

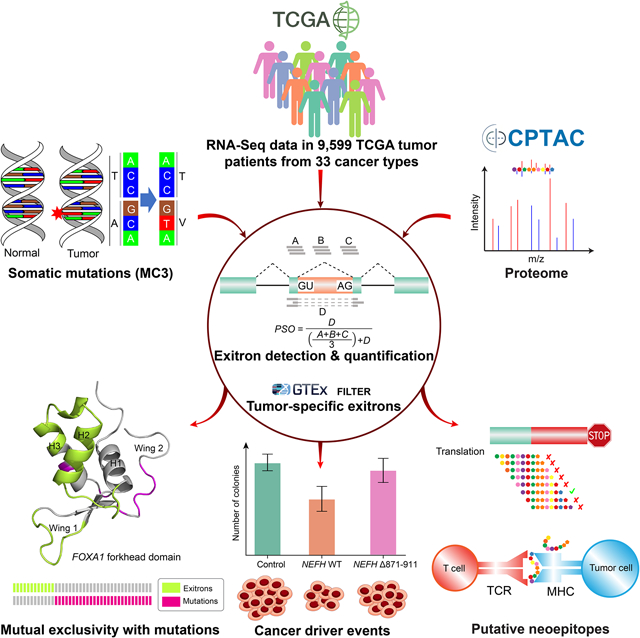
Summary
Exitron splicing (EIS) creates a cryptic intron (termed an exitron) within a protein-coding exon to increase proteome diversity. EIS is poorly characterized, but emerging evidence suggests a role for EIS in cancer. Through a systematic investigation of EIS across 33 cancers from 9,599 tumor transcriptomes, we discovered EIS affected 63% of human coding genes and 95% of those events were tumor-specific. Notably, we observed a mutually exclusive pattern between EIS and somatic mutations in their affected genes. Functionally, we discovered EIS altered known and novel cancer driver genes for causing gain- or loss-of-function, by which promotes tumor progression. Importantly, we identified EIS-derived neoepitopes that bind to MHC class I or II. Analysis of clinical data from a clear cell renal cell carcinoma cohort revealed an association between EIS-derived neoantigen load and checkpoint inhibitor response. Our findings establish the importance of considering EIS alterations when nominating cancer driver events and neoantigens.
Graphical Abstract
eTOC blurb
The comprehensive analysis of exitron splicing events in cancer provides a reference of candidate cancer driver events, potential immunogenic neoantigens, and predictive signatures for immunotherapy response that are missed by genetic alteration analysis alone.
Introduction
Alternative splicing of pre-mRNA plays a vital role in shaping the transcriptome and this process is frequently altered in cancers (Oltean and Bates, 2014). Recently, a type of non-canonical splicing, exitron, was found to be dysregulated between breast cancer and normal tissues (Marquez et al., 2015). Exitrons are cryptic introns with both splice sites inside an annotated exon; that is, an exitron is an internal region of an exon that has both protein-coding (exon) and splicing (intron) potential. Exitrons possess canonical splicing signals, such as 5ʹ and 3ʹ splice-site motifs (e.g. GT-AG). Because exitrons are protein-coding sequences flanked by exonic sequences, they do not contain stop codons or premature termination codons. Such genomic feature distinguishes exitrons from conventional introns (Staiger and Simpson, 2015). Moreover, unlike intron retention as a form of splicing aberration, exitrons are retained under normal conditions. However, when exitrons are spliced, non-canonical and unannotated protein isoforms are produced through translation of the exitron-spliced transcripts, which may link to disease pathogenesis (Sibley et al., 2016).
Although intron retention and several other basic splicing models have been implicated in neoplastic diseases (Dvinge and Bradley, 2015; Jung et al., 2015; Kahles et al., 2018), exitron splicing has received less attention and little is known about its role in human cancers. A pilot transcriptome study of metastatic prostate cancer patients revealed that exitron splicing occurs recurrently in known tumor suppressor genes (Yang et al., 2018). This suggests that the proteins encoded by exitron-spliced mRNAs may contribute to cancer development. Despite the potentially high impact in cancer, the functional consequences of exitron splicing in cancer genes and its clinical relevance remain unknown. This likely reflects the lack of computational tools for de novo exitron detection and annotation on a genome-wide scale.
To date, there are 670 retained introns (in 577 genes) that fulfill the definition of exitrons annotated in the human Ensembl genome database (Marquez et al., 2015). Further, analysis of high-throughput RNA sequencing (RNA-Seq) data facilitated the discovery of approximately 900 exitrons from six human tissues and one breast cancer sample (Marquez et al., 2015). However, this set is not exhaustive, and the significance of these and other exitrons to human pathology is poorly understood. Functionally, exitron splicing can contribute to proteomic diversity by causing inframe internal deletions (in cases where exitrons have nucleotide lengths divisible by 3) or frameshifts (in cases where exitrons have nucleotide lengths not divisible by 3) in encoded proteins. Notably, mRNA splicing has been recognized as a source of neoantigens (Frankiw et al., 2019), suggesting a potential for exitron splicing to generate tumor neoantigens that could form a basis for the development of new cancer vaccines or T-cell therapeutic strategies.
Surveys of cancer driver genes or immunogenic neoantigens have so far largely focused on the impact of DNA mutations. Large-scale sequencing efforts by The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research, 2008) and The Genotype-Tissue Expression (GTEx) (Consortium, 2013) provide unique RNA-Seq datasets to investigate whether knowledge of exitrons could yield additional cancer driver gene or neoantigen candidates. In this study, we systematically detect and characterize exitron splicing events across 33 cancer types. Our findings imply exitron splicing represents an additional cancer-driving mechanism beyond genetic alterations. Further, our analyses reveal exitron splicing-derived neoantigen burden is a candidate predictor for cancer immunotherapy response.
Results
Landscape of exitron splicing events in cancer
To assess the landscape of exitrons across cancer transcriptomes, we collected RNA-Seq data in 9,599 patients across 33 cancer types together with 670 matched normal samples from the TCGA study (Figure 1A and Table S1). We developed a bioinformatic tool, ScanExitron, to analyze RNA-seq data for exitron detection. ScanExitron identified exitrons based on splicing junctions and gene annotations, and filtered out low-confident candidates with a percent spliced out (PSO) metric that measures the percentage of transcripts in which a given exitron is spliced (Figure 1B). As a result, we identified 129,406 exitrons in tumor samples that were contained within 39,755 exons, which accounts for 14.8% of the GENCODE human exome. We selected a panel of exitron splicing events found in TCGA breast cancer samples and validated them in SKBR3 breast cancer cells with different approaches (Figure S1A). Briefly, we obtained long-read isoform sequencing (Iso-Seq) data and found predicted PSOs by RNA-Seq correlated well (r = 0.88, p = 0.02, Pearson correlation) with corresponding isoform fractions from Iso-Seq (Figure S1B). We further validated individual events detected by both RNA-Seq and Iso-Seq using RT-PCR. (Figure S1C).
Figure 1. Samples and workflow for exitron splicing discovery.
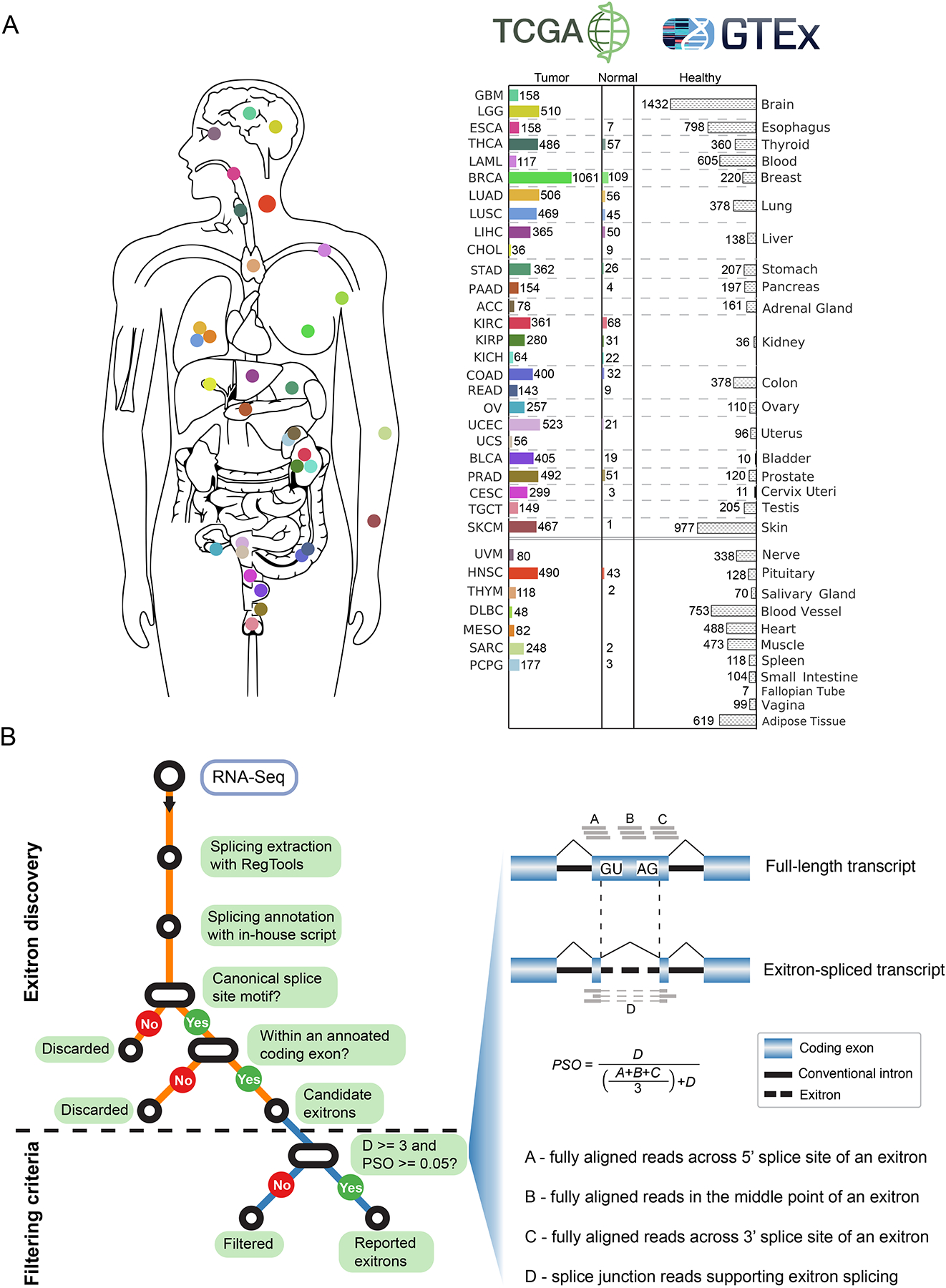
(A) Data source for the 33 cancer types in this study. Bar charts describe numbers of tumor and matched normal samples for each cancer type from TCGA (with color) and healthy samples from GTEx (without color). The number of samples with available RNA-Seq data is indicated. (B) Workflow and criteria of exitron detection in TCGA data. Left, the computational pipeline to detect exitron splicing events within annotated protein-coding exons from TCGA RNA-Seq data. Right, the criteria to report an exitron splicing event including the number of supporting reads (indicated by D) and a percent spliced out (PSO) metric.
As a class of non-canonical splicing, exitrons possessed distinct cis-acting features (weak 5’ and 3’ splice sites, high GC content, and short length) setting them apart from constitutively spliced introns and, more importantly, from retained introns detected in cancer (Figure S1D). In addition, we found the sizes of exitrons and their parent exons were correlated (Figure S1E), and most of the identified exitrons resided in medium-sized exons (e.g. 100bp to 1kbp) (Figure S1F). Next, we sought to estimate the proportion of exitron-bearing genes in cancer and non-cancer genomes. As normal tissues in TCGA are adjacent to the tumor and are only available for a subset of cancer types, we expanded our exitron analysis to include GTEx (v7) RNA-Seq data from 9,636 healthy tissue samples representing 53 tissue types from 30 anatomical sites (Figure 1A). This yielded a total of 7,701 exitrons spanning 5,735 exons in GTEx samples. We found the percentage of genes containing exitrons was remarkably higher in TCGA cancer specimens (62.7%) than in GTEx healthy populations (17.1%) (Figure S1G), indicating that exitron splicing is prevalent across the cancer transcriptomes. To investigate whether the number of exitron-bearing genes has reached saturation, we performed ‘down-sampling’ saturation analysis on random subsets of TCGA samples of various smaller sizes. We observed that the number of exitron-bearing genes increased steadily with increasing sample size (Figure S1H), implying the catalog of exitron-bearing genes remains far from complete.
Across all tumor samples, we observed a median count of 121 exitron splicing events per sample. Ovarian (OV), esophageal (ESCA), stomach (STAD) and acute myeloid leukemia (LAML) cancers were found to have higher exitron splicing burden than other cancer types (Figure 2A and Table S2). Furthermore, we observed exitrons were spliced towards a gender-bias or a tumor stage-bias in certain cancer types (Figure S2A). Next, we compared the exitron splicing burden between tumor and matched normal samples in eight tumor types where RNA-Seq data were available from at least 40 tumor-normal paired samples. In the meta-analysis of the eight cancer types, we observed a significantly higher exitron splicing load in tumor samples than that in matched normal samples (meta p = 5.1e-5) (Figure 2B). These findings suggest that exitron splicing may contribute to cancer phenotypes.
Figure 2. Detection of dysregulated exitron splicing (EIS) events in cancer.
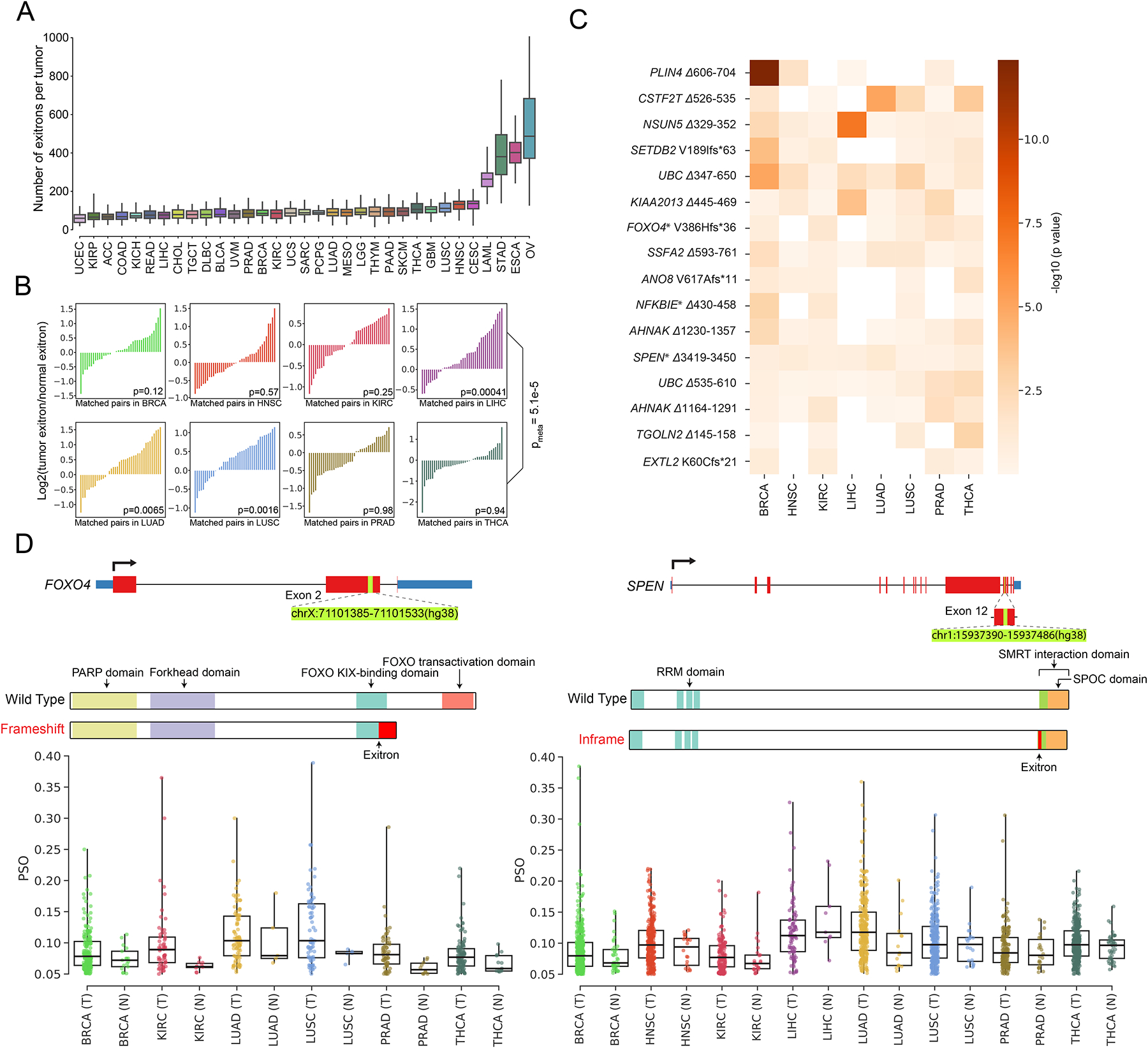
(A) Count of EIS events across 33 cancer types. For each cancer type, we randomly choose 36 samples for EIS burden evaluation to account for cohort size variations. (B) Pairwise comparison of EIS load in 40 randomly selected pairs of tumor specimens (T) and matched adjacent histologically normal tissues (N) for TCGA cancer types with at least 40 T/N matched samples. The p value is calculated using the Wilcoxon signed-ranks test. (C) Results of differential splicing analysis of exitrons between tumor and normal tissues for 8 cancer types. Rows represent 16 dysregulated exitrons that were found to be differentially spliced after FDR correction. Shading corresponds to −log10(p value). Columns represent cancer types. Genes marked with an asterisk are annotated in the COSMIC cancer gene census. (D) Illustration of the dysregulated EIS events identified in FOXO4 (left) and SPEN (right) and comparison of their splicing between tumor and normal samples for the eight TCGA cancer types. Each dot corresponds to the percent spliced out (PSO) value of the selected EIS in one sample.
We next sought to identify exitron splicing events that were dysregulated between tumor and normal samples across different tumor types. To achieve this, we conducted an analysis to identify exitrons that were differentially spliced between tumor and matched normal samples in the eight cancer types. We identified 16 exitron splicing events that displayed recurrent dysregulation in multiple cancers (Figure 2C). Genes impacted by these differentially spliced exitrons were significantly enriched for genes causally implicated in cancer annotated by the Catalogue of Somatic Mutations in Cancer (COSMIC) cancer gene census (3 out of 16, p = 0.019, fold change 5.19, hypergeometric test) (Figure 2C). One prominent example is a frameshift exitron splicing event within exon 2 of FOXO4 (FOXO4 V386Hfs*36), which exhibited increased splicing in tumors compared with normal samples (Figure 2D). FOXO4 is known to be a tumor suppressor gene (Greer and Brunet, 2005). The truncated protein predicted from this exitron-spliced mRNA isoform may cause FOXO4 loss of function due to the lack of FOXO functional domains (Figure 2D). Another example is an inframe exitron splicing event within exon 12 of the tumor suppressor gene SPEN (Legare et al., 2015) (SPEN Δ3419–3450). Splicing of this exitron removed part of the repression domain that interacts with nuclear receptor corepressor 2 (NCOR2) (Ariyoshi and Schwabe, 2003) (Figure 2D). Therefore, increased splicing of this exitron in SPEN in tumor samples (Figure 2D) suggests a potential loss of its transcriptional repression function.
Because exitron splicing has been linked to splicing factor (SF) dysregulation (Marquez et al., 2015), we investigated the extent to which trans-acting factors can explain the differences in the abundance of exitron splicing across tumor types. By clustering 404 literature-curated SF genes according to their mRNA expression profiles, we observed that certain cancer types with high exitron splicing burden, such as ESCA, STAD and LAML, grouped together (Figure S2B), suggesting the large number of exitron splicing events in these cancers may be attributed to a partial breakdown of the splicing machinery that is the result of dysregulated expression of SFs. Supporting this, when examining individual tumor types, we observed the activity of SFs was correlated with exitron splicing burden (Figure S2C). To identify candidate SFs contributing to exitron splicing misregulation in cancer, we correlated exitron splicing changes with the expression differences of SFs in cancer vs. matched normal samples using a generalized additive model (GAM) (Wood, 2011). Remarkably, we found a large portion of exitron splicing dysregulation could be explained by expression alteration of SFs (Figure S2D). Further, linear regression analysis of GAM-derived exitron/SF pairs identified a subset of SFs generally promoting exitron splicing (Figure S2E). Functional annotation revealed these genes encoded protein factors composing the spliceosome (DHX15, DHX16, CDC5L, SF3B4, WBP11, SNRNP200, HNRNPM, ZNF326), proteins regulating RNA transport (EIF3A, THOC2) and mRNA surveillance (MSI2) (Figure S2F).
Tumor-specific exitrons enable novel cancer driver gene discovery
Based on our observation that certain tumors display a higher degree of exitron splicing than normal cells, we sought to identify those exitrons that are predominantly spliced in tumor samples, which we termed tumor-specific exitrons (TSEs). We compared a panel of normal samples (TCGA normal and GTEx) with TCGA tumor data to identify TSEs that 1) were not spliced in GTEx samples and 2) were spliced in no more than three TCGA normal samples. As a result, a total of 123,338 (95.3%) exitrons were qualified as TSEs. We found that OV, ESCA, STAD and LAML had the highest number of TSE splicing events compared with the other cancers (Figure 3A). Given the large number of TSE splicing events observed across tumor types, we sought to evaluate their clinical relevance and functional impact. We first examined the splicing frequency of each TSE in tumor samples, and found 39.5% of them were recurrently spliced (Figure S3A). Next, we defined clinically-informative exitrons as those TSEs that were spliced in ≥ 10 samples and associated with survival for a cancer type. Among the 25 cancer types with clinical information and a cohort size ≥ 100, we found clinically-informative TSE splicing events in 21 cancers (Figure S3B). OV had the highest number of clinically-informative TSE splicing events that correlated with at least one of the three survival endpoints (overall, progression-free and disease-free). Notably, we found 21 TSE splicing events were clinically informative in more than one cancer type (Figure S3C), suggesting that they may play important roles in different tumor contexts. For example, an EWSR1 Δ573–603 exitron splicing event was associated with overall survival in lung squamous cell carcinoma (LUSC) and disease-free survival in uterine corpus endometrial carcinoma (UCEC) (Figure S3D).
Figure 3. Detection of genes enriched with tumor-specific exitrons (TSEs).
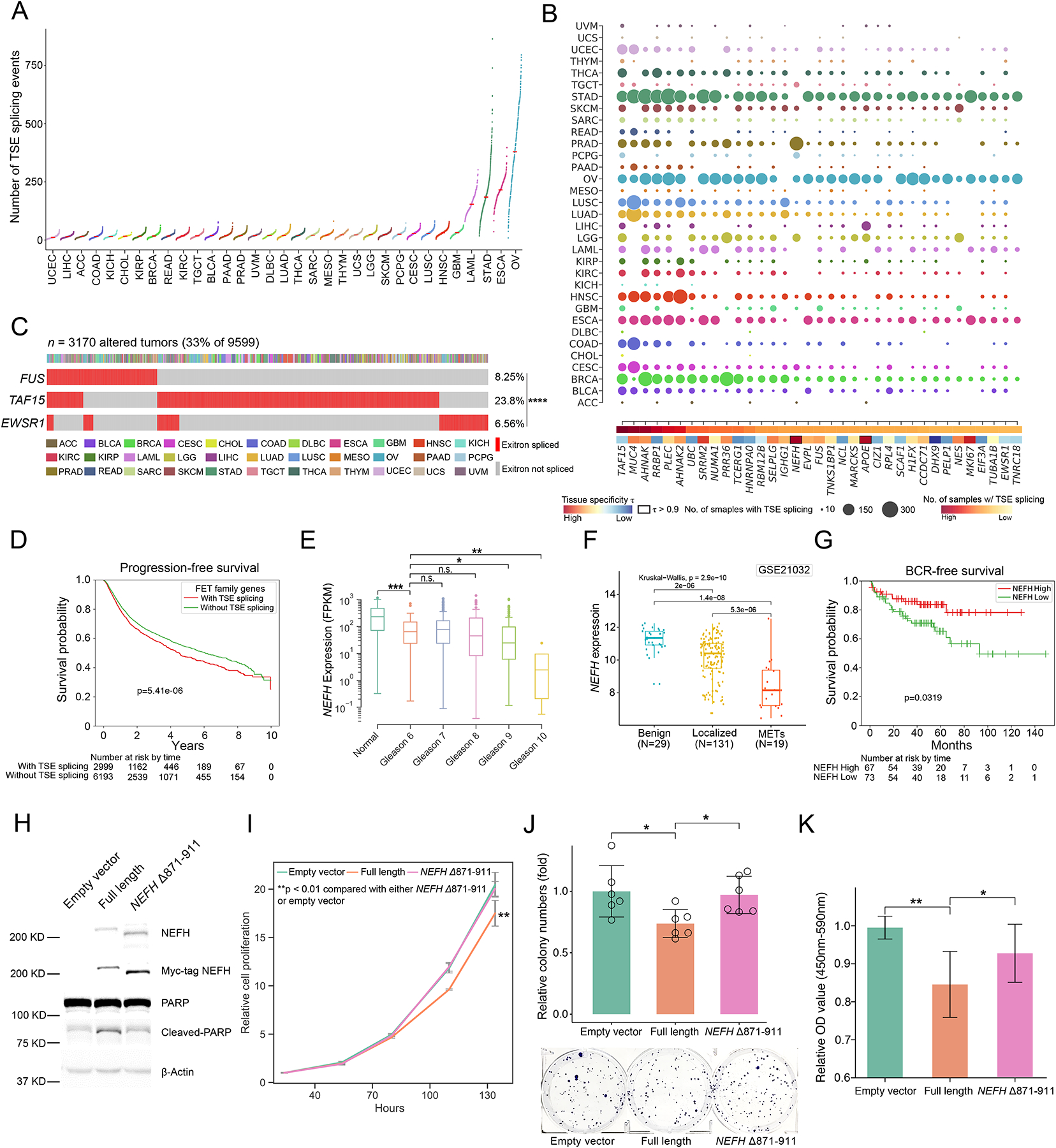
(A) Number of TSE splicing events for TCGA cohorts. Each dot represents the number of TSE splicing events in a TCGA tumor sample. (B) Top 35 significantly exitron-spliced genes (SEGs). Circle size correlates with the number of samples with spliced TSEs and colored by cancer type. Highly tissue specific SEGs (τ > 0.9) are highlighted. (C) Mutual exclusivity of exitron splicing events in FET genes including EWSR1, FUS and TAF15 in TCGA pan-cancer cohort (****p < 0.0001, Fisher’s exact test). (D) Exitron splicing of FET family genes predicts progression-free survival in TCGA pan-cancer cohort. (E) The expression of the SEG gene NEFH is correlated with Gleason grade in PRAD cohort (n.s., not significant (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, Mann-Whitney rank test). (F) NEFH is downregulated in prostate tumors. NEFH mRNA expression from microarray data set (GSE21032) is compared in benign, localized, and metastatic prostate cancer. The p value is calculated by Kruskal-Wallis test. (G) Low NEFH mRNA expression is associated with poor clinical outcome. Kaplan-Meier analysis of prostate cancer outcome using GSE21032 dataset is shown. Prostate cancer cases are stratified based on their NEFH mRNA expression level and analyzed for biochemical recurrence. The p value is calculated by a log-rank test. (H) Representative western blot against C4-2 stable cell lines expressing Myc-tagged wild-type NEFH and exitron-spliced NEFH. Apoptosis was evaluated by western blot analysis for poly (ADP-ribose) polymerase (PARP) cleavage. (I) CellTiter-Glo growth assays indicate that overexpression of wild-type NEFH, but not exitron-spliced NEFH, significantly inhibited cell growth in C4-2 cells. (J) Overexpressing wild-type NEFH significantly decreased colony-formation ability of C4-2 cells. Cells were fixed and stained with crystal violet. n = 6. The figure is a representative of three experiments with similar results. Quantification was performed by manual counting. (K) BrdU ELISA assay of C4-2 cells overexpressing wild-type NEFH and or exitron-spliced NEFH with 24hrs of BrdU label (n=9). Y axis, absorbance of 450–590 nm relative to empty vector. There was less incorporation of BrdU in cells expressing wild-type NEFH. All p values are calculated using unpaired, two-tailed Student’s t-test. Error bars indicate ± s.d. (*p < 0.05, **p < 0.01, ***p < 0.001).
To examine the functional impact of TSE splicing, we implemented a frequency-based method to identify genes that were enriched for TSE splicing events in each tumor type, hereafter termed significantly exitron-spliced genes (SEGs, Figure S3E, Table S3). We ranked SEGs based on their exitron splicing frequency and found that the top 35 ranked SEGs were significantly enriched for genes in the COSMIC cancer gene census, including TAF15, MUC4, NUMA1, FUS and EWSR1 (5 out of 35, p = 0.008, fold change 3.95, hypergeometric test; Figure 3B). The highest-ranked SEG was TAF15, which is a member of the FET protein family involved in transcriptional regulation and RNA processing (Law et al., 2006). Notably, the other members of the FET protein family, FUS and EWSR1 were also among the top-ranked SEGs. Intriguingly, we found mutual exclusivity between exitron splicing in these three genes in tumor samples, implying these exitron splicing events may have similar functional impacts (Figure 3C). Moreover, patients with TSE alterations in FET proteins were associated with impaired progression-free survival (Figure 3D), suggesting that exitron splicing in FET family genes may play a role in promoting cancer progression. Besides, we observed that exitron splicing hotspots in FET family genes mapped to regions encoding the C-terminal domains of FET proteins, which are enriched for post-translational modification sites (Figure S3F). This suggests that exitron splicing has the potential to alter the regulation of FET proteins.
Because we found that SEGs were enriched for known cancer genes, we examined whether identification of SEGs could nominate novel cancer driver genes. As cancer driver genes tend to display a tissue-specific alteration spectrum (Haigis et al., 2019), we performed a tissue-specific analysis of SEGs. We found that NEFH was an SEG in prostate cancer (Figure 3B). NEFH encodes the heavy neurofilament protein, which forms the framework for nerve cells (Hirokawa and Takeda, 1998). Although loss of NEFH expression has been observed in esophageal squamous cell carcinoma (Kim et al., 2010), a role for NEFH in prostate cancer remains unknown. Therefore, we examined NEFH expression in the TCGA prostate adenocarcinoma (PRAD) cohort. NEFH expression levels were significantly higher in benign samples than those in prostate tumor samples. Strikingly, NEFH expression was inversely correlated with Gleason score, which is a scoring system that predicts cancer aggressiveness from tumor histology (Figure 3E). We further evaluated NEFH expression in 150 tumor and 29 normal samples from the MSKCC Prostate Cancer Oncogenome Project (Taylor et al., 2010), which collected array-based gene expression data and comprehensive clinical information. We found that NEFH was significantly downregulated in metastatic prostate cancer when compared with localized prostate tumors and benign tissues (Figure 3F). In addition, low NEFH expression in tumors was associated with high risk of biochemical recurrence (Figure 3G). Collectively, these data strongly suggest that NEFH is a tumor suppressor gene in prostate cancer.
To support this, we sought to validate the function of NEFH using two NEFH-negative prostate cancer cell lines, C4-2 and PC-3 (Figure 3H, Figure S3G). When comparing prostate cancer cells overexpressing NEFH relative to control cells, we observed that ectopic expression of NEFH inhibited cell growth (Figure 3I, Figure S3H), colony formation (Figure 3J, Figure S3I) and DNA replication (Figure 3K, Figure S3J) but promoted apoptosis as measured by cleaved PARP protein levels (Figure 3H), confirming that NEFH has tumor suppressor function in prostate cancer. Next, we investigated whether exitron splicing of NEFH affects this tumor suppressor function. Strikingly, we confirmed that an inframe, exitron-spliced NEFH protein isoform lacking 41 amino acids in the C-terminal domain, NEFH Δ871–911 (Figure S3K), increased cell growth (Figure 3I, Figure S3H), colony formation (Figure 3J, Figure S3I) and DNA replication (Figure 3K, Figure S3J) but reduced apoptosis (Figure 3H) in prostate cancer cells relative to wild-type NEFH. Importantly, NEFH is rarely mutated in prostate cancer (Figure S3L), but NEFH Δ871–911 is the most frequent TSE splicing detected in PRAD and an independent cohort of advanced prostate cancer (Quigley et al., 2018) (Figure S3M). Collectively, these data demonstrate that exitron splicing can functionally inactivate tumor suppressor genes in cancer.
Mutual exclusivity between tumor-specific exitrons and somatic mutations
To ask whether lack of mutations in SEGs such as NEFH is a general feature, we focused on genes affected by somatic mutations and/or TSE splicing. We observed a clear pattern wherein significantly mutated genes (SMGs) such as TP53, PIK3CA and KRAS, and SEGs such as TAF15, RRBP1 and NEFH were bifurcated into two groups (Figure 4A), suggesting SMGs and SEGs are mutually exclusive. When examining genes affected by somatic mutations and/or TSE splicing in an individual cancer type, we confirmed this pattern of mutual exclusivity (Figure S4A). To investigate the relationship between SEGs and SMGs in more detail, we focused on the PRAD cohort. As expected, NEFH was the top-ranked SEG, but did not display mutations (Figure 4B). Conversely, known SMGs in prostate cancer, such as TP53 and SPOP, displayed no exitron splicing. This pattern further supported the notion that SEGs could represent an independent catalog of genes with functionally important exitron splicing alterations in cancer. Notably, when we focused on genes that displayed somatic mutations and exitron splicing in the PRAD cohort (for instance, the epigenetic regulator KMT2D, the pioneer factor FOXA1 and the tumor suppressor gene ZFHX3), the pattern of mutual exclusivity between mutations and exitron splicing was apparent at the patient level (Figure 4B).
Figure 4. Comparison of TSE splicing and somatic mutations.
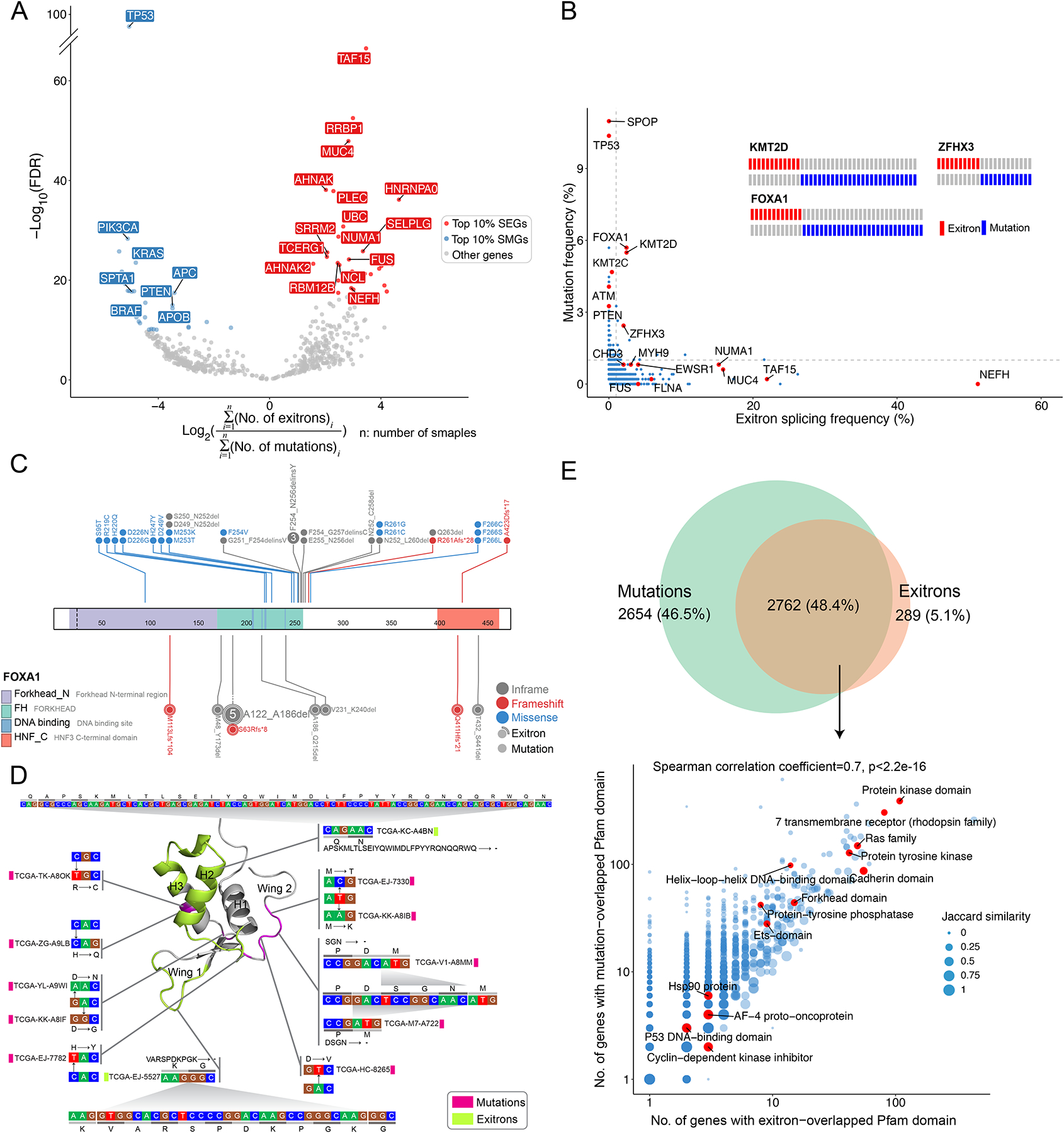
(A) Volcano plot shows mutation and TSE splicing frequency difference separating genes as SMGs and SEGs. (B) The frequencies of mutation and exitron splicing events in genes are inversely correlated in the PRAD cohort. DNA mutations and exitron splicing are mutually exclusive in FOXA1, KMT2D, and ZFHX3. Genes of interest are highlighted. (C) DNA mutations and exitron splicing are clustered in the forkhead DNA binding domain of the FOXA1 gene in PRAD. (D) The nucleotide and amino acid changes caused by exitron splicing and somatic mutations are shown against the 3D structure of the FOXA1 forkhead domain. The α-helix and wing regions are highlighted. (E) Comparison on Pfam protein domains affected by somatic mutations versus exitron splicing events. Venn diagram (top panel) shows that Pfam domains affected by somatic mutations or exitron splicing events share extensive overlap. The scatterplot (bottom panel) shows high correlation (Spearman correlation coefficient = 0.7) in the Pfam domains affected by exitron splicing events and somatic mutations. Pfam domains of interest are highlighted. Jaccard similarity is used to measure the similarity between exitron splicing- and mutation-altered gene sets.
To investigate the consequences of exitron splicing on protein functions, we examined the landscape of FOXA1 TSE splicing events in PRAD. We observed that exitrons clustered in the Forkhead DNA binding domain (FKHD) (Figure 4C). Recently, the Wing 2 region of the FKHD was identified as a FOXA1 mutational hotspot (Adams et al., 2019; Parolia et al., 2019). Mapping mutations and exitrons to the 3D crystal structure of FKHD demonstrated that exitron splicing alterations largely affected non-Wing 2 regions (Figure 4D), suggesting that exitron splicing in FOXA1 could represent a distinct, yet-unexplored mechanism for altered FOXA1 pioneer activity in prostate cancer. To further test this, we cloned two representative, inframe exitron-spliced FOXA1 protein isoforms, each missing a region in the FKHD: Δ186–215 and Δ231–240 (Figure S4B). In reporter assays using luciferase with FOXA1 binding sequences, we observed that ectopically expressed FOXA1 Δ186–215 and Δ231–240 displayed higher transcriptional activities than the wild-type FOXA1 (Figure S4C). Consequently, exitron-spliced FOXA1 resulted in stronger transcriptional activation of oncogenic androgen receptor (AR) signaling (Figure S4D). Further, we confirmed overexpression of these two FOXA1 TSE splicing events in prostate cancer C4-2 cells (Figure S4E) promoted cell growth (Figure S4F) and colony formation (Figure S4G) relative to control cells. Concordantly, transcriptomic analyses of PRAD tumor tissues predicted AR and FOXA1 to be driver transcription factors for upregulated genes in patients with FOXA1 TSE alterations (Figure S4H). We further confirmed that patients with FOXA1 TSE alterations had the highest AR activity score (Figure S4I). When examining TSEs across all TCGA tumors, we found a significant overlap (p < 2.2e-16, hypergeometric test) and a strong correlation in the protein family (Pfam) domains affected by exitron splicing and somatic mutations (Figure 4E), indicating exitron splicing impacts protein functions in a manner similar to genetic alterations.
Besides gene level analysis, we compared somatic mutations and TSE alterations at the patient level. We found that mutation burden and exitron splicing burden were independent across all cancer types (Figure S4J). To further investigate the consequences of mutations and exitron splicing events at the transcriptome level, we calculated the mutational burden, variant allele frequency and variant size of expressed mutations and compared them with TSE alterations. We found that TSE alterations had a higher burden and allele fraction in tumor samples, and had a larger impact by size on transcripts than insertions and deletions (indels) (Figures S4K–M).
Because RTK/RAS and PI3K/AKT signaling pathways are frequently mutated in cancer (Sanchez-Vega et al., 2018), we attempted to investigate whether TSE splicing alterations preferentially occur in cancer hallmark pathways. Hence, we extended the single gene-based SEG analysis into a multi-gene-based analysis to test whether TSE splicing events were significantly enriched in gene sets representing major biological processes. To achieve this, we focused on 50 “hallmark” gene sets from the Molecular Signature Database (MSigDB) (Liberzon et al., 2015) and performed the multi-gene-based analysis for each gene set in a particular tumor type. We identified 20 gene sets that were significantly altered by TSE splicing across different tumor types (Figure 5). “IL2_STAT5_SIGNALING” and “UNFOLDED_PROTEIN_RESPONSE” were the top two gene sets that enriched with TSE alterations across the highest number of cancer types (18 out of 33). Conversely, certain gene sets displayed significant TSE splicing only in a specific malignancy, such as “ANDROGEN_RESPONSE” in PRAD. Within some gene sets, TSE alterations occurred over many genes (e.g., P53 and MYC_V1 gene sets), while in other gene sets TSE alterations only affected a few genes (e.g., HYPOXIA and SPERMATOGENESIS) (Figure S5). Interestingly, frequently mutated pathways, such as PI3K, Notch, Wnt and TGFβ (Sanchez-Vega et al., 2018), were not enriched with TSE alterations in any cancer type (Figure 5), reaffirming the notion that exitron splicing represents a distinct, yet-unexplored mechanism in cancer that could be complementary to genetic alterations.
Figure 5. MsigDB hallmark gene sets affected by exitron splicing in TCGA cohorts.
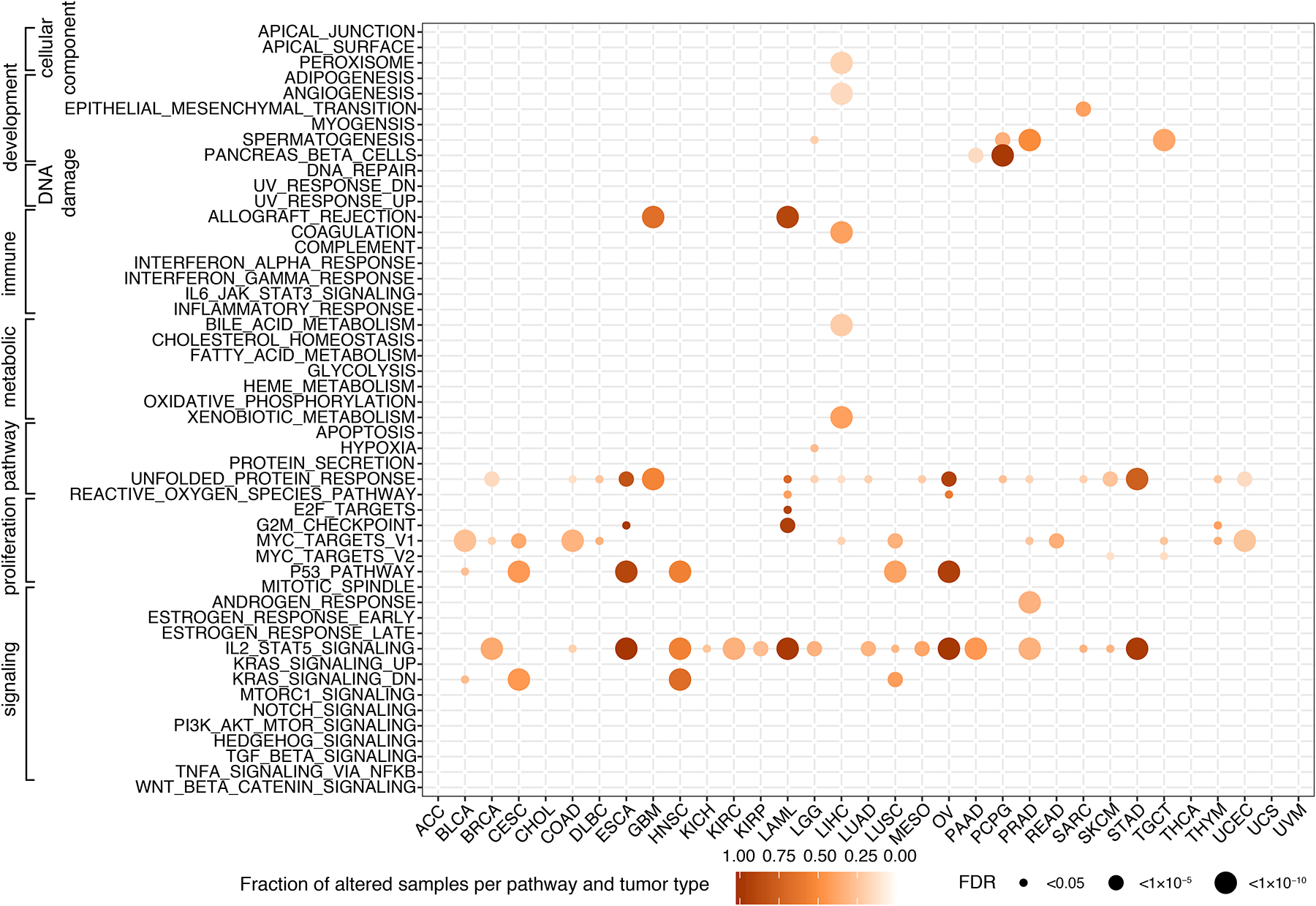
The size of the circles represents the significance of TSE enrichment measured by FDR. Color indicates the fraction of TSE splicing altered samples per gene set and tumor type.
Tumor-specific exitrons represent a source of neoepitopes
Because exitron splicing affects protein-coding exons, they can cause inframe deletions of functional protein domains or generate novel reading frames through frameshifts. We observed 56% of TSE splicing events retained the reading frame (Figure S6A), and 38% of these inframe TSEs were found to overlap with Pfam domains (Figure S6B). Frameshift TSE splicing events may produce novel protein sequences or introduce premature termination codons, trigging nonsense-mediated RNA decay (NMD) to degrade resultant transcripts. Remarkably, we observed the overall expression of exitron-spliced transcripts was higher than those with retained introns (Figure S6C), suggesting exitron splicing is less affected by NMD than intron retention. Indeed, when focusing on frameshifting events that have the potential to trigger NMD, we found exitron splicing events are much more likely to evade NMD than intron retentions according to the canonical rules of NMD (Lindeboom et al., 2016) (Figure S6D). In this regard, exitron splicing alterations are similar to indels, which are known to be a rich source of neoantigens due to frameshifts and those escaping NMD can predict response to checkpoint immunotherapy (Litchfield et al., 2020; Turajlic et al., 2017). Following this, we sought to investigate whether TSE slicing has the potential to produce immunogenic neoantigens.
We applied ScanNeo (Wang et al., 2019) to predict potential TSE neoantigens in TCGA tumors. For a total of 123,338 unique TSE splicing events, we identified 168,206 putative neoantigens with a mean number of 1.4 neoantigens per TSE splicing event, which is higher than the average number of 0.64 for non-synonymous single nucleotide variants (nsSNVs) (Turajlic et al., 2017). Across all tumor types, OV was found to have the highest burden of TSE neoantigens (Figure S6E). To validate the expression of these predicted peptides, we used MS-GF+ (Kim and Pevzner, 2014) to search mass spectrometry (MS) data in 32 OV and 35 breast invasive carcinoma (BRCA) samples available from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) project (Ellis et al., 2013), and identified a total of 28 neoepitopes derived from TSE splicing (Table S4). Comparing to nsSNVs and indels, TSE splicing introduced a higher number of CPTAC-confirmed neoantigens per sample in OV, but a lower number in BRCA (Figure 6A), which may reflect differences in TSE splicing- and mutation-derived neoantigen burdens between the two cancer types. Similarly, when examining MS-confirmed neoantigens derived from TSE splicing or somatic mutations across five cancer types offered by the CPTAC study (phase III), we found TSE splicing introduced a higher number of validated peptides in cancers of low tumor mutation burden, such as clear cell renal cell carcinomas (ccRCC), but a lower number than nsSNVs in tumors with high mutational burden, such as lung adenocarcinoma (LUAD) (Figure S6F).
Figure 6. Putative TSE neoantigens and their correlation with immune response.
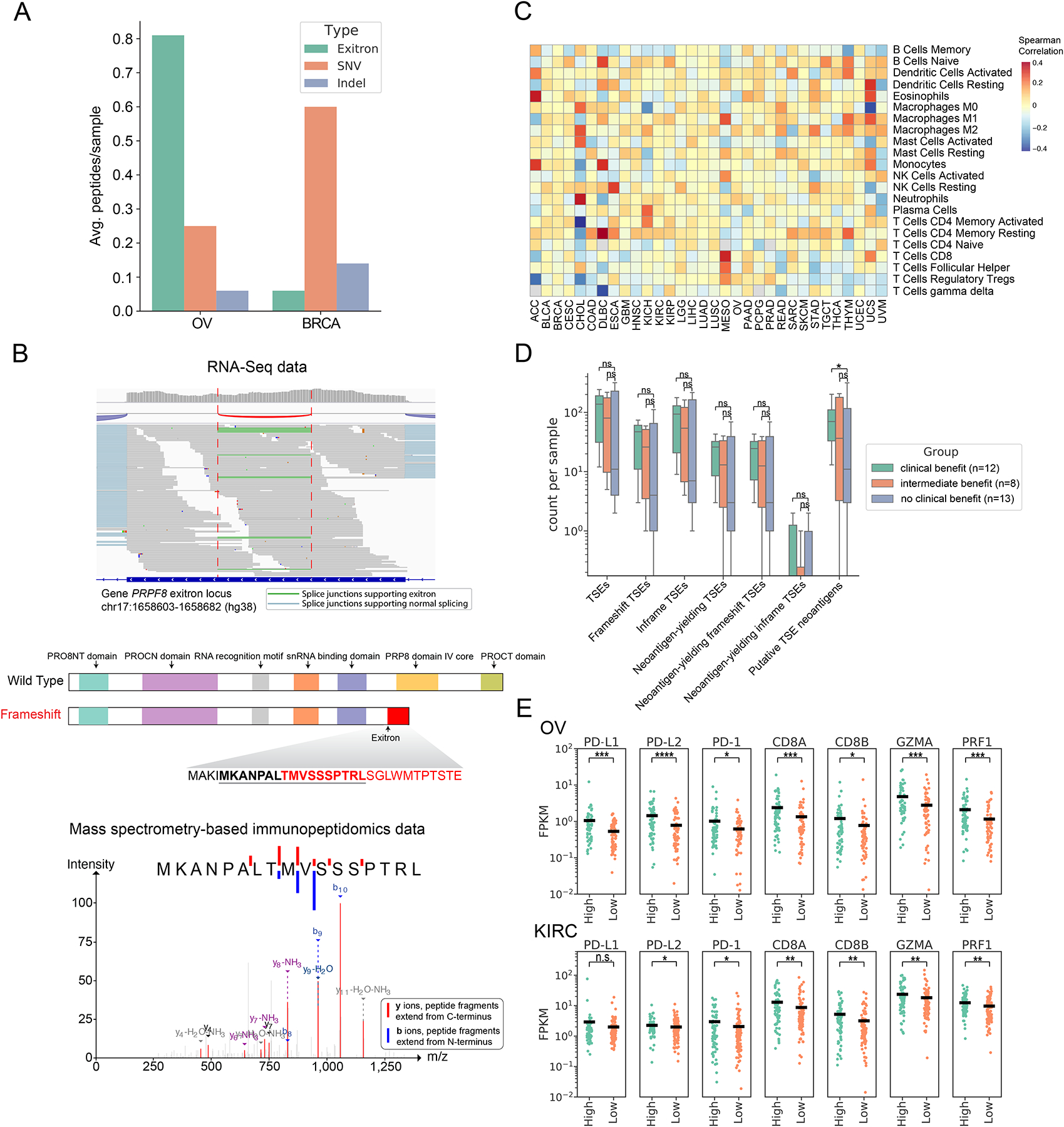
(A) Comparison of the contribution of TSE splicing, somatic SNVs and indels to CPTAC proteomic-confirmed putative neoantigens in BRCA and OV. (B) RNA-Seq data of ovarian cancer patient OvCa65 showed a 79bp exitron in PRPF8 exon 33 (top panel). Predicted functional domains disrupted by this frameshift exitron splicing event in PRPF8 (middle panel). A predicted neoantigen resulting from this frameshift exitron in PRPF8 was found by mass spectrometry to be presented in the corresponding immunopeptidome (bottom panel). (C) TSE neoantigen burden correlates with individual immune cell types in TCGA tumors. Values displayed are the Spearman correlation of immune cell fractions (rows) with neoantigen count within each tumor type (columns). Red indicates positive correlation (increasing proportion of indicated cell type with increasing neoantigen burden), and blue indicates negative correlation. (D) TSE neoantigen burden is associated with checkpoint inhibitor response in clear cell renal cell carcinoma (ccRCC). (E) Expression of T cell markers (PD-1, CD8A, CD8B), cytolytic activity markers (GZMA and PRF1) and immune-regulatory molecules (PD-L1 and PD-L2) in patients between top quartile TSE neoantigen load (named high group) and bottom quartile TSE neoantigen load (named low group) in OV and KIRC (n.s., not significant (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Mann-Whitney rank test).
Although CPTAC proteomic data confirmed the expression of peptides derived from TSE splicing, they were not able to demonstrate whether these peptides were processed and presented on the major histocompatibility complex (MHC). To validate this, we conducted analysis on a separate cohort of 11 ovarian cancer samples (Schuster et al., 2017), where tumors were subjected to transcriptome profiling and immunoaffinity purification of MHC complexes followed by MS analysis. Altogether, 26 TSE neoantigens were confirmed to be presented by MHC class I or II (Figure S6G). In particular, we found that 69% of these neoantigens (18 out of 26) were derived from splicing of TSEs that caused frameshifts, indicating the importance of frameshift-inducing exitron splicing events as a source of neoantigens. For example, PRPF8 displayed recurrent splicing of an exitron (chr17:1658603–1658682) in two patients (OvCa65 and OvCa109, Table S5) that was predicted to cause a frameshift truncating the PRP8 domain. The predicted neoepitope MKANPALTMVSSSPTRL resulting from this frameshift event was confirmed by MS-based immunopeptidome analysis (Figure 6B).
Next, we assessed the potential immunogenicity of TSE neoantigens by comparing them with experimentally validated immunogenic neoantigens and evaluating their association with anti-tumor immune response. We observed that MS-confirmed TSE neoantigens displayed a higher potential to be recognized by CD8+ T cells as measured by the relative hydrophobicity of T-cell receptor contact residues, a hallmark of immunogenic CD8+ T cell epitopes (Chowell et al., 2015), when compared to 295 curated immunogenic nsSNV-derived neoantigens (Figure S6H) and 15 functionally validated immunogenic frameshift indel-derived neoantigens (Figure S6I). Recently, frameshift indels with highly elongated neo open reading frames (neoORF) have demonstrated a high potential to elicit immune response (Litchfield et al., 2020). Accordingly, we found MS-confirmed frameshift TSEs had longer neoORF length than immunogenic frameshift indels (Figure S6J), suggesting frameshift TSEs could be highly immunogenic. Further, we calculated the correlation between the TSE neoantigen load and the immune cellular fractions of TCGA tumors to evaluate the association between TSE neoantigen load and immune responses. We observed that TSE neoantigen load positively correlated with a higher content of CD8+ T cells, M1 macrophages, and CD4+ memory T cells in multiple cancer types (Figure 6C).
Given that indel burden is a known correlate of immune checkpoint inhibitor response, we next sought to examine whether TSE load and TSE neoantigen load were associated with clinical response to immune checkpoint blockade (ICB). To test this, we analyzed data from three cohorts of melanoma patients (n=94) and one cohort of ccRCC patients (n=33) receiving anti-CTLA-4 or anti-PD-1 treatments (Hugo et al., 2016; Miao et al., 2018; Riaz et al., 2017; Van Allen et al., 2015). Although TSE load and TSE neoantigen load had no association with response to ICB in melanoma (Figure S6K), we found a significant association between TSE neoantigen load and clinical benefit from ICB in ccRCC (p = 0.045) (Figure 6D). Moreover, receiver operating characteristic (ROC) analysis demonstrated that TSE neoantigen load was the best predictor of response to ICB in ccRCC compared to known ICB response signatures, such as mutation neoantigen load, CD8+ T cell, PD-L1 and interferon gamma response (Jiang et al., 2018) (Figure S6L). Besides, we found ccRCC patients with clinical benefit had a significantly higher number of NMD-escape exitron splicing events than patients without benefit (Figure S6M). In contrast, no association was found between exitron splicing events predicted to trigger NMD and clinical benefit (Figure S6M), suggesting NMD on exitron splicing may modulate the efficacy of cancer immunotherapy.
To investigate the potential of TSE neoantigen load mediating ICB response in other cancers, we evaluated the correlation between TSE neoantigen load and the expression of T cell markers (PD-1, CD8A and CD8B), immune-regulatory molecules (PD-L1 and PD-L2) and markers of cytolytic activity (GZMA and PRF1) across all TCGA tumor types. We observed that OV and renal clear cell carcinoma (KIRC) were the top two cancer types where patients with a high load of TSE neoantigens expressed higher levels of these immunogenic gene markers (Figure 6E, Figure S6N). Because we have demonstrated the association between TSE neoantigen load and ICB response in ccRCC, our finding suggests that ovarian tumors with a high TSE neoantigen burden would likely benefit from ICB therapy.
Discussion
In this study, we identified widespread exitron splicing in human cancer transcriptomes. By integrating transcriptome sequencing data in nearly ten thousand samples across 33 cancer types, we discovered that 63% of human coding genes display exitron splicing, which expands significantly on previous studies that indicated only 4% of genes display exitron splicing (Marquez et al., 2015). Although this pan-cancer analysis enabled an investigation of exitron splicing at an unprecedented scale and resolution, saturation analysis indicated that the catalog of exitrons likely to exist in the human transcriptome is far from complete. In addition, the ScanExitron method described in this study identifies exitrons only if they are spliced with canonical splice site motifs. Exitrons can be spliced using non-canonical splice motifs, as evidenced by a recent report that exitron EGR1 Δ141–278 used CC-AG splice site motifs (Aliperti et al., 2019). Therefore, future work is needed to provide a more comprehensive portrait of exitron splicing in cancer.
Exitron splicing alterations cause changes in proteins, and functionally mimic the outcome of genetic alterations. Interestingly, we found that genes enriched for exitron splicing (i.e., SEGs) were mutually exclusive with SMGs in cancer. This has importance because analysis of SMGs is an established approach for identifying cancer driver genes (Bailey et al., 2018). The analysis of SEGs in our study led to the discovery of NEFH as a novel tumor suppressor gene in prostate cancer, indicating that analysis of SEGs has the potential to reveal previously undetected cancer driver genes. Beyond SEGs, exitron splicing events that function as cancer drivers (i.e., TSEs) are still largely unknown. Our discovery of functional exitron splicing alterations in FOXA1 supports a model by which proteins frequently altered in cancer through somatic mutations may be affected in a similar way by exitron splicing alterations. Detection and characterizations of these driver exitron splicing events relies on further development of both computational and experimental technologies. For instance, exitrons are usually in a medium- to large-size range that is challenging for existing algorithms to predict their driver potential. High-throughput functional screening technologies such as HiTMMoB (Ng et al., 2018) may provide an alternative approach for assessing the functional impact of exitron splicing on a large-scale.
Indels and alternative splicing events have been recognized as DNA- and RNA-level processes that can contribute to the generation of tumor neoantigens (Kahles et al., 2018; Smart et al., 2018; Turajlic et al., 2017). Exitron splicing is a type of alternative splicing with a proteomic outcome similar to indels in that both can cause a frameshift or an inframe changes of protein sequences. As a result, exitron splicing has the potential to introduce highly immunogenic neoantigens and therefore promotes anti-tumor immune responses. Our discovery of neoantigens derived from exitron splicing expands our knowledge of the tumor immunopeptidome and contributes potential substrates for identifying patients who are most likely to benefit from cancer immunotherapy.
Limitations
The current study reveals that genes enriched with exitron splicing events have the potential to be cancer driver genes. However, most of these genes are understudied in cancer as their overall mutation rates are lower than those of well-known cancer driver genes. An important extension of the current work is to systematically characterize the functions of SEGs with dedicated computational predictions and functional validations. In addition, our current analysis of immunogenicity of exitron splicing-derived neoantigens was mainly driven by availability within the public datasets. Further work to experimentally demonstrate T-cell recognition of identified neoantigens in the tumor and/or peripheral blood will be crucial to prioritize cancer immunotherapy targets in clinical trials.
STAR Methods
Resource Availability
Lead Contact
Further information for resources and data should be directed to and will be fulfilled by the Lead Contact Rendong Yang (yang4414@umn.edu).
Materials Availability
All reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Code for identification and quantification of exitrons is available on GitHub (https://github.com/ylab-hi/ScanExitron). Calculated TSE data and predicted TSE-derived neoantigens data for TCGA are available on Mendeley Data at http://dx.doi.org/10.17632/vdkpfzjjvg.1. RNA-Seq data and processed gene expression data from TCGA cohort, and RNA-Seq data and mutation data from CPTAC cohort (phase3) are available on Genomic Data Commons (GDC) (https://portal.gdc.cancer.gov). TCGA Unified MC3 Variant Calls from Ellrott et al., 2018 analyzed in this manuscript can be found at GDC (https://gdc.cancer.gov/about-data/publications/mc3-2017). HLA types for TCGA samples from Thorsson et al., 2018 is available at GDC (https://gdc.cancer.gov/about-data/publications/panimmune). The clinical outcome endpoints data and gender and tumor stage data for patients were obtained from the TCGA Pan-Cancer Clinical Data Resource (Liu et al., 2018). RNA-Seq data from GTEx cohort are available at GTEx data portal (https://www.gtexportal.org/home/). Protein mass spectrometry data from CPTAC cohort from are available at CPTAC data portal: https://cptac-data-portal.georgetown.edu/cptacPublic/ (Edwards et al., 2015). The RNA-Seq data for ccRCC study (Miao et al., 2018) are available at dbGap: phs001493.v1.p1. The RNA-Seq data for melanoma studies are available from the GEO or dbGaP repository under the following accession codes: GSE78220 (Hugo et al., 2016), phs000452.v2.p1 (Van Allen et al., 2015) and GSE91061 (Riaz et al., 2017). The somatic mutation data from ccRCC and melanoma studies are from their original publications. Illumina RNA-Seq data of SKBR3 are available from the NCBI SRA under accession SRX5414723 (Ghandi et al., 2019). PacBio single-molecular real-time long-read Iso-Seq data of breast cancer cell line SKBR3 are available from the NCBI SRA under accession: SRX4220391 (Nattestad et al., 2018). MS-based immunopeptidomics data and corresponding RNA-Seq data from ovarian cancer samples can be found at PRIDE: PXD007635 and Bioproject: PRJNA398141 (Schuster et al., 2017). RNA-Seq data from 101 metastatic prostate cancer in WC-SU2C cohort are available at dbGap under accession phs001648.v1.p1 (Quigley et al., 2018). Gene expression and clinical annotation data from MSKCC Prostate Cancer Oncogenome Project can be assessed through the MSKCC Prostate Cancer Genomics Data Portal: https://cbio.mskcc.org/cancergenomics/prostate/data (Taylor et al., 2010). nsSNV-derived neoantigens that induce a T-cell response are available from the dbPepNeo database (http://www.biostatistics.online/dbPepNeo). PTM sites for FET family proteins including EWSR1, FUS and TAF15 can be found at the PhosphoSitePlus: https://www.phosphosite.org (Hornbeck et al., 2015).
Experimental Model and Subject Details
Cell lines and cell culture
C4-2 cell line was a kind gift from Dr. Leland W. Chung. All other cell lines (HEK293T, PC-3, and SKBR3) used in this study were obtained from the American Type Culture Collection (Rockville, MD, USA). HEK293T cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% FBS (Gibco) and Penicillin/Streptomycin antibiotics (Gibco). PC-3 and C4-2 cells were cultured in RPMI 1640 medium containing 10% FBS and Penicillin/Streptomycin antibiotics. SKBR3 cells were maintained in McCoy’s 5A medium (Sigma) supplemented with 10% FBS (Millipore), 1% Penicillin/Streptomycin antibiotics (Corning), and 25 μg/mL plasmocin (Invivogen). All cells used in this study were grown 5% CO2 at 37°C and regularly tested as mycoplasma-negative.
Method Details
Clinical and Molecular Data of TCGA study
Gene expression and gene mutation data were obtained for this study. For gene mutation data, we used TCGA Unified MC3 Variant Calls (Ellrott et al., 2018). For gene expression data, the RNA-Seq alignment files in BAM format and HTSeq-Count files in tab-delimited format were downloaded from the GDC (https://gdc.cancer.gov). We only used samples that had available data across these two genomic platforms: gene mutations and mRNA expression. TCGA aliquot barcodes flagged as “do not use” or excluded by pathology review by the PanCancer Atlas Consortium, and annotated according to the Merged Sample Quality Annotation file were removed from the study. For somatic mutations, FILTER values were required to be one of PASS, wga, or native_wga_mix, and only protein-coding mutations were retained (Variant_Classification in one of Frame_Shift_Del, Frame_Shift_Ins, In_Frame_Del, In_Frame_Ins, Missense_Mutation, Nonsense_Mutation, Nonstop_Mutation, Splice_Site, and Translation_Start_Site). Mutations calls were required to be made by two or more mutations callers (NCALLERS > 1), satisfy allele requirements: reference allele count >= 3 and total depth >= 10, allele frequency >= 0.05. For RNA-Seq data, a single representative aliquot was selected per participant for cases where more than one aliquot was available, as follows. When data on more than one tumor sample was available, we used the following rule of priority order (01A (not FFPE), 01B (FFPE), 01C (FFPE), 06A (Metastatic), 02A (Recurrent tumor), 05A (additional primary)) to keep one aliquot, according to the sample identifier of TCGA barcode. The clinical outcome endpoints data and gender and tumor stage data for patients were obtained from the TCGA Pan-Cancer Clinical Data Resource (Liu et al., 2018). The immune cell fraction data were obtained from Thorsson et al. (Thorsson et al., 2018).
Identification and quantification of exitron splicing events
Exitron splicing events were detected from RNA-Seq using ScanExitron pipeline as illustrated in Figure 1B. Briefly, high quality uniquely mapped reads (MAPQ >50) were extracted from BAM files with samtools view -q 50 command. ScanExitron first extracted splicing junctions from these high mapping quality reads using RegTools (version 0.4.0) (Feng et al., 2018). Next, junctions from annotated intron regions were removed based on the GTF annotation files from GENCODE v21 and NCBI RefSeq (GRCh38.p7). The remaining unannotated junctions were further processed using in-house Python scripts to identify their splice site motifs. Splicing junctions with canonical spliced site motifs (GT-AG, GC-AG, and AT-AC) located within protein-coding exons were identified as putative exitron splicing events. We measured exitron splicing expression as the fraction of alternatively spliced junction spanning reads over the total number of reads across the spliced exitron region, which we refer to as percent of spliced-out (PSO) value. We set filters that required exitrons to have: 1) at least three uniquely-mapped reads across the exitron junction and 2) a minimum of 5% PSO in this study.
Detection of differentially spliced exitrons between tumor and normal tissues
The differentially spliced exitrons were detected in tumor types that had at least 40 tumor samples and 40 matched normal samples, including BRCA, HNSC, KIRC, LIHC, LUAD, LUSC, PRAD and THCA. For each tumor type, a linear regression model with PSO as the dependent variable and tumor status as independent variables was employed to identify exitrons that are differentially spliced between tumor and matched normal tissues. The results from different tumor cohorts were aggregated using Stouffer method (Stouffer et al., 1949) for meta-analysis, the meta p values are Benjamini-Hochberg FDR corrected for multiple testing. Those exitron splicing events present in less than four cancer types were discarded.
Effect of cis-acting features
MaxEntScan (Yeo and Burge, 2004) was used to calculate maximum entropy scores for 9-bp 5′ splice sites and 23-bp 3′ splice sites. The length and GC content of constitutive introns, exitrons and retained introns were calculated using the hg38 human genome assembly. Constitutive introns were defined as those present in all child transcripts of a given GENCODE gene. Retained introns of TCGA tumor samples were derived from Kahles et al. (Kahles et al., 2018).
Effect of trans-acting splicing factors
First, 404 SFs were collected from the literature (Seiler et al., 2018). Based on these SFs, we defined the SF gene signature to quantify SF activity for each sample. In brief, Z-scores for 404 SF genes were computed by subtracting the pooled mean from the RNA-Seq expression FPKM values and dividing by the pooled standard deviation. The SF gene signature was defined as the summation of the Z-scores.
To calculate the proportion of the variation in exitron splicing that could be explained by SFs, we used a generalized additive model (GAM) from the R-package mgcv (Wood, 2011) v1.8.3. We selected tumor types that had at least 40 tumor samples and 40 matched normal samples, including BRCA, HNSC, KIRC, LIHC, LUAD, LUSC, PRAD and THCA in the GAM analysis. For each cancer type, we selected the most variable exitrons, defined as having a standard deviation of changes in exitron for tumor-normal pairs across samples exceeding 0.02. For the 404 literature-curated SF genes, we used the log2 fold-change of FPKM normalized gene expression of the matched tumor-normal pairs. GAM models for all individual exitron/gene pairs were calculated using a Gaussian distribution with “identity” as the link function, and the method “GCV.Cp” was used for estimating the smoothing parameters of the log2 fold-changes for the genes. Following GAM predictions, a linear regression model was used to measure the relationship between predictors and responses for GAM-derived significantly associated exitron/SF pairs (FDR<0.05). For each SF in the analysis, the number of exitron/SF pairs was required to be three or more. The positive Pearson Correlation Coefficient (PCC) of exitron/SF pair indicated that the specific SFs generally promoting this exitron splicing.
Characterization of tumor-specific exitrons and their clinical relevance
Due to the lack of matched normal RNA-Seq data for most TCGA tumor samples, GTEx healthy samples were incorporated to identify the tumor-specific exitrons. Raw RNA-Seq reads of GTEx samples were obtained from dbGap (phs000424.v7.p2) and mapped to the human reference genome (hg38) using HISAT2 (Kim et al., 2019). Next, we identified exitron splicing events of TCGA and GTEx project using the criteria as described above. Exitrons that were spliced in GTEx samples or spliced in more than three TCGA normal samples were excluded from the exitrons detected in TCGA tumor samples. The remaining exitrons were considered as the tumor-specific exitrons. We further defined the informative and clinically relevant exitron splicing events if a tumor-specific exitron meets the following criteria:
The exitron splicing recurred in more than ten samples in a cancer type.
The exitron splicing was significantly associated with one of the three clinical endpoints: overall survival, progression-free survival and disease-free survival.
The exitron-containing gene expression was not associated with the same type of survival detected in (b).
Protein domain and post-translational modification sites analysis
The inframe exitron splicing events and somatic mutations (missense mutations and inframe indels) encode whole or parts of Pfam (Finn et al., 2014) protein domains were calculated using Variant Effect Predictor (VEP) (McLaren et al., 2016). Then the number of genes with the affected Pfam domains was obtained for exitron splicing alterations and somatic mutations, respectively. PTM sites for FET family proteins including EWSR1, FUS and TAF15 were obtained from PhosphoSitePlus (Hornbeck et al., 2015) (Version Aug 19, 2019).
Expressed mutation identification
For somatic mutations reported in TCGA Unified MC3 Variant Calls, we firstly filtered out the mutations, where their harboring genes have zero reads count in the corresponding RNA-Seq data. For the remaining mutations, freebayes (Garrison and Marth, 2012) was applied to the RNA-Seq data to call variants in the recorded mutation sites from MC3 to check whether the mutations were expressed or not at transcriptomic level. The MC3 mutations detected by freebayes from RNA-Seq were considered as expressed in RNA. Variant allele fraction (VAF) of expressed mutations were calculated as AO/DP, where AO is the number of RNA-Seq reads supporting the mutations, and DP is the RNA-Seq total depth at the mutation site. VAFs of not expressed MC3 mutations were set to 0 in the expressed mutation list.
NMD efficiency estimation
We used gene expression data and the position of premature termination codon (PTC) to estimate NMD efficiency, respectively. To estimate the NMD efficiency with gene expression data, we adopted the NMD index method (Turajlic et al., 2017). We estimated the extent of NMD for all tumor-specific exitrons and retained introns by comparing mRNA expression in samples with a splicing aberration event to the median mRNA expression of the same gene across all other tumor samples where the splicing aberration was absent. For a gene in one sample with splicing aberration and n samples where the splicing aberration was absent. NMD index (NMDI) for the gene in the sample t is calculated by
Frameshift variants-derived PTCs can result in the degradation of mRNAs by triggering NMD or the production of truncated proteins by escaping NMD. The NMD efficiency is linked to the sequence positions (Lindeboom et al., 2016). We defined NMD-escaped frameshift events if a PTC meets any of the following criteria (Litchfield et al., 2020): (a) first exon within the first 200 nucleotides of coding sequence (CDS), (b) penultimate exon within 50 nucleotides of the 3′ exon junction, and (c) last gene exon (Figure S6D). Tumor-specific exitrons were detected as described above. Tumor-specifically retained introns were obtained from Kahles et al. (Kahles et al., 2018).
HLA typing with OptiType
HLA class I four-digit types of 8,915 out of 9,599 TCGA tumor samples were obtained from Thorsson et al (Thorsson et al., 2018). For the remaining 684 TCGA tumor samples used in this study, OptiType (Szolek et al., 2014) and yara aligner (Siragusa et al., 2013) were employed for HLA class I typing procedure, as follows. The RNA-Seq alignment files in BAM format were first converted to raw reads in FASTQ format using Picard tool (http://broadinstitute.github.io/picard/). Next, the raw reads of each sample were aligned to the HLA class I database provided by OptiType using yara aligner with error rate of 3%. Finally, OptiType was used to predict the HLA class types for each sample under its default parameters for the RNA-Seq data.
Neoantigen prediction for tumor-specific exitron splicing events and somatic mutations
Tumor-specific exitron splicing derived neoantigen candidates were identified using ScanNeo (Wang et al., 2019) based on HLA types derived from RNA-Seq data as described above. In brief, predicted tumor-specific exitron splicing events were first reported in VCF files. Next VEP was used to annotate exitron splicing alterations with the raw VCF file as input. HGVS indel notation rules were used to report exitrons (den Dunnen et al., 2016). Inframe events are coded to start with a delta (means deletion) notion followed by the amino acid positions removed by this exitron splicing event. A frameshift event is coded to contain ‘fs’ indicates this type of change is frameshift, following the length of neo Open Reading Frame (neoORF). Using NetMHC (Lundegaard et al., 2008) and NetMHCpan (Nielsen and Andreatta, 2016), ScanNeo predicted neoantigen candidates that bound to autologous HLA (IC50 < 500 nM) with the VEP annotated VCF files as input. Neoantigens derived from non-synonymous single nucleotide variants (nsSNVs) and indel variants were predicted for TCGA-BRCA, TCGA-OV and five CPTAC tumor types using the ScanNeo method with the same criteria as were used for tumor-specific exitron splicing events.
Exitron splicing-derived neoantigen prediction for patient cohorts treated with ICB
The RNA-Seq data for the clear cell renal cell carcinoma (ccRCC) study (Miao et al., 2018) was download from dbGap: phs001493.v1.p1. The RNA-Seq data for melanoma studies were download from GEO: GSE78220 (Hugo cohort (Hugo et al., 2016)), dbGap: phs000452.v2.p1 (Van Allen cohort (Van Allen et al., 2015)) and GEO: GSE91061 (Riaz cohort (Riaz et al., 2017)). Exitrons and tumor-specific exitrons of these studies were identified using the methods and parameters applied for TCGA cohorts and described above. HLA class I four-digit types were obtained from the original studies or inferred by using OptiType and yara aligner for HLA class I typing from the RNA-Seq data. TSE splicing-derived neoantigen candidates were identified using ScanNeo as described above.
Performance comparison on predicting ICB response
We collected the somatic mutation data from ccRCC (Miao et al., 2018) and melanoma studies (Hugo et al., 2016; Riaz et al., 2017; Van Allen et al., 2015). Mutation-derived neoantigens were identified using ScanNeo as described above. Gene expression profiles (FPKM values) were quantified by featureCounts (Liao et al., 2014) from the RNA-Seq data as described above. In addition to TSE-splicing load, TSE splicing-derived neoantigen load and mutation-derived neoantigen load, we include literature-reported ICB response biomarkers (expression levels of PD-L1, CD8+ T cell, and interferon gamma) (Jiang et al., 2018) for comparison. If the biomarker includes multiple gene members (e.g., CD8+ T cell: CD8A and CD8B), the average expression values among all members will be used as the expression value of this biomarker.
To determine the predictive power, we stratified patients into responders and non-responders, and performed a ROC analysis detailing the true-positive rates versus false-positive rates at various thresholds of predictor values. The area under the curve (AUC) from the ROC analysis was used as the performance measurements of prediction. Smoothed ROC curve was generated by R package pROC (Robin et al., 2011).
Identification of expressed peptides in CPTAC
Proteomics data for TCGA breast (Mertins et al., 2016) (35 samples) and ovarian cancer (Zhang et al., 2016) (32 samples) were downloaded from the CPTAC data portal (Edwards et al., 2015). For each of the 67 TCGA tumor samples, we generated individual polypeptide databases comprising mutant (MT) peptide sequences with ten flanking amino acids on each side of the exitrons/nsSNVs/indels concatenating with UniProt human proteome. OpenMS (Rost et al., 2016) was used to identify polypeptides from a sample’s polypeptide database as follows: 1) decoy sequences were added to the database to control false discovery rates; 2) The sample’s CPTAC mass spectrometry data set was searched against the corresponding polypeptide database using MS-GF+ (Kim and Pevzner, 2014). The peptide-spectrum match (PSM) FDR was set to 5% for the polypeptide identification. Any 9-mer putative neoantigen contained in at least one of the identified polypeptides is considered CPTAC-confirmed. We used the same commands and parameters as described in Kahles, A. et al. (Kahles et al., 2018) to perform the polypeptide identification, as follows:
-
Create decoy sequences database.
DecoyDatabase -in <in.fasta> -out <db.fasta>
-
Search CPTAC mass spectra.
MSGFPlusAdapter -ini <config.ini> -in <spectra.mzML> -out <out.idXML> -database <db.fasta> -executable <MSGFPlus.jar> -java_memory 20000 -threads 16
-
Refresh the mapping of peptides to proteins and add target/decoy information.
PeptideIndexer -in <out.idXML> -fasta <db.fasta> -out <pi_out.idXML> -allow_unmatched - enzyme:specificity ‘semi’
-
Merge peptide identification files from multiple runs.
IDMerger -in <pi_out.idXML files> -out <merged.idXML>
-
Control for false discovery rate
FalseDiscoveryRate -in <merged.idXML> -out <fdr_out.idXML>
IDFilter -in <fdr_out.idXML> -out <fdr_filtered.idXML> -score:pep 0.05
The codes and the configuration file are also available on GitHub (https://github.com/ylab-hi/ScanExitron).
In addition to TCGA tumor samples, five cancer types offered by CPTAC (phase III) have been used for the proteomic analysis. CPTAC samples include ccRCC (Clark et al., 2019) (110 samples), Glioblastoma (99 samples), Head and Neck Squamous Cell Carcinoma (HNSCC) (110 samples), Uterine Corpus Endometrial Carcinoma (Dou et al., 2020) (101 samples) and Lung Adenocarcinoma (Gillette et al., 2020) (111 samples). The corresponding mutation data and the RNA-Seq alignment files were downloaded from the GDC (https://gdc.cancer.gov). Proteomics data were downloaded from the CPTAC data portal (Edwards et al., 2015). For somatic mutations data, mutation calls were required to be made by two or more mutation callers at the position where total depth >= 10. Following the same procedure applied for TCGA samples, neoantigens derived from exitrons/nsSNVs/indels and proteomic-confirmed neoantigens were identified.
Identification of expressed peptides in immunopeptidome data
We obtained the MS-based immunopeptidomics raw data and the corresponding RNA-Seq data from eleven ovarian cancer samples (patient ids: OvCa48, OvCa58, OvCa65, OvCa70, OvCa80, OvCa84, OvCa104, OvCa105, OvCa109, OvCa111, OvCa114) (Schuster et al., 2017). The immunopeptidomics raw data were converted to mzML files with ProteoWizard Toolkit (Chambers et al., 2012). Tumor-specific exitrons of these ovarian cancer samples were identified by ScanExitron from their RNA-Seq data following the same criteria used for tumor-specific exitron analysis for TCGA samples, which requires tumor-specific exitrons were not spliced in GTEx samples and were spliced in no more than three TCGA normal samples. For each ovarian cancer sample, we generated individual polypeptide databases comprising MT peptide sequences with ten flanking amino acids on each side of the exitrons. The decoy sequences were added to the database for false discovery rates control. OpenMS (Rost et al., 2016) was used to perform polypeptide identification. Tandem MS spectra were searched against the target-decoy databases by MS-GF+ with the following settings: (a) No cleavage specificity, (b) one dynamic modification allowed (oxidized methionine), (c) precursor mass tolerance 5 ppm, (d) peptide length allowed: 8–11 amino acids for MHC-I ligands, 12–21 amino acids for MHC-II ligands and (e) a false discovery rate of 5% on the peptide-spectrum match level to filter the identified polypeptides.
Immunogenicity score and neoORF length calculation for exitron splicing events and mutations
We measure the immunogenicity of neoantigens by calculating the relative hydrophobicity of amino acids at T-cell receptor contact residues, which is a strong hallmark of CD8+ T cell-mediated immunity (Chowell et al., 2015). pTuneos (Zhou et al., 2019) was used to calculate a hydrophobicity score for candidate neoantigens, which utilize the amino acid biochemical property (Kyte-Doolittle numeric hydrophobicity) and eXtreme Gradient Boosting (XGBoost) machine learning model to infer the score based on experimentally validated T-cell response epitopes deposited in Immune Epitope Database (IEDB, www.iedb.org ). To evaluate the immunogenic potential of TSE-derived neoantigens, 295 nsSNV-derived neoantigens that induce a T-cell response curated from dbPepNeo (Tan et al., 2020), 15 literature-reported functionally validated immunogenic frameshift (fs) indel-derived neoantigens (Litchfield et al., 2020) and 4 non-immunogenic fs-indel-derived neoantigens from the same study were used for immunogenicity predictions. The length of neoORF was used as another measurement of immunogenicity as it has been reported that long neoORF was associated with immunogenic neoantigens (Litchfield et al., 2020). VEP was used to determine the neoORF lengths as described above.
RNA-Seq and Iso-Seq validation of exitrons in SKBR3
Illumina RNA-Seq data of SKBR3 was downloaded from NCBI SRA (Accession: SRX5414723) (Ghandi et al., 2019). Exitron splicing events were identified using the methods and parameters applied for TCGA cohorts and described above. PacBio single-molecular real-time long-read Iso-Seq data of breast cancer cell line SKBR3 was downloaded from NCBI SRA (Accession: SRX4220391) (Nattestad et al., 2018). The long-read raw data in FASTA format were retrieved using the SRA toolkit (Leinonen et al., 2011). Minimap2 (Li, 2018) was used to align Iso-Seq long-read raw data to hg38 human genome assembly with the parameters for spliced long reads alignment. Exitron splicing events were identified from aligned long reads using ScanExitron as described above. TALON (Wyman et al., 2020) was used for transcript-level quantification to long-read identified exitron-spliced and wild-type transcripts, respectively.
PCR- and Sanger sequencing-based validation of exitrons in SKBR3
Genomic DNA (gDNA) was isolated from SKBR3 human breast cancer cells were using standard methods. Briefly, cells were lysed with 500 μl of cell lysis buffer (10 mM Tris-HCl, 10 mM EDTA, 50 mM NaCl, 10% SDS, pH 7.5) supplemented with 10μl proteinase K (Roche). Following centrifugation, DNA was precipitated from lysates using isopropanol, washed with 70% ethanol, and resuspended in 100 μl of nuclease-free water for subsequent experiments. Alternatively, mRNA was extracted from SKBR3 cells using an RNeasy Plus kit (Qiagen) according to the manufacturer’s instructions. Eluted RNA (1 μg) was used to generate cDNA for subsequent experiments using a cDNA synthesis kit (BioRad) as described in the kit provided protocol. Standard PCR was performed using gDNA and mRNA-derived cDNA templates, Choice-Taq™ DNA Polymerase (Thomas Scientific), and five pairs of gene specific oligonucleotides designed to flank the exitronic deletions identified in SETDB2, MOGS, NOD1 and CHD2 genes. Forward (5’−3’) and reverse (5’−3’) primer sequences used to amplify exitron splice products are as follows: SETDB2-F: TGCCACTGAACTTGAAGGGA, SETDB2-R: CCGAGCCAACTGAACATAGG, MOGS-F: TGACAGATGGCAAGGAAGTC, MOGS-R: CCCTTGTCCGTAGAAGTAGCC, NOD1-F: TGACTCCAAGTTCGTGCTGT, NOD1-R: CTCAGGTCCAAGTCCGAGTG, CHD2-F: CGGATAGCCGAGTGCCTTAAA, CHD2-R: CTCTGCCAGTCTCCTCGATCT. PCR amplified DNA extracted from a 2% agarose gel was purified using QIAEX II® Gel Extraction Kit (Qiagen) according to the manufacturer’s instructions. Gel extracted PCR products were cloned into the pCR®-Blunt II-TOPO® vector (Invitrogen) for subsequent Sanger sequencing. Plasmids isolated using ZymoPURE™ Plasmid Miniprep Kit (Zymo Research) were subjected to Sanger sequencing using the pCR®-Blunt II-TOPO® kit supplied M13-R primer, 5’- CAGGAAACAGCTATGAC-3′.
Transcriptional signature of FOXA1 exitron-spliced tumors
To determine transcriptional signatures associated with FOXA1 exitron splicing events, we identified significantly differentially expressed genes between FOXA1 exitron-spliced and wild-type samples. Wild-type samples were determined as those lacking ETS family gene fusions (ERG, ETV1, ETV4 and FLI1) and SPOP mutations (Parolia et al., 2019) in addition to FOXA1 mutations and exitron splicing events. To identify putative transcription factors regulating differentially expressed genes between FOXA1 exitron-spliced tumors and wild-type patients, BART (Wang et al., 2018) was used to infer the specific transcription factors mediating transcriptional changes.
The AR activity score was calculated as previously described (Cancer Genome Atlas Research, 2015). In brief, Z-scores for 20 androgen-induced genes were computed by subtracting the pooled mean from the RNA-Seq expression FPKM values and dividing by the pooled standard deviation. The sum of the Z-scores for the AR signaling gene signature of 20 androgen-induced genes represents the AR activity score for each sample.
Experimental validation of FOXA1 Δ186–215 and Δ231–240 and NEFH Δ871–911
(a) Antibodies
The following antibodies were used for western blotting: NEFH antibody (ab207176, RRID:AB_2827968); Myc-Tag antibody (9B11; RRID:AB_331783); PARP antibody (46D11, RRID:AB_659884); β-ACTIN antibody (Ab8227, RRID:AB_2305186).
(b) Cell Proliferation assay
Cells were seeded at a density of 1×103 cells/well in a 96-well plate with outer wells left empty for addition of PBS. The proliferation rate was performed with CellTiter-Glo® 2.0 Cell Viability Assay Kit (G9241, Promega). Each assay was performed at least three times with triplicate wells.
(c) BrdU incorporation assay
The BrdU incorporation assay was performed in wild type and mutant NEFH expressing C4-2 and PC-3 cells with BrdU ELISA kit (ab126556, Abcam) according to manufacturer’s recommended conditions. The data are presented as the means and standard deviations of three independent experiments with triplicate wells.
(d) Colony-forming assay
PC-3 and C4-2 cells (500 cell/well) were seeded on 6-well plates and maintained in RPMI1640 cell culture medium supplemented with 10% FBS in a humidified chamber atmosphere comprising 95% air and 5% CO2 at 37°C for 2 weeks. Following PBS wash, cells were stained using 0.1% crystal violet solution at room temperature for 30 min. Stained cells were washed with water gently, pictures were taken after air dried at room temperature.
(e) Luciferase reporter assay
Oligonucleotide fragments containing six tandem FKHD-consensus (canonical or non-canonical) (Adams et al., 2019) motifs with 5-bp spacers (Oligo list) were cloned into pGL3-Promoter Luciferase Reporter Vector (Pomega) between NheI and XhoI restriction sites. Oligonucleotide sequences were verified using Sanger sequencing. The pGL3–6FBS-Luc (600 ng per well) was transiently transfected using Lipofectamine 3000 (Thermo Fisher) into HEK293T cells in 12 well plates along with CMV-Renilla (60 ng per well) (pRL-CMV Renilla, Promega) as an internal control. To test the response of these reporters to various mutants of FOXA1 introduced into the system, the same total amount of DNA was transfected into each well. Response ratios are calculated relative to the signal obtained for the wells transfected with wild-type FOXA1 (1200 ng per well), which was set to 1. In evaluating relative response ratios between FOXA1 (wild type) and various mutants, one concentration of cDNA (1200 ng per well) was used and relative response ratios reflect the activity of the given variant on the reporter. Luminescence measurements were taken 24 hrs after transfection. All results are means and standard deviations from experiments performed in biological triplicates, and luciferase activity of individual well was normalized against Renilla luciferase activity using the Dual-Glo Luciferase assay (Promega; E2980).
HEK293T cells stably overexpressing the wild-type AR protein (HEK293T-AR) were used for the TMPRSS2 reporter assays. 14 hrs after seeding cells in 12-well plate, medium was replaced with 10% CSS-supplemented phenol-free medium (androgen-depleted) and cells were transfected with the TMPRSS2 promoter Firefly luciferase reporter. TMPRSS2 promoter luciferase reporter (Liu et al., 2019) construct was described previously. It was transiently transfected (1200 ng per well) using Lipofectamine 3000 (Thermo Fisher) into HEK293T-AR cells along with CMV-Renilla (60 ng per well) (pRL-CMV Renilla, Promega). 8 hrs after transfection, cells were treated with DHT at 10 nM dosage diluted with fresh androgen-depleted medium (Parolia et al., 2019); and after incubation for additional 24 hrs, dual luciferase activity was recorded for every sample using the Dual-Glo Luciferase assay (Promega; E2980). All results are means and standard deviations from experiments performed in biological triplicates, and luciferase activity of individual well was normalized against Renilla luciferase activity.
(f) Cloning of representative FOXA1 and NEFH mutants
Human wild-type FOXA1 coding sequence was cloned in pLenti-C-Myc-DDK-P2A-Puro (Origene PS100092) by primers (Oligo list). Human NEFH wild type expressing vector was purchased from Origene (RC213487L3). Truncate mutations were engineered from the wild-type FOXA1 and NEFH vector using specific primers (Oligo list). All mutations were confirmed using Sanger sequencing through GENEWIZ. Engineered mutant plasmids were further transfected in HEK293T cells to confirm the expression of the mutant protein. All FOXA1 and NEFH variants had the Myc tag fused on the C terminus, and these lentivirus vectors were further used for generating stable cell lines in PC-3 and C4-2 cells by puromycin selection.
Quantification and Statistical Analysis
Characterization of significantly exitron-spliced genes and gene sets
We adopted the method used in MutSig (Lawrence et al., 2013) to identify significantly exitron-spliced genes (SEGs) that are enriched for exitron splicing events. For a tumor type, a single average genome-wide background exitron splicing rate (BER) was calculated by , where N is the number of exitron splicing events in a specific tumor cohort, L is total length protein-coding exons harboring exitron splicing events in pan-cancer cohorts. A binomial distribution was used to calculate the gene-specific enrichment p value as below:
where k is observed number of exitron splicing events for a gene, n is the length of protein-coding exons for this gene. Benjamini-Hochberg FDR was controlled at 10−5. For each identified SEG, tissue specificity among 33 cancer cohorts was defined using tissue specificity index tau (Yanai et al., 2005), as follows.
where n is the number of cancer cohorts (n=33) and xi is the number of exitron splicing events occurred in a particular SEG divided by the total number of exitron splicing events occurred in the cohort i.
For a gene set {ℊ1, ℊ2 ⋯ ℊm}; that includes m genes, a binomial distribution was used to calculate the gene set-specific enrichment p value as below:
where ki is observed number of exitron splicing events in gene ℊi and ni is the length of protein-coding exons for gene ℊi. Benjamini-Hochberg FDR was controlled at 0.05.
Differential analysis between somatic mutation and tumor specific exitron splicing altered genes
The differential analysis was run on all tumor types that had at least 50 tumor samples available, CHOL, DLBC and UCS were excluded. For each tumor type we randomly selected 50 samples. We then used Wilcoxon signed-rank test to perform a differential test between mutations and exitron splicing, utilizing the event number for every gene in every sample. Formally, the p value of each gene (Pr(ℊ)) is obtained by Wilcoxon signed-rank test ([x1, x2, …, xn], [y1, y2, …, yn]), where n is total number of selected samples, xi is the number of exitron splicing events and yi is the number of mutations for gene ℊ in patient i. Benjamini-Hochberg FDR correction was applied for Pr(ℊ). The effect size was calculated as the log-ratio of total exitron splicing count to total mutation count. We kept the top 300 genes ranking by the average exitron splicing count or average mutation count in all tumor samples. To account for variability in the random selection process, we repeated the testing 10 times, each time on a different random subset. For each gene, the final FDR and effect size was recorded as the median of the ten results.
Statistical analysis
Student’s t-test, Kruskal–Wallis test, Mann-Whitney test, Fisher’s exact test and Hypergeometric test were performed using R v3.2.2 (R Core Team, 2017). Benjamini-Hochberg multiple testing correction was used to estimate the FDR when multiple testing correction was applied, unless specified otherwise. Significance was reported at four levels and is indicated in figure legends: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Kaplan-Meier estimate and log-rank test were performed using Python package lifelines (Davidson-Pilon, 2019). Other details regarding parameters pertaining to results shown in figures can be found in the associated legends, including statistical analysis performed, statistical significance and counts.
Supplementary Material
Table S2. The number of EIS, EIS carrying exons, EIS carrying genes, TSE-derived neoantigens in TCGA cohorts, Related to Figure 2.
Table S3. Significantly exitron-spliced genes (SEGs) in TCGA cohorts, Related to Figure 3.
Highlights.
Large-scale transcriptome analysis compiles a cancer exitron splicing landscape
Exitron splicing disrupts functional protein domains to cause cancer driver effects
Immunopeptidome analysis identifies exitron splicing-derived neoantigens
Exitron splicing neoantigen load predicts response to checkpoint inhibitor therapy
Acknowledgements
We acknowledge the following sources of funding: DoD (W81XWH-19-1-0161), American Cancer Society (#129819-IRG-16-189-58-IRG103), Prostate Cancer Foundation Young Investigator Award and PhRMA Foundation Research Starter Grant to R.Y.; DoD (W81XWH-17-1-0357, W81XWH-19-1-0563 and W81XWH-20-1-0504), American Cancer Society (RSG-15-192-01) and NIH (R01CA208257 and Prostate SPORE P50CA180995 DRP) to Q.C.; NIH (R01CA174777) to S.M.D.; American Cancer Society (#129819-IRG-16-189-58-IRG81) to L.H.H. and Eagles Telethon Postdoctoral Fellowship to T.-Y.W. and S.K.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Adams EJ, Karthaus WR, Hoover E, Liu D, Gruet A, Zhang Z, Cho H, DiLoreto R, Chhangawala S, Liu Y, et al. (2019). FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliperti V, Sgueglia G, Aniello F, Vitale E, Fucci L, and Donizetti A (2019). Identification, Characterization, and Regulatory Mechanisms of a Novel EGR1 Splicing Isoform. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyoshi M, and Schwabe JW (2003). A conserved structural motif reveals the essential transcriptional repression function of Spen proteins and their role in developmental signaling. Genes Dev 17, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 174, 1034–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowell D, Krishna S, Becker PD, Cocita C, Shu J, Tan X, Greenberg PD, Klavinskis LS, Blattman JN, and Anderson KS (2015). TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc Natl Acad Sci U S A 112, E1754–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih TM, Chang HY, et al. (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983 e931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT (2013). The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson-Pilon C (2019). lifelines: survival analysis in Python. Journal of Open Source Software 4 (40), 1317. [Google Scholar]
- den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, and Taschner PE (2016). HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat 37, 564–569. [DOI] [PubMed] [Google Scholar]
- Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, et al. (2020). Proteogenomic Characterization of Endometrial Carcinoma. Cell 180, 729–748 e726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, and Bradley RK (2015). Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards NJ, Oberti M, Thangudu RR, Cai S, McGarvey PB, Jacob S, Madhavan S, and Ketchum KA (2015). The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J Proteome Res 14, 2707–2713. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Gillette M, Carr SA, Paulovich AG, Smith RD, Rodland KK, Townsend RR, Kinsinger C, Mesri M, Rodriguez H, et al. (2013). Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov 3, 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 6, 271–281 e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y-Y, Ramu A, Cotto KC, Skidmore ZL, Kunisaki J, Conrad DF, Lin Y, Chapman WC, Uppaluri R, Govindan R, et al. (2018). RegTools: Integrated analysis of genomic and transcriptomic data for discovery of splicing variants in cancer. bioRxiv, 436634. [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. (2014). Pfam: the protein families database. Nucleic Acids Res 42, D222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankiw L, Baltimore D, and Li G (2019). Alternative mRNA splicing in cancer immunotherapy. Nat Rev Immunol. [DOI] [PubMed] [Google Scholar]
- Garrison E, and Marth G (2012). Haplotype-based variant detection from short-read sequencing. In arXiv e-prints. [Google Scholar]
- Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang WW, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225 e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, and Brunet A (2005). FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 24, 7410–7425. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Cichowski K, and Elledge SJ (2019). Tissue-specificity in cancer: The rule, not the exception. Science 363, 1150–1151. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, and Takeda S (1998). Gene targeting studies begin to reveal the function of neurofilament proteins. J Cell Biol 143, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Lee D, Lee J, Park D, Kim YJ, Park WY, Hong D, Park PJ, and Lee E (2015). Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 47, 1242–1248. [DOI] [PubMed] [Google Scholar]
- Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Cancer Genome Atlas Research, N., et al. (2018). Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34, 211–224 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Chang X, LeBron C, Nagpal JK, Lee J, Huang Y, Yamashita K, Trink B, Ratovitski EA, and Sidransky D (2010). Neurofilament heavy polypeptide regulates the Akt-beta-catenin pathway in human esophageal squamous cell carcinoma. PLoS One 5, e9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, and Pevzner PA (2014). MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law WJ, Cann KL, and Hicks GG (2006). TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomic Proteomic 5, 8–14. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legare S, Cavallone L, Mamo A, Chabot C, Sirois I, Magliocco A, Klimowicz A, Tonin PN, Buchanan M, Keilty D, et al. (2015). The Estrogen Receptor Cofactor SPEN Functions as a Tumor Suppressor and Candidate Biomarker of Drug Responsiveness in Hormone-Dependent Breast Cancers. Cancer Res 75, 4351–4363. [DOI] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M, and International Nucleotide Sequence Database, C. (2011). The sequence read archive. Nucleic Acids Res 39, D19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom RG, Supek F, and Lehner B (2016). The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet 48, 1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield K, Reading JL, Lim EL, Xu H, Liu P, Al-Bakir M, Wong YNS, Rowan A, Funt SA, Merghoub T, et al. (2020). Escape from nonsense-mediated decay associates with anti-tumor immunogenicity. Nat Commun 11, 3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wang G, Li Q, Jiang W, Kim JS, Wang R, Zhu S, Wang X, Yan L, Yi Y, et al. (2019). Polycomb group proteins EZH2 and EED directly regulate androgen receptor in advanced prostate cancer. Int J Cancer 145, 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, and Nielsen M (2008). NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res 36, W509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez Y, Hopfler M, Ayatollahi Z, Barta A, and Kalyna M (2015). Unmasking alternative splicing inside protein-coding exons defines exitrons and their role in proteome plasticity. Genome Res 25, 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bosse D, Wankowicz SM, Cullen D, et al. (2018). Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattestad M, Goodwin S, Ng K, Baslan T, Sedlazeck FJ, Rescheneder P, Garvin T, Fang H, Gurtowski J, Hutton E, et al. (2018). Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res 28, 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PK, Li J, Jeong KJ, Shao S, Chen H, Tsang YH, Sengupta S, Wang Z, Bhavana VH, Tran R, et al. (2018). Systematic Functional Annotation of Somatic Mutations in Cancer. Cancer Cell 33, 450–462 e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, and Andreatta M (2016). NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med 8, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltean S, and Bates DO (2014). Hallmarks of alternative splicing in cancer. Oncogene 33, 5311–5318. [DOI] [PubMed] [Google Scholar]
- Parolia A, Cieslik M, Chu SC, Xiao L, Ouchi T, Zhang Y, Wang X, Vats P, Cao X, Pitchiaya S, et al. (2019). Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM, et al. (2018). Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 174, 758–769 e759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A Language and Environment for Statistical Computing. (Vienna, Austria, R Foundation for Statistical Computing; ). [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, and Muller M (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost HL, Sachsenberg T, Aiche S, Bielow C, Weisser H, Aicheler F, Andreotti S, Ehrlich HC, Gutenbrunner P, Kenar E, et al. (2016). OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat Methods 13, 741–748. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster H, Peper JK, Bosmuller HC, Rohle K, Backert L, Bilich T, Ney B, Loffler MW, Kowalewski DJ, Trautwein N, et al. (2017). The immunopeptidomic landscape of ovarian carcinomas. Proc Natl Acad Sci U S A 114, E9942–E9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, Smith PG, Cancer Genome Atlas Research, N., Buonamici S, and Yu L (2018). Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep 23, 282–296 e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley CR, Blazquez L, and Ule J (2016). Lessons from non-canonical splicing. Nat Rev Genet 17, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragusa E, Weese D, and Reinert K (2013). Fast and accurate read mapping with approximate seeds and multiple backtracking. Nucleic Acids Res 41, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong KK, and Van Allen EM (2018). Intron retention is a source of neoepitopes in cancer. Nat Biotechnol 36, 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger D, and Simpson GG (2015). Enter exitrons. Genome Biol 16, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer SA, Suchman EA, Devinney LC, Star SA, and Williams RM Jr (1949). The American soldier: Adjustment during army life. (Studies in social psychology in World War II), Vol. 1 (Oxford, England: Princeton Univ. Press; ). [Google Scholar]
- Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, and Kohlbacher O (2014). OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30, 3310–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Li D, Huang P, Jian X, Wan H, Wang G, Li Y, Ouyang J, Lin Y, and Xie L (2020). dbPepNeo: a manually curated database for human tumor neoantigen peptides. Database (Oxford) 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YNS, Rowan A, Kanu N, Al Bakir M, et al. (2017). Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 18, 1009–1021. [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TY, Wang L, Alam SK, Hoeppner LH, and Yang R (2019). ScanNeo: identifying indel derived neoantigens using RNA-Seq data. Bioinformatics. [DOI] [PubMed] [Google Scholar]
- Wang Z, Civelek M, Miller CL, Sheffield NC, Guertin MJ, and Zang C (2018). BART: a transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics 34, 2867–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SN (2011). Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 73, 3–36. [Google Scholar]
- Wyman D, Balderrama-Gutierrez G, Reese F, Jiang S, Rahmanian S, Forner S, Matheos D, Zeng W, Williams B, Trout D, et al. (2020). A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. bioRxiv, 672931. [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. (2005). Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659. [DOI] [PubMed] [Google Scholar]
- Yang R, Van Etten JL, and Dehm SM (2018). Indel detection from DNA and RNA sequencing data with transIndel. BMC Genomics 19, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, and Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11, 377–394. [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, et al. (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Wei Z, Zhang Z, Zhang B, Zhu C, Chen K, Chuai G, Qu S, Xie L, Gao Y, et al. (2019). pTuneos: prioritizing tumor neoantigens from next-generation sequencing data. Genome Med 11, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. The number of EIS, EIS carrying exons, EIS carrying genes, TSE-derived neoantigens in TCGA cohorts, Related to Figure 2.
Table S3. Significantly exitron-spliced genes (SEGs) in TCGA cohorts, Related to Figure 3.
Data Availability Statement
Code for identification and quantification of exitrons is available on GitHub (https://github.com/ylab-hi/ScanExitron). Calculated TSE data and predicted TSE-derived neoantigens data for TCGA are available on Mendeley Data at http://dx.doi.org/10.17632/vdkpfzjjvg.1. RNA-Seq data and processed gene expression data from TCGA cohort, and RNA-Seq data and mutation data from CPTAC cohort (phase3) are available on Genomic Data Commons (GDC) (https://portal.gdc.cancer.gov). TCGA Unified MC3 Variant Calls from Ellrott et al., 2018 analyzed in this manuscript can be found at GDC (https://gdc.cancer.gov/about-data/publications/mc3-2017). HLA types for TCGA samples from Thorsson et al., 2018 is available at GDC (https://gdc.cancer.gov/about-data/publications/panimmune). The clinical outcome endpoints data and gender and tumor stage data for patients were obtained from the TCGA Pan-Cancer Clinical Data Resource (Liu et al., 2018). RNA-Seq data from GTEx cohort are available at GTEx data portal (https://www.gtexportal.org/home/). Protein mass spectrometry data from CPTAC cohort from are available at CPTAC data portal: https://cptac-data-portal.georgetown.edu/cptacPublic/ (Edwards et al., 2015). The RNA-Seq data for ccRCC study (Miao et al., 2018) are available at dbGap: phs001493.v1.p1. The RNA-Seq data for melanoma studies are available from the GEO or dbGaP repository under the following accession codes: GSE78220 (Hugo et al., 2016), phs000452.v2.p1 (Van Allen et al., 2015) and GSE91061 (Riaz et al., 2017). The somatic mutation data from ccRCC and melanoma studies are from their original publications. Illumina RNA-Seq data of SKBR3 are available from the NCBI SRA under accession SRX5414723 (Ghandi et al., 2019). PacBio single-molecular real-time long-read Iso-Seq data of breast cancer cell line SKBR3 are available from the NCBI SRA under accession: SRX4220391 (Nattestad et al., 2018). MS-based immunopeptidomics data and corresponding RNA-Seq data from ovarian cancer samples can be found at PRIDE: PXD007635 and Bioproject: PRJNA398141 (Schuster et al., 2017). RNA-Seq data from 101 metastatic prostate cancer in WC-SU2C cohort are available at dbGap under accession phs001648.v1.p1 (Quigley et al., 2018). Gene expression and clinical annotation data from MSKCC Prostate Cancer Oncogenome Project can be assessed through the MSKCC Prostate Cancer Genomics Data Portal: https://cbio.mskcc.org/cancergenomics/prostate/data (Taylor et al., 2010). nsSNV-derived neoantigens that induce a T-cell response are available from the dbPepNeo database (http://www.biostatistics.online/dbPepNeo). PTM sites for FET family proteins including EWSR1, FUS and TAF15 can be found at the PhosphoSitePlus: https://www.phosphosite.org (Hornbeck et al., 2015).