV IN activity leads to enhanced activity in neighboring PyNs. We performed whole-cell recordings to measure spontaneous EPSCs (sEPSCs) in PyNs in the PL-mPFC of PV-Ophn1cKO;Ai14 and control PV-Ophn1WT;Ai14 mice and found that the frequency (but not amplitude) of sEPSCs was significantly increased in PV-Ophn1cKO;Ai14 mice compared to control littermates (Fig. 6C, D). Similarly, an increase in sEPSC frequency was observed in AAV-fDIO-Cre-GFP-injected Ophn1flox/Y;PV-Flp mice (Fig. 6E, F), supporting the idea that ablating Ophn1 in PL-mPFC PV INs leads to increased PyN activity. In accordance with our findings that Ophn1 deficiency in SOM INs or Emx1-expressing PyNs did not affect the animals’ behavioral responses to stress, no difference in the frequency or amplitude of sEPSCs in PL-mPFC PyNs was observed between the SOM-Ophn1cKO;Ai14 and SOM-Ophn1WT;Ai14 groups or the Emx1-Ophn1cKO and Emx1-Ophn1WT groups (Fig. S6A–D). Finally, we also measured sEPSCs in PyNs in the PL-mPFC of ACTB-Ophn1cKO mice, which like PV-Ophn1cKO mice display maladaptive behavioral responses to stress. As seen in PV-Ophn1cKO mice, the frequency (but not amplitude) of sEPSCs was significantly increased in ACTB-Ophn1cKO mice compared to control littermates (Fig. 6G, H). Together, these data indicate that Ophn1 deficiency dampens excitatory synaptic transmission onto PL-mPFC PV INs, leading to less active PV INs and consequently increased PL-mPFC neuronal activity.
Normalizing PL-mPFC Neuronal Activity Alleviates Helpless Behavior in Ophn1-Deficient Mice
Our above findings raise the question: is the increase in PL-mPFC neuronal activity resulting from less active PV INs responsible for the helpless behavior observed in Ophn1-deficient mice? To address this, we first sought to increase the activity of PV INs in the PL-mPFC of PV-Ophn1cKO mice and then test whether such manipulation could alleviate helpless behavior. To increase PV IN activity, we took a chemogenetic approach utilizing the excitatory DREADD hM3D(Gq), a modified form of the human Gq-coupled receptor that can be activated by the clozapine metabolite clozapine-N-oxide (CNO) (Pei et al., 2010; Rogan and Roth, 2011; Smith et al., 2016). We bilaterally injected an AAV that expresses in a Cre-dependent manner hM3D(Gq) tagged with the fluorescent protein mCherry (AAV-DIO-hM3D(Gq)-mCherry) in the PL-mPFC of P21 PV-Ophn1cKO mice (Fig. 7A, S7A). We confirmed functional DREADD hM3D(Gq) expression by whole-cell recordings of mCherry-positive PL-mPFC PV INs in brain slices prepared from PV-Ophn1cKO mice 4–5 weeks after AAV-DIO-hM3D(Gq)-mCherry injection. Local CNO puff application increased spiking activity in all of the mCherry-positive PV cells tested (n=6 cells from 3 animals) (Fig. 7B).
Figure 7. Normalizing PL-mPFC Neuronal Activity Alleviates Helpless Behavior in ACTB-Ophn1cKO mice.

(A) Representative image showing expression of hM3D(Gq)-mCherry in the PL-mPFC of PV-Ophn1cKO mice injected with Cre-dependent excitatory DREADD (AAV-DIO-hM3D(Gq)-mCherry). Scale bar, 200 μm.
(B) Representative traces of spontaneous spikes of hM3D(Gq)-mCherry-positive PV INs (mCherry+) without or with (yellow box) focal/puff application of CNO (20 μM).
(C) Schematic of experimental procedure for D.
(D) LH test of PV-Ophn1cKO (cKO) mice in which the PL-mPFC was injected with AAV-DIO-hM3D(Gq)-mCherry or AAV-DIO-mCherry and subsequently treated with saline or CNO. n = 11–14 mice per group; Kruskal-Wallis with Dunn’s post-hoc test.
(E) Representative image showing expression of hM4Di-mCherry in the PL-mPFC of ACTB-Ophn1cKO mice injected with Cre-dependent inhibitory DREADD (AAV-DIO-hM4Di-mCherry).
Scale bar, 200 μm.
(F) Representative traces of spontaneous spikes of hM4Di-mCherry-positive neurons (mCherry+) without or with (yellow box) focal/puff application of CNO (20 μM).
(G) Schematic of experimental procedure for H.
(H) LH test of ACTB-Ophn1cKO (cKO) mice in which the PL-mPFC was injected with AAV-DIO-hM4Di-mCherry or AAV-DIO-mCherry and subsequently treated with saline or CNO. n = 11–24 mice per group; Kruskal-Wallis with Dunn’s post-hoc test.
For D, H, data are presented with the median and interquartile ranges shown in red. Closed and open circles represent LH and resilient mice, respectively. Percentage of LH+ mice is indicated at the bottom of each group. *p < 0.05, **p < 0.01.
See also Figure S7.
To determine if increasing the activity of PV INs could mitigate/reverse the helpless phenotype of PV-Ophn1cKO mice, we administrated CNO or saline intraperitoneally to AAV-DIO-hM3D(Gq)-mCherry-injected PV-Ophn1cKO mice during the LH procedure (Fig. 7C). Strikingly, we found that escape latency and number of escape failures were significantly reduced in the CNO-treated group compared to the saline-treated group (Fig. 7D). While 63.6% of the saline-treated group displayed LH, only 14.3% of the CNO-treated group exhibited LH (Fig. 7D). Administering CNO to PV-Ophn1cKO mice injected with control AAV-DIO-mCherry did not change the helpless behavior of the animals (Fig. 7D). Thus, increasing the activity of Ophn1-deficient PL-mPFC PV INs is sufficient to alleviate the stress-induced helpless behavior of PV-Ophn1cKO mice.
Next, we examined if suppressing the elevated PL-mPFC neuronal activity with the inhibitory DREADD hM4Di could alleviate the helpless phenotype of ACTB-Ophn1cKO mice. hM4Di is a modified form of the human M4 muscarinic (hM4) receptor that can be activated by CNO (Pei et al., 2010; Rogan and Roth, 2011; Smith et al., 2016). Similar as above, we bilaterally injected AAV-DIO-hM4Di-mCherry in the PL-mPFC of P21 ACTB-Ophn1cKO mice (Fig. 7E, S7B) and first confirmed functional DREADD hM4Di expression by whole-cell recordings of mCherry-positive PL-mPFC neurons in brain slices from ACTB-Ophn1cKO mice 4–5 weeks after AAV-DIO-hM4Di-mCherry injection. Of note, we found by coimmunostaining for the PyN marker neurogranin that 88% of the mCherry-positive cells were PyNs (Fig. S7B, C). Local CNO puff application suppressed spiking activity in the majority of mCherry-positive PyNs (5 of 6 cells from 3 animals) (Fig. 7F). No effect of CNO was observed in mCherry-negative cells (Fig. S7D). We then tested if suppressing neuronal activity in the PL-mPFC of ACTB-Ophn1cKO mice could reverse their helpless phenotype by delivering CNO or saline intraperitoneally to AAV-DIO-hM4Di-mCherry-injected ACTB-Ophn1cKO mice during the LH procedure (Fig. 7G). The escape latency and number of escape failures were significantly reduced in the CNO-treated group compared to the saline-treated group (Fig. 7H). While 77% of the saline-treated group displayed LH, only 17% of the CNO-treated group reached criteria of LH (Fig. 7H). Administering CNO to ACTB-Ophn1cKO mice injected with AAV-DIO-mCherry did not change their helpless behavior (Fig. 7H). Notably, in contrast to administering CNO 30 min prior to the induction sessions of the LH procedure, CNO administration to ACTB-Ophn1cKO mice just before the testing session did not reverse the helpless behavioral phenotype of such animals (Fig. S7E, F), implying that suppressing PL-mPFC activity prevents the emergence of helpless behavior.
Together, these data reveal that the increased PL-mPFC neuronal activity resulting from less active PV INs is responsible for the helpless behavior seen in Ophn1-deficient mice.
OPHN1’s Effect on PL-mPFC Neuronal Activity and Stress-Related Behavior Depends on its Inhibition of the RhoA/Rho-kinase Pathway
Next, we investigated the underlying molecular mechanism by which loss of Ophn1 leads to aberrant/elevated PL-mPFC neuronal activity and stress-induced helpless behavior. OPHN1 is a multi-domain-containing Rho-GAP that interacts with a number of adaptor/signaling molecules, including Homer 1b/c, endophilins, RhoA/Rho-kinase, and nuclear receptor Rev-erbα (Billuart et al., 1998; Govek et al., 2004; Khelfaoui et al., 2009; Nadif Kasri et al., 2009; Nadif Kasri et al., 2011; Nakano-Kobayashi et al., 2009; Nakano-Kobayashi et al., 2014; Valnegri et al., 2011). OPHN1’s interaction with RhoA/Rho-kinase is particularly relevant: OPHN1 through its GAP activity regulates PyN spine/synapse development by suppressing the activities of RhoA and its downstream effector Rho-kinase (Govek et al., 2004; Nadif Kasri et al., 2009). These findings, together with the fact that both RhoA and Rho-kinase are expressed in PV INs (Jiang et al., 2016), prompted us to examine if elevated RhoA/Rho-kinase activity in PV INs lacking OPHN1 could be the primary cause of the reduced number of excitatory synapses on such neurons and the consequent increase in neuronal activity in the PL-mPFC. We reasoned that if this were the case, then antagonizing the RhoA/Rho-kinase pathway should rescue the decrease in excitatory synapse number on PV INs, rectify the decrease in synaptic transmission onto such neurons, and normalize the elevated neuronal activity in the PL-mPFC of Ophn1-deficient mice.
To test this, we chose the isoquinoline derivative fasudil, a potent inhibitor of Rho-kinase that is well-tolerated and brain-penetrable (Koch et al., 2018; Meziane et al., 2016; Takayasu et al., 1986). Fasudil or saline was intraperitoneally injected into P40 PV-Ophn1cKO;Ai14 and PV-Ophn1WT;Ai14 mice twice a day for 10 days. Brain sections were then collected and coimmunostained with antibodies against PV, VGLUT1, and PSD-95, and the density of PSD-95 puncta colocalizing with VGLUT1 on PV IN cell bodies and dendritic processes in the PL-mPFC was quantified. Interestingly, fasudil treatment successfully rescued the reduced number of excitatory synapses on PL-mPFC PV IN cell bodies and dendritic processes in PV-Ophn1cKO; Ai14 mice, while it had no effect on their WT littermates (Fig. S8A–C). Fasudil treatment also did not affect the number of excitatory synapses on PL-mPFC SOM IN cell bodies and dendritic processes in SOM-Ophn1cKO;Ai14 and SOM-Ophn1WT;Ai14 mice (Fig. S8D–F).
We then examined if fasudil could rectify the decrease in synaptic transmission onto PL-mPFC PV INs in PV-Ophn1cKO;Ai14 mice. We recorded mEPSCs in PV INs in the PL-mPFC of PV-Ophn1cKO;Ai14 and PV-Ophn1WT;Ai14 mice treated with fasudil or saline. We found that fasudil was able to rectify the observed decrease in mEPSC frequency and amplitude in PV INs in PV-Ophn1cKO;Ai14 mice, while it had no effect on WT littermates (Fig. 8A, B). Finally, we examined whether inhibiting the RhoA/Rho-kinase pathway could reverse the elevated neuronal activity in the PL-mPFC of Ophn1-deficient mice. We recorded sEPSCs in PyNs in the PL-mPFC of ACTB-Ophn1cKO and ACTB-Ophn1WT mice treated with fasudil or saline. Fasudil effectively reversed the increased sEPSC frequency observed in ACTB-Ophn1cKO mice, while it had no effect on sEPSC frequency in ACTB-Ophn1WT mice (Fig. 8C, D). Thus, suppressing the RhoA/Rho-kinase pathway is able to rectify the decrease in excitatory drive onto PV INs and thereby normalize the elevated neuronal activity in the PL-mPFC of Ophn1-deficient mice.
Figure 8. Suppressing RhoA/Rho-Kinase Pathway Normalizes PL-mPFC Neuronal Hyperactivity and Attenuates Helpless Behavior in Ophn1-Deficient Mice.

(A) Representative traces of mEPSCs recorded from LII/III PV INs in the PL-mPFC of PV-Ophn1WT (left) and PV-Ophn1cKO (right) mice treated with saline or fasudil.
(B) Quantification of mEPSC amplitude and frequency. n = 15–18 cells from 3 mice for each genotype/condition; Two-way ANOVA (Amplitude: genotype x treatment interaction F(1, 62) = 6.539, p = 0.013; Frequency: genotype x treatment interaction F(1, 62) = 4.069, p = 0.048) with Bonferroni post-hoc test.
(C) Representative traces of sEPSCs recorded from LII/III PyNs in the PL-mPFC of ACTB-Ophn1WT (left) and ACTB-Ophn1cKO (right) mice treated with saline or fasudil.
(D) Quantification of sEPSC amplitude and frequency. n = 18–25 cells from 3–4 mice for each genotype/condition; Two-way ANOVA (Amplitude: genotype x treatment interaction F(1, 84) = 1.507, p = 0.223; Frequency: genotype x treatment interaction F(1, 84) = 4.394, p = 0.04) with Bonferroni post-hoc test.
(E) Schematic of experimental procedure for F and G.
(F) LH test of PV-Ophn1WT (WT) and PV-Ophn1cKO (cKO) mice treated with saline or fasudil. n = 11–14 mice per group; Aligned-Rank Transform ANOVA (Escape latencies: genotype x treatment interaction F(1, 48) = 4.097, p = 0.048; Failures: genotype x treatment interaction F(1, 48) = 5.314, p = 0.025) followed by multiple comparisons using Mann-Whitney test with Bonferroni correction.
(G) LH test of ACTB-Ophn1WT (WT) and ACTB-Ophn1cKO (cKO) mice treated with saline or fasudil. n = 10–12 mice per group; Aligned-Rank Transform ANOVA (Escape latencies: genotype x treatment interaction F(1, 40) = 9.092, p = 0.0044; Failures: genotype x treatment interaction F(1, 40) = 11.98, p = 0.0013) followed by multiple comparisons using Mann-Whitney test with Bonferroni correction.
(H) Schematic of experimental procedure for I and J.
(I) LH test of PV-Ophn1WT (WT) and PV-Ophn1cKO (cKO) mice with guide cannulae in the PL-mPFC infused with saline or fasudil. n = 12–18 mice per group; Aligned-Rank Transform ANOVA (Escape latencies: genotype x treatment interaction F(1, 52) = 6.37, p = 0.015; Failures: genotype x treatment interaction F(1, 52) = 7.662, p = 0.0078) followed by multiple comparisons using Mann-Whitney test with Bonferroni correction.
(J) LH test of ACTB-Ophn1WT (WT) and ACTB-Ophn1cKO (cKO) mice with guide cannulae in the PL-mPFC infused with saline or fasudil. n = 12–13 mice per group; Aligned-Rank Transform ANOVA (Escape latencies: genotype x treatment interaction F(1, 46) = 4.689, p = 0.035; Failures: genotype x treatment interaction F(1, 46) = 7.528, p = 0.0086) followed by multiple comparisons using Mann-Whitney test with Bonferroni correction.
For B and D, data are mean ± SEM. For F, G, I, J, data are presented with the median and interquartile ranges shown in red. Closed and open circles represent LH and resilient mice, respectively. Percentage of LH+ mice is indicated at the bottom of each group. n.s., p ≥ 0.05, *p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01.
See also Figures S8 and S9.
Next, we investigated if antagonizing RhoA/Rho-kinase signaling could alleviate the helpless behavioral phenotype of Ophn1-deficient mice. We administrated fasudil or saline intraperitoneally to PV-Ophn1cKO and ACTB-Ophn1cKO mice and corresponding control littermates (Fig. 8E) (Garcia-Rojo et al., 2017; Zhao et al., 2015) and subjected the animals to the LH procedure. We found that escape latency and number of escape failures were significantly reduced in fasudil-treated PV-Ophn1cKO and ACTB-Ophn1cKO mice compared to saline-treated animals (Fig. 8F, G). While 73% and 90%, respectively, of saline-treated PV-Ophn1cKO and ACTB-Ophn1cKO mice exhibited LH, only 14% and 18%, respectively, of fasudil-treated PV-Ophn1cKO and ACTB-Ophn1cKO mice displayed LH. Administering fasudil to corresponding WT littermates did not significantly change their helpless behavior (Fig. 8F, G). Thus, systemic administration of fasudil can alleviate the helpless behavioral phenotype of Ophn1-deficient mice.
To determine if fasudil exerts its effect by acting on PV INs in the PL-mPFC, fasudil or saline was infused via cannulae implanted in the PL-mPFC of PV-Ophn1cKO and ACTB-Ophn1cKO mice (Fig. 8H) (Arita et al., 2009; Swanson et al., 2017). We found that fasudil infusion significantly reduced the escape latency and number of escape failures of both mouse lines (Fig. 8I, J). While 69% and 75%, respectively, of saline-infused PV-Ophn1cKO and ACTB-Ophn1cKO mice displayed LH, only 17% and 25 %, respectively, of fasudil-infused PV-Ophn1cKO and ACTB-Ophn1cKO mice were helpless. Fasudil infusion in the PL-mPFC of PV-Ophn1WT or ACTB-Ophn1WT mice had no effect (Fig. 8I, J). A previous study reported that deregulation of protein kinase A (PKA) in the mPFC of conventional Ophn1 KO mice impairs their working memory, as evaluated in Y-maze spatial working memory tests (Zhang et al., 2017). Hence, we also examined the effect of PKA inhibition on the helpless behavioral phenotype of Ophn1-deficient mice. We used the same PKA-signaling antagonist cAMPS-Rp at the same concentration used by Zhang et al. (Zhang et al., 2017) and a similar delivery protocol (Fig. S9A). Unlike fasudil, the infusion of cAMPS-Rp in the PL-mPFC of ACTB-Ophn1cKO mice did not significantly alter/reduce the escape latency and number of escape failures of the mice, and its infusion in the PL-mPFC of ACTB-Ophn1WT mice also had no effect (Fig. S9B). Thus, while suppressing the RhoA/Rho-kinase pathway can alleviate the helpless behavioral phenotype of Ophn1-deficient mice, suppressing PKA signaling fails to do so, suggesting that different mechanisms mediate behavioral responses to stress than those involved in spatial working memory.
Together, these data demonstrate that OPHN1’s effect on PL-mPFC neuronal activity and stress-related behavior depends on its inhibition of the RhoA/Rho-kinase signaling pathway.
DISCUSSION
In this study, we investigated the role of the syndromic X-linked ID protein OPHN1 in the development of behavioral responses to stressful/aversive events. To do so, we employed the LH procedure, an animal stress model widely used to study neural changes underlying resilient vs. susceptible phenotypes following repeated/uncontrollable stress. We chose this model because people with ID frequently encounter uncontrollable stressful events in their everyday life and, furthermore, individuals with OPHN1 mutations commonly display behavioral problems, including emotional imbalance and frustration intolerance—core symptoms of helplessness/hopelessness. Our results revealed that Ophn1-deficient mice subjected to the LH procedure display a marked increase in helpless behavior compared to their control littermates. In accordance with findings in humans, this phenotype was robust in male, but not female, Ophn1-deficient mice. Importantly, the increase in stress susceptibility in male Ophn1-deficient mice was also observed in a repeated social defeat stress model, with Ophn1-deficient mice displaying strong social avoidance behavior. Thus, our data uncover a previously unrecognized role for OPHN1 in establishing adaptive behavioral responses to stress.
To gain insight into the underlying brain/neural mechanisms, we focused on the mPFC, a structure required for executive function and emotional regulation that plays a central role in stress adaptation. Stress has consistently been found to cause structural and functional changes in multiple subregions of the mPFC in both rodents and humans (Dias-Ferreira et al., 2009; Drevets et al., 2008; Goldwater et al., 2009; McEwen, 2007; McEwen and Morrison, 2013; Ota et al., 2014; Perova et al., 2015; Wang et al., 2014; Yuen et al., 2011), and mPFC dysfunction has been linked to maladaptive behavioral responses to different types of stressors (Amat et al., 2005; Lammel et al., 2014; Ota et al., 2014; Perova et al., 2015; Shrestha et al., 2015; Wang et al., 2014; Warden et al., 2012) and to mood disorders (Drevets et al., 2008; Duman et al., 2019; Price and Drevets, 2012; Rudebeck et al., 2019). Recent studies have begun to dissect the mPFC circuits underlying or contributing to stress-induced behavioral changes, revealing how different circuits within mPFC subregions mediate distinctive behavioral phenotypes/symptoms (reviewed in Ghosal et al., 2017; Lammel et al., 2014; Riga et al., 2014; and see further below). Emerging evidence suggests that the PL and IL areas of the mPFC mediate separate and often opposite effects on stress susceptibility/resilience (Capuzzo and Floresco, 2020; Hamani et al., 2010a; Lammel et al., 2014; McKlveen et al., 2015). In line with this, we found that deleting Ophn1 in the PL-mPFC promotes helpless behavior, while its deletion in the IL-mPFC had no significant effect.
Our data further uncover a critical role for OPHN1 in regulating PV IN activity in the PL-mPFC and present evidence that such regulation is required for shaping adaptive behavioral responses to stress. We observed that Ophn1 deficiency exclusively in PV, but not SOM, INs in the PL-mPFC is sufficient to induce helpless behavior and that Ophn1 deficiency selectively weakens excitatory synaptic transmission onto PL-mPFC PV INs. Notably, the strength of excitatory synaptic transmission onto PL-mPFC PV, but not SOM, INs was similarly found to be weakened in helpless mice compared to resilient or naïve mice (Perova et al., 2015). Our data further revealed that this decrease in excitatory drive leads to less active PV INs and an increase in the frequency of sEPSCs in neighboring PL-mPFC PyNs. Based on these findings, we put forward a model in which decreased PV IN activity causes PyN disinhibition in the PL-mPFC, leading to PL-mPFC hyperactivity (Fig. S9C). An alternative explanation, though, is that decreased PV IN activity triggers some network changes (i.e., the entire cortex is affected), causing homeostatic changes in PyNs that could be reflected by an increase in sEPSCs. Regardless, this alternative explanation would still support the conclusion that Ophn1 deficiency in PL-mPFC PV INs leads to increased PL-mPFC activity.
Evidence from our studies indicates that the enhanced activity in the PL-mPFC resulting from Ophn1 deficiency in PV INs promotes helpless behavior. Pharmacogenetically activating PV INs (with an excitatory DREADD) or suppressing overall neuronal activity (with an inhibitory DREADD) in the PL-mPFC during the LH procedure alleviated the stress-induced helpless behavior phenotype of Ophn1-deficient mice. While it has recently been reported that the DREADD ligand CNO can convert back to its parent compound clozapine, potentially causing behavioral off-target effects (Manvich et al., 2018; Martinez et al., 2019), our data show that CNO administration to non-DREADD-expressing animals does not produce any phenotype in the LH procedure, supporting that normalizing mPFC neuronal activity in Ophn1-deficient mice indeed improves the helpless phenotype.
Our findings, to the best of our knowledge, are the first to show a critical role for OPHN1 in regulating PV INs. Previous studies revealed a central role for OPHN1 in regulating glutamatergic spine/synapse maturation and plasticity in hippocampal PyNs (Khelfaoui et al., 2007; Khelfaoui et al., 2014; Khelfaoui et al., 2009; Nadif Kasri et al., 2009; Nadif Kasri et al., 2011; Nakano-Kobayashi et al., 2009; Nakano-Kobayashi et al., 2014). Silencing OPHN1 in CA1 PyNs was found to largely prevent the increase in mEPSC frequency that normally occurs during the development of CA3–CA1 synapses (Nadif Kasri et al., 2009). While we also observed a decrease in mEPSC frequency (but not amplitude) in Emx1-expressing cortical PyNs lacking OPHN1 (Fig. S6E, F), the overall network activity was not altered, and no increase in helpless behavior was detected in Emx1-Ophn1cKO mice. Moreover, Ophn1 deletion in both inhibitory and excitatory neurons resulted in enhanced excitatory drive within the PL-mPFC, and, accordingly, Ophn1-deficient (ACTB-Ophn1cKO) mice displayed increased LH. Thus, OPHN1, by regulating PV IN activity, maintains the proper balance of excitation and inhibition (E/I) within mPFC microcircuits required for shaping adaptive behavioral responses to stress (Fig. S9C). As to how OPHN1 controls the activity of PV INs, we show that OPHN1 regulates PV IN synapse development and thereby proper E/I balance in the PL-mPFC by suppressing RhoA/Rho-kinase signaling. First, inhibiting the RhoA/Rho-kinase pathway with fasudil rescued the reduced number of excitatory synapses on PV INs, rectified the decrease in synaptic transmission onto these neurons, and normalized the elevated neuronal activity in the PL-mPFC of Ophn1-deficient mice. Second, suppressing the RhoA/Rho-kinase pathway either systemically or locally with fasudil in the PL-mPFC alleviated the stress-induced helpless behavior in PV-Ophn1cKO and ACTB-Ophn1cKO mice. Given that fasudil has been successfully used to treat cerebral vasospasm in Japan and does not appear to produce any major adverse side effects (Olson, 2008; Tanaka et al., 2005), it could be a promising drug for the stress-related behavioral problems of OPHN1 patients.
Our data further support the notion that different subpopulations/types of mPFC neurons or microcircuits play specific roles in a particular behavior(s) or response(s) to stress (Lammel et al., 2014; Maier and Seligman, 2016; Riga et al., 2014). Previous studies have reported that acute inhibition of SOM INs in the PL-mPFC increases anhedonic-like behaviors in mice (Soumier and Sibille, 2014). We found that Ophn1 deficiency in SOM INs neither affects excitatory synaptic transmission onto such neurons nor influences LH behavior. Moreover, as mentioned above, synaptic transmission onto SOM INs was not altered in helpless mice, suggesting that these neurons are not critically involved in moderating helpless behavioral responses to unpredictable/uncontrollable stress. In support of this, inhibiting PV, but not SOM, INs promoted LH (Perova et al., 2015; and data not shown). In a similar vein, stimulating different neuronal subpopulations or circuits were found to produce distinct, sometimes opposite, behavioral responses and/or mediate distinctive behavioral phenotypes (Baratta et al., 2009; Cerniauskas et al., 2019; Covington et al., 2010; Ferenczi et al., 2016; Hamani et al., 2010a; Hamani et al., 2010b; Holz et al., 2019; Kumar et al., 2013; Lammel et al., 2014; Marcus et al., 2020; Riga et al., 2014; Warden et al., 2012). For instance, stimulating the projection of the mPFC to the dorsal raphe nucleus (DRN) caused decreased immobility in a forced swim test, while stimulating the projection to the lateral habenula (LHb) increased immobility (Amat et al., 2005; Dolzani et al., 2016; Li et al., 2011; Warden et al., 2012). Thus, stimulating the mPFC-DRN pathway promotes resilience, while stimulating the mPFC-LHb pathway induces depression-like responses. Of note, in nearly all of the above studies, both the PL and IL regions of the mPFC were stimulated. Future studies could explore if the effect of Ophn1 deficiency in PL-mPFC PV INs, which leads to enhanced activity in neighboring PyNs and increased helpless behavior, is predominated by the PL-mPFC-LHb pathway.
In summary, our study unveils a previously unidentified function of OPHN1 in regulating PV IN activity and thereby E/I balance in mPFC microcircuits and sheds light on the mechanistic links among OPHN1 genetic deficits, mPFC circuit dysfunction, and abnormalities in stress-related behaviors (Fig. S9C). It also further suggests that aberrant activity within specific mPFC microcircuits could contribute to the development of stress-related behavioral problems and mood disorders in individuals with ID.
STAR METHODS
RESOURCE AVAILIBILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Linda Van Aelst (vanaelst@cshl.edu).
Materials availability
All unique reagents generated in this study are available from the Lead Contact with a completed Material Transfer Agreement.
Data and code availability
All data have been presented in Figures and Supplemental Figures and Videos. Original images/data will be made available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All protocols and procedures followed the guidelines of the Animal Care and Use Committee of Cold Spring Harbor Laboratory (CSHL). Ophn1-flox mice were generated using ES cell clones from the European Conditional Mouse Mutagenesis Program (EUCOMM) with knock-out first mutation (EPD0109_5_D11; http://www.mousephenotype.org/data/alleles/MGI:2151070/tm1a (KOMP)Wtsi). ES clones were injected into C57BL/6J blastocysts, and the resulting chimeras were mated to C57BL/6J females to obtain germ-line transmission. Offspring were genotyped by PCR using the following primers: D11–1, 5’-CCAGGTTACCCCCAACAAG; D11–2, 5’-CCAACTGACCTTGGGCAAGAACAT; D11–3, 5’-CACACCTCCCCCTGAACCTGAAA; D11–5, 5’-CATTTTCTACTTCATTGAGCAATTC (see Fig. S1A). Detection of PCR products (272 bp for D11–1 and D11–2 primer pair and 816 bp for D11–3 and D11–5 primer pair) indicated the presence of KO-first reporter-tagged insertion. Heterozygous F1 female mice (Ophn1KO-flox/+) were mated with ACTB-FLPe mice (B6.Cg-Tg(ACTFLPe)9205Dym/J; JAX Stock 005703) to remove the promoter-driven selection cassette of the knock-out first mutation, and the resulting offspring (Ophn1flox/+;FLP/+ and Ophn1flox/Y;FLP/+) were subsequently mated with C57BL/6J mice to segregate the FLPe transgene. Heterozygous female Ophn1flox/+ mice derived from the latter cross were mated to ACTB-Cre (B6N.FVB-Tmem163Tg(ACTB-cre)2Mrt/CjDswJ, JAX Stock 019099), PV-Cre (B6;129P2-Pvalbtm1(cre)Arbr/J, JAX Stock 008069), SOM-Cre (B6N.Cg-Ssttm2.1(cre)Zjh/J JAX Stock 018973), or Emx1-Cre (B6.Cg-Zfp335tm1.2CawEmx1tm1(cre)Krj/J, JAX Stock 022762) mice to generate Ophn1flox/Y;ACTBCre/+ (ACTB-Ophn1cKO) and Ophn1+/Y;ACTBCre/+ (ACTB-Ophn1WT), Ophn1flox/Y;PValbCre/+ (PV-Ophn1cKO) and Ophn1+/Y;PValbCre/+ (PV-Ophn1WT), Ophn1flox/Y;SOMCre/+ (SOM-Ophn1cKO) and Ophn1+/Y;SOMCre/+ (SOM-Ophn1WT), and Ophn1flox/Y;Emx1Cre/+ (Emx1-Ophn1cKO) and Ophn1+/Y;Emx1Cre/+ (Emx1-Ophn1WT) mice, respectively. Female Ophn1flox/+ mice were also mated to ACTB-Cre to generate Ophn1flox/+;ACTBCre/+ (ACTB-Ophn1HET) and Ophn1+/+;ACTBCre/+ (ACTB-Ophn1WT) female mice and to Ophn1flox/Y;ACTBCre/Cre to generate Ophn1flox/+;ACTBCre/+ (ACTB-Ophn1HET) and Ophn1flox/flox;ACTBCre/+ (ACTB-Ophn1HOM) female mice. Ophn1flox/Y;ACTBCre/Cre mice were obtained by crossing male ACTB-Cre mice with Ophn1flox/+;ACTBCre/+ mice. Presence of the Cre allele was in all cases determined by PCR analysis using previously published methods (Hippenmeyer et al., 2005; Taniguchi et al., 2011). To generate Ophn1flox/Y;PValbCre/+;Ai14+/− (PV-Ophn1cKO;Ai14) and Ophn1+/Y;PValbCre/+;Ai14+/− (PV-Ophn1WT;Ai14) mice, we first mated Ophn1flox/+ female mice with the Ai14 Cre-dependent reporter line (Ai14+/+; B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, JAX Stock 007908) to obtain Ophn1flox/+;Ai14+/− female offspring. These mice were crossed with Ai14+/+ mice to generate Ophn1flox/+;Ai14+/+ mice, which were then mated to PV-Cre mice. The presence of the Ai14 allele was analyzed by PCR as previously described (Madisen et al., 2010). Ophn1flox/+;Ai14+/+ mice were also mated to SOM-Cre mice to generate Ophn1flox/Y;SOMCre/+;Ai14+/− (SOM-Ophn1cKO;Ai14) and Ophn1+/Y;SOMCre/+;Ai14+/− (SOM-Ophn1WT;Ai14). For the generation of Ophn1flox/Y;PVFlp+/− (Ophn1flox/Y;PV-Flp) mice, we first crossed Ophn1flox/+ mice to Ophn1flox/Y mice to obtain homozygous female Ophn1flox/flox mice. Offspring were genotyped by PCR using primers: D11–1 and D11–5 (see above). The sizes of the PCR products are 554 bp (WT band) and 649 bp (mutant band). Ophn1flox/flox mice were then mated with PV-Flp mice (gift from Josh Huang, CSHL) (He et al., 2016) to generate Ophn1flox/Y;PV-Flp mice. All mice were maintained in standard housing conditions on a 12 h light/dark cycle with food and water provided ad libitum.
HEK293T cell culture
HEK293T (ATCC, RRID: CVCL_0063) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS) (HyClone), 100 IU/ml penicillin (Thermo Fisher Scientific), and 100 μg/ml streptomycin (Thermo Fisher Scientific) in a humidified incubator at 37°C with 5% CO2. The cell line used in this study was not further authenticated and not found to be on the list of commonly misidentified cell lines (International Cell Line Authentication Committee).
METHOD DETAILS
Experimental Design
Animals were randomly allocated to experimental groups prior to experimental manipulation(s), and the experimenter was blinded to the condition until the experiment and quantifications/analyses were complete. For electrophysiology experiments, analyses were performed on brains from a minimum of 3 animals. For immunostaining and morphometric experiments, quantifications/analyses were performed on cells from at least 3 individual animals. Western blotting experiments were performed in triplicate and depicted images are representative. No statistical methods were used to predetermine sample size in experiments.
Western blotting
Whole brains or cerebral cortices collected from 2-month-old male ACTB-Ophn1WT or ACTB-Ophn1cKO mice were homogenized using micropestles (Eppendorf) in lysis buffer (50 mM Tris-HCL pH 7.5, 1% Triton X-100, 150 mM NaCl, 5% glycerol, 5 mM NaF, 1 mM Na3VO4, and 1x cOmplete™ EDTA-free protease inhibitor cocktail (Sigma-Aldrich)). Total lysates were resolved by SDS-PAGE and transferred to an Immobilon-P PVDF membrane (Millipore). Membranes were blocked in Tris-buffered saline with 0.5% Tween 20 (TBST) containing 5% fat-free milk for 30 min at room temperature (RT) and then incubated in TBST with 5% bovine serum albumin (BSA) (Equitech-Bio, Inc.) and 0.05% NaN3 containing primary antibodies overnight at 4°C. The following day, the membranes were incubated in blocking buffer containing horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hour at RT. The following primary antibodies were used: anti-OPHN1 (rabbit pAb, 1724, 1:500; (Govek et al., 2004)) and anti-GAPDH (rabbit mAb, 1:10,000; Cell Signaling Technology 5174). HRP-conjugated goat anti-rabbit secondary antibody (Cell Signaling Technology 7074) was used at 1:5,000. Pierce™ ECL and Western Blotting Substrate (Thermo Fisher Scientific) was used for detection of HRP activity.
Immunohistochemistry
For immunostaining of mouse brain sections, animals were deeply anesthetized with isoflurane and perfused transcardially with PBS followed by 4% paraformaldehyde (PFA) in PBS. Brains were isolated and post-fixed in 4% PFA in PBS overnight at 4°C and then cryoprotected with a 30% PBS-buffered sucrose solution for 36 h. Brains were subsequently embedded in 3% low melting point agarose (Thermo Fisher Scientific) and coronal brain sections (40–50 μm) were prepared using a vibratome (Leica VT1000S). Brain sections were washed in PBS (3 × 5 min), incubated in PBST (0.2% Triton X-100 in PBS) for 15 min at RT, and washed again with PBS (3 × 5 min). Next, brain sections were blocked in 10% normal goat serum (NGS) in PBST for 30 min at RT, followed by incubation with primary antibodies overnight at 4 °C. Sections were washed with PBS (3 × 15 min) and incubated with fluorescently-conjugated secondary antibodies at RT for 1 h. After washing with PBS (3 × 15 min), sections were mounted onto glass slides with Fluoromount-G (SouthernBiotech 0100–01). Images were taken using a LSM800 or 710 confocal microscope with a Plan-Apochromat 20x/0.8 M27 objective (Zeiss). The primary antibodies used were: anti-OPHN1 (rabbit pAb, 1724, 1:100, (Govek et al., 2004)), anti-NeuN (mouse mAb, 1:100, Millipore MAB377), anti-parvalbumin (mouse mAb, 1:1000, Sigma P3088), anti-somatostatin (rat mAb, 1:500, Millipore MAB354), anti-neurogranin (mouse mAb, 1:500, Biolegend 845602), anti-VGLUT1 (Rabbit pAb, 1:1000, Synaptic Systems 135 302), and anti-PSD-95 (mouse mAb, 1:200, Millipore MAB1598). The secondary antibodies used were: Alexa Fluor (AF) 488 goat anti-rabbit IgG (H+L) (Thermo Fisher Scientific A11008), AF 488 goat anti-mouse IgG2a (Thermo Fisher Scientific A21131), AF 555 goat-anti-mouse IgG (H+L) (Thermo Fisher Scientific A21422), AF 555 goat anti-rat IgG (H+L) (Thermo Fisher Scientific A21434), AF 555 goat-anti-mouse IgG1 (Thermo Fisher Scientific A21127), and AF 647 goat-anti-rabbit IgG (H+L) (Thermo Fisher Scientific A21245).
Cresyl violet/Nissl staining
Post-fixed brains were embedded in 3% low melting point agarose and coronal brain sections (40–50 μm) were generated using a vibratome (Leica VT1000S). Brain sections were immersed in 75% ethanol once and 95% ethanol once for 3 min each time, followed by two immersions in 100% ethanol (Sigma-Aldrich) for 3 min each time. The sections were then immersed in xylene (Poly Scientific R&D Corp) twice for 3 min each time. The previous steps were subsequently repeated in reverse, first by dipping the sections in xylene twice for 3 min each time, then two immersions in 100% ethanol for 3 min each time, followed by dipping in 95% ethanol once and 75% ethanol once for 3 min each time, before they were placed in a container with Milli-Q water for 1 min. The sections were then immersed in 0.1% cresyl violet staining solution (Sigma-Aldrich) for 10 min and washed again in a container with distilled water for another 2 min. Next, the sections were dipped again in 75% ethanol for 3 min, then 95% ethanol for 3 min, followed by two immersions in 100% ethanol for 3 min each time. The final three immersions were in xylene solution for 5 min each. Brain sections were then mounted onto glass slides with Fluoromount-G mounting medium (SouthernBiotech 0100–01).
Behavioral procedures
All behavioral experiments were performed using groups of 4–8 littermate male or female mice of 7–8 weeks of age. Mice were single housed at least for one week before the start of the experiments. When mice were subjected to multiple behavioral procedures, the learned helplessness (LH) procedure was performed last, following the open field, elevated plus maze, and light/dark exploration tests. All experiments were done during the light phase, the second part of the day.
Open field (OF) test. The OF test was performed in a nontransparent box (43 × 43 × 40 cm) at least 3 h before the onset of the dark cycle (Wang et al., 2014). Lighting condition for the OF test is 150–200 lux. Mice were placed in one of the corners of the arena at the start of each session. The center zone was set to 21 × 21 cm in the middle of the arena. Mice explored the arena for 5 min while being monitored using a PC6EX3 infrared camera (SuperCircuits Inc.) connected to a computerized video tracking system (Ethovision XT 5.1, Noldus). The arena was cleaned thoroughly between each trial. Total distance traveled, average velocity, and the frequency of visits, time spent, and distance traveled in the center zone were measured.
Elevated plus maze (EPM) test. The apparatus used for the EPM test consisted of two “open” arms (30 × 5 × 2 cm) and two “closed” arms (30 × 5 × 15 cm) forming a cross. The arms were separated by a central platform (5 × 5 cm), and the maze was elevated 60 cm above the floor. Lighting condition for the EPM test is 150–200 lux. Mice were placed on the central platform facing one of the open arms (File et al., 2005). Behavior was monitored using a PC6EX3 infrared camera connected to a computerized video tracking system (Ethovision XT 5.1, Noldus). The apparatus was cleaned thoroughly between each trial. Total distance traveled, time spent in open and closed arms, and frequencies of entries in open and closed arms were measured.
Light/dark box (L/D) test. The L/D apparatus consisted of a plexiglass box divided into two compartments by a partition with a small opening. One compartment (28 × 21 × 21 cm) was brightly lit (500 lux), whereas the other (14 × 21 × 21 cm) was dark. At the beginning of the test, mice were placed in the lit compartment (light zone) facing away from the opening and allowed to freely explore for 5 min (File et al., 2005; Walsh et al., 2017). Behavior was monitored using a PC6EX3 infrared camera connected to a computerized video tracking system (Ethovision XT 5.1, Noldus). The apparatus was cleaned thoroughly between each trial. The distance traveled and time spent in the light zone, and the number of transitions between the light and dark compartments, were measured.
LH procedure. The LH procedure in mice was conducted based on previously described procedures (Chourbaji et al., 2005; Perova et al., 2015; Wang et al., 2014). In brief, mice were first exposed to two induction sessions that were separated by 24 h. Each session consisted of 360 inescapable, uncontrollable electric foot shocks over a 60 min period. The shock intensity was set at 0.3 mA, the duration of each shock was randomized between 1 and 3 s, and the inter-shock intervals (ITIs) were randomized between 1 and 15 s. 24 h after the second induction session, mice were subjected to a testing session. The testing, which was fully automated using Graphic State 3.0 software (Coulbourn Instruments), was performed in a shuttle box (14 × 7 × 12 inches; Coulbourn Instruments) equipped with an electrical grid floor, a door separating the two halves, and photocell detectors. The shuttle box was placed in a sound-attenuating chamber. Mice explored the shuttle box for 2 min, and behavioral performance was evaluated over 30 trials of escapable foot shocks (0.3 mA intensity, 10 s duration, with ITIs of 30 s). Each trial started with a 5 s cue light, followed by the foot shocks. When an animal shuttled to another compartment of the box during the 5 s cue light presentation (and thus before the shock onset), avoidance was scored. If the animal shuttled during the 10 s shocks (i.e., escaped), escape latency was measured. Failure was recorded if no shuttling was made during the 10 s shock presentation. Shocks were terminated if the animal shuttled to another side of the box (in case of escape) or at the end of the 10 s shock (in case of failure). Animals’ behavior was classified as being “resilient” or “learned helpless” based on their behavioral parameters in the LH testing session. A k-means (k = 2) cluster analysis was applied to a database consisting of 98 wild-type C57BL/6J mice subjected to the LH procedure. We used failures and escape latency, the most commonly reported indices of helplessness (Chourbaji et al., 2005; Seligman, 1978; Wang et al., 2014), as parameters for classification. We further performed a linear discriminant analysis on the clustering results, with the number of failures and escape latency as predictor variables, to obtain classification equations for new cases: R= −2.28185+ (2.17825*escape latency) + (−0.57025*failures); and LH = −26.97824 + (−0.10226*escape latency) + (2.15966*failures), where the escape latency and the number of failures define the classification scores, R (resilience) and LH (learned helplessness). A mouse is classified as being resilient if R > LH, or learned helpless if LH > R (Chourbaji et al., 2005; Wang et al., 2014). For the “test-only” group (Fig. 1B), mice were exposed to one LH testing session (i.e., without prior induction sessions) as described above.
Hot plate test. Mice were subjected to the hot plate test to rule out altered pain sensitivity as a confounding factor for LH (Bannon, 2001; Shin et al., 2015). The surface of the hotplate was heated to a constant temperature of 54 ± 1°C. The mice were then placed on the surface of the hot plate and covered by a transparent plexiglass cylinder. The latency to the first response (licking forepaws, flinching, or jumping) was recorded manually. The duration of the test was a maximum of 30 s, and the test was terminated if the mouse did not respond after 30 s.
Social defeat stress and social interaction. The social defeat procedure was performed largely as described before (Golden et al., 2011; Shin et al., 2015). In brief, each ACTB-Ophn1WT or ACTB-Ophn1cKO male mouse was placed into the home cage of an unfamiliar, aggressive CD1 resident mouse for 5–10 min, which resulted in physical defeat of the intruder ACTB-Ophn1WT or ACTB-Ophn1cKO mouse by the CD1 mouse. After the defeat, both the resident CD1 mouse and the intruder mouse were housed together, separated by a perforated plastic divider that allowed visual, olfactory, and auditory contacts, but prevented physical interaction, for the remainder of the 24 h period. The same ACTB-Ophn1WT or ACTB-Ophn1cKO mouse was then subjected to social defeat from a different resident CD1 mouse each day for 5 consecutive days. Non-defeated control mice were handled daily and housed opposite of another wild type C57BL/6 mouse instead of a CD1 mouse. 24 h after the last social defeat session, the test mice (i.e., defeated ACTB-Ophn1WT and ACTB-Ophn1cKO mice and non-defeated mice) were subjected to the social interaction test. The latter was performed in a 43 × 43 cm arena equipped with a wire mesh cage (10 × 6.5 cm). The approach-avoidance behavior of a test mouse to a CD1 mouse was recorded with a video tracking system (Ethovision XT 5.1, Noldus). The social interaction test consisted of two 5 min sessions. In the first 5 min ‘no target’ session, the test mouse was allowed to explore the open arena freely with an empty wire mesh cage. For the second 5 min ‘target’ session, an unfamiliar CD1 mouse was placed into a new wire mesh cage. The wire mesh cage allowed visual, olfactory and auditory interactions between the test mouse and the target CD1 mouse but prevented direct physical contact. The time spent by the test ACTB-Ophn1WT or ACTB-Ophn1cKO mouse in the ‘interaction zone’ (26.0 × 14.5 cm) and ‘corner zone’ (43 × 8.0 cm) of the arena were measured by the video tracking system.
Adeno-associated virus (AAV) vector construction and production
For the construction of the flippase-dependent pAAV-CAG-fDIO-Cre-GFP vector, a DNA cassette harboring two pairs of incompatible FRT sites (FRT and F5) (FRT: 5’-GAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC; F5: 5’-GAAGTTCCTATTCTTC AAAAGGTATAGGAACTTC) was synthesized and Cre-GFP encoding sequences isolated from pCAG-Cre:GFP (Addgene plasmid # 13776; gift from Connie Cepko; (Matsuda and Cepko, 2007)) were inserted between the two sites in the reverse orientation. The resulting cassette was then cloned into a modified version of pAAV-CAG-GFP (Addgene plasmid #: 37825; gift from Ed Boyden) in which the GFP-encoding sequences were removed. The integrity of the insert was confirmed by DNA sequencing. The pAAV-CAG-fDIO-Cre-GFP plasmid was subsequently packaged into functional viruses that were pseudotyped with AAV-DJ/8 capsid proteins (AAV-DJ/8 Helper Free Expression System, Cell Biolabs, Inc.) to produce AAV-DJ/8 viruses. Pseudotyped viruses were produced by helper virus-free transfection of HEK293T cells (using polyethylenimine method) with three plasmids (pAAV-CAG-fDIO-Cre-GFP, pAAV-DJ/8, and pHelper), and the resultant viruses were purified by CsCl gradient ultracentrifugation, as previously described (Ayuso et al., 2010; Wang et al., 2011). Virus preparations were dialyzed to remove remaining CsCl. The titer of purified viruses was determined using a qRT-PCR-based method and the viruses were then stored in aliquots at −80°C until use. For the construction of the pAAV-CAG-Cre-GFP vector, Cre-GFP (see above) was cloned into pAAV-CAG-GFP in which the GFP-encoding sequences were removed. The pAAV-CAG-Cre-GFP plasmid was subsequently packaged into functional viruses that were pseudotyped with AAV-DJ/8 capsid proteins to produce AAV/DJ8 viruses, as described above. AAV9-CAG-GFP (construct: Edward Boyden, MIT, Addgene plasmid #: 37825) was produced by the University of North Carolina (UNC) Vector Core Facilities. AAV2-hSyn-DIO-mCherry was obtained from Addgene (construct: Bryan Roth, UNC, Addgene viral prep #: 50459-AAV2). AAV8-EF1α-DIO-hM4Di-mCherry was produced by the Viral Gene Transfer Core of the McGovern Institute for Brain Research at MIT (construct: Rachael Neve, Gene Delivery Technology Core, Massachusetts General Hospital). AAV-hSyn-DIO-hM3D(Gq)-mCherry was obtained from Addgene (construct: Bryan Roth (Krashes et al., 2011), UNC, Addgene viral prep # 44361-AAV2). The AAV8-EF1α-fDIO-mCherry was a gift from Josh Huang (CSHL) (Lu et al., 2017).
Stereotactic surgery and viral injection
Standard surgical procedures were followed for stereotaxic injection (Li et al., 2011). Briefly, mice were anaesthetized with ketamine (100 mg/kg of body weight) supplemented with dexmedetomidine hydrochloride (0.4 mg/kg of body weight) and positioned in a stereotaxic injection frame (myNeuroLab.com). A digital mouse brain atlas was linked to the injection frame to guide the identification and targeting of the prelimbic (PL) and infralimbic (IL) regions of the mPFC and hippocampal CA1 region (Angle Two Stereotaxic System, myNeuroLab.com). Viruses were delivered with a glass micropipette through a skull window (1–2 mm2) by pressure application (5–12 psi, controlled by a Picospritzer III, General Valve Corp). The following stereotaxic coordinates were used: PL-mPFC, 1.94 mm anterior from bregma, 0.40 mm lateral to midline, and 2.2–2.5 mm ventral from the bregma level; IL-mPFC, 1.94 mm anterior from bregma, 0.40 mm lateral to midline, and 2.8–3.2 mm ventral from the bregma level; and CA1, 2.18 mm posterior from bregma, two sites per hemisphere with 1.0 and 1.5 mm lateral to midline, and 1.3–1.6 mm ventral from the bregma level. For all viral injections, 0.5–0.8 μl of viral solution (~1013 virus particles/ml) was bilaterally injected into the PL-mPFC, IL-mPFC, or hippocampal CA1. Throughout the entire surgical procedure, animals were kept on a heating pad and brought back to their home cages after regaining movement. For postoperative care, mice were hydrated by intraperitoneal injection with 0.3–0.5 ml of lactated Ringer’s solution. Metacam (meloxicam, 1–2 mg/kg of body weight) was used as an analgesic and to reduce inflammation. To assess the viral infection efficiency (for Fig. S2A and B; S4A and B), the number of GFP-positive cells within the PL-mPFC area was quantified using ImageJ, and the density of virus-infected cells (i.e., GFP-positive cells) was determined by dividing the number of infected cells with the PL-mPFC area. Five slices per brain were chosen every 150 μm for analysis.
Preparation of acute brain slices and electrophysiology
Mice (male, 40–45 d old) were anaesthetized with isoflurane and decapitated, and brains were quickly removed and chilled in ice-cold dissection buffer (110 mM choline chloride, 25 mM NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 0.5 mM CaCl2, 7 mM MgCl2, 25 mM glucose, 11.6 mM ascorbic acid, and 3.1 mM pyruvic acid, gassed with 95% O2 and 5% CO2). Coronal slices (300 μm) containing the mPFC were cut in dissection buffer using a Leica VT1000S vibratome and subsequently transferred to a storage chamber containing artificial cerebrospinal fluid (ACSF; 118 mM NaCl, 2.5 mM KCl, 26.2 mM NaHCO3, 1 mM NaH2PO4, 20 mM glucose, 2 mM MgCl2, and 2 mM CaCl2, pH 7.4, gassed with 95% O2 and 5% CO2) kept at 34°C. After at least 40 min recovery time, slices were transferred to RT and constantly perfused with ACSF.
Whole-cell patch-clamp recordings were performed on PV+ or SOM+ INs or neurons fulfilling the morphological criteria of PyNs in layers II/III in the PL region of the mPFC. The PL region was identified based on landmarks (i.e., forceps minor of the corpus callosum) and by measuring the distance from the midline and the cortical surface. Neurons in the area of 0.4–0.7 mm from the midline and 1.2–1.8 mm from the cortical surface were regarded as belonging to the PL region. In addition, we took advantage of the Ai14 Cre-dependent reporter line to label PV or SOM INs with the red fluorescent protein tdTomato. Recordings were obtained with Multiclamp 700B amplifiers (Molecular Devices) and were under visual guidance using a Zeiss Axioskop microscope equipped with both transmitted light and epifluorescence illumination. Miniature and spontaneous postsynaptic currents were recorded in voltage-clamp mode with borosilicate pipettes (3.5–5 MΩ). For recordings in voltage-clamp mode, the internal solution contained the following: 115 mM caesium methanesulphonate, 20 mM CsCl, 10 mM HEPES, 2.5 mM MgCl2, 4 mM Na2-ATP, 0.4 mM Na3GTP, 10 mM Na-phosphocreatine, and 0.6 mM EGTA (pH 7.2). mEPSCs were recorded at a holding potential of −60 mV (for PV and SOM INs or −70 mV (for Emx1-expressing PyNs) with picrotoxin (PTX; 100 μM) and tetrodotoxin (TTX; 1 μM) added to the ACSF. mIPSCs were recorded at a holding potential of 0 mV with 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX, 20 μM), DL-2-amino-5-phosphonopentanoate (DL-AP5, 100 μM), and TTX (1 μM) added to the ACSF. sEPSCS of PyNs were recorded at a holding potential of −70 mV with PTX (100 μM) added to the ACSF. The PyNs were selected based on their characteristic pyramidal-like morphology. Simultaneous whole-cell patch-clamp recordings were performed on pairs of PV INs and neighboring PyNs located within 50 μm of selected PV cells. For evoked EPSCs in PV INs and IPSCs in PyNs, postsynaptic responses were evoked by injecting under current clamp configuration a brief current pulse (duration, 20 ms; amplitude, 200–300 pA) in the presynaptic neurons sufficient to elicit an action potential. Evoked EPSCs and IPSCs were low-pass filtered at 2 kHz and recorded under voltage clamp mode at holding potentials of −70 mV and −50 mV, respectively. Only the successful events during EPSC or IPSC recordings were calculated (referred to as “unitary” EPSC or IPSC amplitudes (uEPSCs, uIPSCs, respectively)). The internal solution for the paired recordings consisted of 130 mM potassium gluconate, 5 mM KCl, 10 mM HEPES, 2.5 mM MgCl2, 4 mM Na2ATP, 0.4 mM Na3GTP, 10 mM Na-phophocreatine, and 0.6 mM EGTA (pH 7.2). Resting membrane potential was measured in the current-clamp configuration. Input resistance was calculated from hyperpolarizing current injections in the voltage-clamp configuration. For spontaneous firing frequency measurements of PV INs, a modified ACSF solution (5 mM KCl, 0.5 mM MgCl2, 1 mM CaCl2; Yang et al., 2013) was used and PV INs were recorded in a tight-seal cell-attached configuration (0.3–1 GΩ) with pipettes filled with the same solution used for paired recordings. For measurements of paired-pulse ratio of PV INs, a bipolar stimulation electrode was placed in the deep layers of the PL-mPFC about 150–200 μm from the recorded cells, which were kept at a holding potential of −60 mV. Pairs of stimulation with 5 Hz, 10 Hz, 20 Hz, 50 Hz were delivered to the recorded cells and the ratio between the amplitude of the second evoked EPSC to the amplitude of the first evoked EPSC was calculated as the paired-pulse ratio. To validate the effect of the inhibitory DREADD hM4Di and excitatory DREADD hM3D(Gq), recordings were performed in current clamp mode. The internal solution was the same as that for paired recordings. The holding potentials were adjusted to make the cells fire at a rate of ~0.5 Hz. After 5 min of stable recording, CNO (20 μM) was puff applied from another glass pipette placed within 50 μm of the recorded neuron. Electrophysiological data were acquired using pCLAMP 10 software (Molecular Devices) and analyzed using the same software (for evoked EPSCs) or Mini Analysis Program (Synaptosoft) for mEPSCs, mIPSCs, and sEPSCs.
Morphometric analyses
To visualize PV INs, 0.2 μl of AAV-DIO-mCherry was bilaterally injected into the PL-mPFC of 5-week old male PV-Ophn1WT and PV-Ophn1cKO mice to sparsely label PV INs. Two weeks later, brain sections were prepared as described above and subsequently imaged using an LSM 800 confocal laser-scanning microscope (Zeiss) with a 63x oil-immersion objective and sequential acquisition settings applied at resolution of 1024 × 1024 pixels. For all 3D reconstructions, 200 μm x 200 μm images of single mCherry+ PV INs, which were spatially isolated from other mCherry+ PV INs and processes, were collected in the PL-mPFC using a z-series of 80–90 images with a depth interval of 0.3 μm. Sholl analyses were carried out using an Imaris algorithm for branching analysis (Bitplane, Inc). In brief, all processes (dendrites and axon) from each analyzed mCherry+ cell were first traced manually in three dimensions, assisted by the AutoPath protocol, and the soma of each cell was identified as the center for Sholl analysis. The number of process intersections was determined for each ring in the series of concentric spheres spaced at 5 μm intervals from the somal center point. Soma area was quantified using the “create surface” tool in Imaris to generate a solid surface best matching the neuronal anatomy of the cell soma.
Analysis of the density/number of PSD-95 puncta colocalizing with VGLUT1 on PV and SOM IN cell bodies and dendrites
To quantify PSD-95-VGLUT1 puncta colocalization, images of coronal brain slices (50 μm thick) prepared from PV-Ophn1cKO;Ai14, PV-Ophn1WT;Ai14, SOM-Ophn1cKO;Ai14 or SOM-Ophn1WT;Ai14 mice and immunostained for PSD-95, VGLUT1, and PV or SOM were acquired using an LSM 710 or 800 confocal laser-scanning microscope (Zeiss) with a 63x oil-immersion objective and acquisition settings applied at a resolution of 1024 × 1024 pixels. Images of cell bodies and proximal dendritic processes of isolated neurons in layers II/III of the PL-mPFC were collected using a z-series of 30– 50 images with a depth interval of 0.23 μm. Imaris was used to process stacked images, select regions of interest, and quantify PSD-95-VGLUT1 co-localization (Fogarty et al., 2013). The “create surface” tool in Imaris was used to generate a solid surface best matching the neuronal cell body or dendritic processes based on the fluorescence signal. All immunostaining of the postsynaptic protein PSD-95 falling outside of the boundary of the cell surface and all immunostaining of the presynaptic protein VGLUT1 within the boundary of the cell surface were disregarded using the “mask all” feature in Imaris. The spots detection algorithm in Imaris was used to quantify VGLUT1 and PSD-95 puncta using a spot diameter threshold of 0.3 μm. Imaris’ “co-localize spots” algorithm was then used to determine which PSD-95 postsynaptic puncta were within a 1-μm threshold distance of VGLUT1 presynaptic specializations. These co-localized PSD-95-VGLUT1 spots were subsequently superimposed onto the neuronal surface of the cell body or dendrites to provide their location and distribution using Imaris’ “find spots close to surface” algorithm (distance threshold of 1 μm). The density of PSD-95 puncta that co-localized with VGLUT1 (puncta number/μm2) on the somatic surface of the neuron was then calculated by dividing the number of PSD-95-VGLUT1 puncta on the somatic surface by the area of the somatic surface. For dendrite surface rendering, the regions of interest were selected manually with the dendrites chosen to be 10–30 μm away from the cell soma. Dendritic lengths were determined using Imaris’ “auto depth” algorithm. The density of PSD-95 puncta that co-localized with VGLUT1 (puncta number/μm) on dendrites was then calculated by dividing the number of PSD-95-VGLUT1 puncta on the dendrites by the length of the selected dendrites.
Chemogenetic manipulation
ACTB-Ophn1cKO mice were bilaterally injected in the PL-mPFC with AAV-DIO-hM4Di-mCherry, AAV-DIO-hM3D(Gq)-mCherry, or AAV-DIO-mCherry. Four to five weeks later, the mice were injected intraperitoneally with either saline solution or clozapine N-oxide (CNO) (Sigma-Aldrich, C-0832; NIMH Chemical Synthesis and Drug Supply Program, C-929) (5 mg/kg of body weight for hM4Di and 1 mg/kg of body weight for hM3D(Gq)) during the LH procedure (i.e., 30 min before the onset of each of the two LH induction sessions, and again 30 min before the onset of the testing session), 30 min before the onset of each of the two induction sessions, or 30 min before the onset of the testing session.
Cannula implantation
Mice were anaesthetized with ketamine (100 mg/kg of body weight) supplemented with dexmedetomidine hydrochloride (0.4 mg/kg of body weight), placed in a stereotaxic injection frame (myNeuroLab.com), and their skull was exposed. Two stainless-steel guide cannulae (26-gauge; RWD Life Science) were implanted into the PL-mPFC (anterior–posterior (AP): 1.98 mm; mediolateral (ML): ± 0.5 mm; dorsoventral (DV): −2 mm under the skull surface, relative to bregma). One cannula was introduced to this position at 0° while the other was laterally tilted by 30° with respect to the vertical axis. The cannulae were fixed to the skull using dental cement (Metabond, Parkell, Inc.). 33-gauge dummy cannulae (RWD Life Science Inc.) were subsequently inserted into the guide cannulae to prevent clogging and reduce the risk of infection. After surgery, the mice were single housed and allowed to recover for at least 7 days prior to administration of the Rho-kinase inhibitor fasudil or PKA inhibitor cAMPS-Rp.
Fasudil and cAMPS-Rp administration
Fasudil (40 μM, Ki = 0.33 μM) and cAMPS-Rp (22 μM, Ki =11 μM) were dissolved in 0.9% saline. Prior to local fasudil or cAMPS-Rp infusion, tip-sharpened 33-gauge injection cannulae were inserted into each of the guide cannulae to ensure clear passage. They were thereafter removed and replaced with new tip-sharpened 33-gauge injection cannulae connected to a microsyringe. Approximately 0.5 μl of fasudil or 0.4 μl cAMPS-Rp solution was then infused (0.2 μl/min) via the injection cannulae to deliver the drug locally into each of the implantation sites in the PL-mPFC. To allow penetration of the drug, the injection cannulae were left in place for an additional 5 min. Mice were subjected to the LH procedure following the 5th infusion of fasudil (see Fig. 8H). cAMPS-Rp was infused 30 min before the onset of each of the two LH induction sessions and again 30 min before the onset of the testing session (see Fig. S9A). Following completion of all behavioral tests, the mice were injected with 0.5 μl CTB-488 solution via the 33-gauge injection cannulae to verify the site of drug infusion. Mice were euthanized 30 min after CTB injection, brain slices were prepared, and the cannulae positions were verified. Only data from mice with the correct injection sites were used. For intraperitoneal injections, mice were injected with fasudil (10 mg/kg of body weight) twice daily for 10 days (see Fig. 8E).
QUANTIFICATION AND STATISTICAL ANALYSES
Data analysis and statistics
Statistical tests were performed with GraphPad Prism and the R programming environment. Normality was tested with the Shapiro-Wilk normality test and homogeneity of variances with the Fligner-Killen test. Behavioral parameters for LH were analyzed with k-means cluster analysis and linear discriminant analysis followed by a non-parametric Mann-Whitney test for comparison of two experimental groups or by non-parametric Kruskal-Wallis H one-way ANOVA with a Dunn’s post-hoc test for comparison of three experimental groups. For non-parametric tests of multiple factors and their interactions, Aligned-Rank Transform ANOVA from the R ARTool package followed by multiple comparisons using Mann-Whitney test with Bonferroni correction was used. For parametric tests of two experimental groups, two-tailed unpaired Student’s t-tests were used. For parametric tests of multiple factors and their interactions, two-way ANOVA or two-way repeated measures ANOVA followed by Bonferroni (Amat et al., 2005) post-hoc test were used. Behavioral data from animals in which viral injection missed the target area were excluded from the analysis. Correlations between viral infection efficiency in PL-mPFC and the behavioral effect were computed using Pearson’s correlation coefficient.
Supplementary Material
Video S1–3. Ophn1 deficient mice subjected to the LH procedure do not display perseverative behaviors. Related to Figure 1.
ACTB-Ophn1WT and ACTB-Ophn1cKO mice were subjected to the learned helplessness (LH) procedure. In brief, mice were first exposed to two induction sessions consisting of 360 inescapable, uncontrollable electric foot shocks and thereafter subjected to a testing session consisting of escapable foot shocks. The testing session was performed in a shuttle box equipped with an electrical grid floor, a door separating the two halves, and photocell detectors. Behavioral performance was evaluated over 30 trials of escapable foot shocks. Each trial started with a 5 s cue light, followed by the foot shocks. When an animal shuttled to another compartment of the box during the 5 s cue light presentation (and thus before the shock onset), avoidance was scored. If the animal shuttled during the 10 s shock (i.e., escaped), escape latency was measured. Failure was recorded if no shuttling was made during the 10 s shock presentation. Shocks were terminated if the animal shuttled to another side of the box (in case of escape) or at the end of the 10 s shock (in case of failure). Animals’ behavior was classified as being “resilient” or “learned helpless” based on their behavioral parameters in the LH testing session (see Methods). Representative examples (2 trials) of an ACTB-Ophn1WT mouse and an ACTB-Ophn1cKO mouse displaying helpless behavior (Video S1 and Video S2, respectively) and of an ACTB-Ophn1WT mouse exhibiting resilience (Video S3) are shown.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-OPHN1 | This paper | N/A |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat# 5174 RRID: AB_10622025 |
| Mouse monoclonal anti-NeuN | Millipore | Cat#MAB377 RRID:AB_2298772 |
| Mouse monoclonal anti-PSD-95 | Millipore | Cat#MAB1598 RRID:AB_94278 |
| Mouse monoclonal anti-Parvalbumin | Sigma-Aldrich | Cat# P3088 RRID: AB_477329 |
| Rat monoclonal anti-Somatostatin | Millipore | Cat# MAB354 RRID: AB_2255365 |
| Mouse monoclonal anti-Neurogranin | BioLegend | Cat# 845602 RRID: AB_2566622 |
| Rabbit polyclonal anti-VGLUT1 | Synaptic Systems | Cat# 135_302 RRID: AB_887877 |
| Goat anti-Rabbit IgG (H+L) Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A11008 RRID: AB_143165 |
| Goat anti-Mouse IgG2a Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A21131 RRID:AB_141618 |
| Goat Anti-Mouse IgG (H+L) Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A21422 RRID: AB_2535844 |
| Goat anti-Rat IgG (H+L) Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A21434 RRID: AB_2535855 |
| Goat anti-Mouse IgG1 Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A21127 RRID:AB_141596 |
| Goat anti-Rabbit IgG (H+L) Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21245 RRID:AB_141775 |
| Goat anti-Rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074 RRID: AB_2099233 |
| Bacterial and Virus Strains | ||
| AAV/DJ8-CAG-Cre-GFP | This paper | N/A |
| AAV9-CAG-GFP | Viral Vector Core, University of North Carolina | Addgene plasmid # 37825 (gift from Edward Boyden) |
| AAV/DJ8-CAG-fDIO-Cre-GFP | This paper | N/A |
| AAV8-Ef1a-fDIO-mCherry | Gift from Z.J. Huang (Lu et. al., 2017) | N/A |
| AAV8-Ef1a-DIO-hM4Di-mCherry | MIT Viral Core | Generated by Rachael L. Neve |
| AAV2-hSyn-DIO-hM3D(Gq)-mCherry | Addgene | Addgene viral prep # 44361-AAV2 (plasmid # 44361 was a gift from Bryan Roth) |
| AAV2-hSyn-DIO-mCherry | Addgene | Addgene viral prep # 50459-AAV2 (plasmid # 50459 was gift from Bryan Roth) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| cOmplete, EDTA-free protease inhibitor cocktail | Sigma-Aldrich | Cat# 11873580001 |
| Clozapine N-oxide | Sigma-Aldrich/NIMH | Sigma-Aldrich Cat# C-0832/NIMH Cat# C-929 |
| Bovine serum albumin | Equitech-Bio, Inc. | Cat# BAH65 |
| Tetrodotoxin (TTX) | Tocris | Cat# 1078 |
| Picrotoxin (PTX) | Sigma-Aldrich | Cat# 010M1441 |
| 6-cyano-7-nitroquinoxaline-2, 3-dione | Sigma-Aldrich | Cat# C127 |
| DL-2-amino-5-phosphonopentanoic acid | Sigma-Aldrich | Cat# 074K38051 |
| Cresyl violet | Sigma-Aldrich | Cat# C5042 |
| Xylene | Poly Scientific R&D Corp. | Cat# c823 |
| DPX mounting medium | Sigma-Aldrich | Cat# 06522 |
| Fluoromount-G | SouthernBiotech | Cat# 0100–01 |
| Fasudil Mono HCl Salt | LC Laboratories | Cat# F-4660 |
| cAMPS-Rp, triethylammonium salt | TOCRIS Bioscience | Cat# 1337 |
| Critical Commercial Assays | ||
| AAV-DJ/8 Helper Free Expression System | Cell Biolabs, Inc | Cat# VPK-410-DJ-8 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Pierce™ ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat# 32106 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216 RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ophn1-cKO | This paper | N/A |
| Mouse: B6N.FVB-Tmem163Tg(ACTB-cre)2Mrt/CjDswJ | Jackson Laboratory | RRID: IMSR_JAX:019099 |
| Mouse: B6.Cg-Zfp335tm1.2Caw Emx1tm1(cre)Krj/J | Jackson Laboratory | RRID: IMSR_JAX:022762 |
| Mouse: B6N.Cg-Ssttm2.1(cre)Zjh/J | Jackson Laboratory | RRID: IMSR_JAX:018973 |
| Mouse: B6;129P2-Pvalbtm1(cre)Arbr/J | Jackson Laboratory | RRID: IMSR_JAX:008069 |
| Mouse: PV-Flp | Gift from Z.J. Huang (He et al., 2016) | N/A |
| Mouse: B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratory | RRID: IMSR_JAX:007908 |
| Mouse: C57BL/6J | Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: CD1 | Charles River | Cat# CRL:22 RRID: IMSR_CRL:22 |
| Recombinant DNA | ||
| pCAG-Cre:GFP | Gift from Connie Cepko (Matsuda et al., 2007) | Addgene plasmid # 13776 |
| pAAV-CAG-GFP | Gift from Edward Boyden | Addgene plasmid # 37825 |
| pAAV-CAG-Cre-GFP | This paper | N/A |
| pAAV-CAG-fDIO-Cre-GFP | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | http://imagej.nih.gov/ij/; RRID: SCR_003070 |
| LSM 5 Image Browser | Zeiss | http://www.embl.de/eamnet/html/body_image_browser.html; RRID: SCR_014344 |
| Zeiss Zen (Blue Edition) Imaging Software | Zeiss | http://www.zeiss.com/microscopy/en_us/products/microscope-software/zen.html#introduction; RRID: SCR_013672 |
| Axon pCLAMP 10 Electrophysiological Data Acquisition & Analysis Software | Molecular Devices | http://www.moleculardevices.com/products/software/pclamp.html RRID: SCR_011323 |
| Mini Analysis Program | Synaptosoft | http://www.synaptosoft.com/MiniAnalysis/; RRID: SCR_002184 |
| GraphPad Prism 7 | GraphPad | http://www.graphpad.com/scientific-software/prism; RRID: SCR_002798 |
| Adobe Illustrator CS6 | Adobe | http://www.adobe.com/products/illustrator.html RRID:SCR_014198 |
| R Project for Statistical Computing | r-project.org | http://www.r-project.org/ RRID: SCR_001905 |
| Graphic State 3.03 | Coulbourn Instruments | N/A |
| Ethovision XT 5.1 | Noldus | https://www.noldus.com/ethovision RRID: SCR_000441 |
| Imaris 9.6 | Oxford Instruments | http://www.bitplane.com/imaris RRID: SCR_007370 |
Highlights.
Ophn1 deficiency markedly enhances stress-induced helpless/depressive-like behavior
Ophn1 deletion in PV interneurons in PL-mPFC is sufficient to induce helplessness
Ophn1 deficiency in PV interneurons leads to enhanced PL-mPFC neuronal activity
Inhibiting neuronal activity or RhoA/Rho-kinase pathway reverses helpless behavior
ACKNOWLEDGMENTS
We thank members of the Van Aelst lab for helpful discussions. We also thank Y-T Yang for assistance in the generation of Ophn1-flox mice and Z. J. Huang for providing reagents and Cre-driver mouse lines. This work was supported by US NIH grants R01MH119819 and R01NS116897 to L.VA, US NIH fellowship F31MH117871 to N.B.G., and NARSAD Young Investigator grant to M.W.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Al-Owain M, Kaya N, Al-Zaidan H, Al-Hashmi N, Al-Bakheet A, Al-Muhaizea M, Chedrawi A, Basran RK, and Milunsky A (2011). Novel intragenic deletion in OPHN1 in a family causing XLMR with cerebellar hypoplasia and distinctive facial appearance. Clin Genet 79, 363–370. [DOI] [PubMed] [Google Scholar]
- Amat J, Baratta MV, Paul E, Bland ST, Watkins LR, and Maier SF (2005). Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat Neurosci 8, 365–371. [DOI] [PubMed] [Google Scholar]
- Arita R, Hata Y, Nakao S, Kita T, Miura M, Kawahara S, Zandi S, Almulki L, Tayyari F, Shimokawa H, et al. (2009). Rho kinase inhibition by fasudil ameliorates diabetes-induced microvascular damage. Diabetes 58, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF (2009). Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci 10, 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson PA, Martin CR, and Rankin J (2009). Resilience revisited. J Psychiatr Ment Health Nurs 16, 137–145. [DOI] [PubMed] [Google Scholar]
- Ayuso E, Mingozzi F, and Bosch F (2010). Production, purification and characterization of adeno-associated vectors. Curr Gene Ther 10, 423–436. [DOI] [PubMed] [Google Scholar]
- Bagot RC, Labonte B, Pena CJ, and Nestler EJ (2014). Epigenetic signaling in psychiatric disorders: stress and depression. Dialogues Clin Neurosci 16, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman DJ, Sheldon JB, Sherman JA, and Harchik AE (1990). Balancing the right to habilitation with the right to personal liberties: the rights of people with developmental disabilities to eat too many doughnuts and take a nap. J Appl Behav Anal 23, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon AW (2001). Models of pain: hot-plate and formalin test in rodents. Curr Protoc Pharmacol Chapter 5, Unit 5 7. [DOI] [PubMed] [Google Scholar]
- Baratta MV, Zarza CM, Gomez DM, Campeau S, Watkins LR, and Maier SF (2009). Selective activation of dorsal raphe nucleus-projecting neurons in the ventral medial prefrontal cortex by controllable stress. Eur J Neurosci 30, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudewijns L, Ronsse E, Verstraete V, Sabbe B, Morrens M, and Bertelli MO (2018). Problem behaviours and Major Depressive Disorder in adults with intellectual disability and autism. Psychiatry Res 270, 769–774. [DOI] [PubMed] [Google Scholar]
- Beery AK, and Zucker I (2011). Sex bias in neuroscience and biomedical research. Neurosci Biobehav Rev 35, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann C, Zerres K, Senderek J, Rudnik-Schoneborn S, Eggermann T, Hausler M, Mull M, and Ramaekers VT (2003). Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain 126, 1537–1544. [DOI] [PubMed] [Google Scholar]
- Bertelli M, Scuticchio D, Ferrandi A, Lassi S, Mango F, Ciavatta C, Porcelli C, Bianco A, and Monchieri S (2012). Reliability and validity of the SPAID-G checklist for detecting psychiatric disorders in adults with intellectual disability. Res Dev Disabil 33, 382–390. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Der-Sarkissian H, Billuart P, Tissot M, Des Portes V, Bruls T, Chabrolle JP, Chauveau P, Cherry M, Kahn A, et al. (1997). Mapping of the X-breakpoint involved in a balanced X;12 translocation in a female with mild mental retardation. Eur J Hum Genet 5, 105–109. [PubMed] [Google Scholar]
- Billuart P, Bienvenu T, Ronce N, des Portes V, Vinet MC, Zemni R, Roest Crollius H, Carrie A, Fauchereau F, Cherry M, et al. (1998). Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 392, 923–926. [DOI] [PubMed] [Google Scholar]
- Borthwick-Duffy SA (1994). Epidemiology and prevalence of psychopathology in people with mental retardation. J Consult Clin Psychol 62, 17–27. [DOI] [PubMed] [Google Scholar]
- Busa T, Caietta E, Chabrol B, Girard N, Philip N, and Missirian C (2017). Large in-frame intragenic deletion of OPHN1 in a male patient with a normal intelligence quotient score. Clin Dysmorphol 26, 47–49. [DOI] [PubMed] [Google Scholar]
- Capuzzo G, and Floresco SB (2020). Prelimbic and Infralimbic Prefrontal Regulation of Active and Inhibitory Avoidance and Reward-Seeking. J Neurosci 40, 4773–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathomas F, Murrough JW, Nestler EJ, Han MH, and Russo SJ (2019). Neurobiology of Resilience: Interface Between Mind and Body. Biol Psychiatry 86, 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerniauskas I, Winterer J, de Jong JW, Lukacsovich D, Yang H, Khan F, Peck JR, Obayashi SK, Lilascharoen V, Lim BK, et al. (2019). Chronic Stress Induces Activity, Synaptic, and Transcriptional Remodeling of the Lateral Habenula Associated with Deficits in Motivated Behaviors. Neuron 104, 899–915 e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrol B, Girard N, N’Guyen K, Gerard A, Carlier M, Villard L, and Philip N (2005). Delineation of the clinical phenotype associated with OPHN1 mutations based on the clinical and neuropsychological evaluation of three families. Am J Med Genet A 138, 314–317. [DOI] [PubMed] [Google Scholar]
- Chiurazzi P, and Pirozzi F (2016). Advances in understanding - genetic basis of intellectual disability. F1000Res 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourbaji S, Zacher C, Sanchis-Segura C, Dormann C, Vollmayr B, and Gass P (2005). Learned helplessness: validity and reliability of depressive-like states in mice. Brain Res Brain Res Protoc 16, 70–78. [DOI] [PubMed] [Google Scholar]
- Cooper SA, Smiley E, Allan LM, Jackson A, Finlayson J, Mantry D, and Morrison J (2009). Adults with intellectual disabilities: prevalence, incidence and remission of self-injurious behaviour, and related factors. J Intellect Disabil Res 53, 200–216. [DOI] [PubMed] [Google Scholar]
- Cooper SA, Smiley E, Morrison J, Williamson A, and Allan L (2007). An epidemiological investigation of affective disorders with a population-based cohort of 1023 adults with intellectual disabilities. Psychol Med 37, 873–882. [DOI] [PubMed] [Google Scholar]
- Covington HE 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, LaPlant Q, Mouzon E, Ghose S, Tamminga CA, et al. (2010). Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci 30, 16082–16090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb S, Thomas M, and Bright C (2001). Mental disorder in adults with intellectual disability. 1: Prevalence of functional psychiatric illness among a community-based population aged between 16 and 64 years. J Intellect Disabil Res 45, 495–505. [DOI] [PubMed] [Google Scholar]
- des Portes V, Boddaert N, Sacco S, Briault S, Maincent K, Bahi N, Gomot M, Ronce N, Bursztyn J, Adamsbaum C, et al. (2004). Specific clinical and brain MRI features in mentally retarded patients with mutations in the Oligophrenin-1 gene. Am J Med Genet A 124A, 364–371. [DOI] [PubMed] [Google Scholar]
- Dias-Ferreira E, Sousa JC, Melo I, Morgado P, Mesquita AR, Cerqueira JJ, Costa RM, and Sousa N (2009). Chronic stress causes frontostriatal reorganization and affects decision-making. Science 325, 621–625. [DOI] [PubMed] [Google Scholar]
- Dolzani SD, Baratta MV, Amat J, Agster KL, Saddoris MP, Watkins LR, and Maier SF (2016). Activation of a Habenulo-Raphe Circuit Is Critical for the Behavioral and Neurochemical Consequences of Uncontrollable Stress in the Male Rat. eNeuro 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL, and Furey ML (2008). Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct 213, 93–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulin PL, Hanson BL, and King DK (2013). Perceived control as a longitudinal moderator of late-life stressors on depressive symptoms. Aging Ment Health 17, 718–723. [DOI] [PubMed] [Google Scholar]
- Duman RS, Sanacora G, and Krystal JH (2019). Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 102, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embregts PJ, du Bois MG, and Graef N (2010). Behavior problems in children with mild intellectual disabilities: an initial step towards prevention. Res Dev Disabil 31, 1398–1403. [DOI] [PubMed] [Google Scholar]
- Esbensen AJ, and Benson BA (2006). A prospective analysis of life events, problem behaviours and depression in adults with intellectual disability. J Intellect Disabil Res 50, 248–258. [DOI] [PubMed] [Google Scholar]
- Fauchereau F, Herbrand U, Chafey P, Eberth A, Koulakoff A, Vinet MC, Ahmadian MR, Chelly J, and Billuart P (2003). The RhoGAP activity of OPHN1, a new F-actin-binding protein, is negatively controlled by its amino-terminal domain. Mol Cell Neurosci 23, 574–586. [DOI] [PubMed] [Google Scholar]
- Feder A, Nestler EJ, and Charney DS (2009). Psychobiology and molecular genetics of resilience. Nat Rev Neurosci 10, 446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczi EA, Zalocusky KA, Liston C, Grosenick L, Warden MR, Amatya D, Katovich K, Mehta H, Patenaude B, Ramakrishnan C, et al. (2016). Prefrontal cortical regulation of brainwide circuit dynamics and reward-related behavior. Science 351, aac9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- File SE, Lippa AS, Beer B, and Lippa MT (2005). Animal tests of anxiety. Curr Protoc Pharmacol Chapter 5, Unit 5 38. [DOI] [PubMed] [Google Scholar]
- Fogarty MJ, Hammond LA, Kanjhan R, Bellingham MC, and Noakes PG (2013). A method for the three-dimensional reconstruction of Neurobiotin-filled neurons and the location of their synaptic inputs. Front Neural Circuits 7, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin TB, Saab BJ, and Mansuy IM (2012). Neural mechanisms of stress resilience and vulnerability. Neuron 75, 747–761. [DOI] [PubMed] [Google Scholar]
- Gacek M, Smolen T, and Pilecka W (2017). Consequences of Learned Helplessness and Recognition of the State of Cognitive Exhaustion in Persons with Mild Intellectual Disability. Adv Cogn Psychol 13, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rojo G, Fresno C, Vilches N, Diaz-Veliz G, Mora S, Aguayo F, Pacheco A, Parra-Fiedler N, Parra CS, Rojas PS, et al. (2017). The ROCK Inhibitor Fasudil Prevents Chronic Restraint Stress-Induced Depressive-Like Behaviors and Dendritic Spine Loss in Rat Hippocampus. Int J Neuropsychopharmacol 20, 336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal S, Hare B, and Duman RS (2017). Prefrontal Cortex GABAergic Deficits and Circuit Dysfunction in the Pathophysiology and Treatment of Chronic Stress and Depression. Curr Opin Behav Sci 14, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden SA, Covington HE 3rd, Berton O, and Russo SJ (2011). A standardized protocol for repeated social defeat stress in mice. Nat Protoc 6, 1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, and Morrison JH (2009). Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience 164, 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Akerman CJ, Cross JR, Van der Veken L, and Van Aelst L (2004). The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci 7, 364–372. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Protic D, Rajaratnam A, Salcedo-Arellano MJ, Aydin EY, and Schneider A (2018). Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front Psychiatry 9, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamani C, Diwan M, Isabella S, Lozano AM, and Nobrega JN (2010a). Effects of different stimulation parameters on the antidepressant-like response of medial prefrontal cortex deep brain stimulation in rats. J Psychiatr Res 44, 683–687. [DOI] [PubMed] [Google Scholar]
- Hamani C, Diwan M, Macedo CE, Brandao ML, Shumake J, Gonzalez-Lima F, Raymond R, Lozano AM, Fletcher PJ, and Nobrega JN (2010b). Antidepressant-like effects of medial prefrontal cortex deep brain stimulation in rats. Biol Psychiatry 67, 117–124. [DOI] [PubMed] [Google Scholar]
- Han MH, and Nestler EJ (2017). Neural Substrates of Depression and Resilience. Neurotherapeutics 14, 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley SL, and Maclean WE (2008). Coping Strategies of Adults with Mild Intellectual Disability for Stressful Social Interactions. J Ment Health Res Intellect Disabil 1, 109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley SL, and Maclean WE (2009). Depression in adults with mild intellectual disability: role of stress, attributions, and coping. Am J Intellect Dev Disabil 114, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley SL, and MacLean WE Jr. (2005). Perceptions of stress and coping strategies among adults with mild mental retardation: insight into psychological distress. Am J Ment Retard 110, 285–297. [DOI] [PubMed] [Google Scholar]
- He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, et al. (2016). Strategies and Tools for Combinatorial Targeting of GABAergic Neurons in Mouse Cerebral Cortex. Neuron 92, 555. [DOI] [PubMed] [Google Scholar]
- Herman JP (2013). Neural control of chronic stress adaptation. Front Behav Neurosci 7, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, and Arber S (2005). A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol 3, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz A, Mulsch F, Schwarz MK, Hollmann M, Dobrossy MD, Coenen VA, Bartos M, Normann C, Biber K, van Calker D, et al. (2019). Enhanced mGlu5 Signaling in Excitatory Neurons Promotes Rapid Antidepressant Effects via AMPA Receptor Activation. Neuron 104, 338–352 e337. [DOI] [PubMed] [Google Scholar]
- Hurley AD (2006). Mood disorders in intellectual disability. Curr Opin Psychiatry 19, 465–469. [DOI] [PubMed] [Google Scholar]
- Janssen CG, Schuengel C, and Stolk J (2002). Understanding challenging behaviour in people with severe and profound intellectual disability: a stress-attachment model. J Intellect Disabil Res 46, 445–453. [DOI] [PubMed] [Google Scholar]
- Jiang X, Lachance M, and Rossignol E (2016). Involvement of cortical fast-spiking parvalbumin-positive basket cells in epilepsy. Prog Brain Res 226, 81–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Karkowski LM, and Prescott CA (1999). Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry 156, 837–841. [DOI] [PubMed] [Google Scholar]
- Khelfaoui M, Denis C, van Galen E, de Bock F, Schmitt A, Houbron C, Morice E, Giros B, Ramakers G, Fagni L, et al. (2007). Loss of X-linked mental retardation gene oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J Neurosci 27, 9439–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelfaoui M, Gambino F, Houbaert X, Ragazzon B, Muller C, Carta M, Lanore F, Srikumar BN, Gastrein P, Lepleux M, et al. (2014). Lack of the presynaptic RhoGAP protein oligophrenin1 leads to cognitive disabilities through dysregulation of the cAMP/PKA signalling pathway. Philos Trans R Soc Lond B Biol Sci 369, 20130160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelfaoui M, Pavlowsky A, Powell AD, Valnegri P, Cheong KW, Blandin Y, Passafaro M, Jefferys JG, Chelly J, and Billuart P (2009). Inhibition of RhoA pathway rescues the endocytosis defects in Oligophrenin1 mouse model of mental retardation. Hum Mol Genet 18, 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Perova Z, Mirrione MM, Pradhan K, Henn FA, Shea S, Osten P, and Li B (2016). Whole-Brain Mapping of Neuronal Activity in the Learned Helplessness Model of Depression. Front Neural Circuits 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, and Binder EB (2015). Epigenetics of Stress-Related Psychiatric Disorders and Gene x Environment Interactions. Neuron 86, 1343–1357. [DOI] [PubMed] [Google Scholar]
- Koch JC, Tatenhorst L, Roser AE, Saal KA, Tonges L, and Lingor P (2018). ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol Ther 189, 1–21. [DOI] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121, 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, et al. (2007). Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131, 391–404. [DOI] [PubMed] [Google Scholar]
- Kumar S, Black SJ, Hultman R, Szabo ST, DeMaio KD, Du J, Katz BM, Feng G, Covington HE 3rd, and Dzirasa K (2013). Cortical control of affective networks. J Neurosci 33, 1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Tye KM, and Warden MR (2014). Progress in understanding mood disorders: optogenetic dissection of neural circuits. Genes Brain Behav 13, 38–51. [DOI] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, and Malinow R (2011). Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature 470, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Tucciarone J, Padilla-Coreano N, He M, Gordon JA, and Huang ZJ (2017). Selective inhibitory control of pyramidal neuron ensembles and cortical subnetworks by chandelier cells. Nat Neurosci 20, 1377–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macri S (2012). On the incongruity between developmental plasticity and methodological rigidity. Front Behav Neurosci 6, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF (2015). Behavioral control blunts reactions to contemporaneous and future adverse events: medial prefrontal cortex plasticity and a corticostriatal network. Neurobiol Stress 1, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF, and Seligman ME (2016). Learned helplessness at fifty: Insights from neuroscience. Psychol Rev 123, 349–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF, and Watkins LR (2005). Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neurosci Biobehav Rev 29, 829–841. [DOI] [PubMed] [Google Scholar]
- Manvich DF, Webster KA, Foster SL, Farrell MS, Ritchie JC, Porter JH, and Weinshenker D (2018). The DREADD agonist clozapine N-oxide (CNO) is reverse-metabolized to clozapine and produces clozapine-like interoceptive stimulus effects in rats and mice. Sci Rep 8, 3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus DJ, Bedse G, Gaulden AD, Ryan JD, Kondev V, Winters ND, Rosas-Vidal LE, Altemus M, Mackie K, Lee FS, et al. (2020). Endocannabinoid Signaling Collapse Mediates Stress-Induced Amygdalo-Cortical Strengthening. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez VK, Saldana-Morales F, Sun JJ, Zhu PJ, Costa-Mattioli M, and Ray RS (2019). Off-Target Effects of Clozapine-N-Oxide on the Chemosensory Reflex Are Masked by High Stress Levels. Front Physiol 10, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, and Cepko CL (2007). Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A 104, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS (2007). Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev 87, 873–904. [DOI] [PubMed] [Google Scholar]
- McEwen BS, and Morrison JH (2013). The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron 79, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JT, and Botvinick MM (2010). Prefrontal cortex, cognitive control, and the registration of decision costs. Proc Natl Acad Sci U S A 107, 7922–7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKlveen JM, Myers B, and Herman JP (2015). The medial prefrontal cortex: coordinator of autonomic, neuroendocrine and behavioural responses to stress. J Neuroendocrinol 27, 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehregan H, Najmabadi H, and Kahrizi K (2016). Genetic Studies in Intellectual Disability and Behavioral Impairment. Arch Iran Med 19, 363–375. [PubMed] [Google Scholar]
- Meziane H, Khelfaoui M, Morello N, Hiba B, Calcagno E, Reibel-Foisset S, Selloum M, Chelly J, Humeau Y, Riet F, et al. (2016). Fasudil treatment in adult reverses behavioural changes and brain ventricular enlargement in Oligophrenin-1 mouse model of intellectual disability. Hum Mol Genet 25, 2314–2323. [DOI] [PubMed] [Google Scholar]
- Moortgat S, Lederer D, Deprez M, Buzatu M, Clapuyt P, Boulanger S, Benoit V, Mary S, Guichet A, Ziegler A, et al. (2018). Expanding the phenotypic spectrum associated with OPHN1 mutations: Report of 17 individuals with intellectual disability but no cerebellar hypoplasia. Eur J Med Genet 61, 442–450. [DOI] [PubMed] [Google Scholar]
- Morgan VA, Leonard H, Bourke J, and Jablensky A (2008). Intellectual disability co-occurring with schizophrenia and other psychiatric illness: population-based study. Br J Psychiatry 193, 364–372. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, and Caroni P (2018). Infralimbic cortex is required for learning alternatives to prelimbic promoted associations through reciprocal connectivity. Nat Commun 9, 2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadif Kasri N, Nakano-Kobayashi A, Malinow R, Li B, and Van Aelst L (2009). The Rho-linked mental retardation protein oligophrenin-1 controls synapse maturation and plasticity by stabilizing AMPA receptors. Genes Dev 23, 1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadif Kasri N, Nakano-Kobayashi A, and Van Aelst L (2011). Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron 72, 300–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano-Kobayashi A, Kasri NN, Newey SE, and Van Aelst L (2009). The Rho-linked mental retardation protein OPHN1 controls synaptic vesicle endocytosis via endophilin A1. Curr Biol 19, 1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano-Kobayashi A., Tai Y., Nadif Kasri N., and Van Aelst L. (2014). The X-linked mental retardation protein OPHN1 interacts with Homer1b/c to control spine endocytic zone positioning and expression of synaptic potentiation. J Neurosci 34, 8665–8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF (2008). Applications for ROCK kinase inhibition. Curr Opin Cell Biol 20, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota KT, Liu RJ, Voleti B, Maldonado-Aviles JG, Duric V, Iwata M, Dutheil S, Duman C, Boikess S, Lewis DA, et al. (2014). REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat Med 20, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Dong S, and Roth BL (2010). Generation of designer receptors exclusively activated by designer drugs (DREADDs) using directed molecular evolution. Curr Protoc Neurosci Chapter 4, Unit 4 33. [DOI] [PubMed] [Google Scholar]
- Perova Z, Delevich K, and Li B (2015). Depression of excitatory synapses onto parvalbumin interneurons in the medial prefrontal cortex in susceptibility to stress. J Neurosci 35, 3201–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip N, Chabrol B, Lossi AM, Cardoso C, Guerrini R, Dobyns WB, Raybaud C, and Villard L (2003). Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia. J Med Genet 40, 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirozzi F, Di Raimo FR, Zanni G, Bertini E, Billuart P, Tartaglione T, Tabolacci E, Brancaccio A, Neri G, and Chiurazzi P (2011). Insertion of 16 amino acids in the BAR domain of the oligophrenin 1 protein causes mental retardation and cerebellar hypoplasia in an Italian family. Hum Mutat 32, E2294–2307. [DOI] [PubMed] [Google Scholar]
- Plummer JT, Gordon AJ, and Levitt P (2016). The Genetic Intersection of Neurodevelopmental Disorders and Shared Medical Comorbidities - Relations that Translate from Bench to Bedside. Front Psychiatry 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AD, Gill KK, Saintot PP, Jiruska P, Chelly J, Billuart P, and Jefferys JG (2012). Rapid reversal of impaired inhibitory and excitatory transmission but not spine dysgenesis in a mouse model of mental retardation. J Physiol 590, 763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, and Drevets WC (2012). Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn Sci 16, 61–71. [DOI] [PubMed] [Google Scholar]
- Redolfi N, Galla L, Maset A, Murru L, Savoia E, Zamparo I, Gritti A, Billuart P, Passafaro M, and Lodovichi C (2016). Oligophrenin-1 regulates number, morphology and synaptic properties of adult-born inhibitory interneurons in the olfactory bulb. Hum Mol Genet 25, 5198–5211. [DOI] [PubMed] [Google Scholar]
- Riga D, Matos MR, Glas A, Smit AB, Spijker S, and Van den Oever MC (2014). Optogenetic dissection of medial prefrontal cortex circuitry. Front Syst Neurosci 8, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan SC, and Roth BL (2011). Remote control of neuronal signaling. Pharmacol Rev 63, 291–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudebeck PH, Rich EL, and Mayberg HS (2019). From bed to bench side: Reverse translation to optimize neuromodulation for mood disorders. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Murrough JW, Han MH, Charney DS, and Nestler EJ (2012). Neurobiology of resilience. Nat Neurosci 15, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Reboucas CB, Belet S, Guedes de Almeida L, Ribeiro MG, Medina-Acosta E, Bahia PR, Alves da Silva AF, Lima dos Santos F, Borges de Lacerda GC, Pimentel MM, et al. (2014). A novel in-frame deletion affecting the BAR domain of OPHN1 in a family with intellectual disability and hippocampal alterations. Eur J Hum Genet 22, 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MV, Trumbach D, Weber P, Wagner K, Scharf SH, Liebl C, Datson N, Namendorf C, Gerlach T, Kuhne C, et al. (2010). Individual stress vulnerability is predicted by short-term memory and AMPA receptor subunit ratio in the hippocampus. J Neurosci 30, 16949–16958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TS, Wojcik MH, Pelletier RC, Edward HL, Picker JD, Holm IA, Towne MC, Beggs AH, and Agrawal PB (2019). Expanding the phenotypic spectrum associated with OPHN1 variants. Eur J Med Genet 62, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott HM, and Havercamp SM (2014). Mental health for people with intellectual disability: the impact of stress and social support. Am J Intellect Dev Disabil 119, 552–564. [DOI] [PubMed] [Google Scholar]
- Seese RR, Chen LY, Cox CD, Schulz D, Babayan AH, Bunney WE, Henn FA, Gall CM, and Lynch G (2013). Synaptic abnormalities in the infralimbic cortex of a model of congenital depression. J Neurosci 33, 13441–13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seligman ME (1978). Learned helplessness as a model of depression. Comment and integration. J Abnorm Psychol 87, 165–179. [PubMed] [Google Scholar]
- Shin S, Kwon O, Kang JI, Kwon S, Oh S, Choi J, Kim CH, and Kim DG (2015). mGluR5 in the nucleus accumbens is critical for promoting resilience to chronic stress. Nat Neurosci 18, 1017–1024. [DOI] [PubMed] [Google Scholar]
- Shrestha P, Mousa A, and Heintz N (2015). Layer 2/3 pyramidal cells in the medial prefrontal cortex moderate stress induced depressive behaviors. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, and Quirk GJ (2011). Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36, 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley E, Cooper SA, Finlayson J, Jackson A, Allan L, Mantry D, McGrother C, McConnachie A, and Morrison J (2007). Incidence and predictors of mental ill-health in adults with intellectual disabilities: prospective study. Br J Psychiatry 191, 313–319. [DOI] [PubMed] [Google Scholar]
- Smith KS, Bucci DJ, Luikart BW, and Mahler SV (2016). DREADDS: Use and application in behavioral neuroscience. Behav Neurosci 130, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumier A, and Sibille E (2014). Opposing effects of acute versus chronic blockade of frontal cortex somatostatin-positive inhibitory neurons on behavioral emotionality in mice. Neuropsychopharmacology 39, 2252–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwick SM, and Charney DS (2012). The science of resilience: implications for the prevention and treatment of depression. Science 338, 79–82. [DOI] [PubMed] [Google Scholar]
- Southwick SM, Vythilingam M, and Charney DS (2005). The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu Rev Clin Psychol 1, 255–291. [DOI] [PubMed] [Google Scholar]
- Swanson AM, DePoy LM, and Gourley SL (2017). Inhibiting Rho kinase promotes goal-directed decision making and blocks habitual responding for cocaine. Nat Commun 8, 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayasu M, Suzuki Y, Shibuya M, Asano T, Kanamori M, Okada T, Kageyama N, and Hidaka H (1986). The effects of HA compound calcium antagonists on delayed cerebral vasospasm in dogs. J Neurosurg 65, 80–85. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Minami H, Kota M, Kuwamura K, and Kohmura E (2005). Treatment of cerebral vasospasm with intra-arterial fasudil hydrochloride. Neurosurgery 56, 214–223; discussion 214–223. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentler D, Gustavsson P, Leisti J, Schueler M, Chelly J, Timonen E, Anneren G, Willard HF, and Dahl N (1999). Deletion including the oligophrenin-1 gene associated with enlarged cerebral ventricles, cerebellar hypoplasia, seizures and ataxia. Eur J Hum Genet 7, 541–548. [DOI] [PubMed] [Google Scholar]
- Valnegri P, Khelfaoui M, Dorseuil O, Bassani S, Lagneaux C, Gianfelice A, Benfante R, Chelly J, Billuart P, Sala C, et al. (2011). A circadian clock in hippocampus is regulated by interaction between oligophrenin-1 and Rev-erbalpha. Nat Neurosci 14, 1293–1301. [DOI] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, and Quirk GJ (2006). Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem 13, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmayr B, and Henn FA (2001). Learned helplessness in the rat: improvements in validity and reliability. Brain Res Brain Res Protoc 8, 1–7. [DOI] [PubMed] [Google Scholar]
- Walsh RM, Shen EY, Bagot RC, Anselmo A, Jiang Y, Javidfar B, Wojtkiewicz GJ, Cloutier J, Chen JW, Sadreyev R, et al. (2017). Phf8 loss confers resistance to depression-like and anxiety-like behaviors in mice. Nat Commun 8, 15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Blouin V, Brument N, Bello-Roufai M, and Francois A (2011). Production and purification of recombinant adeno-associated vectors. Methods Mol Biol 807, 361–404. [DOI] [PubMed] [Google Scholar]
- Wang M, Perova Z, Arenkiel BR, and Li B (2014). Synaptic modifications in the medial prefrontal cortex in susceptibility and resilience to stress. J Neurosci 34, 7485–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim SY, Adhikari A, Tye KM, Frank LM, and Deisseroth K (2012). A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschbusch DA, Sellers DP, LeBlanc M, and Kelley ML (2003). Helpless attributions and depression in adolescents: the roles of anxiety, event valence, and demographics. J Adolesc 26, 169–183. [DOI] [PubMed] [Google Scholar]
- Wayment HA, and Zetlin AG (1989). Coping responses of adolescents with and without mild learning handicaps. Ment Retard 27, 311–316. [PubMed] [Google Scholar]
- Wu G, Feder A, Cohen H, Kim JJ, Calderon S, Charney DS, and Mathe AA (2013). Understanding resilience. Front Behav Neurosci 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JM, Zhang J, Chen XJ, Geng HY, Ye M, Spitzer NC, Luo JH, Duan SM, and Li XM (2013). Development of GABA circuitry of fast-spiking basket interneurons in the medial prefrontal cortex of erbb4-mutant mice. J Neurosci 33, 19724–19733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, and Yan Z (2011). Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry 16, 156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanni G, Saillour Y, Nagara M, Billuart P, Castelnau L, Moraine C, Faivre L, Bertini E, Durr A, Guichet A, et al. (2005). Oligophrenin 1 mutations frequently cause X-linked mental retardation with cerebellar hypoplasia. Neurology 65, 1364–1369. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Aime M, Laheranne E, Houbaert X, El Oussini H, Martin C, Lepleux M, Normand E, Chelly J, Herzog E, et al. (2017). Protein Kinase A Deregulation in the Medial Prefrontal Cortex Impairs Working Memory in Murine Oligophrenin-1 Deficiency. J Neurosci 37, 11114–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YF, Zhang Q, Xi JY, Li YH, Ma CG, and Xiao BG (2015). Multitarget intervention of Fasudil in the neuroprotection of dopaminergic neurons in MPTP-mouse model of Parkinson’s disease. J Neurol Sci 353, 28–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1–3. Ophn1 deficient mice subjected to the LH procedure do not display perseverative behaviors. Related to Figure 1.
ACTB-Ophn1WT and ACTB-Ophn1cKO mice were subjected to the learned helplessness (LH) procedure. In brief, mice were first exposed to two induction sessions consisting of 360 inescapable, uncontrollable electric foot shocks and thereafter subjected to a testing session consisting of escapable foot shocks. The testing session was performed in a shuttle box equipped with an electrical grid floor, a door separating the two halves, and photocell detectors. Behavioral performance was evaluated over 30 trials of escapable foot shocks. Each trial started with a 5 s cue light, followed by the foot shocks. When an animal shuttled to another compartment of the box during the 5 s cue light presentation (and thus before the shock onset), avoidance was scored. If the animal shuttled during the 10 s shock (i.e., escaped), escape latency was measured. Failure was recorded if no shuttling was made during the 10 s shock presentation. Shocks were terminated if the animal shuttled to another side of the box (in case of escape) or at the end of the 10 s shock (in case of failure). Animals’ behavior was classified as being “resilient” or “learned helpless” based on their behavioral parameters in the LH testing session (see Methods). Representative examples (2 trials) of an ACTB-Ophn1WT mouse and an ACTB-Ophn1cKO mouse displaying helpless behavior (Video S1 and Video S2, respectively) and of an ACTB-Ophn1WT mouse exhibiting resilience (Video S3) are shown.
Data Availability Statement
All data have been presented in Figures and Supplemental Figures and Videos. Original images/data will be made available upon request.