

Abstract
Background & Aims:
Altered bile acid (BA) homeostasis is an intrinsic facet of cholestic liver diseases, but clinical usefulness of plasma BA assesment in primary sclerosing cholangitis (PSC) remains understudied. We performed BA profiling in a large retrospective cohort of PSC patients and matched healthy controls, hypothesizing that plasma BA profiles vary among patients and have clinical utility.
Approach & Results:
Plasma BA profiling was performed in the Clinical Biochemical Genetics Laboratory at Mayo Clinic using a mass spectrometry based assay. Cox proportional hazard (univariate) and gradient boosting machines (multivariable) models were used to evaluate whether BA variables predict 5-year risk of hepatic decompensation (HD: defined as ascites, variceal hemorrhage or encephalopathy). There were 400 PSC patients and 302 controls in the derivation cohort (Mayo Clinic) and 108 PSC patients in the validation cohort (Norwegian PSC Research Center). PSC patients had increased BA levels, conjugated fraction and primary-to-secondary BA ratios relative to controls. UDCA increased total plasma BA level while lowering cholic acid (CA) and chenodeoxycholic acid (CDCA) concentrations. Patients without inflammatory bowel disease (IBD) had primary-to-secondary BA ratios between those of controls and patients with ulcerative colitis. HD risk was associated with increased concentration and conjugated fraction of many BA, whereas higher G:T conjugation ratios were protective. The machine learning model, PSC-BAP (bile acid profile) score (C-statistic, 0.95), predicted HD better than individual measures including alkaline phosphatase and performed well in validation (C-statistic, 0.86).
Conclusions:
PSC patients demonstrated alterations of plasma BA consistent with known mechanisms of cholestasis, UDCA treatment and IBD. Notably, BA profiles predicted future HD, establishing the clinical potential of BA profiling, which may be suited for use in clinical trials.
Keywords: Machine learning, Cholestasis, Ascites, Variceal hemorrhage, Encephalopathy
Primary sclerosing cholangitis (PSC) is a rare, progressive cholestatic liver disease, which leads to stricturing fibrosis of the intra- and extra-hepatic bile ducts and decompensated cirrhosis in the majority of patients over time (1). PSC is highly comorbid with inflammatory bowel disease (IBD) and is associated with increased risk of cholangiocarcinoma (CCA) and colorectal cancer (1). There remain no reliable biomarkers or effective medical therapy for PSC. However, a significant subset of patients are treated with ursodeoxycholic acid (UDCA), a safe and efficacious therapy in improving the liver biochemistries, with no significant impact on hepatic decompensation (HD) or mortality (2). The majority of PSC patients with cirrhosis and HD require orthotopic liver transplantation (OLT) (1).
PSC pathogenesis is complex, involves hereditary and environmental factors, and remains poorly understood. Factors including bile acid (BA) toxicity (3), intestinal dysbiosis (3) and abberant immune activation (4) have been shown to contribute to PSC pathogenesis. Genome-wide association studies suggest the genetic component of PSC lies primarily in immune-inflammatory processes, with little evidence for genetically determined alterations to mechanisms enforcing BA homeostasis (5, 6), However, increasing BA toxicity during the course of progressive cholestasis most certainly impacts disease, and therapeutics focused on altering the BA pool in PSC patients are promising (7).
Bile acids act as signaling molecules regulating hepatic metabolism and contribute to maintaining intestinal microbiota homeostasis. The two primary BA, cholic acid (CA) and chenodeoxycholic acid (CDCA) are synthesized and conjugated with glycine or taurine in the liver. Following secretion into the small intestine, BA are deconjugated and converted to secondary bile acids by the intestinal microbiota. Specifically, CA is converted to deoxycholic acid (DCA) and CDCA can ultimately be converted to UDCA, lithocholic acid (LCA) or hyodeoxycholic acid (HDCA). The majority of BA are reabsorbed in the ileum and undergo enterohepatic circulation that maintains the BA pool (8). In cholestasis, bile flow is compromised and potentially toxic BA accumulate, stimulating mechanisms to reduce BA concentrations in the hepatocyte including increased excretion into the systemic circulation for removal by the kidneys. However, these compensatory mechanisms are not adequate to overcome progressive liver damage in PSC (9).
Recent advancements have brought inexpensive and non-invasive clinical assays to assess circluting BA profiles. Such tests offer promise for better clinical management of patients with cholestatic liver diseases and are of particular interest in evaluating clinical trials of anti-cholestatic drugs. However, there have been no large-scale studies reporting use of these assays in PSC. In this study, we aim to evaluate the clinical utility of BA profiling in PSC using traditional and machine-learning approaches.
PATIENTS AND METHODS
Patients
Patients and healthy controls comprising the derivation cohort were selected from the PSC Scientific Community Resource, an extension of the PSC Resource of Genetic Risk, Environment and Synergy Studies (PROGRESS) resource (10), which actively collects biospecimens and clinical data for use in PSC research. PSC diagnoses were confirmed by qualified physicians using standard criteria (11) and controls were excluded if they had history of chronic liver disease. Clinical records were reviewed and pertinent information such as IBD status, PSC subtype including small duct disease and overlap with autoimmune hepatitis (AIH), UDCA treatment and progression to clinical endpoints was documented. Cirrhotic-stage PSC was defined by the presence of histological stage IV according to the Ludwig staging system (12), hepatic parenchymal changes on cross-sectional imaging consistent with cirrhosis (13) and/or presence of portal hypertension manifested as gastroesophageal varices or splenomegaly. Cirrhosis with HD was defined as one or more clinical episodes of ascites, variceal hemorrhage or hepatic encephalopathy. Patients lacking clinical follow-up or with history of OLT, diagnosis of CCA or HD prior to sample collection were excluded. Controls were selected to proportionally match to patients based on sex and to minimize age differences between the groups to the extent possible. We collaborated with the Norwegian PSC Research Center to obtain a validation cohort of patients, selected using identical criteria, to attempt validation of the BA predictive model. The study was carried out in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants and the studies were approved by the Mayo Clinic Institutional Review Board and the Regional Committee for Medical and Health Research Ethics of South-Eastern Norway.
Bile Acid Profiling
Plasma samples had been previously collected in EDTA tubes under fasting conditions and stored at −80°C. Bile acid profiling was performed in the Clinical Biochemical Genetics Laboratory at Mayo Clinic using the BAPS (Bile Acid Profile, Serum) assay. This method is based on liquid chromatography-tandem mass spectrometry (LC-MS) incorporating isotopically labeled internal standards and is well-calibrated to evaluate bile acid levels in EDTA plasma in the research setting. The assay provides quantitation of primary BA: CA and CDCA; secondary BA: DCA, LCA, UDCA and HDCA; and their glyco- and tauro-conjugates: glycocholic acid (GCA), taurocholic acid (TCA), glycochenodeoxycholic acid (GCDCA), taurochenodeoxycholic acid (TCDCA), glycodeoxycholic acid (GDCA), taurodeoxycholic acid (TDCA), glycolithocholic acid (GLCA), taurolithocholic acid (TLCA), glycoursodeoxycholic acid (GUDCA), tauroursodeoxycholic acid (TUDCA), glycohyodeoxycholic acid (GHDCA), and taurohyodeoxycholic acid (THDCA). Additional information about the BAPS assay is provided in Supplemental Methods. The total bile acid (TBA) value is calculated by summing the values of all evaluated bile acid forms. Bile acid “family” concentrations were calculated by adding the values of unconjugated and conjugated forms (e.g., CA+GCA+TCA). Conjugated fraction was calculated as the sum of the conjugated forms divided by the total (e.g., (GCA+TCA) / (CA+GCA+TCA)); value was set to blank if the denominator was equal to zero. G:T conjugation ratios were calculated by dividing the the glycine conjugated form by the taurine conjugated form (e.g., GCA / TCA); value was set to blank if either form was equal to zero. Finally, ratios of CA:CDCA, CA:DCA and CDCA:(LCA+HDCA+UDCA) were calculated using the BA family values.
Statistical analyses
Continuous variables were expressed as median and interquartile range (IQR) using the Kruskal-Wallis rank sum test, unless otherwise specified. Categorical variables were compared using the Pearson’s chi-squared test. Cox proportional hazard models were run for univariate variables to evaluate their ability to predict risk of HD (defined as ascites, variceal hemorrhage or encephalopathy) within a 5-year time period. Censoring was made at the time of OLT, CCA diagnosis, or at the time of last clinical encounter or death. Probability of HD was estimated using Aalen-Johansen curves based on quartiles of variable values, accounting for death and liver transplantation as competing risks. Hazard ratios were calculated using IQR-normalized values and thus reflect risk of HD in individuals at the 75th percentile relative to the 25th percentile of the variable. The Harrell’s concordance statistic (C-statistic) was used to measure discrimination ability of the variables in the univariate analysis. Correlation between individual BA was evaluated using the Spearman’s rank correlation coefficient. Principle components analyses was performed using concentrations of the 18 individual bile acids.
Gradient boosting machines (GBM) was used to build a multivariable model to predict development of HD in a 5-year time window, utilizing the generalized boosted model package available in the R software environment (R Foundation for Statistical Computing, Vienna, Austria) (14). GBM utilizes an ensemble of weak prediction models, in this case decision trees, building the model in a step-wise, iterative process involving recursive partitioning. Each decision tree is likely to have relatively poor predictive performance; however, when merged in the final model prediction is significantly improved. Censoring was as per the univariate analyses. Discrimination was assessed using the C-statistic and 95% confidence intervals (CIs) were created using bootstrapping. Calibration was assessed by comparing observed and expected values in subjects with low, medium, and high predicted risk as determined by the tertiles of the model’s risk score distribution.
RESULTS
Patient characteristics
Patient characteristics are presented in Table 1. There were 400 PSC patients and 302 controls in the derivation cohort and 108 PSC patients in the validation cohort. Patients and controls in the derivation cohort were well matched on sex and race, with the patients being somewhat younger than controls. The two patient cohorts were similar in terms of race, age at PSC diagnosis and IBD status. Compared to the derivation cohort, the validation group was younger, included fewer female patients and had shorter disease duration before sample collection. In the 5-year post-sample collection observation period there were 51 HD events in the derivation cohort and 8 HD events in the validation cohort. Censoring in time-to-event analyses for both cohorts was primarily based on patients having no clinical event at time of last clinical encounter.
Table 1.
Patient Characteristics
| Derivation | Validation | ||||
|---|---|---|---|---|---|
| Controls | PSC Patients | PSC Patients | |||
| (n=302) | (n=400) | p-valuea | (n=108) | p-valueb | |
| Age (yrs), median (IQR) | 56.4 (48.5-65.3) | 48.4 (33.0-59.7) | <0.001 | 41.2 (33.3-52.0) | 0.006 |
| Sex, %Male | 58.3 | 57.8 | 0.888 | 76.9 | <0.001 |
| Caucasian Race, % | 99.0 | 96.9 | 0.398 | 100 | 0.495 |
| ALP x ULN, median (IQR) | 0.6 (0.5-0.7) | 1.5 (0.9-2.9) | <0.001 | 2.2 (1.2-3.4) | <0.001 |
| Total Bilirubin, median (IQR) | NA | 0.7 (0.5-1.1) | - | 0.2 (0.1-0.4) | <0.001 |
| Age PSC Diagnosis (yrs), median (IQR) | NA | 40.0 (26.8-51.5) | - | 34.7 (26.9-47.1) | 0.113 |
| PSC Duration (yrs), median (IQR) | NA | 4.8 (1.8-9.7) | - | 2.2 (0.1-7.9) | <0.001 |
| PSC subtype, n | 400 | 108 | |||
| Small duct, n (%) | NA | 26 (6.5) | - | 0 | 0.007 |
| AIH overlap, n (%) | NA | 14 (3.5) | - | 0 | 0.049 |
| IBD status, n | 347 | 108 | |||
| Ulcerative colitis, n (%) | NA | 231 (66.6) | - | 69 (63.9) | 0.615 |
| Crohn’s disease, n (%) | 26 (7.5) | 10 (9.3) | |||
| Indeterminate, n (%) | 25 (7.2) | 6 (5.6) | |||
| None, n (%) | 65 (18.7) | 23 (21.3) | |||
| HD Events, n | 51 | 8 | |||
| Ascites, n (%) | NA | 42 (82.4) | - | 8 (100) | 0.435 |
| Encephalopathy, n (%) | 4 (7.8) | 0 | |||
| Variceal bleeding, n (%) | 5 (9.8) | 0 | |||
| Censoring Events, n | 349 | 100 | |||
| Last encounter, n (%) | NA | 321 (92.0) | - | 70 (70.0) | <0.001 |
| CCA, n (%) | 10 (2.9) | 11 (11.0) | |||
| OLT, n (%) | 15 (4.3) | 19 (19.0) | |||
| Death, n (%) | 3 (0.9) | 0 | |||
Controls vs. Derivation PSC,
Derivation PSC vs. Validation PSC; IQR: interquartile range, ALP x ULN: alkaline phosphatase times upper limit of normal
Abbreviations: PSC: primary sclerosing cholangitis; AIH: autoimmune hepatitis; CCA: cholangiocarcinoma; OLT: orthotopic liver transplantation; IBD: inflammatory bowel disease; HD: hepatic decompensation
Bile acid profiles in PSC patients and controls
We compared concentrations, conjugated fractions and G:T conjugation ratio for each BA family and for TBA, as well as concentrations of the individual BA, between PSC patients and controls of the derivation group (Figure 1 and Supplemental Table 1). PSC patients demonstrated markedly increased level of TBA (median (IQR) 16.34 (6.04-44.31) μmol/L) compared to controls (median (IQR) 1.90 (1.02-3.52) μmol/L), which was accompanied by significant increases in CA, CDCA, LCA and UDCA. Notably, levels of the secondary bile acid DCA did not significantly differ. The conjugated fraction of TBA was elevated in PSC patients (median (IQR) 0.91 (0.74-0.98)) relative to controls (median (IQR) 0.64 (0.49-0.79)), with similar increases in all of the other BA families save for HDCA. The G:T conjugation ratio of TBA was similar between PSC patients and controls. However, G:T conjugation ratios of CDCA and DCA were significantly lower, and UDCA significantly higher in PSC patients. Differences in concentration of individual BA broadly reflected that of the BA family, except for DCA, in which the unconjugated form was found to be significantly decreased in patients compared to controls, whereas the taurine-conjugated form (TDCA) was slightly increased in patients. Finally, age and sex were not found to have substantial impact on BA concentrations, conjugated fractions or G:T conjugation ratios in both PSC patients and controls (Supplemental Figures 1 and 2).
Fig. 1. Bile acids in PSC patients and controls.
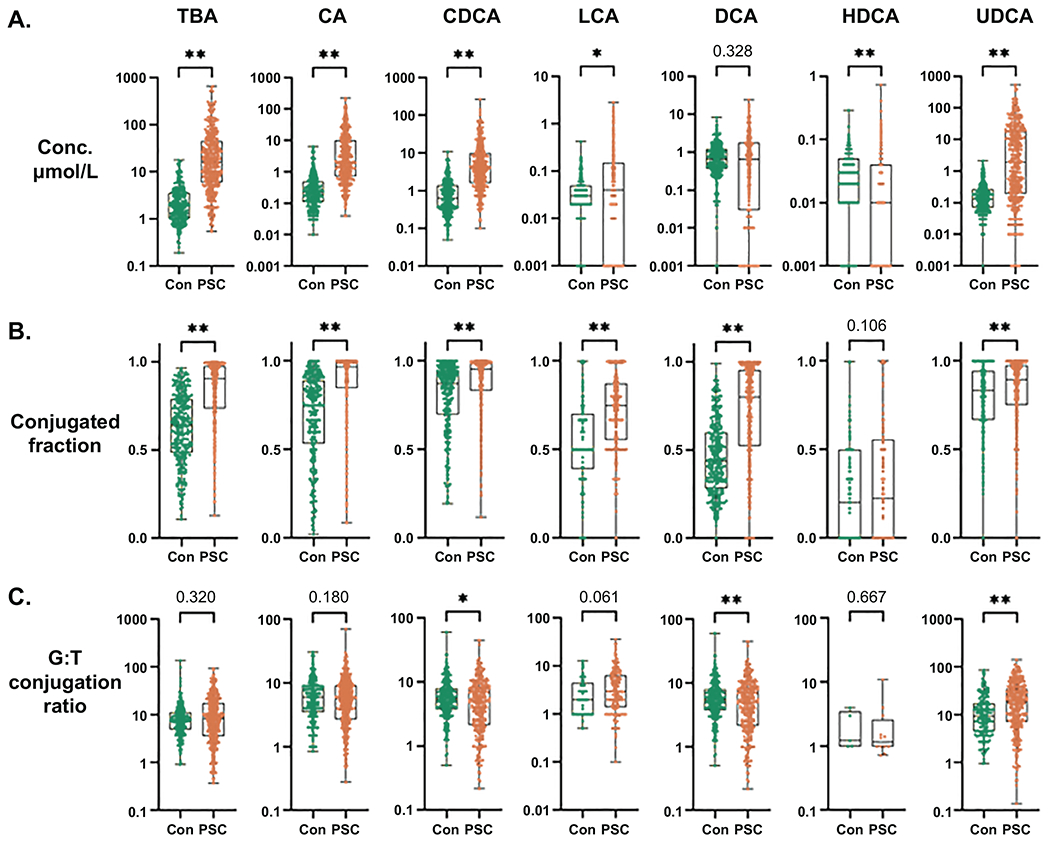
Comparisons of (A) Concentration (μmol/L), (B) Conjugated fraction and (C) G:T conjugation ratio of total bile acids (TBA) and each bile acid family (e.g., the CA family includes CA (unconjugated), GCA and TCA) between PSC patients and controls. Significance level: *p<0.05, **p≤0.001; Kruskal-Wallis rank sum test.
Variabiltiy in BA concentrations could lead to significant alterations to the composition of plasma BA in PSC patients compared to controls. We find that PSC patients have increased proportions of CA and UDCA and decreased proportions of CDCA, DCA, LCA and HDCA relative to controls (Figure 2A and Supplemental Table 1). While the findings suggest that PSC is characterized by increased size and altered compostion of plasma BA, there may be PSC patients with BA profiles similar to controls. To begin addressing this possibility, we performed principle components analysis of the individual BA concentrations. In the plot of the first two principle components it is clear that the controls form a relatively tight cluster intermingled with a subset of the PSC patients (Figure 2B).
Fig. 2. PSC patient subgrouping based on total bile acid levels and UDCA treatment.
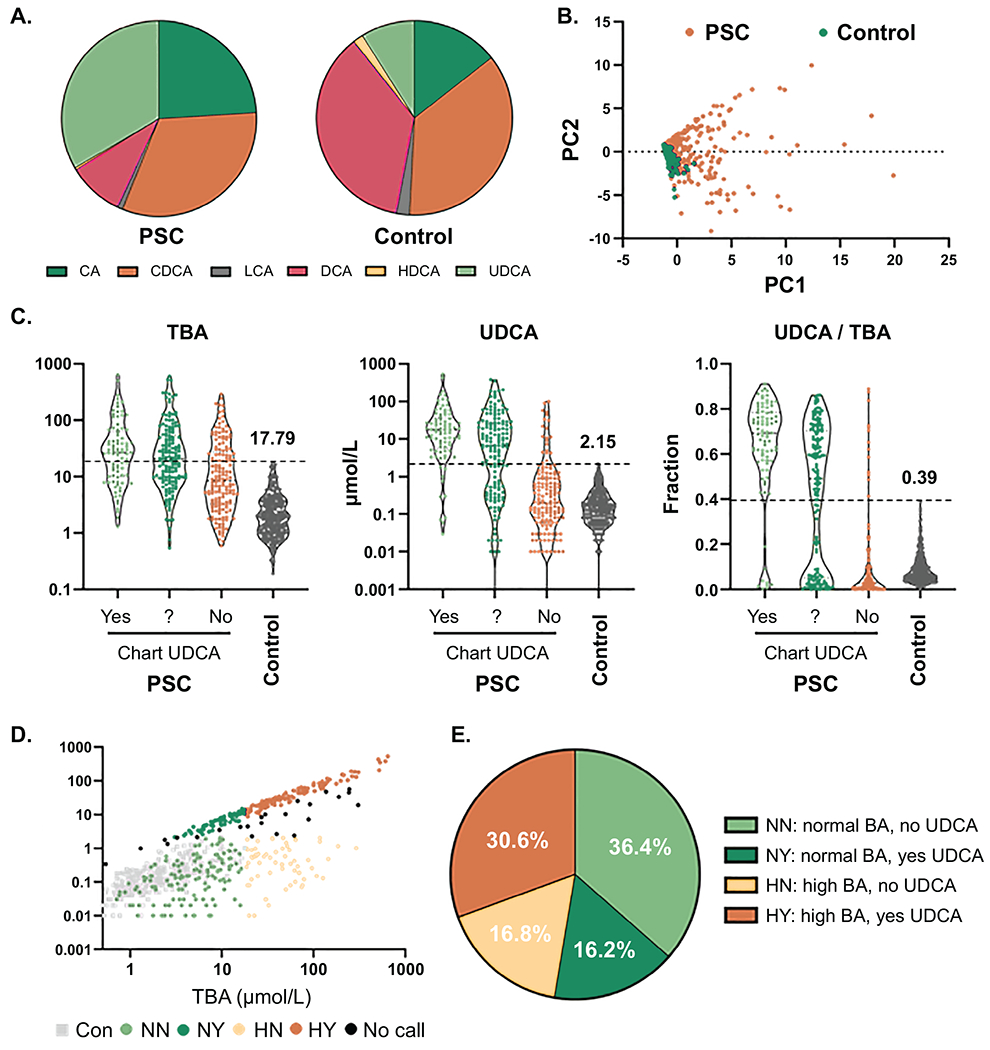
(A) Plasma bile acid (BA) composition by BA family type in PSC patients and controls, (B) Principle components analysis of individual bile acid concentrations in PSC patients and controls, (C) Concentrations of total bile acids (TBA), UDCA and fraction of UDCA in TBA (UDCA/TBA) of PSC patients separated by chart-reviewed determination of UDCA treatment and controls. Values shown are maximum cut-offs observed in controls and were used to establish PSC patient subgroups based on TBA (normal: ≤ 17.79, high: > 17.79 μmol/L) and predicted UDCA treatment (no: UDCA conc. ≤ 2.15 and fraction ≤ 0.39, yes: UDCA conc. > 2.15 and fraction > 0.39). (D) UDCA and TBA concentrations in PSC patients and controls colored by TBA/UDCA groups, (E) proportion of PSC patients in each of the TBA/UDCA groups.
Many PSC patients are treated with UDCA, which may account for increases in TBA and the proportion of UDCA in PSC patients. To begin understanding the potential effect of UDCA treatment on plasma BA, we first performed a retrospective review of clinical records to establish whether our PSC patients were taking UDCA at or around the time of sample collection. For 246 of the 400 PSC patients (61.5%) we were able to establish UDCA treatment status, with 85 of these 246 patients (34.6%) found to be prescribed UDCA. We next looked at TBA concentration, UDCA concentration and UDCA as a fraction of TBA in light of the review-determined UDCA status (Figure 2C). Some of the patients from the “No UDCA” group had very high UDCA levels and were likely on UDCA at the time of sample collection. Conversely, a subset of the “Yes UDCA” group had very low UDCA levels, suggesting they were not taking the therapy or the UDCA had limited bioavailability. In order to include more patients and better compare groups accounting for high BA levels and likely UDCA therapy, we split the patients into 4 groups based on TBA (Normal/High) and predicted UDCA therapy (Yes/No) using the highest levels found in controls for TBA concentration (17.79 μmol/L), UDCA concentration (2.15 μmol/L) and UDCA fraction of TBA (0.39) (Figure 2C). We required UDCA concentration and fraction of TBA to be geater or less than the control cut-offs, so a small subset of patients were classified as “no call” for UDCA therapy. Visualiztion of the groups (NN: normal TBA, no UDCA; NY: normal TBA, yes UDCA; HN: high TBA, no UDCA; and HY: high TBA, yes UDCA) by UDCA and TBA concentration shows the clear separation (Figure 2D), with a relatively small number of patients near the demarcation lines, suggesting misclassification effects in comparative analyses should be minimal. Finally, the NN (36.4%) and HY (30.6%) groups were the most prevalent, with HN (16.8%) and NY (16.2%) making up smaller proportions of the PSC patient population (Figure 2E).
Bile acid profiles considering TBA/UDCA groups
We compared BA data between various TBA/UDCA determined groups to better understand differences compared to controls (control vs. NN), the effect of UDCA therapy (NN vs. NY and HN vs. HY) and the effect of abnormally high levels of BA (NN vs. HN and NY vs. HY). The results of these analyses are presented in Figure 3 and Supplemental Tables 2–6. Compared to controls, the NN PSC group had higher concentration, increased conjugated fraction and similar G:T conjugation ratio of TBA (Figure 3A), consistent with the global case-control analysis. However, unlike the broader analysis, UDCA concentration was very similar between NN patients and controls, with concentrations of primary BA (CA and CDCA) significantly increased and concentrations of secondary BA (DCA, LCA and HDCA) significantly decreased in the NN PSC patients (Supplemental Table 2). UDCA treated groups (NY and HY) had higher concentration and G:T conjugation ratio and lower conjugated fraction of TBA than comparable non-UDCA treated groups (NN and HN). Changes in conjugated fraction and G:T conjugation ratios were consistent across the individual BA in these groups, with variability in concentrations of individual BA and increased concentration of TBA primarily coming from UDCA (Supplemental Tables 3 and 4). Notably, while TBA was increased in UDCA-treated groups, the non-UDCA component of TBA was significantly reduced in UDCA-treated compared to non-treated groups; median (IQR) of TBA-excluding UDCA: 2.93 (1.87-4.28) μmol/L in NY vs. 4.54 (2.37-7.83) μmol/L in NN, p<0.001 and 14.63 (7.75-36.40) μmol/L in HY vs. 38.02 (22.96-62.69) μmol/L in HN, p<0.001). In addition to higher TBA concentration, the high BA groups (HN and HY) had increased conjugated fraction and decreased G:T conjugation ratio relative to their comparable normal BA groups (NN and NY) (Figure 3A and Supplemental Tables 5 and 6).
Fig. 3. Bile acids in controls and PSC patient TBA/UDCA subgroups.
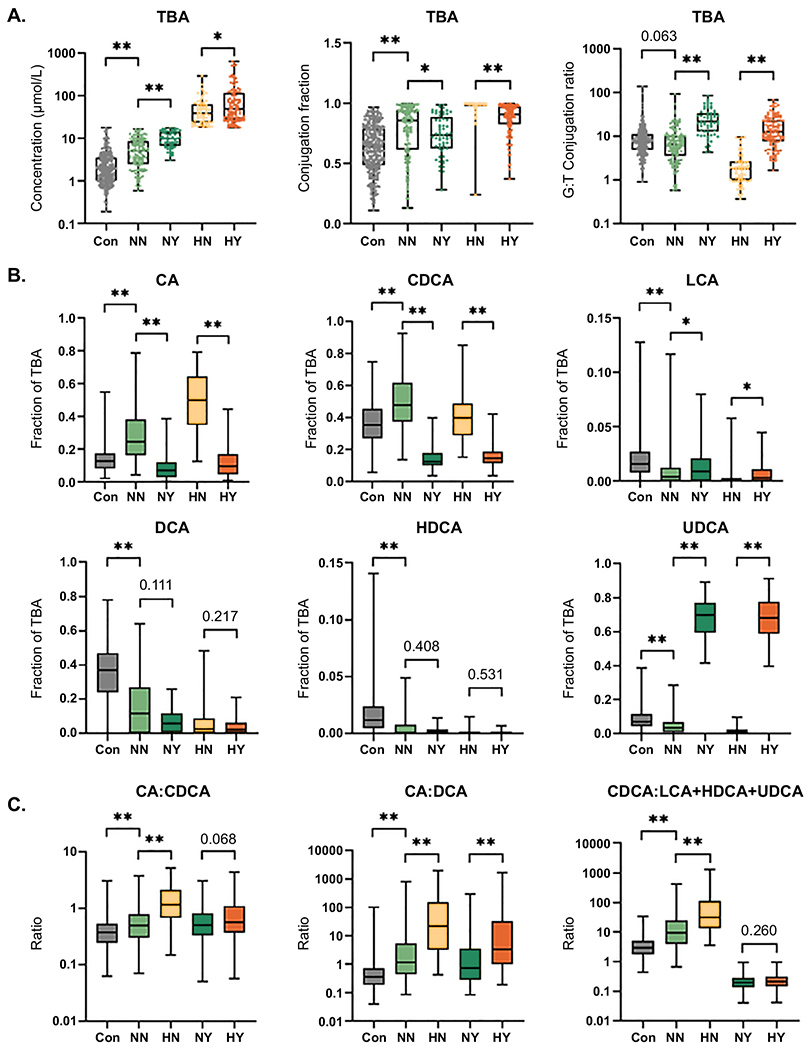
Comparisons of (A) Concentration (μmol/L), Conjugated fraction and G:T conjugation ratio of total bile acids (TBA) and (B) Concentration of each BA family as fraction of TBA between (1) controls and NN PSC patients, (2) NN and NY PSC patients and (3) HN and HY PSC patients. (C) Comparison of CA:CDCA, CA:DCA and CDCA:LCA+HDCA+UDCA ratios between (1) controls and NN PSC patients, (2) NN and HN PSC patients and (3) NY and HY PSC patients. Significance level: *p<0.05, **p≤0.001; Kruskal-Wallis rank sum test.
The NN PSC patient group demonstrated increased fractions of CA and CDCA and decreased fractions of LCA, DCA, HDCA and UDCA compared to controls (Figure 3B), consistent with the observed differences in BA concentrations. As expected, plasma BA of UDCA-treated patient groups (NY and HY) were significantly enriched for UDCA compared to non-treated groups (NN and HN) (Figure 3B). The increased UDCA fraction was reflected by reduced representation of CA and CDCA in UDCA-treated patient groups, although the fraction of DCA and HDCA remained similar despite treatment. Notably, while only a small fraction of TBA, the LCA fraction was significantly increased in the UDCA-treated groups (Figure 3B), suggesting that supplemental UDCA may be available for conversion to LCA. Removal of UDCA from the analyses of NN:NY and HN:HY differences in BA fraction of TBA were mostly consistent with the UDCA-included results, but with DCA comprising an increased fraction of the non-UDCA TBA in UDCA treated vs. non-treated groups (Supplemental Figure 3). Compared to controls, plasma BA of the NN PSC patient group had increases in CA:CDCA ratio, CA:DCA ratio and CDCA:(LCA+HDCA+UDCA) ratio, all of which were further increased in the comparable high BA group (i.e., control < NN < HN) (Figure 3C). Coupled with observed differences in BA concentrations, these findings suggest that alterations in BA metabolism, such as deficiency in conversion of primary to secondary bile acids, increased BA synthesis or decreased BA excretion may play a role in PSC. Comparisons between the two UDCA-treated groups (NY and HY) did not show a difference in CA:CDCA ratio but did demonstrate an increased CA:DCA ratio, supporting the finding in the non-treated groups. Notably, in the high BA groups UDCA treatment lowered both the CA:CDCA and CA:DCA ratios (HN vs. HY, Supplemental Table 4); however, this UDCA effect was not noted in the normal BA groups (NN vs. NY, Supplemental Table 3).
Comorbid inflammatory bowel disease and bile acid profiles
IBD frequently co-occurs with PSC and has been shown to affect the BA pool through disruption of homeostatic enterohepatic circulation. Thus, we next accounted for IBD in our analysis. First, we performed a principle components analysis of BA levels using only the PSC patients, which shows that patients do not cluster based on presence of IBD or the clinical IBD subtype (Figure 4A). We next evaluated plasma BA compositions of PSC patients with comorbid IBD relative to PSC patients without history or evidence of IBD (Figure 4B and Supplemental Tables 7–10). Patients with ulcerative colitis (UC) had significantly increased fraction of CA and decreased fractions of LCA, DCA and HDCA, whereas patients with Crohn’s Disease (CD) and Indeterminate IBD had increased fraction of CDCA, relative to the no-IBD patients. G:T conjugation ratios of TBA and the individual BA did not differ based on IBD status. Patients without IBD had similar concentration and conjugated fraction of TBA relative to patients with UC and Indeterminate IBD, but increased compared to patients with CD (Figure 4C). Finally, CA:CDCA and CA:DCA ratios were increased in no-IBD PSC patients compared to controls and further increased in PSC patients with UC compared to the no-IBD patients (Figure 4D). This suggests that changes to plasma BA in PSC are not dependant on, but are exasperated by, UC comorbidity.
Fig. 4. Inflammatory bowel disease and bile acids in PSC.
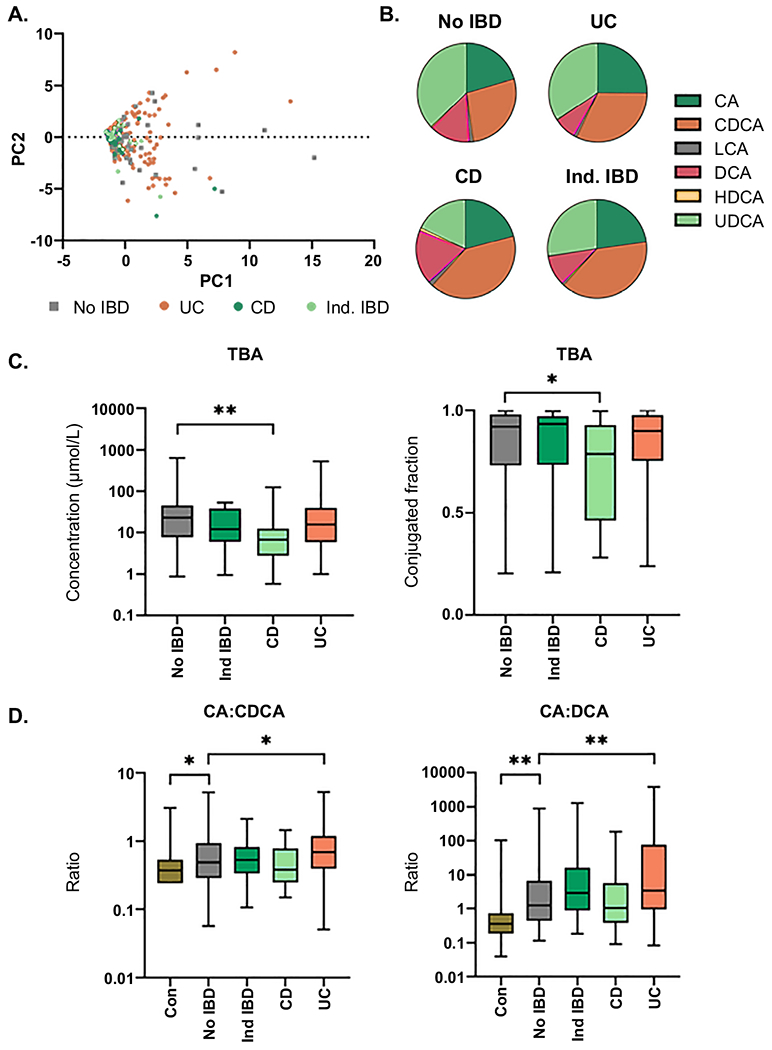
(A) Principle components analysis (PCA) of individual BA concentrations in PSC patients with No IBD, ulcerative colitis (UC), Crohn’s Disease (CD) or indeterminate IBD (Ind IBD); (B) Plasma BA composition by BA family type in PSC patients by IBD group; (C) Comparison of concentration (μmol/L) and conjugated fraction of total bile acids (TBA) between PSC patients with No IBD and PSC patients with Ind IBD, CD or UC; (D) Comparison of CA:CDCA and CA:DCA ratios between (1) Controls and PSC patients with No IBD and (2) PSC patients with No IBD and PSC patients with Ind IBD, CD or UC. Significance level: *p<0.05, **p≤0.001; Kruskal-Wallis rank sum test. Non-significant comparisons (p≥0.05) are not highlighted in this figure.
Bile acid profiles in cirrhotic patients and those with AIH overlap or small duct PSC
Comparison of BA variables between PSC patients with compensated cirrhosis and non-cirrhotic patients is presented in Supplemental Table 11 and Supplemental Figure 4. Not surprisingly, cirrhotic patients demonstrated increase concentrations of BA including CA, CDCA, LCA and TBA; although, some cirrhotic patients did have relatively low BA levels. Conjugated fraction of BA tended to be increased while G:T conjugation ratios were decreased in cirrhotic compared to non-cirrhotic patients. As well, CA:DCA and CDCA:(LCA+HDCA+UDCA) ratios were increased in cirrhotic patients. However, the CA:CDCA ratio was similar.
Patients with AIH overlap did not demonstrate significant differences in BA variables compared to non-overlap PSC (Supplemental Table 12). However, the number of AIH overlap patients was quite small, limiting our ability to detect differences. Patients with small duct PSC were younger than patients with traditional large duct disease and only demonstrated significant differences in a few BA variables including having lower concentrations of CA and lower CA:CDCA and CA:DCA ratios (Supplemental Table 13). As with AIH, there were a limited number of small duct PSC patients.
Bile acids as potential biomarkers of future hepatic decompensation
To evaluate whether BA have utility in prediction of disease complications we performed univariate time-to-event analyses of phenotypic and BA parameters. Hepatic decompensation was selected as the clinical event instead of liver transplantation due to broad variation in listing and transplant criteria and wait times across the United States and the world (15). Results of these analyses are presented in Figure 5 and Supplemental Tables 14 and 15. PSC patients with compensated cirrhosis had elevated risk of HD relative to non-cirrhotic patients (hazard ratio (95% CI) = 5.29 (3.02-9.25), p < 0.001). However, cirrhosis status was not among the most predictive variables with a C-statistic of 0.66. Concentration of TBA and 10 of 18 individually measured BA (LCA, UDCA, GCA, GCDCA, GLCA, GUDCA, TCA, TCDCA, TLCA and TUDCA) were significantly associated with development of HD. Conjugation fraction of TBA, CA, CDCA and UDCA were also associated with development of HD; whereas G:T conjugation ratios generally demonstrated significant protective effects. The four parameters with highest C-statistics were TCDCA (0.83), TCA (0.82), TBA (0.80) and TBA with UDCA removed (0.80). These variables outperformed alkaline phosphatase (expressed as times upper limit of normal) and total bilirubin, commonly used biochemical measures to evaluate PSC, which had C-statistics of 0.70 and 0.77, respectively (Figure 5). Notably, there is a complex correlation structure in plasma BA that is somewhat altered in PSC patients compared to controls (Supplemental Figure 5) and the top BA in the time-to-HD analysis are highly correlated in PSC: Spearman correlation of TCDCA-TCA = 0.94, TCDCA-TBA = 0.76 and TCA-TBA = 0.72.
Fig. 5. Univariate analysis of bile acid variables for time-to-event (hepatic decompensation) in PSC.
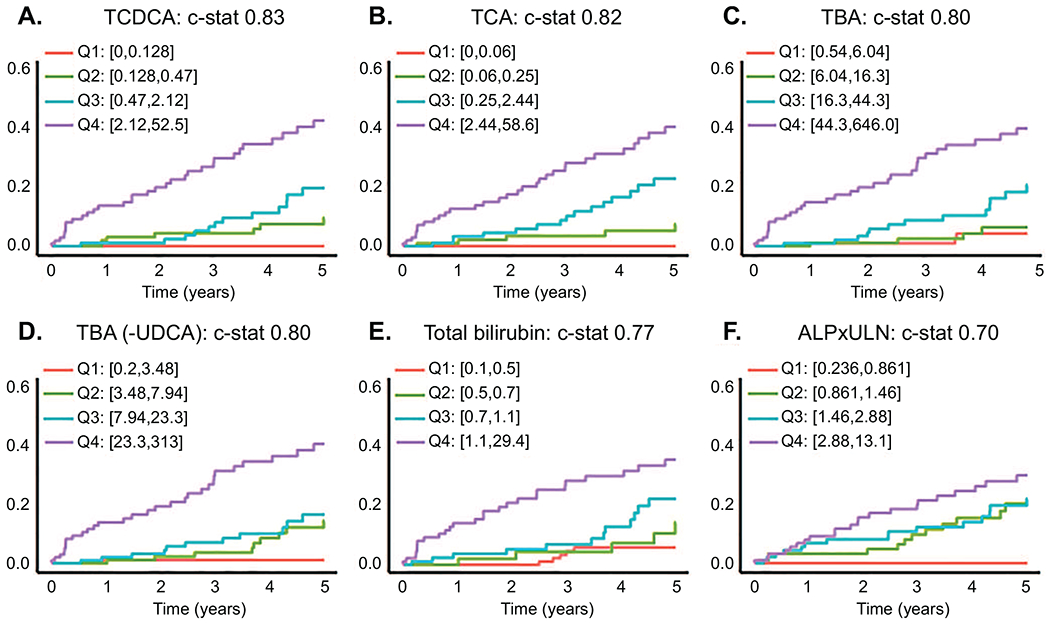
(A) TCDCA, (B) TCA and (C) total bile acids (TBA) were the top 3 variables capable of predicting hepatic decompensation (HD; defined as ascites, esophageal varices or encephalopathy) in PSC over a 5 year window. (D) TBA with UDCA and it’s conjugates subtracted performed similar to TBA. These variables outperformed (E) Total bilirubin and (F) alkaline phosphatase expressed as times upper limit of normal (ALPxULN). Cox proportional hazard models were used to evaluate risk of HD (p<0.001 all variables). The Harrell’s concordance statistic (c-stat) was used to measure discrimination ability of the variables. Data is presented using Aalen-Johansen curves based on quartiles of variable values, accounting for death and liver transplantation as competing risks.
Machine learning of bile acid profile to predict HD events: PSC-bile acid profile score
We next used GBM to investigate 40 potential bile acid based model covariates (Supplemental Table 16) to determine whether the overall BA profile could better predict future HD in PSC than individual BA. A total of 651 decision trees were utilized, decision tree depth was optimized at 3 and the shrinkage parameter was optimized at 0.005. Six covariates were retained in the final model to predict 5-year probability of HD (Figure 6A). The resulting PSC-BAP (bile acid profile) score was median (IQR) 7.1% (5.0-22.3%) in the derivation cohort, performed well in predicting HD (C-statistic = 0.95, 95%CI, 0.92-0.97), and was well calibrated (Figure 6B). Notably, these covariates were also among the top variables in alternate GBM models run to test model sensitivity to removal of PSC patients with small duct disease and/or AIH overlap and addition of variables including cirrhosis status, bilirubin and alkaline phosphatase (Supplemental Figure 6). The validation cohort had similar TBA concentration as the derivation group, but there were notable differences in BA composition and the validation cohort demonstrated higher conjugation fractions and lower G:T ratios than the derivation cohort (Supplemental Table 17). In the validation cohort, median (IQR) of the PSC-BAP score was 7.3% (5.6-23.8) and despite BA differences performed relatively well (C-statistic = 0.86, 95%CI, 0.74-0.96). Moreover, the score was well-calibrated but tended to overestimate risk, particularly in the high-risk tertile (Figure 6C). This overestimation could be a consequence of significantly increased TCDCA and TCA concentrations (Supplemental Table 17), both important components of the model, in the validation relative to derivation cohort. Finally, PREsTo did slightly outperform the PSC-BAP score in the validation cohort (PREsTo C-statistic 0.94, 95% CI 0.87-0.98), suggesting PREsTo may be better calibrated across a wide range of patients.
Fig. 6. Predictive model for hepatic decompensation in PSC using machine learning and the bile acid profile: PSC-BAP score.
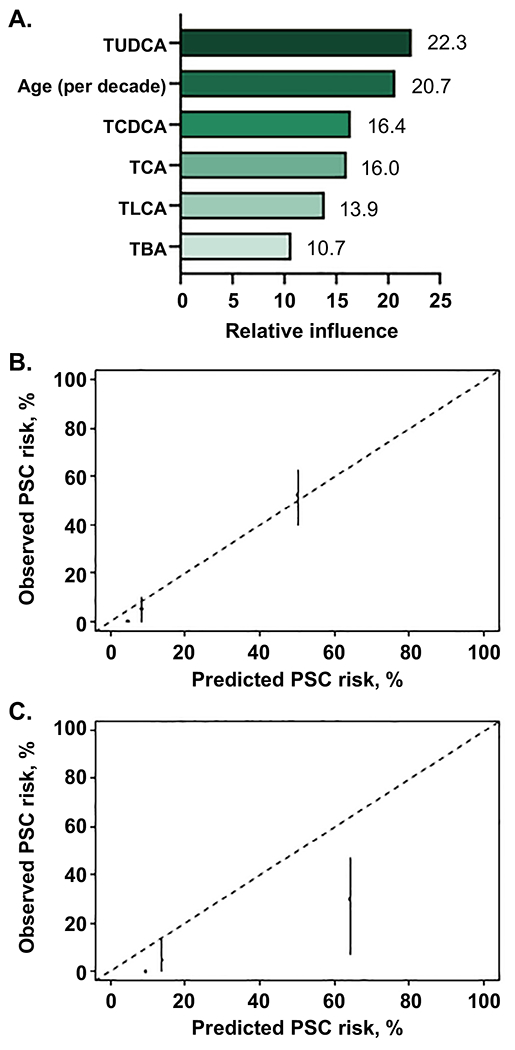
(A) Variables included in the model and their relative importance, (B) derivation cohort and (C) validation cohort model calibration by tertiles (low, medium, high) of predicted risk.
DISCUSSION
We report the first ever large-scale study of plasma BA profiles focused on PSC patients and healthy controls using a non-invasive LC-MS based BA profiling assay. We found that PSC patients have alterations in plasma BA that are consistent with known mechanisms of cholestasis, UDCA treatment and IBD; suggesting that plasma BA are reflective of the overall BA pool. Risk of HD was positively influenced by increased concentration and conjugated fraction of many BA, whereas higher G:T conjugation ratios were generally protective. We developed the PSC-BAP (bile acid profile) score using machine learning of BA variables to create a model capable of predicting future HD events better than individual measures. This model was well-calibrated and validated in an independent cohort of PSC patients; although, overall performance was similar to that of the recently reported PREsTo score (10). Of note, the univariate analyses and GBM model indicate that high levels of taurine-conjugated BA seem to be most predictive of future HD events in PSC. This study provides a solid foundation for persuing BA profiles as a clinical measure in PSC, which may be particularly well suited to use in clinical trials.
The use of UDCA as a therapy for PSC remains controversial, with American and European medical associations offering conflicting advice towards its use (16). It is generally well-tolerated and has demonstrated efficacy in lowering serum liver enzyme levels (17). However, there is no evidence that UDCA improves outcomes in PSC and a study of high-dose UDCA found an increased rate of adverse events in the treated group (18). In our study, we assign patients to UDCA “yes” or “no” treatment groups using a calculation based on maximum values for UDCA concentration and fraction of the BA pool found in controls, as we found medical record review to be an incomplete and somewhat inaccurate means to determine past UDCA treatment. These groups are further stratified by “normal” and “high” levels of TBA, again based on the maximum value seen in controls. While this approach is artificial and leaves room for potential misclassification, it does separate patients into groups with strong evidence for or against UDCA enrichment. UDCA treated groups had increased TBA level compared to non-treated groups, suggesting that therapy may be directly expanding the BA pool or otherwise facilitating transfer of BA from the enterohepatic to systemic circulation. Increased TBA levels in treated groups were driven by increases in UDCA, with concentrations of more-toxic CA and CDCA (conjugated and unconjugated forms) being significantly reduced with treatment. However, UDCA treated groups also had significantly increased concentrations of the toxic BA LCA, a finding that is consistent with a previous study of 56 PSC patients who had received high-dose UDCA and showed marked UDCA enrichment in serum with a significant increase in LCA concentration compared to the placebo group (19). Together these studies highlight the assertion that supplemental UDCA may be available for conversion to LCA in the gut. While hepatotoxic, LCA and other secondary bile acids may have have anti-inflammatory effects mediated through the BA membrane receptor TGR5 (20). Finally, UDCA treatment lowered the conjugated fraction and increased the G:T conjugation ratio of most bile acids. Notably, many of the BA changes observed in the UDCA treatment groups were consistent with beneficial effects in the univariate time-to-HD analysis. However, predicted UDCA treatment as a variable was not found to be protective against or a risk factor for future HD.
IBD is common in PSC, affecting some 70% of patients. Dysbiosis of the gut microbiota is a typical characteristic of IBD and effects the BA pool by disrupted enterohepatic circulation and diminished capacity for conversion of primary to secondary bile acids (21). Recent studies have demonstrated that intestinal dysbiosis is also common in PSC, with effects independent of concomitant IBD (22). However, we are not aware of any studies that evaluate BA in the context of IBD in PSC. The ratio of primary to secondary bile acids was found to be significantly increased in PSC patients compared to controls. As intestinal dysbiosis has been shown to drive similar observations in IBD and IBD is a common comorbidity in PSC, we evaluated whether the observation was due solely to concomitant IBD, focusing on the CA:DCA ratio to avoid inflation in secondary bile acids due to UDCA treatment. Notably, the no-IBD PSC patients had significantly elevated CA:DCA ratio compared to controls, with UC-PSC patients having even greater CA:DCA ratio, suggesting that the increased fraction of primary bile acids is an independent phenomenon in PSC that is further exasperated by IBD. A similar pattern is seen in the CA:CDCA ratio, which has been reported to be increased in intrahepatic cholestasis of preganancy (23) and in animal models of cholestasis (24). This observation highlights the importance of the gut-liver axis in perpetuating cholestasis in PSC.
Modulation of the BA pool forms the basis of several therapeutic approaches to cholestatic liver disease. A number of studies evaluating compounds, which directly- or indirectly-influence BA pathways, have been reported or proposed (25). The majority of these trials rely on using alkaline phosphatase reductions, which are not highly reliable (26), as the primary endpoint and would benefit from using biomarkers that more directly assess the BA changes these therapies attempt to achieve. Our machine learning model, PSC-BAP, includes six BA variables and significantly improved HD prediction relative to individual variables. Notably, four of the six variables included in PSC-BAP are taurine-conjugated bile acids (TUDCA, TCDCA, TCA and TLCA). Levels of TCA are known to be increased in cirrhotic patients and may directly promote liver damage through upregulation of TLR4 and activation of hepatic stellate cells (27). As well, TCDCA and TLCA have been long known to be hepatotoxic (28, 29). However, TUDCA, which is the strongest contributor to the PSC-BAP model, is thought to be hepatoprotective and potentially therapeutic outside of heptatobiliary disease (30). Of note, TUDCA levels are highly correlated with TBA levels in our PSC patients. Thus, the importance of TUDCA in the model could be reflective of metabolic changes driven by high overall bile acid levels and does not neccessarilly suggest a hepatotoxic effect of UDCA. The PSC-BAP model performed quite well in the derivation and validation cohorts, although the model was over-predicitve in validation, due likely to differences in BA pool compositions between the groups, most notably significant increases in TCA, TCDCA and TLCA concentrations in the validation compared to derivation cohort. Comparison between PSC-BAP and PREsTo, another machine-learning based tool for predicting future HD in PSC, showed performance of the two scores to be similar. Unfortunately, we were not able to compare PSC-BAP to the enhanced liver fibrosis score, which is another non-invasive test that has recently been shown to have prognostic value in PSC (31).
Despite the significance of our findings, we recognize the limitations of the study. This is a retrospective, cross sectional study of BA measurements and PSC outcomes. Potentially important BA species, such as sulfated and glucuronidated forms, were not evaluated due to limitations of our profiling assay. Moreover, our assay is quite sensitive but not fully capable of quantifying extremely low BA levels. Thus, interpretation of observed differences in low-concentration BA such as HDCA and LCA should be done with caution. Underlying processes in PSC, which may significantly alter BA levels of individual patients, such as presence of dominant strictures, variation in IBD activity or specific disease location were not accounted for. We assign patients to UDCA therapy groups using a calculation, so some patients may be misclassified. While this is not likely to have a major impact on the findings, an important aspect of BA metabolism, particularly limited UDCA bioavailability as a potential treatment failure is not addressed. Finally, we are not able to directly compare plasma BA profiles to those present in the enterohepatic circulation due to the invasive nature of collecting BA from the liver or gallbladder. Furure prospective and longitutunal studies of BA are needed to further address the limitations of this work.
In conclusion, plasma BA profiles were able to predict future HD in PSC patients, establishing the clinical potential of the PSC-BAP scoring system. Further exploration of BA profiles in PSC, particularly their use in clinical management of patients and as end-points in clinical trials is warranted.
Supplementary Material
ACKNOWLEDEMENTS
We are indebted to all the patients and controls who participated in this study.
Financial Support: This study was supported by RC2 DK118619 (KNL), the Chris M. Carlos and Catharine Nicole Jockisch Carlos Endowment Fund in Primary Sclerosing Cholangitis (PSC) (KNL), and the Department of Laboratory Medicine and Pathology, Mayo Clinic (DO).
List of Abbreviations:
- PSC
Primary sclerosing cholangitis
- IBD
inflammatory bowel disease
- CCA
cholangiocarcinoma
- UDCA
ursodeoxycholic acid
- HD
hepatic decompensation
- OLT
orthotopic liver transplantation
- BA
bile acid
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- DCA
deoxycholic acid
- LCA
lithocholic acid
- HDCA
hyodeoxycholic acid
- AIH
autoimmune hepatitis
- LC-MS
liquid chromatography-tandem mass spectrometry
- GCA
glycocholic acid
- TCA
taurocholic acid
- GCDCA
glycochenodeoxycholic acid
- TCDCA
taurochenodeoxycholic acid
- GDCA
glycodeoxycholic acid
- TDCA
taurodeoxycholic acid
- GLCA
glycolithocholic acid
- TLCA
taurolithocholic acid
- GUDCA
glycoursodeoxycholic acid
- TUDCA
tauroursodeoxycholic acid
- GHDCA
glycohyodeoxycholic acid
- THDCA
taurohyodeoxycholic acid
- TBA
total bile acid
- IQR
interquartile range
- GBM
gradient boosting machines
- CIs
confidence intervals
- UC
ulcerative colitis
- CD
Crohn’s Disease
- PSC-BAP
PSC bile acid profile score
Footnotes
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1002/hep.31652
REFERENCES
- 1.Lazaridis KN, LaRusso NF. Primary Sclerosing Cholangitis. N Engl J Med 2016;375:2501–2502. [DOI] [PubMed] [Google Scholar]
- 2.Poropat G, Giljaca V, Stimac D, Gluud C. Bile acids for primary sclerosing cholangitis. Cochrane Database Syst Rev 2011:CD003626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fickert P, Wagner M. Biliary bile acids in hepatobiliary injury - What is the link? J Hepatol 2017;67:619–631. [DOI] [PubMed] [Google Scholar]
- 4.de Krijger M, Wildenberg ME, de Jonge WJ, Ponsioen CY. Return to sender: Lymphocyte trafficking mechanisms as contributors to primary sclerosing cholangitis. J Hepatol 2019;71:603–615. [DOI] [PubMed] [Google Scholar]
- 5.Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet 2017;49:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang X, Karlsen TH. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat Rev Gastroenterol Hepatol 2017;14:279–295. [DOI] [PubMed] [Google Scholar]
- 7.Cheung AC, Lazaridis KN, LaRusso NF, Gores GJ. Emerging pharmacologic therapies for primary sclerosing cholangitis. Curr Opin Gastroenterol 2017;33:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyer JL. Bile formation and secretion. Compr Physiol 2013;3:1035–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Tang R, Leung PSC, Gershwin ME, Ma X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun Rev 2017;16:885–896. [DOI] [PubMed] [Google Scholar]
- 10.Eaton JE, Vesterhus M, McCauley BM, Atkinson EJ, Schlicht EM, Juran BD, et al. Primary Sclerosing Cholangitis Risk Estimate Tool (PREsTo) Predicts Outcomes of the Disease: A Derivation and Validation Study Using Machine Learning. Hepatology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51:660–678. [DOI] [PubMed] [Google Scholar]
- 12.Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis). Virchows Arch A Pathol Anat Histol 1978;379:103–112. [DOI] [PubMed] [Google Scholar]
- 13.Huber A, Ebner L, Heverhagen JT, Christe A. State-of-the-art imaging of liver fibrosis and cirrhosis: A comprehensive review of current applications and future perspectives. Eur J Radiol Open 2015;2:90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.GBM GR: Generalized Boosted Regression Models. R Package Version 2.1.3. 2007. [Google Scholar]
- 15.Shung DL, Assis DN. Machine Learning in a Complex Disease: PREsTo Improves the Prognostication of Primary Sclerosing Cholangitis. Hepatology 2020;71:8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazaridis KN, LaRusso NF. Primary Sclerosing Cholangitis. N Engl J Med 2016;375:1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med 1997;336:691–695. [DOI] [PubMed] [Google Scholar]
- 18.Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50:808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinakos E, Marschall HU, Kowdley KV, Befeler A, Keach J, Lindor K. Bile acid changes after high-dose ursodeoxycholic acid treatment in primary sclerosing cholangitis: Relation to disease progression. Hepatology 2010;52:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keitel V, Haussinger D. Role of TGR5 (GPBAR1) in Liver Disease. Semin Liver Dis 2018;38:333–339. [DOI] [PubMed] [Google Scholar]
- 21.Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013;62:531–539. [DOI] [PubMed] [Google Scholar]
- 22.Sabino J, Vieira-Silva S, Machiels K, Joossens M, Falony G, Ballet V, et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016;65:1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brites D, Rodrigues CM, Oliveira N, Cardoso M, Graca LM. Correction of maternal serum bile acid profile during ursodeoxycholic acid therapy in cholestasis of pregnancy. J Hepatol 1998;28:91–98. [DOI] [PubMed] [Google Scholar]
- 24.Matsuzaki Y, Bouscarel B, Ikegami T, Honda A, Doy M, Ceryak S, et al. Selective inhibition of CYP27A1 and of chenodeoxycholic acid synthesis in cholestatic hamster liver. Biochim Biophys Acta 2002;1588:139–148. [DOI] [PubMed] [Google Scholar]
- 25.Vesterhus M, Karlsen TH. Emerging therapies in primary sclerosing cholangitis: pathophysiological basis and clinical opportunities. J Gastroenterol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bakhshi Z, Hilscher MB, Gores GJ, Harmsen WS, Viehman JK, LaRusso NF, et al. An update on primary sclerosing cholangitis epidemiology, outcomes and quantification of alkaline phosphatase variability in a population-based cohort. J Gastroenterol 2020;55:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Zhang Z, Huang M, Sun X, Liu B, Guo Q, et al. Taurocholic acid is an active promoting factor, not just a biomarker of progression of liver cirrhosis: evidence from a human metabolomic study and in vitro experiments. BMC Gastroenterol 2018;18:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sokol RJ, Devereaux M, Khandwala R, O’Brien K. Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology 1993;17:869–881. [PubMed] [Google Scholar]
- 29.Scholmerich J, Becher MS, Schmidt K, Schubert R, Kremer B, Feldhaus S, et al. Influence of hydroxylation and conjugation of bile salts on their membrane-damaging properties--studies on isolated hepatocytes and lipid membrane vesicles. Hepatology 1984;4:661–666. [DOI] [PubMed] [Google Scholar]
- 30.Kusaczuk M Tauroursodeoxycholate-Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vesterhus M, Hov JR, Holm A, Schrumpf E, Nygard S, Godang K, et al. Enhanced liver fibrosis score predicts transplant-free survival in primary sclerosing cholangitis. Hepatology 2015;62:188–197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.